

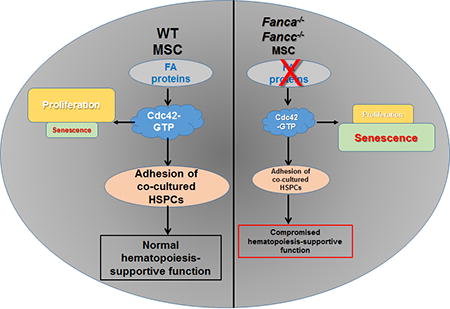
Abstract
Hematopoietic stem cells (HSCs) preserve their ability to self-renew and differentiate to different lineages in the bone marrow (BM) niche, which is composed in large part by BM stromal cells. Studies have shown that altered signaling in the BM niche results in leukemia initiation or progression. Fanconi anemia (FA) is an inherited BM failure syndrome associated with extremely high risk of leukemic transformation. By employing two FA mouse models, here we have investigated the hematopoiesis-supportive function of FA BM mesenchymal stroma cells (MSCs). We found that MSCs deficient for Fanca or Fancc gene are defective in proliferation and prone to undergo senescence in vitro. Mechanistically, we show that the activity of cell division control protein 42 (Cdc42), a Rho GTPase known to be a critical regulator for cytoskeleton organization, is significantly reduced in FA MSCs. Furthermore, we demonstrate that this reduction in Cdc42 activity plays a causal role in defective hematopoiesis-supportive function of the FA MSCs. The progenies of WT HSPCs co-cultured on FA MSCs exhibit compromised self-renewal capacity both in vitro and in vivo. Genetic correction of FA deficiency restores Cdc42 activity and improves hematopoiesis-supportive capacity of FA MSC. Finally, ectopic expression of a constitutively active Cdc42 mutant, Cdc42F28L, or pretreatment with Wnt5a, increases the active Cdc42 level and rescues the hematopoietic supportive defects of FA MSCs. Taken together, our results identify a novel link between Cdc42 activity and hematopoiesis-supportive function of MSCs and suggest that a niche-specific increase of Cdc42 activity may be beneficial for FA therapy.
Keywords: Fanconi anemia, Mesenchymal stromal cells, hematopoietic stem progenitor cells, cell adhesion, hematopoietic reconstitution capacity
Graphical abstract
Introduction
Fanconi anemia (FA) is a rare inherited disease resulted from dysfunctional hematopoietic stem cells [1, 2]. FA is genetically heterogeneous with at least 22 genes (FANCA-W) identified thus far [1–11]. Patients with mutations in any of these genes lead to an FA phenotype manifested by the cellular and phenotypic consequences of genetic instability, growth retardation, congenital malformations, bone marrow (BM) failure, high risk of neoplasia and premature aging [1, 2, 8]. The hematologic complications of FA are nearly universal among patients with FA. In fact, progressive BM failure represents the major causes of morbidity and mortality of FA. To date, allogenic hematopoietic stem cell transplantation (HSCT) from related and unrelated donors is the only known definite therapy for severe BM failure in patients with FA [1, 2].
The BM microenvironment provides unique regions that support the function of hematopoietic stem cells (HSCs) and other hematopoietic lineages, including monocytes and lymphocytes [12, 13]. Cells within these regions constitute the hematopoietic stem cell (HSC) niche, which contributes to the control of HSC quiescence, proliferation, self-renewal and differentiation [12, 13]. Recent studies have revealed that disordered niche function could contribute to disease. For example, altered signaling in the BM stroma/niche results in leukemia initiation or progression [14–16]. Conversely, leukemic cells can reprogram the BM niche, which favors leukemic proliferation while compromising normal hematopoiesis [17]. To date, only a few studies have described the functional characteristics of FA BM mesenchymal stroma cells (MSCs) [18–20], all of which implicate a role of FA BM niche in the pathophysiology of FA BM failure. Defining the role of BM niche in FA may reveal potential therapies not only for FA but also for patients with BM failure and leukemia in general.
Cell division control protein 42 (Cdc42) belongs to the Rho GTPase family, acting as an intracellular signal transducer in response to a variety of extracellular stimuli [21, 22]. It can integrate signals from multiple cell surface receptors, such as c-kit, CXCR4, and β1-integrin, to regulate cytoskeleton dynamics that impacts on cell adhesion and migration. These cell functions are directly relevant to the retention of HSCs in the BM niche. Recent studies of a conditional Cdc42 knockout mouse model and a Cdc42 gain-of activity, Cdc42GAP knockout mouse model demonstrated that Cdc42 is uniquely required for HSC retention and maintenance in the BM niche [23, 24]. Previous study indicated that the cell-autonomous defect of FA HSCs in BM homing and engraftment is associated with a reduction in the activity of Cdc42 [25]. In this study, we employed two FA mouse models to show that MSCs isolated from Fanca−/− and Fancc−/− mice have low Cdc42 activity, which is correlated with compromised hematopoiesis-supportive function. Genetic correction of FA deficiency or activation of Cdc42 improves the capacity of FA MSCs to support hematopoiesis.
Materials and methods
Mice and treatment
Fanca+/+, Fanca−/− and Fancc+/+, Fancc−/− mice were generated by interbreeding the heterozygous Fanca+/− or Fancc+/− mice [26, 27], respectively. 6–8 week-old BoyJ mice were used as bone marrow transplant (BMT) recipients. Animals including BoyJ recipient mice were maintained in the animal barrier facility at West Virginia University (WVU). All experimental procedures conducted in this study were approved by the Institutional Animal Care and Use Committee of WVU.
Mesenchymal stromal cell (MSC) culture and treatment
Whole bone marrow cells (WBMCs) were isolated from wild-type (WT), Fanca−/− and Fancc−/− mice by gently flushing out of tibias and femurs using DPBS + 10% FBS. Cells obtained from two tibias and two femurs were pooled and plated in 100mm culture dish (BD Falcon, Tewksbury, MA) in 10 ml of MSCs media (Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Gaithersburg, MD) supplemented with 15% fetal bovine serum (FBS, Gibco, Gaithersburg, MD) and 1% Penicillin-Streptomycin (Life Technologies # 15140-122 adapted and modified from previous report [27].
