

Abstract
Mitochondrial structure and function are influenced by the unique phospholipid composition of its membranes. While mitochondria contain all the major classes of phospholipids, recent studies have highlighted specific roles of the non-bilayer forming phospholipids phosphatidylethanolamine (PE) and cardiolipin (CL) in the assembly and activity of mitochondrial respiratory chain (MRC) complexes. The non-bilayer phospholipids are cone-shaped molecules that introduce curvature stress in the bilayer membrane and have been shown to impact mitochondrial fusion and fission. In addition to their overlapping roles in these mitochondrial processes, each non-bilayer phospholipid also plays a unique role in mitochondrial function; for example, CL is specifically required for MRC supercomplex formation. Recent discoveries of mitochondrial PE and CL trafficking proteins and prior knowledge of their biosynthetic pathways have provided targets for precisely manipulating non-bilayer phospholipid levels in the mitochondrial membranes in vivo. Thus, the genetic mutants of these pathways could be valuable tools in illuminating molecular functions and biophysical properties of non-bilayer phospholipids in driving mitochondrial bioenergetics and dynamics.
Keywords: Cardiolipin, phosphatidylethanolamine, mitochondria
Introduction
Biological membranes enclosing cells and organelles not only act as a physical barrier from the surrounding environment, but also define their contents and provide a matrix for many essential biochemical reactions and physiological processes. For example, the mitochondrial inner membrane not only provides the ion impermeable barrier necessary for oxidative phosphorylation but also harbours respiratory chain complexes that catalyze redox reactions to power energy production. Different subcellular membranes are defined by their unique protein and lipid compositions, which are tailored to their specialized tasks [1,2]. In the case of mitochondrial membranes, the bulk of the lipid matrix is composed of phospholipids [3], which are primarily responsible for determining the biochemical and biophysical properties of the membrane.
Phospholipids can assume different structures in an aqueous environment depending on their overall shape. For example, when the area of the cross-section of the lipid head group is similar to the area of the cross-section of the acyl chains, the phospholipid has a cylindrical shape (Fig. 1), as in the case of the most abundant mitochondrial membrane phospholipid, phosphatidylcholine (PC). These phospholipids aggregate to form a bilayer by self–assembling into a liquid crystalline lamellar phase (Fig. 1) [4,5]. On the other hand, when the cross-sectional area of the phospholipid head group is smaller than the cross-sectional area of its acyl chains, the phospholipids have a conical shape (Fig. 1). These conical shaped phospholipids have the propensity to aggregate into non-bilayer structures such as the inverted hexagonal phase (HII) (Fig. 1). Phosphatidylethanolamine (PE), the second most abundant mitochondrial phospholipid, is an example of this non-bilayer class of phospholipids [4,5]. The overall shape of phospholipids is also influenced by environmental parameters such as pH, salt concentration, temperature, and the presence of divalent cations [5]. Accordingly, cardiolipin (CL), a mitochondria-specific phospholipid, acquires a conical shape in the presence of divalent cations and thus promotes non-bilayer structures.
Figure 1. Schematic representation of shape-structure concept of lipid polymorphism.
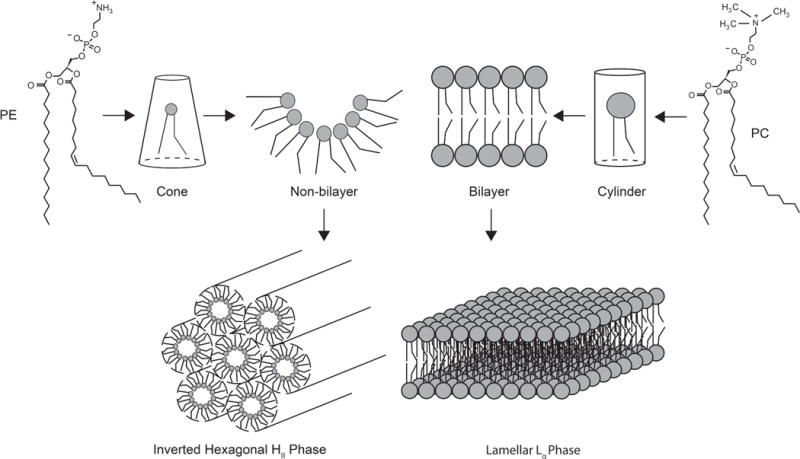
Cone-shaped molecules like phosphatidylethanolamine (PE) form non-bilayer structures, which assemble into inverted hexagonal (HII) phase. The cylinder-shaped molecules like phosphatidylcholine (PC) form bilayer structures, which assemble into liquid crystalline lamellar (Lα) phase.
Typically, the lipids in biomembranes are organized into bilayers despite the presence of the non-bilayer phospholipids, raising questions about their role in membrane structure. Non-bilayer phospholipids, by virtue of their shape, are proposed to form local, transient structures that are thought to play an important role in vital cellular processes, such as membrane fusion, vesicle formation, and cell division [4]. Apart from these processes, non-bilayer phospholipids impact membrane function by influencing the bulk properties of membranes, which in turn affect the insertion, folding, and function of certain integral membrane proteins [5,6]. Thus, it is necessary to consider the physical properties of membrane lipids to understand the molecular basis of membrane protein structure, function, and biogenesis.
In this article, we will summarize the recent findings that highlight distinct roles of non-bilayer phospholipids in the maintenance of mitochondrial structural and functional homeostasis by regulating different processes that include mitochondrial dynamics and bioenergetics. We will mainly focus on the specific roles of the non-bilayer forming phospholipids PE and CL, which together constitute almost 40% of all mitochondrial membrane phospholipids and have been shown to be the key determinants of mitochondrial structure and function. We will discuss both the biosynthetic pathways and regulation of PE and CL as well as highlight the latest discoveries of protein complexes involved in inter- and intra-organellar phospholipid trafficking.
Mitochondrial membrane phospholipid composition
Mitochondria contain all the major classes of phospholipids including phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), and phosphatidic acid (PA) [3]. In addition to these ubiquitous phospholipids, mitochondria contain cardiolipin (CL), an evolutionarily conserved dimeric phospholipid that is specifically found in this organelle [3]. Owing to their large head group PC, PI, and PS have a cylindrical shape, which promotes bilayer formation. On the other hand, the conically shaped phospholipids PE, CL, and PA impose negative curvature stress on the membrane, which increases its tendency to form non-bilayer structures [1]. The ratio of bilayer to non–bilayer phospholipids in the mitochondria is roughly equal and highly conserved across eukaryotic organisms (Fig. 2), suggesting its importance for mitochondrial structure and function [7]. Compared to other subcellular membranes, the mitochondrial inner membrane is highly enriched in non-bilayer forming phospholipids [8]. Thus, the mitochondrial inner membrane represents an excellent system to dissect the role of non-bilayer phospholipids in membrane structure and dynamics.
Figure 2. Phospholipid composition of mitochondrial membranes.
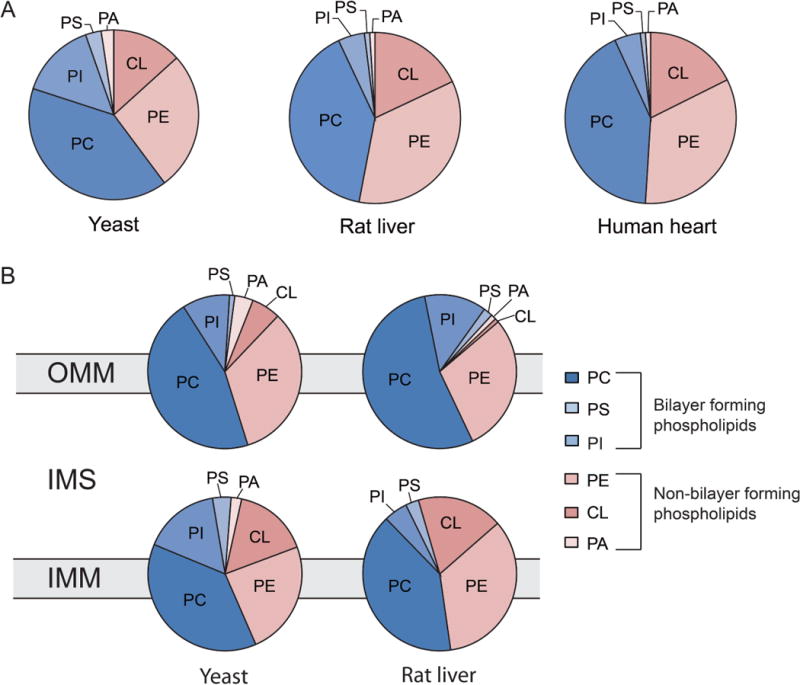
(A) Mitochondrial phospholipid composition is evolutionarily conserved. Data is taken from [7,8]. (B) Mitochondrial phospholipid composition of the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM) of yeast mitochondria. Data is taken from [8]. PC, phosphatidylcholine; PS, phosphatidylserine; PI, phosphatidylinositol; PE, phosphatidylethanolamine; CL, cardiolipin; PA, phosphatidic acid; IMS, intermembrane space.
Phospholipids make up 75–95% of total mitochondrial membrane lipids in yeast and higher eukaryotes [7]. Within the mitochondrial phospholipid milieu, PC, PE, and CL together make up ~90% of total phospholipids while the less abundant PI, PS, and PA make up the rest (Fig. 2A) [3,7,8]. In Saccharomyces cerevisiae, PC is the most abundant phospholipid, comprising ~45% of all mitochondrial phospholipids, followed by PE, which comprises ~25% of total mitochondrial phospholipids. CL is found almost exclusively in mitochondria and constitutes ~15% of total mitochondrial phospholipids [9]. The phospholipid composition of the outer mitochondrial membrane (OMM) is significantly different from that of the inner mitochondrial membrane (IMM) (Fig. 2B). In yeast, PC comprises ~50% and ~40% of all phospholipids in the OMM and IMM, respectively, whereas PE and CL are enriched in the IMM (Fig. 2B) [3,8]. Furthermore, phospholipids are not evenly distributed across the lipid bilayer leaflets of the mitochondrial membranes, but are instead arranged asymmetrically. For example, in yeast, 76% of total PE is found on the inner leaflet of the OMM facing the intermembrane space (IMS) [10]. In the case of rat liver mitochondria, the majority of PE and CL in the OMM are found in the cytosolic leaflet, whereas PI and PS are preferentially oriented towards the IMS [11]. Data from the mammalian system indicates asymmetric phospholipid distribution in the IMM leaflets as well, with specific enrichment of PI and CL in the matrix-facing leaflet of the IMM [3]. A number of earlier studies reported tissue-specific and method-specific differences in the asymmetric distribution of phospholipids within the IMM of mammalian cells [12,13]. Regardless of these discrepancies, the unique composition and asymmetric phospholipid distribution of the mitochondrial membranes suggests a specialized physiological role of phospholipids.
