

Abstract
Bone marrow derived mesenchymal stromal cells (BM-MSCs) have been shown to have significant therapeutic effects in experimental models of pneumonia and lung injury. The current study examined the roles of the toll like receptor 4 (TLR4) and protease activated receptor 1 (PAR1) pathways on MSC survival and therapeutic activity in a murine model of pneumonia. MSCs from TLR4 −/− and R41Q-PAR1 mutated mice were isolated to test the effect of mutating these specific pathways on MSC survival when exposed to cytotoxic stimuli in vitro. An E. coli pneumonia model was utilized to assess the effect of these specific pathways on MSC therapeutic activity in vivo. Our results showed that mutation of either the TLR4 or PAR1 pathways in MSCs impaired cell survival under conditions of inflammatory stress in vitro, and eliminated their therapeutic efficacy in vivo. Also, stimulation of the TLR4 pathway on MSCs led to secretion of low levels of prothrombin by MSCs, while disrupting the TLR4 pathway impaired canonical signaling through PAR1 in response to thrombin. Therefore, this study demonstrates that both TLR4 and PAR1 are required for MSC survival under inflammatory conditions in vitro and therapeutic capacity in vivo, and that the TLR4 pathway regulates signaling through PAR1 on MSCs.
Keywords: mesenchymal stem cells, lung injury, pneumonia, protease-activated receptor, toll-like receptor
Graphical Abstract
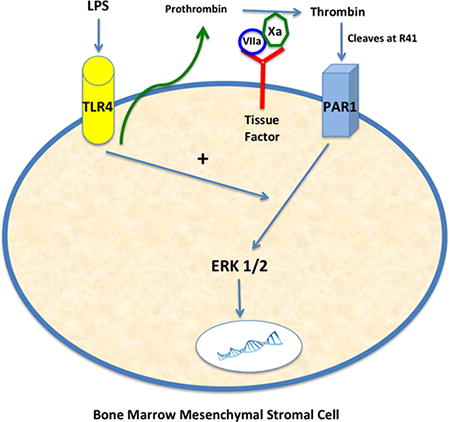
Figure. Schematic demonstrating interaction between TLR4 and PAR1 pathways on MSCs. LPS stimulation of TLR4 on MSCs leads to low levels of prothrombin secretion, which can then be converted to active thrombin by factor Xa bound by tissue factor (expressed by MSCs). Thrombin can then cleave PAR1 on MSCs and lead to ERK1/2 activation, in the presence of an intact TLR4 pathway, and a pro-survival phenotype.
Introduction
Bone marrow mesenchymal stromal cells (BM-MSCs) make up the supportive environment for hematopoietic stem cells and have the capacity to differentiate into bone, fat and cartilage. MSCs have been extensively studied as a potential therapy for lung injury and sepsis, and have been shown to have several properties that facilitate recovery from lung infection and injury such as immunomodulation, secretion of reparative growth factors, and augmentation of host defense to infection [1–11]. Based on these promising pre-clinical data, efforts have begun to translate MSCs to patients with lung injury [12,13]. However, further investigation is needed to determine how MSC survival is regulated in an inflammatory environment in order to optimize therapeutic capacity, since prior work has demonstrated non-viable MSCs exert no protective effect [2].
The goal of this study was to investigate the role of the toll like receptor-4 (TLR4) and protease activated receptor-1 (PAR1) pathways on the therapeutic capacity of MSCs in an experimental model of Gram-negative bacterial pneumonia. These particular pathways were chosen as the focus of investigation given the principal role of TLR4 and PAR1 in inflammation and coagulation, respectively, in response to infectious stimuli. Furthermore, since inflammation and coagulation, in response to infection, are often geographically coupled within the host, we hypothesized that TLR4 and PAR1 would be co-regulated on MSCs. There have been published reports linking different PARs and TLRs in various experimental models, but the TLR4-PAR1 axis on MSCs has not been reported on to date [14–20]. There was a particular focus on understanding how these pathways regulate MSC survival under inflammatory conditions.
TLRs are expressed by MSCs and have been shown to regulate a range of functions including migration and immunomodulation, as well as cell survival [21–29]. The role of TLR4, in particular, was studied since it is the primary TLR responsible for recognizing LPS on Gram-negative bacteria. In order to investigate this pathway, MSCs from TLR4 −/− mice were isolated and tested in both an in vivo infection model as well as in vitro assays of cell death.
PARs are G-protein coupled receptors that are cleaved predominantly by coagulation-based serine proteases. PAR1 is classically cleaved by thrombin near the N-terminus of the extracellular tail at residue 41, leaving a self-tethered ligand that binds to the active site of the receptor. The role of PAR1 on MSC survival and therapeutic activity has been minimally investigated to date [30, 31]. In the current study, the PAR1 pathway was interrogated in vivo and in vitro using a combination of genetically modified cells (MSCs from R41Q-PAR1 mutated mice) as well as pharmacological agents specific for PAR1 [32]. PAR1 can be cleaved by several enzymes, however, as mentioned, thrombin cleaves specifically at residue 41 [33]. The use of the genetically modified R41Q-PAR1 mutant MSCs allowed for specific analysis of the role of thrombin cleavage of PAR1 on MSC survival and activity.
Since MSCs are reparative in response to inflammatory stimuli and the activation of inflammation and coagulation are often co-regulated, our hypotheses were that both TLR4 and PAR1 would be required for MSC survival and that TLR4 and PAR1 signaling would be coupled. Our results substantiated these hypotheses by showing that both TLR4 and PAR1 mediate MSC survival and therapeutic capacity, and that TLR4 stimulation controls secretion of prothrombin as well as PAR1 signaling in MSCs.
Methods
Isolation and culture of bone marrow derived mesenchymal stem cells
MSCs were isolated from WT, TLR4 −/−, and R41Q-PAR1 mutated mice as described before. Characterization was done as previously described to determine if cells met minimal, essential criteria for MSCs (see supplementary material for differentiation assays and flow cytometry methods) [2,5,34].
MSCs were cultured in α-MEM media (Gibco, catalog #12561) supplemented with 15% FBS (Gibco, catalog #12662-029) and 1% Pen-Strep and L-Glutamine. Cells were split at 70–80% confluency and seeded into T-150cm2 flasks at approximately 150,000 cells per flask. Media was changed every 3–4 days and cells were split every 6–7 days. MSCs were used for experiments between passages 5–10.
In vivo E. coli pneumonia model and experimental design
All mice used for these experiments were male C57BL/6J (Jackson Labs) between the ages of 10–15 weeks of age. All experiments were approved by the University of California, San Diego (UCSD) Institutional Animal Care and Use Committee (IACUC), and mice were housed in a UCSD facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) before and during the experiment. The survival experiments, using death as the endpoint, were approved by the UCSD IACUC. In non-survival experiments, mice were euthanized with CO2 inhalation and then cervical dislocation.
