

Abstract
G1/G2 variants in the Apolipoprotein L1 (APOL1) gene are associated with end stage renal disease (ESRD) in people with African ancestry. Plasma biomarkers may have utility for risk stratification in APOL1 high-risk individuals of African ancestry. To evaluate this, we measured tumor necrosis factor receptor 1/2 (TNFR1/2) and kidney injury molecule-1 (KIM1) in baseline plasma specimens from individuals of African ancestry with high-risk APOL1 genotype. Biomarker association with a composite renal outcome of ESRD or 40% sustained decline in estimated glomerular filtration rate (eGFR) was then determined and then assessed as improvement in area under curve. Among the 498 participants, the median age was 56 years, 67.7% were female and the baseline eGFR was 83.3-ml/min/1.73 m2 with 80 reaching outcome over 5.9 years. TNFR1, TNFR2, and KIM1 at enrollment were independently associated with renal outcome continuously (adjusted hazard ratio 2.0 (95% confidence interval 1.3-3.1); 1.5 (1.2-1.9); and 1.6 (1.3-1.9) per doubling in levels, respectively) or by tertiles. The area under the curve significantly improved from 0.75 with the clinical model to 0.79 with the biomarkerenhanced model. The event rate was 40% with all 3 biomarkers elevated (adjusted odds ratio of 5.3 (2.3-12.0) vs. 17% (adjusted odds ratio 1.8 (0.9-3.6) with 1 or 2 elevated, and 7% with no biomarkers elevated. Thus, plasma concentrations of TNFR1, TNFR2, and KIM1 are independently associated with renal outcome and improve discrimination or reclassification of African ancestry individuals with a high-risk APOL1 genotype and preserve renal function. Elevation of all markers had higher risk of outcome and can assist with better clinical prediction and improved clinical trial efficiency by enriching event rates.
Keywords: APOL1, Biomarkers, Chronic kidney disease, Inflammation
INTRODUCTION
Rates of end stage renal disease (ESRD) are higher among persons with African ancestry (AA) compared to European Americans (EAs) across all baseline estimated glomerular filtration rate (eGFR) levels.1 Genetic admixture studies demonstrated that two distinct alleles in the Apolipoprotein L1 (APOL1) gene on chromosome 22 confer substantially increased risk for a number of kidney diseases in AA, including focal segmental glomerulosclerosis, human immunodeficiency virus-associated nephropathy, and hypertensionattributable kidney disease.2,3,4,5 The APOL1 high-risk genotypes (two copies of the APOL1 renal risk variants; G1/G1; G2/G2 or G1/G2) are associated with increased ESRD risk, chronic kidney disease (CKD) progression,6 eGFR decline,7 and incident CKD.8 Thus, ancestry differences in APOL1 risk prevalence could partly explain disparities in kidney disease between AA’s and EA’s.
While the relative risks for incident or progressive CKD for the APOL1 risk variants are important and highly significant, there is wide variation in the absolute rates of these outcomes.9 Thus, improved methods to further predict those individuals with high-risk APOL1 genotype, who will have kidney disease progression, will be helpful for clinicians, patients and investigators. As plasma biomarkers representing inflammation (soluble tumor necrosis factor receptor 1/2 [TNFR1/2]) and renal tubular injury (kidney injury molecule-1 [KIM1]) significantly improve risk prognostication when added to clinical markers (eGFR and albuminuria) in diabetic patients with both preserved and impaired renal function,10 we sought to explore whether these biomarkers improve risk prognostication for longitudinal renal functional decline in a large cohort of AA individuals with the APOL1 high-risk genotype and preserved baseline renal function.
RESULTS
Baseline Characteristics of Participants
There were follow-up data and biospecimens available on 498 participants in BioMe with two APOL1 risk alleles. Median age was 56 years, 337 (67.6%) were female, and median eGFR was 83.3 ml/min. Of them, 80 (16.1%) reached a renal outcome over 5.9 years. Participants reaching the endpoint, on average, were older (66 vs. 55 years) and had a lower baseline eGFR (75.5 vs. 85.5 ml/min) compared to those without the endpoint. Participants with the renal endpoint also had higher systolic (132.5 vs. 128 mm of Hg), diastolic (80 vs. 76 mm of Hg) and mean arterial pressure (96.8 vs. 93.3 mm of Hg). In addition, they had higher proportion of type 2 diabetes (31.3% vs. 12.7%), hypertension (68.8% vs. 39.5%), coronary artery disease (13.8% vs. 6.7%) and heart failure (8.8% vs 1.9%) and were more likely to be on an ACEI/ARB (36.3 vs. 22.3%). There were no significant differences in baseline BMI, hemoglobin A1c, UACR or urine protein to creatinine ratio. (Table 1)
Table 1.
