

Abstract
Selective hydrogenation of dienes and trienes is an important process in the pharmaceutical and chemical industries. Our group previously reported that the thiosulfate protocol using a sodium S-alkylthiosulfate ligand could generate catalytically active Pd nanoparticles (PdNP) capped with a lower density of alkanethio-late ligands. This homogeneously soluble PdNP catalyst offers several advantages such as little contamination via Pd leaching and easy separation and recycling. In addition, the high activity of PdNP allows the reactions to be completed under mild conditions, at room temperature and atmospheric pressure. Herein, a PdNP catalyst capped with octanethiolate ligands (C8 PdNP) is investigated for the selective hydrogenation of conjugated dienes into monoenes. The strong influence of the thiolate ligands on the chemical and electronic properties of the Pd surface is confirmed by mechanistic studies and highly selective catalysis results. The studies also suggest two major routes for the conjugated diene hydrogenation: the 1,2-addition and 1,4-addition of hydrogen. The selectivity between two mono-hydrogenation products is controlled by the steric interaction of substrates and the thermodynamic stability of products. The catalytic hydrogenation of trienes also results in the almost quantitative formation of mono-hydrogenation products, the isolated dienes, from both ocimene and myrcene.
Introduction
Many different homogeneous or heterogeneous catalysts have been studied for various organic reactions including hydrogenation and carbon–carbon coupling reaction.1–3 Group VIII transition metals are usually the most common catalysts for these reactions. Moreover, palladium is more accessible and suitable for selective hydrogenation compared to other metal catalysts such as platinum and rhodium.4,5 It is because the lower metal–hydrogen bonding energy of platinum or rhodium makes these metal catalysts readily hydrogenate multiene substrates to alkanes.6–8 Therefore, more attention has been paid to developing palladium nanoparticles as selective hydrogenation catalysts.9–13
Based on previous research work from our group, alkanethiolate-capped palladium nanoparticles generated using the thiosulfate protocol have shown good catalytic reactivity for the hydrogenation or isomerization of alkenes.14–17 The lower surface coverage of alkanethiolate ligands on palladium nanoparticles could provide controlled poisoning for highly selective catalytic reactions. Palladium nanoparticles are still found to be stable enough to maintain homogeneous solubility for multiple recycling. Ligand-capped colloidal Pd nanoparticles are considered as semi-heterogeneous catalysts because the core contains various surface adsorption sites with different activities such as corners, edges, steps, and terraces. This characteristic of colloidal Pd nanoparticles led to their good reactivity, selectivity, facile separation and reusability, displaying the advantages of both homogenous and heterogeneous catalysts.
Freund et al. found out that the catalytic reaction of a terminal alkene would produce an alkane, the hydrogenation product, in a higher yield by increasing the size of Pd nano-particles.18,19 This positive correlation strongly indicates that the hydrogenation reaction is affected by the number of terrace sites in Pd nanoparticles, which also relates to the formation of a di-σ bonded Pd–alkyl intermediate. The poisoning effect of Pd nanoparticles by thiolate ligands would play an important role in the adsorption of alkene on the metal surface. This is because the available sites in the terrace surface would decrease more for ligand-passivated nano-particles, making the hydrogenation of alkenes to alkanes more difficult to occur.
Selective hydrogenation of small dienes is an important reaction for the petroleum industry, because dienes could act as monomers for polymerization that would deteriorate fuels during pyrolysis and storage processes.20–22 Fully saturated alkane petroleum products in a pure form are also not the most ideal since their lower boiling points and higher gas pressures would require containers with larger storage volumes. The naturally occurring trienes, ocimene and myrcene, are frequently used in the perfume industry, and the selective hydrogenation of natural triene products is also important in the pharmaceutical industry. It is because the primary product has the potential to be the precursor for functionalized compounds such as oxygenated derivatives.23,24 Many research groups have utilized different metal complexes and hydrogen sources to reduce diene and triene compounds in various solvents using either homogeneous or heterogeneous conditions.