The mitochondrial phospholipid composition of yeast varies with growth conditions and growth media (Table 1) [9,14,15]. Typically, the levels of the mitochondrial non-bilayer phospholipids PE and CL are increased when yeast are cultured in a non-fermentable carbon source like lactate (Table 1) [9,15]. The increased levels of non-bilayer phospholipids in non-fermentable growth conditions correlate with increased biogenesis of MRC components, reflecting their specific requirement for respiration. It is also important to keep in mind that complex media containing yeast extract consists of a number of soluble lipid precursors, including ethanolamine (Etn), choline, and inositol. These phospholipid precursors are incorporated into cells and contribute to phospholipid biosynthesis via alternate pathways, masking any defects caused by mutations in genes encoding core phospholipid biosynthetic pathways [15,16].
Table 1. Mitochondrial phospholipid levels of the yeast Saccharomyces cerevisiae cultured in different growth media.
Phospholipid levels are expressed as percentage (%) of total phospholipid. The data for YPD and YPLactate have been taken from [9]. The data for SCLactate has been taken from [15]. Y: yeast extract, P: bactopeptone; D:Dextrose; SC: synthetic complete.
| Growth Media | PC | PE | CL | PI | PS | PA | Others |
|---|---|---|---|---|---|---|---|
| YPD | 33.4±0.8 | 22.7±2.7 | 7.2±0.2 | 20.6±0.9 | 3.3±0.5 | 1.7±0.5 | 11.1±0.7 |
| YPLactate | 39.8±0.6 | 26.4±0.8 | 15.7±0.9 | 10.9±0.5 | 2.3±0.8 | 1.7±0.3 | 3.2±0.7 |
| SCLactate | 48.5±2.0 | 24.4±1.2 | 15.6±1.8 | 5.6±1.2 | 1.3±0.3 | 2.4±0.7 | 2.1±1.0 |
Biosynthetic and trafficking pathways of CL and PE
CL biosynthesis takes place exclusively in mitochondria from the PA imported from ER (Fig. 3). The mechanism by which PA is imported from the ER to the OMM is not known, however, the transport of PA from the OMM to the IMM has been shown to be carried out by the IMS protein Ups1 (Fig. 3) [17]. The enzymes of the CL biosynthetic pathway are localized to the IMM. Tam41 mediates the first reaction of the CL biosynthetic pathway by catalyzing the conversion of PA to CDP-diacyl glycerol (CDP-DAG) [18]. In the subsequent steps, CDP-DAG is converted to nascent CL via sequential action of Pgs1, Gep4 and Crd1 [19–21]. The topology of all these reactions has been determined to be in the matrix-facing leaflet of the IMM [22]. An acyl chain of newly synthesized CL is remodelled through the sequential action of cardiolipin-specific deacylase, Cld1, and a transacylase, Taz1, to generate remodelled CL [23,24]. Cld1 is also a matrix-facing enzyme associated with the IMM [25]. However, unlike the other enzymes of the CL biosynthetic pathway, Taz1 has been shown to be present in both the OMM and IMM, with its active site facing the IMS in both cases [26,27]. This topology of Taz1 necessitates the flipping of monolyso cardiolipin (MLCL) from the inner leaflet of the IMM to the IMS-facing leaflet of the IMM. So far, proteins required for mediating CL redistribution in the mitochondrial membranes, including flippases, have not been reported, although a mitochondrial phospholipid scramblase (PLS3) has been suggested to facilitate transbilayer lipid trafficking in mammalian systems [28]. Since CL levels of the OMM are distinct from those of the IMM, regulated mechanisms of CL transport from its site of synthesis in the IMM to the OMM must exist. Phospholipid transport between the IMM and OMM has been proposed to occur at membrane contact sites, specialized structures where two mitochondrial membranes (IMM and OMM) are in close vicinity. The discovery of the Mitochondria Contact Site and Cristae Organizing System (MICOS) complex offers a powerful genetic tool to rigorously test the role of contact-sites in mediating inter-membrane phospholipid transport within mitochondria [29–31]. Details of intra-mitochondrial phospholipid trafficking and the topology of phospholipid biosynthetic enzymes have been reviewed elsewhere [22,32].
Figure 3. Biosynthetic and trafficking pathways of non-bilayer forming phospholipids.
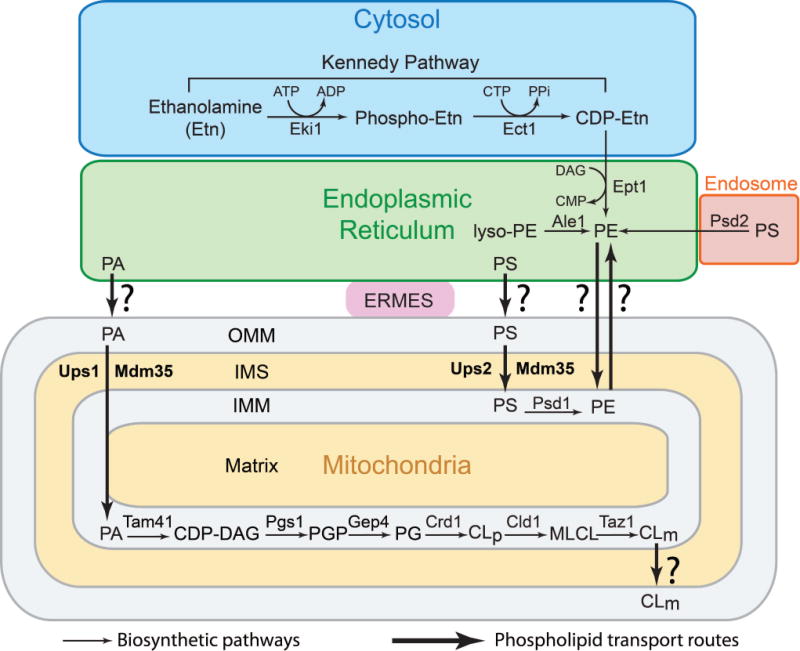
PA required for CL biosynthesis is imported from the ER to the OMM by an unknown mechanism. Ups1-Mdm35 mediates transport of PA from OMM to the IMM. CL biosynthesis occurs exclusively in the mitochondria via four enzyme-catalysed reactions carried out by IMM-associated Tam41, Pgs1, Gep4, and Crd1. The resulting nascent CL (CLp) undergoes remodeling via sequential deacylation and reacylations reactions catalysed by Cld1 and Taz1, respectively, to form mature CL (CLm). The mechanism by which CLm is transported from the IMM to the OMM remains to be elucidated. PE biosynthesis in yeast is accomplished by four different pathways: 1) Psd1-catalyzed decarboxylation of PS in the mitochondria, 2) Psd2-catalyzed decarboxylation of PS in the endosome, 3) incorporation of Etn in to PE via the cytosolic/ER Kennedy pathway enzymes Eki1, Ect1, and Ept1, 4) Ale1 catalysed conversion of lysoPE to PE in the ER or the mitochondrial associated membranes. The mechanism by which PS is transported from ER to mitochondria remains unclear, although membrane contact site structures like the ER-mitochondria encounter structure (ERMES) has been proposed to play a role in this process. Ups2 and Mdm35 transport PS from the outer mitochondrial membrane (OMM) to the inner mitochondrial membrane (IMM). PE biosynthesized in the ER can also be trafficked to IMM by an unknown mechanism. The mechanism of PE export is also currently unknown. ATP, adenosine triphosphate; ADP, adenosine diphosphate; CTP, cytidine triphosphate; PPi, pyrophosphate; CDP, cytidine diphosphate; DAG, diacylglycerol; CMP, cytidine monophosphate; PA, phosphatidic acid; PS, phosphatidylserine; PE, phosphatidylethanolamine; CLp, precursor cardiolipin; CLm, mature cardiolipin; MLCL, monolysocardiolipin; PG, phosphatidylglycerol; PGP, phosphatidylglycerol phosphate.
PE biosynthesis in S. cerevisiae can be accomplished by the decarboxylation of PS [33,34], acylation of lyso-PE [35], and by the cytidine diphosphate-ethanolamine (CDP-Etn) branch of the Kennedy pathway (Fig. 3) [36]. An IMM associated phosphatidylserine decarboxylase 1 (Psd1) that has its active site exposed to IMS catalyses the biosynthesis of PE by decarboxylation of PS [37]. Because PS is synthesized in the endoplasmic reticulum (ER), it has to be transported to the IMM to be accessible to the active site of Psd1. Recent studies have identified a protein complex comprised of Ups2 and Mdm35 in the mitochondrial IMS that is involved in PS transport from the OMM to the IMM (Fig. 3) [38,39]. Interestingly, it was also shown that Psd1 localized to the IMM can work in trans by decarboxylating PS present in the OMM, obviating the requirement of Ups2-Mdm35 for PE biosynthesis in the mitochondria [39]. For Psd1 to act on PS in the OMM, a close apposition of the OMM and IMM is required, which is achieved by MICOS [39]. While the mitochondrial PE biosynthesis pathway is the primary source of cellular PE, a second yeast specific phosphatidylserine decarboxylase 2 (Psd2) found in an endosomal compartment also contributes to the cellular PE content, albeit to a lesser extent (Fig. 3) [34,40].
In addition to these de novo biosynthetic pathways, PE can be synthesized from its precursor, Etn, through the Kennedy pathway enzymes that are localized in the cytosol and ER compartments (Fig. 3) [36,41]. Exogenously added Etn is converted to PE by three sequential reactions catalysed by the enzymes ethanolamine kinase (Eki1), ethanolamine-phosphate cytidylyltransferase (Ect1), and ethanolamine phosphotransferase (Ept1), respectively (Fig. 3). Another route of PE biosynthesis is through the acylation of lyso-PE, catalyzed by the acyl-CoA-dependent acyltransferase Ale1, in the mitochondria-associated membranes [35]. The contribution of this pathway towards cellular PE under basal conditions remains to be elucidated. A functional difference between PE produced by the mitochondrial and Kennedy pathways can be presumed from the observation that each pathway is essential in mammals, as shown by the embryonic lethality of the knockouts of mouse homologues of PSD1 (PISD) and ECT1 (PCYT2) [42,43]. Moreover, each pathway preferentially generates PE with different acyl species, implying their compartment specific roles, although rapid equilibration between PE formed in the ER with that of mitochondria was observed [44].
