

Abstract
Neuroblastoma is the most common extra-cranial solid tumors of childhood and the most common in the first year of life. It is a unique malignancy in that infants often present with either localized or metastatic disease that can spontaneously regress without intervention while older children can succumb to the disease after months to years of arduous therapy. Given this wide range of outcomes, the International Neuroblastoma Risk Group was created to stratify patients based on presenting characteristics and tumor biology in order to guide intensity of treatment strategies. The goal has been to decrease therapy for low risk patients to avoid long-term complications while augmenting and targeting therapies for high risk patients to improve overall survival. The international risk stratification depends on age, stage, histology, MYCN gene amplification status, tumor cell ploidy and segmental chromosomal abnormalities. Treatment for asymptomatic low risk patients with an estimated survival of >98% is often observation or surgical resection alone, whereas intermediate risk patients, with an estimated survival of >90% require moderate doses of response-adjusted chemotherapy along with resection. High risk patients undergo multiple cycles of combination chemotherapy before surgery, followed by consolidation with myeloablative autologous hematopoietic stem cell transplantation and local radiation, and finally immunotherapy with differentiation therapy as maintenance phase. With this approach, outcome for patients with neuroblastoma has improved, as the field continues to expand efforts in more targeted therapies for high risk patients.
Keywords: Neuroblastoma, Pediatric Oncology, Clinical Presentation, Treatment, Risk Classification
Introduction
Neuroblastoma is the most common extracranial solid tumor of childhood (Maris, 2010). It arises from the developing sympathetic nervous system from neural crest cells, usually resulting in tumors in the adrenal glands or the sympathetic ganglia (Irwin and Park, 2015). The age-standardized annual incidence in North America is 5.5 to 11.5 cases per million people (Stiller and Parkin, 1992). It is the most common malignancy overall in the first year of life with a median age at diagnosis of 18 months and 90% of cases diagnosed by 10 years of age (London, et al., 2005).
The clinical presentation can be quite heterogeneous, ranging from asymptomatic incidental tumors to widespread metastases with systemic manifestations. The clinical and biological heterogeneity leads to differences in outcome, ranging from spontaneous regression to inexorable progression, metastasis and death despite intensive therapy (Matthay, et al., 2016). In addition to the usual prognostic importance of disease stage, many biologic factors help to explain the clinical behavior in neuroblastoma, including histologic features, cytogenetic features, and the molecular changes, particularly amplification of the MYCN oncogene (Brodeur, et al., 1984, Seeger, et al., 1985). The better understanding of the prognostic importance and role of these clinical and biologic features has allowed for more precise risk stratification to guide therapy, in order to improve outcome for high risk patients by intensification of treatment and addition of novel agents while decreasing doses of chemotherapy for lower risk patients to reduce late effects. Recent advances of molecular changes involved in tumor initiation and progression have led to early phase clinical trials to test new inhibitors of potential tumor and tumor microenvironment targets.
This review will focus on the clinical aspects of this disease: the clinical presentation, diagnosis, risk stratification, and therapy for children with neuroblastoma.
Clinical Presentation
Clinical presentation of neuroblastoma varies widely by age and stage. The location of the primary tumor and any metastatic sites dictates the symptomatology. Neuroblastoma arises from the sympathetic nervous system, most often from the adrenal medulla (Vo, et al., 2014). The remainder arise from the paraspinal or other sympathetic ganglia and can present anywhere from the neck to the pelvis.
Neuroblastoma is prone to involve surrounding nerve roots due to the paraspinal location of most of the sympathetic ganglia; thus, tumors arising in the neck frequently cause Horner syndrome. Thoracic tumors usually present in the posterior mediastinum and paraspinal ganglia, frequently with invasion of the neural foramina. A localized abdominal or pelvic masses can be noted by the caregivers without any symptoms or can cause significant distention with or without pain. Pelvic tumors can also cause neurologic symptoms such as bladder dysfunction, constipation, or lower extremity pain or weakness due to nerve root involvement. Any of these locations has the potential to invade the neural foramina and cause spinal cord compression symptoms.
Metastases are present at diagnosis in about 50% of patients, with the bone marrow, bone and regional lymph nodes being the most common sites while involvement of central nervous system and lungs are rare, present in less than 5% of metastatic patients at diagnosis (DuBois, et al., 1999, Morgenstern, et al., 2016). Extensive liver involvement can be seen in infants and can cause liver disease such as a coagulopathy, and renal and lung dysfunction due to abdominal distention. Also more common in infants, metastases in the skin can appear as painless subcutaneous nodules anywhere on the body with a blue hue (Figure 1). Bone marrow infiltration, present in 80% of metastatic patients, can cause anemia and thrombocytopenia. Metastases to the bone can be very painful causing limping or refusal to bear weight. Bone lesions are commonly seen in the skull, and if they are in the periorbital region, cause proptosis or periorbital bruising (known commonly as “raccoon eyes” or Hutchinson syndrome).
Figure 1. Skin Metastasis in Infant with Stage MS Neuroblastoma.

A) Skin metastasis in neuroblastoma can be seen in stage MS disease, consisting of subcutaneous nodules with a bluish hue. B) Fine needle aspirate of a skin metastasis showing histology consistent with neuroblastoma, here showing a Homer-Wright rosette pattern.
Neuroblastoma can also present with systemic symptoms such as fever or weight loss. Neuroblastomas can release catecholamines with patients experiencing flushing, tachycardia or hypertension. In a very rare paraneoplastic phenomenon, the tumor can secrete vasoactive intestinal peptide (VIP), which causes profuse watery diarrhea. Paraneoplastic presentation is also possible with opsoclonus myoclonus syndrome (OMS), in which patients can have an array of neurologic symptoms including opsoclonus which is spontaneous saccades of the eyes in all directions, myoclonus (involuntary muscle twitching), and occasionally ataxia or other cerebellar signs. A child that presents with this constellation of symptoms, even without overt signs of malignancy, should be evaluated for neuroblastoma, as it has been diagnosed in 50–80% of patients with OMS, but only 2–3% of children with neuroblastoma overall are affected by OMS (Hero and Schleiermacher, 2013). The neurologic symptoms are probably attributable to anti-neuronal antibodies cross-reacting with cerebellum (Antunes, et al., 2000, Rudnick, et al., 2001).
Evaluation, Staging and Screening
When neuroblastoma is suspected, a variety of laboratory tests, imaging studies and pathologic examinations are required to confirm the diagnosis and staging. Initially a complete blood count, prothrombin time and partial thromboplastin time, uric acid, electrolytes, creatinine, liver function tests, ferritin and lactate dehydrogenase should be tested; the latter two tests indicate a lower survival when elevated (Cohn, et al., 2009, Hann, et al., 1985, Hann, et al., 1980, Shuster, et al., 1992). Urine should also be collected for vanillylmandelic acid (VMA), homovanillic acid (HVA) and dopamine levels. These catecholamines and catecholamine metabolites are found in 90% of children with neuroblastoma. Plasma-free and total normetadrenaline, metadrenaline and methoxytyramine can be drawn in the serum as a replacement if urine cannot be collected from a young child (Franscini, et al., 2015). Bilateral bone marrow biopsies may be used to confirm the diagnosis if tumor is seen and urine catecholamines are elevated, and these are also required to complete staging, with immunohistochemistry to increase sensitivity of tumor detection (Beiske, et al., 2009).
Extensive imaging is required for staging, tumor characterization and surgical planning. Ultrasound is an accessible way without requiring sedation to confirm the presence and location of a mass. However, cross sectional imaging with either computed tomography (CT) or magnetic resonance imaging (MRI) is necessary for accurate localization and characterization (Figure 2A). The primary site should be included as well as the chest, abdomen and pelvis to check for lymph node spread or further extension. MRI is preferred if possible as it does not use ionizing radiation and can provide superior imaging, especially of the spinal cord, but has the disadvantage of more often requiring sedation in young children. For neuroblastoma, an 123I-metaiodobenzylguanidine (MIBG) scan is essential for staging and evaluation of response (Figure 2B) (Matthay, et al., 2010). MIBG is a radiolabeled molecule similar in structure to norepinephrine, that is taken up by the norepinephrine transporter in 90% of neuroblastomas (Dubois, et al., 2012). Thus, an MIBG scan provides an extremely specific method of discovering and following sites of metastases, which is more specific and sensitive than technetium bone scan (Gauguet, et al., 2017). MIBG can be combined with other imaging modalities such as is done in a single-photon emission computed tomography (SPECT) to provide more exact anatomical localization of the MIBG uptake (Figure 2C) (Biermann, et al., 2013). Multiple semi-quantitative scoring systems for MIBG uptake in neuroblastoma exist to track metastatic disease burden and have been shown to be associated with prognosis (Decarolis, et al., 2013, Yanik, et al., 2013). In the 10% of neuroblastomas that are MIBG non-avid, or if MIBG is not available, 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET) CT is an acceptable alternative (Sharp, et al., 2009). Newer radiologic techniques are also under investigation for utility in neuroblastoma such as 18F-l-dihydroxyphenylalanine-PET (18F-DOPA-PET) and gallium-68 (68Ga)-DOTATATE-PET (Gains, et al., 2011, Liu, et al., 2016).
Figure 2. Imaging in Neuroblastoma.

Imaging in neuroblastoma must be multimodal to accurately locate and characterize the primary tumor with cross-sectional imaging and locate metastatic disease with MIBG. A) An MRI showing a paraspinal mass invading the spinal canal across many thoracic levels, causing spinal cord compression. Also note the metastatic involvement of multiple vertebral bodies. B) 123I-metaiodobenzylguanidine (MIBG) scan showing the primary thoracic tumor and revealing wide spread metastatic disease involving the bones. C) Single-photon emission computed tomography (SPECT) combining MIBG and CT to better localize the MIBG uptake.
Biopsy of the tumor confirms the diagnosis and provides important information on prognosis. Neuroblastoma is a small round blue cell tumor that can be differentiated from other tumors such as Ewing sarcoma and other sarcomas, lymphoma, and Wilms tumor by the appropriate immunohistochemical stains (Hachitanda, et al., 1989, Miettinen, 1987). These tumors can show differing levels of maturation. Neuroblastoma is primarily immature while a ganglioneuroma is composed of cells that have matured and completed differentiation as ganglion cells. A ganglioneuroblastoma has components of both of these types of tumors. The International Neuroblastoma Pathology Committee further subtyped these immunohistochemical classifications by the amount of Schwannian stroma present in the tumor (Shimada, et al., 1999, Shimada, et al., 2001). These subtypes are neuroblastoma which is Schwannian stroma-poor, ganglioneuroblastoma intermixed which is Schwannian stroma-rich, ganglioneuroblastoma nodular which is a combination of Schwannian stroma-rich and poor, and ganglioneuroma which is Schwannian stroma-dominant. Ganglioneuroblastoma nodular is divided into favorable and unfavorable, depending on the mitosis-karyorrexis index (MKI) and patient age (Peuchmaur, et al., 2003). Generally, poorly differentiated or undifferentiated histology portend a worse prognosis, but age is also an important indicator. In an infant <18 months, a poorly differentiated neuroblastoma is still considered favorable if the MKI is not high, but in a patient >18 months, a poorly differentiated neuroblastoma is always unfavorable (Shimada, et al., 1999). In the same International Neuroblastoma Pathology Classification, a child >5 years of age with neuroblastoma is always considered unfavorable, but ganglioneuroblastoma can still be considered favorable.