An octanethiolate-passivated palladium nanoparticle (C8 PdNP) catalyst has shown good reactivity for the selective hydrogenation of penta-1,4-diene, an isolated diene, to monoenes under mild reaction conditions in our previous work.25 Building upon this previous work, in this article we investigate the catalytic applications of C8 PdNP for the selective hydrogenation of various conjugated dienes and trienes. The mechanism for the high selectivity of C8 PdNP is proposed based on kinetic studies and catalysis results for various substrates. Comprehensive comparisons with other catalytic systems on the selective hydrogenation of conjugated dienes and trienes are also presented.12,26,27
Results and discussion
An octanethiolate-capped Pd nanoparticle (C8 PdNP) catalyst with an average core size of ~2.3 nm is synthesized and its composition and structure are confirmed using various instruments as shown in our previous article and in the ESI.†25 C8 PdNP is used as the catalyst for the hydrogenation of various conjugated diene and triene substrates as shown in Table 1. The catalytic activity of C8 PdNP is directly compared with other Pd-based heterogeneous catalysts (Pd/C and Pd/Al2O3).
Table 1.
Catalysis results of various dienes and trienes with the octanethiolate-capped Pd nanoparticle catalyst, Pd on carbon, and Pd on alumina in CDCl3 at 1 atm H2 after 24 ha
| Substrate | C8 PdNP | Pd/C | Pd/Al2O3 | |||
|---|---|---|---|---|---|---|
|
|
|
|
||||
| MHb | DHb | MHb | DHb | MHb | DHb | |
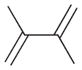 1 |
100% | 0% | 0% | 100% | 0% | 100% |
 2 |
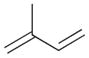
100% | 0% | 0% | 100% | 0% | 100% |
|
3 |
100% | 0% | 0% | 100% | —c | —c |
|
4 |
100% | 0% | 0% | 100% | 0% | 100% |
|
5 |
97% | 3% | 9% | 91% | 6% | 94% |
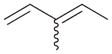 6 |
95% | 0% | 0% | 100% | 0% | 100% |
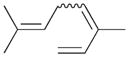 7 |
100% | 0% | 0% | 100% | 0% | 100% |
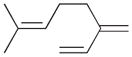 8 |
92% | 0% | 4% | 96% | 10% | 90% |
Reaction conditions: 0.5 mmol diene, 5 mol% Pd catalyst, 2.5 mL CDCl3, 1 atm H2, 24 h reaction. Other heterogeneous catalysts, Pd/C and Pd/Al2O3, were also examined under the same reaction conditions. Yields were obtained by 400 MHz NMR.
Mono-hydrogenation (MH), double-hydrogenation (DH).
Not attempted.
The catalytic reactions of conjugated dienes with different substitution patterns around C=C bonds (1–6) using C8 PdNP produce almost exclusively mono-hydrogenation products (MH), without having double hydrogenation products (DH). In addition, the hydrogenation of trienes (7 and 8) using C8 PdNP also results in high yields of isolated dienes, the mono-hydrogenation products (MH). This high selectivity of C8 PdNP for mono-hydrogenation reaction is clearly distinguishable from the catalytic results of other Pd-based heterogeneous catalysts, Pd/C and Pd/Al2O3, that produce mostly double-hydrogenation products (DH) from both dienes and trienes. This different reactivity confirms the important role of alkanethiolate ligands in controlling the activity of the PdNP catalyst surface. With its surface passivated by alkane-thiolate ligands, the PdNP has increased stability, so that it avoids aggregation and keeps the large surface area intact. The alkanethiolate surface ligand on the PdNP would block more active sites (terrace surfaces) for hydrogenation and promote the selective hydrogenation of dienes.
