

Abstract
Cancer patients in pain require high doses of opioids and quickly become opioid-tolerant. Previous studies have shown that chronic cancer pain as well as high-dose opioid use lead to mu-opioid receptor downregulation. In this study we explore downregulation of the mu-opioid receptor gene (OPRM1), as a mechanism for opioid tolerance in the setting of opioid use for cancer pain. We demonstrate in a cohort of 84 cancer patients that high-dose opioid use correlates with OPRM1 hypermethylation in peripheral leukocytes of these patients. We then reverse-translate our clinical findings by creating a mouse cancer pain model; we create opioid tolerance in the mouse cancer model to mimic opioid tolerance in the cancer patients. Using this model we determine the functional significance of OPRM1 methylation on cancer pain and opioid tolerance. We focus on 2 main cells within the cancer microenvironment: the cancer cell and the neuron. We show that targeted re-expression of mu-opioid receptor on cancer cells inhibits mechanical and thermal hypersensitivity, and prevents opioid tolerance, in the mouse model. The resultant analgesia and protection against opioid tolerance are likely due to preservation of mu-opioid receptor expression on the cancer-associated neurons.
Keywords: Methylation, cancer pain, mu-opioid receptor, OPRM1, opioid tolerance
Cancer patients with pain experience a poor quality of life.1,3,7,10 A meta-analysis of 52 studies that evaluated the prevalence of cancer pain found that more than 50% of patients, regardless of cancer type, reported inadequately controlled pain.30 Opioids remain the most effective treatment for cancer pain. Unfortunately, some patients become opioid-tolerant and require escalating doses of opioids. On the basis of the definition of opioid tolerance by Portenoy and Hagen22 the prevalence of opioid tolerance in cancer patients ranges between 40% and 80%.9,15 High-dose opioids in cancer patients is associated with decreased survival.23 To improve pain management, a need exists for a biomarker of opioid tolerance and an increased understanding of a mechanism to prevent its development.
Inadequate pain management is partially due to a poor understanding of the mechanisms of cancer pain and opioid tolerance. One proposed mechanism for opioid tolerance is downregulation of the mu-opioid receptor on the neuronal cell membrane through receptor internalization.27 Two well known mechanisms that cause mu-opioid receptor downregulation are chronic pain and opioid administration.17 Studies of heroin users found that high opioid use correlates with downregulation of the mu-opioid receptor on circulating leukocytes.2 In addition, we showed in a cancer mouse model that the mu-opioid receptor is downregulated on the dorsal root ganglia (DRG) that innervate the cancer, even in opioid-naive animals.31 Moreover, we demonstrated that epigenetic silencing of the mu-opioid receptor gene (OPRM1) in cancer cells exacerbates cancer pain, and that reversal of OPRM1 gene silencing in the cancer produces antinociception. Antinociception is produced through endogenous opioid secretion by cancer cells into the cancer microenvironment. In addition, analgesia from the endogenous opioids maintains mu-opioid receptor expression on the corresponding DRG.31 On the basis of these findings we hypothesized that epigenetic silencing of the OPRM1 would correlate with opioid tolerance in the setting of opioid use for cancer pain and that forced expression of OPRM1 leads to antinociception.
Methods
To test our hypothesis, we quantified OPRM1 methylation in 84 cancer patients who were prescribed opioids for pain control. Because a neuronal biopsy was not possible in these patients, we determined whether OPRM1 methylation in their peripheral leukocytes could be used as a surrogate marker for increased risk of opioid tolerance and inadequate pain control. We then reverse-translated our findings by developing a cancer mouse model with opioid tolerance, which mimicked cancer patients who were opioid-tolerant. To establish our cancer mouse model we chose a head and neck squamous cell carcinoma (HNSCC) cell line, because HNSCC produces the highest pain prevalence and severity in patients.16,30 Last, we re-expressed OPRM1 in the HNSCC tumors of our mouse model, which is normally devoid of OPRM1 expression, and determined the effect on cancer-induced mechanical and thermal hypersensitivity, and opioid tolerance, in the mouse model.
Patient Recruitment and Blood Collection
This study was part of a large, multicenter randomized clinical trial to evaluate 2 doses of a psychoeducational intervention to improve cancer pain management. The study was approved by the University of California San Francisco (UCSF) Committee for Human Research, the Protocol Review Committee of the Helen Diller Comprehensive Cancer Center, and the institutional review board at each clinical site. Oversight of the qualitative analysis was provided by the UCSF Committee for Human Research and the institutional review board at the University of Nebraska Medical Center, where the qualitative analysis was conducted. Potential participants were approached at each clinical site by a staff member, who introduced the study and inquired about interest in enrolling. Interested patients were referred to a research nurse, who assessed eligibility, and conducted the informed consent process. When written informed consent was provided, patients were randomized and an appointment was made for the first home visit. Patients were eligible if they were at least 18 years of age; able to read, write, and understand English; gave informed consent; had a Karnofsky Performance Status Score ≥ 50; had an average pain intensity score of at least 3 on a 0 to 10 numeric rating scale; had a life expectancy of at least 6 months; were receiving outpatient treatment for cancer (not AIDS-related) with any single or combination therapy; and had a telephone line. From the 222 patients, data from a subsample of 84 patients were used in the current study. Before the first home visit, patients were sent a number of questionnaires to obtain information on demographic and clinical characteristics. During the first home visit, the intervention nurse performed a comprehensive pain assessment and taught the patient to complete a Pain Management Diary on a daily basis. In the diary, the patients recorded pain intensity scores, as well as around the clock and as needed intake of nonopioid and opioid analgesics. For this study, all opioid analgesic intake for the first 2 days of the study was converted to oral morphine equivalent dosage. The opioid conversion table published by the American Pain Society was used to determine the morphine equivalent dose (MED) for all opioids used by patients in this study.
Blood was collected before starting the randomized clinical trial. DNA was extracted from buffy coat using Puregene Blood Core Kit C (Qiagen, Valencia, CA).
Cell Culture
Cancer Cells
The human tongue squamous cell carcinoma (SCC) cell line, HSC-3 (ATCC), was cultivated in Dulbecco’s Modified Eagle’s Medium (DMEM) with 4.5 g/L glucose, L-glutamine, and sodium pyruvate, supplemented with 10% fetal bovine serum, 25 μg/mL fungizone, 100 μg/mL streptomycin sulfate, and 100 U/mL penicillin G and cultivated at 37°C in 5% CO2.
Re-Expression of OPRM1 Using Adenoviral Transduction
Human cDNA of OPRM1 containing a C-terminal green-fluorescent protein (GFP) tag (OriGene, Rockville, MD) was subcloned into a shuttle plasmid as described previously.33 Viral particle purification was completed through Viraquest (North Liberty, IA). Adenovirus (Ad) containing GFP (Ad-GFP) was used as a transduction control.
The human tongue SCC cell line, HSC-3, was transduced with recombinant Ad (Ad containing OPRM1 [Ad-OPRM1] or Ad-GFP) at increasing multiplicities of infection (MOI; number of viral particles per cell). Transduction was performed in DMEM with 4.5 g/L glucose, L-glutamine, and sodium pyruvate, supplemented with 2% fetal bovine serum, 25 μg/mL fungizone, 100 μg/mL streptomycin sulfate, and 100 U/mL penicillin G. Twenty-four hours after transduction the cell medium was changed to DMEM containing 10% fetal bovine serum with supplements. Transduction efficiency was determined using visualization of GFP-expressing cells.
Morphine-Tolerant Mouse Cancer Model
To create the soft tissue malignancy mouse model we used the tongue SCC cell line HSC-3 because it produces more profound mechanical and thermal nociception than cell lines from other soft tissue malignancies.33 Experiments were performed on 4-week-old female BALB/c, athymic mice weighing 16 to 20 g at the time of SCC inoculation. All the procedures were approved by the New York University Committee on Animal Research. Researchers were trained under the Animal Welfare Assurance Program. Mice were housed in a temperature-controlled room on a 12:12-hour light cycle (7:00 AM to 7:00 PM light), with ad libitum access to food and water; estrous cycles were not monitored. The mice were divided into 2 inoculation groups and inoculated with the respective cell types: 1) HSC-3 transduced with Ad-GFP at 100 MOI, and 2) HSC-3 transduced with Ad-OPRM1 at 100 MOI. Five × 106 cells from each group were suspended in Matrigel (Becton Dickinson & Co, Franklin Lakes, NJ) to a total volume of 50 μL and inoculated into the plantar surface of the right hind paw. Isoflurane inhalational anesthesia was used for inoculation. From our preliminary immunofluorescence experiments we had determined that HSC-3 cells and Ad-GFP-transduced HSC-3 cells did not express the mu-opioid receptor, whereas HSC-3 transduced with Ad-OPRM1 at 100 MOI maximally re-expressed the mu-opioid receptor with minimal cell death.