Molecular testing of the tumor has become increasingly important as well to determine overall prognosis. MYCN gene amplification is one of the most important markers of aggressive disease and poor prognosis in neuroblastoma (Bagatell, et al., 2009, Brodeur, et al., 1984, Campbell, et al., 2017, Seeger, et al., 1985). MYCN amplification is most commonly measured by fluorescent in situ hybridization (FISH) as a fourfold increase in the MYCN signal number compared with the reference probe located on chromosome 2q (Ambros, et al., 2009). Cell ploidy is also an important prognostic marker in neuroblastoma with triploidy or hyperdiploidy having a better prognosis than diploidy (Ambros, et al., 2009). This is measured by flow cytometry and reports a DNA index of 1 for diploidy, thus a value greater than 1 is associated with better outcome. Segmental chromosomal copy number alterations are also seen in neuroblastoma and are commonly measured by array comparative genomic hybridization. The most common of these are gain of 17q, loss of 1p and loss of 11q, with multiple other segmental chromosomal alterations that are less common, but all are associated with worse outcome (Attiyeh, et al., 2005, Bown, et al., 1999, Schleiermacher, et al., 2012).
Mutations in specific genes in neuroblastoma are not common but the presence of these alterations are being discovered at an increasing rate with the next generation sequencing becoming more accessible (Molenaar, et al., 2012, Pugh, et al., 2013). One such mutation that was discovered in the germline DNA in families with heritable neuroblastoma is in the ALK gene, which has since also been shown to be somatically mutated spontaneously in a subset of neuroblastomas (Chen, et al., 2008, George, et al., 2008, Janoueix-Lerosey, et al., 2008, Mosse, et al., 2008). Familial neuroblastoma is rare, found in approximately 1% of patients, but ALK germline testing should be considered in this context. Tumor testing should also be considered in newly diagnosed non-heritable cases, where 8–15% of patients are estimated to have somatic mutations in ALK, as there are emerging therapies targeting this constitutively active kinase (Mosse, et al., 2013). PHOX2B germline mutations are another cause for familial neuroblastoma but have not been found commonly as a somatic mutation (Trochet, et al., 2004). It has also been observed that germline mutations in other cancer associated genes such as TP53, NF1, BRCA1/2, NRAS, APC and PTPN11 can predispose to neuroblastoma as well. Somatic mutations are not common in neuroblastoma but have been seen repeatedly in a few genes such as loss of function alterations in ATRX (which is more common in adolescents) (Cheung, et al., 2012, Molenaar, et al., 2012, Pugh, et al., 2013) and promoter rearrangements in TERT (Peifer, et al., 2015). Both of these alterations have been shown to elongate telomeres, which is a known mechanism of survival in cancer cells.
Given that metastatic neuroblastoma can be quite advanced at diagnosis, attempts to screen for this disease in infants were undertaken. Urine catecholamines are rather sensitive and specific so they were used for screening trials across the world. In Japan, mass screening led to possible over-diagnosis of neuroblastoma, but overall mortality was lower in the screened group (Hisashige and Group, 2014, Hiyama, et al., 2008, Sawada, et al., 1982, Yamamoto, et al., 1995). The neuroblastomas discovered by screening, however, tended to have more favorable molecular characteristics (Nakagawara, et al., 1991), and when screening was terminated, Japan did not see an increase in mortality from neuroblastoma (Kerbl, et al., 2003, Shinagawa, et al., 2017). Screening in North America showed similar results (Woods, et al., 2002). A large German study also showed an over-diagnosis of low risk patients and no improvement in the detection or mortality of high risk patients even when screening was performed at 12 months of age instead of in the first 3 months (Schilling, et al., 2002, Schilling, et al., 2003). In aggregate, screening studies for neuroblastoma did not decrease mortality and likely led to over-diagnosis of patients who did not benefit from earlier intervention, so wide-spread screening has not been implemented.
Risk Stratification
Prior to the formation of the International Neuroblastoma Risk Group (INRG) Task Force, multiple staging systems were used across the world, making research difficult to perform and compare. The most recent and frequently used was the International Neuroblastoma Staging System (INSS), used from about 1989–2010, was based, in part, on the extent of surgical resection (Brodeur, et al., 1993). This was problematic as it did not allow for pretreatment classification of stage, making comparison across clinical groups, especially for research purposes, more complex and not well standardized.
In 2009, the INRG Task Force published their recommendations both for new staging and risk group stratification (Table 1 and Figure 3) (Cohn, et al., 2009, Monclair, et al., 2009). INRG staging is based on image-defined risk factors (IDRFs) pre-therapy rather than post-surgical. These IDRFs were chosen based on elements that represent that that the tumor is more threatening to the patient or could make surgical resection more dangerous, thus less successful. IDRFs include: tumor extension into a second body compartment, encasement of any large blood vessels, tracheal or large bronchial compression, involvement of major nerve roots (such as the brachial plexus), invasion of the spinal canal, or infiltration of the nearby kidneys, mesentery, pericardium, liver, diaphragm or pancreas. These are predictive of worse event-free and overall survival.
Table 1. International Neuroblastoma Risk Group Staging System.
Reprinted with permission. ©2009 American Society of Clinical Oncology. All rights reserved. Monclair, T et al: J Clin Oncol 27(2), 2009: 298–303.
| Stage | Description |
|---|---|
| L1 | Localized tumor not involving vital structures as defined by the list of image-defined risk factors and confined to one body compartment |
| L2 | Locoregional tumor with presence of one or more image-defined risk factors |
| M | Distant metastatic disease (except stage MS) |
| MS | Metastatic disease in children younger than 18 months with metastases confined to skin, liver, and/or bone marrow |
Figure 3. International Neuroblastoma Risk Group (INRG) Consensus Pretreatment Classification Schema.

Reprinted with permission. ©2009 American Society of Clinical Oncology. All rights reserved. Cohn, S et al: J Clin Oncol 27(2), 2009: 289–297. This is the original published INRG risk classification. All blank fields represent any value. GN, ganglioneuroma; GNB, ganglioneuroblastoma; Amp, amplified; NA, not amplified.
There are four INRG stages (Table 1): L1, L2, M and MS (Monclair, et al., 2009). L1 is local disease only, not meeting criteria for any of the IDRFs. L2 is locoregional tumors with one or more IDRFs. Local lymph node status is not included in the new INRG staging. M encompasses all patients with metastatic disease, except for MS. MS is analogous to the old stage 4S and includes metastatic tumor in children less than 18 months of age that is restricted to the liver, skin and/or bone marrow. INSS stage 4S only included infants less than 12 months, but data showing excellent outcome in these patients with the same metastatic pattern out to 18 months, providing their tumor biology was favorable (Taggart, et al., 2011), led to the increase in this age limit. Furthermore, the MS stage does not exclude patients with large unresectable primary tumors, unlike INSS 4S, where the primary tumor must be only stage 1 or 2. With the complex nature of risk factors influencing outcome in patients with neuroblastoma, however, treatment decisions cannot be based solely on these staging criteria, but further pathologic and molecular risk stratification is necessary to assign therapy in this heterogeneous disease.
The INRG Task Force then derived these risk stratifications including not only stage, but tumor biology (Cohn, et al., 2009). They collected data from over 8000 patients from cooperative groups in North America, Europe and Japan. When available, they explored 35 different potential risk factors and compared these to event-free and overall survival. They made Kaplan-Meier survival curves for the condensed risk factors and assessed which were most significantly associated with outcome in order to make survival trees with branch points that could form prognostic categories. Using the risk factors that were the most significant as well as clinically relevant, they chose INRG staging, age, histologic category, grade of tumor differentiation, MYCN amplification, 11q aberration and ploidy to divide patients into pretreatment risk groups (see Figure 3). Generally speaking, poorly differentiated or undifferentiated, MYCN amplified, presences of 11q aberration and diploid are considered unfavorable biologic characteristics. Very low risk is defined as an event-free survival of >85%, low risk is 75–85%, intermediate risk is 50–75% and high risk is <50%.
These risk stratifications help in understanding the amount that each risk factor contributes to survival in a way that separate survival curves cannot. For example, in a patient with MS disease that is MYCN non-amplified, the presence of an 11q aberration would increase them from very low risk to high risk, whereas with other stages of disease, it does not have such a dramatic effect on outcome. This system is complex but allows for very specific homogenous grouping of patients with neuroblastoma, and simplifies how researchers and clinicians alike can communicate about patients and consider the intensity of therapy needed. Event-free survival curves from Children’s Oncology Group (COG) studies by these risk groups are shown in Figure 4 (Park, et al., 2013).
Figure 4. Event Free Survival Based on Risk Stratification (reproduced with permission from (Park, et al., 2013)).
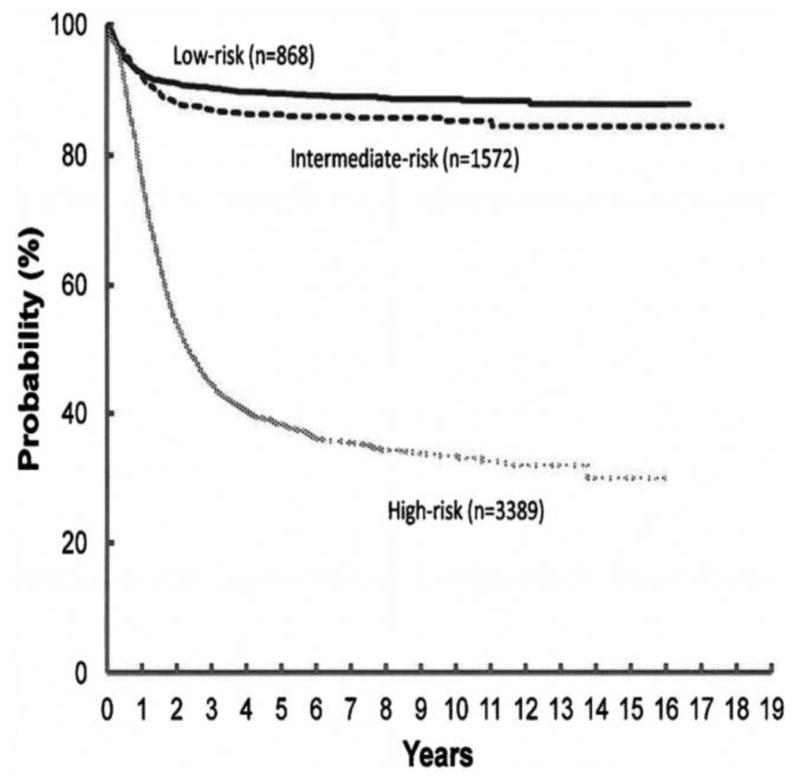
Patients with high risk disease have significantly worse event free survival than those with low or intermediate risk disease. Event free Kaplan-Meier survival curves from the time of diagnosis for children enrolled on Children’s Oncology Group, Children’s Cancer Group or Pediatric Oncology Group studies between 1990 and 2010. Risk stratification based on INRG risk classification.