The ability of extra-mitochondrial PE to functionally substitute for the lack of mitochondrial PE has long been debated [41,45]. Recently, our group showed that in yeast, non-mitochondrial PE biosynthesized via the Kennedy pathway is transported to mitochondria, where it functionally compensates for the loss of mitochondrial PE biosynthesis [15]. This finding implies the existence of mitochondrial PE import machinery. However, the molecular players of this mitochondrial PE import pathway are currently unknown. Membrane contact sites between mitochondria and the ER [46] or mitochondria and vacuoles [47,48] have been suggested to play a role in transport of lipids between these organelles. The contact sites between the ER and mitochondria, termed Endoplasmic Reticulum-Mitochondria Encounter Structure (ERMES) (46), form an ER-mitochondria tether, but ERMES’s role in phospholipid transport remains controversial [49]. Our own work has shown that deletion of the subunits of the ERMES complex in psd1Δ yeast cells did not stop Kennedy pathway synthesized PE from being imported into the mitochondria, thus ruling out ERMES as potential PE import machinery [15]. The vacuole-mitochondria tethering complex, termed vacuole and mitochondria patch (vCLAMP), provides a molecular basis of communication between the mitochondria and vacuoles [47,48]. It has been suggested that vCLAMP might facilitate lipid transport between vacuoles and mitochondria, but this has not been rigorously tested.
Regulation of CL and PE levels in the mitochondria
The steady state levels of CL and PE in mitochondrial membranes are regulated by the combined activities of biosynthetic, trafficking, and degrading enzymes. As described above, the molecular identities of biosynthetic and trafficking proteins in the CL and PE pathways are known, but the same cannot be said for the enzymes involved in the turnover of these phospholipids. Therefore, the regulation of CL and PE levels has been mostly studied from the perspective of biosynthesis and trafficking. A number of factors govern the expression and activity of CL biosynthetic enzymes. For example, expression and activity of Crd1 is regulated by mitochondrial biogenesis factors, including the carbon source, growth phase, presence of mitochondrial DNA, and functionality of the MRC [50,51]. In each case, factors that promote mitochondrial biogenesis stimulate expression and activity of Crd1 and other enzymes of this pathway. One unique mode of regulation reported for Crd1 is its response to mitochondrial matrix pH, such that an alkaline pH stimulates Crd1 activity [52]. An increase in matrix pH is expected in respiring mitochondria, as protons are being pumped out of the matrix into the IMS. Thus, CL biosynthesis is co-ordinately regulated with mitochondrial energy production.
In addition to mitochondrial biogenesis factors, inositol, a phospholipid precursor, regulates the activity of a rate-limiting enzyme of the CL biosynthetic pathway, Pgs1, by phosphorylation [53]. Inositol-induced phosphorylation of Pgs1 reduces its activity by 50% [53]. However, the identity of the kinase mediating the phosphorylation and the physiological relevance of this observation has not been determined. More importantly, the effect of inositol on steady-state levels of CL has also not been determined.
Nascent CL undergoes rapid remodelling by Taz1 so that its substrate MLCL does not accumulate in wild type cells, and only deletion of Taz1 leads to the accumulation of MLCL. Schlame and colleagues sought to explain the mechanism underlying MLCL accumulation in TAZ deficient cells [54]. Their work led to an interesting finding that MLCL accumulation is observed when there is an increased turnover of CL, implying that MLCL is formed during CL degradation. In an elegant study, they showed that CL turnover is slower than other phospholipids due to its sequestration by supercomplexes in the mitochondrial membranes. Thus, enhanced supercomplex assembly decreased both the turnover of CL and the concentration of MLCL. In TAZ deficient cells where MRC supercomplexes are destabilized, CL is subjected to rapid turnover with the accumulation of MLCL.
PE levels in mitochondria are regulated by PS import, Psd1 activity, and PE export pathways. The biochemical pathway by which PE is exported from the mitochondria is currently unknown. It would be expected that blocking the PE export pathway would elevate mitochondrial PE levels. Interestingly, mitochondria can also import PE synthesized via the Etn-Kennedy pathway or the Psd2 pathway (Fig. 3). The relative contribution of each of these pathways to mitochondrial PE levels is relatively minor [41] unless the Etn-Kennedy pathway is stimulated by addition of exogenous Etn [15]. Finally, the maturation and activation of Psd1 in mitochondria has been shown to involve multiple proteolytic processes involving autocatalysis and mitochondrial proteases that include Oxa1 and Yme1 [55]. Indeed, deletion of Oxa1 has been shown to decrease mitochondrial PE levels [55]. Psd1 activity has also been shown to be growth-phase dependent, with maximum activity in cells entering the stationary phase [56]. Similar to biosynthetic regulation of CL, the phospholipid precursor inositol has been shown to supress Psd1 expression [57]. Thus, perturbation in Psd1 maturation or expression leads to a decrease in mitochondrial PE levels [58].
Another mode of maintaining mitochondrial CL and PE levels is by regulating the transport of their phospholipid precursors, PA and PS respectively, from the ER to their biosynthetic machinery localized to the IMM. A number of key transport proteins in this process were discovered by an unbiased genome-wide yeast screen designed to understand the function of prohibitins, multimeric protein complexes of mitochondrial membranes with a proposed role in cell proliferation and cristae morphogenesis [59]. The screen identified a set of genes termed Genetic interactors of Prohibitins (GEP) that were involved in regulating the levels of CL, PE, or both. Biochemical investigations on the role of GEP proteins led to the discovery of Ups1, an IMS protein that was shown to be required for PA transport from the OMM to the IMM [17]. The loss of Ups1 leads to decreased CL levels [17]. However, deletion of UPS1 does not completely abrogate CL biosynthesis, suggesting alternative modes of PA transport to the IMM. Ups2, also known as Gep1, another IMS protein, was recently shown to be involved in PS transport from the OMM to the IMM, such that deletion of UPS2 would result in reduced mitochondrial PE levels [38,39]. Interestingly, both Ups1 and Ups2 work in conjunction with Mdm35 in the IMS to facilitate PA and PS transport, respectively. In addition to these proteins, the same screen also identified a number of GEP proteins, including the ERMES components that regulate PE and/or CL levels in mitochondrial membranes [59]. The precise biochemical function of some of these GEP proteins in the regulation of mitochondrial non-bilayer phospholipids is yet to be delineated.
Analysis of phospholipid profiles of psd1Δ, crd1Δ, and pem2Δ cells showed that loss of one phospholipid is compensated by an increase in absolute amount of other phospholipids such that the overall phospholipid content of mitochondria and cells is maintained at a constant level [15]. For example, depletion of PE in psd1Δ is always accompanied by an elevation in the levels of PC, whereas loss of CL in crd1Δ is accompanied with increased PE and PG levels [15,60]. Currently, the molecular basis for homeostatic mechanisms that strictly maintain mitochondrial phospholipid levels remains unknown. Such homeostatic mechanisms could either involve a “sensor” protein or could simply be explained by feedback inhibition of phospholipid biosynthetic enzymes. Future research will be needed to distinguish between these possibilities. The coordinate regulation of mitochondrial CL and PE levels suggests that a critical amount of these non-bilayer forming phospholipids is required for mitochondrial function.
Role of CL in mitochondrial structure and function
The availability of CL deficient cells in different model systems, including yeast, fly, mouse, and human patient cell lines, has proved invaluable in uncovering the role of CL in mitochondrial function in different organisms [21,61–63]. Below we provide key examples from the studies performed using these model systems as well as in vitro experiments showing how CL influences various aspects of mitochondrial structure and function. CL can impact mitochondrial function either through specific molecular interactions between itself and target proteins or by modulating the bulk properties of membranes due to its propensity to form non-bilayer structures. While in many cases it is hard to distinguish between these two possibilities, we have highlighted the instances where strong evidence for one or the other exists.
Role of CL in mitochondrial inner membrane architecture
The IMM can be divided into two distinct regions: the inner boundary membrane, which runs parallel to the OMM, and the cristae – tube-like invaginations of the IMM that project into the matrix [64]. While cristae architecture is variable and depends on the energetic state of the organelle, the overall structure of cristae is very much conserved – a dome-shaped tip caps the tubular cristae [64]. The enrichment of non-bilayer lipids in the negatively curved monolayer leaflet facing the cristae lumen would favour dome-shaped tip formation and makes for a compelling argument for the asymmetric distribution of IMM phospholipids. But are the non-bilayer phospholipids necessary to produce this distinctive cristae architecture? This question can be answered by electron microscopic observations of mitochondrial ultrastructure from CL and PE deficient mitochondria. Interestingly, in the yeast model, the loss of CL seems to have a very small effect on cristae length and the gross mitochondrial ultra structure [65, Basu Ball, unpublished results]. However, disruption of CL remodelling in a Drosophila model of Barth syndrome (BTHS) resulted in structurally abnormal mitochondria with hyper-dense cristae that formed swirls, curls, or rings [61]. Similarly, enlarged mitochondria with abnormal, circularly-stacked cristae were observed in BTHS patient cardiac cells [66] and in the muscle of a murine model of BTHS [62]. These findings support a critical role of CL in cristae architecture. In contrast, our own work has shown that the gross mitochondrial ultra structure in the yeast mutants of PE, PC, and CL biosynthesis is highly resistant to perturbations in mitochondrial phospholipid composition [15, Basu Ball unpublished results]. One possible explanation of this divergent observation between different species is that CL deficient yeast accumulates PG and PE, which may compensate for CL [14]. In support of this argument, the loss of both CL and PG in yeast pgs1Δ cells results in aberrant mitochondria containing ring-like cristae, which are reminiscent of the MICOS mutants [17]. It has also been shown that CL is required for the assembly of the MICOS sub-complex, thus it may mediate its impact on mitochondrial architecture in part via its effect on assembly of the MICOS sub-complex [67]. Notably, the loss of PE is not compensated for by an increase in CL in psd1Δ cells, yet they exhibit typical cristae morphology, albeit with reduced cristae length [15]. To conclusively determine the role of non-bilayer forming phospholipids in cristae architecture a psd1Δcrd1Δ double mutant would be required. However, this mutant is lethal [60]; therefore, it can only be useful when the levels of mitochondrial PE and or CL in this mutant can be exogenously controlled, as has been previously demonstrated [68].