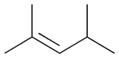
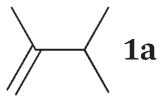
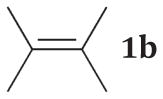
The product ratios of the 1,4- and 1,2-addition products in addition to the overall conversion yields during the kinetic study are summarized in Table 2. The catalytic reaction of C8 PdNP with 2,3-dimethyl-buta-1,3-diene 1 exhibits excellent selectivity toward the mono-hydrogenation products with 100% conversion yield. The analysis of the final monoene composition showed that the major product is 2,3-dimethyl-2-butene 1b, the 1,4-addition product, and the minor product is 2,3-dimethyl-1-butene 1a, the 1,2-addition product. The high yield of 1b is the result of both the initial 1,4-addition reaction and the subsequent isomerization of the terminal alkene, the 1,2-addition product, into an internal alkene. This is clearly evidenced by the kinetic study of dienes to monoenes, which is monitored by 1H NMR as shown in Fig. 1. The decreasing signals at 1.9 and 4.9 ppm in Fig. 1 indicate the consumption of compound 1, and the increased signals at 1.7 (or 1.0) and 1.6 ppm correspond to two mono-hydrogenation products, which are the chemical shifts for 1a and 1b, respectively. After the first hour of reaction, the conversion of 1 reaches over 50% with the ratio of the 1,4-/1,2-addition products at 3.43. The TOF at the first hour is calculated to be 375.1 molecules per Pd per h. The consumption of reactant 1 is almost complete after 5 h of reaction with the ratio of the 1,4-/1,2-addition products at 4.9. The result of the 24 h reaction indicates that the yield of 1b is still increasing after 5 h of reaction with the ratio of the 1,4-/1,2-addition products reaching 10.4. This is clear evidence suggesting that the isomerization of the terminal alkene 1a to the internal alkene 1b continuously takes place after the 5th hour of reaction. The good activity of C8 PdNP for the isomerization of terminal alkenes to internal alkenes driven by a thermodynamic effect has previously been reported by our group.25 The mechanistic explanation for the high selectivity to the 1,4-addition product in the 2,3-dimethyl-buta-1,3-diene 1 hydrogenation reaction is provided in Scheme 1.
Table 2.
Hydrogenation of different diene and triene substrates using C8 PdNPa
| Substrate | 1,2-Add | Yield (%) | 1,4-Add | Yield (%) | 1,4-Add/1,2-add |
|---|---|---|---|---|---|
|
1 |
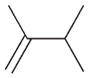 1a |
9 |
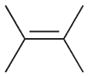 1b |
91 | 10.4 |
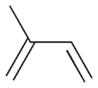 2 |
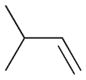
 2a |
7 |
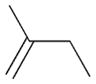
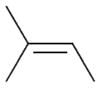 2c |
93 | 14.2 |
 2b |
0 | ||||
|
3b |
3a |
92c E/Z = 4.0 |
3a |
— | — |
|
3b |
8 | ||||
|
4 |
4a |
29 |
4c |
71 E/Z = 3.5 | 2.4 |
|
4b |
0 | ||||
|
5b |
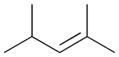 5a |
90c |
 5a |
— | — |
 5b |
7 | ||||
|
6b |
 6a |
90c |
 6a |
— | — |
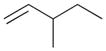 6b |
5 | ||||
|
7 |
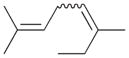 7a |
41 |
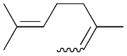 7c |
59 | 1.5 |
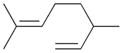 7b |
0 | ||||
|
8 |
8a |
3 |
7c |
69 | 3.0 |
 8b |
20 |
Reaction conditions: 0.5 mmol diene, 5 mol% Pd catalyst, 2.5 mL CDCl3, 1 atm H2, 24 h reaction. Yields were obtained by 400 MHz 1H NMR.
The catalytic reaction of this substrate yields the same product after 1,2- and 1,4-addition reactions.
This yield is the combination of both 1,2- and 1,4-addition reactions.
Fig. 1.

(A) Kinetic study of 2,3-dimethylbuta-1,3-diene 1 hydrogenation using C8 PdNP at room temperature under 1 atm H2 in CDCl3. (B) The zoom is for the area of the 5 and 24 h spectra. Chemical shifts of compound 1a are at 1.02 ppm (–CH3) and 1.71 ppm (allylic-CH3). Chemical shift for compound 1b is at 1.64 ppm (–CH3).
Scheme 1.