Baseline Characteristics of AA participants with APOL1 risk variants with and without renal endpoint
| Overall (n=498) | Without Renal Endpoint (n=418) | With Renal Endpoint (n=80) | p | |
|---|---|---|---|---|
| Clinical Characteristics | ||||
| Age in years, Median (IQR) | 56 [46-66] | 55 [45-64] | 66 [58.5-72] | <0.01 |
| Female, n (%) | 337 (67.6) | 282 (67.5) | 55 (68.8) | 0.8 |
| Body Mass Index in kg/m2, Median [IQR] | 30.5 [26.2-35.8] | 30.7 [26.3-35.5] | 30.5 [25.2-35.3] | 0.1 |
| Type 2 diabetes, n (%) | 78 (15.7) | 53 (12.7) | 25 (31.3) | <0.01 |
| Hypertension, n (%) | 220 (44.2) | 165 (39.5) | 55 (68.8) | <0.01 |
| Coronary Artery Disease, n (%) | 39 (7.8) | 28 (6.7) | 11 (13.8) | <0.01 |
| Heart Failure, n (%) | 15 (3) | 8 (1.9) | 7 (8.8) | <0.01 |
| Systolic Blood Pressure in mm Hg, Median [IQR] | 129 [117-140.5] | 128 [115-140] | 132.5 [121.5-146.8] | 0.006 |
| Diastolic Blood Pressure in mm Hg, Median [IQR] | 77 [69.5-84.5] | 76 [69-84] | 80 [72.5-86] | 0.03 |
| Mean Arterial Pressure in mm Hg, Median [IQR] | 93.7 [86-102] | 93.3 [84.7-101.7] | 96.8 [90-104.5] | 0.007 |
| Follow up Time in years, Median [IQR] | 5.9 [3.9-7.1] | 5.9 [3.6-7.0] | 6.9 [5.6-8.4] | <0.01 |
| Laboratory Characteristics | ||||
| Baseline eGFR, Median [IQR} | 83.3 [68.9-99.4] | 85.5 (70.3 - 100.1) | 75.5 (55.2 - 96.3) | 0.05 |
| Baseline Hemoglobin A1C, Median [IQR] | 5.9 [5.5-6.4] | 5.8 (5.5 - 6.4) | 6.0 (5.6 - 7.0) | 0.08 |
| Baseline urine albumin/creatinine, Median [IQR] | 11 [4.5-55] | 10.5 [4-42.3] | 16 [5-206] | 0.09 |
| Medications | ||||
| ACE/ARB, n (%) | 122 (24.5) | 93 (22.3) | 29 (36.3) | 0.01 |
| Plasma Biomarker Concentrations | ||||
| TNFR1, in pg/ml, Median [IQR] | 2465 [1988-3266] | 2394 [1910-3085] | 3110 [2327-4212] | <0.01 |
| TNFR2, in pg/ml, Median [IQR] | 4215 [3234-5654] | 4075 [3142-5328] | 5392 [4008-7913] | <0.01 |
| KIM1, in pg/ml, Median [IQR] | 154 [96-269] | 139 [92-226] | 278 [158-464] | <0.01 |
Values are presented as mean (SD) for normally distributed continuous values, median (interquartile range) for skewed continuous values, and N (%) for categorical values. NA, not applicable or not available. Renal Endpoint is defined as ESRD (defined by the initiation of maintenance dialysis or receipt of kidney transplant) or a sustained (on two or more time intervals ≥3 months apart) decline in eGFR of ≥40% from baseline eGFR
Correlations between Plasma Biomarkers and other risk factors
TNFR1 was positively correlated with age (0.3;p=0.01), hemoglobin A1c (0.18; p=0.02) and UACR (0.22;p=0.02) (Table 2). TNFR1 was also correlated with TNFR2 (0.23;p=0.01) and KIM1 (0.28;p=0.01). KIM1 correlated with UACR (0.28;p=0.01). In addition, TNFR1 correlated with KIM1 (0.28;p=0.01).
Table 2.
Correlation Coefficients of Biomarkers and Clinical Variables
| TNFR1 | TNFR2 | KIM1 | Age | eGFR | HbA1c | UACR | BMI | |
|---|---|---|---|---|---|---|---|---|
| TNFR1 | NA (NA) | 0.23 (0.01) | 0.28 (0.01) | 0.3 (0.01) | - 0.37 (0.01) | 0.18 (0.02) | 0.22 (0.02) | 0.005 (0.94) |
| TNFR2 | 0.23 (0.01) | NA (NA) | 0.05 (0.31) | 0.01 (0.79) | - 0.07 (0.14) | 0.05 (0.36) | 0.03 (0.71) | - 0.008 (0.89) |
| KIM1 | 0.28 (0.01) | 0.05 (0.31) | NA | 0.06 (0.21) | - 0.09 (0.07) | 0.05 (0.33) | 0.28* (0.01) | - 0.009 (0.89) |
Values within parentheses indicate p-values for that pairwise correlation UACR, urine albumin creatinine ratio; HbA1C, hemoglobin A1C; BMI, Body mass Index
Association of Biomarkers with Composite Renal Outcome
Participants with the renal endpoint also had higher levels of TNFR1 (3110 vs. 2394 pg/ml); TNFR2 (5392 vs. 4075 pg/ml) and KIM1 (278 vs. 139 pg/ml), compared with participants without the endpoint. After adjustment for all covariates, including baseline eGFR, log transformed TNFR1, TNFR2 and KIM1 were significantly associated with composite renal endpoint. The adjusted hazard ratio (aHR) for renal endpoint per doubling of biomarker was 2.0 (95% CI 1.3-3.1); 1.5 (95% CI 1.2-1.9) and 1.6 (95% CI 1.3-1.9) for TNFR1, TNFR2 and KIM1, respectively (Table 3). These estimates were significant even after correcting for false discovery rate using the Benjamini-Hochberg procedure.11
Table 3.