We produced morphine tolerance in these mice on postinoculation day (PID) 9 when tumors were visible on the hind paws. One 25-mg morphine pellet or 1 vehicle pellet was implanted subcutaneously into the dorsum of each mouse. Morphine and vehicle pellets were obtained from the National Institute on Drug Abuse. Release of morphine from the pellet was continuous and was expected to be 3.7 mg during the first 24 hours and 2 mg each subsequent day until day 7, at an average rate of approximately 180 mg/kg/d.4 Continuous morphine exposure for 7 days with this method is superior to intermittent morphine injection in producing a morphine-tolerant mouse model.4 A total of 24 mice were divided into 4 groups: 1) Ad-GFP vehicle, 2) Ad-GFP morphine, 3) Ad-OPRM1 vehicle, and 4) Ad-OPRM1 morphine. We did not observe any signs of respiratory depression, change in locomotor activity, or other deviations from normal behavior in the groups that were implanted with morphine pellets.
Paw Volume Measurement
Paw volume measurements were performed with a plethysmometer (IITC Life Sciences, Woodland Hills, CA). Triplicate measurements were taken. Each mouse was used as its own control and relative changes in paw volume were calculated on the basis of day 0 baseline.
Mechanical Allodynia Measurement
Paw withdrawal testing was performed as described previously.21 Testing was performed between 9:00 AM and noon by an observer blinded to the experimental groups. Mice were placed in a plastic cage with a wire mesh floor, which allowed access to the paws. One hour was allowed for acclimation before testing. The probe was applied to the midplantar right hind paw. Paw withdrawal thresholds were determined in response to pressure from an electronic von Frey anesthesiometer (2390 series, IITC Life Sciences). The amount of pressure (g) needed to produce a paw withdrawal response was measured 6 times on each paw separated by 3-minute intervals to allow resolution of previous stimuli. The results of the 6 values were averaged for each paw for that day. Each mouse was used as its own control, and relative changes in paw volume were calculated on the basis of day 0 baseline.
Thermal Hyperalgesia Measurement
Thermal hyperalgesia of the hind paw was assessed according to Hargreaves et al.8 Testing was performed by an observer blinded to the experimental groups. Mice were acclimated to the test room and chamber for 30 minutes twice a week for 2 weeks. Mice were then acclimated in the plantar test apparatus for 30 minutes before actual testing. A thermal stimulus originating from a focused projection bulb was positioned under the right foot pad of each mouse. The time required for a paw withdrawal response was considered an index of the heat nociceptive threshold. Paw withdrawal to heat was calculated as a mean of 6 measurements, carried out at 5-minute intervals. Each mouse was used as its own control, and relative changes in heat nociception threshold were calculated on the basis of day 0 baseline.
Cumulative Morphine Dosing
On PID 16 we determined the shift in morphine dose required to produce complete antinociception in cumulative dosing studies. Tolerance to morphine is manifest by requiring a higher dose of morphine to produce the same analgesic effect. An “analgesic effect” was defined as return to the thermal withdrawal threshold on day 0, before cancer inoculation. The cumulative dosing protocol was on the basis of previous studies.5 Mice were injected subcutaneously with a starting dose of morphine (.5 mg/kg; Sigma Aldrich, St. Louis, MO) and tested for antinociception 30 minutes after injection. If the mouse was analgesic, it was not tested further. Otherwise, mice were immediately injected again with another dose of morphine and tested again (at increments of 1 mg/kg up to a cumulative dose of 8.5 mg/kg, then 2 mg/kg thereafter) yielding cumulative doses of .5, 1.5, 2.5, 3.5, 4.5, 5.5, 6.5, 7.5, 8.5, 10.5, and 12.5 mg/kg.
Quantitative Methylation Analysis
Quantitative methylation analysis of the human OPRM1 promoter was performed through the Genome Analysis Core Facility at UCSF, using the EpiTYPER assay (Sequenom, San Diego, CA) in conjunction with the MassARRAY (Sequenom) system, by a technician who was blinded to the treatment groups. The target region in the human promoter spanned from −232 to +109 relative to the transcription start site. The target region on the mouse promoter was −304 to +71 relative to the transcription start site. Primers were designed using EpiDesigner (Sequenom) software.
At least 1 μg of DNA from each sample was treated with sodium bisulfite, amplified with polymerase chain reaction, and excess dNTP (deoxyribonucleotide triphosphate) was treated with shrimp alkaline phosphatase, as described previously.33 The samples were then spotted on a 384-pad Spectro-CHIP (Sequenom) and analyzed using a MassARRAY analyzer compact MALDI-TOF MS (matrix-assisted laser desorption/ionization time-of-flight mass spectrometry). Methylation calls were analyzed using EpiTyper software version 1.0 (Sequenom) to produce quantitative results for each CpG unit, which consists of a single CpG (5′-C-phosphate-G-3′) site or aggregate of adjacent CpG sites. Fully methylated DNA was used as a positive control and water was used as a negative control.
Quantitative Reverse Transcription Polymerase Chain Reaction Analysis
mRNA was reverse transcribed with Random Hexamers (Applied Biosystems, Foster City, CA). A 2-μL cDNA aliquot was amplified with the Taqman gene expression assay (Applied Biosystems) for OPRM1, which does not detect residual genomic DNA. Human β-glucuronidase and mouse β-actin were used as the endogenous controls.
Immunohistochemistry
Animals were euthanized with 4% isoflurane and perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde. Paw tissues and DRG were harvested after behavioral experiments, fixed in 10% formalin and embedded in paraffin. Immunohistochemical staining was performed by a blinded observer. Sections were heated and deparaffinized and then pretreated with Target Retrieval Solution (Dako, Carpinteria, CA). Endogenous peroxidases were quenched by immersing sections in 3% hydrogen peroxide and Tris-Buffered Saline (Sigma-Aldrich, St. Louis, MO) for 5 minutes, followed by placement in a Tris-Buffered Saline (Sigma-Aldrich) bath for 5 minutes. Sections were incubated for 2 hours with monoclonal mu-opioid receptor antibody (Epitomics, Burlingame, CA) at a 1:500 dilution. The primary antibody was replaced with nonspecific antibody (Epitomics) for the negative control. After a 45-minute incubation with a goat anti-rabbit secondary antibody, DAB substrate (Abcam, Cambridge, MA) was placed on the sections for 2 minutes, and the slides were then rinsed in water, dehydrated in alcohol, and coverslipped.
In Situ Hybridization
Paraffin-embedded tumor tissue sections (5 μm) were deparaffinized, rehydrated with graded alcohol solution, and washed in PBS. These tissue samples were subjected to proteinase-K treatment (18 μg/mL) at 37°C for 10 minutes. Sections were washed twice in PBS with Tween-20 (PBST) and preheated for 1 hour at 57°C in pre-hybridization buffer (50% formaldehyde, 5X saline-sodium citrate [SSC], .1% Tween, 9.2 mM citric acid pH 6, 50 μg/mL heparin, 500 μg/mL yeast RNA). Hybridization was carried out overnight at 57°C with 30 nM custom-designed digoxygenin (DIG)-labeled LNA probes (Exiqon, Vedbaek, Denmark). The sequence of the OPRM1 probe was TTAGGGCAACGGAGCAGTT. A scrambled probe with no complementary sequence target was used as a negative control. All LNA oligos (Exiqon) were digoxigenin-labeled at the 5′ end. Sections were washed stringently with 5 × SSC, 1 × SSC and .2 × SSC buffer at 55°C over 60 minutes, and in PBST for 5 minutes at room temperature (RT). The samples were then blocked for 1 hour at RT in blocking buffer (2% sheep serum, 2 mg/mL bovine serum albumin in PBST), and incubated at 4°C overnight in alkaline phosphatase-conjugated anti-digoxigenin (Roche, Indianapolis, IN) diluted to 1:4000 in blocking reagent. Enzymatic development was carried out at RT for 5 minutes to 16 hours using 4-nitro-blue tetrazolium and 5-brom-4-chloro-3′-indolylphosphate, or Vector Lab AP substrate (Burlingame, CA). Finally, sections were stained with hematoxylin counterstain (Roche). The sections were mounted in aqueous mounting medium (Santa Cruz Biotechnology, Santa Cruz, CA).
