

Abstract
Multiple sclerosis (MS) is a metabolically demanding disease involving immune-mediated destruction of myelin in the central nervous system. We previously demonstrated a significant alteration in disease course in the experimental autoimmune encephalomyelitis (EAE) preclinical model of MS due to diet. Based on the established crosstalk between metabolism and gut microbiota, we took an unbiased sampling of microbiota, in the stool, and metabolites, in the serum and stool, from mice (Mus musculus) on the two different diets, the Teklad global soy protein-free extruded rodent diet (irradiated diet) and the Teklad sterilisable rodent diet (autoclaved diet). Within the microbiota, the genus Lactobacillus was found to be inversely correlated with EAE severity. Therapeutic treatment with Lactobacillus paracasei resulted in a significant reduction in the incidence of disease, clinical scores and the amount of weight loss in EAE mice. Within the metabolites, we identified shifts in glycolysis and the tricarboxylic acid cycle that may explain the differences in disease severity between the different diets in EAE. This work begins to elucidate the relationship between diet, microbiota and metabolism in the EAE preclinical model of MS and identifies targets for further study with the goal to more specifically probe the complex metabolic interaction at play in EAE that may have translational relevance to MS patients.
Keywords: experimental autoimmune encephalomyelitis, myelin oligodendrocyte glycoprotein, metabolomics, microbiome
1. Introduction
In light of recent reports demonstrating a distinct gut microbiota in multiple sclerosis (MS) patients when compared to healthy individuals, as well as MS patients receiving treatment compared to those that are not, there is a clear link between commensal gut bacteria and MS (Chen et al., 2016; Jangi et al., 2016; Miyake et al., 2015; Tremlett et al., 2016) reviewed in (Tremlett et al., 2017). The gut microbiota is a critical modulator of host immunity through a variety of mechanisms including secreted metabolites and bacterial antigens (reviewed in Rooks and Garrett, 2016). Conversely, disease modifying MS therapies are largely immunosuppressive in nature and can lead to iatrogenic dysbiosis/dysbacteriosis (a microbial imbalance caused by treatment) (reviewed in Winkelmann et al., 2016). Previously, we demonstrated that altering diet can have marked effects on central nervous system (CNS) disease severity and outcome (Libbey et al., 2016). We examined the effects of two different food sources, the Teklad global soy protein-free extruded rodent diet (irradiated diet) and the Teklad sterilisable rodent diet (autoclaved diet), on CNS disease in three preclinical models for human CNS disease: two different experimental autoimmune encephalomyelitis (EAE) models (MS) and Theiler’s murine encephalomyelitis virus (TMEV)-induced seizure model (epilepsy). We found that mice fed the irradiated diet had more severe clinical disease and enhanced seizures compared to animals provided the autoclaved diet in both forms of EAE examined and in TMEV-induced seizures (Libbey et al., 2016). We have extended these results in this study by focusing on one of the two EAE models: the C57BL/6J myelin oligodendrocyte glycoprotein (MOG)35-55-induced EAE, by performing histological analysis at the peak of clinical disease and by analysing both the gut microbiota and the metabolic profile of the gut and the serum of the host (Supplementary Figure S1). We show that diet is sufficient to alter gut microbial community composition in EAE and leads to significant differences in disease severity. This work highlights the role of diet in optimising therapy for MS patients and the need for caution when managing infection in MS patients. Diet may provide a subtle mechanism for promoting beneficial gut microbial communities.
The microbiota is the complex communities of microorganisms that inhabit the body surfaces, including the lower intestine of vertebrates (reviewed in Hooper et al., 2012). The host diet, and specific nutrients derived from it, have been shown to play a dominant role in determining the composition of host-associated gut microbial communities, due in part to the diverse intrinsic capabilities of the individual microbes to use the available nutrients (Ley et al., 2006; Sonnenburg et al., 2010; Turnbaugh et al., 2006). Our rationale for examining the microbiota of the lower intestine is that the two different diets may result in an alteration in the composition of these microbial communities, which, in turn, have distinct effects on host immunity. Differing compositions of gut microbiota could regulate the balance between protection and disease induction (Ochoa-Reparaz et al., 2010). It has previously been shown that oral antibiotic treatment to reduce and modify the composition of the gut microbiota resulted in protection against the development of EAE, an autoimmune animal model of MS (Ochoa-Reparaz et al., 2009; Yokote et al., 2008) reviewed in (Rothhammer and Quintana, 2016; Wang and Kasper, 2014; Wekerle, 2016). Germ-free mice were also protected against the development of spontaneous EAE (Berer et al., 2011). Additionally, the presence of a single organism, segmented filamentous bacteria, in otherwise germ-free mice was sufficient to drive the development of EAE (Lee et al., 2011).
In addition to influencing immune function, the microbiota can also influence host metabolism (Hooper et al., 2012; Sonnenburg et al., 2010). Metabolomics is the unbiased survey of metabolites found within a biological sample. Metabolomics is a comparative science allowing us to analyse the relative differences found between biological samples subjected to a treatment. Our rationale for performing metabolic profiling studies, using gas chromatography-mass spectrometry (GC-MS) to measure the metabolic composition of biological samples, is that the different diets may differentially influence metabolic signalling in the microbiota (reviewed in Louis et al., 2007) and, through microbial-mammalian cometabolism, the host (reviewed in Holmes et al., 2011).
2. Materials and methods
In vivo animal studies
Four wk old, male C57BL/6J mice (cat. #000664) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). All animal experiments were reviewed and approved by the University of Utah Institutional Animal Care and Use Committee (Protocols #12-09006, #15-08004) and conducted in accordance with the guidelines prepared by the Committee on Care and Use of Laboratory Animals, Institute of Laboratory Animals Resources, National Research Council. Animals were maintained on a 12 h light/12 h dark cycle at 22 °C. Food and water was available ad libitum. All efforts were made to minimise suffering. Mice were randomly divided into two experimental groups, one of which received Teklad global soy protein-free extruded rodent diet (irradiated diet) (cat. #2920X; Harlan Laboratories, Indianapolis, IN, USA) and the other of which received Teklad sterilisable rodent diet (autoclaved diet) (cat. #8656; Harlan Laboratories). Mice were euthanised through an overdose of isoflurane.
Experimental autoimmune encephalomyelitis induction
C57BL/6J mice were injected subcutaneously in the flanks with 200 μl of 1 mM MOG35-55 peptide (Peptide Synthesis Core Facility, University of Utah) emulsified with reconstituted complete Freund’s adjuvant (CFA), composed of incomplete Freund’s adjuvant (Pierce Biotechnology, Rockford, IL, USA) containing Mycobacterium tuberculosis H37 Ra (2 mg/ml) (Difco Laboratories, Detroit, MI, USA). Mice were injected intravenously with 100 μl Bordetella pertussis (BP) toxin (List Biological Laboratories, Inc., Campbell, CA, USA) at 0.2 μg per mouse on days 0 and 2 following sensitisation. To control for adjuvant-induced inflammation, mice sensitised with CFA (without peptide) and injected with BP toxin were used as controls. Naïve mice were also included as a control. MOG35-55 EAE mice developed a monophasic disease course.
Experimental autoimmune encephalomyelitis scoring
C57BL/6J mice injected with MOG35-55, plus CFA/BP injected and naïve control mice, were weighed and scored daily for clinical signs. Clinical scoring was as follows: 0, no clinical disease; 1, loss of tail tonicity; 2, presents with mild hind leg paralysis with no obvious gait disturbance; 3, mild leg paralysis with gait disturbance and paralysis; 4, hind limbs are paralysed; and 5, moribund or dead. If the mice were paralysed to the point where they could not feed or groom themselves (moribund), or animals lost 20% of their body weight, the mice were euthanised via inhaled anaesthetic.
Sample collection
On the day that mice were randomly divided into the two diet groups (day-2), serum was collected via cheek bleed and faecal pellets were collected. On the day of sacrifice (day 16 post sensitisation), serum was collected via heart puncture and faeces were collected directly from the bowel. All samples were stored at −80 °C until analysis.