E. coli K1 strain (originally isolated from the blood of a patient with biliary sepsis; provided by Xiao Su, MD, PhD, Pasteur Institute-Shanghai) was used for all in vivo experiments, and the experimental design is as we have previously described [2,5]. To briefly summarize, E. coli was grown in LB broth for 16–17 h overnight at 37°C, and the following morning the E. coli bacteria were washed and diluted first to OD = 1 at 600 nM. The E. coli was then diluted to 1 × 106 colony-forming units (cfu) in 25 µl of PBS. Subsequently, mice were anesthetized with ketamine and xylazine (90 mg/kg and 10 mg/kg i.p., respectively), and placed on an intubating stand with a fiberoptic light over their neck to transilluminate the trachea. Then, the mouth and oropharynx were opened with a forceps, and the E. coli instillate (1 × 106 cfu/25 µl PBS) was injected through the vocal cords into the trachea. The mice were then recovered until they regained activity. After 4–5 h, mice were reanesthetized at half the usual dose of ketamine/xylazine and either MSC or PBS (control) was administered intratracheally (IT) using the method outlined above. MSCs were harvested and resuspended at a concentration of 500,000 cells / 40 µl PBS; PBS was given at a volume of 40 µl. Mice were then followed for 12 – 48 hrs before experiments were terminated to collect samples for analysis.
Bronchoalveolar lavage (BAL) analyses
Mice were euthanized at predetermined time points (12–24 hr), and a 20-gauge luer stub adapter was inserted into the trachea. Then, 1 ml of cold PBS without Ca++ or Mg++ was flushed through the luer adapter into the trachea and lungs and then aspirated back into the syringe. This was done a total of 3 times. Cell counts were measured using a cell counter (Invitrogen), and a differential was determined using a cytospin to pellet cells on a slide, which was then stained with hematoxylin and eosin. The supernatant from the BAL fluid was collected and analyzed for total protein using the BCA method (Pierce) as well as for macrophage inflammatory protein-2 (MIP-2) measurement using an ELISA (R&D, catalog #MM200). MIP-2 was chosen since it is an important neutrophil chemokine and correlates with neutrophil recruitment to the lung. Supernatants were stored at −80°C until analysis.
Measurement of lung injury, histology and bacterial burden
Lung injury was quantified by calculating extravascular lung water (ELW) using the method outlined in our previous studies [2,5]. For histological analysis, mice were euthanized, a luer adapter was placed into the trachea, and 1.5 ml of a 10% zinc formalin solution was gently instilled into the lungs for fixation and inflation, ensuring a distention pressure less than 25 cm H2O, in order to avoid injury. The trachea was tied off with a suture, and the lungs were removed from the thoracic cavity and immersed in a vial of 10% zinc formalin. The lungs were then processed and sectioned in a standard manner and stained with hematoxylin and eosin.
In order to measure the bacterial burden, lungs were removed from the thoracic cavity, immersed in 1 ml of PBS, and homogenized thoroughly. Dilutions of the solution were made and plated on LB Agar plate overnight at 37°C. Colonies were counted the next morning.
Measurement of Caspase 3/7 Activity
Cell death was assessed using a plate-based assay that measures caspase 3/7 activity (Promega, catalog #G7790). MSCs were plated at approximately 10,000 cells per well and allowed to grow to confluence overnight. Then, MSCs were serum starved in 0.5% FBS the next night to prepare them for induction for cell death. Cell death was induced with a combination of TNF-α (100 ng/ml) and cycloheximide (20 µg/ml) for approximately 4 h. This combination of compounds was chosen as it resulted in the most reproducible quantity of cell death for MSCs, and it has been published as an in vitro method to model cell death in an inflammatory environment [35, 36]. After 4 h of exposure, the caspase 3/7-substrate was added to the wells and fluorescence was measured at different time points using a plate reader.
Assessment of MSC barrier resistance
Using an electrical impedance system (ACEA biosciences, iCelligence) to assess the barrier resistance of monolayer of MSCs, we determined the sensitivity of different populations of MSCs to cytotoxic stimuli (TNF-α and cycloheximide). MSCs were plated at a density of 30,000 cells per well (8 well chamber) and allowed to grow to confluency. Cells were then serum starved overnight in 0.5% FBS and then were exposed to TNF-α (100 ng/ml) and cycloheximide (20 µg/ml). Barrier resistance was then continuously monitored in real-time with the iCelligence device. While MSCs do not classically form monolayers in the alveolar space in vivo, this system provided a unique method in which to monitor cytotoxicity in real time over the course of several hours to days.
RNA isolation and Reverse Transcriptase-PCR
RNA was isolated from MSCs using standard procedures (Qiagen Mini-Kit), and purity and quantity were assessed using a Nano-drop system. RNA was then reverse transcribed into cDNA (Applied Biosystems), and cDNA was amplified using standard PCR procedure (Applied Biosystems, Eppendorf Thermocycler). The resulting product was then loaded and run on a 1% agarose gel, stained with ethidium bromide, and developed using a standard gel imaging system (Bio-Rad). Primer sequences for reverse-transcriptase PCR for PAR1 and GAPDH gene expression were as follows:
PAR1 forward: CCTATGAGCCAGCCAGAATC, PAR1 reverse: TAGACTGCCCTACCCTCCAG (GenBank: BC031516.1, PAR1 forward from 131–150, PAR1 reverse from 173–192). GAPDH forward: AACTTTGGCATTGTGGAAGG, GAPDH reverse: ACACATTGGGGGTAGGAACA (GenBank: GU214026.1, GAPDH forward from 564–583, GAPDH reverse from 767–786).
For the detection of prothrombin gene expression in MSCs, cells were stimulated with a range of LPS concentrations (0.1 – 1 µM, Sigma, catalog #L6529) for 6 h, and then cells were lysed for RNA extraction and cDNA production as above. The following primers sequences were then used for PCR: F2 forward: GGACGCTGAGAAGGGTATCG, F2 reverse: CTTGTCTGGCAAACACACGG (GenBank: NM_010168.3, F2 forward from 1156–1175, F2 reverse from 1441–1460).
Protein Isolation and Western Blotting
MSCs were lysed and protein isolated using RIPA buffer (ThermoFisher). The quantity of protein in each sample was determined using the BCA method (Pierce, ThermoFisher). Approximately 30 µg of protein was mixed with loading buffer and reducing agent and then denatured at 90°C for 10 min. Samples were loaded onto a 4–12% Bis-Tris gel (NuPage, Invitrogen) along with a molecular weight marker and run at 120 volts for 60 min. The gel was transferred to a nitrocellulose membrane, blocked and incubated with primary (Actin, Neomarkers, Fremont, CA, catalog #MS-1295-P0; PAR1, Abbiotec, San Diego, CA, catalog #251324; Tissue Factor, American Diagnostics, Stamford, CT, catalog #4515) and secondary antibodies (LiCor) in standard, sequential fashion. After appropriate washing with PBS-0.1% Tween-20 buffer, the membrane was visualized using a LiCor machine.