Current Treatment Regimens
In the low and intermediate risk patients, high overall survival of greater than 90% has been achieved while minimizing therapy (Baker, et al., 2010, Rubie, et al., 2011, Strother, et al., 2012). While significant progress has been made in high risk patients, the long-term survival is still below 50%, so the focus has been on intensifying treatment and complementing standard chemotherapy with targeted therapies. A patient with newly diagnosed neuroblastoma can be treated with a wide range of therapies including observation only, surgery, chemotherapy, radiation, immunotherapy, differentiation therapy and autologous stem cell transplant. The INRG classification has been valuable in better understanding prognosis and thus guiding systematic therapy decisions suggested by the international cooperative groups, mainly the Children’s Oncology Group (COG) and the International Society of Paediatric Oncology European Neuroblastoma (SIOPEN) (Figure 5).
Figure 5. Treatment Overview for Neuroblastoma by Risk Classification.

Patients with low risk disease are often managed with surgical resection or observation alone with tumors likely to spontaneously regress that are not causing symptoms. Intermediate patients are treated with chemotherapy with the number of cycles dependent on their response as well as surgical resection of the primary tumor. High risk disease requires intensive multimodal therapy, including chemotherapy, surgery, myeloablation, radiation, immunotherapy and differentiation therapy.
The mainstay of treatment for low risk L1/L2 patients has been complete surgical resection if possible. More recently, however, observation alone, even without a biopsy to confirm the diagnosis, has been shown to be an appropriate approach in infants less than six months of age with an adrenal tumor less than 16 ml in volume without symptoms, as reported in a COG pilot study (Nuchtern, et al., 2012). Observation is appropriate, as a large proportion of these tumors will spontaneously regression without therapy, and surgical complications are not insignificant in these young patients. Recommended monitoring initially is every six weeks, with physical exams, imaging (MRI or ultrasound) and urine catecholamines. After observing for rapid tumor growth for the first 12 weeks, recommended monitoring can be spaced to every three months for the first year, then every six months. Repeating the metastatic evaluation in these cases is not necessary unless there is evidence of local tumor progression (Nuchtern, et al., 2012). Studies are ongoing in the COG (ClinicalTrials.gov Identifier NCT02176967) to assess if these recommendations can be expanded to infants up to 12 months with L1 tumors, including non-adrenal tumors if urine catecholamines are elevated or MIBG uptake demonstrated. Generally, the other infants who have INRG very low or low risk disease are eligible for a trial of observation after biopsy confirms favorable histologic and molecular subtypes, including MYCN non-amplification, if the patient has no symptoms related to their tumor or concerning IDRFs, but confirmatory studies are ongoing (De Bernardi, et al., 2009, Hero, et al., 2008, Rubie, et al., 2011, Strother, et al., 2012). If, however, there are symptoms, problematic IDRFs or the patients are older than 12 months, either surgical resection or limited chemotherapy to decrease tumor burden is recommended. While gross total resection is preferable, localized L2 tumors with favorable histology and molecular findings can be safely observed even if not completely removed (Iehara, et al., 2013, Marachelian, et al., 2012, Strother, et al., 2012).
For low risk MS patients, if they are asymptomatic and have favorable biology (MYCN non-amplified, no 11q aberration and hyperdiploid), observation is also appropriate without surgery or chemotherapy, given the excellent survival and frequent spontaneous regression (Strother, et al., 2012, Taggart, et al., 2011). The exceptions are those infants in the first few months of life with massive hepatomegaly, who require emergent chemotherapy to avoid life-threatening consequences of respiratory impairment or abdominal compression syndrome (Nickerson, et al., 2000). Stage MS patients with unfavorable biologic features, such as diploidy or segmental chromosome abnormalities also require chemotherapy, while those with MYCN gene amplification should be treated as high risk (Taggart, et al., 2011).
For intermediate risk neuroblastoma, treatment can vary depending on individual response to therapy. This intermediate group is largely comprised of L2 tumors that are not MYCN amplified but are either histologically or genetically unfavorable (either undifferentiated or presence of 11q aberration), stage M disease in patients less than 18 months, and stage MS with unfavorable biology. Usually patients are treated with two to eight cycles of chemotherapy, depending on response to treatment. This chemotherapy may be given in an outpatient setting and is less dose intensive than that prescribed to the high risk patients. Surgery to remove the primary tumor is indicated when possible, but as in the low risk patients, complete resection is not essential (Baker, et al., 2010, Defferrari, et al., 2015, Kohler, et al., 2013, Marachelian, et al., 2012). However, the patients over 18 months with L2 tumors that cannot be resected have a lower overall survival, thus more intense therapy including radiation therapy for local control is recommended (Baker, et al., 2010, Defferrari, et al., 2015, Kohler, et al., 2013, Matthay, et al., 1998).
High risk disease has overall poor long-term outcome, despite the improvement over the past two decades from 29% to 50% 5-year survival (Pinto, et al., 2015). Therefore, the focus has been on continually increasing therapy intensity, with a resulting prolonged and intensive course for patients and families, with significant in-patient hospital stays (see Figure 5). The high risk patients include stage M with age greater than 18 months at diagnosis and patients with any age and stage with MYCN amplification. L2 patients with unfavorable histology and age greater than 18 months are often included in the high risk group as well, though the benefit of myeloablative therapy in this subgroup has not been unequivocally demonstrated (Meany, et al., 2014, Park, et al., 2009). Treatment starts with multiple cycles of induction chemotherapy to reduce tumor burden, making it more amenable to surgical resection. Surgery is then done to remove the primary tumor. This is followed by one or two courses of myeloablative therapy with autologous stem cell transplantation, then radiation to the primary tumor bed. Maintenance therapy is comprised of differentiation therapy with isotretinoin and immunotherapy with anti-GD2 monoclonal antibody and cytokines. Treatment duration is approximately 18 months or longer if there are delays.
The purpose of induction chemotherapy, which typically includes a platinum drug, anthracyclines, and alkylating agents, is to decrease tumor burden, eliminate metastatic deposits, and to allow for safer surgical removal of the primary tumor. The primary tumor often encases important blood vessels, such as the aorta or renal arteries, or invade neural foramina, making complete surgical resection dangerous or impossible at presentation. Patients with complete or very good partial response to induction, which, have a significantly improved event free survival than those who responded less robustly, meaning that response to induction is an important prognostic indicator (Ladenstein, et al., 1998, Matthay, et al., 1999, Yanik, et al., 2015, Yanik, et al., 2017, Yanik, et al., 2013). In practice, if a patient has not had a complete response, other therapies, such as additional chemotherapy with or without immunotherapy (Modak, et al., 2017), or radiopharmaceutical therapy with 131I-MIBG (Matthay, et al., 2007), are often utilized to attempt to achieve complete response before stem cell transplantation. In North America, common regimens used to improve metastatic response include immunochemotherapy (Mody, et al., 2017), 131I-MIBG therapy (Matthay, et al., 2007), or simply irinotecan with temozolomide (Bagatell, et al., 2011). The International Society of Paediatric Oncology European Neuroblastoma (SIOPEN) group in their current high risk protocol add a combination of topotecan, vincristine, and doxorubicin to try to achieve metastatic response (Amoroso, et al., 2017), and previously used a protocol combining 131I-MIBG with topotecan (Gaze, et al., 2005).
The INRG recently published new guidelines on measuring treatment response which remains based on the Response Evaluation Criteria in Solid Tumors (RECIST) criteria, but includes semi-quantitative scoring criteria for MIBG scans (Matthay, et al., 2010) as well as evaluation of percent tumor in bone marrow biopsies (Burchill, et al., 2017, Park, et al., 2017).
Surgical resection is typically scheduled for the end or near the end of induction chemotherapy and removes the remaining primary tumor. If it is safe, complete resection should be attempted. However, in patients with metastatic disease, it has not yet been proven to be beneficial to overall survival to entirely resect the tumor (Simon, et al., 2013, von Allmen, et al., 2017). In contrast, gross total resection of the high risk L2 tumors is associated with an improved prognosis (Adkins, et al., 2004, Matthay, et al., 1998, Mullassery, et al., 2014, Park, et al., 2009). Thus, the benefit must be weighed against the risk of surgery. Surgical resection is often accompanied by a number of complications, some severe and long lasting. Patients can have life threatening hemorrhage during surgery, and later can have bowel injury with nausea, vomiting and severe diarrhea, preventing enteral feeds for a prolonged period of time after surgery. There are often renal complications given the tumor proximity to the kidneys and renal vasculature, infrequently requiring dialysis. These complications can often delay the next cycle of chemotherapy or stem cell transplantation, leaving the patient susceptible to tumor recurrence while healing.
Myeloablative consolidation therapy after surgery has been shown to significantly improve outcome (Berthold, et al., 2005, Matthay, et al., 1999, Pritchard, et al., 2005). For autologous stem cell transplantation, the peripheral blood stem cells are usually collected by apheresis from the patients after two cycles of chemotherapy, as studies have shown satisfactory yield without significant contamination at that time point (Kreissman, et al., 2013, Park, et al., 2011). Some groups, however, prefer to collect after induction chemotherapy, based on the premise that there would likely be a lower risk of tumor cell contamination, although overall yields of CD34 cells would be lower. Purging these collected stem cells of neuroblastoma cells did not change the event free survival in a randomized trial, and is no longer routinely recommended (Kreissman, et al., 2013). The optimal conditioning regimen for myeloablation is still under study. Total body irradiation was initially used but did not provide apparent benefit over dose intensive chemotherapy regimens and has significantly more long-term effects so it is no longer routinely used (Saarinen-Pihkala, et al., 2012). A single transplant with carboplatin, etoposide and melphalan (CEM) conditioning regimen was studied, with SIOPEN comparing CEM to a single busulfan and melphalan (BuMel) transplant while the COG studied adding a tandem transplant with thiotepa and cyclophosphamide, followed six weeks later by CEM. Both BuMel and tandem transplant were shown to be superior to CEM alone and are now the standard of care in their respective regions (George, et al., 2006, Ladenstein, et al., 2017, Park, et al., 2016, Seif, et al., 2013).
For maintenance therapy, patients are treated with a combination of isotretinoin and anti-GD2 antibody immunotherapy. The former has been shown to promote tumor cell differentiation and slow growth of neuroblastoma cells and has improved event free survival in a randomized trial (Matthay, et al., 1999). Fenretinide, a synthetic retinoid derivative, is under investigation as well (Maurer, et al., 2013). GD2 is a disialoganglioside that is expressed on neuroectodermal tumors including neuroblastoma. The addition of a chimeric monoclonal antibody targeting GD2, known as ch14.18 or dinutuximab, along with GM-CSF and interleukin-2 (IL-2) has been shown to significantly improve event free survival and overall survival over isotretinoin alone, and the combination is now the accepted maintenance therapy (Yu, et al., 2010). The short-term side effects, however, can be significant with common toxicities of fever, allergic reactions, and fluid-overload causing respiratory distress and hypotension. Pain is also a common manifestation, as GD2 is found on peripheral pain fibers, and parenteral opiates are usually required. Further study is ongoing to determine if IL-2, which contributes to this side effect profile, is necessary for the beneficial effect of anti-GD2 antibodies (Siebert, et al., 2016). Anti-GD2 therapy has also been promising earlier in therapy (Talleur, et al., 2017), thus will be included in induction in an upcoming COG trial. In the relapsed/refractory setting, anti-GD2 immunotherapy has recently shown beneficial responses in over half of patients when combined with chemotherapy (Mody, et al., 2017) and when combined with natural killer cell infusions (Federico, et al., 2017). Other methods of targeting GD2 such as a vaccine (Kushner, et al., 2014), humanized antibodies or immunocytokines (Navid, et al., 2014, Shusterman, et al., 2010) and chimeric antigen receptor (CAR) T-cells (Louis, et al., 2011) are under investigation.