Statistical Analysis
Statistical analysis was performed using Sigma Plot, version 11.0 (Systat Software Inc, San Jose, CA). Data were analyzed using the Student t-test and analysis of variance (ANOVA) with Holm-Sidak post hoc testing. In the experiments using the mouse cancer model, groups were compared with the Ad-GFP vehicle control group. Results are presented as mean ± standard error of the mean.
Results
High-Dose Opioid Use Correlates With OPRM1 Hypermethylation
We enrolled 84 cancer patients. Thirty-two patients had breast cancer, 14 had prostate cancer, 5 had lung cancer, and the remaining had melanoma, colon cancer, or non-Hodgkin lymphoma. The mean age of the patients was 57.9 years (range, 28–88 for female patients and 35–80 for male patients). Twenty-seven of the patients were male.
On the basis of the opioid intake for the week before the intervention, the 84 patients were then divided into either: 1) no opioid use, 2) low-dose, 3) moderate-dose, or 4) high-dose opioid users. The no opioid use group consisted of 19 patients who were prescribed opioids, but did not take them for pain during the evaluation period. The low-dose group (n = 18) used <40 mg MED for days 1 and 2 after enrollment. The mid-dose group (n = 32) used between 40 and 200 mg MED during the days 1 and 2 of enrollment. The high-dose group (n = 15) used >200 mg MED.
First, we compared the mean methylation value between the high-dose opioid group and the no opioid use group (Fig 1A). The high-dose opioid group had higher methylation values than the no opioid use group in 18 of 20 queried methylation (CpG) sites of our OPRM1 amplicon. The high-dose group had significantly higher methylation than the no opioid use group across the entire OPRM1 amplicon (P = .032, 2-way ANOVA, Holm-Sidak post hoc test; Table 1). In addition, we analyzed the differences in methylation between the high-dose opioid group and no opioid use group using an alternative area under the curve approach, which compared the area under the curve of the 2 groups (Supplementary Fig 1). This alternative approach yielded the same findings as those in Fig 1A. We then compared the percent change in methylation value of the low-, moderate-, and high-dose groups by using the mean values of the no opioid use group as the baseline. On the basis of our previous work32 a 10% change in methylation value at a CpG site was defined as a significant change. The percentage of patients in each group who had a >10% methylation value increase at a CpG site was determined and compared with the no opioid use group mean. The high-dose group had more patients with a >10% increase in methylation than the low-dose group at 19 of 20 CpG sites (Fig 1B). Compared on a single CpG site basis, there was a dose-dependent relationship between opioid use and methylation level. This dose-dependent relationship was present in 17 of the 20 queried CpG sites, such that low-dose patients had the lowest methylation level, and high-dose patients had the highest methylation level (Fig 1C).
Figure 1.
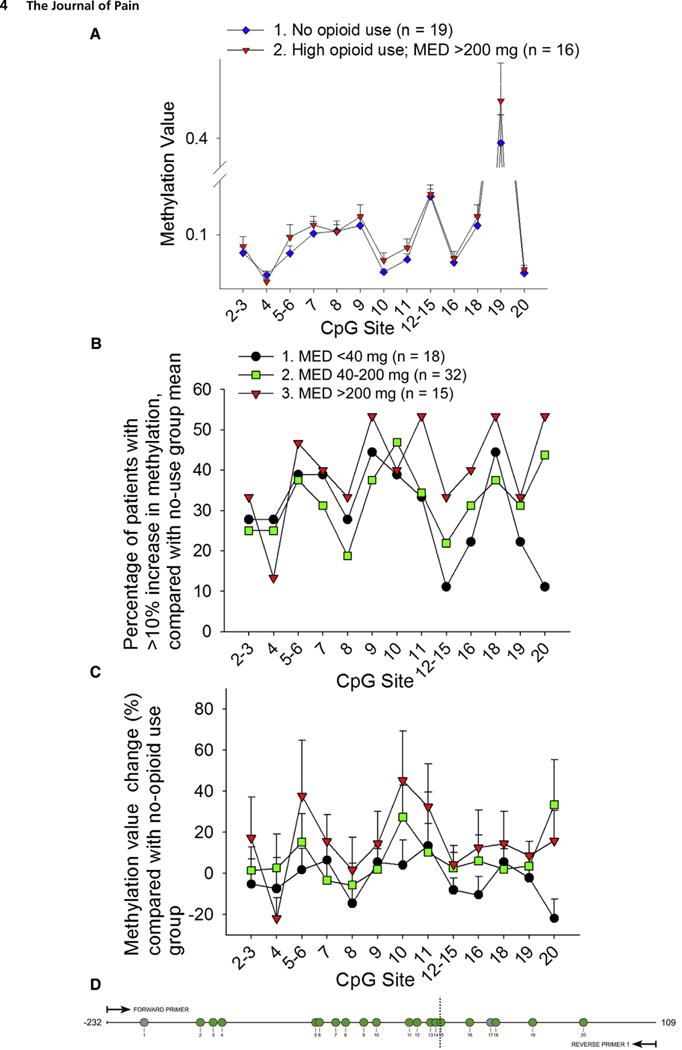
High-dose opioid use correlated with OPRM1 hypermethylation. Patients were divided according to their opioid use on the first 2 days of study enrollment into low-dose (<40 mg MED, n = 18), moderate-dose (40–200 mg MED, n = 32), and high-dose (>200 mg MED, n = 15) groups. Patients who were prescribed but did not take opioids (n = 19) were used as the control group. The queried CpG sites within the amplified region (−232 to +109 relative to the transcription start site) are shown. Methylation results from the EpiTYPER (Sequenom) assay are expressed from 0 to 1.0, with 1.0 representing 100% methylation at the CpG site of interest. CpG sites 1 and 17 could not be quantified because of inability to design primers and probes, and are shown in the diagram as gray circles. The remaining quantified CpG sites are shown as green circles and are quantified either as single sites or a cluster of sites. (A) This graph shows methylation values of patients in the no opioid use control group and patients in the high-dose opioid use group. The methylation status of the amplicon was significantly different between the 2 groups (P = .032, 2-way ANOVA, Holm-Sidak post hoc test, see Table 1 for statistics). (B) The graph depicts the percentage of patients in each opioid use group with a >10% increase in methylation compared with the control group methylation mean, at each CpG site. The high-dose group had the highest percentage of patients with a >10% increase in methylation at all but 1 CpG unit. (C) The graph shows the mean methylation change (% + standard error of the mean) compared with the no-opioid use group mean. The high-dose group had the greatest methylation change at all but 2 CpG units. (D) The diagram shows the queried OPRM1 amplicon, which includes 20 CpG sites as numbered. CpG sites 1 and 17 could not be quantified because of primer and probe design, and are shown in the diagram as gray circles. The remaining quantified CpG sites are shown as green circles.
Table 1.
The Methylation Status of the Amplicon Was Significantly Different Between the High-Dose Opioid and No Opioid Groups
|
TWO-WAY ANOVA OR 2-WAY
RM ANOVA
|
HOLM-SIDAK POST HOC
TEST
|
||||
|---|---|---|---|---|---|
| EFFECTS | DF | F | P | GROUPS | P |
| Fig 1A | |||||
| Opioid use | 1 | 4.62 | .032 | 1 versus 2 | .032 |
| CpG | 12 | 112.91 | <.001 | ||
| Opioid use × CpG | 12 | .325 | |||
| Fig 1C | |||||
| Opioid use | 2 | 4 | .019 | 1 versus 2 | .063 |
| CpG | 12 | 1.282 | .224 | 1 versus 3 | .005 |
| Opioid use × CpG | 24 | .56 | .957 | 2 versus 3 | .172 |
| Fig 2B | |||||
| Opioid use | 1 | 14.903 | <.001 | 1 versus 2 | <.001 |
| CpG | 12 | 64.053 | <.001 | ||
| Opioid use × CpG | 12 | .992 | .992 | ||
| Fig 3A | |||||
| Treatment | 3 | 424.902 | <.001 | 1 versus 2 | <.001 |
| Time | 4 | 500.381 | <.001 | 1 versus 3 | <.001 |
| Treatment × Time | 12 | 80.796 | <.001 | 1 versus 4 | <.001 |
| Fig 3B | |||||
| Treatment | 3 | 150.074 | <.001 | 1 versus 2 | <.001 |
| Time | 4 | 2317.49 | <.001 | 1 versus 3 | <.001 |
| Treatment × Time | 12 | 338.532 | <.001 | 1 versus 4 | <.001 |
Abbreviation: RM, repeated measures.