Plastic adherent cells were passaged 3 times followed by staining with fluorescently labelled antibodies for mesenchymal (CD29, CD44, CD73, SCA-1, CD106) and hematopoietic markers (CD45, CD11b) using the mouse multipotent mesenchymal stromal cell marker antibody panel (Cat # SC018; R&D systems; Minneapolis, MN) [28–30]. We confirmed that at least 98% purity of MSCs was obtained with this culture method.
For Wnt5a treatment, MSCs at passages three were plated to obtain 95% confluence. Cells were pre-treated with different doses of Wnt5a (R&D System, Minneapolis, MN) for 16 hrs [31], followed by co-culture in fresh media with WT LSK (Lin−Sca1+c-kit+) cells.
Senescence-associated-β-galactosidase activity analysis
Senescence-associated-β-galactosidase (SA-β-gal) activity was determined using an SA-β-gal staining kit from Cell Signaling Technology (Beverly, MA) in accordance to the manufacturer’s instruction. Briefly, cells were washed in phosphate-buffered saline (PBS) and fixed in 2% formaldehyde-0.2% glutaraldehyde. Then the cells were washed and incubated at 37°C incubator overnight with fresh SA-β-gal stain solution [1 mg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside, 40 mM citric acid-sodium phosphate (pH 6.0), 150 mM NaCl, 2 mM MgCl2, and 5 mM potassium ferricyanide]. Senescent cells were identified as blue-stained cells by standard light microscopy, and a total of 500 cells were counted in random fields to determine the percentage of SA-β-gal-positive cells.
MSC colony forming efficiency (CFE)
MSC CFE assay was performed following the manufacturer’s instruction (CELLnTEC, Ontario, Canada). Briefly, 100 MSCs were seeded into 100 mm diameter dish in 10 ml of MSC medium. Cells were grown for 12 days at 37 °C without further medium changes. After 12 days, culture media were removed. Cells were rinsed with PBS then fixed using 100% MeOH for 5 min at room temperature. Cells were then stained with Stain Solution I and II subsequently. Colonies were visualized and enumerated under microscopy.
Cell adhesion assay
Cell-cell adhesion was performed by adding sorted LSK cells from WT mice onto confluent WT, Fanca−/− or Fancc−/− BM-derived MSCs and incubated at 37°C for 4 hours. Non-adherent cells were then subjected to colony forming cell (CFC) assay. Progenitor adhesion was shown as a percentage of the input CFCs.
Western blot and Cdc42-GTPase effector domain pull-down assays
Relative levels of GTP-bound Cdc42 were determined by an effector pull-down assay [25]. Briefly, 1×106 of lineage-depleted bone-marrow cells were lysed in a Mg2+ lysis/wash buffer (Upstate cell signaling solutions, Boston, MA) containing 10% glycerol, 25 mM sodium fluoride, 1 mM sodium orthovanadate and a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Samples were incubated with PAK-1 binding domain/agarose beads and bound (activated) as well as unbound (non-activated) Cdc42 fractions were probed by immunoblotting with antibody against Cdc42 (rabbit polyclonal, Millipore, Billerica, MA). Activated protein was normalized to total protein and/or β-actin (Sigma-Aldrich, St. Louis, MO) and the relative amount was quantified using ImageJ software (NIH). To detect Wnt5a protein levels, cells were lysed as described above and directly blotted with a goat anti-mouse Wnt5a antibody (R&D Systems, Minneapolis, MN). Wnt5a protein levels were normalized based on actin protein levels in the same blotted samples.
Cobble stone Area Forming Cell assay (CAFC)
WT bone marrow cells (BMCs) were seeded on top of confluent WT, Fanca−/− and Fancc−/− MSCs in 35 mm culture dish (BD Falcon, Tewksbury, MA) and allow the precursor cells forming hematopoietic clones under the stromal layers [19, 32]. The cells were cocultured in 37°C incubator supplemented with 5% CO2 and fed weekly by half medium change. Phase-dark hematopoietic clone was imaged under phase contrast images were taken at 20× objective. The identified area was analyzed with ImageJ software (NIH).
Limited dilution assay (LDA)
Limited dilution assay (LDA) of a LSK cells was conducted by the use of five dilutions (0, 10, 20, 90, 270, and 810) differing with a factor of 3 and 10 wells per cell concentration [19]. Three different LDA experiments were performed with independently derived MSCs from WT, Fanca−/− and Fancc−/− mice. A well was scored as “positive” if contained one or more cobblestone areas and “negative” if contained no cobblestone areas. Cobblestone area is at least 6 cells (in proximity of each other) growing underneath the stroma. Although cobblestone-like cells appear as phase dark, these cells appear as nonregractile in 96-well plates because of the deflection of light. Only dilution with both negative and positive wells are informative for frequent analysis.
Bone marrow transplantation (BMT)
In the competitive repopulation study, 1×105 output cells (CD45.2+) collected from co-cultures were mixed with 3×105 competitor cells (CD45.1+), and injected into lethally irradiated (split dose of 700 Rad + 475 Rad with 3 hours apart) BoyJ mice (CD45.1+). After 16 weeks, the recipient mice were sacrificed, and nucleated cells from peripheral blood and the BM were analyzed were stained with CD45.2 and CD45.1 for donor-derived chimeras. For secondary BMT, 3 million of WBMCs from primary recipients were pooled and injected into sublethally irradiated (7.0 Gy) BoyJ recipients.
Molecular cloning and materials
For retroviral vector construction, full-length FANCA or FANCC cDNA were cloned into MIEG3-GFP vector [25, 33]. The Cdc42(F28L) cDNA, a Cdc42 mutant that constitutively exchanges GDP for GTP but still hydrolyzes GTP [33], was subcloned into the BamH1 and EcoR1 sites of the MIEG3 retroviral vector. The plasmids (10 µg each) were used to produce retroviral supernatant.