Microbiota
Microbiota was examined with the assistance of Dr Kael Fischer, University of Utah. Sixty total experimental faecal samples (1 pellet each), collected on days-2 and 16 post sensitisation, were examined (Table 1). Controls included: (1) faecal samples collected on days-2 and 16 from mice maintained on autoclaved and irradiated diet that were sensitised with CFA (without peptide) and injected with BP toxin (6 mice per group, 24 samples total), and (2) faecal samples collected from naïve mice on day 16 that were maintained on autoclaved and irradiated diet (6 mice per group, 12 samples total). The faecal pellets were added to ZR BashingBead lysis tubes containing a mixture of 0.1 and 0.5 mm garnet beads (Zymo Research, Irvine, CA, USA). DNA was extracted and purified using the Zymo Research Quick-DNA™ Faecal/Soil Microbe Miniprep Kit. Bacterial 16S rRNA was amplified with modified primers as previously described (Kubinak et al., 2015). Briefly, PCR cycling conditions were: 98 °C for 2 min; 30 cycles of 98 °C for 30 s, 53.6 °C for 20 s, 72 °C for 30 s; followed by 72 °C for 2 min, using Phusion HotStartII enzyme (Thermo Scientific, Waltham, MA, USA) with the supplied GC buffer and 200 nM of each primer in a 20 μl reaction volume. PCR products were cleaned up using SequalPrep Normalization Plate (Thermo Scientific), mixed together and analysed on a 4200 TapeStation Instrument (Agilent, Santa Clara, CA, USA) and then mixed with PhiX control (5% of final library) (Illumina, San Diego, CA, USA), and sequenced on an Illumina MiSeq at the University of Utah’s High-Throughput Genomics core facility with paired-end 300 cycle sequencing. De-multiplexed sequences were quality filtered (Sickle 1.33, Univ. of California), and overlapping sequence pairs were joined, using pandaseq (Masella et al., 2012). QIIME (Caporaso et al., 2010) (v1.9.1) was used to make taxonomic calls. Sequence data have been deposited in the SRA archive (Leinonen et al., 2011) (www.ncbi.nlm.nih.gov/sra, Accession Number: SRP107023).
Table 1.
Mouse groupings for microbiota and metabolomics analysis.1
| Autoclaved diet
|
Irradiated diet
|
||
|---|---|---|---|
| Clinical score (Max)2 | Cage number | Clinical score (Max)2 | Cage number |
| High (4) | 5 | High (4) | 7 |
| High (3.5) | 3 | High (4) | 10 |
| High (3) | 2 | High (4) | 7 |
| High (3) | 1 | High (4) | 8 |
| High (3) | 2 | High (3.5) | 6 |
| High (3) | 3 | High (3)3 | 9 |
| Low (1) | 1 | Low (1) | 9 |
| Low (1) | 1 | Low (2) | 8 |
| Low (1) | 3 | Low (2) | 8 |
| Low (1) | 3 | Low (3) | 9 |
| Low (1) | 4 | Low (3) | 7 |
| Low (2) | 5 | Low (3) | 6 |
| Zero | 5 | Zero | 6 |
| Zero | 4 | Zero | 10 |
| Zero | 4 | ||
| Zero | 2 | ||
Microbiota = stool samples (n=60) on days-2 and 16; Metabolomics = serum (n=60) and stool (n=60) samples on days-2 and 16.
Maximum clinical score achieved per mouse over entire time course.
A mouse with a maximum clinical score of 3 (selected randomly) was included in the high category as there were no more mice with clinical scores higher than this and a group of 6 was desired for statistical analysis.
Metabolomics
Metabolomics analysis was performed by the Metabolomics Core Facility at the University of Utah with the assistance of Dr James Cox. Sixty total experimental samples of serum (30 μl each) and 60 total experimental samples of faeces (2 pellets each), collected on days-2 and 16 post-sensitisation, were examined via GC-MS (Table 1). Two additional samples analysed by GC-MS were samples of both the autoclaved and irradiated diet. Inflammation controls included serum and faecal samples collected on days-2 and 16 from mice maintained on autoclaved and irradiated diet that were sensitised with CFA (without peptide) and injected with BP toxin (6 mice per group, 48 samples total). Metabolite extraction from serum samples entailed allowing the samples to thaw at 4 °C, adding 270 μl of cold 90% methanol solution containing the following internal standards: 1 μg of D4-succinic acid and 5 μg of labelled amino acids (13C-, 15N-labelled) mixture per sample, vortexing the samples, incubating the samples at −20 °C for 1 h, centrifuging the samples at 20,000×g for 5 min at 4 °C, after which the supernatants were transferred to new microcentrifuge tubes. Quality control samples were generated by removing 10% volume of the collected supernatant from each sample and adding these into a single tube. From this mixture aliquots of an amount equal to the final volume of the actual samples were used for quality control. Finally, the samples were dried in a speed-vac. Metabolite extraction from faeces entailed weighing the samples (Hintze et al., 2014), adding 5 ml of 75% ethanol solution containing the same internal standards as above, heating the samples in boiling water for 10 min, letting the samples cool at room temperature for 2 min, followed by centrifugation at 4,000×g for 5 min. The supernatant was then transferred to a new tube and dried in a speed-vac. All GC-MS analysis was performed with a Waters (Milford, MA, USA) GCT Premier mass spectrometer fitted with an Agilent 6890 gas chromatograph and a Gerstel MPS2 autosampler (Mülheim an der Ruhr, Germany). Dried samples were suspended in 40 μl of a 40 mg/ml O-methoxylamine hydrochloride in pyridine solution and incubated for 1 h at 30 °C. To autosampler vials was added 25 μl of this solution. N-methyl-N-trimethylsilyltrifluoracetamide (40 μl) was added automatically via the autosampler and incubated for 60 min at 37 °C with shaking. After incubation 3 μl of a fatty acid methyl ester standard solution was added via the autosampler, then 1 μl of the prepared sample was injected to the gas chromatograph inlet in the split mode with the inlet temperature held at 250 °C. A 10:1 split ratio was used for analysis. The gas chromatograph had an initial temperature of 95 °C for 1 min followed by a 40 °C/min ramp to 110 °C and a hold time of 2 min. This was followed by a second 5 °C/min ramp to 250 °C, a third ramp to 350 °C, then a final hold time of 3 min. A 30 meter Phenomex ZB5-5 MSi column (Torrance, CA, USA) with a 5 meter long guard column was employed for chromatographic separation. Helium was used as the carrier gas at 1 ml/min. Due to the high amounts of several metabolites, including valine, leucine, isoleucine, proline, phosphate and inositol, the samples were analysed once more at a 10-fold dilution. Data was collected using MassLynx 4.1 software (Waters). All GC-MS chromatograms were processed using QuanLynx (Waters) for non-targeted metabolite identification and peak area analysis. Metabolite identification was established using a combination of an in-house metabolite library developed using pure purchased standards and the commercially available NIST library. Further analysis was performed using the MetaboAnalyst 3.0 software and a targeted approach. To characterise and visualise the metabolomic data, principal component analysis, an unsupervised visualisation approach for multidimensional data sets, was used. Analysis identified potential metabolites that were altered with a fold-change of >1.5 and a P-value less than 0.05 (t test).
Histology
Mice were euthanised, on day 16 post sensitisation, and perfused with phosphate-buffered saline (PBS). Spinal cords were harvested, fixed in 4% paraformaldehyde phosphate-buffered solution, divided into 12 transverse portions, embedded in paraffin and cut into 4 μm thick tissue sections. To visualise myelin, sections were stained with Luxol fast blue. For scoring of perivascular cuffing (PVC), meningitis and demyelination in spinal cord sections, each spinal cord segment was divided into four quadrants: the anterior and posterior funiculi, and each lateral funiculus. Any quadrant containing PVC, meningitis or demyelination was given a score of 1 in that pathologic class. The overall score was determined by giving a quadrant a score of 1 if the quadrant had any PVC, meningitis or demyelination within the quadrant. The total number of positive quadrants for each pathologic class and overall was determined, then divided by the total number of quadrants scored on the slide and multiplied by 100 to give the percent involvement for each pathologic score and overall.
Lactobacillus treatment
Lactobacillus paracasei, subspecies paracasei, was obtained from ATCC (cat. #27092, Manassas, VA, USA). L. paracasei was cultured aerobically in De Man, Rogosa and Sharpe (MRS) broth (VWR, Denver, CO, USA) at 37 °C in 5% CO2. Growing cultures were frozen at −80 °C in 20% glycerol until use. Immediately prior to gavage, cultures were centrifuged, washed, and resuspended in PBS.
