

Abstract
The “dysconnection hypothesis” of psychosis suggests that a disruption of functional integration underlies cognitive deficits and clinical symptoms. Impairments in the P300 potential are well documented in psychosis. Intrinsic (self‐)connectivity in a frontoparietal cortical hierarchy during a P300 experiment was investigated. Dynamic Causal Modeling was used to estimate how evoked activity results from the dynamics of coupled neural populations and how neural coupling changes with the experimental factors. Twenty‐four patients with psychotic disorder, twenty‐four unaffected relatives, and twenty‐five controls underwent EEG recordings during an auditory oddball paradigm. Sixteen frontoparietal network models (including primary auditory, superior parietal, and superior frontal sources) were analyzed and an optimal model of neural coupling, explaining diagnosis and genetic risk effects, as well as their interactions with task condition were identified. The winning model included changes in connectivity at all three hierarchical levels. Patients showed decreased self‐inhibition—that is, increased cortical excitability—in left superior frontal gyrus across task conditions, compared with unaffected participants. Relatives had similar increases in excitability in left superior frontal and right superior parietal sources, and a reversal of the normal synaptic gain changes in response to targets relative to standard tones. It was confirmed that both subjects with psychotic disorder and their relatives show a context‐independent loss of synaptic gain control at the highest hierarchy levels. The relatives also showed abnormal gain modulation responses to task‐relevant stimuli. These may be caused by NMDA‐receptor and/or GABAergic pathologies that change the excitability of superficial pyramidal cells and may be a potential biological marker for psychosis. Hum Brain Mapp 38:3262–3276, 2017. © 2017 Wiley Periodicals, Inc.
Keywords: psychosis, schizophrenia, unaffected relatives, genetic risk, effective connectivity, intrinsic connectivity, dynamic causal modeling, DCM, synaptic gain, cortical excitability, self‐inhibition, NMDA, GABA, P300
INTRODUCTION
Psychotic disorders are severe mental illnesses characterized not only by a broad range of clinical symptoms and cognitive dysfunctions, but also by underlying neurophysiological abnormalities. Patients with psychotic disorder have well‐replicated changes in several electroencephalography (EEG) event‐related potential (ERP) components such as the P300 [Bramon et al., 2008; Ford, 1999; Jeon and Polich, 2003]. Their unaffected relatives also show these alterations, albeit to a lesser extent, suggesting that the P300 might be a biological marker of genetic vulnerability to develop psychosis [Bramon et al., 2005; Hall et al., 2009; Thaker, 2008; Turetsky et al., 2007].
The P300 potential is elicited during an oddball paradigm after the onset of task‐relevant infrequent targets amid frequent task‐irrelevant stimuli, and it is thought to reflect high‐level cognitive processes, such as selective attention and working memory [Bledowski et al., 2006; Polich and Criado, 2006]. The P300 has been thoroughly studied in healthy and clinical populations, and a frontoparietal attentional network seems to be involved in its generation [Polich, 2007]. Frontal and parietal regions are robustly coupled during auditory attention [Dietz et al., 2014; Auksztulewicz and Friston, 2015], and working memory paradigms [Ma et al., 2012]. Functional frontoparietal disconnection has been found both in patients with psychotic disorder [Kim et al., 2003; Roiser et al., 2013] and individuals at high genetic risk [Deserno et al., 2012; Whalley et al., 2005].
There is broad evidence that abnormal neural oscillations contribute to cognitive dysfunction and clinical symptoms in psychosis [Uhlhaas and Singer, 2010], which may reflect alterations in synchronous gain and effective connectivity [Chawla et al., 1999]. For instance, in previous work we found an inefficient increase of frontal activity, in this case in the gamma band, related to abnormal P300 and working memory deficits in patients with schizophrenia and their unaffected relatives [Díez et al., 2013, 2014]. The “dysconnection hypothesis” suggests that not only focal brain abnormalities but also a disruption of synaptic plasticity and hence functional integration are responsible for psychosis [Friston, 1998; Stephan et al., 2006, 2009]. Although this hypothesis is widely accepted, the underlying architecture of dysfunctional coupling is not yet well understood.
EEG ERPs can be modeled as perturbations of cortical networks. Dynamic Causal Modeling (DCM) [Friston et al., 2003] is a Bayesian inference‐based method for estimating changes in the effective connectivity—that is, directed coupling within or between cortical sources—in a hierarchical network given changes in its inputs. DCM for EEG data [David et al., 2006; Kiebel et al., 2006] uses biologically constrained spatiotemporal generative models of ERPs. This requires the specification of a neurobiological—or neural mass—model that makes predictions about the ensemble dynamics of interacting inhibitory and excitatory subpopulations [David et al., 2005; David and Friston, 2003]; and involves a forward mapping of source to sensor activity that can generate predictions of electrophysiological responses [David et al., 2006; Kiebel et al., 2006; Moran et al., 2013; Pinotsis et al., 2012]. These predictions are compared with recorded EEG data to explore different hypotheses about how brain connectivity generates observed responses. In brief, given a particular model, Bayesian model inversion is used to estimate the probability of the data by optimizing the marginal likelihood or model evidence. DCM uses this evidence to compare alternative connectivity models, allowing inferences about the activity of cortical pathways and investigating how connectivity parameters are influenced by experimental factors such as task condition or sample group.
Disordered brain connectivity in psychosis is thought to result from abnormal regulation of N‐methyl‐D‐aspartate (NMDA) receptor‐dependent synaptic plasticity [Stephan et al., 2006, 2009], for example, by a loss of cortical dopamine release [Slifstein et al., 2015]. Pyramidal cells are also directly influenced by inhibitory interneurons transmitting gamma‐amino butyric acid (GABA), which have also been strongly associated with the pathology of psychosis [Corlett et al., 2011; Gonzalez‐Burgos and Lewis, 2012]. Hence, abnormal NMDA receptor, dopaminergic or GABAergic interneuron function, would have profound effects on synaptic gain—that is, the excitability or responsiveness of neurons to their inputs—both directly through a failure of neuromodulation and indirectly through a failure of oscillatory coordination; and abnormal synaptic gain control has been proposed to underlie key phenomena in psychosis such as thalamocortical dysconnectivity, abnormal EEG responses, smooth pursuit deficits, loss of sensory attenuation, and psychotic symptoms themselves [Adams et al., 2013; Fletcher and Frith, 2009; Frith and Friston, 2013]. Crucially, synaptic gain is parameterized as the intrinsic—or self‐inhibitory—connectivity of superficial pyramidal cell populations in DCM [Bastos et al., 2012; Friston, 2008; Pinotsis et al., 2014; Stephan et al., 2006].
We recently reported DCM evidence of altered synaptic gain control in a frontal source in patients with psychotic disorder and their unaffected relatives during the sensory mismatch negativity potential [Ranlund et al., 2016]. Here, we used DCM to study, in the same sample, the effect of diagnosis and genetic liability to psychosis on P300‐related intrinsic connectivity—dependent on higher cognitive demands—in the frontoparietal network. We hypothesized that, just as in our mismatch negativity DCM analysis, synaptic gain control of superficial pyramidal cells differs between groups (patients, relatives, or controls) in both condition‐specific and condition‐general ways.
MATERIALS AND METHODS
Participants
The total sample comprised 24 patients with a psychotic illness, 24 of their first‐degree relatives without a personal history of psychosis, and 25 unrelated controls without personal or family history of psychosis (see Table 1 for demographic, diagnostic, and clinical details). Patients were significantly younger than relatives (t = −2.641, P = 0.011) but were matched in age to controls (t = −1.694, P = 0.097). As frequent in family studies of psychosis, the proportion of males was significantly higher in patients than controls (χ2 = 3.989, P = 0.046) and relatives (χ2 = 12.084, P = 0.002). There were no significant differences in age (t = −0.915; P = 0.365) or gender (χ2 = 2.643; P = 0.104) between relatives and controls. All participants were of European Caucasian ethnicity.
Table 1.