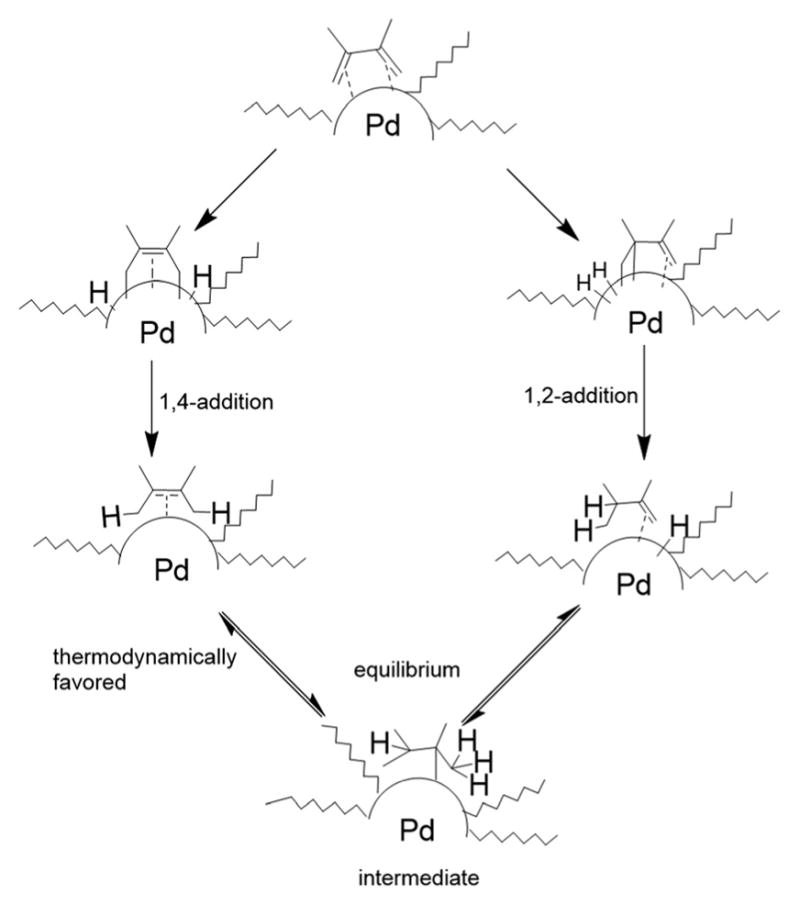
Proposed mechanism for the catalytic reaction of 2,3-dimethylbuta-1,3-diene 1 with octanethiolate-capped Pd nanoparticles.
In order to prove that the direct 1,4-addition is the major reaction route instead of the isomerization of the 1,2-addition product, the catalytic hydrogenation of commercially available compound 1a is studied. The result shows the low conversion efficiency of isomerization from 1a to 1b with only 52% of 1a being isomerized to 1b after 24 h of reaction (Table 3). The slow conversion of 1a to 1b is partly due to the lower number of α hydrogen atoms in the mono-σ-bonded palladium alkyl intermediate generated from the adsorption of alkene 1a on the PdNP surface. Since the terminal carbon has more hydrogen atoms than the internal α carbon, the mono-σ-bonded intermediate would most likely take the hydrogen from the terminal carbon. It would cause the intermediate to equilibrate back to the starting material, the terminal alkene. The kinetic study showing the low conversion rate of 1a to 1b proves that the initial direct 1,4-addition route for 1 would be the major route for the formation of 1b and supplemented by the isomerization of the 1,2-addition product 1a.
Table 3.
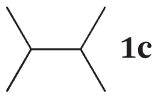
Hydrogenation of 2,3-dimethylbut-1-ene with the C8 PdNP catalysta
| Time |
|
|
|
|---|---|---|---|
|
|
|
|
|
| Terminal alkene (%) | Internal alkene (%) | Alkane (%) | |
| 1 h | 88% | 12% | 0% |
| 3 h | 78% | 22% | 0% |
| 6 h | 66% | 34% | 0% |
| 24 h | 48% | 52% | 0% |
Reaction conditions: 0.5 mmol diene, 5 mol% Pd catalyst, 2.5 mL CDCl3, 1 atm H2, 24 h reaction. Yields were obtained by 400 MHz 1H NMR.
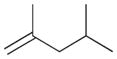
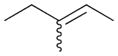
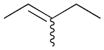
The selectivity between the 1,4- and 1,2-addition products is also summarized for other diene and triene substrates in Table 2. Due to the structural characteristics of substrates 3, 5, and 6, generating the same products for the 1,2-addition and 1,4-addition reactions, the major reaction route for these substrates could not be identified by analysing the products. However, C8 PdNP clearly shows high selectivity for the 1,4-addition product from the reactions of 2-methylbuta-1,3-diene 2 and hexa-1,3-diene 4. After the direct formation of both 1,4-addition product 2c and 1,2-addition product 2a, the isomerization of 2a would further take place readily because of the presence of two hydrogens that can be removed from its mono-σ-bonded Pd alkyl intermediate. Though a trace amount of 2b is observed during the first few hours of reaction, the thermodynamic stability of the isomerized product 2c makes 2b even easier to isomerize, completely depleting the presence of 2b after 24 h of reaction.