Overall 5-year survival from high risk neuroblastoma has improved significantly over the past 20 years, from 29% for patients diagnosed from 1990 to 1994 to 50% for patients diagnosed from 2005 to 2010 (Pinto, et al., 2015). It is thought that the intensification of therapy through myeloablative therapy and immunotherapy have led to this progress. Patients with refractory or relapsed neuroblastoma, however, can rarely be cured. Nevertheless, current salvage therapies can offer partial or even complete response with improvement in symptoms and quality of life. The most commonly used conventional cytotoxic chemotherapies in this setting are topotecan with either cyclophosphamide or temozolomide (Di Giannatale, et al., 2014) or irinotecan and temozolomide (Bagatell, et al., 2011, Kushner, et al., 2006, London, et al., 2010). Radiation therapy can be used locally to provide symptomatic relief, especially at sites of boney disease.
Another form of salvage therapy is 131I-mIBG therapy. This has the same localizing properties as 123I-mIBG used for imaging, but has higher amounts of the radioactive isotope, which also has a longer half-life, thus delivering a focal dose of radiation to all the tumor sites. A 30–40% response rate has been observed in refractory and relapsed neuroblastoma (Matthay, et al., 2007, Wilson, et al., 2014, Zhou, et al., 2015), and studies using it in upfront therapy are underway. A randomized COG trial will study the inclusion of 131I-mIBG during induction therapy. This therapy can be beneficial but logistically difficult as patients must be kept in isolation for multiple days until they are no longer radioactive, and only certain centers can provide this safely.
Targeted small molecule inhibitors are also being developed in neuroblastoma, some showing activity in the relapsed setting, which can provide an oral life-prolonging option. ALK inhibitors are appropriate in patients whose tumors harbor activating ALK mutations (Mosse, et al., 2013). Some of these mutations, such as F1174L, however, are resistant to the current ALK inhibitors (such as crizotinib) and a new inhibitor formulated to overcome this resistance, lorlatinib, (Infarinato, et al., 2016) is actively being evaluated in the New Approaches to Neuroblastoma Therapy (NANT) consortium. Aurora kinase inhibitors are known to cause cell cycle arrest, but they have also been found to destabilize MYCN which is particularly appealing given that there are no direct inhibitors available targeting this driver in neuroblastoma (Gustafson, et al., 2014). Indeed, an aurora kinase inhibitor, alisertib, combined with chemotherapy showed promise in a phase I trial and further studies are ongoing (DuBois, et al., 2016, Mosse, et al., 2012). In an attempt to block the Ras pathway, sorafenib, which is a Raf kinase inhibitor, (Kakodkar, et al., 2012) and newer PI3K inhibitors are being studied in children with neuroblastoma (Chanthery, et al., 2012, Erdreich-Epstein, et al., 2017). VEGF inhibition paired with inhibition of Nutlin-3A showed promising effects in vivo (Patterson, et al., 2011), thus bevacizumab is now in clinical trials in neuroblastoma combined with chemotherapy. TRK fusions have been discovered in many tumor types, including neuroblastoma (Vaishnavi, et al., 2015), and TRK inhibitors have shown partial responses and stable disease in a Phase I NANT trial (Minturn, et al., 2011) so remains under intense study. Checkpoint inhibitors are an exciting new target in cancer therapy (Cole, et al., 2011, Russell, et al., 2013), and phase I study with a CHK1/2 inhibitor in children with relapsed solid tumors is ongoing. Another interesting target in neuroblastoma is ornithine decarboxylase (ODC) which is the rate-limiting enzyme in polyamine synthesis, as it is a downstream target of MYC. Difluoromethylornitine (DFMO), an ODC inhibitor, is being studied in neuroblastoma in the relapsed setting as well as for maintenance (Evageliou, et al., 2016, Rounbehler, et al., 2009, Saulnier Sholler, et al., 2015). Other inhibitors in various stages of investigation include bromodomain inhibitors (Henssen, et al., 2016, Puissant, et al., 2013), and histone deacetylase inhibitors (DuBois, et al., 2015, Fouladi, et al., 2010). Further immunotherapy is also understudy in neuroblastoma in early phases with PD1 inhibitors (Wagner and Adams, 2017), CAR-T cells (Heczey, et al., 2017), and NK cells (Talleur, et al., 2017). As the safety and efficacy of these inhibitors are elucidated, the more promising could eventually be incorporated in upfront therapy, ideally improving survival before relapse.
Summary
Detailed, evidence-based risk stratification in neuroblastoma has allowed for intensification of therapy for the highest risk patients while decreasing therapy in the low risk patients. Overall outcomes have been improving as a result, but further work is needed to continue this progress. The treatments discussed here for relapsed and refractory patients are targeted therapies and immunotherapy, which, as further research develops, could potentially be included in initial therapy of newly diagnosed patients, with fewer complications and better efficacy.
Acknowledgments
Funding Information:
This work was supported by National Cancer Institute P01 CA217959 and the Alex Scott Lemonade Foundation Center of Excellence, Dougherty Foundation, Mildred V. Strouss Chair, and the Conner Fund.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- Adkins ES, Sawin R, Gerbing RB, London WB, Matthay KK, Haase GM. Efficacy of complete resection for high-risk neuroblastoma: a Children's Cancer Group study. J Pediatr Surg. 2004;39:931–936. doi: 10.1016/j.jpedsurg.2004.02.041. [DOI] [PubMed] [Google Scholar]
- Ambros PF, Ambros IM, Brodeur GM, Haber M, Khan J, Nakagawara A, Schleiermacher G, Speleman F, Spitz R, London WB, Cohn SL, Pearson AD, Maris JM. International consensus for neuroblastoma molecular diagnostics: report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br J Cancer. 2009;100:1471–1482. doi: 10.1038/sj.bjc.6605014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoroso L, Erminio G, Makin G, Pearson AD, Brock P, Valteau-Couanet D, Castel V, Pasquet M, Laureys G, Thomas C, Luksch R, Ladenstein R, Haupt R, Garaventa A, Group S. Topotecan-Vincristine-Doxorubicin in Stage 4 High Risk Neuroblastoma Patients Failing to Achieve a Complete Metastatic Response to Rapid COJEC - a SIOPEN Study. Cancer Res Treat. 2017 doi: 10.4143/crt.2016.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes NL, Khakoo Y, Matthay KK, Seeger RC, Stram DO, Gerstner E, Abrey LE, Dalmau J. Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus. J Pediatr Hematol Oncol. 2000;22:315–320. doi: 10.1097/00043426-200007000-00007. [DOI] [PubMed] [Google Scholar]
- Attiyeh EF, London WB, Mosse YP, Wang Q, Winter C, Khazi D, McGrady PW, Seeger RC, Look AT, Shimada H, Brodeur GM, Cohn SL, Matthay KK, Maris JM Children's Oncology G. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005;353:2243–2253. doi: 10.1056/NEJMoa052399. [DOI] [PubMed] [Google Scholar]
- Bagatell R, Beck-Popovic M, London WB, Zhang Y, Pearson AD, Matthay KK, Monclair T, Ambros PF, Cohn SL International Neuroblastoma Risk G. Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol. 2009;27:365–370. doi: 10.1200/JCO.2008.17.9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagatell R, London WB, Wagner LM, Voss SD, Stewart CF, Maris JM, Kretschmar C, Cohn SL. Phase II study of irinotecan and temozolomide in children with relapsed or refractory neuroblastoma: a Children's Oncology Group study. J Clin Oncol. 2011;29:208–213. doi: 10.1200/JCO.2010.31.7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DL, Schmidt ML, Cohn SL, Maris JM, London WB, Buxton A, Stram D, Castleberry RP, Shimada H, Sandler A, Shamberger RC, Look AT, Reynolds CP, Seeger RC, Matthay KK Children's Oncology G. Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med. 2010;363:1313–1323. doi: 10.1056/NEJMoa1001527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiske K, Burchill SA, Cheung IY, Hiyama E, Seeger RC, Cohn SL, Pearson AD, Matthay KK International neuroblastoma Risk Group Task F. Consensus criteria for sensitive detection of minimal neuroblastoma cells in bone marrow, blood and stem cell preparations by immunocytology and QRT-PCR: recommendations by the International Neuroblastoma Risk Group Task Force. Br J Cancer. 2009;100:1627–1637. doi: 10.1038/sj.bjc.6605029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold F, Boos J, Burdach S, Erttmann R, Henze G, Hermann J, Klingebiel T, Kremens B, Schilling FH, Schrappe M, Simon T, Hero B. Myeloablative megatherapy with autologous stem-cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with high-risk neuroblastoma: a randomised controlled trial. Lancet Oncol. 2005;6:649–658. doi: 10.1016/S1470-2045(05)70291-6. [DOI] [PubMed] [Google Scholar]
- Biermann M, Schwarzlmuller T, Fasmer KE, Reitan BC, Johnsen B, Rosendahl K. Is there a role for PET-CT and SPECT-CT in pediatric oncology? Acta Radiol. 2013;54:1037–1045. doi: 10.1258/ar.2012.120616. [DOI] [PubMed] [Google Scholar]
- Bown N, Cotterill S, Lastowska M, O'Neill S, Pearson AD, Plantaz D, Meddeb M, Danglot G, Brinkschmidt C, Christiansen H, Laureys G, Speleman F, Nicholson J, Bernheim A, Betts DR, Vandesompele J, Van Roy N. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med. 1999;340:1954–1961. doi: 10.1056/NEJM199906243402504. [DOI] [PubMed] [Google Scholar]
- Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, Favrot M, Hedborg F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11:1466–1477. doi: 10.1200/JCO.1993.11.8.1466. [DOI] [PubMed] [Google Scholar]
- Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- Burchill SA, Beiske K, Shimada H, Ambros PF, Seeger R, Tytgat GA, Brock PR, Haber M, Park JR, Berthold F. Recommendations for the standardization of bone marrow disease assessment and reporting in children with neuroblastoma on behalf of the International Neuroblastoma Response Criteria Bone Marrow Working Group. Cancer. 2017;123:1095–1105. doi: 10.1002/cncr.30380. [DOI] [PubMed] [Google Scholar]