For the detection of prothrombin in MSC conditioned media, MSCs were stimulated with LPS for 6 h using DMEM:F12 50:50 with no added FBS or phenol red. The conditioned media was then concentrated with a 50-kDa filter (Amicon, Millipore) and subsequently run on a gel using standard technique as above. Prothrombin was detected using a sheep polyclonal antibody (Haematologic Technologies Inc., catalog #PAHFII-S) along with a corresponding secondary antibody (LiCor). The positive control for these studies was recombinant human prothrombin (Haematologic Technologies Inc., catalog #HCP-0010).
Signaling Assays
On-Cell Western assays were done using a 96-well black, with clear bottom, plate (Corning) to determine intracellular signaling pathways activated by thrombin. MSCs were plated at approximately 10,000 cells per well, allowed to adhere overnight, serum starved in 0.5% FBS the next night, and then stimulated with thrombin (Enzyme Research Laboratories, catalog #MIIa) at 5–10 nM for 0 – 120 min. MSCs were subsequently fixed with 4% formaldehyde, permeabilized with 0.1% Triton X-100, and blocked with appropriate buffer (LiCor). MSCs were then incubated with an anti-pERK1/2 (Cell Signaling, rabbit polyclonal, catalog #9102) or anti-pAKT antibody (Cell Signaling, rabbit polyclonal, catalog #9271) overnight at 4°C, washed, and incubated with the appropriate secondary antibody (LiCor) before developing on the LiCor machine. DRAQ5 (ThermoFisher, catalog #62251) was added with the secondary antibody solution to quantify and normalize for the total number of cells per well.
A phospho-protein signaling array (Cell Signaling, catalog #7323) was done according to manufacturer’s instructions in order to characterize the differences between WT and TLR4 −/− MSC intracellular pathway activation (see supplementary material for details on methods).
ERK1/2 inhibition of MSCs
An ERK1/2 inhibitor, FR180204 (Sigma, catalog #SML0320), was incubated with MSCs in tissue culture at a concentration of 5 µM for 30–45 min before washing and harvesting the cells. MSCs were then administered IT to mice previously injured with E. coli as outlined in the experimental design above. The effect of ERK1/2 inhibition on MSC function was ascertained by assessing the therapeutic effect of the MSCs in vivo.
Blockade of PAR1 on Human MSCs with Vorapaxar
Bone marrow derived-human MSCs (Texas A&M) were incubated with vorapaxar (Axon Medchem, catalog #1755) 0.3 µM for 30 min to block PAR1, and then cells were washed, harvested, and administered in vivo to mice using the pneumonia model outlined above. Mouse survival was measured to determine the effect of PAR1 blockade on the therapeutic value of human MSCs.
Statistical analysis
Survival data was analyzed using a log-rank test, while the majority of the other data presented was presented as mean ± SD for each group analyzed. An unpaired, two sided student’s t test was used for most comparisons between two sets of data, however, for sets of data with a small sample size (total n < 20) a Mann-Whitney U test was used. If multiple groups of data were compared simultaneously, an ANOVA was used. A p-value < 0.05 was used for statistical significance.
Results
Characterization of MSCs
MSCs from WT, TLR4 −/− and R41Q-PAR1 mutated mice were isolated and characterized as we have described [2,5]. Both differentiation assays and flow cytometry were done as specified by the previously prescribed criteria for defining MSCs [34]. WT and TLR4 −/− MSCs demonstrated the ability to differentiate into bone, fat and cartilage, whereas R41Q-PAR1 mutated MSCs retained the ability to differentiate into bone and fat (Supplementary Figure 1A – 1H). Flow cytometry showed that WT, TLR4 −/− and R41Q-PAR1 mutated MSCs met the majority of the specified criteria for MSC cell surface marker expression with the exception of CD90 (Supplementary Figure 2A – 2F). In addition, TLR4 −/− MSCs did not show expression of CD105 (Supplementary Figure 2C).
TLR4 −/− MSCs displayed increased susceptibility to cell death
When exposed to cycloheximide and TNF-α, TLR4 −/− MSCs exhibited increased levels of cell death compared to WT MSCs as assessed by caspase 3/7 release (Figure 1A). In addition, measurement of transcellular resistance of an MSC monolayer also suggested increased cellular toxicity of TLR4 −/− MSCs, as compared to WT MSC, when exposed to cycloheximide and TNF-α (Figure 1B, 1C). Histological analysis of WT and TLR4 −/− MSCs qualitatively showed more disruption of normal cellular architecture and cell death in TLR4 −/− MSCs in these assays (Supplementary Figure 3).
Figure 1. TLR4 −/− MSCs show increased susceptibility to cell death when exposed to cytotoxic stimuli in vitro.

After exposure to TNF-α (100 ng/ml) and cycloheximide (20 µg/ml) (TNF/CHX), TLR4 −/− MSC showed significantly higher caspase 3/7 activity (A) and a significant reduction in monolayer resistance (B, C) (panel A, **p < 0.01 WT vs TLR4 −/− MSC, n = 6 per group; panel B and C, **p < 0.01 TLR4 −/− MSC + TNF/CHX vs unstim TLR4 −/− MSC and WT MSC + TNF/CHX, n = 4 per group).
A signaling array was done to determine potential pathways involved in the increased susceptibility to cell death observed in TLR4 −/− MSCs. As shown in Supplementary Figures 4A – 4D, WT MSCs exhibited qualitatively higher levels of phosphorylated pro-survival proteins, such as ERK1/2, PRAS40 and p38, when compared to TLR4 −/− MSCs under basal and TNF-α stimulated conditions. Meanwhile, TLR4 −/− MSCs demonstrated greater relative phosphorylation of pro-apoptotic proteins such as p53 and Bad when exposed to TNF-α.
MSCs lacking TLR4 Confer No Protective Effect In Vivo
When TLR4−/− MSCs were administered IT to mice 4 h after IT instillation of E. coli K1, there was no evidence of survival benefit, reduction in lung injury, or enhancement of bacterial clearance 48 h after infection (Figure 2A – 2C). Hematoxylin and eosin staining of lung sections qualitatively demonstrated no improvement in lung damage in mice treated with TLR4 −/− MSCs, supporting the quantitative analysis (Figure 2D).
Figure 2. TLR4 −/− MSCs exhibit no therapeutic effects in the in vivo E. coli pneumonia model.