Association of Plasma TNFR1, TNFR2, KIM1 with the Renal Outcome in AA with APOL1 risk genotype
| Biomarker | N | N (%) with outcome | Model 1 | Model 2 | Model 3 |
|---|---|---|---|---|---|
| HR (95% CI) | HR (95% CI) | HR (95% CI) | |||
| TNFR1 in pg/ml | |||||
| Continuous (per doubling) | 498 | 80 (16.1) | 2.1 (1.5-3.1) | 2.1 (1.4-3.1) | 2.0 (1.3-3.1) |
| Tertiles | |||||
| ≤2139 pg/ml | 166 | 11 (6.6) | 1.0 (ref) | 1.0 (ref) | 1.0 (ref) |
| 2140-2936 pg/ml | 166 | 24 (14.5) | 2.0 (1.1-4.2) | 1.7 (0.8-3.5) | 1.6 (0.8-3.4) |
| ≥2937pg/ml | 166 | 45 (27.1) | 4.1 (2.1-8.0) | 3.0 (1.5-6.1) | 2.4 (1.1-4.9) |
| TNFR2 in pg/ml | |||||
| Continuous (per doubling) | 498 | 80 | 1.6 (1.4-1.9) | 1.6 (1.3-1.9) | 1.5 (1.2-1.9) |
| Tertiles | |||||
| ≤ 3532 pg/ml | 166 | 15 (9) | 1.0 (ref) | 1.0 (ref) | 1.0 (ref) |
| 2533-5018 pg/ml | 166 | 23 (13.9) | 1.6 (0.8-3.1) | 1.3 (0.7-2.6) | 1.3 (0.7-2.5) |
| ≥5019 pg/ml | 166 | 42 (25.3) | 3.1 (1.7-5.6) | 2.3 (1.2-4.4) | 1.8 (0.9-3.5) |
| KIM1 in pg/ml | |||||
| Continuous (per doubling) | 498 | 80 | 1.8 (1.5-2.1) | 1.7 (1.4-2.1) | 1.6 (1.3-1.9) |
| ≤ 111 pg/ml | 166 | 11 (6.6) | 1.0 (ref) | 1.0 (ref) | 1.0 (ref) |
| 112-215 pg/ml | 166 | 18 (10.8) | 1.8 (0.8-3.8) | 1.4 (0.6-3) | 1.2 (0.5-2.8) |
| ≥216 pg/ml | 166 | 51 (30.7) | 6.3 (3.3-12.1) | 4.1 (1.9-8.4) | 3.1 (1.4-6.8) |
| Variance Inflation Factor | NA | 1.31; 1.32; 1.33 | 1.31; 1.42; 1.43 | ||
Model 1 = unadjusted analysis.
Model 2 = Adjusted for age, sex, baseline eGFR
Model 3 = Model 2 + DM2, HTN, coronary disease, heart failure, baseline MAP and ACEI/ARB use
For TNFR1;
For TNFR2;
For KIM1
When participants were stratified by tertiles of biomarker concentration, the event rates were higher over follow-up for the top tertiles of TNFR1, TNFR2 and KIM1 compared to the bottom two tertiles (Figures 1A, 1B, 1C). On further adjustment for potential confounders, the highest tertiles of TNFR1 and KIM1 were significantly associated with composite renal endpoint. The aHR for renal endpoint for the top tertile was 2.4 (95 % CI 1.1-4.9) for TNFR1 and 3.1 (95% CI 1.4-6.8) for KIM1. The aHR for the top tertile of TNFR2 was 2.3 (95% CI 1.2-4.4) after adjusting for demographics and baseline eGFR, but the lower bound of the 95% CI marginally covered 1 (aHR 1.8; 95% 0.9-3.5) with further adjustment (Table 3).
Figure 1.
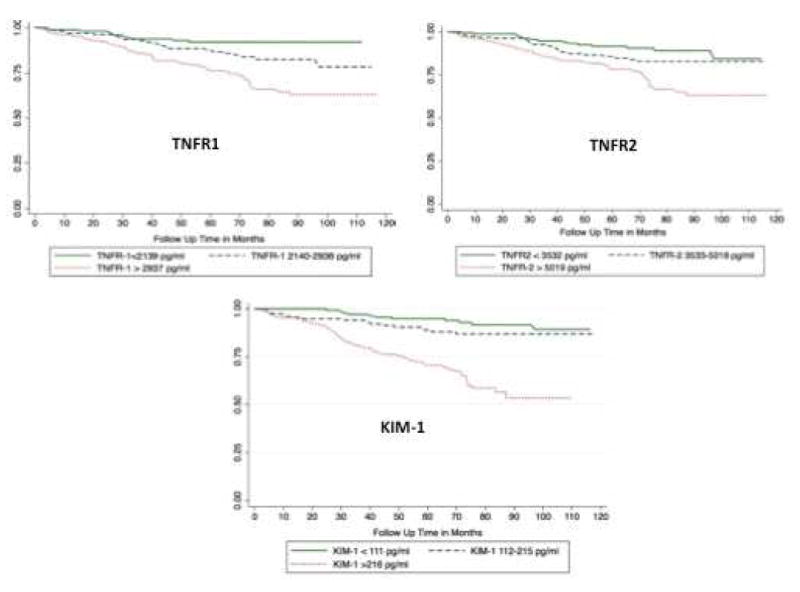
Kaplan Meier estimates of the rates of the composite renal outcome by A. Plasma TNFR1, B. Plasma TNFR2, and C. Plasma KIM1
This figure shows the Kaplan-Meier survival curves for composite renal outcome by biomarker tertiles. The time to event was calculated as time from biomarker measurement to ESRD or 40% sustained decline.