Demographic, clinical, and task‐related data
| Patients with psychotic disease (N = 24) |
Unaffected relatives (N = 24) |
Unaffected controls (N = 25) |
|
|---|---|---|---|
| Age (mean, SD) | 34.0 (9.4) | 43.2 (14.3) | 39.6 (13.3) |
| Age range (min–max) | 23–54 | 16–59 | 19–69 |
| Females (N, %) * | 5 (20.8%) | 17 (70.8%) | 13 (52.0%) |
| Education (mean years, SD) | 13.6 (2.8) | 13.4 (2.5) | 14.8 (4.0) |
| Diagnosis (N, %) | |||
| Schizophrenia | 19 (79.2%) | – | – |
| Schizoaffective disorder | 4 (16.6%) | – | – |
| Psychoses NOS | 1 (4.2%) | – | – |
| Mayor depression | – | 4 (16.6%) | 1 (4.0%) |
| No psychiatric illness | – | 20 (83.3%) | 24 (96.0%) |
| Illness duration (mean years, SD) | 11.8 (8.3) | – | – |
| Medication (N, %) | |||
| No medication | 2 (8.3%) | – | – |
| Clozapine | 4 (16.7%) | – | – |
| Flupentixol | 3 (12.5%) | – | – |
| Haloperidol | 1 (4.2%) | – | – |
| Olanzapine | 5 (20.8%) | – | – |
| Quetiapine | 3 (12.5%) | – | – |
| Risperidone | 5 (20.8%) | – | – |
| Sulpiride | 2 (8.3%) | – | – |
| Thioridazine | 2 (8.3%) | – | – |
| Trifluoperazine | 1 (4.2%) | – | – |
| Lithium or Sodium Valproate | 1 (4.2%) | – | – |
| Antiepileptic | 6 (25.0%) | – | – |
| Benzodiazepine | 4 (16.7%) | – | – |
| Antidepressant | 4 (16.7%) | 1 (4.2%) | – |
| CPZ equivalent (mean, min–max) | 564.2 (30–1100) | – | – |
| Years medicated (mean, SD) | 10.5 (8.6) | – | – |
| First medicated (mean years, SD) | 24.8 (7.2) | – | – |
| PANSS (mean, SD) | |||
| Positive ** | 12.3 (4.6) | 7.2 (0.6) | 7.0 (0.0) |
| Negative ** | 15.1 (5.4) | 7.2 (0.6) | 7.0 (0.0) |
| General ** | 23.8 (4.8) | 17.4 (2.3) | 16.1 (0.5) |
| Total ** | 51.2 (13.0) | 31.8 (2.8) | 30.1 (0.5) |
| Relationship to proband (N, %) | |||
| Mother | – | 8 (33.3%) | – |
| Father | – | 4 (17.7%) | – |
| Sister | – | 8 (33.3%) | – |
| Brother | – | 3 (12.5%) | – |
| Daughter | – | 1 (4.2%) | – |
| P300 correct targets (%, SD) | 98.2% (3.0) | 99.3% (1.0) | 99.3% (0.9) |
| P300 rejected epochs (mean, SD) | 36.8 (4.6) | 58 (7.3) | 45.2 (3.0) |
Differences between groups are presented in the first column.
SD, standard deviation; NOS, not otherwise specified; CPZ equivalent, average chlorpromazine equivalent dosage (mg).
Patients versus controls: * P < 0.05, ** P < 0.001; there were no significant difference between relatives and controls (T‐test for independent samples or χ2 test when corresponding).
Patients and relatives were recruited through National voluntary organizations, advertisements in the press and from referrals by clinicians. Controls were recruited by advertisements in the press and local job centers. Participants were excluded if they had a diagnosis of alcohol or substance dependence in the last 12 months, neurological disorders or a previous head injury with loss of consciousness. A personal history of nonpsychotic psychiatric illnesses did not constitute an exclusion criterion for relatives or controls, provided they were well and not taking any psychotropic medication at the time of testing and for the preceding 12 months. This was to avoid recruiting biased control groups unrepresentative of the general population [Bramon et al., 2005, 2008].
All participants were clinically interviewed in order to confirm or exclude a Diagnostic and Statistical Manual of Mental Disorders—Fourth Edition (DSM‐IV) [American Psychiatric Association, 1994] diagnosis. All patients were interviewed by an experienced clinician to confirm their diagnosis using the Schedule for Affective Disorders and Schizophrenia—Lifetime version [SADS‐L; Endicott and Spitzer, 1978] and psychopathology was assessed using the Positive and Negative Syndrome Scale (PANSS) [Kay et al., 1987]. All participants gave informed written consent to participate, and the study was approved by the Institute of Psychiatry (King's College London) Research Ethics Committee, conforming to the standards set by the Declaration of Helsinki.
EEG Methods
Data acquisition
Data were collected from seventeen scalp sites (Fp1, Fp2, F3, F4, F7, F8, C3, C4, P3, P4, T3, T4, T5, T6, Fz, Cz, and Pz) according to the 10/20 International System, using silver/silver‐chloride electrodes and a Nihon Kohden amplifier. Vertical and horizontal bipolar electrooculographs (EOGs) monitored eye movements, and the left ear lobe served as reference. Data were continuously digitized at 500 Hz with a 0.03–120 Hz band‐pass filter (24 dB/octave roll‐off). Impedances were kept below 5 kΩ.
We used an auditory two‐tone oddball task to elicit the P300 response. The stimuli were four hundred 80 dB tones (2 s inter‐stimulus interval and 5 ms rise/fall time), presented through bilateral earphones. About 80% of the tones were “standards” (1,000 Hz; 25 ms duration) and 20% were “targets” (1,500 Hz; 50 ms duration) presented in a random sequence. Subjects were instructed to press a button in response to targets only, and to maintain their eyes open looking at a fixation point. These methods have been described in previous articles [Bramon et al., 2005, 2008; Hall et al., 2006; Schulze et al., 2008].
Data pre‐processing
Using Fieldtrip [Oostenveld et al., 2011], EEG data were re‐referenced to the average of all EEG sensors, and further filtered with a 0.5–70 Hz band‐pass and a 50 Hz notch. We divided the continuous recording into 900 ms epochs starting 100 ms before stimulus onset. This pre‐stimulus interval was used for baseline correction.
Independent Component Analysis (ICA) was used to correct for ocular artifacts in the data. EEG activity was decomposed in 17 independent components, of which a maximum of 2 that clearly corresponded to eye blinks were removed from the data. Additional automatic artifact rejection was then conducted, removing any trials exceeding ±70 µV across all channels. A participant was included if 60 or more epochs per task condition remained. Overall, the mean rate of rejected segments per participant was 11.7% (Table 1). The resulting waveforms after artifact correction were averaged per task condition and grand‐averaged independently per group.
There were no significant group differences in behavioral accuracy (Table 1). We defined and calculated the P300 as the average amplitude at Pz for the oddball condition and the time window 300–600 ms (Fig. 1). Patients (t = 2.047, P = 0.047) but not relatives (t = 0.110, P = 0.913) showed a significant lower P300 component than controls.
Figure 1.
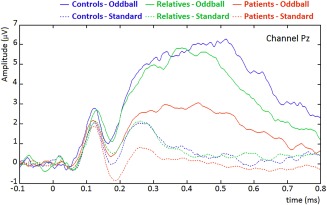
EEG signal to standard and oddball tones for each group (grand‐averages across subjects), at channel PZ. [Color figure can be viewed at http://wileyonlinelibrary.com]
Dynamic Causal Modeling
Spatial model selection
Bayesian inference is used in DCM to optimize neural source dipoles based on a priori information about their locations. In order to obtain a plausible a priori spatial model, we performed a literature review of functional magnetic resonance imaging (fMRI) studies of equivalent auditory oddball tasks (i.e., including a frequent standard and an infrequent target condition), and in which the coordinates of the main sources involved were reported. We focused on the auditory frontoparietal network and our final selection comprised bilateral primary auditory, superior parietal and superior frontal cortices (see Table 2). We did not include other regions with fMRI evidence (e.g., superior temporal, supramarginal or cingulate cortices) and we omitted ventral sources such as inferior parietal and frontal cortices in order to focus on frontoparietal connectivity and keep the model space as simple as possible. Some of the reviewed studies [Benar et al., 2007; Friston, 2012; Juckel et al., 2012; Mulert et al., 2004; Muller et al., 2003; Walz et al., 2013] crucially supported our anatomical model by using combined fMRI‐ERP source reconstruction.