Mice, on the autoclaved diet, received either 109 cfu of L. paracasei (13 mice per group) or an equivalent volume of PBS (15 mice per group) via daily oral gavage for 14 days prior to EAE induction. After disease induction, mice were weighed and scored for clinical signs of EAE daily. Therapeutic treatment, with either L. paracasei or PBS, resumed on an individual basis, beginning at the onset of disease, defined as a clinical score of 1 or higher. The earliest day on which treatment resumed was day 7 post sensitisation. Once therapeutic treatment was reinitiated, mice continued to receive daily gavage until the experimental endpoint (day 19 post sensitisation).
Metabolic flux assay
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using the Seahorse XFe96 metabolic extracellular flux analyser (Seahorse Bioscience, North Billerica, MA, USA). Spleens were harvested from C57BL/6J mice and enriched for T cells using the EasySep Mouse T cell Isolation Kit (Stemcell Technologies, Vancouver, Canada). Cells were stimulated with anti-CD3/CD28 beads (Thermo Scientific), in RPMI-1640 media (Mediatech, Herndon, VA, USA) supplemented with 10% Cosmic calf serum (Hyclone, Logan, UT, USA), 1% pen/strep (Mediatech) and 1% L-glutamine (Mediatech), for 24 h at 37 °C before being plated onto Seahorse cell plates (6×105 cells per well) coated with Cell-Tak (VWR). The ratio of OCR to ECAR was calculated by dividing the oxidative phosphorylation (OXPHOS) capacity, the OCR reading obtained after injection of 1 μM oligomycin (Sigma Aldrich, St. Louis, MO, USA), by the glycolytic capacity, the ECAR reading obtained after injection of 1 μM oligomycin.
Statistical analysis
The program StatView (SAS Institute Inc., Cary, NC, USA) was used for statistical analyses. P-values of less than 0.05 were considered significant in all cases. The Student’s t test was performed for pairwise comparison (clinical score and weight change). The Chi-Square test was utilised for nominal data (survival: yes or no; disease: yes or no). The unpaired two-group Mann-Whitney U test was performed for all nonparametric analyses (pathology score). Correlation analysis was performed using the Spearman correlation coefficient. The numbers of mice per group (n) can be found in the figure legends and in Table 1.
3. Results
MOG35-55-induced experimental autoimmune encephalomyelitis
The effects of diet on disease were compared using the C57BL/6J MOG35-55-induced EAE mouse model. Mice were sacrificed on day 16 post sensitisation, a time point nearing the peak of disease for the mice receiving the irradiated diet and a day for which there was a significant difference in clinical score between the two groups (Figure 1A). The average clinical score for mice given the irradiated diet was significantly higher on day 16 (P<0.05, t-test) post sensitisation, compared to animals fed the autoclaved diet (Figure 1A). Although the differences did not reach significance for days 12-15, the animals given the irradiated diet tended to have higher average clinical scores for those days, compared to animals fed the autoclaved diet (Figure 1A). In terms of weight change, the animals on the irradiated diet tended to have a precipitous loss in weight over days 13-16, compared to the animals on the autoclaved diet (Figure 1B), although these differences did not reach significance (P=0.08-0.14, t-test). The percentage of mice that were either found dead or were moribund and needed to be sacrificed was greater for the mice receiving the irradiated diet (4/20, 20%) compared to the mice on the autoclaved diet (2/20, 10%). Of the mice that become moribund, mice on the irradiated diet tended to become moribund earlier in the clinical course than mice on the autoclaved diet (Figure 1C). Mice from the two diet groups were selected for further metabolomic and microbiota analysis, see below, based on clinical scores. These mice were grouped into mice with high clinical scores, mice with low clinical scores and mice with clinical scores equal to 0 (Table 1). Note that the groupings by clinical score did not correlate with any single cage, but rather mice with similar clinical scores were dispersed throughout all of the cages on a particular diet (Table 1). The number of mice that presented with no clinical signs of disease (clinical score of 0) was greater for the mice on the autoclaved diet (4/20, 20%) compared to the mice on the irradiated diet (2/20, 10%) (Table 1). Analysis of the high and low groupings shows that the maximum clinical scores for the high groups tended to be higher in the mice receiving the irradiated diet (4/6 scored a 4), compared to the mice receiving the autoclaved diet (4/6 scored a 3), and, likewise, the maximum clinical scores for the low groups were significantly higher in the mice receiving the irradiated diet (5/6 scored a 2 or 3), compared to mice receiving the autoclaved diet (5/6 scored a 1) (P<0.05, t-test) (Table 1). Examination, at the peak of disease (day 16 post sensitisation), of spinal cord pathology revealed that mice receiving the irradiated diet had higher pathology scores for all categories examined: demyelination, PVC, meningitis and overall pathology, and that the pathology scores for meningitis and overall pathology were significantly higher in the mice receiving the irradiated diet compared to the mice receiving the autoclaved diet (P<0.05, Mann-Whitney U test) (Figure 1D). Overall, these results demonstrate that the animals on the irradiated diet were generally sicker than those animals on the autoclaved diet.
Figure 1.

Effects of diet on disease course and pathology in C57BL/6J mice sensitised with MOG35-55 and maintained on irradiated or autoclaved diet. (A) Clinical scores given as the mean ± standard error of the mean (SEM) for groups of 18 mice (autoclaved) and 16 mice (irradiated). * P<0.05, t-test. (B) Weight changes represented as percent of daily weight in comparison to weight at day-1, given as mean ± SEM for groups of 18 mice (autoclaved) and 16 mice (irradiated). (C) Mortality represented as percent daily survival of animals in comparison to day 0 (20 mice per group). (D) Demyelination (Demyl.), perivascular cuffing (PVC), meningitis (Menin.) and overall spinal cord pathology scores for mice sacrificed on day 16 post sensitisation. Data is given as the mean + SEM for groups of 18 mice (autoclaved) and 16 mice (irradiated). * P<0.05, Mann-Whitney U test.
Microbiota
To investigate whether the gut microbial composition differed as a function of diet and/or disease severity, we analysed the microbiota in faeces by high-throughput sequencing. Analysis was based on grouping mice by clinical score: high clinical scores (Hi), low clinical scores (Lo) and clinical scores equal to 0 (Zero) (Table 1). The microbial composition of control mice, sensitised with CFA (without peptide) and injected with BP toxin, and naïve mice was also analysed. We felt it was important to include the adjuvant only treated group to control for changes induced by injection of the CFA/BP. Examination of the bacterial population to the genus level showed that Lactobacillus (Family Lactobacillaceae) was present as a greater percentage of total assignments in the control (CFA) and naïve animals maintained on the autoclaved diet than control and naïve animals maintained on the irradiated diet (Table 2). The percentages of Lactobacillus in the bacterial populations of the peptide sensitised mice (Experimental), sampled at day 16 post sensitisation, were reduced compared to the control and naïve mice and inversely correlated (Spearman correlation coefficient =−0.67) with disease within each respective diet (Table 2). Finally, the percentages of Lactobacillus in the bacterial populations of the peptide sensitised mice sampled at day 16 post sensitisation maintained on the irradiated diet were lower than those for peptide sensitised mice maintained on the autoclaved diet, when comparing groups by clinical score (Table 2). Lactobacillus was the only genus to display these characteristics.
Table 2.
Percentage of Lactobacillus in bacterial populations in faecal samples of peptide sensitised mice.1
| Autoclaved diet
|
Irradiated diet
|
||||||
|---|---|---|---|---|---|---|---|
| Experimental2 | CFA | Naïve | Experimental | CFA | Naïve | ||
| Day-2 | Hi | 19.0% | 18.7% | ND | 20.4% | 12.8% | ND |
| Lo | 21.2% | 21.3% | |||||
| Zero | 20.4% | ND | |||||
| Day 16 | Hi | 6.5% | 24.1% | 28.6% | 4.2% | 17.2% | 13.8% |
| Lo | 12.9% | 7.4% | |||||
| Zero | 14.5% | ND | |||||
CFA = complete Freund’s adjuvant; ND = not determined.
Experimental autoimmune encephalomyelitis mice grouped by clinical score as high (Hi)/low (Lo)/Zero.
To assess the validity of our microbiota screen, mice on the autoclaved diet were treated therapeutically with either L. paracasei or PBS via oral gavage (Supplementary Figure S1). Mice treated with L. paracasei exhibited significantly lower clinical scores, on days 10-19 post sensitisation (P<0.05, day 10; P<0.001, day 11; P<0.0001, days 12-19, t-test), than mice treated with PBS (Figure 2A). L. paracasei-treated mice also exhibit significantly less weight loss, on days 10-19 post sensitisation (P<0.05, days 10 and 18; P<0.01, days 11, 16-17, and 19; P<0.001, day 13; P<0.0001, days 12 and 14-15, t-test), than mice treated with PBS (Figure 2B). Additionally, while all 15 mice treated with PBS developed EAE, only nine of the 13 mice treated with L. paracasei developed EAE, which is a significant difference (P<0.05, Chi-Square test).