RNA Isolation, Reverse Transcriptase (RT)-PCR
Total RNA isolated from cells of the indicated genotypes was prepared by using RNeasy kit (Qiagen, Valencia, CA) following the manufacturer’s procedure. Reverse transcription was performed with random hexamers and Superscript II RT (Invitrogen, Grand Island, NY) and was carried out at 42 °C for 60 min and stopped at 95 °C for 5 min. First-strand cDNA was used for real-time PCR using primers listed in Table S1. Samples were normalized to the level of GAPDH mRNA.
Determination of Wnt5a protein level
Protein levels of Wnt5a in WT, Fanca−/− or Fancc−/− MSCs were measured by ELISA assay following manufacturer’s instruction (Cloud-Clone Corp., TX, USA).
Statistical analysis
Paired or unpaired student’s t-test was used for two-group comparisons. Survival data were plotted by the Kaplan-Meier curve method and analyzed by the log-rank test. Values of p<0.05 were considered statistically significant. Results are presented as mean ± SD. * indicates p<0.05; ** indicates p<0.01.
Results
Reduced Cdc42 activity and adhesion defect in Fanca−/− and Fancc−/− MSC
The bone marrow (BM) niche represents the supportive environment for hematopoietic stem cells (HSCs) [34, 35]. As a major component of BM niche, mesenchymal stroma cells (MSCs) being precursors to osteoblasts, adipocytes and chrondrocytes, are crucial for maintenance of quiescent HSC [35]. To investigate the effect of FA deficiency on MSCs, we first measured in vitro proliferating capacity of MSCs from mice deficient for the Fanca and Fancc genes, which encode two essential components of the FA core complex (1–5). By using a well-established MSC colony forming efficiency (CFE) assay [36], we observed a significantly lower rate of proliferation of FA MSCs compared to that of wild-type (WT) MSCs (Fig 1A). In addition, MSCs from Fanca−/− or Fancc−/− mice were more prone to undergo senescence after four times passaging in culture as detected by SA-β-gal staining (Fig 1B).
Fig. 1. Reduced Cdc42 activity and adhesion defect in Fanca−/− and Fancc−/− MSCs.

(A) Fanca−/− and Fancc−/− MSCs exhibit defective proliferation in vitro. MSCs isolated from wild-type (WT), Fanca−/− or Fancc−/− mice were cultured in MSC medium followed by MSC colony forming efficiency (CFE) assay. Numbers of colonies formed were enumerated on day 12 in triplicate from five individual WT, Fanca−/− or Fancc−/− mice. Quantification are shown. (B) Fanca−/− and Fancc−/− MSCs are prone to senescence. Cells described in (A) were passaged for 3 times in vitro followed by SA-β-gal staining. Percentages of the cells stained positive for SA-β-gal were quantified by counting a total of 100 cells in random fields per well. (C) Decreased Cdc42 activity in Fanca−/− and Fancc−/− MSCs. MSCs isolated from WT, Fanca−/− or Fancc−/− mice were cultured in MSC medium and passaged for 3 times. Whole cell lysates (WCL) was then extracted from the cells and subjected to Western blotting using antibodies against Cdc42. The level of active, GTP-bound Cdc42, total Cdc42 and β-actin were determined. The relative levels of active Cdc42 are indicated below the blot. (D) WT HSPCs co-cultured on Fanca−/− or Fancc−/− MSCs display decreased adhesion. Sorted LSK (Lin−Sca1+c-kit+) cells from WT mice were added onto confluent WT, Fanca−/− or Fancc−/− BM-derived MSCs and incubated for 4 hours at 37°C; non-adherent cells were then subjected to colony forming cell (CFC) assay. Numbers are given as the percentage of input CFCs with three independent experiments in triplicates in each experiments (n=5). *p<0.05.
The Rho GTPase, Cdc42 plays a critical role in mediating morphologic polarity in many cell types and in regulating HSC differentiation [23, 24]. Hematopoietic stem and progenitor cells (HSPCs) from FA patients and Fanca−/− mice have been shown to be defective in cell adhesion and BM homing, and these defects are attributable, at least in part, to a reduced Cdc42 GTPase activity that is subjected to FANCA regulation [25; unpublished data]. Given the role of MSCs in regulating HSC function, we attempted to determine Cdc42 activity in FA MSCs. We expanded the MSCs at passage 3 with a characteristic MSC surface phenotype: CD45−CD11b−CD29+CD44+CD73+SCA-1+CD105+CD106+ (29; Fig S1A). The MSCs also showed negligible levels of the Fanca or Fancc mRNA, thus confirming the knockout genotypes of the cells (Fig S1B). Consistent with the decreased Cdc42 activity observed in FA HSPCs [23], the active form of Cdc42-GTP in Fanca−/− and Fancc−/− MSCs was reduced by more than 50% compared to that in WT MSCs (Fig 1C). Notably, Rac1-GTP content of the Fanca−/− and Fancc−/− MSCs showed comparable level to that of WT cells (Fig S1C).
It is known that nestin-expressing MSCs constitute an essential HSC niche component [3]. One of the most important features of MSC is hematopoiesis-supportive function. We then asked whether the decreased Cdc42 activity in FA MSCs affected the adhesion of HSC to MSCs. To this end, we performed a cell-cell adhesion assay by adding 5×105 BM cells isolated from WT mice onto confluent MSC monolayers [19, 25]. After 4 hours of co-culture, adherent cells were subjected to colony-forming cell (CFC) assay. The results showed that adhesion of WT BM progenitor cells to Fanca−/− or Fancc−/− MSCs was reduced by approximately 50% compared to those to WT MSCs (Fig 1D). These results indicate a potential link between impaired adhesion of co-cultured WT progenitors and decreased Cdc42 activity in FA MSCs.