Table 2.
Literature review of auditory P300 regions of interest in healthy adults
| Reviewed article | Neuroimaging technique | N | SF | SP |
|---|---|---|---|---|
| Walz et al., 2013 | Simultaneous fMRI/EEG | 17 | R | – |
| Juckel et al., 2012 | Simultaneous fMRI/EEG | 32 | L/R | – |
| Friedman et al., 2009 | Event‐related fMRI | 15 | L/R | – |
| Goldman et al., 2009 | Simultaneous fMRI/EEG | 11 | L/R | L/R |
| Benar et al., 2007 | Simultaneous fMRI/EEG | 12 | L/R | – |
| Liddle et al., 2006 | Event‐related fMRI | 28 | L/R | L/R |
| Stevens et al., 2006 | Event‐related fMRI | 20 | L/R | L/R |
| Kiehl et al., 2005 | Event‐related fMRI | 100 | L/R | L/R |
| Stevens et al., 2005 | Event‐related fMRI | 100 | L/R | L/R |
| Mulert et al., 2004 | Simultaneous fMRI/EEG | 9 | L/R | – |
| Horn et al., 2003 | Non‐simultaneous fMRI/iEEG | 15 | L/R | – |
| Muller et al., 2003 | Simultaneous fMRI/EEG | 16 | L/R | – |
| Horovitz et al., 2002 | Non‐simultaneous fMRI/EEG | 7 | R | – |
| Downar et al., 2001 | Event‐related fMRI | 5 | L | – |
| Kiehl et al., 2001 | Event‐related fMRI | 10 | L/R | L/R |
| Kiehl and Liddle, 2001 | Event‐related fMRI | 11 | R | L/R |
| Stevens et al., 2000 | Event‐related fMRI | 10 | R | – |
| Linden et al., 1999 | Non‐simultaneous fMRI/EEG | 5 | – | L/R |
| Yoshiura et al., 1999 | Event‐related fMRI | 13 | L/R | – |
Previous studies using an equivalent P300 auditory oddball task (i.e., including at least a frequent standard and an infrequent oddball condition), and any neuroimaging method to report the coordinates of selected regions (see references below).
SF, superior or middle frontal gyri; SP, superior parietal lobules; N, sample size; L, left hemisphere; R, right hemisphere; fMRI, functional magnetic resonance imaging; EEG, electroencephalography; iEEG, intracranial electroencephalography.
Before the DCM study, we used SPM12 [Litvak et al., 2011] to perform our own source reconstruction (multiple sparse priors’ algorithm [Friston et al., 2008]) during the first 600 ms for the standard and target conditions and including all participants. This confirmed the engagement of the selected sources by our paradigm (Fig. 2). Other DCM studies of the frontoparietal attention network during alternative auditory oddball tasks used more inferior parietal sources [Auksztulewicz and Friston, 2015; Dietz et al., 2014], but our source localization clearly indicated a more superior parietal source. Occipital and precentral areas, present in our source reconstruction, were not included in our network model, as we wished to focus on the frontoparietal network, rather than response execution in precentral (motor) areas. We, therefore, assumed occipital responses were of secondary importance in our auditory task, and that they were likely due to participants keeping their eyes open.
Figure 2.
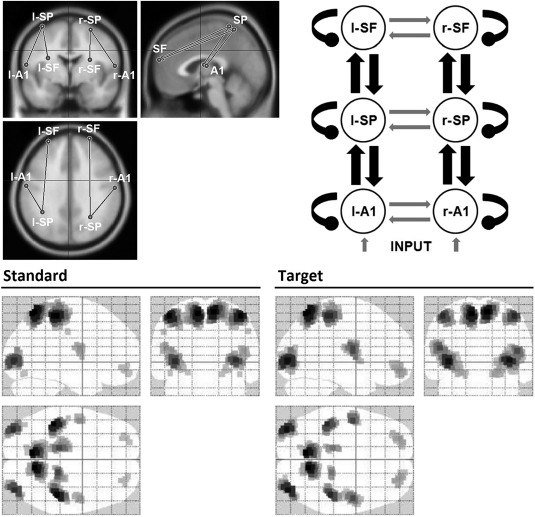
Selected dipoles composing the DCM spatial model. Top‐left: studied regions over a MRI head model template; left (−59, −10, 13) and right (61, −13, 11) primary auditory cortices (l‐/r‐A1), left (−37 −48 68) and right (28 −56 63) superior parietal lobules (l‐/r‐SP), and left (−29 55 22) and right (27 60 20) superior frontal gyri (l‐/r‐SF). Coordinates reported in the Montreal Neurological Institute (MNI) system. Top right: structural model presenting the studied extrinsic (black pointed arrows) and intrinsic (black oval arrows) connections. Bottom: source reconstruction of the evoked activity for standard and target conditions, 0–600 ms time window, including all participants.
Prior coordinates for the parietal and frontal sources were taken from Kiehl et al. [2001]. Bilateral primary auditory cortices were selected as the initial processing step and their coordinates taken from Yoshiura et al. [1999]. Talairach to Montreal Neurological Institute (MNI) coordinate transformation was carried out using BrainMap GingerALE 2.3 software [Eickhoff et al., 2009]. MNI coordinates are reported in Figure 2. Importantly, an accurate a priori activity localization is not essential as the DCM inversion algorithm will provide efficient Bayesian estimates of dipole source locations [Kiebel et al., 2009].
Bayesian model inversion
Condition‐specific grand‐averaged data were converted into Statistical Parametric Mapping (SPM) format separately for patients, relatives and controls. SPM12 was used to perform DCM at the group level [Fogelson et al., 2014; Ranlund et al., 2016] by creating cells of a 2 × 3 factorial design; with two levels of “task condition” (standard and oddball tones) and three levels of “group” (patients, relatives, and controls) [see Ranlund et al., 2016]. Studied group effects were: (1) “diagnosis” (patients vs. relatives and controls combined), and (2) “genetic risk” (relatives vs. controls). We tested for a main effect of diagnosis and genetic risk on intrinsic connectivity, and their interactions with the effect of task condition.
Sources of cortical activity were modeled as single equivalent current dipoles (ECD) under bilateral symmetry assumptions [Kiebel et al., 2006]. We used the Canonical Microcircuit neural mass model [Bastos et al., 2012; Pinotsis et al., 2013], in which each neural source comprises four cell populations: superficial and deep pyramidal cells, spiny stellate cells, and inhibitory interneurons. Within this model, extrinsic—that is, between‐sources—connections are excitatory: forward connections originate from superficial pyramidal cells and target spiny stellate cells, and backward extrinsic connections originate from deep pyramidal cells and target superficial pyramidal cells. All subpopulations have also intrinsic—that is, within‐source inhibitory—self‐connections, which essentially parameterize their synaptic gain or responsiveness to their own inputs [Bastos et al., 2012; Pinotsis et al., 2013].
A Boundary Elements model (BEM) [Fuchs et al., 2001] was used as an approximation to the brain, cerebrospinal fluid, and skull and scalp surfaces. A structural magnetic resonance imaging (MRI) head model was used for the co‐registration of electrode positions. The time window modeled was 0–600 ms post stimulus onset to ensure full‐length modeling of the P300 response.
Bayesian model selection
We used Bayesian Model Selection (BMS) [Penny et al., 2004] to identify which model, per studied effect and interaction, was a better explanation of the data. This method finds the model with the largest log‐evidence—a free energy approximation—among those tested, assuming equal prior probabilities for all models considered. It balances model accuracy and complexity, thereby selecting the most generalizable model. A difference in log‐evidence of three or more is considered strong evidence in favor of the more likely model in comparison to the second best model, which corresponds to an odds ratio of about 20:1 [Friston and Penny, 2011].
Dynamic causal modeling procedure
Importantly, before testing for diagnosis and genetic risk effects, we established the best model explaining the task condition effects across groups. Here we considered eight candidate models differing in forward, backward, and/or intrinsic connectivity effects of condition. The model allowing for forward connections only had the highest evidence (Fig. 3), and was used as task condition coupling in subsequent modeling steps.