Role of CL in mitochondrial membrane dynamics
Although bilayer-forming PC accounts for ~50% of the phospholipids in the membranes, the inclusion of non-bilayer forming phospholipids like CL in bilayers imposes a curvature stress onto the membrane, which facilitates budding, fission and fusion. Thus, these biophysical properties of CL have the potential to impact mitochondrial membrane dynamics. Consistent with its biophysical characteristics, CL is critical for fusion of the inner membrane via its interaction with the dynamin-related protein OPA1, a GTPase, which mediates inner membrane fusion. A recent study has shown that the membrane fusion of OPA1-containing liposomes is dependent on the presence and abundance of CL [69]. In this elegant study, the authors demonstrated that when donor and acceptor liposomes containing high concentrations of CL (~25% of total phospholipids) and Opa1 are mixed, maximum lipid mixing and membrane fusion is observed. In trials when both the liposome partners either lack Opa1 machinery or contain only a minimal amount of CL, no membrane fusion was observed. Interestingly, in this experimental set up, the presence of Opa1 in one fusion partner and high CL levels in the other is sufficient for membrane fusion. Thus, besides defining the minimal fusion machinery, these results suggest that a critical level of CL alone primes the membrane for fusion. This finding is an important advancement in our understanding of membrane fusion because, in contrast to previously reported fusion mechanisms, no trans-protein complexes for membrane tethering are required. Furthermore, the authors went on to show that the acyl chain length and saturation of CL also influences membrane fusion, with longer length and higher unsaturation enhancing fusion. These in vitro findings were confirmed in vivo using mitochondria from OPA1 and CLS1 (cardiolipin synthase) knockdown HeLa cells. The yeast homolog of OPA1, Mgm1, also requires CL for dimerization and activation of its GTPase activity for membrane fusion [70]. However, unlike OPA1, Mgm1 works in trans [70]. Thus, the role of CL in mitochondrial fusion appears to be evolutionarily conserved.
The most obvious role for CL in the fission pathway involves Drp1, the protein that directly mediates the fission process. CL has been shown to mediate both Drp1 recruitment to membrane surfaces and to activate Drp1’s GTPase activity through protein-lipid interactions [71,72]. A recent study by Stepanyants et al., [73] showed that Drp1 and CL function cooperatively in effecting fission in three distinct steps. First Drp1 preferentially associates with CL localized at a high spatial density in the membrane bilayer. Second, CL molecules reorganize around Drp1. Third, Drp1 promotes phase transition of CL containing membranes from a lamellar, bilayer arrangement to an inverted hexagonal, nonbilayer configuration resulting in the creation of localized membrane constrictions that are primed for fission [73]. On the other hand phosphatidic acid (PA), a relatively minor non-bilayer constituent of the mitochondrial membranes, has recently been shown to be a negative regulator of mitochondrial fission [74]. In this elegant work, Adachi et al., showed that Drp1 interacts with the head group of saturated PA in the OMM. Drp1 finds PA in the OMM by physically interacting with mitochondrial phospholipase D (MitoPLD), an enzyme that catalyses the formation of PA from CL. The interaction of Drp1 with MitoPLD places it in PA rich membrane microdomains. Once bound to the PA head group, the GTPase activity of Drp1 is inhibited, which in turn prevents mitochondrial division [74]. Thus, two non-bilayer phospholipids of mitochondria, PA and CL, antagonistically regulate mitochondrial fission by differentially interacting with Drp1 GTPase. Taken together, mitochondrial membrane fusion and fission are modulated by local non-bilayer phospholipid concentrations and are likely dependent on both specific phospholipid:protein interactions as well as curvature stress induced by the promotion of local non-bilayer structures.
Role of CL in mitochondrial bioenergetics
The exclusive presence of CL within membranes associated with energy transduction reactions suggests an intimate association of CL with bioenergetics. The most striking evidence for the role of CL in influencing mitochondrial bioenergetics comes from the studies involving respiratory supercomplexes in yeast and mammals. The individual components of mitochondrial respiratory chain (MRC) complexes assemble into supramolecular structures called supercomplexes. These respiratory supercomplexes are proposed to stabilize individual MRC complexes, minimize the generation of reactive oxygen species, and enhance catalytic efficiency by substrate channeling [75]. In mammals, complex I, III, and IV assemble in different stoichiometries to form ‘respirasomes’ [76–78]. In the yeast S. cerevisiae, which lack complex I, the respiratory supercomplexes are composed of complex III and IV assembled in different stoichiometries- III2IV2 (large supercomplex) and III2IV1 (small supercomplex) [76]. CL has been shown to stabilize the respiratory supercomplexes in both yeast and mammals [79–81]. Structural studies with purified yeast and bovine MRC supercomplexes employing cryoelectron microscopy have estimated the presence of 50 and 200 molecules of CL in yeast and bovine MRC supercomplexes, respectively [82,83]. The most direct evidence for the role of CL in MRC supercomplex formation comes from an in vitro reconstitution experiment with the purified yeast complexes, which showed that supercomplex formation specifically required the presence of CL in liposomes [84]. These data together suggest that specific molecular interactions of CL with the MRC proteins are essential for the supramolecular assembly of the MRC.
Apart from maintaining the overall structural integrity of the MRC supercomplexes, CL has been shown to directly interact with individual MRC complexes and influence their activities. Previous studies have shown reduced MRC complex III activity in BTHS patient mitochondria [85] and diminished complex IV activity in yeast crd1Δ cells [79], although this could be due to reduced expression of the MRC components, as has been shown in yeast cells devoid of both CL and PG [86]. The presence of CL is observed in the cryo-electron microscopic structures of complex III and complex IV [82,83], and the topology of CL in complex III and IV suggests that it is required for proton conductance [87–89]. Although CL has never been associated with complex II previously, in a recent study complex II subunits reconstituted on phospholipid nanodiscs were shown to require CL for assembly and activity [90]. These interactions of CL with MRC complexes define the role of CL in regulating mitochondrial bioenergetics. The physiological consequences of the loss of CL and, therefore, CL-MRC interactions, have been studied by classical bioenergetics experiments utilizing mitochondria isolated from CL deficient crd1Δ and taz1Δ yeast cells [91–93]. Mitochondria from CL deficient cells show reduced energetic coupling (i.e. decreased ATP formation per molecule of oxygen consumed). This phenotype is exacerbated during high respiratory rates and by temperature and osmotic stress [91,92]. Consistent with these observations in the yeast system, ATP levels are reduced in TAZ deficient cardiac fibroblasts [94]. Consistent with its role in maintaining MRC assembly and activity, CL deficient mitochondria exhibit diminished mitochondrial membrane potential [95].
In addition to directly influencing assembly and activity of the MRC complexes, CL has also been shown to modulate the activities of a number of IMM carrier proteins. We refer readers to a more detailed review on specific interactions of CL with mitochondrial integral membrane proteins published recently [96]. Here we provide key examples of specific interactions of CL with mitochondrial integral membrane proteins that may indirectly influence mitochondrial bioenergetics. The ADP/ATP carrier (AAC) mediates transport of ATP from the matrix to the IMS in exchange for ADP, and the activity of this protein requires tetralinoleoyl-CL. This species of CL cannot be replaced by either other species of CL or other phospholipids, emphasizing the requirement for specific molecular interactions between tetralinoleoyl-CL and AAC [97]. The absence of CL destabilizes AAC complex and its interaction with MRC supercomplexes, reducing the efficiency of mitochondrial ATP synthesis and its transport outside of mitochondria [98]. A recent study has uncovered a role of CL in acetyl CoA biosynthesis via possible interaction with OMM localized acetyl CoA synthetase [99]. Interestingly, CL deficiency also leads to defects in mitochondrial protein import pathways. CL has been shown to be involved in the proper functioning of the mitochondrial translocase of outer membrane (TOM) and sorting and assembly machinery (SAM), compromising the efficient import of cytoplasmic proteins to mitochondria [100]. CL also impacts the translocation of proteins across the IMM via translocase of the inner membrane (Tim23) and the presequence translocase-associated motor (PAM) protein. A decrease in CL, caused by the loss of Ups1, alters the conformation of Tim23, reducing its functional interaction with PAM and thereby decreasing mitochondrial protein import [101]. Additionally, reduced membrane potential observed in CL deficient cells also contributes to decreased mitochondrial protein import [95]. Details on how CL impacts mitochondrial protein import have been recently reviewed [102].
Role of CL in mitophagy
A number of studies have demonstrated the importance of mitochondrial CL in influencing the mitochondrial quality control process through mitophagy, a selective degradation of mitochondria through autophagy. In an elegant study, Chu and colleagues showed that externalization of CL to the OMM of neurons serves as a signal for mitophagy to eliminate damaged mitochondria [103]. They further showed that CL on the OMM is recognized by microtubule-asociated-protein-1 light chain 3 (LC3), which promotes engulfment of mitochondria by the autophagic system. The role of CL in mitophagy is likely dependent on its specific interaction with LC3, which has CL binding sites required for mediating mitochondrial degradation by mitophagy [103,104]. A more recent study has identified the surprising role of nucleoside diphosphate kinase D, an IMS protein, in transport of CL from the IMM to the OMM, priming mitochondria for mitophagy [105]. Consistent with these observations, a decrease in CL in TAZ deficient primary mouse embryonic fibroblasts, resulted in defective mitophagosome biogenesis [106]. This finding provides new insights into the possible role of mitophagy in BTHS disease pathogenesis and suggests that pharmacological stimulation of mitophagy may ameliorate this disease condition. The role of CL in mitophagy appears to be conserved because a recent study showed that CL deficiency perturbs mitophagy in yeast cells via decreased activation of protein kinase c pathway [107].
Thus, genetic studies in different model systems have uncovered important mechanisms underlying mitochondrial dysfunction and disease pathologies associated with CL depletion. Going forward, developing a physiologically relevant small molecule that can promote supercomplex formation, increase MRC complex activities, and curb mitochondrial oxidative stress will be required to ameliorate CL-associated mitochondrial dysfunction. Towards this goal, novel approaches and promising compounds have emerged [108].