For substituted buta-1,3-dienes 1 and 2, the hydrogenation results in a higher 1,4-/1,2-addition ratio because the formation of a 1,4-di-σ-bonded intermediate for these substrates is sterically more accessible than that of other substrates with substituent groups on the 1,4-position. In addition, the isomerization of the 1,2-addition product occurs more easily because the equilibrium of isomerization would be driven by forming a more thermodynamically stable product. For the catalytic reaction of hexa-1,3-diene 4, however, the 1,4-/1,2-addition product ratio is relatively lower than that of substituted buta-1,3-dienes. The 1,4-di-σ-bonded intermediate generated from 4 is less accessible due to the presence of an ethyl substituent group causing steric hindrance for the initial hydrogen addition. Moreover, the isomerization of 4a, one internal di-substituted alkene, to 4c, another internal di-substituted alkene, does not provide any additional thermodynamic enthalpy reduction eliminating a driving force for the isomerization.
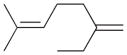
The analyses of the mono-hydrogenation products generated from the hydrogenation of trienes 7 and 8 are also shown in Table 2. The presence of a large alkyl substituent group at the diene part of ocimene 7 decreases the ratio of the 1,4- and 1,2-addition products. The yield for 7a, the 1,2-addition product, is relatively high due to the good accessibility of the terminal alkene group to the Pd surface. Another possible 1,2-addition product 7b does not form because of the steric interference provided by the surrounding substituent groups. The lack of a thermodynamic driving force from products 7a to 7c as seen from the reaction of 4 is another main reason for the decreased yields of the 1,4-addition product. The ratio of the 1,4-/1,2-addition products for the catalytic reaction of myrcene 8 is, however, more than twice higher than that of ocimene 7. This is because both terminal carbons of the diene group do not possess any substituent group resulting in less steric interference for direct 1,4-hydrogen addition compared to ocimene 7. The thermodynamic stability of 7c is also higher than that of 8a and 8b, the more likely 1,2-addition products, making the isomerization more likely to occur. As shown in Table 2, C8 PdNP is highly selective towards the mono-hydrogenation products for both ocimene and myrcene and would not hydrogenate the highly substituted and isolated C6═C7 double bond.
C8 PdNP clearly exhibits excellent selectivity to internal alkenes, the mono-hydrogenation and 1,4-addition products, compared to other reported catalytic systems summarized in Table 4 for diene hydrogenation.12,21,27–32 Not only are the conversion yields and selectivity superior but also the reaction conditions (room temperature and atmospheric pressure) are much friendlier than any known homogenous and heterogeneous catalysts tested for diene hydrogenation. Since both dienes and terminal alkenes are potential monomers for the polymerization reaction, C8 PdNP could either hydrogenate the dienes by a 1,4-addition reaction or isomerize terminal alkenes to internal alkenes under mild reaction conditions, so that the deterioration of petroleum products can be effectively prevented by limiting the potential polymerization in the petroleum pyrolysis process. The comparison of C8 PdNP with other catalysts used for ocimene and myrcene hydrogenation is also summarized in Table 4.23,33 The results again confirmed the excellent activity and selectivity of C8 PdNP for the selective mono-hydrogenation of trienes to internal isolated dienes.
Table 4.