Inadequate Pain Relief Correlated With OPRM1 Hypermethylation
We performed a linear regression plot to determine the association between level of pain relief and amount of opioid used as indicated; an r2 value of .02 from a linear regression plot (Fig 2A) showed that there was no association between these 2 factors. Furthermore, additional analysis showed that there was also no relationship between average pain score and the amount of opioid used (r = .179), no relationship between worst pain and opioid use (r = .205) or between the number of hours a patient had pain relief and opioid use (r = .115).
Figure 2.
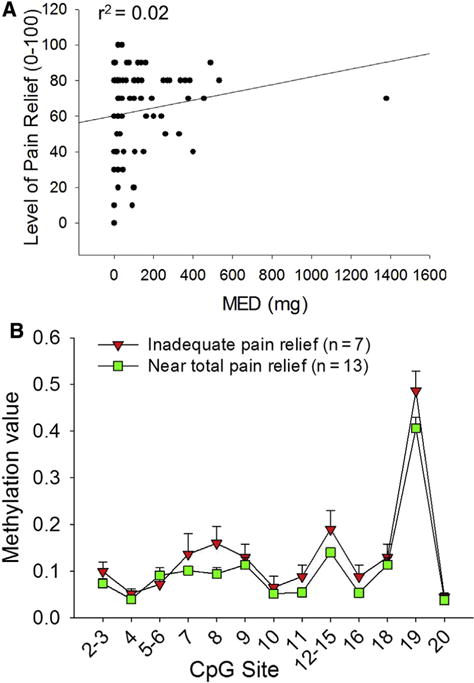
Inadequate pain relief correlated with OPRM1 hypermethylation and was unrelated to opioid dosage. (A) No relationship was found between opioid use (x-axis) versus pain relief (y-axis), as shown by this regression plot with r2 = .02. (B) Patients were divided according to their pain relief score into those that had inadequate pain relief (≤20) or those that had near total pain relief (≥90). Patients with inadequate pain relief had significantly higher methylation values in the −232 to +109 amplicon than patients with near total pain relief (P < .001, 2-way ANOVA, Holm-Sidak post hoc test; Table 1).
We did show, however, that pain relief correlated to methylation of the OPRM1 promoter. Pain relief was characterized as a percentage from 0% to 100%, with 100% representing complete pain relief. Patients who reported inadequate pain relief (relief score ≤20%) and patients who reported near total pain relief (relief score ≥ 90%) had significantly different OPRM1 promoter methylation patterns (P < .001, 2-way ANOVA, Holm-Sidak post hoc test; Table 1). Patients with inadequate pain relief had higher methylation at 19 of the 20 queried CpG sites within the amplicon (−232 to +109; see Table 2 for CpG positions).
Table 2.
Patients With Inadequate Pain Relief Had Higher Methylation at 19 of the 20 Queried CpG Sites Within the Amplicon
| CPG IN AMPLICON | CORRELATING POSITION IN HUMAN GENOME (RELATIVE TO OPRM1 TRANSCRIPTION START SITE) |
|---|---|
| 1 | −197 |
| 2 | −169 |
| 3 | −159 |
| 4 | −151 |
| 5 | −93 |
| 6 | −90 |
| 7 | −80 |
| 8 | −71 |
| 9 | −60 |
| 10 | −50 |
| 11 | −32 |
| 12 | −25 |
| 13 | −18 |
| 14 | −14 |
| 15 | −10 |
| 16 | +12 |
| 17 | +23 |
| 18 | +27 |
| 19 | +53 |
| 20 | +84 |
Ad-Mediated OPRM1 Re-Expression and Morphine Treatment Did Not Affect Cancer Growth
We next generated a cancer pain mouse model to replicate soft tissue cancer pain and opioid tolerance as seen in the patients. We created a mouse paw cancer model by inoculating the hind paw with HSC-3, a tongue SCC line. Whereas our cancer patient cohort did not include head and neck cancer patients, we needed a preclinical model of cancer pain and this model using HSC-3 predictably produces soft tissue cancer with profound nociception. The goal of the experiment was to determine whether manipulating OPRM1 expression in the cancer microenvironment of this model would decrease cancer pain. In our previous studies we demonstrated that cancer pain results from an interaction between the cancer cell and the primary afferent neuron within the cancer microenvironment.31,37 In addition we found that targeted re-expression of OPRM1 on cancer cells with adenoviruses decreases cancer pain. In this study we determined whether re-expression of OPRM1 on cancer cells could also treat opioid tolerance. We chose to target the cancer cell, rather than neurons, because this approach is more clinically translatable with intratumor injection. We used adenoviruses to re-express either OPRM1 or a control plasmid with GFP (hereafter referred to as Ad-OPRM1 or Ad-GFP groups).
To produce opioid tolerance we implanted morphine pellets subcutaneously into the dorsum of mice with cancer. Upon creation of the mouse paw cancer model we quantified the increase in paw volume as an index of cancer growth. The mean paw volume at baseline was .147 mL, .143 mL, .143 mL, and .147 mL for the Ad-GFP vehicle, Ad-GFP morphine, Ad-OPRM1 vehicle, and Ad-OPRM1 morphine groups, respectively. On PID 14, the increase in paw volume was 106%, 90%, 92%, and 88% for the Ad-GFP vehicle, Ad-GFP morphine, Ad-OPRM1 vehicle, and Ad-OPRM1 morphine groups, respectively. Cancer growth (ie, paw volume increase) was not significantly different between these 4 groups. These results indicate that neither OPRM1 re-expression nor morphine treatment affected cancer growth.
OPRM1 Re-Expression in Cancer Combined With Subcutaneous Morphine Pellets Resulted in Complete Antinociception to Thermal Stimuli
Next, we determined whether OPRM1 re-expression in cancer cells affected cancer pain by quantifying thermal and mechanical nociception. Thermal hyperalgesia was quantified on PID 4, 7, 11, and 14. Fig 3A shows the mean changes in thermal hyperalgesia relative to day 0 baseline. All groups had an initial decrease from baseline, signifying increased thermal hyperalgesia with tumor growth. The Ad-OPRM1 groups, which were inoculated with HSC-3 re-expressing OPRM1 by adenoviral transduction, had significantly less thermal hyperalgesia than the Ad-GFP vehicle control group, and this effect was seen on PID 4 as well as 7. To model morphine tolerance, morphine (or vehicle) pellets were implanted on PID 9, depicted by the red dotted line in Fig 3A. The pellets induced tolerance after 7 days. However, before the seventh day, on PID 11 and 14, we noted that morphine pellets augmented the thermal antinociceptive effects seen in Ad-OPRM1 mice, such that the Ad-OPRM1 morphine group had maximal antinociception at these 2 time points. Formal morphine tolerance testing was performed on PID 16, 7 days after pellet implantation. Results are discussed below in the section titled, “Morphine Pellet Implantation Produced Morphine Tolerance That Was Prevented by OPRM1 Re-Expression in Cancer.”
Figure 3.
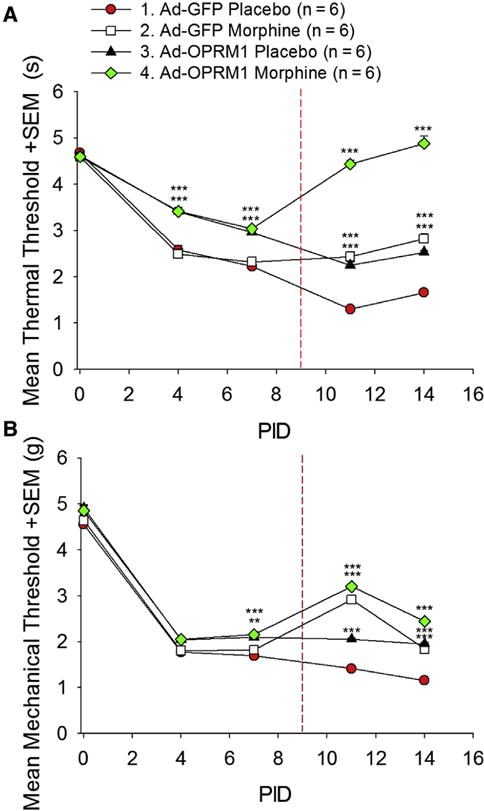
OPRM1 re-expression in HNSCC combined with morphine treatment inhibited thermal and mechanical hypersensitivity in a mouse HNSCC model. (A) The graph depicts the thermal threshold in seconds + standard error of the mean (SEM). Mouse orthotopic HNSCC tumors re-expressing OPRM1 had significantly less thermal hyperalgesia compared with HNSCC tumors not expressing OPRM1 (P < .001, 2-way RM ANOVA, Holm-Sidak post hoc test). After subcutaneous implantation of morphine pellets on PID 9 (indicated by dashed red line), mice with HNSCC tumors that expressed OPRM1 displayed reversal of thermal hyperalgesia back to day 0 baseline. (B) The graph depicts mechanical threshold in grams + SEM. OPRM1 expression in orthotopic HNSCC tumors produced mechanical antinociception, and morphine significantly increased this anti-nociceptive effect (P < .001, 2-way RM ANOVA, Holm-Sidak post hoc test; Table 1). ***P < .001, Holm-Sidak post hoc test, main effect of treatment at each time point, Ad-GFP vehicle group as control group). Abbreviation: RM, repeated measures.