Role of PE in mitochondrial structure and function
PE is an ancient and ubiquitous phospholipid that is present in all membranes of the cell. Unlike CL, the specific role of PE in mitochondrial structure and function has not been examined until recently. This is because multiple biochemical pathways can synthesize PE and deletion of the mitochondrial pathway of PE alone is not sufficient to completely deplete mitochondrial PE levels [109]. Furthermore, PE has an essential role in cell growth independent of its ability to form non-bilayer structures [110]. Therefore, it is not feasible to construct a yeast strain completely lacking PE. Thus, yeast psd1Δ cells, which are unable to synthesize mitochondrial PE, are the best possible model system to investigate the role of PE in mitochondrial function. However, when cultured in glucose-containing growth medium, this mutant exhibits a rapid loss of mitochondrial DNA, confounding the interpretation of its role in specific aspects of mitochondrial function [15,41]. The loss of mitochondrial DNA would disrupt multiple complexes of the MRC, which would mask a subtler role of PE in mitochondrial bioenergetics. In a mouse model, it was shown that loss of the mammalian homolog of PSD1, termed PISD, results in embryonic lethality, preventing any biochemical or physiological studies on PE deficient mammalian mitochondria [42]. This limitation was overcome by using RNAi silencing of PISD [111]. In the case of yeast, the loss of mitochondrial DNA can be overcome by using a defined synthetic growth media with a non-fermentable carbon source. Both of these strategies were employed recently in uncovering the role of mitochondrial PE. Below we discuss key findings from these studies and refer readers to recent reviews for a more detailed description of the role of PE in cellular physiology [56,58].
Role of PE in mitochondrial ultrastructure
Mitochondrial ultrastructure in PE deficient mitochondria has been determined in both yeast and mammalian cell lines. Like CL, in psd1Δ yeast cells mitochondrial ultrastructure was largely unperturbed even with an almost five-fold reduction in mitochondrial PE levels [15], although this reduction in PE levels was associated with a small but significant decrease in cristae length [15]. In PISD knockdown mammalian CHO-K1 cells, the reduction in mitochondrial PE levels was associated with a more pronounced increase in aberrant mitochondria, which were more rounded and swollen compared to the control mitochondria [111]. The apparent discrepancy between yeast and higher eukaryotic cells could be due to the ability of yeast cells to alter their phospholipid composition in order to maintain optimal intrinsic membrane curvature. For example, the loss of mitochondrial PE is accompanied by an increase in PC levels and vice-versa, such that the overall phospholipid content of mitochondria remains unaltered [15]. The alteration in phospholipid content in phospholipid mutants is typically accompanied by changes in the fatty acyl species, such that the intrinsic membrane curvature is maintained. For example, it was previously shown that the depletion of bilayer forming PC in yeast cells results in PE molecules with shorter and more saturated acyl chains, increasing its bilayer-forming propensity. This molecular adaptation maintains intrinsic membrane curvature in an optimal range [112]. Therefore, it would not be surprising to find that phospholipid species in PE deficient cells have a different fatty acid composition that has a higher propensity to form non-bilayer structures.
Role of PE in mitochondrial dynamics
Mitochondria are dynamic organelles that frequently fuse and divide to maintain their function and morphology. As mentioned previously, members of the dynamin-like GTPase protein family mediate mitochondrial fission and fusion. IMM fusion involves OPA1 in mammals and its homolog, Mgm1, in yeast. Light microscopy-based studies on psd1Δ yeast cells have shown increased accumulation of both fragmented and aggregated mitochondria, suggesting defects in mitochondrial dynamics [45]. Further analysis of these mitochondria revealed a defect in fusion. The molecular basis for this observation was traced to a decrease in s-Mgm1, a short isoform of Mgm1, in psd1Δ yeast cells [59,68]. How exactly PE impacts Mgm1 processing remains to be elucidated, though this relationship appears to be PE-specific and unrelated to its propensity to form non-bilayer structures because Mgm1 processing is unaltered in CL deficient mitochondria. The role of PE in membrane fusion is also supported by in vitro experiments that demonstrate decreased rates of lipid mixing in protein-free liposomes that mimic mitochondrial membranes with reduced PE levels [45]. Interestingly, a yeast mutant with the combined loss of PE and CL exhibited highly fragmented mitochondria, loss of mitochondrial DNA, and reduced membrane potential, which is characteristic of fusion mutants [68]. Remarkably, deletion of the fission promoting protein Dnm1 in this mutant restored the tubular mitochondrial morphology. These results suggest that PE has an important role in maintaining mitochondrial dynamics that becomes essential in the absence of CL. Based on these findings it can be assumed that PE, in addition to its specific role in Mgm1 processing, has an additional role as a non-bilayer promoting phospholipid in mitochondrial dynamics. Another example of the role PE and CL play in mitochondrial fission comes from a yeast genetic study on Mdm33, a protein essential for normal mitochondrial structure. Defective mitochondrial fission with a concomitant decrease in PE and CL levels is observed in Mdm33 overexpressing cells [113]. Furthermore, strong genetic interactions were reported between Mdm33, phospholipid biosynthetic genes, and prohibitin genes, indicating a complex genetic network that maintains mitochondrial architecture by regulating fusion and fission machinery [113].
Role of PE in mitochondrial bioenergetics
The identification and characterization of the yeast PSD1 provided the first clue for the requirement of PE in MRC function [33]. The psd1Δ cells exhibited impaired growth in non-fermentable growth medium and showed increased petite formation, a hallmark of MRC dysfunction in yeast [33,41]. While these phenotypes of psd1Δ cells were reported over two decades ago, the biochemical basis for MRC dysfunction in PE-depleted mitochondria has only been recently investigated. The decreased growth of psd1Δ cells in non-fermentable media has been attributed to reduced respiration and a decrease in ATP levels caused by a reduction in MRC complex III and IV activities [15,114]. While these findings were consistent between three independent groups, there is less consensus on the role of PE in MRC supercomplex formation. One of the reports suggested that mitochondrial PE deficiency leads to larger oligomeric assembly of MRC supercomplexes [114], whereas another report suggested that PE deficiency neither promotes higher-order MRC complexes nor perturbs MRC supercomplex assembly [15]. Notably, neither study reported a loss of supercomplex formation in PE deficient cells, highlighting a specific requirement of CL for supercomplex formation. Furthermore, it was shown that a decrease in the bilayer-forming PC did not disrupt supercomplex formation but rather shifted the supercomplex distribution towards the larger form [15]. Consistent with the role of PE in MRC function in yeast, depletion of mitochondrial PE in Arabidopsis thaliana resulted in decreased respiration and decreased complex IV activity without any alteration in individual MRC subunit levels, suggesting a specific role of PE in the catalytic activity of MRC complexes [115]. PE requirement for the optimal activity of complex IV could be explained from its crystal structure analysis. The crystal structure of complex IV from bovine heart identified a number of phospholipids, with PE being one of the most abundant, suggesting its possible role in enzyme catalysis [116]. PE has been shown to act as a molecular chaperone that guides the topology of integral membrane proteins in bacteria [117]; however, this phenomenon has not been demonstrated in eukaryotic cells. Because mitochondrial structure and function of PE deficient mitochondria is not grossly affected, it is unlikely that PE will have a wide spread effect on the membrane topology of multiple mitochondrial proteins.
Consistent with yeast psd1Δ cells, inhibition of mitochondrial PE biosynthesis in mammalian cell lines by RNAi based knockdown of PISD resulted in reduced respiration, decreased ATP production, and diminished activities of MRC complexes [111]. The abundance of respiratory chain supercomplexes in PE depleted CHO cells was reduced, likely due to an overall reduction in subunits of individual complexes [111]. An interesting role of PE in hepatocyte mitochondrial membranes has been proposed for glucose metabolism and oxidative phosphorylation in mouse liver, where about 20–30% of PC is synthesized by the methylation of PE via the enzymatic action of phosphatidylethanolamine methyltransferase (PEMT). The hepatocytes of pemt−/− mice have increased mitochondrial PE levels with concomitant increase in MRC complex IV activity, respiration, and ATP production [118]. This increase in respiratory function correlated with an increase in the mitochondrial PE to PC ratio. Although the molecular mechanism underlying the increase in mitochondrial function with an increased PE/PC ratio remained unclear, this study highlighted a novel mechanism of stimulating MRC function.
Role of PE in mitochondrial protein import
PE deficient mitochondria are specifically impaired in the biogenesis of β-barrel proteins, but not α-helical proteins of the OMM [119]. PE mediates this effect by compromising the function of the protein import machinery of the OMM rather than affecting its stability. In PE-depleted mitochondria, the translocase of outer membrane (TOM) complex binds precursor proteins with reduced efficiency, affecting protein import at an early stage. Subsequent translocation of β-barrel precursors through the TOM complex into the IMS is also reduced in PE-depleted yeast cells. In addition, mitochondrial PE is also required for the import of IMM proteins [114]. The import of pre-proteins into and across the IMM through the translocases of the inner membrane (TIM) complexes (TIM22 and TIM23) is driven by the mitochondrial membrane potential across the IMM, which is generated by the optimal functioning of the MRC. Because MRC activity is reduced in psd1Δ yeast cells, the membrane potential is also reduced, resulting in mitochondrial protein import defects [114]. Interestingly, the mitochondrial protein import defect is not specific to the non-bilayer forming phospholipids. A recent report has shown that a depletion in PC levels impairs mitochondrial protein import by destabilization of TIM23 [120]. Thus, both bilayer and non-bilayer phospholipids impact mitochondrial protein import, albeit by different mechanisms. Even though in vitro protein import assays highlight the role of phospholipids in mitochondrial protein import, it is not clear how these findings translate in vivo. Clearly, CL, PE, or PC deficiency does not completely abrogate protein import because these cells are viable, do not show any fitness defect under optimal growth conditions, and do not exhibit gross mitochondrial morphology defects.
Conclusions and Perspectives
The emerging picture of the role of phospholipids is that of specific requirements of the non-bilayer forming phospholipids in mitochondrial membrane biogenesis. Any perturbation in the levels of these conical-shaped lipids decreases MRC function, whereas a decrease in bilayer forming phospholipid is well tolerated. Moreover, even among the non-bilayer phospholipids, each phospholipid has been shown to have a specific role, with PE being required for the enzymatic activities of MRC complex III and IV and CL required for MRC supercomplex formation. Because the genomes of the majority of bacterial species do not contain PC biosynthetic enzymes yet harbour all the key components of the respiratory chain, it is not too surprising that the MRC has evolved to function without PC [121]. On the contrary, PE and CL are ubiquitously found across the prokaryotic kingdom. Thus, it is tempting to speculate that the MRC components have evolved with specific requirements for these non-bilayer forming phospholipids. Interestingly, the balance between bilayer and non-bilayer phospholipids is tightly regulated in prokaryotes and has been shown to be essential for their viability [122]. Given their prokaryotic ancestry, it is not surprising that mitochondria have similar requirements for non-bilayer phospholipids.