Comparison table for the catalytic hydrogenation selectivity of dienes and trienes
| Substrate | Catalyst | Reaction pathwayb | Convb | MHb | DHb | 1,2-Add | 1,4-Add | Internal alkene | Reaction conditions |
|---|---|---|---|---|---|---|---|---|---|
|
|
C8 PdNP | Semi-HET | 100% | 100% | 0% | 8.8% | 91.2% | 91.2% | R.T., CDCl3, 1 atm H2 |
| PhCOOMeCr(CO)3 (ref. 27) | HOM | 100% | 100% | 0% | 18% | 82% | 82% | 160 °C, n-heptane, 30 atm H2 | |
 |
Pd/sgPF6 (ref. 12) | Semi-HET | >99% | 100% | 0% | 19% | 81% | 81% | 40 °C, CH2Cl2, 4 bar H2 |
| C8 PdNP | Semi-HET | 100% | 100% | 0% | 6.6% | 93.4% | 93.6% | R.T., CDCl3, 1 atm H2 | |
| Silica polyamine PdNP composite21 | HET | 100% | 94% | 6% | 46.5% 2a: 24.5% 2b: 22% | 47.5% | 47.5% | 70 °C, toluene, 3 MPa H2 | |
| Pd/sgPF6 (ref. 12) | Semi-HET | >99% | 100% | 0% | 25% 2a: 18% 2b: 7% | 75% | 75% | 40 °C, CH2Cl2, 4 bar H2 | |
|
|
C8 PdNP | Semi-HET | 100% | 100% 3a: 92% 3b: 8% | 0% | — | — | 98.2% | R.T., CDCl3, 1 atm H2 |
| Co3O4 (ref. 28) | HET | 39.3% | 39.3% 3a: 31.1% 3b: 8.2% | 0% | — | — | 31.1% | R.T., P = 50–60 C5H8 mmHg, P/P = 2–3 H2 C5H8 | |
| [Cp2ZrH(CH2PPh2)]n (ref. 29) | HOM | 100% | 0% | 100% | — | — | 0%a | 80 °C, hexane/THF, 40 bar H2 | |
|
|
C8 PdNP | Semi-HET | 100% | 100% | 0% | 29.3% 4a only | 70.7% | 100% | R.T., CDCl3, 1 atm H2 |
| PhCOOMeCr(CO)3 (ref. 27) | HOM | 100% | 100% | 0% | 24% 4a: 18% 4b: 6% | 76% | 94% | 160 °C, n-heptane, 30 atm H2 | |
|
|
C8 PdNP | Semi-HET | 100% | 96.6% 5a: 90.3% 5b: 6.3% | 3.4% | — | — | 90.3% | R.T., CDCl3, 1 atm H2 |
| Polystyrene coated PdNP on SiO2 (ref. 30) | HET | >99.7% | >99.6% 5a: 80.1% 5b: 19.8% | 0.1% | — | — | 80.1% | Supercritical CO2, 3 MPa H2 | |
| Dendrimer Pd complex31 | HOM | >99% | >99% 5a: 57% 5b: 43% | 0% | — | — | 57% | 25 °C, MeOH, 14 p.s.i. H2 | |
|
|
C8 PdNP | Semi-HET | 95% | 95% 6a: 90.6% 6b: 4.1% | 0% | — | — | 90.6% | R.T., CDCl3, 1 atm H2 |
| Pd ferrocenyl complex32 | — | 87.1% | 82.4% 6a: 82.4% 6b: 4% | 0.7% | — | — | 82.4% | R.T., acetone, 80 p.s.i. H2 | |
 |
C8 PdNP | Semi-HET | 100% | 100% | 0% | 40.8% 7a only | 59.2% | 100% | R.T., CDCl3, 1 atm H2 |
| Copper–chromium oxide33 | HET | 100% | 85% | 15% | 78% 7a: 58% 7b: 20% | 7% | 65% | 140 °C, trans-decalin, 1 atm pressure | |
|
|
C8 PdNP | Semi-HET | 92.2% | 92.2% | 0% | 23.2% 8a: 3.1% 8b: 20% | 69% | 92.2% | R.T., CDCl3, 1 atm H2 |
| Sol/gel 3% Pd/SiO2 (ref. 23) | HET | 96% | 93.1% | 2.9% | 33.6% 8a: 19.2% 8b: 14.4% | 59.5% | 59.5% | 80 °C, 20 atm, H2 cyclohexane | |
| Copper–chromium oxide33 | HOM | 100% | 100% | 0% | 92% 8a: 23% 8b: 69% | 8% | 8% | 140 °C, trans-decalin, 1 atm pressure |
The data are chosen from the best selectivity results for each type of catalyst.
Abbreviations for the reaction pathway: semi-heterogeneous (Semi-HET), homogeneous (HOM), heterogeneous (HET). Conversion (Conv). Mono-hydrogenation (MH), double-hydrogenation (DH).
The stability and recyclability of C8 PdNP are tested using the catalytic reaction of 2,3-dimethylbuta-1,3-diene, 1. C8 PdNP is precipitated and separated from the reaction mixture. The PdNP is further dried without any solvent before being recycled for additional catalysis reactions. As shown in Fig. 2, the activity and selectivity of C8 PdNP are mostly maintained especially without generating any double hydrogenation product even after 7 cycles. The conversion of the reactant after 7 cycles is still above 95% and the selectivity for the 1,4-addition product remains over 80% after 7 catalytic cycles. To examine the leaching of Pd atoms from C8 PdNP during the catalytic reaction, the presence of Pd2+ in the crude solution is determined using ICP-AES. The results suggest that the digested product solution has only ~0.0546 ppm Pd2+, which shows the overall high stability of C8 PdNP during the hydrogenation reaction.