Figure 3.
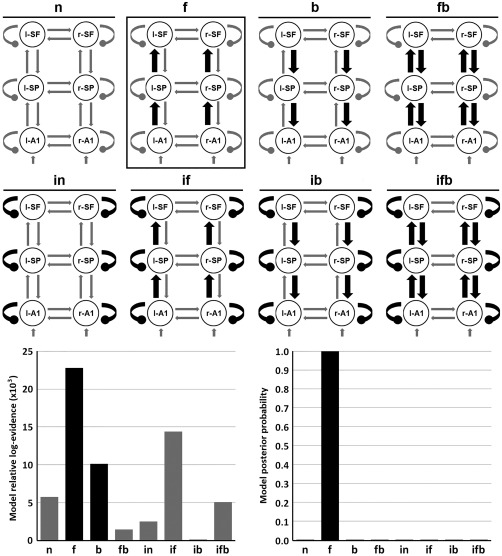
Preliminary DCM study for studying task condition. Top: eight studied models including bilateral intrinsic (black oval arrows) and/or forward extrinsic (black pointed arrows) modulation. These models included four combinations of extrinsic connectivity: null (n; no extrinsic), forward (f), backward (b) and forward‐backward (fb); and two intrinsic combinations: with and without intrinsic (i) modulation at all levels in the cortical hierarchy. Bottom: relative log‐evidences and posterior probabilities for each model. The winning model “f” included forward extrinsic modulation at the three hierarchy levels. l‐/r‐A1: left/right primary auditory cortices; l‐/r‐SP: left/right superior parietal lobules; l‐/r‐SF: left/right superior frontal gyri.
Secondly, by studying diagnosis and genetic risk effects we established where in the hierarchy intrinsic connectivity—that is, synaptic gain—was modulated by these between‐subject factors and their interaction with the within‐subject task condition factor. Additionally, forward extrinsic connectivity was also studied in this step due its involvement in the task condition effect. Our final model space comprised sixteen models (see Fig. 4).
Figure 4.

DCM study for studying diagnosis and genetic risk. Top: sixteen studied models including bilateral intrinsic (black oval arrows) and/or forward extrinsic (black pointed arrows) modulation. These models included eight bilateral combinations of intrinsic connectivity (i) and two extrinsic combinations: with and without forward (f) modulation. Bottom: relative log‐evidences and posterior probabilities for each model. The winning model “i8” included intrinsic modulation at the three hierarchy levels. l‐/r‐A1: left/right primary auditory cortices; l‐/r‐SP: left/right superior parietal lobules; l‐/r‐SF: left/right superior frontal gyri.
Finally, having established the model with the greatest evidence, we examined its posterior estimates of intrinsic connectivity to identify differences between patients, relatives and controls. We considered a connectivity difference of 20% or above to be a nontrivial effect size [Ranlund et al., 2016].
RESULTS
Bayesian Model Selection
DCM analysis showed that the best model of group effects was “i8,” which allowed intrinsic modulation bilaterally at all three hierarchical levels (primary auditory, superior parietal, and superior frontal cortices). Log‐evidences for all models relative to the worst performing are presented in Figure 4. We obtained a highly significant difference in log‐evidence between the winning model and the runner‐up, corresponding to almost 100% posterior probability. Posterior estimates and probabilities of changes in intrinsic connectivity for the winning model are shown in Figure 5 per source, due to group effects (diagnosis and genetic risk) and their interaction with task condition effect (standard vs. target). Figure 6 shows posterior estimates per group and task condition.
Figure 5.
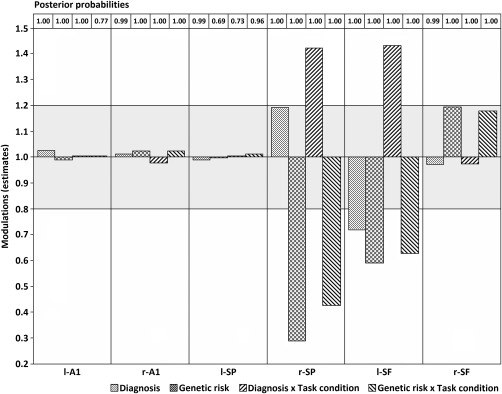
Posterior estimates of the intrinsic connections under the winning model for each source and experimental effect. Posterior probabilities are presented in the top for each posterior estimate bar. Bars lying outside the grey area show relevant changes of greater than 20%. l‐/r‐A1: left/right primary auditory cortices; l‐/r‐SP: left/right superior parietal lobules; l‐/r‐SF: left/right superior frontal gyri.
Figure 6.
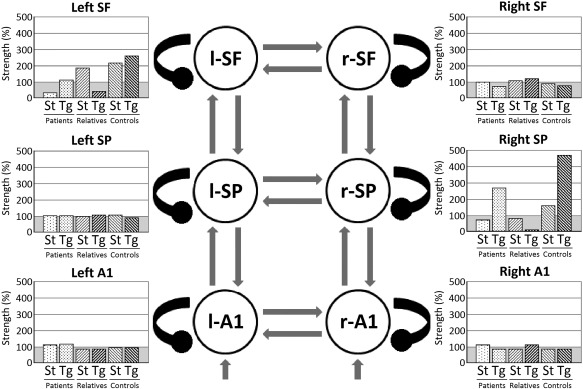
Intrinsic connectivity strengths under the winning model per source, group and task condition. l‐/r‐A1: left/right primary auditory cortices; l‐/r‐SP: left/right superior parietal lobules; l‐/r‐SF: left/right superior frontal gyri; St: standard; Tg: target.
Diagnosis and Genetic Effects
The largest effects are observed at parietal and frontal levels of the hierarchy (Figs. 5 and 6). Firstly, patients show reduced intrinsic—or self‐inhibitory—connectivity (i.e., greater excitability) in left superior frontal gyrus across task conditions compared with relatives and controls combined (a diagnosis effect). Secondly, unaffected relatives show a reduction in intrinsic connectivity across task conditions in left superior frontal and right superior parietal sources compared with controls (a genetic risk effect).
Interactions Between Clinical Group and Task Condition
There is an interaction between diagnosis and task condition in both left superior frontal and right superior parietal sources (Fig. 5). This corresponds to patients having an increased intrinsic or self‐inhibition (i.e., decreased excitability) in response to targets compared with standard tones (Fig. 6), whereas relatives and controls combined (unaffected participants) exhibit the opposite pattern. Finally, there is also an interaction between genetic risk and task condition at the same sources. In this case (Fig. 6), relatives show a decreased change in intrinsic excitability in response to targets compared with standard tones, whereas controls show the opposite response pattern.
DISCUSSION
We investigated whether patients with psychotic disorder (diagnosis effect) and/or their unaffected relatives (genetic risk effect) show alterations in intrinsic—or self‐inhibitory—connectivity during the evocation of the P300 potential.
Patients showed reduced P300‐related intrinsic connectivity within left superior frontal cortex across task conditions, which suggests a context‐independent dysfunction of synaptic gain control at the highest hierarchical level. The loss of recurrent inhibition in superficial pyramidal cells corresponds to local hyperexcitability. We recently used a similar approach to study intrinsic connectivity at three hierarchical levels during a mismatch negativity experiment [Ranlund et al., 2016] and found lower right inferior frontal self‐inhibition in psychosis. Our previous and current results give further support to the hypothesis of a context‐independent frontal hyperexcitability in psychosis at higher cortical levels, present both during a sensory mismatch negativity and the more cognitively demanding P300 experiments. Other DCM studies are consistent with our findings too. For example, a recent fMRI‐DCM study of the default mode network in first‐episode schizophrenia [Bastos‐Leite et al., 2015] demonstrated weaker frontal self‐inhibition, concluding that there is greater prefrontal excitability even during the resting state.