Compromised hematopoietic supportive capacity of FA MSCs
To further characterize the hematopoiesis-supportive function of FA MSCs, we established an ex vivo co-culture assay to examine the supportive capacity of Fanca−/− or Fancc−/− MSCs on WT HSPCs. Limiting-dilution cobblestone area-forming cell (CAFC) assay [19, 32] (Fig S2) with graded numbers of flow sorted WT LSK cells showed that the frequencies of CAFC, at both week 1 and week 2, was significantly reduced in co-cultures on Fanca−/− or Fancc−/− MSCs compared to those on WT MSCs (Fig 2A), indicating that co-culture with Fanca−/− and Fancc−/− MSCs compromises self-renewal capacity of WT HSPCs.
Fig. 2. Compromised hematopoietic supportive capacity of FA MSCs.

(A) Co-cultured WT HSCs exhibit reduced Cobblestone area-forming capacity (CAFC). Sorted LSK (Lin−Sca1+c-kit+) cells from WT mice were added onto confluent WT, Fanca−/− or Fancc−/− BM-derived MSCs and incubated for 4 hours at 37°C followed by Limited dilution analysis (LDA) of CAFC. Assay was conducted in a flat bottom 96 well plate with confluent MSCs before plating the sorted LSK cells. Cultures were maintained in 40% methylcellulose medium for two weeks and colonies were counted on week 1 and 2. Group of at least 6 phase dim cells were counted as one colony. (B) Reduced in vivo repopulating capacity of WT HSPCs co-cultured with FA MSCs. Co-cultured WT LSK cells were transplanted into lethally irradiated BoyJ recipients. Donor-derived chimera were determined at 4 and 16 weeks post-transplant. (C, D) FA MSCs fail to support long-term hematopoiesis. Whole bone marrow cells (WBMCs) isolated from recipients described in (B) at 16 weeks post-transplant were transplanted into sublethally irradiated BoyJ recipients. Donor-derived chimera (Total CD45.2+; C) and percentages of LSK, SLAM cells in the donor-derived (Total CD45.2+) cell compartment (D) were determined at 16-weeks post-transplant. Representative flow plots and quantifications were shown. Results are means ± standard deviation (SD) of 3 independent experiments (n=9 per group). *p<0.05; ** p<0.01.
We next evaluate the repopulating ability of the co-cultured HSPCs in vivo by competitive repopulation assay using 1×105 output cells that had been co-cultured for one week on WT, Fanca−/− or Fancc−/− MSCs, along with 3×105 fresh BM competitor cells into lethally irradiated BoyJ recipients. At 16 weeks post-transplantation, the frequency of donor-derived cells, presumably the progenies of the output HSPCs, in the peripheral blood (PB) of mice that injected with cells co-cultured on Fanca−/− or Fancc−/− MSCs was significantly lower compared to that in the recipient mice that transplanted with cells co-cultured on WT MSCs (Fig 2B), suggesting a defective hematopoiesis-supportive capacity of FA MSCs.
To substantiate these findings, we next performed secondary transplantation by injecting whole bone marrow cells (WBMCs) from primary recipients to sublethally irradiated BoyJ recipients. Flow cytometry analysis at 16 weeks post BM transplantation (BMT) confirmed a decrease in donor-derived chimera in the secondary recipient mice transplanted with WBMCs from the primary recipients of co-cultured cells on Fanca−/− or Fancc−/− MSCs (Fig 2C). In addition, we observed a significantly decreased proportion of progenitor cells (Lin−Sca1+c-kit+, LSK) and phenotypic HSCs (LSKCD150+CD48−, SLAM; 37) in the donor-derived compartment of these secondary recipient mice compared to those receiving co-cultured cells on WT MSCs (Fig 2D). Together, these results indicate that FA MSCs are defective in supporting long-term repopulation of co-cultured HSPCs.
Genetic correction of FA deficiency restores Cdc42 activity and hematopoietic supportive capacity of FA MSCs
Since genetic correction of Fanca and Fancc deficiency can rescue FA HSPC function [38–40], we asked whether complementation of FA deficiency could correct the hematopoietic supportive defect of FA MSCs. To this end, we transduced MSCs isolated from Fanca−/− or Fancc−/− mice as well as their WT littermates with retroviral vectors expressing eGFP, eGFP-FANCA, or eGFP-FANCC. Sorted GFP+ cells (Fig S3) were then subjected to in vitro co-culture experiments. Significantly, we noticed that FA gene correction restored Cdc42 activity to nearly the level observed in WT MSCs (Fig 3A) and reversed the senescent phenotype of FA MSCs (Fig S4A). Concomitantly, complementation of Fanca−/− or Fancc−/− MSCs with FANCA or FANCC almost completely rescued self-renewal defects of the co-cultured WT HSC as determined by in vitro CFAC assay (Fig 3B). In addition, the output progenies from the co-culture on the gene-corrected Fanca−/− or Fancc−/− MSCs exhibited significantly improved hematopoietic reconstitution in both primary transplanted recipients (Fig 3C) and secondary recipients (Fig 3D), as compared to those co-cultured on the vector-alone (eGFP)-transduced Fanca−/− or Fancc−/− MSCs. These data suggest that the compromised supportive capacity of FA MSC is, at least in part, attribute to the decreased Cdc42 activity.
Fig. 3. Genetic correction of FA deficiency restores Cdc42 activity and supportive function of FA MSC.

(A) Genetic correction of FA deficiency restores Cdc42 activity in FA MSCs. MSCs isolated from WT, Fanca−/− or Fancc−/− mice were transduced with retroviral vector expressing eGFP, eGFP-FANCA, or eGFP-FANCC. Sorted GFP+ cells were subjected to in vitro Cdc42 pull down assay. The level of active, GTP-bound Cdc42, total Cdc42 and β-actin were determined. (B) Complementation of FANCA or FANCC improves CFAC of FA MSCs. Sorted GFP+ MSC cells described in (A) were co-cultured with graded numbers of flow sorted WT LSKs. Cultures were maintained in 40% methyl cellulose medium for two weeks and the colonies were counted on week 1 and 2. Group of at least 6 phase dim cells were counted as one colony. (C) Genetic correction of FA deficiency improves reconstitution of co-cultured WT HSCs in irradiated recipients. Co-cultured WT LSK cells described in (B) were transplanted into lethally irradiated BoyJ recipients. Donor-derived chimera was detected by flow cytometry analysis at 4 and 16 weeks post-transplant. (D) Complementation of FA deficiency improves long-term repopulating capacity of co-culture cells. WBMCs from the primary recipients were transplanted into sublethally irradiated BoyJ recipients. Donor-derived chimera were determined 16 week post transplant. Representative flow plots and quantifications were shown. Results are means ± standard deviation (SD) of 3 independent experiments (n=9 per group). *p<0.05; ** p<0.01.