Fig. 2.
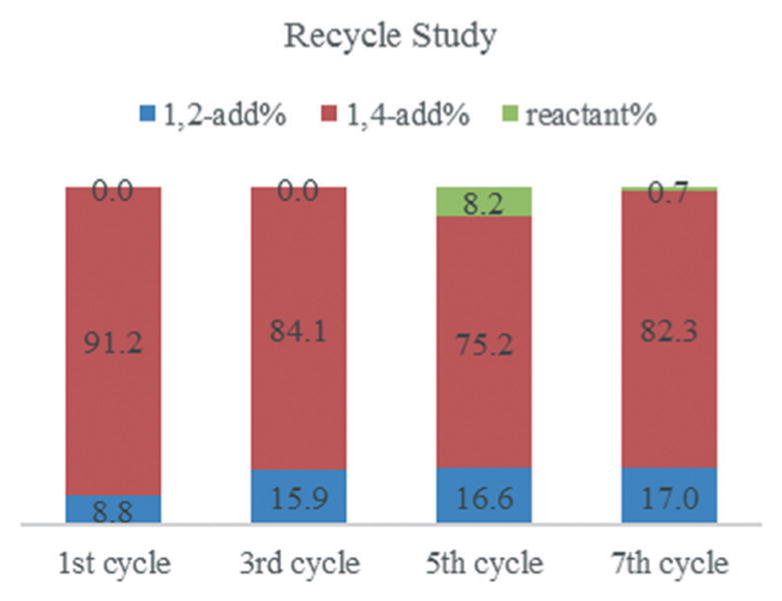
The recycle study of 2,3-dimethylbuta-1,3-diene 1 hydrogenation using C8 PdNP. Reaction conditions: 0.5 mmol diene, 5 mol% Pd catalyst, 2.5 mL CDCl3, 1 atm H2, 24 h reaction. Yields were obtained by 400 MHz NMR.
Conclusions
Octanethiolate-capped palladium nanoparticles show excellent activity and selectivity for the mono-hydrogenation of dienes into internal alkenes and trienes into isolated internal dienes. The kinetic study and comparison of various catalysis reactions indicate that the conjugated diene hydrogenation reactions mainly involve the direct 1,4-addition reaction in addition to the isomerization of the 1,2-addition product that further isomerizes terminal alkenes into internal alkenes. The leaching examination and recycling study of the octanethiolate-capped palladium nanoparticles show the high stability of the catalyst. This octanethiolate-capped palladium nanoparticle catalyst, therefore, has high technological potential for effective elimination of reactive monomers such as dienes and terminal alkenes for polymerization and prevention against the deterioration of crude oil storage.
Supplementary Material
Acknowledgments
This research was supported by the W. M. Keck Foundation and the National Institute of General Medical Sciences (SC3GM089562).