These findings, including ours, are consistent with the hypothesis of impaired modulation of synaptic efficacy in psychosis. Neurobiological research supports a hypofunction of NMDA receptors in psychosis [Corlett et al., 2011; Stephan et al., 2006, 2009], alongside reductions in cortical dopaminergic function [Slifstein et al., 2015] and in parvalbumin‐positive GABAergic interneuron‐mediated inhibition of pyramidal cells, especially in prefrontal cortex [Gonzalez‐Burgos and Lewis, 2012; Lewis and Gonzalez‐Burgos, 2006]. This pathophysiology could result in a loss of prefrontal excitation/inhibition balance—for example, during working memory [Murray et al., 2014]—and hence hyperexcitability. This is also in line with our previous work in a different sample of patients with psychotic disorder [Díez et al., 2013, 2014], in whom we found an abnormal P300‐related increase of frontal gamma activity, a frequency range related to fast GABAergic firing during cognitive processing [Lewis et al., 2012]. Although the relation between structural and functional connectivity is still inconclusive [Stam et al., 2016], these findings may also be related to the frontoparietal white matter abnormalities reported in schizophrenia, if these alterations affect synaptic gain control within cortical areas.
The second key finding in this article is that unaffected relatives of patients also show decreased intrinsic connectivity across conditions within the left superior frontal and right superior parietal cortices. Thus people with genetic vulnerability show similar prefrontal synaptic gain abnormalities to those seen in psychosis. Likewise, during a basic mismatch negativity pre‐attentional auditory discrimination experiment, these same unaffected relatives also showed decreased intrinsic connectivity in the right inferior frontal gyrus [Ranlund et al., 2016]. These findings are important for two reasons: first, they indicate that the similar frontal hyperexcitability in subjects with psychotic disorder is unlikely to be a medication effect; and second, impaired prefrontal synaptic gain control might reflect a core neurobiological marker of increased vulnerability for psychosis. The use of endophenotypes [Gottesman and Gould, 2003] might help to understand the pathophysiological mechanisms underlying illness onset and the functional effects of identified genetic risk loci [Bramon et al., 2014; Hall and Smoller, 2010]. For instance, Dima et al. [2013] found that CACNA1C and ANK3 genetic variants, which modulate GABAergic interneuron function, are associated with frontolimbic effective connectivity alterations in bipolar disorder.
On the other hand, DCM studies of fMRI data in people with “at‐risk mental states” predisposing to psychosis revealed backward connectivity attenuation from frontal sources during working memory [Crossley et al., 2009] and verbal fluency tasks [Dauvermann et al., 2013]. Compared with controls, there were connectivity deficits in the frontoparietal network in the at‐risk mental state group, with greater severity in unmedicated first episode schizophrenia cases. Interestingly, this abnormal modulation of connectivity normalized after antipsychotic treatment [Schmidt et al., 2013, 2014]. Thus, different alterations of brain connectivity may be better “state” (prefrontal hyperexcitability) or “trait” (backward connectivity attenuation) markers of psychotic illness.
Our third and last key finding is that relatives and controls show an opposite pattern of responses to standard and target stimuli (i.e., a genetic risk by task condition interaction) at both left superior frontal and right superior parietal sources. While controls respond to targets with an increase in self‐inhibition in these sources, the relatives show a decrease of self‐inhibition in response to task‐relevant stimuli. Unexpectedly, when analyzing the interaction between diagnosis and task condition effects; we found, as seen in Figure 6, that the standard/target response pattern seems the same in patients and controls. We did not predict this pattern and any interpretation of it must be speculative. Interestingly, Schmidt et al. [2013] demonstrated using fMRI‐DCM that abnormal reduction in working memory‐induced frontoparietal modulation in first episode patients was normalized by treatment with antipsychotics. Thus it may be that the abnormal context‐dependent aspect of synaptic gain control seen in relatives is normalized by antipsychotic medication in patients. We did not see such a normalization in our mismatch negativity study, however, in which both patients and relatives showed context‐dependent abnormalities [Ranlund et al., 2016].
Our results might also potentially be explained by confounding variables. Firstly, effects of antipsychotic medication have been demonstrated to modulate prefrontal brain activity during cognitive tasks [Artigas, 2010]. However, as discussed above, effective connectivity seems to become normalized in patients after initial pharmacological treatment [Schmidt et al., 2013]. Secondly, as is typical in family studies of psychosis, the relatives were older and included more females than the patient group. This should be taken into consideration when interpreting our results, as there is evidence of working memory network differences between genders [Hill et al., 2014] and ages [Steffener et al., 2009] that can affect effective connectivity. On the other hand, patients and controls were matched and the group differences can be more reliably interpreted.
How do these results relate to predictive coding accounts of psychosis? Predictive coding considers the brain as a hierarchical Bayesian inference engine that optimizes top‐down predictions based on prior beliefs of the causes of sensory data by minimizing bottom‐up—that is, sensory‐driven—prediction errors throughout the cortical hierarchy [Bastos et al., 2012; Friston, 2008]. In this scheme, ascending prediction errors are encoded by superficial pyramidal cells, which send projections up the cortical hierarchy; and, importantly, are weighted in proportion to their expected precision, which is an inverse variance. This weighting is thought to be implemented by the synaptic gain or excitability of superficial pyramidal cells, such that the prediction errors in which there is greatest confidence—or highest precision—are broadcast with greater “volume” [Adams et al., 2013; Bastos et al., 2012; Friston, 2008]. The optimization of precision—that is, the boosting of channels that encode reliable information—corresponds to attentional gain. In this P300 paradigm, this would enable the amplification of prediction errors that are considered to convey precise information—that is, targets—in a given context [Feldman and Friston, 2010]. Importantly, Auksztulewicz and Friston [2015] showed in a similar cortical network that attention has exactly this enhancing effect on synaptic gain in A1 [Auksztulewicz and Friston, 2015]. Note that due to the non‐linear interactions among neuronal subpopulations in DCM, changes in the gain of superficial pyramidal cells can have a non‐intuitive effect on the P300 waveform. In this case, increased excitability of pyramidal cells in frontoparietal areas results in a lower—not higher—amplitude waveform in patients. An intuitive explanation for this effect rests upon the fact that neuronal transients have faster time courses when synaptic efficacy is higher; thereby attenuating later, slow endogenous components such as the P300. This follows from the fact that synaptic efficacy or excitability plays the role of a rate constant from a dynamical perspective.
There is considerable evidence that psychosis involves abnormalities of synaptic plasticity: NMDA receptors and GABAergic interneurons crucial for sustaining oscillations; and hence message‐passing, and dopamine release in striatum and cortex, are all implicated in the disorder [Adams et al., 2013]. A loss of cortical gain control would lead to aberrant precision‐weighting of prediction errors (e.g., just as overestimating the precision of the data inflates the t‐statistic), abnormalities of selective attention and a predisposition to false perceptual and conceptual inference (e.g., hallucinations and delusions). Problems with predictive coding and selective attention would result in context (i.e., prediction)‐dependent effects in paradigms that exploit these processes, such as the mismatch negativity—in which prediction but not attention is important—and the P300 paradigm used here.
DCM analysis of electrophysiological data allows one to estimate the connectivity differences between patients and controls that contribute to these context‐dependent and invariant effects. Given synaptic gain is abnormal in psychosis, one would expect to see consistent differences in intrinsic connectivity between patients and controls, and this is indeed the case: for example, Dima et al. [2012] demonstrated reduced intrinsic connectivity in right auditory cortex in patients in response to oddballs during a mismatch negativity paradigm, as did Ranlund et al. [2016]. Crucially, Ranlund et al. [2016] also demonstrated both context‐dependent and invariant effects on intrinsic connectivity in a right prefrontal source in both patients and their relatives. Likewise, Fogelson et al. [2014] reported a striking loss of intrinsic connectivity modulation by stimulus predictability in occipital, temporal and parietal sources in patients during visual oddball detection. According to the authors, while controls were able to modulate ascending prediction errors, patients failed to exploit predictability in a context‐dependent fashion, processing both predictable and unpredictable stimuli in the same way [Fogelson et al., 2014]. Our findings are thus commensurate with this growing literature demonstrating alterations in cortical synaptic gain in both patients and, importantly, their relatives.