Discrimination with Plasma Biomarkers
The area under the curve (AUC) for the clinical model alone for the composite renal endpoint was 0.75, was 0.77 with the addition of TNFR1 and TNFR2. The clinical model plus KIM1 yielded an AUC of 0.78, and addition of all three biomarkers resulted in an AUC of 0.79. The AIC/BIC did not change with further sequential adjustment (Table 4).
Table 4.
Discrimination and Reclassification of the Biomarkers for the Renal Outcome
| Model | Renal Endpoint | ||
|---|---|---|---|
| AUC (SEM) | Akaike’s Information Criteria | Bayesian Information Criteria | |
| Clinical model alone | 0.75 (0.02) | 381.3 | 422.8 |
| Clinical model with each individual biomarker | |||
| TNFR1 | 0.77 (0.02 | 370 | 416.1 |
| TNFR2 | 0.77 (0.02) | 374.4 | 420 |
| KIM1 | 0.78 (0.02)* | 371.1 | 416.7 |
| Clinical model with all biomarkers | 0.79 (0.02)* | 368.8 | 422.7 |
Clinical Model = age, sex, baseline eGFR, DM2, HTN, coronary disease, heart failure, baseline MAP and ACEI/ARB use
Delong p value < 0.05
The proportion of individuals with 0, 1, 2, or 3 biomarkers in the highest tertile was 47.2, 52.8, 30.7, and 16.5%, respectively. There was a large gradient of risk for the renal endpoint: only 7.2% of participants in the 0 biomarkers group elevated experienced an event compared to 17% (adjusted OR 1.8, 9% CI 0.9-3.6) for 1 or 2 biomarkers elevated, and 40.2% (adjusted odds ratio 5.3, 95% CI 2.3-12.0) in those with all three biomarkers elevated (Figure 2). Participants with all biomarkers elevated comprised 16.5% of the cohort but accounted for 41.3% of events. Similarly, in the participants without composite outcome 52.2% of all participants had no biomarkers elevated compared to 11.7% with all biomarkers elevated (Supplementary Table 1).
Figure 2.
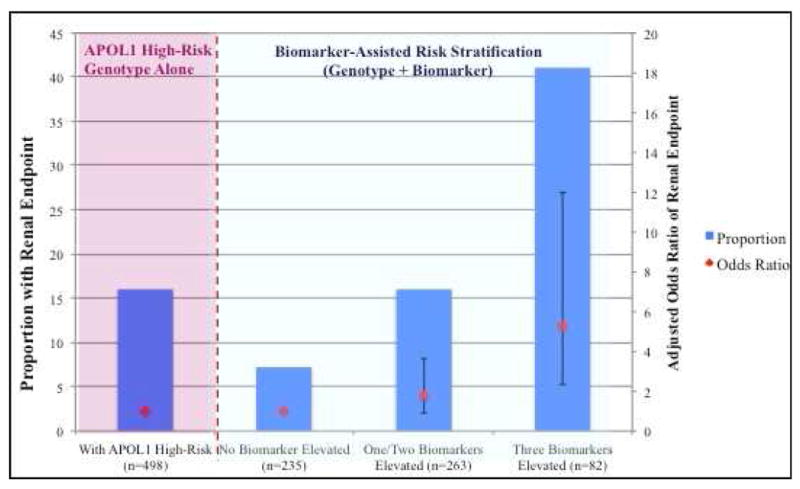
Proportion Reaching the Renal Endpoint Stratified by Number of Elevated Biomarkers
This figure shows the proportion of renal events in three groups: 1) With all biomarker values in bottom two tertiles; 2) With either one/two biomarkers in the top tertile and 3) With all biomarkers in the top tertile. For comparison purposes, the event rate in the cohort with APOL1 high-risk genotype without biomarker stratification is shown on the left
Sensitivity Analyses
In participants with available UACR/UPCR measurements at baseline (n=209), the biomarkers remained significantly associated with renal outcome even after full adjustment for all confounders including UACR or UPCR. The adjusted hazard ratios for the renal endpoint per doubling of biomarker were 2.0 (95% CI 1.2-3.4); 1.5 (95% CI 1.2-2.1) and 1.7 (95% CI 1.3-2.2) for TNFR1, TNFR2 and KIM1, respectively (Supplementary Table 2). In this subgroup, the AUC improved from 0.75 for the clinical model (including UACR) to 0.80 with addition of biomarkers (p<0.01).