TLR4 −/− MSCs did not improve survival (A), lung edema (B), or bacterial burden (C) as compared to PBS treated mice at 48 h (panels A and B, *, √ p < 0.05 for WT MSC vs PBS and TLR4 −/− MSC groups, respectively; panel C, **, # p < 0.01 for TLR4 −/− MSC vs WT MSC and PBS treated groups, respectively; n = 15 – 20 per group). Lung histology also demonstrated that TLR4 −/− MSC treated mice had no appreciable decrease in lung damage versus PBS treated mice (D). BAL at 12 h post-infection demonstrated that TLR4 −/− MSCs did not reduce inflammatory cell influx into the alveolar space (panels E and F, *, √ p < 0.05 for WT MSC vs PBS and TLR4 −/− MSC groups, respectively, n = 4 per group). At 24 h post-infection, BAL studies showed that TLR4 −/− MSCs did not reduce the levels of the inflammatory cytokine, MIP-2, or the albumin concentration (G and H, respectively, *, √ p < 0.05 for WT MSC vs PBS and TLR4 −/− MSC groups, n = 6–9 per group).
12–24 h BAL fluid from mice treated with TLR4 −/− MSCs exhibited no reduction in the influx of inflammatory cells (predominantly neutrophils), the neutrophil chemokine MIP-2, or the total level of protein when compared to PBS treated mice (Figure 2E – 2H). WT MSC treated mice showed an improvement in all of these parameters.
MSCs express PAR1 and TLR4 pathway regulates MSC secretion of prothrombin and signaling through PAR1
Given that inflammation and coagulation are coupled during infection and the published reports linking TLRs and PARs [14–20], we sought to examine the interaction between the TLR4 and PAR1 pathways in MSCs. Specifically, we studied how stimulation and disruption of the TLR4 pathway affects activation and signaling through PAR1, respectively. We postulated that MSCs may be a source of prothrombin themselves, and that this may be resulting in an autocrine, pro-survival signaling pathway for MSCs. This might explain the increased susceptibility of R41Q-PAR1 mutated MSCs to cell death as seen in Figure 5A. In fact, we found that stimulation of MSCs with LPS led to upregulation of the prothrombin gene transcript at 6 h as well as detection of the prothrombin protein in the conditioned media of LPS stimulated MSCs at 6 h (Figure 3A, 3B). The slight difference in the size of the prothrombin protein between the positive control (recombinant human prothrombin) and experimental conditions (mouse MSC conditioned media) may reflect species and cell processing alternations in the prothrombin protein. We also determined that MSCs do express the PAR1 receptor at the gene and protein levels, but that TLR4 stimulation with LPS did not seem to lead to significant changes in PAR1 expression (Figure 3C, 3D). Furthermore, we found that MSCs express tissue factor as determined by Western blotting (Figure 3E), which could provide the scaffolding for factor Xa to cleave the self-generated prothrombin into thrombin and thereby lead to PAR1 activation.
Figure 5. R41Q-PAR1 mutated MSCs demonstrate increased cell death when exposed to inflammatory stimuli in vitro and no therapeutic capacity in vivo.

When exposed to TNF-α and cycloheximide, R41Q-PAR1 mutated MSCs demonstrated increased cell death compared to WT MSCs as measured by caspase 3/7 activity (A, **p < 0.01 for R41Q MSCs vs WT MSCs, n = 6 per group). Pre-treatment of R41Q-PAR1 mutated MSCs with 10 µM of TRAP for 1 h reduced the quantity of cell death upon exposure to TNF-α and cycloheximide (B, **p < 0.01 for R41Q MSC + TRAP vs R41Q MSC, n = 6 per group). R41Q-PAR1 mutated MSCs exhibited significantly less ERK1/2 phosphorylation than WT MSCs after 5 min of 5 nM of thrombin stimulation (C, **p < 0.01 for R41Q MSC vs WT MSC, n = 3 per group). In vivo, R41Q-PAR1 mutated MSCs showed no evidence of therapeutic benefit in terms of 48 h survival or whole lung bacterial burden (D, E, *, # p < 0.05 for WT MSCs vs PBS and R41Q MSCs, respectively, n = 17–22 per group). Human MSCs were incubated with vorapaxar at 0.3 µM for 30 min and then administered as a treatment in the in vivo model. Vorapaxar pre-incubation significantly inhibited the therapeutic capacity of human MSCs in vivo when compared to non-pre-treated cells (F, *p < 0.05 for hMSCs vs hMSCs + vorapaxar, n = 10–18 per group).
Figure 3. TLR4 stimulation induces prothrombin secretion by MSCs and regulates signaling through PAR1.

WT MSCs demonstrated evidence of prothrombin gene expression upon stimulation with 100 nM of LPS, which was not seen in TLR4 −/− MSC or 3T3 fibroblasts. Mouse liver cDNA was used as a positive control for prothrombin (A). Conditioned media from MSCs stimulated with LPS demonstrated the presence of prothrombin protein as detected by Western blotting. Recombinant human prothrombin was used as the positive control for these studies (B). Reverse transcriptase-PCR demonstrated that MSCs expressed the transcript for PAR1 (C), and Western blotting confirmed that MSCs expressed the PAR1 protein (D). There was no appreciable change in PAR1 expression by MSCs with LPS stimulation (3T3, mouse embryonic fibroblast cell line; bEND.3, mouse brain endothelial cells line used as positive control). MSCs also were shown to express tissue factor by Western blotting (E, bEND.3 cell line used as positive control). Thrombin stimulation (10 nM) led to significant ERK1/2 phosphorylation at 5 min in WT MSCs but not in TLR4 −/− MSCs (F, G, **p < 0.01 for TLR4 −/− MSCs vs WT MSCs; n = 3 per group per time point), despite both WT and TLR4 −/− MSC qualitatively expressing similar amounts of PAR1 protein by Western blotting (H). Using the in vivo model, we determined that inhibition of ERK1/2 with FR180204 eliminated the therapeutic efficacy of WT MSCs, suggesting that this central signaling pathway is required for MSC-derived protection (I, *p < 0.05 for MSC vs PBS treated group, n = 18 – 20 per group).
In order to investigate the effect of the TLR4 pathway on PAR1 signaling, we compared the quantity of ERK1/2 phosphorylation in WT and TLR4 −/− MSCs in response to thrombin stimulation. ERK1/2 is part of the mitogen-activated protein kinase (MAPK) family and is classically activated with thrombin cleavage of PAR1 [33]. Stimulation with thrombin only led to ERK1/2 phosphorylation in WT MSCs (Figure 3F, 3G), even though both WT and TLR4 −/− MSCs expressed the PAR1 protein (Figure 3H). These data suggest that the TLR4 pathway at least partly controls stimulation and signaling through PAR1 on MSCs.