Activation of Cdc42 improves the hematopoietic supportive function of FA MSCs
Since we observed reduced Cdc42 activity in FA MSCs, we next asked whether genetically increased Cdc42 activation could improve the hematopoiesis-supportive function of Fanca−/− and Fancc−/− MSCs. We took the approach of ectopically expressing the constitutively active form of Cdc42 mutant (Cdc42F28L in Fanca−/− or Fancc−/− MSCs) by retroviral gene transfer (Fig 4A). We found that forced expression of the constitutively active Cdc42F28L reversed the senescent phenotype (Fig S4B) and significantly increased the frequency of CAFC generated by WT HSPCs co-cultured on the transduced FA MSCs both at week 1 and week 2 (Fig 4B). When transplanted into irradiated mice, donor hematopoietic repopulation was profoundly increased in both primary and secondary recipients transplanted with the progenies of the HSPCs co-cultured on Cdc42F28L-expressing Fanca−/− or Fancc−/− MSCs (Fig 4C). Furthermore, analysis of secondary transplant recipients showed that ectopic expression of the active Cdc42F28L significantly increased the numbers of donor-derived progenitors (LSK) and HSCs (SLAM) (Fig 4D). These data further support the link of Cdc42 activity of FA MSCs to their hematopoiesis-supportive function.
Fig. 4. Expression of the constitutively active Cdc42 improves hematopoietic supportive function of FA MSCs.

(A) Ectopic expression of Cdc42F28L in FA MSCs. MSCs isolated from WT, Fanca−/− or Fancc−/− mice were transduced with retroviral vector expressing the constitutively active Cdc42 mutant, Cdc42F28L. Sorted GFP+ MSC cells were subjected to Western blot analysis. Expression of the constitutive active mutant of Cdc42 (Cdc42-F28L) containing an N-terminal HA tag in WT, Fanca−/− or Fancc−/− MSCs was analyzed by anti-HA western blotting. (B) Expression of Cdc42F28L improves Cobblestone forming of co-cultured WT HSPCs. Sorted GFP+ MSC cells described in (A) were co-cultured with graded numbers of flow sorted WT LSKs. Scoring of Cobblestone area as endpoint was determined after 7 days. (C) Expression of the constitutive active Cdc42 in FA MSCs improves repopulating ability of co-cultured WT HSPCs in vivo. Co-cultured WT HSPCs described in (B) along with 3×105 congenic BM cells isolated from BoyJ mice were transplanted into lethally irradiated BoyJ recipients. Donor-derived chimera were analyzed at the indicated time points. (D) Ectopic expression of Cdc42F28L improves long-term repopulating of co-culture WT HSPCs in the recipients. Secondary BMT was performed by injecting WBMCs from recipients described in (C) at 16 weeks post transplant. Donor-derived chimera, LSK and SLAM cells were analyzed at 16 weeks post transplant. *p<0.05; ** p<0.01.
Wnt5a improves FA MSCs function in supporting hematopoiesis of WT co-cultures
Recent studies have shown that stem-cell-intrinsic elevation of non-canonical Wnt5a plays a causative role in Cdc42 activation and HSC ageing [31]. We next asked whether Wnt5a could improve FA MSC function. Treatment of Fanca−/− or Fancc−/− MSCs with Wnt5a restored the active form of Cdc42 (Cdc42-GTP) to the normal WT level (Fig 5A) and reversed the senescent phenotype (Fig S4C). Pre-treatment Fanca−/− or Fancc−/− MSCs with Wnt5a also significantly increased cobblestone formation of the co-cultured WT HSPCs (Fig 5B). Furthermore, we observed a significant increase of donor-derived chimera in both primary and secondary recipient mice transplanted with the progenies cultured on Fanca−/− or Fancc−/− MSCs pre-treated with Wnt5a (Fig 5C). Finally, analysis of showed that Wnt5a there was a significant increase in the numbers of donor-derived LSK and SLAM cells in the secondary transplant recipients of the progenies cultured on Fanca−/− or Fancc−/− MSCs pre-treated with Wnt5a (Fig 5D). Thus, non-canonical Wnt5a improves FA MSC function through restoring Cdc42 activity.
Fig. 5. Activation of Cdc42 by Wnt5a improves hematopoietic supportive function of FA MSCs.

(A) Wnt5a activates Cdc42 in FA MSCs. MSCs isolated from WT, Fanca−/− or Fancc−/− mice were treated with Wnt5a followed by Western blot analysis using antibodies against Cdc42. The level of active, GTP-bound Cdc42, total Cdc42 and β-actin were determined. The relative levels of active Cdc42 are indicated below the blot. (B) Wnt5a treatment improves Cobblestone forming of co-cultured WT HSPCs. MSCs described in (A) were co-cultured with graded numbers of flow sorted WT LSKs. Scoring of Cobblestone area as endpoint was determined after 7 days. Results are means ± standard deviation (SD) of 3 independent experiments (n=6 per group). (C) Pretreatment of FA MSCs with Wnt5a improves reconstituting hematopoiesis of co-cultured WT HSPC in irradiated recipient. Co-cultured WT HSPCs described in (B) along with 3×105 congenic BM cells isolated from BoyJ mice were transplanted into lethally irradiated BoyJ recipients. Donor-derived chimera were analyzed at the indicated time points. (D) Wnt5a treatment improves long-term repopulating of co-cultured WT HSPCs in secondary recipients. Secondary BM transplant was performed by injecting WBMCs from the primary recipients described in (C). Donor-derived chimera, LSK and SLAM cells were analyzed at 16 weeks post-secondary transplant. Representative flow plots and quantifications were shown. Results are means ± standard deviation (SD) of 3 independent experiments (n=9 per group). *p<0.05; ** p<0.01.