- Campbell K, Gastier-Foster JM, Mann M, Naranjo AH, Van Ryn C, Bagatell R, Matthay KK, London WB, Irwin MS, Shimada H, Granger MM, Hogarty MD, Park JR, DuBois SG. Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children's Oncology Group. Cancer. 2017 doi: 10.1002/cncr.30873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanthery YH, Gustafson WC, Itsara M, Persson A, Hackett CS, Grimmer M, Charron E, Yakovenko S, Kim G, Matthay KK, Weiss WA. Paracrine signaling through MYCN enhances tumor-vascular interactions in neuroblastoma. Sci Transl Med. 2012;4:115ra113. doi: 10.1126/scitranslmed.3002977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, Mano H, Ogawa S. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, Heguy A, Pappo AS, Federico S, Dalton J, Cheung IY, Ding L, Fulton R, Wang J, Chen X, Becksfort J, Wu J, Billups CA, Ellison D, Mardis ER, Wilson RK, Downing JR, Dyer MA St Jude Children's Research Hospital-Washington University Pediatric Cancer Genome P. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012;307:1062–1071. doi: 10.1001/jama.2012.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, Mosseri V, Simon T, Garaventa A, Castel V, Matthay KK. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:289–297. doi: 10.1200/JCO.2008.16.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, Diskin SJ, Attiyeh EF, Sennett R, Norris G, Laudenslager M, Wood AC, Mayes PA, Jagannathan J, Winter C, Mosse YP, Maris JM. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci U S A. 2011;108:3336–3341. doi: 10.1073/pnas.1012351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bernardi B, Gerrard M, Boni L, Rubie H, Canete A, Di Cataldo A, Castel V, Forjaz de Lacerda A, Ladenstein R, Ruud E, Brichard B, Couturier J, Ellershaw C, Munzer C, Bruzzi P, Michon J, Pearson AD. Excellent outcome with reduced treatment for infants with disseminated neuroblastoma without MYCN gene amplification. J Clin Oncol. 2009;27:1034–1040. doi: 10.1200/JCO.2008.17.5877. [DOI] [PubMed] [Google Scholar]
- Decarolis B, Schneider C, Hero B, Simon T, Volland R, Roels F, Dietlein M, Berthold F, Schmidt M. Iodine-123 metaiodobenzylguanidine scintigraphy scoring allows prediction of outcome in patients with stage 4 neuroblastoma: results of the Cologne interscore comparison study. J Clin Oncol. 2013;31:944–951. doi: 10.1200/JCO.2012.45.8794. [DOI] [PubMed] [Google Scholar]
- Defferrari R, Mazzocco K, Ambros IM, Ambros PF, Bedwell C, Beiske K, Benard J, Berbegall AP, Bown N, Combaret V, Couturier J, Erminio G, Gambini C, Garaventa A, Gross N, Haupt R, Kohler J, Jeison M, Lunec J, Marques B, Martinsson T, Noguera R, Parodi S, Schleiermacher G, Tweddle DA, Valent A, Van Roy N, Vicha A, Villamon E, Tonini GP. Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer. 2015;112:290–295. doi: 10.1038/bjc.2014.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giannatale A, Dias-Gastellier N, Devos A, Mc Hugh K, Boubaker A, Courbon F, Verschuur A, Ducassoul S, Malekzadeh K, Casanova M, Amoroso L, Chastagner P, Zwaan CM, Munzer C, Aerts I, Landman-Parker J, Riccardi R, Le Deley MC, Geoerger B, Rubie H. Phase II study of temozolomide in combination with topotecan (TOTEM) in relapsed or refractory neuroblastoma: a European Innovative Therapies for Children with Cancer-SIOP-European Neuroblastoma study. Eur J Cancer. 2014;50:170–177. doi: 10.1016/j.ejca.2013.08.012. [DOI] [PubMed] [Google Scholar]
- Dubois SG, Geier E, Batra V, Yee SW, Neuhaus J, Segal M, Martinez D, Pawel B, Yanik G, Naranjo A, London WB, Kreissman S, Baker D, Attiyeh E, Hogarty MD, Maris JM, Giacomini K, Matthay KK. Evaluation of Norepinephrine Transporter Expression and Metaiodobenzylguanidine Avidity in Neuroblastoma: A Report from the Children's Oncology Group. Int J Mol Imaging. 2012;2012:250834. doi: 10.1155/2012/250834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBois SG, Groshen S, Park JR, Haas-Kogan DA, Yang X, Geier E, Chen E, Giacomini K, Weiss B, Cohn SL, Granger MM, Yanik GA, Hawkins R, Courtier J, Jackson H, Goodarzian F, Shimada H, Czarnecki S, Tsao-Wei D, Villablanca JG, Marachelian A, Matthay KK. Phase I Study of Vorinostat as a Radiation Sensitizer with 131I-Metaiodobenzylguanidine (131I-MIBG) for Patients with Relapsed or Refractory Neuroblastoma. Clin Cancer Res. 2015;21:2715–2721. doi: 10.1158/1078-0432.CCR-14-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBois SG, Kalika Y, Lukens JN, Brodeur GM, Seeger RC, Atkinson JB, Haase GM, Black CT, Perez C, Shimada H, Gerbing R, Stram DO, Matthay KK. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J Pediatr Hematol Oncol. 1999;21:181–189. doi: 10.1097/00043426-199905000-00005. [DOI] [PubMed] [Google Scholar]
- DuBois SG, Marachelian A, Fox E, Kudgus RA, Reid JM, Groshen S, Malvar J, Bagatell R, Wagner L, Maris JM, Hawkins R, Courtier J, Lai H, Goodarzian F, Shimada H, Czarnecki S, Tsao-Wei D, Matthay KK, Mosse YP. Phase I Study of the Aurora A Kinase Inhibitor Alisertib in Combination With Irinotecan and Temozolomide for Patients With Relapsed or Refractory Neuroblastoma: A NANT (New Approaches to Neuroblastoma Therapy) Trial. J Clin Oncol. 2016;34:1368–1375. doi: 10.1200/JCO.2015.65.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdreich-Epstein A, Singh AR, Joshi S, Vega FM, Guo P, Xu J, Groshen S, Ye W, Millard M, Campan M, Morales G, Garlich JR, Laird PW, Seeger RC, Shimada H, Durden DL. Association of high microvessel alphavbeta3 and low PTEN with poor outcome in stage 3 neuroblastoma: rationale for using first in class dual PI3K/BRD4 inhibitor, SF1126. Oncotarget. 2017;8:52193–52210. doi: 10.18632/oncotarget.13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evageliou NF, Haber M, Vu A, Laetsch TW, Murray J, Gamble LD, Cheng NC, Liu K, Reese M, Corrigan KA, Ziegler DS, Webber H, Hayes CS, Pawel B, Marshall GM, Zhao H, Gilmour SK, Norris MD, Hogarty MD. Polyamine Antagonist Therapies Inhibit Neuroblastoma Initiation and Progression. Clin Cancer Res. 2016;22:4391–4404. doi: 10.1158/1078-0432.CCR-15-2539. [DOI] [PubMed] [Google Scholar]
- Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P, Meagher M, Shafer A, Ng CY, Leung W, Janssen WE, Wu J, Mao S, Brennan RC, Santana VM, Pappo AS, Furman WL. A Pilot Trial of Humanized Anti-GD2 Monoclonal Antibody (hu14.18K322A) with Chemotherapy and Natural Killer Cells in Children with Recurrent/Refractory Neuroblastoma. Clin Cancer Res. 2017;23:6441–6449. doi: 10.1158/1078-0432.CCR-17-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouladi M, Park JR, Stewart CF, Gilbertson RJ, Schaiquevich P, Sun J, Reid JM, Ames MM, Speights R, Ingle AM, Zwiebel J, Blaney SM, Adamson PC. Pediatric phase I trial and pharmacokinetic study of vorinostat: a Children's Oncology Group phase I consortium report. J Clin Oncol. 2010;28:3623–3629. doi: 10.1200/JCO.2009.25.9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franscini LC, Vazquez-Montes M, Buclin T, Perera R, Dunand M, Grouzmann E, Beck-Popovic M. Pediatric reference intervals for plasma free and total metanephrines established with a parametric approach: relevance to the diagnosis of neuroblastoma. Pediatr Blood Cancer. 2015;62:587–593. doi: 10.1002/pbc.25385. [DOI] [PubMed] [Google Scholar]
- Gains JE, Bomanji JB, Fersht NL, Sullivan T, D'Souza D, Sullivan KP, Aldridge M, Waddington W, Gaze MN. 177Lu-DOTATATE molecular radiotherapy for childhood neuroblastoma. J Nucl Med. 2011;52:1041–1047. doi: 10.2967/jnumed.110.085100. [DOI] [PubMed] [Google Scholar]
- Gauguet JM, Pace-Emerson T, Grant FD, Shusterman S, DuBois SG, Frazier AL, Voss SD. Evaluation of the utility of 99m Tc-MDP bone scintigraphy versus MIBG scintigraphy and cross-sectional imaging for staging patients with neuroblastoma. Pediatr Blood Cancer. 2017;64 doi: 10.1002/pbc.26601. [DOI] [PubMed] [Google Scholar]
- Gaze MN, Chang YC, Flux GD, Mairs RJ, Saran FH, Meller ST. Feasibility of dosimetry-based high-dose 131I-meta-iodobenzylguanidine with topotecan as a radiosensitizer in children with metastatic neuroblastoma. Cancer Biother Radiopharm. 2005;20:195–199. doi: 10.1089/cbr.2005.20.195. [DOI] [PubMed] [Google Scholar]
- George RE, Li S, Medeiros-Nancarrow C, Neuberg D, Marcus K, Shamberger RC, Pulsipher M, Grupp SA, Diller L. High-risk neuroblastoma treated with tandem autologous peripheral-blood stem cell-supported transplantation: long-term survival update. J Clin Oncol. 2006;24:2891–2896. doi: 10.1200/JCO.2006.05.6986. [DOI] [PubMed] [Google Scholar]
- George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, Webb TR, Gray NS, Gilliland DG, Diller L, Greulich H, Morris SW, Meyerson M, Look AT. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, Simonds EF, Seeger R, Matthay KK, Hertz NT, Eilers M, Shokat KM, Weiss WA. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell. 2014;26:414–427. doi: 10.1016/j.ccr.2014.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachitanda Y, Tsuneyoshi M, Enjoji M. Expression of pan-neuroendocrine proteins in 53 neuroblastic tumors. An immunohistochemical study with neuron-specific enolase, chromogranin, and synaptophysin. Arch Pathol Lab Med. 1989;113:381–384. [PubMed] [Google Scholar]
- Hann HW, Evans AE, Siegel SE, Wong KY, Sather H, Dalton A, Hammond D, Seeger RC. Prognostic importance of serum ferritin in patients with Stages III and IV neuroblastoma: the Childrens Cancer Study Group experience. Cancer Res. 1985;45:2843–2848. [PubMed] [Google Scholar]
- Hann HW, Levy HM, Evans AE. Serum ferritin as a guide to therapy in neuroblastoma. Cancer Res. 1980;40:1411–1413. [PubMed] [Google Scholar]
- Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, Liu H, Wu MF, Mei Z, Gee A, Mehta B, Zhang H, Mahmood N, Tashiro H, Heslop HE, Dotti G, Rooney CM, Brenner MK. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther. 2017;25:2214–2224. doi: 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henssen A, Althoff K, Odersky A, Beckers A, Koche R, Speleman F, Schafers S, Bell E, Nortmeyer M, Westermann F, De Preter K, Florin A, Heukamp L, Spruessel A, Astrahanseff K, Lindner S, Sadowski N, Schramm A, Astorgues-Xerri L, Riveiro ME, Eggert A, Cvitkovic E, Schulte JH. Targeting MYCN-Driven Transcription By BET-Bromodomain Inhibition. Clin Cancer Res. 2016;22:2470–2481. doi: 10.1158/1078-0432.CCR-15-1449. [DOI] [PubMed] [Google Scholar]
- Hero B, Schleiermacher G. Update on pediatric opsoclonus myoclonus syndrome. Neuropediatrics. 2013;44:324–329. doi: 10.1055/s-0033-1358604. [DOI] [PubMed] [Google Scholar]
- Hero B, Simon T, Spitz R, Ernestus K, Gnekow AK, Scheel-Walter HG, Schwabe D, Schilling FH, Benz-Bohm G, Berthold F. Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol. 2008;26:1504–1510. doi: 10.1200/JCO.2007.12.3349. [DOI] [PubMed] [Google Scholar]
- Hisashige A, Group NBSE. Effectiveness of nationwide screening program for neuroblastoma in Japan. Glob J Health Sci. 2014;6:94–106. doi: 10.5539/gjhs.v6n4p94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiyama E, Iehara T, Sugimoto T, Fukuzawa M, Hayashi Y, Sasaki F, Sugiyama M, Kondo S, Yoneda A, Yamaoka H, Tajiri T, Akazawa K, Ohtaki M. Effectiveness of screening for neuroblastoma at 6 months of age: a retrospective population-based cohort study. Lancet. 2008;371:1173–1180. doi: 10.1016/S0140-6736(08)60523-1. [DOI] [PubMed] [Google Scholar]
- Iehara T, Hamazaki M, Tajiri T, Kawano Y, Kaneko M, Ikeda H, Hosoi H, Sugimoto T, Sawada T Japanese Infantile Neuroblastoma Cooperative Study G. Successful treatment of infants with localized neuroblastoma based on their MYCN status. Int J Clin Oncol. 2013;18:389–395. doi: 10.1007/s10147-012-0391-y. [DOI] [PubMed] [Google Scholar]
- Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, Li Y, Zou HY, Lee NV, Smeal T, Lemmon MA, Mosse YP. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov. 2016;6:96–107. doi: 10.1158/2159-8290.CD-15-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin MS, Park JR. Neuroblastoma: paradigm for precision medicine. Pediatr Clin North Am. 2015;62:225–256. doi: 10.1016/j.pcl.2014.09.015. [DOI] [PubMed] [Google Scholar]
- Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, Michon J, Lyonnet S, Amiel J, Delattre O. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- Kakodkar NC, Peddinti RR, Tian Y, Guerrero LJ, Chlenski A, Yang Q, Salwen HR, Maitland ML, Cohn SL. Sorafenib inhibits neuroblastoma cell proliferation and signaling, blocks angiogenesis, and impairs tumor growth. Pediatr Blood Cancer. 2012;59:642–647. doi: 10.1002/pbc.24004. [DOI] [PubMed] [Google Scholar]
- Kerbl R, Urban CE, Ambros IM, Dornbusch HJ, Schwinger W, Lackner H, Ladenstein R, Strenger V, Gadner H, Ambros PF. Neuroblastoma mass screening in late infancy: insights into the biology of neuroblastic tumors. J Clin Oncol. 2003;21:4228–4234. doi: 10.1200/JCO.2003.10.168. [DOI] [PubMed] [Google Scholar]
- Kohler JA, Rubie H, Castel V, Beiske K, Holmes K, Gambini C, Casale F, Munzer C, Erminio G, Parodi S, Navarro S, Marquez C, Peuchmaur M, Cullinane C, Brock P, Valteau-Couanet D, Garaventa A, Haupt R. Treatment of children over the age of one year with unresectable localised neuroblastoma without MYCN amplification: results of the SIOPEN study. Eur J Cancer. 2013;49:3671–3679. doi: 10.1016/j.ejca.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Kreissman SG, Seeger RC, Matthay KK, London WB, Sposto R, Grupp SA, Haas-Kogan DA, Laquaglia MP, Yu AL, Diller L, Buxton A, Park JR, Cohn SL, Maris JM, Reynolds CP, Villablanca JG. Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol. 2013;14:999–1008. doi: 10.1016/S1470-2045(13)70309-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner BH, Cheung IY, Modak S, Kramer K, Ragupathi G, Cheung NK. Phase I trial of a bivalent gangliosides vaccine in combination with beta-glucan for high-risk neuroblastoma in second or later remission. Clin Cancer Res. 2014;20:1375–1382. doi: 10.1158/1078-0432.CCR-13-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner BH, Kramer K, Modak S, Cheung NK. Irinotecan plus temozolomide for relapsed or refractory neuroblastoma. J Clin Oncol. 2006;24:5271–5276. doi: 10.1200/JCO.2006.06.7272. [DOI] [PubMed] [Google Scholar]
- Ladenstein R, Philip T, Lasset C, Hartmann O, Garaventa A, Pinkerton R, Michon J, Pritchard J, Klingebiel T, Kremens B, Pearson A, Coze C, Paolucci P, Frappaz D, Gadner H, Chauvin F. Multivariate analysis of risk factors in stage 4 neuroblastoma patients over the age of one year treated with megatherapy and stem-cell transplantation: a report from the European Bone Marrow Transplantation Solid Tumor Registry. J Clin Oncol. 1998;16:953–965. doi: 10.1200/JCO.1998.16.3.953. [DOI] [PubMed] [Google Scholar]
- Ladenstein R, Potschger U, Pearson ADJ, Brock P, Luksch R, Castel V, Yaniv I, Papadakis V, Laureys G, Malis J, Balwierz W, Ruud E, Kogner P, Schroeder H, de Lacerda AF, Beck-Popovic M, Bician P, Garami M, Trahair T, Canete A, Ambros PF, Holmes K, Gaze M, Schreier G, Garaventa A, Vassal G, Michon J, Valteau-Couanet D, Group SEN. Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): an international, randomised, multi-arm, open-label, phase 3 trial. Lancet Oncol. 2017;18:500–514. doi: 10.1016/S1470-2045(17)30070-0. [DOI] [PubMed] [Google Scholar]
- Liu YL, Lu MY, Chang HH, Lu CC, Lin DT, Jou ST, Yang YL, Lee YL, Huang SF, Jeng YM, Lee H, Miser JS, Lin KH, Liao YF, Hsu WM, Tzen KY. Diagnostic FDG and FDOPA positron emission tomography scans distinguish the genomic type and treatment outcome of neuroblastoma. Oncotarget. 2016;7:18774–18786. doi: 10.18632/oncotarget.7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London WB, Castleberry RP, Matthay KK, Look AT, Seeger RC, Shimada H, Thorner P, Brodeur G, Maris JM, Reynolds CP, Cohn SL. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children's Oncology Group. J Clin Oncol. 2005;23:6459–6465. doi: 10.1200/JCO.2005.05.571. [DOI] [PubMed] [Google Scholar]
- London WB, Frantz CN, Campbell LA, Seeger RC, Brumback BA, Cohn SL, Matthay KK, Castleberry RP, Diller L. Phase II randomized comparison of topotecan plus cyclophosphamide versus topotecan alone in children with recurrent or refractory neuroblastoma: a Children's Oncology Group study. J Clin Oncol. 2010;28:3808–3815. doi: 10.1200/JCO.2009.27.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marachelian A, Shimada H, Sano H, Jackson H, Stein J, Sposto R, Matthay KK, Baker D, Villablanca JG. The significance of serial histopathology in a residual mass for outcome of intermediate risk stage 3 neuroblastoma. Pediatr Blood Cancer. 2012;58:675–681. doi: 10.1002/pbc.23250. [DOI] [PubMed] [Google Scholar]
- Maris JM. Recent advances in neuroblastoma. The New England Journal of Medicine. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, Weiss WA. Neuroblastoma. Nat Rev Dis Primers. 2016;2:16078. doi: 10.1038/nrdp.2016.78. [DOI] [PubMed] [Google Scholar]
- Matthay KK, Perez C, Seeger RC, Brodeur GM, Shimada H, Atkinson JB, Black CT, Gerbing R, Haase GM, Stram DO, Swift P, Lukens JN. Successful treatment of stage III neuroblastoma based on prospective biologic staging: a Children's Cancer Group study. J Clin Oncol. 1998;16:1256–1264. doi: 10.1200/JCO.1998.16.4.1256. [DOI] [PubMed] [Google Scholar]
- Matthay KK, Shulkin B, Ladenstein R, Michon J, Giammarile F, Lewington V, Pearson AD, Cohn SL. Criteria for evaluation of disease extent by (123)I-metaiodobenzylguanidine scans in neuroblastoma: a report for the International Neuroblastoma Risk Group (INRG) Task Force. Br J Cancer. 2010;102:1319–1326. doi: 10.1038/sj.bjc.6605621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeur GM, Gerbing RB, Reynolds CP. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N Engl J Med. 1999;341:1165–1173. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- Matthay KK, Yanik G, Messina J, Quach A, Huberty J, Cheng SC, Veatch J, Goldsby R, Brophy P, Kersun LS, Hawkins RA, Maris JM. Phase II study on the effect of disease sites, age, and prior therapy on response to iodine-131-metaiodobenzylguanidine therapy in refractory neuroblastoma. J Clin Oncol. 2007;25:1054–1060. doi: 10.1200/JCO.2006.09.3484. [DOI] [PubMed] [Google Scholar]
- Maurer BJ, Kang MH, Villablanca JG, Janeba J, Groshen S, Matthay KK, Sondel PM, Maris JM, Jackson HA, Goodarzian F, Shimada H, Czarnecki S, Hasenauer B, Reynolds CP, Marachelian A. Phase I trial of fenretinide delivered orally in a novel organized lipid complex in patients with relapsed/refractory neuroblastoma: a report from the New Approaches to Neuroblastoma Therapy (NANT) consortium. Pediatr Blood Cancer. 2013;60:1801–1808. doi: 10.1002/pbc.24643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meany HJ, London WB, Ambros PF, Matthay KK, Monclair T, Simon T, Garaventa A, Berthold F, Nakagawara A, Cohn SL, Pearson AD, Park JR. Significance of clinical and biologic features in Stage 3 neuroblastoma: a report from the International Neuroblastoma Risk Group project. Pediatr Blood Cancer. 2014;61:1932–1939. doi: 10.1002/pbc.25134. [DOI] [PubMed] [Google Scholar]
- Miettinen M. Synaptophysin and neurofilament proteins as markers for neuroendocrine tumors. Arch Pathol Lab Med. 1987;111:813–818. [PubMed] [Google Scholar]