OPRM1 Re-Expression in Cancer Stabilized Mechanical Allodynia Threshold, Whereas Morphine Pellets Provided Additional Mechanical Antinociception
Mechanical allodynia was quantified on PID 4, 7, 11, and 14. Fig 3B shows the mean change in mechanical allodynia relative to day 0 baseline. All groups had an initial decrease from baseline, signifying increased mechanical allodynia with tumor growth. By PID 7, the Ad-OPRM1 groups had significantly less mechanical allodynia than the Ad-GFP vehicle control group (P < .001, 2-way RM (repeated measures) ANOVA, Holm-Sidak post hoc test). Similar to the thermal antinociception we measured with Ad-OPRM1 morphine mice, we noted that morphine pellet implantation provided additional mechanical antinociception, such that Ad-OPRM1 mice implanted with morphine had an increase in mechanical baseline. Ad-OPRM1 mice treated with vehicle pellets also had mechanical antinociception compared with control. However this effect was not as strong as that seen in the combination Ad-OPRM1 morphine group. Morphine also produced mechanical antinociception in the Ad-GFP group. Ad-mediated OPRM1 re-expression did not have a profound effect on mechanical antinociception as it did on thermal antinociception, although re-expression stabilized the mechanical allodynia threshold of Ad-OPRM1 groups at a level above the control Ad-GFP group.
Morphine Pellet Implantation Produced Morphine Tolerance That Was Prevented by OPRM1 Re-Expression in Cancer
Morphine pellet implantation produced tolerance after 7 days.4 On PID 16, 7 days after implantation, we determined the extent of morphine tolerance using a cumulative dosing assay. The assay was performed by determining the total morphine dose required to produce complete antinociception. Complete antinociception was defined as a withdrawal response to the same thermal threshold as day 0 baseline. We used thermal rather than mechanical stimuli because previous studies of morphine tolerance have used thermal stimuli.4,5 The Ad-GFP vehicle control group required 6.5 mg of morphine for complete anti-nociception. The Ad-GFP morphine group required 11.7 mg, which was 180% that of the Ad-GFP vehicle group (Fig 4A). This increase in morphine requirement signified morphine tolerance, and was comparable with previous studies using the same cumulative dosing methods.5 The Ad-OPRM1 vehicle group required 3.3 mg of morphine, which was only 51% of the Ad-GFP vehicle group dose. The Ad-OPRM1 morphine group required .8 mg of morphine, which was only 12% of the Ad-GFP vehicle group dose. These findings suggest that the Ad-OPRM1 mice did not develop morphine tolerance when implanted with morphine pellets. The total morphine dose used versus the level of thermal antinociception is shown in Fig 4B with 0% on the y-axis representing a return to thermal threshold baseline. The Ad-GFP morphine group required a higher morphine dose than the Ad-GFP vehicle group, signifying morphine tolerance in the Ad-GFP morphine group. However, this trend was not observed when the Ad-OPRM1 groups were compared with each other, signifying lack of morphine tolerance in mice with cancer that re-expressed OPRM1. In fact, the Ad-OPRM1 morphine group only required 24% of the morphine dose used by the Ad-OPRM1 vehicle group to achieve complete antinociception.
Figure 4.
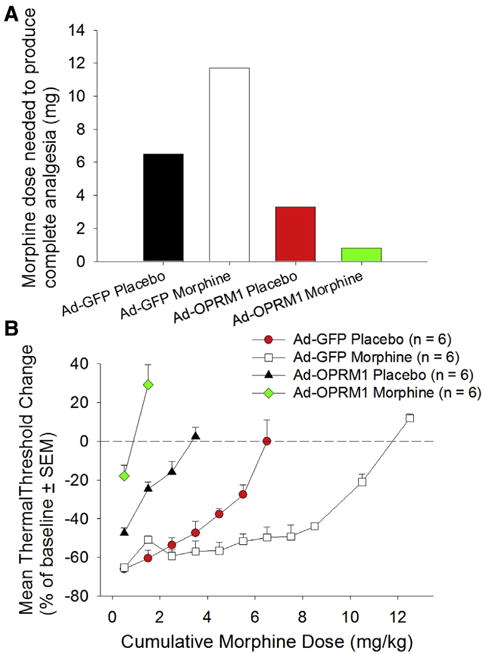
OPRM1 expression prevented morphine tolerance after morphine pellet implantation. (A) The bar graph depicts morphine doses required to achieve complete thermal antinociception. Although morphine pellet implantation in the Ad-GFP group increased morphine requirement from 6.5 mg to 11.7 mg, signifying morphine tolerance, the reverse trend was seen in the Ad-OPRM1 group. OPRM1 expression in the HNSCC tumor prevented development of morphine tolerance after morphine implantation. (B) The graph depicts total morphine dosage used versus the level of thermal antinociception, with 0% on the y-axis representing a return to thermal threshold baseline. Abbreviation: SEM, standard error of the mean.
Morphine Tolerance Correlated With Oprm1 Methylation and Silenced Expression in Mouse Peripheral Neural Tissue
We determined the effect of chronic morphine treatment on mouse DRG in the right lumbar region. We chose these neural tissues because nociceptive afferents from the hind paw, the site of HNSCC inoculation, terminate in the lumbar DRG. We quantified expression of Oprm1 in the right DRG (L4–L5 ganglia). Consistent with our previous findings,31 Oprm1 expression was higher in DRG from Ad-OPRM1 mice than Ad-GFP mice (Fig 5). The Ad-GFP morphine group had significantly reduced Oprm1 expression compared with the control Ad-GFP vehicle group (P = .033, 1-way ANOVA, Holm-Sidak post hoc test). To determine the possible mechanism for silenced Oprm1 expression, we quantified Oprm1 promoter methylation (Fig 5). We determined that the Ad-GFP morphine group was hypermethylated at 3 separate CpG sites in the mouse Oprm1 promoter, compared with the control Ad-GFP vehicle groups, and that this trend was significant (P = .005, 2-way ANOVA, Holm-Sidak post hoc test).
Figure 5.
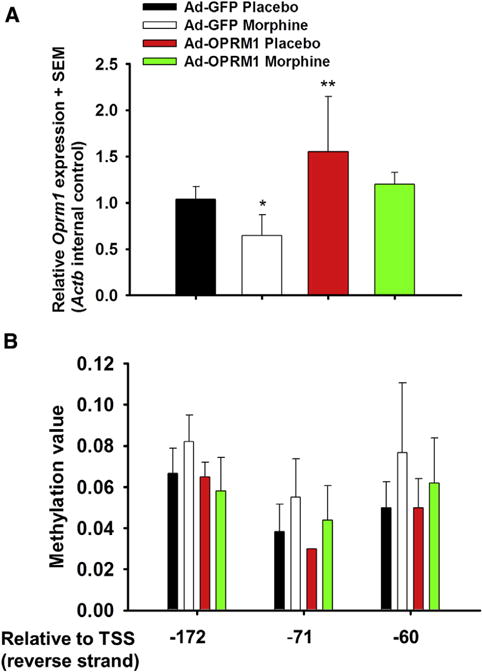
Morphine tolerance correlated with Oprm1 methylation and silenced expression. (A) Relative expression and (B) methylation value for each of the treatment groups (6 mice per group) are shown. Oprm1 expression was higher in DRG from Ad-OPRM1 mice than in the Ad-GFP mice. The Ad-GFP morphine group had significantly reduced Oprm1 expression compared with the control Ad-GFP vehicle group (P = .033, 1-way ANOVA, Holm-Sidak post hoc test). Accordingly, the Ad-GFP morphine group was hypermethylated at 3 separate CpG sites in the mouse Oprm1 promoter, compared with the control Ad-GFP vehicle groups (P = .005, 2-way ANOVA, Holm-Sidak post hoc test). Abbreviations: SEM, standard error of the mean; Actb, mouse β-actin; TSS, transcription start site.