Furthermore, since our data demonstrate that both the TLR4 and PAR1 pathways are linked to ERK1/2 phosphorylation in MSCs, we carried out experiments with an ERK1/2 inhibitor (FR180204) to determine if phosphorylation of this signaling protein is required for MSC derived therapeutic efficacy. We found that pre-treatment of WT MSCs with FR180204 negated the therapeutic effect of the cells in vivo as determined by survival, suggesting that ERK1/2 phosphorylation is required for MSC activity in vivo (Figure 3I).
Thrombin induces a pro-survival phenotype in MSCs
Given the links between the TLR4 and PAR1 pathways we observed, we further explored the effect of thrombin, the principal PAR1 agonist, on MSC survival when cells were exposed to inflammatory stimuli in vitro. Initially, we investigated whether thrombin activated other pro-survival pathways such as Akt, but as seen in Figures 4A and 4B, only ERK1/2 was rapidly phosphorylated. This is consistent with previous literature demonstrating that PAR1 cleavage by thrombin shows biased agonism towards ERK1/2 signaling [33]. Thrombin also enhanced MSC barrier resistance, when compared to unstimulated MSCs, as measured by transcellular electrical impedance (Figure 4C, 4D). In addition, thrombin reduced MSC death, as assessed by caspase 3/7-release, when cells were exposed to the cytotoxic stimuli of TNF-α and cycloheximide (Figure 4E).
Figure 4. Thrombin leads to ERK1/2 biased signaling and exerts a pro-survival phenotype in MSCs.

Thrombin (IIa) led to phosphorylation of ERK1/2, 5 min after it was added to MSCs as seen in panels A and B, but there was no evidence of Akt phosphorylation (**p < 0.01 for 5 min phosphorylation versus 0 and 120 min; n = 3 per group per time point). Thrombin also increased MSC monolayer barrier resistance under basal conditions (C, D, **p < 0.01 for WT + IIa vs Unstim WT MSCs, n = 4 per group). When added 30 min before TNF + CHX, thrombin reduced caspase 3/7 activity in MSCs (E, *p < 0.05 for IIa + TNF + CHX vs TNF + CHX, n = 5 per group).
R41Q-PAR1 mutated MSCs exhibit greater sensitivity to cytotoxic stimuli and diminished therapeutic capacity
Since thrombin activation of PAR1 leads to pro-survival signaling and phenotype in MSCs, we next examined the effects of mutating PAR1 on MSC survival and therapeutic activity. R41Q-PAR1 mutated MSCs exhibited increased susceptibility to cell death from TNF-α and cycloheximide compared to WT MSCs as measured by caspase 3/7-release (Figure 5A). Pre-stimulation of the R41Q-PAR1 mutated MSCs with thrombin receptor activating peptide (TRAP) at 10 µM for 1 h, which specifically activates PAR1, reduced the amount of caspase 3/7-release compared to unstimulated PAR1-mutated MSCs (Figure 5B). In addition, we showed that R41Q-PAR1 mutated MSCs have impaired ERK1/2 phosphorylation in response to thrombin stimulation, which is consistent with mutation of the canonical thrombin cleavage site in these cells (Figure 5C).
In order to ascertain the role of thrombin activation of PAR1 on MSCs in vivo, we tested the ability of the PAR1-mutated MSCs to rescue mice with E. coli pneumonia. We found that R41Q-PAR1 mutated MSCs conferred no beneficial effect in vivo in terms of survival and bacterial burden at 48 h post-infection (Figure 5D, 5E). In contrast, WT MSCs did significantly improve survival and bacterial clearance when compared to the PBS control treated group.
To determine whether our findings in mouse MSCs could be extrapolated to human MSCs, we used a newer generation, clinically tested PAR1 antagonist, vorapaxar, to block PAR1 activation on human MSCs (obtained from an NIH repository at Texas A&M School of Medicine, Dr. Darwin Prockop). Pre-treatment of human MSCs with vorapaxar completely eliminated the therapeutic effect of human MSCs in a mouse model of E. coli pneumonia. Untreated human MSCs conferred a trend towards improved survival versus PBS treated mice, which did not reach a level of statistical significance (p = 0.08). This finding suggests that PAR1 activation on human MSCs is necessary for their therapeutic ability in the E. coli pneumonia model (Figure 5F).
Discussion
BM-MSCs have been extensively studied in the last decade given their potential promise as cell based immunotherapy for a wide range of disease states that are due to dysregulated inflammation. The literature has demonstrated that MSCs have potent, reproducible, beneficial effects in acute, inflammatory lung injury due to infection, and these results have been attributed to a number of properties of MSCs including immunomodulation, secretion of reparative growth factors, and augmentation of host defense mechanisms [1–11]. The current study specifically aimed to determine the roles of the TLR4 and PAR1 pathways in a Gram-negative pneumonia model in mice, and the following results were demonstrated: (a) TLR4 and PAR1 are both required for the therapeutic effect of MSCs in vivo; (b) disruption of TLR4 and PAR1 signaling lends MSCs more susceptible to cell death under inflammatory conditions in vitro; and (c) TLR4 regulates MSC secretion of prothrombin as well as signaling through PAR1. To our knowledge, this is the first study to demonstrate the coupling of TLR4 and PAR1 signaling on MSCs as well as the secretion of a coagulation factor by MSCs.
Inflammation and coagulation are spatially coupled at the sites of infection as part of the host’s response to bacterial pathogens [37–41]. Since we administered MSCs directly into E. coli infected lungs in this study, we were particularly interested in the roles of TLR4 and PAR1 on the therapeutic effects of MSCs and the potential crosstalk between these pathways. TLR4 has been shown to regulate several MSC functions including migration, differentiation, and immunomodulation [21–29]. In this study, we used TLR4−/− MSCs to investigate the role of the TLR4 pathway on cell survival under inflammatory conditions. Using a combination of TNF-α and cycloheximide to simulate the cytotoxic milieu of the alveolar space in pneumonia, we were able to demonstrate that TLR4−/− MSCs were more susceptible to cell death than WT MSCs. This was determined by measuring caspase 3/7-activity in the cells as well as by assessing the barrier resistance of MSC monolayers. Mechanistically, this may be due to the finding that the signaling profile of TLR4 −/− MSCs has less activation of pro-survival pathways such as ERK1/2, p38, and PRAS40 and greater activation of pro-apoptotic pathways such as p53 and Bad, when compared to WT MSCs. In vivo, TLR4 −/− MSCs exerted no therapeutic effect in terms of survival, lung injury or bacterial clearance. Of note, the characterization of the different lines of MSCs used in this study demonstrated that TLR4 −/− MSCs did not express detectable CD105. This finding may also impact the therapeutic capacity of TLR4 −/− MSCs, since CD105 is known to play a role in transforming growth factor-beta (TGF-β) signaling, immunomodulation and differentiation [42,43].