In participants with preserved eGFR (eGFR> 60 ml/min/1.73 m2), all biomarkers were associated with composite outcome after adjusting for age, sex and eGFR but the lower bound of the 95% CI for TNFR1 covered 1 after further adjusting for additional risk factors. The AUC improved from 0.72 for clinical model alone to 0.76 with addition of biomarkers (p<0.01). In the subset of participants with the median eGFR> 83-ml/min/1.73 m2 all biomarkers except KIM1 lost significance however the effect sizes were consistently in the same direction. Finally, in the 420 participants without type 2 diabetes at baseline, all biomarkers were strongly associated with outcome in unadjusted and adjusted analyses (Supplementary Table 2).
DISCUSSION
In this study, we demonstrated that three plasma biomarkers with known prognostic utility in patients with diabetic kidney disease, were strongly and independently associated with the risk for the composite renal outcome in this cohort of AA with the APOL1 high-risk genotype and largely preserved eGFR at baseline in the BioMe cohort with approximately 6 years of median follow-up. The biomarkers added to discrimination and provided fairly robust reclassification metrics. Importantly, simple tallying of the number of biomarkers elevated was able to segregate the supposedly “high-risk” genotype participants into low risk for progression (7% with no biomarkers elevated) to very high risk (> 40% probability of progression with all 3 biomarkers elevated). Thus, the approximate 6-fold difference in risk, if validated in other cohorts, could be leveraged to improve risk-prediction for AA with the high-risk APOL1 genotype.
CKD is a public health problem, with few interventions that have been proven to prevent development or slow progression. The genomic revolution that has occurred over the past decade, with falling costs of both genotyping and sequencing has been a boon to many fields in medicine. The discovery of the APOL1 genotype has been one of the most important discoveries in the nephrology field to date. It has renewed focus and vigor and inspired a race toward possible new therapies for CKD in AA. Investigators,7,12,13,14 regulatory and funding agencies, are actively applying massive efforts towards determination of pathogenesis of APOL1, its modifiers, and trying to discover and validate therapeutics to combat APOL1-associated renal disease. There are ongoing clinical trials are ongoing that are testing AA for the APOL1 high-risk genotype and are randomized to “intervention”, which involves reporting of the results to patients and their providers.15
The ever-burgeoning expansion of genetic data from biobanks from various institutions, clinical genetic testing and direct to consumer testing can potentially provide great benefit, but paradoxically, may also cause some harm. First, one’s genotype is very distant from what will ultimately become the final expressed phenotype, especially in the case of complex disease such as kidney disease.16 Second, while knowledge of one’s own genotype may be positive information for some, by inducing lifestyle change or taking their risk factors more seriously.17 In contrast, knowledge of genotype, particularly when risk cannot be modified, may serve to cause anxiety. These fears may be burdensome, despite the fact that the presence of the high-risk genotype does not guarantee the outcome. Thus, further risk stratification in the setting of known high-risk genotype can serve as a powerful tool in the clinic, and for counseling patients.
APOL1 high risk genotypes are the ideal setting in which further risk stratification, via various tools, is needed especially in patients with relatively preserved renal function. In addition, since APOL1 high-risk genotypes account for ethnic differences in renal disease, targeting this very high-risk population may be of benefit in reducing disparities. In our cohort, 16.1% of participants reached the renal outcome. This is lower than AA cohorts with prevalent CKD, where approximately half of the patients with the APOL1 high-risk genotype progressed over 4.4 to 9 years but is comparable to rates of faster renal decline in young AA participants with preserved eGFR with an incidence rate of 15.6/1000 person years.6,18 However, in AA participants with high-risk APOL1 genotype, there was substantial heterogeneity in trajectory of renal deterioration over time.9 This underscores the importance to find additional determinants and predictors of renal progression in this population to enhance the impact of APO1 testing, in targeted populations, and before widespread screening of the black general population is justified.9
Prognostic biomarkers can serve another purpose in this high-risk population. Several investigators and industry sponsors are in the process of testing agents to prevent or treat APOL1 related kidney disease. Better selection of the approximately 5 million of AA (14% of the 37 million AA’s in the US) with APOL1 high-risk genotype would improve the speed and efficiency of determining which novel agents will be effective. For example, if the panel of the three biomarkers (TNFR1, TNFR2, KIM1) with an AUC of 0.79 for predicting renal endpoints, was added as an enrichment criteria for a new trial, feasibility would be improved through enhanced selection of those patients most at risk for progression. With an event rate of 40% in the group with all three biomarkers elevated, compared with an event rate of 16% in a non-enriched cohort with APOL1 high risk genotype, the sample size needed would be reduced by over 50%, resulting in substantial cost savings and ultimately, shorter, more-efficient trials (www.prognosticenrichment.com).19
Finally, another area where these biomarkers could be tested is the field of renal transplantation. Kidney transplant recipients, who receive APOL1 high-risk kidneys from deceased donors, have worse graft outcomes.20 These biomarkers could be involved in pre-transplantation decision making as well as posttransplantation monitoring for graft outcomes. The APOL1 Long-term Kidney Transplantation Outcomes Network (APOLLO) consortium, which has been formed to study APOL1 in kidney transplantation, is expected to assess the utility of these and other biomarkers in renal transplantation.21
There are several limitations of our study. First, 60% lacked baseline UACR/UPCR measurement in this cohort, however this is likely due to preserved renal function at baseline. However, it should be noted that in the 209 with UACR/UPCR available at the time of enrollment, the independent association between the biomarkers and outcomes was maintained despite adjustment for albuminuria or proteinuria as a key confounder. Second, due to the fact that we used EMR-based lab data, there was potential for ascertainment bias for the renal outcome in those with more comorbidities or worse kidney function. However, the median number of creatinine values by biomarker tertile was relatively equal and the range of creatinine values was 31-99, and range of follow-up was 3.9-7.1 years in the cohort. We also mitigated some ascertainment bias for the renal endpoint via linkage with USRDS. Third, the three plasma markers we measured are not specific for APOL1-mediated pathophysiology of CKD progression. Rather, these markers seem to segregate patients at risk for GFR decline in type 1 DM, 22-24type 2 DM,10,25,26 lupus nephritis,27 general population,28 and, for the first time in the literature, those with high-risk APOL1 genotypes. While the median eGFR of our cohort was relatively normal at 83 ml/min/1.73 m2, some participants had renal function that would not be considered normal, or had albuminuria, indicating presence of CKD. Despite the size of the cohort with double risk alleles, we still lacked sufficient statistical power to test the association of these biomarkers in persons with completely normal renal function. Further studies should focus on associations of these biomarkers in APOL1 high-risk patients with higher eGFR levels and in the absence of proteinuria in order to discern whether these biomarkers still have predictive power in patients with normal kidney function.