The non-bilayer phospholipids are known to stimulate the activity of both peripheral and integral membrane proteins by imposing curvature stress on the membrane. The curvature stress may act as a source of energy that influences binding of peripheral membrane proteins and provides energy for conformational changes of integral membrane proteins. Therefore, while elucidating the molecular mechanisms of particular functions of CL and PE, it is important to not only consider specific molecular interactions of these phospholipids with their protein partners but to also consider the possibility that they may be mediating their function by altering biophysical properties of the membrane such as curvature stress. In the same light, it would be interesting to see if non-bilayer phospholipids form membrane microdomains in the mitochondrial membranes akin to CL clusters observed in bacteria [123]. In fact, a model showing prohibitins as organizers of non-bilayer phospholipid clusters in the IMM has already been proposed, though experimental evidence for the same is lacking [59]. The discovery of a number of key intra-mitochondrial phospholipid trafficking proteins has provided important targets to investigate the roles of individual phospholipids within mitochondrial sub-compartments. However, key questions about how CL is trafficked to the OMM, how PE is exported from mitochondria, and how non-mitochondrial PE is imported into mitochondria are yet to be fully answered. Finally, understanding the homeostatic mechanism by which mitochondrial phospholipid composition is maintained will be a major breakthrough in understanding mitochondrial membrane biogenesis.
Acknowledgments
We thank members of the Gohil lab for their valuable comments in the preparation of this manuscript. This work was supported by the Welch Foundation Grant (A-1810), the American Heart Association Award (16GRNT31020028) and the National Institutes of Health award (R01GM111672) to V.M.G. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- ATP
Adenosine triphosphate
- MRC
Mitochondrial respiratory chain
- CDP-DAG
Cytidinediphospho-diacylglycerol
- CL
Cardiolipin
- MLCL
monolyso-cardiolipin
- PE
Phosphatidylethanolamine
- PA
Phosphatidic acid
- PC
Phosphatidylcholine
- PG
Phosphatidylglycerol
- PI
Phosphatidylinositol
- PS
Phosphatidylserine
- ER
Endoplasmic reticulum
- ERMES
Endoplasmic reticulum-mitochondria encounter structure
- Etn
Ethanolamine
References
- 1.Holthuis JC, Menon AK. Lipid landscapes and pipelines in membrane homeostasis. Nature. 2014;510:48–57. doi: 10.1038/nature13474. [DOI] [PubMed] [Google Scholar]
- 2.Dowhan W. Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu Rev Biochem. 1997;66:199–232. doi: 10.1146/annurev.biochem.66.1.199. [DOI] [PubMed] [Google Scholar]
- 3.Horvath SE, Daum G. Lipids of mitochondria. Prog Lipid Res. 2013;52:590–614. doi: 10.1016/j.plipres.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 4.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–24. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van den Brink-van der Laan E, Killian JA, de Kruijff B. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim Biophys Acta. 2004;1666:275–88. doi: 10.1016/j.bbamem.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 6.Lee AG. How lipids affect the activities of integral membrane proteins. Biochim Biophys Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 7.Daum G. Lipids of mitochondria. Biochim Biophys Acta. 1985;822:1–42. doi: 10.1016/0304-4157(85)90002-4. [DOI] [PubMed] [Google Scholar]
- 8.Zinser E, Sperka-Gottlieb CD, Fasch EV, Kohlwein SD, Paltauf F, Daum G. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J Bacteriol. 1991;173:2026–34. doi: 10.1128/jb.173.6.2026-2034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuller G, Nemec T, Hrastnik C, Daum G. Lipid composition of subcellular membranes of an FY1679-derived haploid yeast wild-type strain grown on different carbon sources. Yeast. 1999;15:1555–64. doi: 10.1002/(SICI)1097-0061(199910)15:14<1555::AID-YEA479>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 10.Sperka-Gottlieb CD, Hermetter A, Paltauf F, Daum G. Lipid topology and physical properties of the outer mitochondrial membrane of the yeast, Saccharomyces cerevisiae. Biochim Biophys Acta. 1988;946:227–34. doi: 10.1016/0005-2736(88)90397-5. [DOI] [PubMed] [Google Scholar]
- 11.Hovius R, Thijssen J, van der Linden P, Nicolay K, de Kruijff B. Phospholipid asymmetry of the outer membrane of rat liver mitochondria. Evidence for the presence of cardiolipin on the outside of the outer membrane. FEBS Lett. 1993;330:71–6. doi: 10.1016/0014-5793(93)80922-h. [DOI] [PubMed] [Google Scholar]
- 12.Crain RC, Marinetti GV. Phospholipid topology of the inner mitochondrial membrane of rat liver. Biochemistry. 1979;18:2407–14. doi: 10.1021/bi00578a041. [DOI] [PubMed] [Google Scholar]
- 13.Harb JS, Comte J, Gautheron DC. Asymmetrical orientation of phospholipids and their interactions with marker enzymes in pig heart mitochondrial inner membrane. Arch Biochem Biophys. 1981;208:305–18. doi: 10.1016/0003-9861(81)90153-3. [DOI] [PubMed] [Google Scholar]
- 14.Zhong Q, Gohil VM, Ma L, Greenberg ML. Absence of cardiolipin results in temperature sensitivity, respiratory defects, and mitochondrial DNA instability independent of pet56. J Biol Chem. 2004;279:32294–300. doi: 10.1074/jbc.M403275200. [DOI] [PubMed] [Google Scholar]
- 15.Baker CD, Basu Ball W, Pryce EN, Gohil VM. Specific requirements of nonbilayer phospholipids in mitochondrial respiratory chain function and formation. Mol Biol Cell. 2016;27:2161–71. doi: 10.1091/mbc.E15-12-0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahto KK, Singh A, Khandelwal NK, Bhardwaj N, Jha J, Prasad R. An assessment of growth media enrichment on lipid metabolome and the concurrent phenotypic properties of Candida albicans. PLoS One. 2014;9:e113664. doi: 10.1371/journal.pone.0113664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Connerth M, Tatsuta T, Haag M, Klecker T, Westermann B, Langer T. Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science. 2012;338:815–8. doi: 10.1126/science.1225625. [DOI] [PubMed] [Google Scholar]
- 18.Tamura Y, Harada Y, Nishikawa S, Yamano K, Kamiya M, Shiota T, Kuroda T, Kuge O, Sesaki H, Imai K, Tomii K, Endo T. Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metab. 2013;17:709–18. doi: 10.1016/j.cmet.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang SC, Heacock PN, Clancey CJ, Dowhan W. The PEL1 gene (renamed PGS1) encodes the phosphatidylglycero-phosphate synthase of Saccharomyces cerevisiae. J Biol Chem. 1998;273:9829–36. doi: 10.1074/jbc.273.16.9829. [DOI] [PubMed] [Google Scholar]
- 20.Osman C, Haag M, Wieland FT, Brügger B, Langer T. A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. EMBO J. 2010;29:1976–87. doi: 10.1038/emboj.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang F, Rizavi HS, Greenberg ML. Cardiolipin is not essential for the growth of Saccharomyces cerevisiae on fermentable or non-fermentable carbon sources. Mol Microbiol. 1997;26:481–91. doi: 10.1046/j.1365-2958.1997.5841950.x. [DOI] [PubMed] [Google Scholar]
- 22.Baile MG, Lu YW, Claypool SM. The topology and regulation of cardiolipin biosynthesis and remodeling in yeast. Chem Phys Lipids. 2014;179:25–31. doi: 10.1016/j.chemphyslip.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beranek A, Rechberger G, Knauer H, Wolinski H, Kohlwein SD, Leber R. Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J Biol Chem. 2009;284:11572–8. doi: 10.1074/jbc.M805511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y, Kelley RI, Blanck TJ, Schlame M. Remodeling of cardiolipin by phospholipid transacylation. J Biol Chem. 2003;278:51380–5. doi: 10.1074/jbc.M307382200. [DOI] [PubMed] [Google Scholar]
- 25.Baile MG, Whited K, Claypool SM. Deacylation on the matrix side of the mitochondrial inner membrane regulates cardiolipin remodeling. Mol Biol Cell. 2013;24:2008–20. doi: 10.1091/mbc.E13-03-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandner K, Mick DU, Frazier AE, Taylor RD, Meisinger C, Rehling P. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth Syndrome. Mol Biol Cell. 2005;11:5202–14. doi: 10.1091/mbc.E05-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Claypool SM, McCaffery JM, Koehler CM. Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J Cell Biol. 2006;174:379–90. doi: 10.1083/jcb.200605043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, Epand RF, Durrant D, Grossman D, Chi NW, Epand RM, Lee RM. Role of phospholipid scramblase 3 in the regulation of tumor necrosis factor-alpha-induced apoptosis. Biochemistry. 2008;47:4518–29. doi: 10.1021/bi701962c. [DOI] [PubMed] [Google Scholar]
- 29.Harner M, Körner C, Walther D, Mokranjac D, Kaesmacher J, Welsch U, Griffith J, Mann M, Reggiori F, Neupert W. The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 2011;30:4356–70. doi: 10.1038/emboj.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoppins S, Collins SR, Cassidy-Stone A, Hummel E, Devay RM, Lackner LL, Westermann B, Schuldiner M, Weissman JS, Nunnari J. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol. 2011;195:323–40. doi: 10.1083/jcb.201107053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von der Malsburg K, Müller JM, Bohnert M, Oeljeklaus S, Kwiatkowska P, Becker T, Loniewska-Lwowska A, Wiese S, Rao S, Milenkovic D, Hutu DP, Zerbes RM, Schulze-Specking A, Meyer HE, Martinou JC, Rospert S, Rehling P, Meisinger C, Veenhuis M, Warscheid B, van der Klei IJ, Pfanner N, Chacinska A, van der Laan M. Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev Cell. 2011;21:694–707. doi: 10.1016/j.devcel.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 32.Tatsuta T, Langer T. Intramitochondrial phospholipid trafficking. Biochim Biophys Acta. 2017;1862:81–89. doi: 10.1016/j.bbalip.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Trotter PJ, Pedretti J, Voelker DR. Phosphatidylserine decarboxylase from Saccharomyces cerevisiae. Isolation of mutants, cloning of the gene, and creation of a null allele. J Biol Chem. 1993;268:21416–24. [PubMed] [Google Scholar]
- 34.Trotter PJ, Voelker DR. Identification of a non-mitochondrial phosphatidylserine decarboxylase activity (PSD2) in the yeast Saccharomyces cerevisiae. J Biol Chem. 1995;270:6062–70. doi: 10.1074/jbc.270.11.6062. [DOI] [PubMed] [Google Scholar]
- 35.Riekhof WR, Wu J, Jones JL, Voelker DR. Identification and characterization of the major lysophosphatidylethanolamine acyltransferase in Saccharomyces cerevisiae. J Biol Chem. 2007;282:28344–52. doi: 10.1074/jbc.M705256200. [DOI] [PubMed] [Google Scholar]