FA proteins regulate Cdc42 activity via modulating Wnt5a expression
We next attempted to investigate the potential mechanism by which the FA pathway regulates Cdc42 activity. To this end, we transduced WT, Fanca−/−, Fancc−/− MSCs with retroviral vectors expressing eGFP, eGFP-FANCA, or eGFP-FANCC. Sorted GFP+ cells (Fig S3) were then subjected to analysis for the expression of Wnt5a. Significantly, we noticed that both mRNA and protein levels of Wnt5a were profoundly lower in Fanca−/− and Fancc−/− MSCs than those in their WT counterparts (Fig 6A). Gene correction of FA deficiency significantly increased the levels of Wnt5a mRNA and protein (Fig 6A). Consistently, pretreatment of Wnt5a restored Wnt5a expression in FA MSCs to almost the levels seen in WT cells (Fig 6B). These results suggest that FA proteins play an important role in regulating Cdc42 activity at least in part through modulating Wnt5a expression.
Fig. 6. FA proteins regulate Cdc42 activity via modulating Wnt5a expression.

(A) FA gene correction rescues Wnt5a mRNA and protein expression. MSCs from WT, Fanca−/− or Fancc−/− mice were transduced with retroviral vector expressing eGFP, eGFP-FANCA, or eGFP-FANCC. Sorted GFP+ cells were subjected to RNA extraction and qPCR analysis using the primers listed in Table S1 or ELISA assay. Samples were normalized to the level of GAPDH mRNA. (B) Wnt5a treatment upregulates Wnt5a mRNA and protein expression. MSCs isolated from WT, Fanca−/− or Fancc−/− mice were treated with Wnt5a followed by qPCR analysis for Wnt5a mRNA (Left) and ELISA analysis for Wnt5a protein (Right). *p<0.05; ** p<0.01.
Discussion
HSCs reside in the BM microenvironment (niche) where they preserve the capacity to self-renew and to continue producing all types of blood lineages throughout a prolonged lifespan [12]. The niche is composed mostly by MSCs, endothelial cells and additional factors [17, 42]. The molecular crosstalk between HSCs and the cellular constituents of these niches controls the balance between HSC self-renewal and differentiation [42]. Thus, defining niche components and how they work in concert to regulate hematopoiesis will provide the opportunity to improve the regeneration of HSCs, and reveal potential therapies for patients with BM failure and leukemia as well as other hematologic diseases. In this study, we demonstrated a deficit hematopoiesis-supportive function of FA MSCs and identified Cdc42 as a critical niche component in regulating this supportive function of FA MSCs. We have provided several lines of evidence to support our conclusion: (1) MSCs deficient for the Fanca or Fancc gene are defective in proliferating and prone to undergo senescence. (2) Cdc42 activity is significantly decreased in Fanca−/− and Fancc−/− MSCs. (3) WT HSPCs co-cultured on Fanca−/− or Fancc−/− MSCs exhibit compromised proliferating capacity both in vitro and in vivo. (4) Genetically correction of FA deficiency restores Cdc42 activity in FA MSCs. (5) Ectopic expression of a constitutively activating form of Cdc42 rescues hematopoiesis-supportive defects of FA MSCs. (6) Activation of Cdc42 by Wnt5a improves the hematopoiesis-supportive function of FA MSCs.
FA is a genomic instability syndrome associated with progressive BM failure and cancer susceptibility [6]. Common to all subtypes is cellular sensitivity to endogenously or exogenously produced interstrand cross links (ICLs), implicating spontaneous and induced chromosomal breakage, cell-cycle arrest and reduced cell survival [43, 44]. Consistent with these cellular phenotypes, we found that MSCs deficient for Fanca or Fancc gene are defective in proliferating when cultured in vitro by colony forming efficiency (CFE) assay (Fig 1A). In addition, compared with WT MSCs, FA MSCs tend to undergo senescence at earlier passages in culture (Fig 1B). These results are in line with the previous studies that show that FA MSCs are defective in their ability to survive in vitro and display spontaneous chromosome breakages [45].
Cdc42 is a member of Rho GTPase family. Previous studies have shown that conditional knockout of Cdc42 in murine HSCs led to defective F-actin polymerizarion, reduced adhesion to fibronectin matrix or stroma cells, dampened directional migration in response to SDF1α, and a massive egress of HSPCs from BM to PB, liver and spleen [23]. In the context of FA, a recent study used primary BM samples from FA patients and patient-derived FA lymphoblast cell lines to demonstrate that the human FA hematopoietic cells have an intrinsic decrease in the activity of Cdc42 [25]. Another recent study shows that Cdc42 is a cell polarity determinant and controls cell division symmetry to block leukemia cell differentiation [46]. Our unpublished data indicate that FA murine HPSCs also exhibit lower Cdc42 activity compared to the WT counterparts and that Cdc42 is de-polarized in FA HSCs (unpublished data). In this study, we show that MSCs from mice deficient for Fanca or Fancc have a reduced Cdc42 activity compared to WT MSCs (Fig 1C). Interestingly, FA deficiency does not affect Rac1-GTP content of Fanca−/− and Fancc−/− MSCs (Fig S1B), suggesting that the defect may be Cdc42-specific. Although it is currently not clear how Cdc42 activity is regulated in FA cells, our findings that genetic correction of FA deficiency (Fig 3), ectopic expression of a constitutively activating form of Cdc42 (Fig 4), or treatment with the non-canonical Wnt5a (Fig 5) can restore Cdc42 activity in FA MSCs shed some light on understanding the underlying regulatory mechanism of Cdc42 activity.