To confirm our gene expression findings we performed in situ hybridization of Oprm1 mRNA and immunohistochemical staining of mu-opioid receptor in paw SCC tissue and L4 to L5 ganglia. Expression levels corresponded with methylation trends of DRG tissues. DRG tissue from mice in the Ad-GFP vehicle and Ad-GFP morphine groups had less positive staining of mu-opioid receptor in immunohistochemical as well as in situ hybridization experiments compared with Ad-OPRM1 vehicle and Ad-OPRM1 morphine groups (Fig 6). This finding was consistent with our previous publication31 in which we showed that although cancer pain tends to dampen mu-opioid receptor expression on the associated sensory neurons, mu-opioid receptor expression in these neurons is maintained in the presence of cancers that are transduced with Ad-OPRM1 to re-express mu-opioid receptors. Furthermore, we showed that morphine treatment, which was generally thought to dampen mu-opioid receptor expression in central nervous system tissue,14 did not dampen mu-opioid receptor expression in neurons from mice transduced with Ad-OPRM1. In other words, Ad-OPRM1 treatment rescued mu-opioid expression on neurons.
Figure 6.
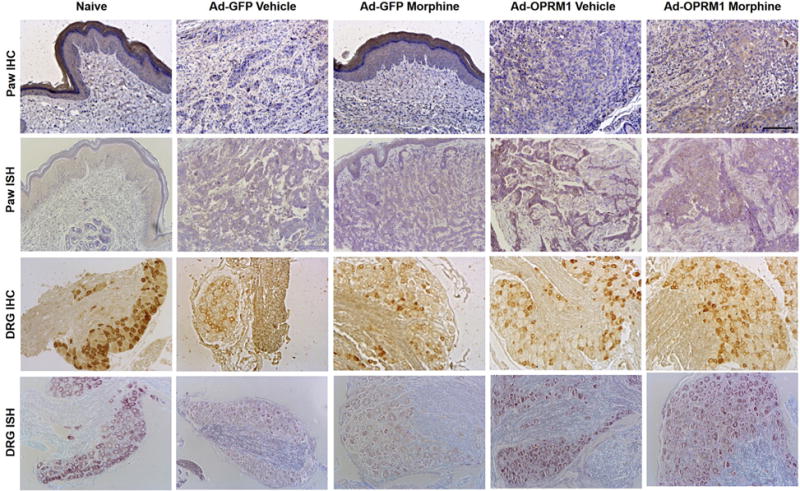
Ad-OPRM1 treatment rescued mu-opioid receptor expression in both paw cancer and corresponding DRG. Representative sections of immunohistochemical (IHC) staining and in situ hybridization (ISH) from paw cancer and DRG (L4–L5) for each treatment group are shown. The treatment group is indicated at the top of each column. Rows 1 and 2 show IHC and ISH stains of the paw cancer, which represent the expression of mu-opioid receptor protein and Oprm1 transcript, respectively. The full-thickness paw section is shown in the naive group and Ad-GFP morphine group. The dermal layer typically expresses mu-opioid receptor and is shown in this figure with positive brown staining. The cancer was inoculated directly below the dermal layer, as shown in the sections from the Ad-GFP morphine group. Brown staining within the cancer represents positive mu-opioid receptor protein expression (row 1) and Oprm1 transcript expression (row 2). Rows 3 and 4 show IHC and ISH stains from the DRG. DRG sections from the Ad-GFP vehicle and Ad-GFP morphine group have muted expression of mu-opioid receptor protein compared with the naive, Ad-OPRM1 vehicle, and Ad-OPRM1 morphine group (row 3, positive expression is represented by brown stain). Transcript expression is also muted in the Ad-GFP vehicle and Ad-GFP morphine groups compared with the other 3 groups (row 4, positive expression is represented by purple stain). Black bar = 100 μm.
Discussion
Peripheral Leukocyte OPRM1 Methylation Status as a Biomarker for Opioid Requirement in Cancer Patients
In this study we analyzed the possible role of OPRM1 methylation in opioid tolerance and cancer pain. We evaluated the status of OPRM1 expression in 2 separate tissues: blood in patients and neurons in a cancer mouse model. Our finding of OPRM1 hypermethylation in peripheral leukocytes from cancer patients receiving opioids is consistent with previous studies that found OPRM1 hypermethylation in peripheral leukocytes of opioid-addicted patients.2 The statistical methods for the analysis of DNA methylation are evolving. A persistent challenge in estimating biological differences in DNA methylation is that CpG sites rarely act independently, but rather neighboring clusters of CpG are jointly methylated. Evaluating each site independently would falsely inflate the multiple testing penalty and result in the rejection of true positive results (ie, false negatives). There exists no agreed-upon method for modeling this joint effect. Our approach to modeling this joint effect was by using the repeated measures approach to account for the nonindependence, which allows for dynamic changes in methylation across sites. On the basis of our previous work32 we defined a 10% change in methylation value at a CpG site as a significant change. Although this difference in methylation may seem small, methylation value differences do not need to be large to confer a biologically relevant effect. As a comparison in colorectal cancer studies where methylation is used as a biomarker for clinically adverse features such as poor differentiation, a methylation value of 4% is considered as a positive biomarker that correlates with adverse clinical features.19 Furthermore, in our case, CpG sites −18 and +12 (relative to the transcription start site, which corresponds to CpG units 13 and 16 in the queried OPRM1 amplicon in this study) are binding sites for the transcription factor Sp1,18 so methylation changes at these sites likely affect mu-opioid receptor level expression. The clinical significance of the association between mu-opioid receptor downregulation and increased opioid requirements is that the latter is associated with shorter progression-free and overall survival in patients with metastatic prostate cancer.38 In addition, inadequate pain relief is linked to depression in patients with lung, prostate, and head and neck cancer.6 To determine opioid requirements in the future, we could potentially quantify OPRM1 methylation in peripheral leukocytes.
Mu-Opioid Receptor Expression on Mouse DRG is Downregulated With Chronic Pain
We created a mouse model of cancer pain to explore the effect of opioid tolerance on Oprm1 methylation. We chose the oral SCC HSC-3 cell line because it predictably produces cancer pain and we have extensively published using this cancer pain mouse model. We have used other cancer cell lines, including melanoma and breast cancer, which do not produce the same profound nociceptive effects in the mouse model.20,33 We analyzed neuronal tissue in the mice rather than peripheral leukocytes. Although the discrepancy in tissues used between human subjects and animals might seem to weaken the translational potential of the study, a direct comparison between the human peripheral leukocyte OPRM1 methylation and mouse peripheral leukocyte Oprm1 methylation is not possible. In humans 70% of circulating leukocytes are T cells. Human leukocytes are the most common cell type used as a marker of a person’s epigenetic status because the circulating T cells contact all peripheral tissues. Our mouse model was established with athymic mice because they are able to grow human cancers, however, because they lack T cells their peripheral leukocytes cannot be compared with that of patients. The only reason we are using peripheral leukocytes as a marker in patients is because we do not have access to DRGs in patients. In mice we can easily harvest DRG neurons, which are the fundamental units of pain processing. Furthermore, the functional significance of OPRM1 is best analyzed in neuronal tissue. Cancer pain in a mouse model induces Oprm1 downregulation in the associated primary afferent neuron (ie, the DRG in our paw cancer model).31 Similarly, the mu-opioid receptor is downregulated in the DRG of a bone cancer mouse model.35 In additional studies of chronic pain (ie, cancer pain from peritoneal carcinomatosis or neuropathic pain from nerve injury) mu-opioid receptors were down-regulated on associated neurons.13,28 Mu-opioid receptor downregulation with persistent cancer pain necessitates higher doses of opioids to achieve analgesia. We induced opioid tolerance in our cancer mouse model to determine the relationship between Oprm1 methylation and opioid tolerance. We showed that opioid tolerance in the cancer model induced Oprm1 methylation and silenced mRNA expression in the DRG. One potential mechanism leading to opioid tolerance is the loss of the mu-opioid receptor on the neuronal cell membrane.27 In this study, persistent cancer pain as well as prolonged opioid exposure resulted in downregulation of mu-opioid receptors on DRGs. The mouse Oprm1 promoter region evaluated in this study encompasses a known nucleosome binding region. Of note, in mouse neuronal tissues nucleosome binding renders the promoter inactive.11
Mu-Opioid Receptor Re-Expression on Cancer Cells Reverses Cancer Pain and Prevents Opioid Tolerance
In the current study, mu-opioid receptor expression on cancer cells produced analgesia and inhibited opioid tolerance. This finding is related to our previous study, which shows that mu-opioid receptor expression on cancer cells induces the secretion of opioids into the cancer microenvironment.31 A high local opioid concentration would prevent the persistent pain state that would down-regulate mu-opioid receptors on the associated neurons (ie, the DRG at L4 and L5 for the paw cancer model). In mouse groups with Ad-GFP treatment, mu-opioid receptors were not expressed on cancer cells and the persistent pain state resulted in downregulation of the mu-opioid receptor on the neuron.31 In addition, opioid tolerance developed in these mice after insertion of morphine pellets, consistent with studies that showed chronic opioid administration led to opioid tolerance via mu-opioid receptor downregulation on neurons.14,27 Our finding that these 2 collective mechanisms, persistent pain and chronic opioid administration, lead to mu-opioid receptor downregulation on neurons in our mouse model is consistent with previous studies demonstrating that these are 2 well known mechanisms of mu-opioid receptor downregulation.17 We attempted to reverse the effect of mu-opioid receptor downregulation through adenoviral gene therapy. We found that re-expression of mu-opioid receptor in the cancer microenvironment produced antinociception as well as prevented opioid tolerance. In our animal experiments mice with cancer expressing mu-opioid receptor did not have opioid tolerance as evidenced by a lower morphine requirement to produce complete antinociception compared with control vehicle animals, which did not express mu-opioid receptor on their cancer cells and had opioid tolerance in response to morphine pellet insertion. Furthermore, the combination of morphine treatment and mu-opioid receptor expression resulted in significant thermal and mechanical antinociception; however, the level of thermal antinociception was more profound than mechanical antinociception. The difference might be attributed to differences in neuronal responses to morphine. Previous studies have shown dissociation of opioid receptors in controlling thermal and mechanical pain, with neurons expressing mu-opioid receptor predominantly controlling thermal pain, whereas neurons expressing delta-opioid receptor control mechanical pain.25 These 2 neuronal populations can overlap in mouse DRG.34 These collective findings explain why the mu-opioid receptor antagonist morphine would produce profound thermal antinociception, but only partial mechanical antinociception.