Figure 2.
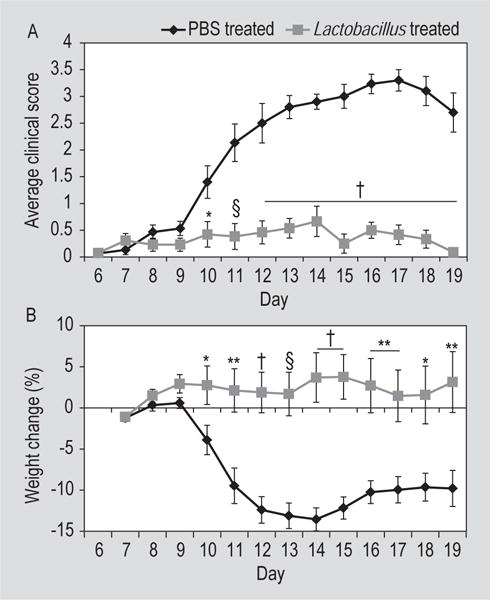
Effects of Lactobacillus paracasei or PBS treatment on disease course in C57BL/6J mice on autoclaved diet and sensitised with MOG35-55. (A) Clinical score is given as the mean ± standard error of the mean (SEM) for groups of 15 mice (PBS) and 13 mice (L. paracasei). (B) Weight change represented as percent of daily weight in comparison to weight at day 6 post sensitisation, given as mean ± SEM for groups of 15 mice (PBS) and 13 mice (L. paracasei). * P<0.05, ** P<0.01, § P<O.001, † P<0.0001, t-test.
Conversely, examination of the bacterial population to the genera level showed several bacterial populations that increased in the presence of disease. These included Bacteroides (Family Bacteroidaceae) (Spearman correlation coefficient = 0.69), Turicibacter (Family Turicibacteraceae) (Spearman correlation coefficient = 0.23), and both the Ruminococcus genus (Spearman correlation coefficient = 0.63) and sequences not assigned a genus of the Family Ruminococcaceae (Table 3). The percentages of these four genera in the bacterial populations of the peptide sensitised mice (Experimental), sampled at day 16 post sensitisation, were increased compared to the control and naïve mice within each respective diet. Finally, the percentages of these four genera in the bacterial populations of the peptide sensitised mice sampled at day 16 post sensitisation maintained on the irradiated diet were lower than those for peptide sensitised mice maintained on the autoclaved diet, when comparing groups by clinical score, in all cases except one (Bacteroides, Hi) (Table 3).
Table 3.
Bacterial genera (percentage of bacterial populations) that increased in faecal samples of mice with disease.1
| Autoclaved diet
|
Irradiated diet
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Experimental2 | CFA | Naïve | Experimental2 | CFA | Naïve | |||
| Bacteroides | Day-2 | Hi | 0% | 0% | ND | 0.8% | 0% | ND |
| Lo | 0% | 0.6% | ||||||
| Zero | 0% | ND | ||||||
| Day 16 | Hi | 5.2% | 0% | 0% | 8.4% | 0% | 0% | |
| Lo | 5.4% | 1.3% | ||||||
| Zero | 2.6% | ND | ||||||
| Turicibacter | Day-2 | Hi | 4.4% | 3.6% | ND | 5.4% | 5.5% | ND |
| Lo | 5.1% | 7.5% | ||||||
| Zero | 6.4% | ND | ||||||
| Day 16 | Hi | 9.6% | 4.3% | 3.8% | 5.1% | 5.1% | 4.3% | |
| Lo | 6.5% | 5.6% | ||||||
| Zero | 8.8% | ND | ||||||
| Ruminococcus | Day-2 | Hi | 0.7% | 0.3% | ND | 0.4% | 0.7% | ND |
| Lo | 0.8% | 0.5% | ||||||
| Zero | 0.7% | ND | ||||||
| Day 16 | Hi | 2.6% | 0.3% | 1.3% | 1.3% | 0.5% | 0.3% | |
| Lo | 2.3% | 1.4% | ||||||
| Zero | 2.2% | ND | ||||||
| Ruminococcaceae (unassigned) | Day-2 | Hi | 3.1% | 1.8% | ND | 1.7% | 3.0% | ND |
| Lo | 2.3% | 1.9% | ||||||
| Zero | 2.9% | ND | ||||||
| Day 16 | Hi | 2.8% | 1.2% | 2.1% | 2.0% | 1.0% | 0.4% | |
| Lo | 2.4% | 1.9% | ||||||
| Zero | 2.1% | ND | ||||||
CFA = complete Freund’s adjuvant; ND = not determined.
Experimental autoimmune encephalomyelitis mice grouped by clinical score as high (Hi)/low (Lo)/Zero.
Metabolomics
To investigate whether individual metabolites differed as a function of diet and/or disease severity, we analysed the metabolic profile of the mouse serum and faeces by GC-MS. Analysis was again based on grouping mice by clinical score: high clinical scores (Hi), low clinical scores (Lo) and clinical scores equal to 0 (Zero) (Table 1). Initially, as a means of uncoupling diet effects from disease effects on the metabolic profile of the serum and faeces, first mice with high clinical scores, regardless of diet, were compared to mice with clinical scores equal to 0; and second mice with any disease at all [high and low clinical scores (HiLo)], again regardless of diet, were compared to mice with clinical scores equal to 0. Zero, in these comparisons, is the immunisation control where mice received all treatments (including peptide) but did not develop clinical disease. For day-2 serum, targeted analysis showed that no metabolites were altered with significance for either comparison. Day-2 is prior to immunisation, so these samples reflected the background control for the experiments. For day 16 serum, targeted analysis showed that 3 metabolites, found for the Hi vs 0 comparison, were altered with significance (P<0.05): glucose-6-phosphate (G6P), stearic acid and 3-(3-hydroxyphenyl)propanoic acid, and 1 metabolite, G6P, was found for the HiLo vs 0 comparison. Upon examination, G6P was found to be higher in the groups of mice with clinical disease (Hi > HiLo) compared to mice with clinical scores equal to 0. For day-2 faeces, 4 known (rhamnose, glycolic acid, 2-hydroxybutyric acid, 22:0 fatty acid) and 2 unknown metabolites were altered with significance for the Hi vs 0 comparison, and 3 known (rhamnose, 3-hydroxybutyrate, 22:0 fatty acid) and 2 unknown (1 of which was the same as for the Hi vs 0 comparison) metabolites were altered with significance for the HiLo vs 0 comparison. The 3 common metabolites, rhamnose, 22:0 fatty acid and un14.96_133.0501 (where un = unknown, 14.96 = retention time, 133.0501 = a characteristic mass), were all higher in the mice with clinical scores equal to 0 compared to the groups of mice with clinical disease (rhamnose & 22:0 fatty acid: Hi = HiLo; un14.96_133.0501: HiLo > Hi). For day 16 faeces, 19 known and 3 unknown metabolites were altered with significance for the Hi vs 0 comparison, and 15 known and 3 unknown metabolites were altered with significance for the HiLo vs 0 comparison; 14 of these metabolites were found in both the Hi vs 0 and the HiLo vs 0 comparisons. The 14 common metabolites were all higher in the mice with clinical scores equal to 0 compared to the groups of mice with clinical disease (lauric acid, 3-hydroxyanthranilic acid, AMP, m-hydroxyphenylproprionic acid, kynurenic acid, succinic acid, 1-monooleoylglycerol, palmitelaidic acid, 2-hydroxyphenylproprionic acid, 9H-purin-6-amine, 1-monolinoleoylglycerol: HiLo > Hi; 20:0 fatty acid: Hi = HiLo; un16.50_117.0729, un30.81_387: Hi > HiLo). Analysis of faeces is expected to result in many more metabolites than analysis of serum.