- Minturn JE, Evans AE, Villablanca JG, Yanik GA, Park JR, Shusterman S, Groshen S, Hellriegel ET, Bensen-Kennedy D, Matthay KK, Brodeur GM, Maris JM. Phase I trial of lestaurtinib for children with refractory neuroblastoma: a new approaches to neuroblastoma therapy consortium study. Cancer Chemother Pharmacol. 2011;68:1057–1065. doi: 10.1007/s00280-011-1581-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modak S, Kushner BH, Basu E, Roberts SS, Cheung NK. Combination of bevacizumab, irinotecan, and temozolomide for refractory or relapsed neuroblastoma: Results of a phase II study. Pediatr Blood Cancer. 2017;64 doi: 10.1002/pbc.26448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody R, Naranjo A, Van Ryn C, Yu AL, London WB, Shulkin BL, Parisi MT, Servaes SE, Diccianni MB, Sondel PM, Bender JG, Maris JM, Park JR, Bagatell R. Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol. 2017;18:946–957. doi: 10.1016/S1470-2045(17)30355-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, Ebus ME, Haneveld F, Lakeman A, Schild L, Molenaar P, Stroeken P, van Noesel MM, Ora I, Santo EE, Caron HN, Westerhout EM, Versteeg R. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483:589–593. doi: 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, Kaneko M, London WB, Matthay KK, Nuchtern JG, von Schweinitz D, Simon T, Cohn SL, Pearson AD. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:298–303. doi: 10.1200/JCO.2008.16.6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern DA, London WB, Stephens D, Volchenboum SL, Simon T, Nakagawara A, Shimada H, Schleiermacher G, Matthay KK, Cohn SL, Pearson AD, Irwin MS. Prognostic significance of pattern and burden of metastatic disease in patients with stage 4 neuroblastoma: A study from the International Neuroblastoma Risk Group database. Eur J Cancer. 2016;65:1–10. doi: 10.1016/j.ejca.2016.06.005. [DOI] [PubMed] [Google Scholar]
- Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, Hou C, Hakonarson H, Torkamani A, Schork NJ, Brodeur GM, Tonini GP, Rappaport E, Devoto M, Maris JM. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, Rolland D, Balis FM, Maris JM, Weigel BJ, Ingle AM, Ahern C, Adamson PC, Blaney SM. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14:472–480. doi: 10.1016/S1470-2045(13)70095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosse YP, Lipsitz E, Fox E, Teachey DT, Maris JM, Weigel B, Adamson PC, Ingle MA, Ahern CH, Blaney SM. Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: a Children's Oncology Group Phase I Consortium study. Clin Cancer Res. 2012;18:6058–6064. doi: 10.1158/1078-0432.CCR-11-3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullassery D, Farrelly P, Losty PD. Does aggressive surgical resection improve survival in advanced stage 3 and 4 neuroblastoma? A systematic review and meta-analysis. Pediatr Hematol Oncol. 2014;31:703–716. doi: 10.3109/08880018.2014.947009. [DOI] [PubMed] [Google Scholar]
- Nakagawara A, Zaizen Y, Ikeda K, Suita S, Ohgami H, Nagahara N, Sera Y, Akiyama H, Kawakami K, Uchino J. Different genomic and metabolic patterns between mass screening-positive and mass screening-negative later-presenting neuroblastomas. Cancer. 1991;68:2037–2044. doi: 10.1002/1097-0142(19911101)68:9<2037::aid-cncr2820680932>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Navid F, Sondel PM, Barfield R, Shulkin BL, Kaufman RA, Allay JA, Gan J, Hutson P, Seo S, Kim K, Goldberg J, Hank JA, Billups CA, Wu J, Furman WL, McGregor LM, Otto M, Gillies SD, Handgretinger R, Santana VM. Phase I trial of a novel anti-GD2 monoclonal antibody, Hu14.18K322A, designed to decrease toxicity in children with refractory or recurrent neuroblastoma. J Clin Oncol. 2014;32:1445–1452. doi: 10.1200/JCO.2013.50.4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson HJ, Matthay KK, Seeger RC, Brodeur GM, Shimada H, Perez C, Atkinson JB, Selch M, Gerbing RB, Stram DO, Lukens J. Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: a Children's Cancer Group study. J Clin Oncol. 2000;18:477–486. doi: 10.1200/JCO.2000.18.3.477. [DOI] [PubMed] [Google Scholar]
- Nuchtern JG, London WB, Barnewolt CE, Naranjo A, McGrady PW, Geiger JD, Diller L, Schmidt ML, Maris JM, Cohn SL, Shamberger RC. A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg. 2012;256:573–580. doi: 10.1097/SLA.0b013e31826cbbbd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JR, Bagatell R, Cohn SL, Pearson AD, Villablanca JG, Berthold F, Burchill S, Boubaker A, McHugh K, Nuchtern JG, London WB, Seibel NL, Lindwasser OW, Maris JM, Brock P, Schleiermacher G, Ladenstein R, Matthay KK, Valteau-Couanet D. Revisions to the International Neuroblastoma Response Criteria: A Consensus Statement From the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol. 2017;35:2580–2587. doi: 10.1200/JCO.2016.72.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JR, Bagatell R, London WB, Maris JM, Cohn SL, Mattay KK, Hogarty M, Committee COGN. Children's Oncology Group's 2013 blueprint for research: neuroblastoma. Pediatr Blood Cancer. 2013;60:985–993. doi: 10.1002/pbc.24433. [DOI] [PubMed] [Google Scholar]
- Park JR, Kreissman SG, London WB, Naranjo A, Cohn SL, Hogarty MD, Tenney SC, Haas-Kogan D, Shaw PJ, Geiger JD, Doski JJ, Gorges SW, Khanna G, Voss SD, Maris JM, Grupp SA, Diller L. A phase III randomized clinical trial (RCT) of tandem myeloablative autologous stem cell transplant (ASCT) using peripheral blood stem cell (PBSC) as consolidation therapy for high-risk neuroblastoma (HR-NB): A Children's Oncology Group (COG) study. Journal of Clinical Oncology. 2016;34:LBA3–LBA3. [Google Scholar]
- Park JR, Scott JR, Stewart CF, London WB, Naranjo A, Santana VM, Shaw PJ, Cohn SL, Matthay KK. Pilot induction regimen incorporating pharmacokinetically guided topotecan for treatment of newly diagnosed high-risk neuroblastoma: a Children's Oncology Group study. J Clin Oncol. 2011;29:4351–4357. doi: 10.1200/JCO.2010.34.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JR, Villablanca JG, London WB, Gerbing RB, Haas-Kogan D, Adkins ES, Attiyeh EF, Maris JM, Seeger RC, Reynolds CP, Matthay KK Children's Oncology G. Outcome of high-risk stage 3 neuroblastoma with myeloablative therapy and 13-cis-retinoic acid: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2009;52:44–50. doi: 10.1002/pbc.21784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson DM, Gao D, Trahan DN, Johnson BA, Ludwig A, Barbieri E, Chen Z, Diaz-Miron J, Vassilev L, Shohet JM, Kim ES. Effect of MDM2 and vascular endothelial growth factor inhibition on tumor angiogenesis and metastasis in neuroblastoma. Angiogenesis. 2011;14:255–266. doi: 10.1007/s10456-011-9210-8. [DOI] [PubMed] [Google Scholar]
- Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, Kramer A, Roncaioli JL, Sand F, Heuckmann JM, Ikram F, Schmidt R, Ackermann S, Engesser A, Kahlert Y, Vogel W, Altmuller J, Nurnberg P, Thierry-Mieg J, Thierry-Mieg D, Mariappan A, Heynck S, Mariotti E, Henrich KO, Gloeckner C, Bosco G, Leuschner I, Schweiger MR, Savelyeva L, Watkins SC, Shao C, Bell E, Hofer T, Achter V, Lang U, Theissen J, Volland R, Saadati M, Eggert A, de Wilde B, Berthold F, Peng Z, Zhao C, Shi L, Ortmann M, Buttner R, Perner S, Hero B, Schramm A, Schulte JH, Herrmann C, O'Sullivan RJ, Westermann F, Thomas RK, Fischer M. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. 2015;526:700–704. doi: 10.1038/nature14980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peuchmaur M, d'Amore ES, Joshi VV, Hata J, Roald B, Dehner LP, Gerbing RB, Stram DO, Lukens JN, Matthay KK, Shimada H. Revision of the International Neuroblastoma Pathology Classification: confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer. 2003;98:2274–2281. doi: 10.1002/cncr.11773. [DOI] [PubMed] [Google Scholar]
- Pinto NR, Applebaum MA, Volchenboum SL, Matthay KK, London WB, Ambros PF, Nakagawara A, Berthold F, Schleiermacher G, Park JR, Valteau-Couanet D, Pearson AD, Cohn SL. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J Clin Oncol. 2015;33:3008–3017. doi: 10.1200/JCO.2014.59.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard J, Cotterill SJ, Germond SM, Imeson J, de Kraker J, Jones DR. High dose melphalan in the treatment of advanced neuroblastoma: results of a randomised trial (ENSG-1) by the European Neuroblastoma Study Group. Pediatr Blood Cancer. 2005;44:348–357. doi: 10.1002/pbc.20219. [DOI] [PubMed] [Google Scholar]
- Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J, Lawrence MS, Lichenstein L, McKenna A, Pedamallu CS, Ramos AH, Shefler E, Sivachenko A, Sougnez C, Stewart C, Ally A, Birol I, Chiu R, Corbett RD, Hirst M, Jackman SD, Kamoh B, Khodabakshi AH, Krzywinski M, Lo A, Moore RA, Mungall KL, Qian J, Tam A, Thiessen N, Zhao Y, Cole KA, Diamond M, Diskin SJ, Mosse YP, Wood AC, Ji L, Sposto R, Badgett T, London WB, Moyer Y, Gastier-Foster JM, Smith MA, Guidry Auvil JM, Gerhard DS, Hogarty MD, Jones SJ, Lander ES, Gabriel SB, Getz G, Seeger RC, Khan J, Marra MA, Meyerson M, Maris JM. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, Kung AL, Weiss WA, Bradner JE, Stegmaier K. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rounbehler RJ, Li W, Hall MA, Yang C, Fallahi M, Cleveland JL. Targeting ornithine decarboxylase impairs development of MYCN-amplified neuroblastoma. Cancer Res. 2009;69:547–553. doi: 10.1158/0008-5472.CAN-08-2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubie H, De Bernardi B, Gerrard M, Canete A, Ladenstein R, Couturier J, Ambros P, Munzer C, Pearson AD, Garaventa A, Brock P, Castel V, Valteau-Couanet D, Holmes K, Di Cataldo A, Brichard B, Mosseri V, Marquez C, Plantaz D, Boni L, Michon J. Excellent outcome with reduced treatment in infants with nonmetastatic and unresectable neuroblastoma without MYCN amplification: results of the prospective INES 99.1. J Clin Oncol. 2011;29:449–455. doi: 10.1200/JCO.2010.29.5196. [DOI] [PubMed] [Google Scholar]
- Rudnick E, Khakoo Y, Antunes NL, Seeger RC, Brodeur GM, Shimada H, Gerbing RB, Stram DO, Matthay KK. Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: clinical outcome and antineuronal antibodies-a report from the Children's Cancer Group Study. Med Pediatr Oncol. 2001;36:612–622. doi: 10.1002/mpo.1138. [DOI] [PubMed] [Google Scholar]
- Russell MR, Levin K, Rader J, Belcastro L, Li Y, Martinez D, Pawel B, Shumway SD, Maris JM, Cole KA. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013;73:776–784. doi: 10.1158/0008-5472.CAN-12-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarinen-Pihkala UM, Hovi L, Koivusalo A, Jahnukainen K, Karikoski R, Sariola H, Wikstrom S. Thiotepa and melphalan based single, tandem, and triple high dose therapy and autologous stem cell transplantation for high risk neuroblastoma. Pediatr Blood Cancer. 2012;59:1190–1197. doi: 10.1002/pbc.24173. [DOI] [PubMed] [Google Scholar]