- 36.Kennedy EP, Weiss SB. The function of cytidine coenzymes in the biosynthesis of phospholipides. J Biol Chem. 1956;222:193–214. [PubMed] [Google Scholar]
- 37.Horvath SE, Böttinger L, Vögtle FN, Wiedemann N, Meisinger C, Becker T, Daum G. Processing and topology of the yeast mitochondrial phosphatidylserine decarboxylase 1. J Biol Chem. 2012;287:36744–55. doi: 10.1074/jbc.M112.398107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyata N, Watanabe Y, Tamura Y, Endo T, Kuge O. Phosphatidylserine transport by Ups2-Mdm35 in respiration-active mitochondria. J Cell Biol. 2016;214:77–88. doi: 10.1083/jcb.201601082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aaltonen MJ, Friedman JR, Osman C, Salin B, di Rago JP, Nunnari J, Langer T, Tatsuta T. MICOS and phospholipid transfer by Ups2-Mdm35 organize membrane lipid synthesis in mitochondria. J Cell Biol. 2016;213:525–34. doi: 10.1083/jcb.201602007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gulshan K, Shahi P, Moye-Rowley WS. Compartment-specific synthesis of phosphatidylethanolamine is required for normal heavy metal resistance. Mol Biol Cell. 2010;21:443–55. doi: 10.1091/mbc.E09-06-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birner R, Bürgermeister M, Schneiter R, Daum G. Roles of phosphatidylethanolamine and of its several biosynthetic pathways in Saccharomyces cerevisiae. Mol Biol Cell. 2001;12:997–1007. doi: 10.1091/mbc.12.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steenbergen R, Nanowski TS, Beigneux A, Kulinski A, Young SG, Vance JE. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J Biol Chem. 2005;280:40032–40. doi: 10.1074/jbc.M506510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fullerton MD, Hakimuddin F, Bakovic M. Developmental and metabolic effects of disruption of the mouse CTP:phosphoethanolamine cytidylyltransferase gene (Pcyt2) Mol Cell Biol. 2007;27:3327–36. doi: 10.1128/MCB.01527-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bleijerveld OB, Brouwers JF, Vaandrager AB, Helms JB, Houweling M. The CDP-ethanolamine pathway and phosphatidylserine decarboxylation generate different phosphatidylethanolamine molecular species. J Biol Chem. 2007;282:28362–72. doi: 10.1074/jbc.M703786200. [DOI] [PubMed] [Google Scholar]
- 45.Chan EY, McQuibban GA. Phosphatidylserine decarboxylase 1 (Psd1) promotes mitochondrial fusion by regulating the biophysical properties of the mitochondrial membrane and alternative topogenesis of mitochondrial genome maintenance protein 1 (Mgm1) J Biol Chem. 2012;287:40131–9. doi: 10.1074/jbc.M112.399428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–81. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hönscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, van der Laan M, Cabrera M, Reggiori F, Ungermann C. Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell. 2014;30:86–94. doi: 10.1016/j.devcel.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 48.Elbaz-Alon Y, Rosenfeld-Gur E, Shinder V, Futerman AH, Geiger T, Schuldiner M. A dynamic interface between vacuoles and mitochondria in yeast. Dev Cell. 2014;30:95–102. doi: 10.1016/j.devcel.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 49.Nguyen TT, Lewandowska A, Choi JY, Markgraf DF, Junker M, Bilgin M, Ejsing CS, Voelker DR, Rapoport TA, Shaw JM. Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic. 2012;13:880–90. doi: 10.1111/j.1600-0854.2012.01352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao M, Schlame M, Rua D, Greenberg ML. Cardiolipin synthase is associated with a large complex in yeast mitochondria. J Biol Chem. 1998;273:2402–8. doi: 10.1074/jbc.273.4.2402. [DOI] [PubMed] [Google Scholar]
- 51.Jiang F, Gu Z, Granger JM, Greenberg ML. Cardiolipin synthase expression is essential for growth at elevated temperature and is regulated by factors affecting mitochondrial development. Mol Microbiol. 1999;31:373–9. doi: 10.1046/j.1365-2958.1999.01181.x. [DOI] [PubMed] [Google Scholar]
- 52.Gohil VM, Hayes P, Matsuyama S, Schägger H, Schlame M, Greenberg ML. Cardiolipin biosynthesis and mitochondrial respiratory chain function are interdependent. J Biol Chem. 2004;279:42612–8. doi: 10.1074/jbc.M402545200. [DOI] [PubMed] [Google Scholar]
- 53.He Q, Greenberg ML. Post-translational regulation of phosphatidylglycerolphosphate synthase in response to inositol. Mol Microbiol. 2004;53:1243–9. doi: 10.1111/j.1365-2958.2004.04202.x. [DOI] [PubMed] [Google Scholar]
- 54.Xu Y, Phoon CK, Berno B, D’Souza K, Hoedt E, Zhang G, Neubert TA, Epand RM, Ren M, Schlame M. Loss of protein association causes cardiolipin degradation in Barth syndrome. Nat Chem Biol. 2016;12:641–7. doi: 10.1038/nchembio.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nebauer R, Schuiki I, Kulterer B, Trajanoski Z, Daum G. The phosphatidylethanolamine level of yeast mitochondria is affected by the mitochondrial components Oxa1p and Yme1p. FEBS J. 2007;274:6180–90. doi: 10.1111/j.1742-4658.2007.06138.x. [DOI] [PubMed] [Google Scholar]
- 56.Di Bartolomeo F, Wagner A, Daum G. Cell biology, physiology and enzymology of phosphatidylserine decarboxylase. Biochim Biophys Acta. 2017;1862:25–38. doi: 10.1016/j.bbalip.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 57.Griac P. Regulation of yeast phospholipid biosynthetic genes in phosphatidylserine decarboxylase mutants. J Bacteriol. 1997;179:5843–8. doi: 10.1128/jb.179.18.5843-5848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Calzada E, Onguka O, Claypool SM. Phosphatidylethanolamine Metabolism in Health and Disease. Int Rev Cell Mol Biol. 2016;321:29–88. doi: 10.1016/bs.ircmb.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Osman C, Haag M, Potting C, Rodenfels J, Dip PV, Wieland FT, Brügger B, Westermann B, Langer T. The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J Cell Biol. 2009;184:583–96. doi: 10.1083/jcb.200810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gohil VM, Thompson MN, Greenberg ML. Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine and cardiolipin biosynthetic pathways in Saccharomyces cerevisiae. J Biol Chem. 2005;280:35410–6. doi: 10.1074/jbc.M505478200. [DOI] [PubMed] [Google Scholar]
- 61.Xu Y, Condell M, Plesken H, Edelman-Novemsky I, Ma J, Ren M, Schlame M. A Drosophila model of Barth syndrome. Proc Natl Acad Sci U S A. 2006;103:11584–8. doi: 10.1073/pnas.0603242103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Acehan D, Vaz F, Houtkooper RH, James J, Moore V, Tokunaga C, Kulik W, Wansapura J, Toth MJ, Strauss A, Khuchua Z. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J Biol Chem. 2011;286:899–908. doi: 10.1074/jbc.M110.171439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu Y, Sutachan JJ, Plesken H, Kelley RI, Schlame M. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab Invest. 2005;85:823–30. doi: 10.1038/labinvest.3700274. [DOI] [PubMed] [Google Scholar]
- 64.Ikon N, Ryan RO. Cardiolipin and mitochondrial cristae organization. Biochim Biophys Acta. 2017;1859:1156–1163. doi: 10.1016/j.bbamem.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baile MG, Sathappa M, Lu YW, Pryce E, Whited K, McCaffery JM, Han X, Alder NN, Claypool SM. Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J Biol Chem. 2014;289:1768–78. doi: 10.1074/jbc.M113.525733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clarke SL, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, Martin RP, Tsai-Goodman B, Garratt V, Ashworth M, Bowen VM, McCurdy KR, Damin MK, Spencer CT, Toth MJ, Kelley RI, Steward CG. Barth syndrome. Orphanet J Rare Dis. 2013;8:23. doi: 10.1186/1750-1172-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Friedman JR, Mourier A, Yamada J, McCaffery JM, Nunnari J. MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. Elife. 2015;4:e07739. doi: 10.7554/eLife.07739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Joshi AS, Thompson MN, Fei N, Hüttemann M, Greenberg ML. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J Biol Chem. 2012;287:17589–97. doi: 10.1074/jbc.M111.330167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, Oka T, Mihara K, Ishihara N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol. 2017;19:856–863. doi: 10.1038/ncb3560. [DOI] [PubMed] [Google Scholar]
- 70.DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol. 2009;186:793–803. doi: 10.1083/jcb.200906098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Francy CA, Clinton RW, Fröhlich C, Murphy C, Mears JA. Cryo-EM Studies of Drp1 Reveal Cardiolipin Interactions that Activate the Helical Oligomer. Sci Rep. 2017;7:10744. doi: 10.1038/s41598-017-11008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bustillo-Zabalbeitia I, Montessuit S, Raemy E, Basañez G, Terrones O, Martinou JC. Specific interaction with cardiolipin triggers functional activation of Dynamin-Related Protein 1. PLoS One. 2014;9:e102738. doi: 10.1371/journal.pone.0102738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stepanyants N, Macdonald PJ, Francy CA, Mears JA, Qi X, Ramachandran R. Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol Biol Cell. 2015;26:3104–16. doi: 10.1091/mbc.E15-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Adachi Y, Itoh K, Yamada T, Cerveny KL, Suzuki TL, Macdonald P, Frohman MA, Ramachandran R, Iijima M, Sesaki H. Coincident Phosphatidic Acid Interaction Restrains Drp1 in Mitochondrial Division. Mol Cell. 2016;63:1034–43. doi: 10.1016/j.molcel.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Acin-Perez R, Enriquez JA. The function of the respiratory supercomplexes: the plasticity model. Biochim Biophys Acta. 2014;1837:444–450. doi: 10.1016/j.bbabio.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 76.Schägger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000;19:1777–83. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Letts JA, Fiedorczuk K, Sazanov LA. The architecture of respiratory supercomplexes. Nature. 2016;537:644–648. doi: 10.1038/nature19774. [DOI] [PubMed] [Google Scholar]