Next, to investigate the effects of disease on the metabolic profile of both the serum and the faeces within each of the diets, first mice with high clinical scores, on the irradiated diet, were compared to mice with low clinical scores, on the irradiated diet, for day-2; second mice with high clinical scores, on the irradiated diet, were compared to mice with low clinical scores, on the irradiated diet, for day 16; third mice with high clinical scores, on the autoclaved diet, were compared to mice with low clinical scores, on the autoclaved diet, for day-2; and fourth mice with high clinical scores, on the autoclaved diet, were compared to mice with low clinical scores, on the autoclaved diet, for day 16. The metabolites that were found to be altered with significance via targeted analysis for these 4 comparisons are presented in Table 4. Only one metabolite, chlorogenic acid (bold), appeared in more than 1 of the above 4 comparisons. It was found in day 16 faeces regardless of diet. No altered metabolites were found in the serum on day 16, regardless of diet.
Table 4.
Metabolites found to be altered from examination of the effects of disease within each diet.1
| (1) Hi vs Lo, irradiated, day-2
|
(2) Hi vs Lo, irradiated, day 16
|
(3) Hi vs Lo, autoclaved, day-2
|
(4) Hi vs Lo, autoclaved, day 16
|
||||
|---|---|---|---|---|---|---|---|
| Serum | Faeces | Serum | Faeces | Serum | Faeces | Serum | Faeces |
| urea | 3-hydroxybutyrate b-hydroxybutyric acid un16.50_117.0729 |
no altered metabolites | kynurenic acid 5-hydroxyindoleacetic acid tryptophan valine 5-aminopentanoic acid n-methylalanine isoleucine pyruvic acid chlorogenic acid p-hydroxyphenyl acetic acid oleic acid tyrosine linoleic acid palmitelaidic acid 19:0 fatty acid threonine 2-hydroxybutyric acid mannitol |
1-monooleoyl-glycerol | un18.55_204.1001 cysteine 2-hydroxyphenyl proprionic acid |
no altered metabolites | phosphate chlorogenic acid glucose-6-phosphate indolecarboxylic acid fructose-6-phosphate |
Clinical disease for experimental autoimmune encephalomyelitis grouped as high (Hi) and low (Lo).
To investigate the effects of diet on the metabolic profile of both the serum and the faeces, first mice with high clinical scores, on the irradiated diet, were compared to mice with high clinical scores, on the autoclaved diet, for day-2; second mice with high clinical scores, on the irradiated diet, were compared to mice with high clinical scores, on the autoclaved diet, for day 16; third mice with low clinical scores, on the irradiated diet, were compared to mice with low clinical scores, on the autoclaved diet, for day-2; and fourth mice with low clinical scores, on the irradiated diet, were compared to mice with low clinical scores, on the autoclaved diet, for day 16. The metabolites that were found to be altered with significance via targeted analysis for these 4 comparisons are presented in Table 5. We noticed that many of the metabolites present in the day 16 (comparisons 2 and 4) serum and faeces were components of glycolysis and the citric acid cycle [tricarboxylic acid (TCA) cycle]. Therefore, we generated separate figures for the serum (Figure 3) and faeces (Figure 4) with the metabolites of interest designated on the glycolysis pathway and TCA cycle. The shading of Table 5 carries over to Figure 3 and 4, with the addition of grey shading in the figures for those metabolites that only occur in either comparison 2 or 4, not both. The boxes that accompany the pathway designate alterations that were significant (*, P<0.05) both for day 16 (comparisons 2 and 4) and for cross-time point comparisons (day-2 vs day 16) that were also performed. Arrows, and their positioning, indicate where the metabolite is the highest for each comparison. The metabolites palmitelaidic acid (16:1 fatty acid) and aspartic acid were also altered with significance in comparison 3 (day-2, faeces), so this information was added to these boxes (blue) accompanying Figure 4. Examination of the boxes accompanying Figure 3 (serum) shows that, for day 16 (comparisons 2 and 4), the majority of these metabolites are higher in mice receiving autoclaved food than in mice receiving irradiated food. The metabolites 2-deoxyglucose (2-DG) and ribose are the exceptions. Also, examination shows that, for cross-time point comparisons, all of these metabolites are higher in the day-2 samples than the day 16 samples. Examination of the boxes accompanying Figure 4 (faeces) shows that, for day 16 (comparisons 2 and 4), the majority of the metabolites are higher in the mice receiving autoclaved food (36/45 significant alterations) than in mice receiving irradiated food. The exceptions where the metabolites are higher in the mice receiving irradiated food (9/45 significant alterations) are pyruvate, isocitric acid, sedoheptulose, uric acid, sarcosine and 5-hydroxyindoleacetic acid. Also, examination shows that, for cross-time point comparisons, the majority of these metabolites are higher in the day 16 samples (49/57 significant alterations). The exceptions where the metabolites are higher in the day-2 samples (8/57 significant alterations) are 3-phosphoglycerate, isocitric acid, malic acid, aspartic acid and 5-hydroxyindoleacetic acid. For day-2, 16:1 fatty acid was higher in the mice receiving irradiated food; aspartic acid was higher in the mice receiving autoclaved food. Correlation analyses of select metabolites that showed the same significance for the same comparison for both serum and faeces are presented in Supplementary Figure S2.
Table 5.
| (1) Irradiated vs autoclaved, Hi, day-2
|
(2) Irradiated vs autoclaved, Hi, day 16
|
(3) Irradiated vs autoclaved, Lo, day-2
|
(4) Irradiated vs autoclaved, Lo, day 16
|
||||
|---|---|---|---|---|---|---|---|
| Serum | Faeces | Serum | Faeces | Serum | Faeces | Serum | Faeces |
| 1-monooleoyl-glycerol | trans-cinnamic acid | citric acid | citric acid | glucose-6-phosphate | palmitelaidic acid | citric acid | citric acid |
| arachidonic acid | isocitric acid | isocitric acid | urea | aspartic acid | isocitric acid | isocitric acid | |
| 2-deoxy-glucose | malic acid | inositol | 2-deoxy-glucose | malic acid | |||
| 4-hydroxy-proline | fumaric acid | oleic acid | malic acid | fumaric acid | |||
| 2-hydroxyphenylproprionic acid aminopimelic acid dihydroxyquinone sedoheptulose-7-P 2-phosphoglycerol 9H-purin-6 amine arabinofuranose 5-hydroxyindoleacetic acid pyruvic acid orotateN lauric acid sorbitol 1-monopalmitoylglycerol 3-phosphoglycerate rhamnose ergosterol phosphate |
linoleic acid glycerol 5-aminovaleric acid adenosine Un12.13_263 |
fumaric acid | 2-hydroxyphenylproprionic acid aminopimelic acid dihydroxyquinone sedoheptulose-7-P 2-phosphoglycerol 9H-purin-6 amine arabinofuranose 5-hydroxyindoleacetic acid pyruvic acid orotateN lauric acid sorbitol 1-monopalmitoylglycerol 3-phosphoglycerate rhamnose ergosterol phosphate |
||||
| aconitate 2-ketoglutaric acid ribose 3-hydroxy-anthranilic acid 3-(3-hydroxy-phenyl) pro-panoic acid | |||||||
| palmitelaidic acid aspartic acid |
sedoheptulose sarcosine |
||||||
| glucose-6-phosphate | mannose | ||||||
| succinic acid glycolic acid D-erythro-2-pentulose 2-hydroxybutyric acid 3-hydroxybutyrate |
galacitol 2-aminoadipic acid 1-monostearylglycerol mannitol mevalonic acid |
||||||
| B-hydroxybutyric acid deoxyinositol pyridine-2-6-dicarboxylic acid |
gluconic acid glucose fructose uric acid |
||||||
| glucose-1-phosphate m-hydroxyphenylproprionic acid fructose-6-phosphate AMP |
Un13.58_204.1001 Un20.83_333 Un20.83_333.1338 Un30.81_387 Un18.55_204.1001 |
||||||
| Un13.58_204.1001 Un20.83_333 Un20.83_333.1338 Un30.81_387 Un18.55_204.1001 |
|||||||
| Un20.99_333.1338 Un30.94_387.1141 Un5.79_211.0033 Un16.50_117.0729 Un6.55_174.1128 |
|||||||
Figure 3.

Several serum metabolites, altered with significance on day 16 with disease in targeted analysis, share the common glycolysis/tricarboxylic acid (TCA) pathway. Grey arrows indicate multi-step reactions. The data presented in table format are discussed in the Metabolomics portion of the Results section; metabolites that were altered In 2 or more of the comparisons are similarly shaded in Table 5. * P<0.05.