PCR and Western blotting confirmed that MSCs express PAR1, and stimulation with thrombin led to rapid activation of the pro-survival signaling protein ERK1/2, reduction in cell death when exposed to cytotoxic stimuli, and enhancement in barrier resistance. In order to specifically ascertain the effect of thrombin cleavage of PAR1 on MSC survival and function, we utilized MSCs derived from R41Q-PAR1 mutant mice generated by our group. PAR1-mutated MSCs exhibited defective phosphorylation of ERK1/2 in response to thrombin (as expected), and an increased susceptibility to cell death upon exposure to cytotoxic stimuli that was reduced by TRAP pre-stimulation. In vivo, R41Q-PAR1 mutated MSCs showed no therapeutic properties in terms of survival or bacterial clearance suggesting that thrombin cleavage of PAR1 is required for MSC-derived protection in bacterial pneumonia. The importance of PAR1 on the activity of MSCs was extended to human MSCs in our study by the use of the clinically tested PAR1 antagonist, vorapaxar. While vorapaxar will block PAR1 activation and signaling non-selectively, the finding that vorapaxar treated human MSCs have no therapeutic activity in vivo provides some evidence that PAR1 is required for human MSC-derived protection as well. Of note, the therapeutic effect seen with untreated human MSCs was not as robust as seen with mouse MSCs as the difference in survival between human MSC and PBS treated mice did not reach a level of statistical significance. This finding may be due to species differences that lend human MSCs less effective in a mouse experimental model.
The finding that PAR1 mutated MSCs were more susceptible to TNF-α and cycloheximide induced cell death, when compared to WT MSCs, despite the absence of exogenous thrombin was the impetus for postulating that MSCs may be secreting prothrombin themselves. Our data supported this hypothesis by demonstrating that LPS-stimulated MSCs do express and secrete low levels of prothrombin. Other pro-inflammatory stimuli such as TNF-α also led to detectable levels of prothrombin in the conditioned media (data not shown). The relative contributions of self-generated versus exogenous prothrombin on MSC signaling and survival during infection in an in vivo system are unclear. Nonetheless, this finding provides a connection between the TLR4 and PAR1 pathways in MSCs. Furthermore, the finding that MSCs express tissue factor, as demonstrated by our data and others [44], provides a platform on which MSCs can bring factor Xa in close proximity to the cell membrane in order to cleave prothrombin into thrombin and thereby activate PAR1.
In addition to the above-described link between TLR4 and PAR1 on MSCs, TLR4 −/− MSCs did not display activation of ERK1/2 in response to thrombin stimulation, suggesting that disrupting the TLR4 pathway impairs PAR1 signaling. Therefore, PAR1 activation and signaling in MSCs is directly coupled to and dependent upon an intact TLR4 pathway. In this fashion, MSCs can integrate pro-inflammatory signals such as LPS and thrombin at sites of infection in order to maximize survival and thereby produce a sustained reparative effect. Furthermore, both pathways are linked to ERK1/2 phosphorylation based on our in vitro studies, and therefore we tested how inhibition of ERK1/2 would influence MSC-derived protection in vivo. Using FR180204, we were able to show that ERK1/2 phosphorylation is required for therapeutic activity in bacterial pneumonia. Given that ERK1/2 is a central regulator of cell survival and migration, this finding is not unexpected but does confirm the importance of this particular MAPK pathway in MSCs.
The results of this study may be used to improve the therapeutic capacity of MSCs designed for clinical administration. Testing for ERK1/2 phosphorylation in response to TRAP or thrombin, for example, would validate the integrity of both the PAR1 and TLR4 pathways in MSCs and could serve as a “potency assay” prior to approval for use. In addition, the TLR4 and/or PAR1 pathways in MSCs could be pharmacologically activated ex-vivo to initiate pro-survival signaling and potentially augment MSC-derived therapeutic activity in vivo.
While we focused on MSC survival and signaling as the underlying mechanisms to account for the therapeutic effects in vivo, it is possible that TLR4 and PAR1 are also controlling the function of MSCs in this experimental system. This may include secretion of antibacterial, growth, and immunomodulatory factors that are important to MSC-derived protection. In addition, we concentrated on using one model of Gram-negative pneumonia, but we cannot definitively extrapolate these findings to other models of pneumonia, particularly ones that use Gram-positive pathogens. Gram-positive bacteria usually stimulate other TLRs such as TLR2, so further studies will need to be done using these organisms and investigating the interaction between TLR2 and PAR1. Furthermore, we did not test for interactions between TLR4 and other PARs, such as PAR2, for which there is some existing literature to show cooperative signaling [14–16]. This interaction could occur by direct association between TLR4 and PAR2 or possibly indirectly by transactivation of other PARs, such as PAR1, as is known to occur in other systems [17].
In conclusion, the current study demonstrates that the TLR4-PAR1 axis is important in promoting MSC survival under inflammatory conditions, and that disruption of these pathways abrogates MSC-derived therapeutic effect in experimental pneumonia. In addition, the results show that TLR4 regulates signaling through PAR1 on MSCs, and in particular, TLR4 stimulation leads to expression and secretion of prothrombin by MSCs. Continued pursuit of these pathways, and their pharmacological or genetic manipulation, may provide a fruitful method to enhance the in vivo therapeutic effect of MSCs.
Supplementary Material
Acknowledgments
The authors thank Patty Lee, MD (Yale University) for her generous donation of bone marrow from TLR4 −/− mice in order to isolate TLR4 −/− MSCs. We also thank Xiao Su, MD, PhD (Pasteur Institute-Shanghai) for his training in the in vivo techniques utilized in this study. The authors would also like to thank the following institutions for funding support: National Institutes of Health, National Heart, Lung and Blood Institute (HL092059, N.G.; HL031950, HL052246, J.H.G.; HL125352, V.N.; R37HL51856, M.A.M.) and the University of California, San Diego, Department of Medicine (N.G.).
MAM declare research funding from Bayer Pharmaceuticals, Amgen, Glaxo Smith Kline.
Footnotes
Author Contributions:
All authors participated in planning experiments
NG, RS, AK and XX carried out experiments
NG wrote the manuscript, all authors participated in editing the manuscript
NG, MAM, VN and JHG funded the studies
Disclosures
All other authors declared no financial conflicts of interest.