It has been shown that FA proteins interact with CtBP1 (the transcriptional corepressor protein C-terminal binding protein-1) and modulate the expression of the Wnt antagonist Dickkopf-1 (DKK1) [41]. Knocking down FANCD2 in Hela cells significantly upregulates DKK1 expression. It is also known that Rho GTPases integrate Wnt-induced signals spatially and temporally to promote morphological and transcriptional changes affecting cell behavior [47]. Specifically, Cdc42 and non-canonical Wnt signal transduction pathways coordinate to promote cell polarity [48]. Recent study demonstrated that Wnt5a activates Cdc42 to induce an ageing-like phenotype in young HSCs [31]. Our qPCR and ELISA analysis show that both mRNA and protein levels of Wnt5a are significantly lower in MSCs deficient for the Fanca or Fancc gene (Fig 6). Gene correction of FA deficiency (Fig 6A) or pretreatment of Wnt5a (Fig 6B) restores both mRNA and protein levels of Wnt5a in Fanca−/− and Fancc−/− MSCs. Our studies, taken together with the other referenced reports, suggest that FA proteins regulate Cdc42 activity possibly via modulating Wnt5a expression.
Another intriguing finding of the present study is our observation that loss of FA deficiency compromises the hematopoiesis-supportive function of MSCs. The hematopoietic supportive function is one of the important features of BM MSCs. It has been well established that the stromal feeder layer can be used to support HSC expansion and maintain stem cell quiescence both in vivo and in vitro [12, 42, 49]. It is also speculated that the environment beneath and/or niche atmosphere created by healthy MSC layer are critical for maintaining HSCs in an immature state [49]. Recent studies on the cellular characteristics of MSCs derived from the BM of certain FA patients implicate a role of the BM niche in the pathophysiology of FA BM failure [18–20]. In addition, a recent study using primary BM samples from FA patients and patient-derived FA lymphoblast cell lines demonstrates that the cell-autonomous defects of FA HSPCs in BM homing and engraftment are associated with a decrease in the activity of Cdc42 [25]. More recently, a study using Fancc and Fancg double knockout mouse model describes a phenotypic hematopoiesis-supportive defects of MSCs [50]. Our studies, taken together with these reports, suggest that FA gene deficiency compromises the function of MSC in supporting hematopoiesis, which is, at least in part, responsible for the hematopoietic defects observed in FA. Our study goes further to link this compromised hematopoiesis- supporting function to decreased Cdc42 activity in FA MSCs. In this context, our results suggest that targeted manipulation of Cdc42 activity in MSCs could be a therapeutic strategy to improve hematopoiesis and stem cell transplantation for FA patients.
Supplementary Material
Acknowledgments
We thank Dr. Madeleine Carreau (Laval University) for Fanca+/− mice, Dr. Manuel Buchwald (University of Toronto) for Fancc+/− mice. This work was supported by the National Natural Science Foundation of China (NNSFC #81470288, 81370608 and U1401221). W.D. is supported by West Virginia University (WVU) Health Science Center (HSC) and School of Pharmacy (SOP) startup funds, a NIH/NIGMS WV CTSI Award (U54 GM104942), a WVU General International Grant, and a Leukemia Research Foundation (LRF) Award.
Footnotes
Author Disclosure Statement
There is no conflict of interest to disclose on the part of any authors.
References
- 1.Bagby GC., Jr Genetic basis of Fanconi anemia. Curr Opin Hematol. 2003;10(1):68–76. doi: 10.1097/00062752-200301000-00011. [DOI] [PubMed] [Google Scholar]
- 2.Tischkowitz MD, Hodgson SV. Fanconi anaemia. J Med Genet. 2003;40(1):1–10. doi: 10.1136/jmg.40.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bluteau D, Masliah-Planchon J, Clairmont C, et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest. 2016;126:3580–3584. doi: 10.1172/JCI88010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11(7):467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V. Update of the human and mouse Fanconi anemia genes. Hum Genomics. 2015;9:32. doi: 10.1186/s40246-015-0054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Du W, Erden O Pang Q. TNF-a signaling in Fanconi anemia. Blood Cells Mol Dis. 2013;52(1):2–11. doi: 10.1016/j.bcmd.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kennedy RD, D’Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19(24):2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 8.Meyer S, Neitzel H, Tönnies H. Chromosomal aberrations associated with clonal evolution and leukemic transformation in Fanconi anemia: clinical and biological implications. Anemia. 2012;349837:2012. doi: 10.1155/2012/349837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JY, Virts EL, Jankowska A, et al. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi anaemia gene. J Med Genet. 2016;53:672–680. doi: 10.1136/jmedgenet-2016-103847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sawyer SL, Tian L, Kähkönen M, et al. Biallelic Mutations in BRCA1 Cause a New Fanconi Anemia Subtype. Cancer Discov. 2015;5(2):135–142. doi: 10.1158/2159-8290.CD-14-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knies K, Inano S, Ramírez MJ, et al. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest. 2017;127(8):3013–3027. doi: 10.1172/JCI92069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6(2):93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- 13.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–334. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walkley CR, Olsen GH, Dworkin S, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor γ deficiency. Cell. 2007;129:1097–1110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang B, Ho YW, Huang Q, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell. 2012;21:577–592. doi: 10.1016/j.ccr.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raaijmakers MH, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kode A, Manavalan JS, Mosialou I, et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature. 2014;506:240–244. doi: 10.1038/nature12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lecourt S, Vanneaux V, Leblanc T, et al. Bone marrow microenvironment in fanconi anemia: a prospective functional study in a cohort of fanconi anemia patients. Stem Cells Dev. 2010;19(2):203–208. doi: 10.1089/scd.2009.0062. [DOI] [PubMed] [Google Scholar]