Abbreviations: 2-DG = 2-deoxyglucose; Glucose/Fructose-6-P = glucose/fructose-6-phosphate; Fru-1,6-diP = fructose-1,6-diphosphate; 3/2-PGA = 3/2-phosphoglycerate; PEP = phosphoenolpyruvate; 3-HPP Acid = 3-(3-hydroxyphenyl)propanoic acid; 3-HA Acid = 3-hydroxyanthranilic acid; Hyp = 4-hydroxyproline; 2-KG Acid = 2-ketoglutaric acid; Succ-CoA = succinyl-CoA; Auto = autoclaved; Irrad = irradiated.
Figure 4.


Several faeces metabolites, altered with significance on day 16 with disease in targeted analysis, share the common glycolysis/tricarboxylic acid (TCA) pathway. Grey arrows indicate multi-step reactions. The data presented in table format are discussed in the Metabolomics portion of the Results section; metabolites that were altered In 2 or more of the comparisons are similarly shaded in Table 5. * P<0.05.
Abbreviations: AMP = adenosine monophosphate; ADP = adenosine diphosphate; ATP = adenosine triphosphate; Pi = phosphate; Glucose/Fructose-6-P = glucose/fructose-6-phosphate; Glucose/Fructose-1-P = glucose/fructose-1-phosphate; Fru-1,6-diP = fructose-1,6-diphosphate; SH-7-P = sedoheptulose-7-phosphate; SH = sedoheptulose; 3/2-PGA = 3/2-phosphoglycerate; PEP = phosphoenolpyruvate; 12:0 FA = lauric acid; 16:1 FA = palmitelaidic acid; 5-HIAA = 5-hydroxyindoleacetic acid; 1-MPG = 1-monopalmitoylglycerol; 1-MSG = 1-monostearylglycerol; 2/3-HB Acid = 2/3-hydroxybutyric acid; Free FA = free fatty acids; 2-KG Acid = 2-ketoglutaric acid; Succ-CoA= succinyl-CoA; a-KB Acid = α-ketobutyric acid; GDP = guanosine diphosphate; GTP = guanosine triphosphate; Auto = autoclaved; Irrad = irradiated.
To assess the validity of our metabolomics screen, we performed metabolic flux analysis on enriched, activated T cell populations isolated from spleens from mice receiving the different diets, stratifying by the severity of disease within the diets. The ratio of OCR, a measure of OXPHOS, to ECAR, a measure of glycolysis, was calculated. The OCR/ECAR ratio was significantly higher for T cells enriched from mice receiving the autoclaved diet with low disease score compared to T cells from mice receiving the autoclaved diet with high disease (P<0.05, t-test) (Figure 5A). However, there was no significant difference in OCR/ECAR between T cells from mice receiving the irradiated diet with low disease compared to T cells from mice receiving the irradiated diet with high disease (Figure 5A). Correlation analysis shows a significance when plotting OCR/ECAR versus disease (clinical score) (Figure 5B).
Figure 5.

Metabolic flux assay. (A) Ratio of oxygen consumption rate (OCR) to extracellular acidification rate (ECAR) in enriched T cell populations stimulated with anti-CD3/CD28 beads for 24 h. T cells were enriched from the spleens of C57BL/6J mice receiving either the irradiated (Irrad) or autoclaved (Auto) diet and exhibiting either high (Hi) or low (Lo) clinical disease, as indicated. Data is given as the mean + standard error of the mean for samples run in triplicate. * P<0.05, ** P<0.01, t-test. (B) Correlation graph plotting the ratio of OCR to ECAR against disease (clinical score), Spearman’s rank correlation.
To further analyse the metabolomics data from serum and faeces, we performed various comparisons with grouped data. Supplementary Table S1 shows comparisons between day-2 vs day 16 for: first mice receiving autoclaved food with clinical scores equal to 0; and second all mice (receiving autoclaved and irradiated food) with clinical scores equal to 0. A similar comparison for mice receiving irradiated food with clinical scores equal to 0 could not be performed due to too few data points. Supplementary Table S2 shows comparisons between day-2 and day 16 for: first mice receiving autoclaved food with any disease (HiLo); second mice receiving irradiated food with any disease (HiLo); and third for all mice (receiving autoclaved and irradiated food) with any disease (HiLo). Supplementary Table S3 shows comparisons between day-2 and day 16 for: first all mice (receiving autoclaved and irradiated food) with high clinical scores (Hi); second all mice (receiving autoclaved and irradiated food) with any disease (HiLo); third all mice (receiving autoclaved and irradiated food) with low clinical scores (Lo); and fourth all mice (receiving autoclaved and irradiated food) with clinical scores equal to 0. For all 3 tables, metabolites that were altered in 2 or more of the comparisons are similarly shaded. While some metabolites are altered with significance in both serum and faeces, many more metabolites are either serum or faeces specific. Yet other metabolites are unique (no shading on tables).
4. Discussion
Genetically identical animals in the vivariums of different institutions and sourced from different commercial breeders have variable microbiota compositions that have likely contributed to conflicting reports of disease susceptibility with various models of autoimmune disease (Ivanov et al., 2008, 2009). The rate of disease development (incidence) in one spontaneous model of EAE, transgenic SJL/J mice expressing a MOG92-106 myelin peptide-recognising T cell receptor (TCR) on the majority of the CD4+ T cells, has been noted to vary from 35-90%, depending on the facility (Berer et al., 2011). This phenomenon was also noted for another spontaneous model of EAE, B10.PL mice expressing a myelin basic protein-specific TCR (Goverman et al., 1993). A direct comparison of SJL/J transgenic mice reared under specific pathogen-free conditions to SJL/J transgenic mice reared under completely germ-free conditions showed that commensal microbiota are required for the development of spontaneous EAE (Berer et al., 2011). The gut microbiota can effect remote organs such as the brain through chemical signalling (reviewed in Holmes et al., 2011).
For these experiments we grouped mice by clinical score: high clinical scores (Hi), low clinical scores (Lo) and clinical scores equal to 0 (Zero). Variation in disease severity, presented as clinical scores, is common for this model and is separate from disease incidence, the number of animals that present with clinical signs of disease. Robust disease was induced as disease incidence was 90% in mice receiving the irradiated diet and 8/20 mice receiving the irradiated diet had a clinical score of 4 or above on a scale of 0-5. Additionally, we are consistently able to induce disease as seen by comparing Figure 1A, here, to Figure 7 in Libbey et al. (2016).
Of particular interest in our microbiota analysis is the inverse correlation of the genus Lactobacillus and the severity of EAE. Lactobacillus is a lactic acid bacteria that is known to be a commensal coloniser in various sites of the human body (Walter, 2008). As a commonly used probiotic, much work has been done to characterise the effects of Lactobacillus on disease. Oral supplementation with Lactobacillus has been previously shown to ameliorate EAE in a strain-specific manner associated with increases in regulatory T cells (Tregs) and increased anti-inflammatory cytokines in the serum (Kwon et al., 2013; Lavasani et al., 2010). However, conflicting reports of no effect or a deleterious effect of Lactobacillus on EAE have also been reported depending on the species and the EAE model under investigation. Even the growth phase, either log phase or stationary phase, of Lactobacillus has been shown to have differential effects on the immune response of their commensal hosts (Maassen et al., 2003). Our results, in the context of previous work, highlight the need to investigate the effects of microbiota using a strain, growth phase and model specific methodology (Kobayashi et al., 2010; Maassen and Claassen, 2008). In validating Lactobacillus sp. as the most promising, negatively correlated, microbiota hit in the MOG35-55-induced EAE model in our hands, we showed that simple supplementation with L. paracasei significantly reduced both the incidence and severity of EAE. Further work is being done to characterise the mechanisms of protection conferred by L. paracasei in EAE.