The Role of Peripheral Opioids and Pain Modulation a Clinical Setting
Part of our study findings hinges on the concept that re-expression of mu-opioid receptor results in opioid production, which contributes to antinociception. We have demonstrated in our previous studies that peripheral endogenous opioid production results in antinociception that is blocked by peripheral naloxone administration. The role of peripheral opioids in pain modulation is controversial, with different clinical trials drawing conflicting conclusions. In a randomized, double-blind clinical trial published in 2014, Jagla et al evaluated 50 patients undergoing knee replacement surgery. They studied whether peripheral opioid receptor blockade with methylnaltrexone, which is commonly used to treat opioid-induced constipation, would affect a patient’s demand for morphine to achieve satisfactory postoperative analgesia. The authors injected patients with methylnaltrexone at .9 mg/kg; their primary end point was cumulative amount of intravenous morphine administered during the first 8 hours. They found a significant increase in morphine dose required by patients to achieve analgesia after methylnaltrexone administration, and concluded that peripheral opioid receptors played a significant role in analgesia.12 In contrast to the study of Jagla et al, other clinical trials found no relationship between peripheral opioid blockade and analgesic requirement. A study by Thomas et al used methylnaltrexone at .15 mg/kg to treat opioid-induced constipation in patients with advanced illness and found a significant effect on laxation without an effect on pain scores. However the pain scores were not recorded at consistent times after methylnaltrexone administration.29 The conflicting conclusions drawn by these 2 studies could be attributed to differences in dosages of methylnaltrexone, but also to differences in the primary end point, with the primary end point of the study of Jagla et al12 being morphine dose requirement to achieve analgesia and that of Thomas et al29 being laxation. The finding that a peripherally restricted opioid antagonist does not completely block analgesia does not mean that central opioid agonism is solely driving analgesia. Parallel and redundant opioid-mediated analgesic mechanisms in the central and peripheral nervous systems might be active.
Conclusions
Our results from this study and previous studies31,33,36 highlight the role of peripheral opioid receptors in mediating cancer pain.
Potential Clinical Applications in Cancer Pain
Effective treatment of cancer pain often requires increasing doses of opioids because of disease progression and/or opioid tolerance. We addressed the issue of poor pain control and opioid tolerance by: 1) development of a biomarker using blood samples to identify cancer patients who are prone to high-dose opioid use and inadequate pain relief, and 2) development of a treatment approach to inhibit opioid tolerance in a cancer model with adenovirus-mediated OPRM1 expression. The main discrepancy in our reverse translational approach of creating a mouse model to replicate opioid tolerance and pain in cancer patients is that we created a head and neck cancer mouse model, whereas none of the patients in our cohort had head and neck cancer. The patients were selected from an existing cohort of an ongoing, large-scale study of a psychoeducational intervention for cancer pain management.24 All of the patients in this study had a solid soft tissue malignancy (ie, not hematologic malignancies or malignancies metastatic to bone). Currently most mouse cancer pain models are bone cancer models. For the study of soft tissue cancer pain certain head and neck cancer cell lines reliably produce cancer pain.26 Head and neck cancer cell lines produce more pain than other cell lines created from soft tissue malignancies.31 Despite the difference in cancer subtype between our patient cohort and mouse model, we demonstrated the potential of OPRM1 methylation as a biomarker for higher opioid requirements, and the functional role of OPRM1 expression in the cancer microenvironment in cancer pain and opioid tolerance.
Supplementary Material
{kind=link}
Perspective.
We demonstrate that epigenetic regulation of OPRM1 contributes to opioid tolerance in cancer patients, and that targeted gene therapy could treat cancer-induced nociception and opioid tolerance in a mouse cancer model.
Acknowledgments
This work was supported by the Oral and Maxillofacial Surgery Foundation and NCI R01 CA116423.
Footnotes
The authors have no conflicts of interest to declare.
Supplementary data accompanying this article are available online at www.jpain.org and www.sciencedirect.com.
Supplementary Data
Supplementary data related to this article can be found online at http://dx.doi.org/10.1016/j.jpain.2017.04.001
References
- 1.Chaplin JM, Morton RP. A prospective, longitudinal study of pain in head and neck cancer patients. Head Neck. 1999;21:531–537. doi: 10.1002/(sici)1097-0347(199909)21:6<531::aid-hed6>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 2.Chorbov VM, Todorov AA, Lynskey MT, Cicero TJ. Elevated levels of DNA methylation at the oprm1 promoter in blood and sperm from male opioid addicts. J Opioid Manag. 2011;7:258–264. doi: 10.5055/jom.2011.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Connelly ST, Schmidt BL. Evaluation of pain in patients with oral squamous cell carcinoma. J Pain. 2004;5:505–510. doi: 10.1016/j.jpain.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Dighe SV, Madia PA, Sirohi S, Yoburn BC. Continuous morphine produces more tolerance than intermittent or acute treatment. Pharmacol Biochem Behav. 2009;92:537–542. doi: 10.1016/j.pbb.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Duttaroy A, Kirtman R, Farrell F, Phillips M, Philippe J, Monderson T, Yoburn BC. The effect of cumulative dosing on the analgesic potency of morphine in mice. Pharmacol Biochem Behav. 1997;58:67–71. doi: 10.1016/s0091-3057(96)00463-7. [DOI] [PubMed] [Google Scholar]
- 6.Fischer DJ, Villines D, Kim YO, Epstein JB, Wilkie DJ. Anxiety, depression, and pain: Differences by primary cancer. Support Care Cancer. 2010;18:801–810. doi: 10.1007/s00520-009-0712-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammerlid E, Bjordal K, Ahlner-Elmqvist M, Boysen M, Evensen JF, Biorklund A, Jannert M, Kaasa S, Sullivan M, Westin T. A prospective study of quality of life in head and neck cancer patients. Part I: At diagnosis. Laryngoscope. 2001;111:669–680. doi: 10.1097/00005537-200104000-00021. [DOI] [PubMed] [Google Scholar]
- 8.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 9.Haugen DF, Hjermstad MJ, Hagen N, Caraceni A, Kaasa S. European Palliative Care Research Collaborative (EPCRC): Assessment and classification of cancer breakthrough pain: A systematic literature review. Pain. 2010;149:476–482. doi: 10.1016/j.pain.2010.02.035. [DOI] [PubMed] [Google Scholar]