- 78.Wu M, Gu J, Guo R, Huang Y, Yang M. Structure of Mammalian Respiratory Supercomplex I1III2IV1. Cell. 2016;167:1598–1609. doi: 10.1016/j.cell.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 79.Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schägger H. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278:52873–80. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- 80.Zhang M, Mileykovskaya E, Dowhan W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem. 2002;277:43553–6. doi: 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- 81.McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol. 2006;361:462–9. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 82.Mileykovskaya E, Penczek PA, Fang J, Mallampalli VK, Sparagna GC, Dowhan W. Arrangement of the respiratory chain complexes in Saccharomyces cerevisiae supercomplex III2IV2 revealed by single particle cryo-electron microscopy. J Biol Chem. 2012;287:23095–103. doi: 10.1074/jbc.M112.367888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Althoff T, Mills DJ, Popot JL, Kühlbrandt W. Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 2011;30:4652–64. doi: 10.1038/emboj.2011.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bazán S, Mileykovskaya E, Mallampalli VK, Heacock P, Sparagna GC, Dowhan W. Cardiolipin-dependent reconstitution of respiratory supercomplexes from purified Saccharomyces cerevisiae complexes III and IV. J Biol Chem. 2012;288:401–11. doi: 10.1074/jbc.M112.425876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barth PG, Van den Bogert C, Bolhuis PA, Scholte HR, van Gennip AH, Schutgens RB, Ketel AG. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): respiratory-chain abnormalities in cultured fibroblasts. J Inherit Metab Dis. 1996;19:157–60. doi: 10.1007/BF01799418. [DOI] [PubMed] [Google Scholar]
- 86.Ostrander DB, Zhang M, Mileykovskaya E, Rho M, Dowhan W. Lack of mitochondrial anionic phospholipids causes an inhibition of translation of protein components of the electron transport chain. A yeast genetic model system for the study of anionic phospholipid function in mitochondria. J Biol Chem. 2001;276:25262–72. doi: 10.1074/jbc.M103689200. [DOI] [PubMed] [Google Scholar]
- 87.Klingen AR, Palsdottir H, Hunte C, Ullmann GM. Redox-linked protonation state changes in cytochrome bc1 identified by Poisson-Boltzmann electrostatics calculations. Biochim Biophys Acta. 2007;1767:204–21. doi: 10.1016/j.bbabio.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 88.Arnarez C, Mazat JP, Elezgaray J, Marrink SJ, Periole X. Evidence for cardiolipin binding sites on the membrane-exposed surface of the cytochrome bc1. J Am Chem Soc. 2013;135:3112–20. doi: 10.1021/ja310577u. [DOI] [PubMed] [Google Scholar]
- 89.Arnarez C, Marrink SJ, Periole X. Identification of cardiolipin binding sites on cytochrome c oxidase at the entrance of proton channels. Sci Rep. 2013;3:1263. doi: 10.1038/srep01263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schwall CT, Greenwood VL, Alder NN. The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim Biophys Acta. 2012;1817:1588–96. doi: 10.1016/j.bbabio.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 91.Koshkin V, Greenberg ML. Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria. Biochem J. 2000;347:687–91. [PMC free article] [PubMed] [Google Scholar]
- 92.Koshkin V, Greenberg ML. Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem J. 2002;364:317–22. doi: 10.1042/bj3640317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ma L, Vaz FM, Gu Z, Wanders RJ, Greenberg ML. The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Delta mutant. Implications for Barth syndrome. J Biol Chem. 2004;279:44394–9. doi: 10.1074/jbc.M405479200. [DOI] [PubMed] [Google Scholar]
- 94.He Q, Wang M, Harris N, Han X. Tafazzin knockdown interrupts cell cycle progression in cultured neonatal ventricular fibroblasts. Am J Physiol Heart Circ Physiol. 2013;305:H1332–43. doi: 10.1152/ajpheart.00084.2013. [DOI] [PubMed] [Google Scholar]
- 95.Jiang F, Ryan MT, Schlame M, Zhao M, Gu Z, Klingenberg M, Pfanner N, Greenberg ML. Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J Biol Chem. 2000;275:22387–94. doi: 10.1074/jbc.M909868199. [DOI] [PubMed] [Google Scholar]
- 96.Musatov A, Sedlák E. Role of cardiolipin in stability of integral membrane proteins. Biochimie. 2017;9084:30222–5. doi: 10.1016/j.biochi.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 97.Klingenberg M. Cardiolipin and mitochondrial carriers. Biochim Biophys Acta. 2009;1788:2048–58. doi: 10.1016/j.bbamem.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 98.Claypool SM, Oktay Y, Boontheung P, Loo JA, Koehler CM. Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J Cell Biol. 2008;182:937–50. doi: 10.1083/jcb.200801152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Raja V, Joshi AS, Li G, Maddipati KR, Greenberg ML. Loss of Cardiolipin Leads to Perturbation of Acetyl-CoA Synthesis. J Biol Chem. 2017;292:1092–1102. doi: 10.1074/jbc.M116.753624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gebert N, Joshi AS, Kutik S, Becker T, McKenzie M, Guan XL, Mooga VP, Stroud DA, Kulkarni G, Wenk MR, Rehling P, Meisinger C, Ryan MT, Wiedemann N, Greenberg ML, Pfanner N. Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr Biol. 2009;19:2133–9. doi: 10.1016/j.cub.2009.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tamura Y, Endo T, Iijima M, Sesaki H. Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J Cell Biol. 2009;185:1029–45. doi: 10.1083/jcb.200812018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Böttinger L, Ellenrieder L, Becker T. How lipids modulate mitochondrial protein import. J Bioenerg Biomembr. 2016;48:125–35. doi: 10.1007/s10863-015-9599-7. [DOI] [PubMed] [Google Scholar]
- 103.Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Wang KZQ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayir H, Kagan VE. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chu CT, Bayır H, Kagan VE. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy. 2014;10:376–8. doi: 10.4161/auto.27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kagan VE, Jiang J, Huang Z, Tyurina YY, Desbourdes C, Cottet-Rousselle C, Dar HH, Verma M, Tyurin VA, Kapralov AA, Cheikhi A, Mao G, Stolz D, St Croix CM, Watkins S, Shen Z, Li Y, Greenberg ML, Tokarska-Schlattner M, Boissan M, Lacombe ML, Epand RM, Chu CT, Mallampalli RK, Bayır H, Schlattner U. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ. 2016;23:1140–51. doi: 10.1038/cdd.2015.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hsu P, Liu X, Zhang J, Wang HG, Ye JM, Shi Y. Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy. 2015;11:643–52. doi: 10.1080/15548627.2015.1023984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shen Z, Li Y, Gasparski AN, Abeliovich H, Greenberg ML. Cardiolipin Regulates Mitophagy through the Protein Kinase C Pathway. J Biol Chem. 2017;292:2916–23. doi: 10.1074/jbc.M116.753574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171:2029–50. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bürgermeister M, Birner-Grünberger R, Nebauer R, Daum G. Contribution of different pathways to the supply of phosphatidylethanolamine and phosphatidylcholine to mitochondrial membranes of the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2004;1686:161–8. doi: 10.1016/j.bbalip.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 110.Storey MK, Clay KL, Kutateladze T, Murphy RC, Overduin M, Voelker DR. Phosphatidylethanolamine has an essential role in Saccharomyces cerevisiae that is independent of its ability to form hexagonal phase structures. J Biol Chem. 2001;276:48539–48. doi: 10.1074/jbc.M109043200. [DOI] [PubMed] [Google Scholar]
- 111.Tasseva G, Bai HD, Davidescu M, Haromy A, Michelakis E, Vance JE. Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J Biol Chem. 2013;288:4158–73. doi: 10.1074/jbc.M112.434183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boumann HA, Gubbens J, Koorengevel MC, Oh CS, Martin CE, Heck AJ, Patton-Vogt J, Henry SA, de Kruijff B, de Kroon AI. Depletion of phosphatidylcholine in yeast induces shortening and increased saturation of the lipid acyl chains: evidence for regulation of intrinsic membrane curvature in a eukaryote. Mol Biol Cell. 2006;2:1006–17. doi: 10.1091/mbc.E05-04-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Klecker T, Wemmer M, Haag M, Weig A, Böckler S, Langer T, Nunnari J, Westermann B. Interaction of MDM33 with mitochondrial inner membrane homeostasis pathways in yeast. Sci Rep. 2015;5:18344. doi: 10.1038/srep18344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Böttinger L, Horvath SE, Kleinschroth T, Hunte C, Daum G, Pfanner N, Becker T. Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J Mol Biol. 2012;423:677–86. doi: 10.1016/j.jmb.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Otsuru M, Yu Y, Mizoi J, Kawamoto-Fujioka M, Wang J, Fujiki Y, Nishida I. Mitochondrial phosphatidylethanolamine level modulates Cyt c oxidase activity to maintain respiration capacity in Arabidopsis thaliana rosette leaves. Plant Cell Physiol. 2013;54:1612–9. doi: 10.1093/pcp/pct104. [DOI] [PubMed] [Google Scholar]
- 116.Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 2007;26:1713–25. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bogdanov M, Dowhan W. Lipid-dependent generation of dual topology for a membrane protein. J Biol Chem. 2012;287:37939–48. doi: 10.1074/jbc.M112.404103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van der Veen JN, Lingrell S, da Silva RP, Jacobs RL, Vance DE. The concentration of phosphatidylethanolamine in mitochondria can modulate ATP production and glucose metabolism in mice. Diabetes. 2014;63:2620–30. doi: 10.2337/db13-0993. [DOI] [PubMed] [Google Scholar]
- 119.Becker T, Horvath SE, Böttinger L, Gebert N, Daum G, Pfanner N. Role of phosphatidylethanolamine in the biogenesis of mitochondrial outer membrane proteins. J Biol Chem. 2013;288:16451–9. doi: 10.1074/jbc.M112.442392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schuler MH, Di Bartolomeo F, Mårtensson CU, Daum G, Becker T. Phosphatidylcholine Affects Inner Membrane Protein Translocases of Mitochondria. J Biol Chem. 2016;36:18718–29. doi: 10.1074/jbc.M116.722694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sohlenkamp C, López-Lara IM, Geiger O. Biosynthesis of phosphatidylcholine in bacteria. Prog Lipid Res. 2003;42:115–162. doi: 10.1016/s0163-7827(02)00050-4. [DOI] [PubMed] [Google Scholar]
- 122.Rietveld AG, Chupin VV, Koorengevel MC, Wienk HL, Dowhan W, de Kruijff B. Regulation of lipid polymorphism is essential for the viability of phosphatidylethanolamine-deficient Escherichia coli cells. J Biol Chem. 1994;269:28670–5. [PubMed] [Google Scholar]
- 123.Mileykovskaya E, Dowhan W. Visualization of phospholipid domains in Escherichia coli by using the cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J Bacteriol. 2000;182:1172–5. doi: 10.1128/jb.182.4.1172-1175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]