CONCLUSION
In summary, our DCM study of the P300 effect found that patients with psychotic disorder have an abnormal decrease in frontal intrinsic inhibitory connections—resulting in increased cortical excitability—across target and standard conditions. This result was also seen in unaffected relatives at frontal and parietal sources. Additionally, relatives show a loss of the normally increased self‐inhibition in frontal and parietal areas during target trials. Our results suggest that there is decreased inhibitory synaptic gain control and hyperexcitability of superficial pyramidal cells in sufferers of psychosis and those at genetic risk. This is consistent with recent neurobiological findings pointing to NMDA receptor hypofunction compromising GABAergic inhibition in psychosis. Abnormalities in relatives suggest that synaptic gain disruption might be a potential endophenotype for psychosis. Our findings support the “dysconnection hypothesis,” which proposes that an impairment of functional integration—dependent upon synaptic efficacy and gain control—underlies both the cognitive deficits and clinical symptoms characterizing psychosis [Friston, 2002].
ACKNOWLEDGMENTS
We would like to thank all the patients, relatives and controls who took part in this research, as well as the clinical staff who facilitated their involvement. Further support: Elvira Bramon's MRC New Investigator Award and a MRC Centenary Award; National Institute of Health Research UK (post‐doctoral fellowship), the Psychiatry Research Trust, the Schizophrenia Research Fund, the Brain and Behavior Research foundation's (NARSAD's) Young Investigator Award, a Wellcome Trust Research Training Fellowship and the NIHR Biomedical Research Centre for Mental Health at the South London and Maudsley NHS Foundation Trust and Institute of Psychiatry Kings College London. Álvaro Díez was supported by a European Commission's Marie Curie Fellowship (grant 330156/CODIP) and a Spanish Ministry of Economy and Competitiveness’ Juan de la Cierva Fellowship (FJCI‐2014‐19800). All authors declare that they have no conflicts of interest.
Rick A. Adams and Elvira Bramon jointly supervised this work.
REFERENCES
- Adams RA, Stephan KE, Brown HR, Frith CD, Friston KJ (2013): The computational anatomy of psychosis. Front Psychiatry 4:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association (1994): Diagnostic and statistical manual of mental disorders : DSM‐IV, 4th ed Washington DC: American Psychiatric Association. [Google Scholar]
- Artigas F (2010): The prefrontal cortex: A target for antipsychotic drugs. Acta Psychiatr Scand 121:11–21. [DOI] [PubMed] [Google Scholar]
- Auksztulewicz R, Friston K (2015): Attentional enhancement of auditory mismatch responses: A DCM/MEG study. Cereb Cortex 25:4273–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos‐Leite AJ, Ridgway GR, Silveira C, Norton A, Reis S, Friston KJ (2015): Dysconnectivity within the default mode in first‐episode schizophrenia: A stochastic dynamic causal modeling study with functional magnetic resonance imaging. Schizophr Bull 41:144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos AM, Usrey WM, Adams RA, Mangun GR, Fries P, Friston KJ (2012): Canonical microcircuits for predictive coding. Neuron 76:695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benar CG, Schon D, Grimault S, Nazarian B, Burle B, Roth M, Badier JM, Marquis P, Liegeois‐Chauvel C, Anton JL (2007): Single‐trial analysis of oddball event‐related potentials in simultaneous EEG‐fMRI. Hum Brain Mapp 28:602–613. [DOI] [PubMed] [Google Scholar]
- Bledowski C, Cohen Kadosh K, Wibral M, Rahm B, Bittner RA, Hoechstetter K, Scherg M, Maurer K, Goebel R, Linden DE (2006): Mental chronometry of working memory retrieval: A combined functional magnetic resonance imaging and event‐related potentials approach. J Neurosci 26:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramon E, McDonald C, Croft RJ, Landau S, Filbey F, Gruzelier JH, Sham PC, Frangou S, Murray RM (2005): Is the P300 wave an endophenotype for schizophrenia? A meta‐analysis and a family study. Neuroimage 27:960–968. [DOI] [PubMed] [Google Scholar]
- Bramon E, Dempster E, Frangou S, Shaikh M, Walshe M, Filbey FM, McDonald C, Sham P, Collier DA, Murray R (2008): Neuregulin‐1 and the P300 waveform–a preliminary association study using a psychosis endophenotype. Schizophr Res 103:178–185. [DOI] [PubMed] [Google Scholar]
- Bramon E, Pirinen M, Strange A, Lin K, Freeman C, Bellenguez C, Su Z, Band G, Pearson R, Vukcevic D, Langford C, Deloukas P, Hunt S, Gray E, Dronov S, Potter SC, Tashakkori‐Ghanbaria A, Edkins S, Bumpstead SJ, Arranz MJ, Bakker S, Bender S, Bruggeman R, Cahn W, Chandler D, Collier DA, Crespo‐Facorro B, Dazzan P, de Haan L, Di Forti M, Dragovic M, Giegling I, Hall J, Iyegbe C, Jablensky A, Kahn RS, Kalaydjieva L, Kravariti E, Lawrie S, Linszen DH, Mata I, McDonald C, McIntosh A, Myin‐Germeys I, Ophoff RA, Pariante CM, Paunio T, Picchioni M, Ripke S, Rujescu D, Sauer H, Shaikh M, Sussmann J, Suvisaari J, Tosato S, Toulopoulou T, Van Os J, Walshe M, Weisbrod M, halley H, Wiersma D, Blackwell JM, Brown MA, Casas JP, Corvin A, Duncanson A, Jankowski JA, Markus HS, Mathew CG, Palmer CN, Plomin R, Rautanen A, Sawcer SJ, Trembath RC, Wood NW, Barroso I, Peltonen L, Lewis CM, Murray RM, Donnelly P, Powell J, Spencer CC (2014): A genome‐wide association analysis of a broad psychosis phenotype identifies three loci for further investigation. Biol Psychiatry 75:386–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla D, Lumer ED, Friston KJ (1999): The relationship between synchronization among neuronal populations and their mean activity levels. Neural Comput 11:1389–1411. [DOI] [PubMed] [Google Scholar]
- Corlett PR, Honey GD, Krystal JH, Fletcher PC (2011): Glutamatergic model psychoses: Prediction error, learning, and inference. Neuropsychopharmacology 36:294–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley NA, Mechelli A, Fusar‐Poli P, Broome MR, Matthiasson P, Johns LC, Bramon E, Valmaggia L, Williams S, McGuire PK (2009): Superior temporal lobe dysfunction and frontotemporal dysconnectivity in subjects at risk of psychosis and in first‐episode psychosis. Hum Brain Mapp 30:4129–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauvermann MR, Whalley HC, Romaniuk L, Valton V, Owens DG, Johnstone EC, Lawrie SM, Moorhead TW (2013): The application of nonlinear Dynamic Causal Modeling for fMRI in subjects at high genetic risk of schizophrenia. Neuroimage 73:16–29. [DOI] [PubMed] [Google Scholar]
- David O, Friston KJ (2003): A neural mass model for MEG/EEG: Coupling and neuronal dynamics. NeuroImage 20:1743–1755. [DOI] [PubMed] [Google Scholar]
- David O, Harrison L, Friston KJ (2005): Modeling event‐related responses in the brain. Neuroimage 25:756–770. [DOI] [PubMed] [Google Scholar]
- David O, Kiebel SJ, Harrison LM, Mattout J, Kilner JM, Friston KJ (2006): Dynamic causal modeling of evoked responses in EEG and MEG. Neuroimage 30:1255–1272. [DOI] [PubMed] [Google Scholar]
- Deserno L, Sterzer P, Wustenberg T, Heinz A, Schlagenhauf F (2012): Reduced prefrontal‐parietal effective connectivity and working memory deficits in schizophrenia. J Neurosci 32:12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz MJ, Friston KJ, Mattingley JB, Roepstorff A, Garrido MI (2014): Effective connectivity reveals right hemisphere dominance in audiospatial perception: Implications for models of spatial neglect. J Neurosci 34:5003–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díez Á, Suazo V, Casado P, Martin‐Loeches M, Molina V (2013): Spatial distribution and cognitive correlates of gamma noise power in schizophrenia. Psychol Med 43:1175–1185. [DOI] [PubMed] [Google Scholar]
- Díez Á, Suazo V, Casado P, Martin‐Loeches M, Perea MV, Molina V (2014): Frontal gamma noise power and cognitive domains in schizophrenia. Psychiatry Res 221:104–113. [DOI] [PubMed] [Google Scholar]