In conclusion, plasma TNFR1, TNFR2 and KIM1 are independently associated with renal outcomes in AA with high-risk APOL1 genotype and improve risk discrimination. These markers can be valuable for risk stratification in AA with APOL1 risk genotype in observational cohort studies, for enrichment of clinical trials, for assessing outcomes in renal transplantation, and ultimately, for clinical-decision making.
METHODS
Study Participants
Study participants were recruited from the BioMe Biobank Program of The Charles Bronfman Institute for Personalized Medicine at the Icahn School of Medicine at Mount Sinai (ISMMS) from 2007 to 2017. The BioMe Biobank is an Institutional Review Board (IRB)- approved, a consented electronic medical record (EMR)-linked medical care setting biorepository in an ancestrally diverse local community of upper Manhattan.29,30 BioMe operations are fully integrated in clinical care processes, including direct recruitment from over 30 broadly selected clinical sites’ waiting areas and phlebotomy stations by dedicated recruiters. For the purpose of this study, BioMe participants with the APOL1 high-risk genotype were analyzed. The Mount Sinai Institutional Review Board approved this study.
Study Design
This was a retrospective cohort study with EMR clinical data linked to biomarker and genetic data.
APOL1 genotyping
We genotyped AA BioMe participants using direct genotyping to determine APOL1 ancestral (G0), G1 and G2 allele status.31 To validate this genotyping method, we performed intra- and inter- assay variation studies that include 48 positive and 10 negative control samples. Sanger sequencing was used to confirm all of genotypes. Among 58 representative samples with all four haplotypes on G1 and G2 loci, the Sanger sequencing results were in complete agreement with the APOL1 direct genotyping results.
Clinical Data
Age, gender, and AA race were obtained from an enrollment questionnaire administered to BioMe participants. We extracted clinical data for all continuous variables (serum creatinine, hemoglobin A1c, urine protein or albumin to creatinine ratios), from the EHR with concurrent time stamps. We defined the baseline period as 1 year before the BioMe enrollment date. We determined eGFR using the CKD-EPI creatinine equation,32 calculated median values per 3 month period of follow up and utilized these for covariate and outcome ascertainment. Body mass indices (BMI) were calculated as the ratio between weight and the square of height in kg/m2. Hypertension and type 2 diabetes status at baseline was determined using the Electronic Medical Records and Genomics (eMERGE) Network phenotyping algorithms.33 Coronary artery disease and heart failure were determined by a validated algorithm and ICD-9/10 codes respectively. We considered a participant to be on an angiotensin converting enzyme-inhibitor (ACE-i) or angiotensin receptor blocker (ARB) if they had a concurrent prescription during the BioMe enrollment. We calculated follow up time from BioMe enrollment date to latest visit in the EHR.
Biomarker Measurements
Plasma samples taken at the time of BioMe enrollment and stored at −80°C were used to derive the baseline biomarker measures. Plasma concentrations of TNFR1, TNFR2, and KIM1 were measured via prototype cytokine arrays from Mesoscale Diagnostics (Meso Scale Discovery, Gaithersburg, MD). The intra- and inter-assay CVs for the quality control samples were 3.5%, 3.9%, and 4.5%, and 12.4%, 10.8%, and 7.7%, for TNFR1, TNFR2, and KIM1, respectively. The average lower limit of detection obtained from multiple runs was 12.5 pg/ml for TNFR1, 7.8 pg/ml for TNFR2, and 9.0 pg/ml for KIM1. The laboratory personnel performing the biomarker assays were blinded to clinical information about the participants.