References
- 1.Xu J, Woods CR, Mora AL, Joodi R, Brigham KL, Iyer S, Rojas M. Prevention of endotoxin-induced systemic response by bone marrow-derived mesenchymal stem cells in mice. Am J Physiol Lung Cell Mol Physiol. 2007;(293):L131–141. doi: 10.1152/ajplung.00431.2006. [DOI] [PubMed] [Google Scholar]
- 2.Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol. 2007;(179):1855–1863. doi: 10.4049/jimmunol.179.3.1855. [DOI] [PubMed] [Google Scholar]
- 3.Lee JW, Fang X, Gupta N, Serikov V, Matthay MA. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc Natl Acad Sci USA. 2009;(106):16357–16362. doi: 10.1073/pnas.0907996106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mei SH, Haitsma JJ, Dos Santos CC, Deng Y, Lai PF, Slutsky AS, Liles WC, Steward DJ. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. Am J Respir Crit Care Med. 2010;(182):1047–1057. doi: 10.1164/rccm.201001-0010OC. [DOI] [PubMed] [Google Scholar]
- 5.Gupta N, Krasnodembskaya A, Kapetanaki M, Mouded M, Tan X, Serikov V, Matthay MA. Mesenchymal stem cells enhance survival and bacterial clearance in murine Escherichia coli pneumonia. Thorax. 2012;(67):533–539. doi: 10.1136/thoraxjnl-2011-201176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JW, Krasnodembskaya A, McKenna DH, Song Y, Abbot J, Matthay MA. Therapeutic effects of human mesenchymal stem cells in ex vivo human lungs injured with live bacteria. Am J Respir Crit Care Med. 2013;(187):751–760. doi: 10.1164/rccm.201206-0990OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krasnodembskaya A, Song Y, Fang X, Gupta N, Serikov V, Lee JW, Matthay MA. Antibacterial effect of human mesenchymal stem cells is mediated in part from secretion of the antimicrobial peptide LL-37. Stem Cells. 2010;(28):2229–2238. doi: 10.1002/stem.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devaney J, Horie S, Masterson C, Elliman S, Barry F, O’Brien T, Curley GF, O’Toole D, Laffey JG. Human mesenchymal stromal cells decrease the severity of acute lung injury induced by E. coli in the rat. Thorax. 2015;(70):625–635. doi: 10.1136/thoraxjnl-2015-206813. [DOI] [PubMed] [Google Scholar]
- 9.Chan MC, Kuok DI, Leung CY, Hui KP, Valkenburg SA, Lau EH, Nicholls JM, Fang X, Guan Y, Lee JW, Chan RW, Webster RG, Matthay MA, Peiris JS. Human mesenchymal stromal cells reduce influenza A H5N1-associated acute lung injury in vitro and in vivo. Proc Natl Acad Sci USA. 2016;(113):3621–3626. doi: 10.1073/pnas.1601911113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, Mezey E. Bone marrow stromal cells attenuate sepsis via prostaglandin E2-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;(15):42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McIntyre LA, Moher D, Fergusson DA, Sullivan KJ, Mei SH, Lalu M, Marshall J, Mcleod M, Griffin G, Grimshaw J, Turgeon A, Avey MT, Rudnicki MA, Jazi M, Fishman J, Stewart DJ Canadian Critical Care Translational Biology Group. Efficacy of Mesenchymal stromal cell therapy for acute lung injury in preclinical animal models: a systematic review. PLoS One. 2016;(11):e0147170. doi: 10.1371/journal.pone.0147170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson JG, Liu KD, Zhuo H, Caballero L, McMillan M, Fang X, Cosgrove K, Vojnik R, Calfee CS, Lee JW, Rogers AJ, Levitt J, Wiener-Kronish J, Bajwa EK, Leavitt A, McKenna D, Thompson BT, Matthay MA. Mesenchymal stem (stromal) cells for treatment of ARDS: a phase 1 clinical trial. Lancet Respir Med. 2015;(3):24–32. doi: 10.1016/S2213-2600(14)70291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simonson OE, Mougiakakos D, Heldring N, Bassi G, Johansson HJ, Dalen M, Jitschin R, Rodin S, Corbascio M, El Andaloussi S, Wiklander OP, Nordin JZ, Skog J, Romain C, Koestler T, Hellgren-Johansson L, Schiller P, Joachimsson PO, Hagglund H, Mattsson M, Lehtio J, Faridani OR, Sandberg R, Korsgren O, Krampera M, Weiss DJ, Grinnemo KH, Le Blanc K. In vivo effects of mesenchymal stromal cells in two patients with severe acute respiratory distress syndrome. Stem Cells Transl Med. 2015;(4):1199–1213. doi: 10.5966/sctm.2015-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rallabhandi P, Nhu QM, Toshchakov VY, Piao W, Medvedev AE, Hollenberg MD, Fasano A, Vogel SN. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: a novel paradigm for receptor cooperativity. J Biol Chem. 2008;(283):24314–24325. doi: 10.1074/jbc.M804800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nhu QM, Shirey K, Teijaro JR, Farber DL, Netzel-Arnett S, Antalis TM, Fasano A, Vogel SN. Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunol. 2010;(3):29–39. doi: 10.1038/mi.2009.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bucci M, Vellecco V, Harrington L, Brancaleone V, Roviezzo F, Mattace Raso G, Ianaro A, Lungarella G, De Palma R, Meli R, Cirino G. Cross-talk between toll-like receptor 4 (TLR4) and proteinase-activated receptor (PAR2) is involved in vascular function. Br J Pharmacol. 2013;(168):411–420. doi: 10.1111/j.1476-5381.2012.02205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R. Proteinase-activated receptors (PARs) – focus on receptor-receptor interactions and their physiological and pathophysiological impact. Cell Commun Signal. 2013;(11):86. doi: 10.1186/1478-811X-11-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antoniak S, Mackman N. Multiple roles of the coagulation protease cascade during virus infection. Blood. 2014;(123):2605–2613. doi: 10.1182/blood-2013-09-526277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palm E, Demirel I, Bengtsson T, Khalaf H. The role of toll-like and protease-activated receptors in the expression of cytokines by gingival fibroblasts stimulated with the periodontal pathogen Porphyromonas gingivalis. Cytokine. 2015;(76):424–432. doi: 10.1016/j.cyto.2015.08.263. [DOI] [PubMed] [Google Scholar]
- 20.Mhatre MV, Potter JA, Lockwood CJ, Krikun G, Abrahams VM. Thrombin Augments LPS-Induced Human Endometrial Endothelial Cell Inflammation via PAR1 Activation. Am J Reprod Immunol. 2016;(76):29–37. doi: 10.1111/aji.12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liotta F, Angeli R, Cosmi L, Fili L, Manuelli C, Frosali F, Mazzinghi B, Maggi L, Pasini A, Lisi V, Santarlasci V, Consoloni L, Angelotti ML, Romagnani P, Parronchi P, Krampera M, Maggi E, Romagnani S, Annunziato F. Toll-like receptors 3 and 4 are expressed by human bone marrow-derived mesenchymal stem cells and can inhibit their T-cell modulatory activity by impairing Notch signaling. Stem Cells. 2008;(26):279–289. doi: 10.1634/stemcells.2007-0454. [DOI] [PubMed] [Google Scholar]