- 10.Hodder SC, Edwards MJ, Brickley MR, Shepherd JP. Multiattribute utility assessment of outcomes of treatment for head and neck cancer. Br J Cancer. 1997;75:898–902. doi: 10.1038/bjc.1997.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hwang CK, Kim CS, Kim do K, Law PY, Wei LN, Loh HH. Up-regulation of the mu-opioid receptor gene is mediated through chromatin remodeling and transcriptional factors in differentiated neuronal cells. Mol Pharmacol. 2010;78:58–68. doi: 10.1124/mol.110.064311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jagla C, Martus P, Stein C. Peripheral opioid receptor blockade increases postoperative morphine demands–a randomized, double-blind, placebo-controlled trial. Pain. 2014;155:2056–2062. doi: 10.1016/j.pain.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 13.Lee CY, Perez FM, Wang W, Guan X, Zhao X, Fisher JL, Guan Y, Sweitzer SM, Raja SN, Tao YX. Dynamic temporal and spatial regulation of mu opioid receptor expression in primary afferent neurons following spinal nerve injury. Eur J Pain. 2011;15:669–675. doi: 10.1016/j.ejpain.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Z, Xu J, Xu M, Pasternak GW, Pan YX. Morphine regulates expression of mu-opioid receptor mor-1a, an intron-retention carboxyl terminal splice variant of the mu-opioid receptor (oprm1) gene via mir-103/mir-107. Mol Pharmacol. 2014;85:368–380. doi: 10.1124/mol.113.089292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mercadante S. Breakthrough pain in cancer patients: Prevalence, mechanisms and treatment options. Curr Opin Anaesthesiol. 2015;28:559–564. doi: 10.1097/ACO.0000000000000224. [DOI] [PubMed] [Google Scholar]
- 16.Mercadante S, Dardanoni G, Salvaggio L, Armata MG, Agnello A. Monitoring of opioid therapy in advanced cancer pain patients. J Pain Symptom Manage. 1997;13:204–212. doi: 10.1016/s0885-3924(96)00302-8. [DOI] [PubMed] [Google Scholar]
- 17.Mohan S, Davis RL, DeSilva U, Stevens CW. Dual regulation of mu opioid receptors in sk-n-sh neuroblastoma cells by morphine and interleukin-1beta: Evidence for opioid-immune crosstalk. J Neuroimmunol. 2010;227:26–34. doi: 10.1016/j.jneuroim.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen DA, Hamon S, Yuferov V, Jackson C, Ho A, Ott J, Kreek MJ. Ethnic diversity of DNA methylation in the oprm1 promoter region in lymphocytes of heroin addicts. Hum Genet. 2010;127:639–649. doi: 10.1007/s00439-010-0807-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogino S, Odze RD, Kawasaki T, Brahmandam M, Kirkner GJ, Laird PW, Loda M, Fuchs CS. Correlation of pathologic features with CPG island methylator phenotype (CIMP) by quantitative DNA methylation analysis in colorectal carcinoma. Am J Surg Pathol. 2006;30:1175–1183. doi: 10.1097/01.pas.0000213266.84725.d0. [DOI] [PubMed] [Google Scholar]
- 20.Pickering V, Jay Gupta R, Quang P, Jordan RC, Schmidt BL. Effect of peripheral endothelin-1 concentration on carcinoma-induced pain in mice. Eur J Pain. 2008;12:293–300. doi: 10.1016/j.ejpain.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pickering V, Jordan RC, Schmidt BL. Elevated salivary endothelin levels in oral cancer patients–a pilot study. Oral Oncol. 2007;43:37–41. doi: 10.1016/j.oraloncology.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 22.Portenoy RK, Hagen NA. Breakthrough pain: Definition, prevalence and characteristics. Pain. 1990;41:273–281. doi: 10.1016/0304-3959(90)90004-W. [DOI] [PubMed] [Google Scholar]
- 23.Portenoy RK, Sibirceva U, Smout R, Horn S, Connor S, Blum RH, Spence C, Fine PG. Opioid use and survival at the end of life: A survey of a hospice population. J Pain Symptom Manage. 2006;32:532–540. doi: 10.1016/j.jpainsymman.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Ritchie C, Dunn LB, Paul SM, Cooper BA, Skerman H, Merriman JD, Aouizerat B, Alexander K, Yates P, Cataldo J, Miaskowski C. Differences in the symptom experience of older oncology outpatients. J Pain Symptom Manage. 2014;47:697–709. doi: 10.1016/j.jpainsymman.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O’Donnell D, Kieffer BL, Basbaum AI. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell. 2009;137:1148–1159. doi: 10.1016/j.cell.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt BL, Pickering V, Liu S, Quang P, Dolan J, Connelly ST, Jordan RC. Peripheral endothelin a receptor antagonism attenuates carcinoma-induced pain. Eur J Pain. 2007;11:406–414. doi: 10.1016/j.ejpain.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Stafford K, Gomes AB, Shen J, Yoburn BC. Mu-opioid receptor downregulation contributes to opioid tolerance in vivo. Pharmacol Biochem Behav. 2001;69:233–237. doi: 10.1016/s0091-3057(01)00525-1. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki M, Narita M, Hasegawa M, Furuta S, Kawamata T, Ashikawa M, Miyano K, Yanagihara K, Chiwaki F, Ochiya T, Suzuki T, Matoba M, Sasaki H, Uezono Y. Sensation of abdominal pain induced by peritoneal carcinomatosis is accompanied by changes in the expression of substance p and mu-opioid receptors in the spinal cord of mice. Anesthesiology. 2012;117:847–856. doi: 10.1097/ALN.0b013e31826a4ac8. [DOI] [PubMed] [Google Scholar]
- 29.Thomas J, Karver S, Cooney GA, Chamberlain BH, Watt CK, Slatkin NE, Stambler N, Kremer AB, Israel RJ. Methylnaltrexone for opioid-induced constipation in advanced illness. N Engl J Med. 2008;358:2332–2343. doi: 10.1056/NEJMoa0707377. [DOI] [PubMed] [Google Scholar]
- 30.van den Beuken-van Everdingen MH, de Rijke JM, Kessels AG, Schouten HC, van Kleef M, Patijn J. Prevalence of pain in patients with cancer: A systematic review of the past 40 years. Ann Oncol. 2007;18:1437–1449. doi: 10.1093/annonc/mdm056. [DOI] [PubMed] [Google Scholar]
- 31.Viet CT, Dang D, Ye Y, Ono K, Campbell RR, Schmidt BL. Demethylating drugs as novel analgesics for cancer pain. Clin Cancer Res. 2014;20:4882–4893. doi: 10.1158/1078-0432.CCR-14-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viet CT, Schmidt BL. Methylation array analysis of preoperative and postoperative saliva DNA in oral cancer patients. Cancer Epidemiol Biomarkers Prev. 2008;17:3603–3611. doi: 10.1158/1055-9965.EPI-08-0507. [DOI] [PubMed] [Google Scholar]
- 33.Viet CT, Ye Y, Dang D, Lam DK, Achdjian S, Zhang J, Schmidt BL. Re-expression of the methylated EDNRB gene in oral squamous cell carcinoma attenuates cancer-induced pain. Pain. 2011;152:2323–2332. doi: 10.1016/j.pain.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang HB, Zhao B, Zhong YQ, Li KC, Li ZY, Wang Q, Lu YJ, Zhang ZN, He SQ, Zheng HC, Wu SX, Hokfelt TG, Bao L, Zhang X. Coexpression of delta- and mu-opioid receptors in nociceptive sensory neurons. Proc Natl Acad Sci U S A. 2010;107:13117–13122. doi: 10.1073/pnas.1008382107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto J, Kawamata T, Niiyama Y, Omote K, Namiki A. Down-regulation of mu opioid receptor expression within distinct subpopulations of dorsal root ganglion neurons in a murine model of bone cancer pain. Neuroscience. 2008;151:843–853. doi: 10.1016/j.neuroscience.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 36.Yamano S, Viet CT, Dang D, Dai J, Hanatani S, Takayama T, Kasai H, Imamura K, Campbell R, Ye Y, Dolan JC, Kwon WM, Schneider SD, Schmidt BL. Ex vivo nonviral gene delivery of mu-opioid receptor to attenuate cancer-induced pain. Pain. 2017;158:240–251. doi: 10.1097/j.pain.0000000000000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye Y, Dang D, Zhang J, Viet CT, Lam DK, Dolan JC, Gibbs JL, Schmidt BL. Nerve growth factor links oral cancer progression, pain, and cachexia. Mol Cancer Ther. 2011;10:1667–1676. doi: 10.1158/1535-7163.MCT-11-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zylla D, Gourley BL, Vang D, Jackson S, Boatman S, Lindgren B, Kuskowski MA, Le C, Gupta K, Gupta P. Opioid requirement, opioid receptor expression, and clinical outcomes in patients with advanced prostate cancer. Cancer. 2013;119:4103–4110. doi: 10.1002/cncr.28345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.