Outcomes
We defined the primary outcome as a composite of ESRD or a sustained 40% decline in eGFR over the follow up period. We defined ESRD status as requirement for dialysis or transplant and ascertained it by linkage to the United States Renal Data System (USRDS). We defined the sustained 40% decline was defined as a decrease in eGFR by 40% from baseline on two or more separate 3-month intervals.
Statistical Analyses
We expressed descriptive results for the participant baseline characteristics and biomarkers as either mean with standard deviation (SD) or as median with interquartile range depending on skewness. We made univariable statistical comparisons between groups by t tests for data that were normally distributed, Wilcoxon tests for skewed continuous data, and chi-squared tests for categorical data. We assessed correlation between the markers and with baseline characteristics using Pearson’s partial correlations. The association of each biomarker with the composite renal endpoint was evaluated both continuously (log base 2–transformed) and by biomarker tertile. We utilized Cox regression with a time-to-event analysis where the composite renal outcome occurring any time during the follow-up was counted as an event, and otherwise counted as a non-event. We computed time to event as the time to first dialysis/transplant for ESRD or the time to the first episode of eGFR decline for the 40% sustained decline. For participants without an event, the follow-up time was computed until the end of follow up. Sequential adjustment was employed to assess the independent association of the biomarkers with the composite end point. We first evaluated unadjusted associations by including only individual biomarkers. We then calculated adjusted association after adjusting for age, sex, baseline eGFR, BMI diabetes, hypertension, heart failure, baseline mean arterial pressure and ACEI/ARB use. To ensure goodness of fit for the multivariable models, we calculated Akaike’s Information Criteria (AIC) and Bayesian information criteria (BIC). Finally, to evaluate multicollinearity, we calculated the variance inflation factor (VIF) for the entire model.
We calculated the proportion of events by groups according to number of biomarkers elevated. Our reference group was zero biomarkers elevated (participants with all biomarker values in bottom two tertiles). We compared event rates in one/two biomarker elevated (one/two biomarkers in top tertile) vs. all three biomarkers elevated (all three markers in top tertile). We then calculated adjusted odds ratios for these groups compared to the reference group.
We also conducted four sensitivity analyses. First, we assessed association of biomarkers with renal outcome in the subset of participants with non-missing baseline urine albuminuria/proteinuria measurements. We also analyzed only that subset of participants without stage 3 CKD at baseline (defined as baseline eGFR<60 ml/min/1.73 m2), and participants without type 2 diabetes at baseline.
We used area under the curve (AUC) to evaluate biomarker discriminative performance. We calculated the AUC of a clinical model comprising of demographics, baseline eGFR, comorbidities and medication use.34 We then calculated the AUC using the clinical model, clinical model with biomarkers one at a time and the clinical mode with all biomarkers. We then repeated this for all analyses and used the Delong test to evaluate the significance of improvements in AUC. We bootstrapped the AUC with 1000 iterations with resampling to adjust for optimism bias.
We considered two-sided p values of <0.05 with adjustment for false discovery rate using the Benjamini-Hochberg procedure to indicate statistical significance. All analyses were performed using STATA version 13 (College Station, TX).
Supplementary Material
Participants with and without composite outcome by number of biomarkers elevated
Association of Plasma TNFR1, TNFR2, KIM1 with the Renal Outcome in AA with APOL1 risk genotype in Subgroups A. with Available UACR/UPCR and B. with Preserved eGFR (> 60 ml/min/1.73 m2 ) C. In participants without type 2 diabetes at baseline.
Acknowledgments
The BioMe health care delivery cohort at Mount Sinai was established and maintained with a generous gift from the Andrea and Charles Bronfman Philanthropies. The authors are fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and approved the final version.
Funding: This research was supported by a grant from the National Institute of Diabetes Digestive and Kidney Diseases (grant no. R01DK096549 to S.G.C). G.N.N is supported by a career development award from the NIH (K23DK107908). C.R.P. is supported by the NIH (K24-DK090203) and P30-DK079310-07 O’Brien Center Grant. S.G.C, G.N.N. and C.R.P are members and are supported in part by the Chronic Kidney Disease Biomarker Consortium (1U01DK106962-01). C.R.H is supported by a grant from the National Human Genome Research Institute (grant no. U01HG007278) to conduct the genetic testing to understand and address renal disease disparities (GUARDD) randomized controlled trial. The BioMe health care delivery cohort at Mount Sinai was established and maintained with a generous gift from the Andrea and Charles Bronfman Philanthropies.
Footnotes
Disclosure:
S.G.C has received consulting fees from Goldfinch Bio, Janssen Pharmaceuticals and is on the scientific advisory board of pulseData LLC. G.N.N has received operational funding from Goldfinch Bio and is on the scientific advisory board of pulseData LLC.