- 22.Romieu-Mourez R, Francois M, Boivin MN, Bouchentouf M, Spaner DE, Galipeau J. Cytokine modulation of TLR expression and activation in mesenchymal stromal cells leads to a proinflammatory phenotype. J Immunol. 2009;(182):7963–7973. doi: 10.4049/jimmunol.0803864. [DOI] [PubMed] [Google Scholar]
- 23.Waterman RS, Tomchuck SL, Henkle SL, Betancourt AM. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an immunosuppressive MSC2 phenotype. PLoS One. 2010;(5):e10088. doi: 10.1371/journal.pone.0010088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cassatella MA, Mosna F, Micheletti A, Lisi V, Tamassia N, Cont C, Calzetti F, Pelletier M, Pizzolo G, Krampera M. Toll-like receptor-3 activated human mesenchymal stromal cells significantly prolong the survival and function of neutrophils. Stem Cells. 2011;(29):1001–1011. doi: 10.1002/stem.651. [DOI] [PubMed] [Google Scholar]
- 25.Giuliani M, Bennaceur-Griscelli A, Nanbakhh A, Oudrhiri N, Chouaib S, Azzarone B, Durrbach A, Lataillade JJ. TLR ligands stimulation protects MSC from NK killing. Stem Cells. 2014;(32):290–300. doi: 10.1002/stem.1563. [DOI] [PubMed] [Google Scholar]
- 26.He X, Wang H, Jin T, Xu Y, Mei L, Yang J. TLR4 activation promotes bone marrow MSC proliferation and osteogenic differentiation via Wnt3a and Wnt5a signaling. PLoS One. 2016;(11):e0149876. doi: 10.1371/journal.pone.0149876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ti D, Hao H, Tong C, Liu J, Dong L, Zheng J, Zhao Y, Liu H, Fu X, Han W. LPS-preconditioned mesenchymal stromal cells modify macrophage polarization for resolution of chronic inflammation via exosome-shuttled let-7b. J Transl Med. 2015;(13):308. doi: 10.1186/s12967-015-0642-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sung DK, Chang YS, Sung SI, Yoo H, Ahn SY, Park WS. Antibacterial effect of mesenchymal stem cells against Escherichia coli is mediated by secretion of beta-defensin-2 via toll-like receptor 4 signaling. Cell Microbiol. 2016;(18):424–436. doi: 10.1111/cmi.12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rashedi I, Gomez-Aristizabal A, Wang XH, Viswanathan S, Keating A. TLR3 or TLR4 activation enhances mesenchymal stromal cell mediated Treg induction via notch signaling. Stem Cells. 2017;(35):265–275. doi: 10.1002/stem.2485. [DOI] [PubMed] [Google Scholar]
- 30.Ho IA, Chan KY, Ng WH, Guo CM, Hui KM, Cheang P, Lam PY. Matrix metalloproteinase 1 is necessary for the migration of human bone marrow-derived mesenchymal stem cells toward human glioma. Stem Cells. 2009;(27):1366–1375. doi: 10.1002/stem.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, Ma Y, Wang Z, Wang H, Wang L, Xiao F, Wang H, Tan J, Guo Z. Thrombin promotes fibronectin secretion by bone marrow mesenchymal stem cells via the protease-activated receptor mediated signaling pathways. Stem Cell Res Ther. 2014;(5):36. doi: 10.1186/scrt424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta N, Sinha RK, Xu X, Griffin JH. R41Q-PAR1 Mutation Eliminates the Therapeutic Capacity of Bone Marrow Derived Mesenchymal Stem Cells in Murine E. coli Pneumonia. Am J Respir Crit Care Med. 2015;(191):A6152. (abstract) [Google Scholar]
- 33.Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood. 2012;(26):5237–46. doi: 10.1182/blood-2012-08-452169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop DJ, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells: The International Society for Cellular Therapy position statement. Cytotherapy. 2006;(8):315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 35.Polunovsky VA, Wendt CH, Ingbar DH, Peterson MS, Bitterman PB. Induction of endothelial cell apoptosis by TNF alpha: modulation by inhibitors of protein synthesis. Exp Cell Res. 1994;(214):584–594. doi: 10.1006/excr.1994.1296. [DOI] [PubMed] [Google Scholar]
- 36.Tsuchida H, Takeda Y, Takei H, Shinzawa H, Takahashi T, Sendo F. In vivo regulation of rat neutrophil apoptosis occurring spontaneously or induced with TNF-alpha or cycloheximide. J Immunol. 1995;(154):2403–2412. [PubMed] [Google Scholar]
- 37.Esmon CT, Xu J, Lupu F. Innate immunity and coagulation. J Thromb Haemost. 2011;(9):182–188. doi: 10.1111/j.1538-7836.2011.04323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;(131):417–430. doi: 10.1111/j.1365-2141.2005.05753.x. [DOI] [PubMed] [Google Scholar]
- 39.Foley JH, Conway EM. Cross Talk Pathways Between Coagulation and Inflammation. Circ Res. 2016;(118):1392–1408. doi: 10.1161/CIRCRESAHA.116.306853. [DOI] [PubMed] [Google Scholar]
- 40.Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;(38):S26–34. doi: 10.1097/CCM.0b013e3181c98d21. [DOI] [PubMed] [Google Scholar]
- 41.Levi M, van der Poll T. Coagulation and sepsis. Thromb Res. 2017;(149):38–44. doi: 10.1016/j.thromres.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 42.Anderson P, Carrillo-Galvez AB, Garcia-Perez A, Cobo M, Martin F. CD105 (Endoglin)-Negative Murine Mesenchymal Stromal Cells Define a New Multipotent Subpopulation with Distinct Differentiation and Immunomodulatory Capacities. PLoS ONE. 2013;(8):e76979. doi: 10.1371/journal.pone.0076979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levi B, Wan DC, Glotzbach JP, Hyun J, Januszyk M, Montoro D, Sorkin M, James AW, Nelson ER, Li S, Quarto N, Lee M, Gurtner GC, Longaker MT. CD105 Protein Depletion Enhances Human Adipose-derived Stromal Cell Osteogenesis through Reduction of Transforming Growth Factor β1 (TGF-β1) Signaling. J Biol Chem. 2011;(286):39497–39509. doi: 10.1074/jbc.M111.256529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tatsumi K, Ohashi K, Matsubara Y, Kohori A, Ohno T, Kakidachi H, Horil A, Kanegae K, Utoh R, Iwata T, Okano T. Tissue factor triggers procoagulation in transplanted mesenchymal stem cells leading to thromboembolism. Biochem Biophys Res Commun. 2013;(431):203–209. doi: 10.1016/j.bbrc.2012.12.134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.