- Saulnier Sholler GL, Gerner EW, Bergendahl G, MacArthur RB, VanderWerff A, Ashikaga T, Bond JP, Ferguson W, Roberts W, Wada RK, Eslin D, Kraveka JM, Kaplan J, Mitchell D, Parikh NS, Neville K, Sender L, Higgins T, Kawakita M, Hiramatsu K, Moriya SS, Bachmann AS. A Phase I Trial of DFMO Targeting Polyamine Addiction in Patients with Relapsed/Refractory Neuroblastoma. PLoS One. 2015;10:e0127246. doi: 10.1371/journal.pone.0127246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada T, Todo S, Fujita K, Iino S, Imashuku S, Kusunoki T. Mass screening of neuroblastoma in infancy. Am J Dis Child. 1982;136:710–712. doi: 10.1001/archpedi.1982.03970440054015. [DOI] [PubMed] [Google Scholar]
- Schilling FH, Spix C, Berthold F, Erttmann R, Fehse N, Hero B, Klein G, Sander J, Schwarz K, Treuner J, Zorn U, Michaelis J. Neuroblastoma screening at one year of age. N Engl J Med. 2002;346:1047–1053. doi: 10.1056/NEJMoa012277. [DOI] [PubMed] [Google Scholar]
- Schilling FH, Spix C, Berthold F, Erttmann R, Sander J, Treuner J, Michaelis J. Children may not benefit from neuroblastoma screening at 1 year of age. Updated results of the population based controlled trial in Germany. Cancer Lett. 2003;197:19–28. doi: 10.1016/s0304-3835(03)00077-6. [DOI] [PubMed] [Google Scholar]
- Schleiermacher G, Mosseri V, London WB, Maris JM, Brodeur GM, Attiyeh E, Haber M, Khan J, Nakagawara A, Speleman F, Noguera R, Tonini GP, Fischer M, Ambros I, Monclair T, Matthay KK, Ambros P, Cohn SL, Pearson AD. Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the INRG project. Br J Cancer. 2012;107:1418–1422. doi: 10.1038/bjc.2012.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. 1985;313:1111–1116. doi: 10.1056/NEJM198510313131802. [DOI] [PubMed] [Google Scholar]
- Seif AE, Naranjo A, Baker DL, Bunin NJ, Kletzel M, Kretschmar CS, Maris JM, McGrady PW, von Allmen D, Cohn SL, London WB, Park JR, Diller LR, Grupp SA. A pilot study of tandem high-dose chemotherapy with stem cell rescue as consolidation for high-risk neuroblastoma: Children's Oncology Group study ANBL00P1. Bone Marrow Transplant. 2013;48:947–952. doi: 10.1038/bmt.2012.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp SE, Shulkin BL, Gelfand MJ, Salisbury S, Furman WL. 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. J Nucl Med. 2009;50:1237–1243. doi: 10.2967/jnumed.108.060467. [DOI] [PubMed] [Google Scholar]
- Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, Stram DO, Gerbing RB, Lukens JN, Matthay KK, Castleberry RP. The International Neuroblastoma Pathology Classification (the Shimada system) Cancer. 1999;86:364–372. [PubMed] [Google Scholar]
- Shimada H, Umehara S, Monobe Y, Hachitanda Y, Nakagawa A, Goto S, Gerbing RB, Stram DO, Lukens JN, Matthay KK. International neuroblastoma pathology classification for prognostic evaluation of patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer. 2001;92:2451–2461. doi: 10.1002/1097-0142(20011101)92:9<2451::aid-cncr1595>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Shinagawa T, Kitamura T, Katanoda K, Matsuda T, Ito Y, Sobue T. The incidence and mortality rates of neuroblastoma cases before and after the cessation of the mass screening program in Japan: A descriptive study. Int J Cancer. 2017;140:618–625. doi: 10.1002/ijc.30482. [DOI] [PubMed] [Google Scholar]
- Shuster JJ, McWilliams NB, Castleberry R, Nitschke R, Smith EI, Altshuler G, Kun L, Brodeur G, Joshi V, Vietti T, et al. Serum lactate dehydrogenase in childhood neuroblastoma. A Pediatric Oncology Group recursive partitioning study. Am J Clin Oncol. 1992;15:295–303. doi: 10.1097/00000421-199208000-00004. [DOI] [PubMed] [Google Scholar]
- Shusterman S, London WB, Gillies SD, Hank JA, Voss SD, Seeger RC, Reynolds CP, Kimball J, Albertini MR, Wagner B, Gan J, Eickhoff J, DeSantes KB, Cohn SL, Hecht T, Gadbaw B, Reisfeld RA, Maris JM, Sondel PM. Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a Children's Oncology Group (COG) phase II study. J Clin Oncol. 2010;28:4969–4975. doi: 10.1200/JCO.2009.27.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert N, Eger C, Seidel D, Juttner M, Zumpe M, Wegner D, Kietz S, Ehlert K, Veal GJ, Siegmund W, Weiss M, Loibner H, Ladenstein R, Lode HN. Pharmacokinetics and pharmacodynamics of ch14.18/CHO in relapsed/refractory high-risk neuroblastoma patients treated by long-term infusion in combination with IL-2. MAbs. 2016;8:604–616. doi: 10.1080/19420862.2015.1130196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon T, Haberle B, Hero B, von Schweinitz D, Berthold F. Role of Surgery in the Treatment of Patients With Stage 4 Neuroblastoma Age 18 Months or Older at Diagnosis. Journal of Clinical Oncology. 2013;31:752–758. doi: 10.1200/JCO.2012.45.9339. [DOI] [PubMed] [Google Scholar]
- Stiller CA, Parkin DM. International variations in the incidence of neuroblastoma. Int J Cancer. 1992;52:538–543. doi: 10.1002/ijc.2910520407. [DOI] [PubMed] [Google Scholar]
- Strother DR, London WB, Schmidt ML, Brodeur GM, Shimada H, Thorner P, Collins MH, Tagge E, Adkins S, Reynolds CP, Murray K, Lavey RS, Matthay KK, Castleberry R, Maris JM, Cohn SL. Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol. 2012;30:1842–1848. doi: 10.1200/JCO.2011.37.9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart DR, London WB, Schmidt ML, DuBois SG, Monclair TF, Nakagawara A, De Bernardi B, Ambros PF, Pearson AD, Cohn SL, Matthay KK. Prognostic value of the stage 4S metastatic pattern and tumor biology in patients with metastatic neuroblastoma diagnosed between birth and 18 months of age. J Clin Oncol. 2011;29:4358–4364. doi: 10.1200/JCO.2011.35.9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talleur AC, Triplett BM, Federico S, Mamcarz E, Janssen W, Wu J, Shook D, Leung W, Furman WL. Consolidation Therapy for Newly Diagnosed Pediatric Patients with High-Risk Neuroblastoma Using Busulfan/Melphalan, Autologous Hematopoietic Cell Transplantation, Anti-GD2 Antibody, Granulocyte-Macrophage Colony-Stimulating Factor, Interleukin-2, and Haploidentical Natural Killer Cells. Biol Blood Marrow Transplant. 2017;23:1910–1917. doi: 10.1016/j.bbmt.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trochet D, Bourdeaut F, Janoueix-Lerosey I, Deville A, de Pontual L, Schleiermacher G, Coze C, Philip N, Frebourg T, Munnich A, Lyonnet S, Delattre O, Amiel J. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet. 2004;74:761–764. doi: 10.1086/383253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5:25–34. doi: 10.1158/2159-8290.CD-14-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo KT, Matthay KK, Neuhaus J, London WB, Hero B, Ambros PF, Nakagawara A, Miniati D, Wheeler K, Pearson AD, Cohn SL, DuBois SG. Clinical, biologic, and prognostic differences on the basis of primary tumor site in neuroblastoma: a report from the international neuroblastoma risk group project. J Clin Oncol. 2014;32:3169–3176. doi: 10.1200/JCO.2014.56.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Allmen D, Davidoff AM, London WB, Van Ryn C, Haas-Kogan DA, Kreissman SG, Khanna G, Rosen N, Park JR, La Quaglia MP. Impact of Extent of Resection on Local Control and Survival in Patients From the COG A3973 Study With High-Risk Neuroblastoma. J Clin Oncol. 2017;35:208–216. doi: 10.1200/JCO.2016.67.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner LM, Adams VR. Targeting the PD-1 pathway in pediatric solid tumors and brain tumors. Onco Targets Ther. 2017;10:2097–2106. doi: 10.2147/OTT.S124008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JS, Gains JE, Moroz V, Wheatley K, Gaze MN. A systematic review of 131I-meta iodobenzylguanidine molecular radiotherapy for neuroblastoma. Eur J Cancer. 2014;50:801–815. doi: 10.1016/j.ejca.2013.11.016. [DOI] [PubMed] [Google Scholar]
- Woods WG, Gao RN, Shuster JJ, Robison LL, Bernstein M, Weitzman S, Bunin G, Levy I, Brossard J, Dougherty G, Tuchman M, Lemieux B. Screening of infants and mortality due to neuroblastoma. N Engl J Med. 2002;346:1041–1046. doi: 10.1056/NEJMoa012387. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Hayashi Y, Hanada R, Kikuchi A, Ichikawa M, Tanimura M, Yoshioka S. Mass screening and age-specific incidence of neuroblastoma in Saitama Prefecture, Japan. J Clin Oncol. 1995;13:2033–2038. doi: 10.1200/JCO.1995.13.8.2033. [DOI] [PubMed] [Google Scholar]
- Yanik G, Naranjo A, Parisi MT, Shulkin BL, Nadel H, Gelfand MJ, Ladenstein R, Boubaker A, Poetschger U, Valteau-Couanet D, Kreissman SG, Park JR, Matthay KK. Impact of Post-Induction Curie Scores in High-Risk Neuroblastoma. Biology of Blood and Marrow Transplantation. 2015;21:S107–S107. [Google Scholar]
- Yanik GA, Parisi MT, Naranjo A, Nadel H, Gelfand MJ, Park JR, Ladenstein RL, Poetschger U, Boubaker A, Valteau-Couanet D, Lambert B, Castellani MR, Bar-Sever Z, Oudoux A, Kaminska A, Kreissman SG, Shulkin BL, Matthay KK. Validation of post-induction Curie scores in high risk neuroblastoma. A Children's Oncology Group (COG) and SIOPEN group report on SIOPEN/HR-NBL1. J Nucl Med. 2017 doi: 10.2967/jnumed.117.195883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanik GA, Parisi MT, Shulkin BL, Naranjo A, Kreissman SG, London WB, Villablanca JG, Maris JM, Park JR, Cohn SL, McGrady P, Matthay KK. Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med. 2013;54:541–548. doi: 10.2967/jnumed.112.112334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, Shimada H, Grupp SA, Seeger R, Reynolds CP, Buxton A, Reisfeld RA, Gillies SD, Cohn SL, Maris JM, Sondel PM Children's Oncology G. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou MJ, Doral MY, DuBois SG, Villablanca JG, Yanik GA, Matthay KK. Different outcomes for relapsed versus refractory neuroblastoma after therapy with (131)I-metaiodobenzylguanidine ((131)I-MIBG) Eur J Cancer. 2015;51:2465–2472. doi: 10.1016/j.ejca.2015.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]