Footnotes
Electronic supplementary information (ESI) available.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Tatumi R, Akita T, Fujihara H. Chem Commun. 2006:3349–3351. doi: 10.1039/b605390b. [DOI] [PubMed] [Google Scholar]
- 2.Guo XW, Hao CH, Wang CY, Sarina S, Guo XN, Guo XY. Catal Sci Technol. 2016;6:7738–7743. [Google Scholar]
- 3.Dostert KH, O’Brien CP, Ivars-Barceló F, Schauermann S, Freund HJ. J Am Chem Soc. 2015;137:13496–13502. doi: 10.1021/jacs.5b04363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tourillon G, Cassuto A, Jugnet Y, Massardier J, Bertolini JC. J Chem Soc, Faraday Trans. 1996;92:4835–4841. [Google Scholar]
- 5.Boitiaux JP, Cosyns J, Robert E. Appl Catal. 1987;35:193–209. [Google Scholar]
- 6.Guo XC, Madix RJ. J Catal. 1995;155:336–344. [Google Scholar]
- 7.Christmann K. Surf Sci Rep. 1988;9:1–163. [Google Scholar]
- 8.Ouchaib T, Massardier J, Renouprez A. J Catal. 1989;119:517–520. [Google Scholar]
- 9.Ooe M, Murata M, Mizugaki T, Ebitani K, Kaneda K. J Am Chem Soc. 2004;126:1604–1605. doi: 10.1021/ja038455m. [DOI] [PubMed] [Google Scholar]
- 10.Wu L, Li BL, Huang YY, Zhou HF, He YM, Fan QH. Org Lett. 2006;8:3605–3608. doi: 10.1021/ol0614424. [DOI] [PubMed] [Google Scholar]
- 11.Gavia DJ, Koeppen J, Sadeghmoghaddam E, Shon YS. RSC Adv. 2013;3:13642–13645. doi: 10.1039/c3ra42119h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luza L, Rambor CP, Gual A, Bernardi F, Domingos JB, Grehl T, Brüner P, Dupont J. ACS Catal. 2016;6:6478–6486. [Google Scholar]
- 13.Mitsudome T, Urayama T, Yamazaki K, Maehara Y, Yamasaki J, Gohara K, Maeno Z, Mizugaki T, Jitsukawa K, Kaneda K. ACS Catal. 2016;6:666–670. [Google Scholar]
- 14.Sadeghmoghaddam E, Gu H, Shon YS. ACS Catal. 2012;2:1838–1845. doi: 10.1021/cs300270d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gavia DJ, Shon YS. Langmuir. 2012;28:14502–14508. doi: 10.1021/la302653u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadeghmoghaddam E, Gaïeb K, Shon YS. Appl Catal, A. 2011;405:137–141. [Google Scholar]
- 17.Gavia DJ, Maung MS, Shon YS. ACS Appl Mater Interfaces. 2013;5:12432–12440. doi: 10.1021/am4035043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doyle AM, Shaikhutdinov SK, Freund HJ. Angew Chem, Int Ed. 2005;44:629–631. doi: 10.1002/anie.200461614. [DOI] [PubMed] [Google Scholar]
- 19.Doyle AM, Shaikhutdinov SK, Jackson SD, Freund HJ. Angew Chem, Int Ed. 2003;42:5240–5243. doi: 10.1002/anie.200352124. [DOI] [PubMed] [Google Scholar]
- 20.Lin TB, Chou TC. Appl Catal, A. 1994;108:7–19. [Google Scholar]
- 21.Karakhanov E, Maximov A, Kardasheva Y, Semernina V, Zolotukhina A, Ivanov A, Abbott G, Rosenberg E, Vinokurov V. ACS Appl Mater Interfaces. 2014;6:8807–8816. doi: 10.1021/am501528a. [DOI] [PubMed] [Google Scholar]
- 22.Yardimci D, Serna P, Gates BC. ACS Catal. 2012;2:2100–2113. [Google Scholar]
- 23.Robles-Dutenhefner PA, Speziali MG, Sousa EMB, dos Santos EN, Gusevskaya EV. Appl Catal, A. 2005;295:52–58. [Google Scholar]
- 24.Speziali MG, Moura FCC, Robles-Dutenhefner PA, Araujo MH, Gusevskaya EV, dos Santos EN. J Mol Catal A: Chem. 2005;239:10–14. [Google Scholar]
- 25.Zhu JS, Shon YS. Nanoscale. 2015;7:17786–17790. doi: 10.1039/c5nr05090a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schrock RR, Osborn JA. J Am Chem Soc. 1976;98:4450–4455. [Google Scholar]
- 27.Frankel EN, Butterfield RO. J Org Chem. 1969;34:3930–3936. [Google Scholar]
- 28.Okuhara T, Tanaka K. J Chem Soc, Chem Commun. 1978;53–54 [Google Scholar]
- 29.Raoult Y, Choukroun R, Basso-Bert M, Gervais D. J Mol Catal. 1992;72:47–58. [Google Scholar]
- 30.Wu T, Jiang T, Hu B, Han B, He J, Zhou X. Green Chem. 2009;11:798–803. [Google Scholar]
- 31.Zweni PP, Alper H. Adv Synth Catal. 2006;348:725–731. [Google Scholar]
- 32.Naiini AA, Lai CK, Ward DL, Brubaker CH. J Organomet Chem. 1990;390:73–90. [Google Scholar]
- 33.Hubaut R, Bonnelle JP, Daage M. J Mol Catal. 1989;55:170–183. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.