Conversely, our analysis identified the following genera that positively correlated with the severity of EAE: Bacteroides (Family Bacteroidaceae), Turicibacter (Family Turicibacteraceae) and Ruminococcus (Family Ruminococcaceae). Others have also seen increases in both Turicibacter and Ruminococcaceae using the NOD model of EAE (Colpitts et al., 2017). This is in agreement with previous studies in which germ-free mice and mice treated with antibiotics were protected in EAE, suggesting that microbiota are required for the development of disease (Berer et al., 2011; Lee et al., 2011; Ochoa-Reparaz et al., 2009; Yokote et al., 2008) reviewed in (Rothhammer and Quintana, 2016; Wang and Kasper, 2014; Wekerle, 2016). Specifically, segmented filamentous bacteria have been found to exacerbate EAE, at least in part due to induction of a Th17 response (Lee et al., 2011). Segmented filamentous bacteria (Candidatus Arthromitus) were found to be absent from or below the limit of detection in the gut microbiota of the mice used in this study. Thus, as is being appreciated in other fields, the interaction between resident microbiota and immunity is a complex interaction. With respect to human disease, recent reports have begun to characterise the microbiota of MS patients compared to healthy controls (Chen et al., 2016; Jangi et al., 2016; Miyake et al., 2015; Tremlett et al., 2016) reviewed in (Tremlett et al., 2017)]. In work by Chen and colleagues, MS patients were found to have a different microbiota composition compared to healthy controls, and in concordance with our work, they see a reduction of Lactobacillus in MS patients compared to controls (Chen et al., 2016). Thus, our results have parallels in human disease. However, whether changes in microbiota are a cause or a result of disease is unknown. The fact that we observed the initial population of Lactobacillus as being predictive of EAE severity suggests a causal role of Lactobacillus in disease. This agrees with previous work supplementing Lactobacillus in EAE (Kwon et al., 2013; Lavasani et al., 2010). However, the therapeutic mechanism of Lactobacillus is not fully understood. It is tempting to speculate that depletion of Lactobacillus will exacerbate the EAE phenotype, but whether it is due to metabolite production or the presence of a Lactobacillus antigen or another mechanism is not known.
Our metabolomics data begins to give hints as to the interplay between diet, microbiota and host metabolism in the context of EAE. Specific to the disease process, we found G6P to be increased in the serum of diseased mice compared to mice immunised with myelin peptide that did not develop disease at day 16 post-sensitisation. G6P is a glycolytic/glycogenic intermediate that is also a substrate for G6P dehydrogenase, a critical enzyme in the pentose phosphate pathway (reviewed in Mathur et al., 2014). Thus, the observed increase in G6P may be reflective of increased glucose metabolism, decreased glycogenesis or dysfunctional G6P dehydrogenase activity. This is in agreement with findings in Th17 T cells, which in part drive EAE pathology, that increase glycolysis when activated (reviewed in Wahl et al., 2012). Our additional finding that levels of 2-DG are higher at day-2 than day 16 in diseased mice also suggests that glycolysis is upregulated during the EAE disease process. Indeed, inhibiting glycolysis using 2-DG decreases Th17 T cell differentiation and reduces the severity of EAE (Shi et al., 2011). Another metabolite specific to the disease process of interest is a decrease in 22:0 fatty acid in diseased mice compared to non-diseased mice at day-2 in faeces before the onset of disease. Tregs, which are protective in EAE, have higher rates of fatty acid oxidation than effector T cells (reviewed in Lochner et al., 2015). It is then tempting to speculate that low levels of long-chain fatty acids in diseased mice predispose these mice to developing disease as less substrate is available for Treg differentiation. However, dietary studies suggest a pathogenic role of long chain fatty acids that skew towards a Th1/Th17 response and a protective role of short chain fatty acids that skew towards a Treg response (Haghikia et al., 2015).
Activated T cells upregulate glycolysis and OXPHOS, however glycolysis is increased many fold over the increase in OXPHOS (reviewed in Wahl et al., 2012). This agrees with our findings that the TCA substrates citric acid and isocitric acid are increased in diseased mice at day-2 compared to day 16 regardless of diet, indicative of T cell activation. Interestingly, citric acid and isocitric acid are significantly increased in mice receiving the autoclaved diet compared to the irradiated diet. This suggests a diet specific effect that alters metabolism in a beneficial manner, as mice on the autoclaved diet exhibit less severe disease on average.
While our serum metabolomics data may be indicative of tissue damage and not necessarily direct metabolic changes of circulating cells, our metabolic flux analysis provides support for metabolic changes identified by the metabolomics screen specifically in T cells. The OCR/ECAR ratio was significantly increased in T cells from mice on the autoclaved diet with low disease compared to T cells from mice on the autoclaved diet with high disease, suggesting a shift away from OXPHOS and towards glycolysis being associated with disease. However, there was no difference in OCR/ECAR in T cells from mice on the irradiated diet with low disease compared to T cells from mice on the irradiated diet with high disease. This may be due to the fact the mice on the irradiated diet exhibit more severe disease on average compared to mice on the autoclaved diet. Still, these results support a model in which the severity of EAE is determined by a metabolic increase in glycolysis, more so than OXPHOS, and it will be interesting to modulate metabolism towards OXPHOS or towards glycolysis with targeted diets and also to probe which specific subsets of T cells are key in modulating metabolism for this phenotype.
While dietary interventions in MS patients have had mixed results, studies in EAE have shown an effect of diet on disease progression (Choi et al., 2016). A fasting mimicking diet has beneficial effect on both autoimmunity and remyelination (Choi et al., 2016). Our current study begins to elucidate the relationship between food, microbiota and metabolism in the context of EAE. In the C57BL/6J MOG35-55-induced EAE mouse model, we found that animals on the irradiated diet had increased severity of disease through the peak of disease, compared to animals on the autoclaved diet, as determined by clinical score, weight loss, morbidity and scoring of spinal cord pathology (demyelination, PVC, meningitis and overall pathology). Here we demonstrated that the distal gut microbiota is significantly altered in EAE and, by stratifying our mouse cohort based on disease severity, we identified specific bacterial genera that significantly correlate with disease severity. Using a non-biased approach, we found that the genus Lactobacillus was inversely correlated with EAE severity and the genera Bacteroides, Turicibacter and Ruminococcus were directly correlated with EAE severity. Therapeutic treatment with L. paracasei resulted in a significant reduction in the incidence of disease as well as a significant reduction in both the clinical scores and the amount of weight loss observed in EAE mice on the autoclaved diet. Furthermore, our metabolomics analysis of serum and faeces identified a litany of metabolites that are significantly altered in disease and between diets. Shifts in glycolysis and the TCA cycle, observed in serum and faeces, may explain the differences in disease severity we observed between diets in the EAE model. Similar alterations in metabolism, i.e. disturbed glucose metabolism, have been reported for MS pathogenesis (Mathur et al., 2014) and our data could help explain these changes. These targets are currently being investigated to more specifically probe the complex metabolic interaction at play in EAE that may have translational relevance to MS patients.
Supplementary Material
Figure S1. Flow chart detailing the treatment groups.
Figure S2. Correlation analyses of select metabolites.
Table S1. Grouped x-timept comparisons day-2 vs day 16 with mice with clinical score 0.
Table S2. Grouped x-timept comparisons day-2 vs day 16 with mice having any disease (HiLo).
Table S3. Grouped x-timept comparisons day-2 vs day 16 for different clinical score groups.
Acknowledgments
We would like to thank Mitchell A. Wilson, Michael J. O’Connell, Allyssa B. Tomlin and Samantha P. Duzy, for excellent technical assistance, Ana B. DePaula-Silva, PhD, and Tyler J. Hanak, BS, for many helpful discussions. We would like to acknowledge Garrett Brown for his technical assistance and work done by Dr Anil Laxman from the Metabolic Phenotyping Core, a part of the Health Sciences Cores at the University of Utah. This work was supported by NIH T32AI055434 (M.F.C.) and NIH 1R01NS082102 (R.S.F.) and the National Multiple Sclerosis Society Collaborative Center Grant #CA-1607-25040. Mass spectrometry equipment was obtained through NCRR Shared Instrumentation Grants # 1 S10 RR020883-01 and 1 S10 RR025532-01A1.
Footnotes
Supplementary material
Supplementary material can be found online at https://doi.org/10.3920/BM2017.0116.
There are no conflicts of interest to report by any of the authors.