- 19.Amarachintha S, Sertorio M, Wilson A, et al. Fanconi Anemia Mesenchymal Stromal Cells-Derived Glycerophospholipids Skew Hematopoietic Stem Cell Differentiation Through Toll-Like Receptor Signaling. Stem Cells. 2015;33(11):3382–3396. doi: 10.1002/stem.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantelli M, Avanzini MA, Rosti V, et al. Comprehensive characterization of mesenchymal stromal cells from patients with Fanconi anaemia. Br J Haematol. 2015;170(6):826–836. doi: 10.1111/bjh.13504. [DOI] [PubMed] [Google Scholar]
- 21.Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 22.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 23.Yang L, Wang L, Kalfa T, et al. Cdc42 critically regulates the balance between myelopoiesis and erythropoiesis. Blood. 2007;110:3853–3861. doi: 10.1182/blood-2007-03-079582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Yang L, Filippi MD, Williams DA, Zheng Y. Genetic deletion of Cdc42GAP reveals a role of Cdc42 in erythropoiesis and hematopoietic stem/progenitor cell survival, adhesion, and engraftment. Blood. 2006;107:98–105. doi: 10.1182/blood-2005-05-2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Shang X, Guo F, et al. Defective homing is associated with altered Cdc42 activity in cells from patients with Fanconi anemia group A. Blood. 2008;112(5):1683–1686. doi: 10.1182/blood-2008-03-147090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen M, Tomkins DJ, Auerbach W, et al. Inactivation of Fac in mice produces inducible chromosomal instability and reduced fertility reminiscent of Fanconi anaemia. Nat Genet. 1996;12(4):448–451. doi: 10.1038/ng0496-448. [DOI] [PubMed] [Google Scholar]
- 27.Wong JC, Alon N, Mckerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 2003;12:2063–2076. doi: 10.1093/hmg/ddg219. [DOI] [PubMed] [Google Scholar]
- 28.Huang S, Xu L, Sun Y, Wu T, Wang K, Li G. An improved protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Journal of Orthopaedic Translation. 2015;3(1):26–33. doi: 10.1016/j.jot.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delorme B, Ringe J, Gallay N, et al. Specific plasma membrane protein phenotype of culture-amplified and native human bone marrow mesenchymal stem cells. Blood. 2008;111(5):2631–2635. doi: 10.1182/blood-2007-07-099622. [DOI] [PubMed] [Google Scholar]
- 30.Houlihan DD, Mabuchi Y, Morikawa S, et al. Isolation of mouse mesenchymal stem cells on the basis of expression of Sca-1 and PDGFR-α. Nat Protoc. 2012;7(12):2103–2111. doi: 10.1038/nprot.2012.125. [DOI] [PubMed] [Google Scholar]
- 31.Florian MC, Nattamai KJ, Dörr K, et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature. 2013;503(7476):392–396. doi: 10.1038/nature12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Haan G, Ploemacher R. The Cobblestone-Area-Forming Cell Assay. Methods Mol Med. 2002;63:143–151. doi: 10.1385/1-59259-140-X:143. [DOI] [PubMed] [Google Scholar]
- 33.Guo F, Zheng Y. Involvement of Rho family GTPases in p19Arf or p53 mediated proliferation of primary mouse embryonic fibroblasts. Mol Cell Biol. 2004;24:1426–1428. doi: 10.1128/MCB.24.3.1426-1438.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mendez-Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morikawa S, Mabuchi Y, Kubota Y, et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med. 2009;206(11):2483–2496. doi: 10.1084/jem.20091046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugiyama T, Kohara H, Noda M, et al. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 37.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 38.Kee Y, D'Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest. 2012;122(11):3799–3806. doi: 10.1172/JCI58321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Du W, Maynard S, Andreassen PR, Pang Q. Oxidative stress-specific interaction between FANCD2 and FOXO3a. Blood. 2010;115(8):1545–1548. doi: 10.1182/blood-2009-07-234385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sejas DP, Rani R, Qiu Y, et al. Inflammatory reactive oxygen species-mediated hematopoietic suppression in Fancc-deficient mice. J Immunol. 2007;178:5277–5287. doi: 10.4049/jimmunol.178.8.5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huard CC, Tremblay CS, Helsper K, et al. Fanconi anemia proteins interact with CtBP1 and modulate the expression of the Wnt antagonist Dickkopf-1. Blood. 2013;121(10):1729–1739. doi: 10.1182/blood-2012-02-408997. [DOI] [PubMed] [Google Scholar]
- 42.Voog J, Jones DL. Stem cells and the niche: a dynamic duo. Cell Stem Cell. 2010;6(2):103–115. doi: 10.1016/j.stem.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Du W, Adam Z, Rani R, Zhang X, Pang Q. Oxidative stress in Fanconi anemia hematopoiesis and disease progression. Antioxid Redox Signal. 2008;10(11):1909–1921. doi: 10.1089/ars.2008.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668(1–2):4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mantelli M, Avanzini MA, Rosti V, et al. Comprehensive characterization of mesenchymal stromal cells from patients with Fanconi anaemia. Br J Haematol. 2015;170(6):826–836. doi: 10.1111/bjh.13504. [DOI] [PubMed] [Google Scholar]
- 46.Mizukawa B, O'Brien E, Moreira DC, et al. The cell polarity determinant CDC42 controls division symmetry to block leukemia cell differentiation. Blood. 2017;14(11):1336–1346. doi: 10.1182/blood-2016-12-758458. 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schlessinger K, Hall A, Tolwinski N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009;23(3):265–277. doi: 10.1101/gad.1760809. [DOI] [PubMed] [Google Scholar]
- 48.Schlessinger K, McManus EJ, Hall A. Cdc42 and noncanonical Wnt signal transduction pathways cooperate to promote cell polarity. J Cell Biol. 2007;178(3):355–361. doi: 10.1083/jcb.200701083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schepers K, Campbell TB, Passegué E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell. 2015;16(3):254–267. doi: 10.1016/j.stem.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Y, He Y, Xing W, et al. An abnormal bone marrow microenvironment contributes to hematopoietic dysfunction in Fanconi anemia. Haematologica. 2017;102(6):1017–1027. doi: 10.3324/haematol.2016.158717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.