- Dima D, Frangou S, Burge L, Braeutigam S, James AC (2012): Abnormal intrinsic and extrinsic connectivity within the magnetic mismatch negativity brain network in schizophrenia: A preliminary study. Schizophr Res 135:23–27. [DOI] [PubMed] [Google Scholar]
- Dima D, Jogia J, Collier D, Vassos E, Burdick KE, Frangou S (2013): Independent modulation of engagement and connectivity of the facial network during affect processing by CACNA1C and ANK3 risk genes for bipolar disorder. JAMA Psychiatry 70:1303–1311. [DOI] [PubMed] [Google Scholar]
- Downar J, Crawley AP, Mikulis DJ, Davis KD (2001): The effect of task relevance on the cortical response to changes in visual and auditory stimuli: An event‐related fMRI study. Neuroimage 14:1256–1267. [DOI] [PubMed] [Google Scholar]
- Eickhoff SB, Laird AR, Grefkes C, Wang LE, Zilles K, Fox PT (2009): Coordinate‐based activation likelihood estimation meta‐analysis of neuroimaging data: A random‐effects approach based on empirical estimates of spatial uncertainty. Hum Brain Mapp 30:2907–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott J, Spitzer RL (1978): A diagnostic interview. The schedule for affective disorders and schizophrenia. Arch Gen Psychiatry 35:837–844. [DOI] [PubMed] [Google Scholar]
- Feldman H, Friston KJ (2010): Attention, uncertainty, and free‐energy. Front Hum Neurosci 4:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher PC, Frith CD (2009): Perceiving is believing: A Bayesian approach to explaining the positive symptoms of schizophrenia. Nat Rev Neurosci 10:48–58. [DOI] [PubMed] [Google Scholar]
- Fogelson N, Litvak V, Peled A, Fernandez‐Del‐Olmo M, Friston K (2014): The functional anatomy of schizophrenia: A dynamic causal modeling study of predictive coding. Schizophr Res 158:204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford JM (1999): Schizophrenia: The broken P300 and beyond. Psychophysiology 36:667–682. [PubMed] [Google Scholar]
- Friedman D, Goldman R, Stern Y, Brown TR (2009): The brain's orienting response: An event‐related functional magnetic resonance imaging investigation. Hum Brain Mapp 30:1144–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friston KJ (1998): The disconnection hypothesis. Schizophr Res 30:115–125. [DOI] [PubMed] [Google Scholar]
- Friston KJ (2002): Dysfunctional connectivity in schizophrenia. World Psychiatry 1:66–71. [PMC free article] [PubMed] [Google Scholar]
- Friston K (2008): Hierarchical models in the brain. PLoS Comput Biol 4:e1000211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friston K (2012): Self‐organisation, inference and cognition. Comment on “Consciousness, crosstalk, and the mereological fallacy: An evolutionary perspective” by Rodrick Wallace. Phys Life Rev 9:456–457. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Harrison L, Daunizeau J, Kiebel SJ, Phillips C, Trujillo‐Bareto N, Henson RNA, Flandin G, Mattout J (2008): Multiple sparse priors for the m/eeg inverse problem. NeuroImage 39:1104–1120. [DOI] [PubMed] [Google Scholar]
- Friston K, Penny W (2011): Post hoc Bayesian model selection. Neuroimage 56:2089–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friston KJ, Harrison L, Penny W (2003): Dynamic causal modeling. Neuroimage 19:1273–1302. [DOI] [PubMed] [Google Scholar]
- Frith CD, Friston KJ (2013): False perceptions & false beliefs: Understanding schizophrenia. Neurosciences and the Human Person: New Perspectives on Human Activities Pontifical Academy of Sciences, Scripta Varia 121.
- Fuchs M, Wagner M, Kastner J (2001): Boundary element method volume conductor models for EEG source reconstruction. Clin Neurophysiol 112:1400–1407. [DOI] [PubMed] [Google Scholar]
- Goldman RI, Wei CY, Philiastides MG, Gerson AD, Friedman D, Brown TR, Sajda P (2009): Single‐trial discrimination for integrating simultaneous EEG and fMRI: Identifying cortical areas contributing to trial‐to‐trial variability in the auditory oddball task. Neuroimage 47:136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Burgos G, Lewis DA (2012): NMDA receptor hypofunction, parvalbumin‐positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull 38:950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman I, Gould T (2003): The endophenotype concept in psychiatry: Etymology and strategic intentions. Am J Psychiatry 160:636–645. [DOI] [PubMed] [Google Scholar]
- Hall M, Smoller J (2010): A new role for endophenotypes in the GWAS era: Functional characterization of risk variants. Harv Rev Psychiatry 18:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MH, Schulze K, Rijsdijk F, Picchioni M, Ettinger U, Bramon E, Freedman R, Murray RM, Sham P (2006): Heritability and reliability of P300, P50 and duration mismatch negativity. Behav Genet 36:845–857. [DOI] [PubMed] [Google Scholar]
- Hall MH, Schulze K, Rijsdijk F, Kalidindi S, McDonald C, Bramon E, Murray RM, Sham P (2009): Are auditory P300 and duration MMN heritable and putative endophenotypes of psychotic bipolar disorder? A Maudsley Bipolar Twin and Family Study. Psychol Med 39:1277–1287. [DOI] [PubMed] [Google Scholar]
- Hill AC, Laird AR, Robinson JL (2014): Gender differences in working memory networks: A BrainMap meta‐analysis. Biol Psychiatry 102:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn H, Syed N, Lanfermann H, Maurer K, Dierks T (2003): Cerebral networks linked to the event‐related potential P300. Eur Arch Psychiatry Clin Neurosci 253:154–159. [DOI] [PubMed] [Google Scholar]
- Horovitz SG, Skudlarski P, Gore JC (2002): Correlations and dissociations between BOLD signal and P300 amplitude in an auditory oddball task: A parametric approach to combining fMRI and ERP. Magn Reson Imaging 20:319–325. [DOI] [PubMed] [Google Scholar]
- Jeon YW, Polich J (2003): Meta‐analysis of P300 and schizophrenia: Patients, paradigms, and practical implications. Psychophysiology 40:684–701. [DOI] [PubMed] [Google Scholar]
- Juckel G, Karch S, Kawohl W, Kirsch V, Jager L, Leicht G, Lutz J, Stammel A, Pogarell O, Ertl M, Reiser M, Hegerl U, Moller HJ, Mulert C (2012): Age effects on the P300 potential and the corresponding fMRI BOLD‐signal. Neuroimage 60:2027–2034. [DOI] [PubMed] [Google Scholar]
- Kay SR, Fiszbein A, Opler LA (1987): The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull 13:261–276. [DOI] [PubMed] [Google Scholar]
- Kiebel SJ, David O, Friston KJ (2006): Dynamic causal modelling of evoked responses in EEG/MEG with lead field parameterization. Neuroimage 30:1273–1284. [DOI] [PubMed] [Google Scholar]
- Kiebel SJ, Garrido MI, Moran R, Chen CC, Friston KJ (2009): Dynamic causal modeling for EEG and MEG. Hum Brain Mapp 30:1866–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehl KA, Laurens KR, Duty TL, Forster BB, Liddle PF (2001): Neural sources involved in auditory target detection and novelty processing: An event‐related fMRI study. Psychophysiology 38:133–142. [PubMed] [Google Scholar]
- Kiehl KA, Liddle PF (2001): An event‐related functional magnetic resonance imaging study of an auditory oddball task in schizophrenia. Schizophr Res 48:159–171. [DOI] [PubMed] [Google Scholar]
- Kiehl KA, Stevens MC, Laurens KR, Pearlson G, Calhoun VD, Liddle PF (2005): An adaptive reflexive processing model of neurocognitive function: Supporting evidence from a large scale (n = 100) fMRI study of an auditory oddball task. Neuroimage 25:899–915. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Kwon JS, Park HJ, Youn T, Kang DH, Kim MS, Lee DS, Lee MC (2003): Functional disconnection between the prefrontal and parietal cortices during working memory processing in schizophrenia: A[15(O)]H2O PET study. Am J Psychiatry 160:919–923. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Gonzalez‐Burgos G (2006): Pathophysiologically based treatment interventions in schizophrenia. Nat Med 12:1016–1022. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW (2012): Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci 35:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddle PF, Laurens KR, Kiehl KA, Ngan ET (2006): Abnormal function of the brain system supporting motivated attention in medicated patients with schizophrenia: An fMRI study. Psychol Med 36:1097–1108. [DOI] [PubMed] [Google Scholar]