References
- Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–541. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Chia N, Kalari KR, Yao JZ, Novotna M, Soldan MM, Luckey DH, Marietta EV, Jeraldo PR, Chen X, Weinshenker BG, Rodriguez M, Kantarci OH, Nelson H, Murray JA, Mangalam AK. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Scientific Reports. 2016;6:28484. doi: 10.1038/srep28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IY, Piccio L, Childress P, Bollman B, Ghosh A, Brandhorst S, Suarez J, Michalsen A, Cross AH, Morgan TE, Wei M, Paul F, Bock M, Longo VD. A diet mimicking fasting promotes regeneration and reduces autoimmunity and multiple sclerosis symptoms. Cell Reports. 2016;15:2136–2146. doi: 10.1016/j.celrep.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colpitts SL, Kasper EJ, Keever A, Liljenberg C, Kirby T, Magori K, Kasper LH, Ochoa-Reparaz J. A bidirectional association between the gut microbiota and CNS disease in a biphasic murine model of multiple sclerosis. Gut Microbes. 2017;8:561–573. doi: 10.1080/19490976.2017.1353843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- Haghikia A, Jorg S, Duscha A, Berg J, Manzel A, Waschbisch A, Hammer A, Lee DH, May C, Wilck N, Balogh A, Ostermann AI, Schebb NH, Akkad DA, Grohme DA, Kleinewietfeld M, Kempa S, Thone J, Demir S, Muller DN, Gold R, Linker RA. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity. 2015;43:817–829. doi: 10.1016/j.immuni.2015.09.007. [DOI] [PubMed] [Google Scholar]
- Hintze KJ, Cox JE, Rompato G, Benninghoff AD, Ward RE, Broadbent J, Lefevre M. Broad scope method for creating humanized animal models for animal health and disease research through antibiotic treatment and human fecal transfer. Gut Microbes. 2014;5:183–191. doi: 10.4161/gmic.28403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E, Li JV, Athanasiou T, Ashrafian H, Nicholson JK. Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends in Microbiology. 2011;19:349–359. doi: 10.1016/j.tim.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Frutos RL, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host and Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangi S, Gandhi R, Cox LM, Li N, Von Glehn F, Yan R, Patel B, Mazzola MA, Liu S, Glanz BL, Cook S, Tankou S, Stuart F, Melo K, Nejad P, Smith K, Topcuolu BD, Holden J, Kivisakk P, Chitnis T, De Jager PL, Quintana FJ, Gerber GK, Bry L, Weiner HL. Alterations of the human gut microbiome in multiple sclerosis. Nature Communications. 2016;7:12015. doi: 10.1038/ncomms12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Kato I, Nanno M, Shida K, Shibuya K, Matsuoka Y, Onoue M. Oral administration of probiotic bacteria, Lactobacillus casei and Bifidobacterium breve, does not exacerbate neurological symptoms in experimental autoimmune encephalomyelitis. Immunopharmacology and Immunotoxicology. 2010;32:116–124. doi: 10.3109/08923970903200716. [DOI] [PubMed] [Google Scholar]
- Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O’Connell RM, Round JL. MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host and Microbe. 2015;17:153–163. doi: 10.1016/j.chom.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HK, Kim GC, Kim Y, Hwang W, Jash A, Sahoo A, Kim JE, Nam JH, Im SH. Amelioration of experimental autoimmune encephalomyelitis by probiotic mixture is mediated by a shift in T helper cell immune response. Clinical Immunology. 2013;146:217–227. doi: 10.1016/j.clim.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Lavasani S, Dzhambazov B, Nouri M, Fak F, Buske S, Molin G, Thorlacius H, Alenfall J, Jeppsson B, Westrom B. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS ONE. 2010;5:e9009. doi: 10.1371/journal.pone.0009009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the USA. 2011;108(Suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Research. 2011;39:D19–21. doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Libbey JE, Doty DJ, Sim JT, Cusick MF, Round JL, Fujinami RS. The effects of diet on the severity of central nervous system disease: One part of lab-to-lab variability. Nutrition. 2016;32:877–883. doi: 10.1016/j.nut.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends in Immunology. 2015;36:81–91. doi: 10.1016/j.it.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Louis P, Scott KP, Duncan SH, Flint HJ. Understanding the effects of diet on bacterial metabolism in the large intestine. Journal of Applied Microbiology. 2007;102:1197–1208. doi: 10.1111/j.1365-2672.2007.03322.x. [DOI] [PubMed] [Google Scholar]
- Maassen CB, Claassen E. Strain-dependent effects of probiotic lactobacilli on EAE autoimmunity. Vaccine. 2008;26:2056–2057. doi: 10.1016/j.vaccine.2008.02.035. [DOI] [PubMed] [Google Scholar]
- Maassen CB, Laman JD, Van Holten-Neelen C, Hoogteijling L, Groenewegen L, Visser L, Schellekens MM, Boersma WJ, Claassen E. Reduced experimental autoimmune encephalomyelitis after intranasal and oral administration of recombinant lactobacilli expressing myelin antigens. Vaccine. 2003;21:4685–4693. doi: 10.1016/s0264-410x(03)00522-x. [DOI] [PubMed] [Google Scholar]
- Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur D, Lopez-Rodas G, Casanova B, Marti MB. Perturbed glucose metabolism: insights into multiple sclerosis pathogenesis. Frontiers in Neurology. 2014;5:250. doi: 10.3389/fneur.2014.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake S, Kim S, Suda W, Oshima K, Nakamura M, Matsuoka T, Chihara N, Tomita A, Sato W, Kim SW, Morita H, Hattori M, Yamamura T. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS ONE. 2015;10:e0137429. doi: 10.1371/journal.pone.0137429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa-Reparaz J, Mielcarz DW, Ditrio LE, Burroughs AR, Begum-Haque S, Dasgupta S, Kasper DL, Kasper LH. Central nervous system demyelinating disease protection by the human commensal Bacteroides fragilis depends on polysaccharide A expression. Journal of Immunology. 2010;185:4101–4108. doi: 10.4049/jimmunol.1001443. [DOI] [PubMed] [Google Scholar]
- Ochoa-Reparaz J, Mielcarz DW, Ditrio LE, Burroughs AR, Foureau DM, Haque-Begum S, Kasper LH. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. Journal of Immunology. 2009;183:6041–6050. doi: 10.4049/jimmunol.0900747. [DOI] [PubMed] [Google Scholar]
- Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nature reviews Immunology. 2016;16:341–352. doi: 10.1038/nri.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhammer V, Quintana FJ. Environmental control of autoimmune inflammation in the central nervous system. Current Opinion in Immunology. 2016;43:46–53. doi: 10.1016/j.coi.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. Journal of Experimental Medicine. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, Sonnenburg JL. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141:1241–1252. doi: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremlett H, Bauer KC, Appel-Cresswell S, Finlay BB, Waubant E. The gut microbiome in human neurological disease: a review. Annals of Neurology. 2017;81:369–382. doi: 10.1002/ana.24901. [DOI] [PubMed] [Google Scholar]
- Tremlett H, Fadrosh DW, Faruqi AA, Zhu F, Hart J, Roalstad S, Graves J, Lynch S, Waubant E. Gut microbiota in early pediatric multiple sclerosis: a case-control study. European Journal of Neurology. 2016;23:1308–1321. doi: 10.1111/ene.13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Wahl DR, Byersdorfer CA, Ferrara JL, Opipari AW, Jr, Glick GD. Distinct metabolic programs in activated T cells: opportunities for selective immunomodulation. Immunological Reviews. 2012;249:104–115. doi: 10.1111/j.1600-065X.2012.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J. Ecological role of lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical research. Applied and Environmental Microbiology. 2008;74:4985–4996. doi: 10.1128/AEM.00753-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kasper LH. The role of microbiome in central nervous system disorders. Brain, Behavior, and Immunity. 2014;38:1–12. doi: 10.1016/j.bbi.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wekerle H. The gut-brain connection: triggering of brain autoimmune disease by commensal gut bacteria. Rheumatology. 2016;55:ii68–ii75. doi: 10.1093/rheumatology/kew353. [DOI] [PubMed] [Google Scholar]
- Winkelmann A, Loebermann M, Reisinger EC, Hartung HP, Zettl UK. Disease-modifying therapies and infectious risks in multiple sclerosis. Nature reviews Neurology. 2016;12:217–233. doi: 10.1038/nrneurol.2016.21. [DOI] [PubMed] [Google Scholar]
- Yokote H, Miyake S, Croxford JL, Oki S, Mizusawa H, Yamamura T. NKT cell-dependent amelioration of a mouse model of multiple sclerosis by altering gut flora. American Journal of Pathology. 2008;173:1714–1723. doi: 10.2353/ajpath.2008.080622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Flow chart detailing the treatment groups.
Figure S2. Correlation analyses of select metabolites.
Table S1. Grouped x-timept comparisons day-2 vs day 16 with mice with clinical score 0.
Table S2. Grouped x-timept comparisons day-2 vs day 16 with mice having any disease (HiLo).
Table S3. Grouped x-timept comparisons day-2 vs day 16 for different clinical score groups.