- Linden DE, Prvulovic D, Formisano E, Vollinger M, Zanella FE, Goebel R, Dierks T (1999): The functional neuroanatomy of target detection: An fMRI study of visual and auditory oddball tasks. Cereb Cortex 9:815–823. [DOI] [PubMed] [Google Scholar]
- Litvak V, Mattout J, Kiebel S, Phillips C, Henson R, Kilner J, Barnes G, Oostenveld R, Daunizeau J, Flandin G, Penny W, Friston K (2011): EEG and MEG data analysis in SPM8. Comput Intell Neurosci 2011:852961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Steinberg JL, Hasan KM, Narayana PA, Kramer LA, Moeller FG (2012): Working memory load modulation of parieto‐frontal connections: Evidence from dynamic causal modeling. Hum Brain Mapp 33:1850–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran R, Pinotsis DA, Friston K (2013): Neural masses and fields in dynamic causal modeling. Front Comput Neurosci 7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulert C, Jager L, Schmitt R, Bussfeld P, Pogarell O, Moller HJ, Juckel G, Hegerl U (2004): Integration of fMRI and simultaneous EEG: Towards a comprehensive understanding of localization and time‐course of brain activity in target detection. Neuroimage 22:83–94. [DOI] [PubMed] [Google Scholar]
- Muller BW, Stude P, Nebel K, Wiese H, Ladd ME, Forsting M, Jueptner M (2003): Sparse imaging of the auditory oddball task with functional MRI. Neuroreport 14:1597–1601. [DOI] [PubMed] [Google Scholar]
- Murray JD, Anticevic A, Gancsos M, Ichinose M, Corlett PR, Krystal JH, Wang XJ (2014): Linking microcircuit dysfunction to cognitive impairment: Effects of disinhibition associated with schizophrenia in a cortical working memory model. Cereb Cortex 24:859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostenveld R, Fries P, Maris E, Schoffelen JM (2011): FieldTrip: Open source software for advanced analysis of MEG, EEG, and invasive electrophysiological data. Comput Intell Neurosci 2011:156869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny WD, Stephan KE, Mechelli A, Friston KJ (2004): Comparing dynamic causal models. Neuroimage 22:1157–1172. [DOI] [PubMed] [Google Scholar]
- Pinotsis DA, Brunet N, Bastos A, Bosman CA, Litvak V, Fries P, Friston KJ (2014): Contrast gain control and horizontal interactions in V1: A DCM study. NeuroImage 92:143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinotsis DA, Moran RJ, Friston KJ (2012): Dynamic causal modeling with neural fields. Neuroimage 59:1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinotsis DA, Schwarzkopf DS, Litvak V, Rees G, Barnes G, Friston KJ (2013): Dynamic causal modelling of lateral interactions in the visual cortex. NeuroImage 66:563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polich J (2007): Updating P300: an integrative theory of P3a and P3b. Clin Neurophysiol 118:2128–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polich J, Criado JR (2006): Neuropsychology and neuropharmacology of P3a and P3b. Int J Psychophysiol 60:172–185. [DOI] [PubMed] [Google Scholar]
- Ranlund S, Adams RA, Díez Á, Constante M, Dutt A, Hall MH, Maestro Carbayo A, McDonald C, Petrella S, Schulze K, Shaikh M, Walshe M, Friston K, Pinotsis D, Bramon E (2016): Impaired prefrontal synaptic gain in people with psychosis and their relatives during the mismatch negativity. Hum Brain Mapp 37:351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roiser JP, Wigton R, Kilner JM, Mendez MA, Hon N, Friston KJ, Joyce EM (2013): Dysconnectivity in the frontoparietal attention network in schizophrenia. Front Psychiatry 4:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Smieskova R, Aston J, Simon A, Allen P, Fusar‐Poli P, Riecher‐Rössler A, Stephan KE, Borgwardt S (2013): Brain connectivity abnormalities predating the onset of psychosis: Correlation with the effect of medication. JAMA Psychiatry 70:903–912. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Smieskova R, Simon A, Allen P, Fusar‐Poli P, McGuire PK, Bendfeldt K, Aston J, Lang UE, Walter M, Radue EW, Riecher‐Rossler A, Borgwardt SJ (2014): Abnormal effective connectivity and psychopathological symptoms in the psychosis high‐risk state. J Psychiatry Neurosci 39:239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze KK, Hall MH, Mcdonald C, Marshall N, Walshe M, Murray RM, Bramon E (2008): Auditory P300 in patients with bipolar disorder and their unaffected relatives. Bipolar Disorders 10:377–386. [DOI] [PubMed] [Google Scholar]
- Slifstein M, van de Giessen E, Van Snellenberg J, Thompson JL, Narendran R, Gil R, Hackett E, Girgis R, Ojeil N, Moore H, D'Souza D, Malison RT, Huang Y, Lim K, Nabulsi N, Carson RE, Lieberman JA, Abi‐Dargham A (2015): Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: A positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry 72:316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam CJ, van Straaten ECW, Van Dellen E, Tewarie P, Gong G, Hillebrand A, Mieier J, Van Mieghem P (2016): The relation between structural and functional connectivity patterns in complex brain networks. Int J Psychophysiol 103:149–160. [DOI] [PubMed] [Google Scholar]
- Steffener J, Brickman AM, Rakitin BC, Gazes Y, Stern Y (2009): The impact of age‐related changes on working memory functional activity. Brain Imaging Behav 3:142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan KE, Baldeweg T, Friston KJ (2006): Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry 59:929–939. [DOI] [PubMed] [Google Scholar]
- Stephan KE, Friston KJ, Frith CD (2009): Dysconnection in schizophrenia: From abnormal synaptic plasticity to failures of self‐monitoring. Schizophr Bull 35:509–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens AA, Skudlarski P, Gatenby JC, Gore JC (2000): Event‐related fMRI of auditory and visual oddball tasks. Magn Reson Imaging 18:495–502. [DOI] [PubMed] [Google Scholar]
- Stevens MC, Calhoun VD, Kiehl KA (2005): fMRI in an oddball task: Effects of target‐to‐target interval. Psychophysiology 42:636–642. [DOI] [PubMed] [Google Scholar]
- Stevens MC, Laurens KR, Liddle PF, Kiehl KA (2006): The hemodynamics of oddball processing during single‐tone and two‐tone target detection tasks. Int J Psychophysiol 60:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker GK (2008): Neurophysiological endophenotypes across bipolar and schizophrenia psychosis. Schizophr Bull 34:760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turetsky BI, Calkins ME, Light GA, Olincy A, Radant AD, Swerdlow NR (2007): Neurophysiological endophenotypes of schizophrenia: The viability of selected candidate measures. Schizophr Bull 33:69–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W (2010): Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci 11:100–113. [DOI] [PubMed] [Google Scholar]
- Walz JM, Goldman RI, Carapezza M, Muraskin J, Brown TR, Sajda P (2013): Simultaneous EEG‐fMRI reveals temporal evolution of coupling between supramodal cortical attention networks and the brainstem. J Neurosci 33:19212–19222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalley HC, Simonotto E, Marshall I, Owens DGC, Goddard NH, Johnstone EC, Lawrie SM (2005): Functional disconnectivity in subjects at high genetic risk of schizophrenia. Brain 128:2097–2108. [DOI] [PubMed] [Google Scholar]
- Yoshiura T, Zhong J, Shibata DK, Kwok WE, Shrier DA, Numaguchi Y (1999): Functional MRI study of auditory and visual oddball tasks. Neuroreport 10:1683–1688. [DOI] [PubMed] [Google Scholar]