Supplementary Material
Supplementary information is available at KI Report’s website. Supplementary material is uploaded as a PDF file.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Choi AI, Rodriguez RA, Bacchetti P, et al. White/Black Racial Differences in Risk of End-Stage Renal Disease and Death. Am J Med. 2009;122:672–678. doi: 10.1016/j.amjmed.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lipkowitz MS, Freedman BI, Langefeld CD, et al. Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int. 2013;83:114–120. doi: 10.1038/ki.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol JASN. 2011;22:2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tzur S, Rosset S, Shemer R, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet. 2010;128:345–350. doi: 10.1007/s00439-010-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parsa A, Kao WHL, Xie D, et al. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med. 2013;369:2183–2196. doi: 10.1056/NEJMoa1310345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayek SS, Koh KH, Grams ME, et al. A tripartite complex of suPAR, APOL1 risk variants and αvβ3 integrin on podocytes mediates chronic kidney disease. Nat Med. 2017 doi: 10.1038/nm.4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foster MC, Coresh J, Fornage M, et al. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol JASN. 2013;24:1484–1491. doi: 10.1681/ASN.2013010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grams ME, Rebholz CM, Chen Y, et al. Race, APOL1 Risk, and eGFR Decline in the General Population. J Am Soc Nephrol JASN. 2016;27:2842–2850. doi: 10.1681/ASN.2015070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coca SG, Nadkarni GN, Huang Y, et al. Plasma Biomarkers and Kidney Function Decline in Early and Established Diabetic Kidney Disease. J Am Soc Nephrol JASN. 2017 doi: 10.1681/ASN.2016101101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9:811–818. doi: 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- 12.Beckerman P, Bi-Karchin J, Park ASD, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med. 2017;23:429–438. doi: 10.1038/nm.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olabisi OA, Zhang J-Y, VerPlank L, et al. APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinases. Proc Natl Acad Sci U S A. 2016;113:830–837. doi: 10.1073/pnas.1522913113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lan X, Jhaveri A, Cheng K, et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol. 2014;307:F326–336. doi: 10.1152/ajprenal.00647.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horowitz CR, Abul-Husn NS, Ellis S, et al. Determining the effects and challenges of incorporating genetic testing into primary care management of hypertensive patients with African ancestry. Contemp Clin Trials. 2016;47:101–108. doi: 10.1016/j.cct.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horowitz CR, Ferryman K, Negron R, et al. Race, Genomics and Chronic Disease: What Patients with African Ancestry Have to Say. J Health Care Poor Underserved. 2017;28:248–260. doi: 10.1353/hpu.2017.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peralta CA, Bibbins-Domingo K, Vittinghoff E, et al. APOL1 Genotype and Race Differences in Incident Albuminuria and Renal Function Decline. J Am Soc Nephrol JASN. 2016;27:887–893. doi: 10.1681/ASN.2015020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kerr KF, Roth J, Zhu K, et al. Evaluating biomarkers for prognostic enrichment of clinical trials. Clin Trials Lond Engl. 2017 doi: 10.1177/1740774517723588. 1740774517723588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freedman BI, Locke JE, Reeves-Daniel AM, et al. Apolipoprotein L1 Gene Effects on Kidney Transplantation. Semin Nephrol. 2017;37:530–537. doi: 10.1016/j.semnephrol.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.APOL1 Long-term Kidney Transplantation Outcomes Network (APOLLO) Clinical Centers (Collaborative U01) RFA-DK-16-025. [Google Scholar]
- 22.Gohda T, Niewczas MA, Ficociello LH, et al. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J Am Soc Nephrol JASN. 2012;23:516–524. doi: 10.1681/ASN.2011060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowak N, Skupien J, Niewczas MA, et al. Increased plasma kidney injury molecule-1 suggests early progressive renal decline in non-proteinuric patients with type 1 diabetes. Kidney Int. 2016;89:459–467. doi: 10.1038/ki.2015.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabbisetti VS, Waikar SS, Antoine DJ, et al. Blood kidney injury molecule-1 is a biomarker of acute and chronic kidney injury and predicts progression to ESRD in type I diabetes. J Am Soc Nephrol JASN. 2014;25:2177–2186. doi: 10.1681/ASN.2013070758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pavkov ME, Nelson RG, Knowler WC, et al. Elevation of circulating TNF receptors 1 and 2 increases the risk of end-stage renal disease in American Indians with type 2 diabetes. Kidney Int. 2015;87:812–819. doi: 10.1038/ki.2014.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niewczas MA, Gohda T, Skupien J, et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J Am Soc Nephrol JASN. 2012;23:507–515. doi: 10.1681/ASN.2011060627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parodis I, Ding H, Zickert A, et al. Serum soluble tumour necrosis factor receptor-2 (sTNFR2) as a biomarker of kidney tissue damage and long-term renal outcome in lupus nephritis. Scand J Rheumatol. 2016:1–10. doi: 10.1080/03009742.2016.1231339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carlsson AC, Nordquist L, Larsson TE, et al. Soluble Tumor Necrosis Factor Receptor 1 Is Associated with Glomerular Filtration Rate Progression and Incidence of Chronic Kidney Disease in Two Community-Based Cohorts of Elderly Individuals. Cardiorenal Med. 2015;5:278–288. doi: 10.1159/000435863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tayo BO, Teil M, Tong L, et al. Genetic Background of Patients from a University Medical Center in Manhattan: Implications for Personalized Medicine. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nadkarni GN, Galarneau G, Ellis SB, et al. Apolipoprotein L1 Variants and Blood Pressure Traits in African Americans. J Am Coll Cardiol. 2017;69:1564–1574. doi: 10.1016/j.jacc.2017.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Fedick A, Wasserman S, et al. Analytical Validation of a Personalized Medicine APOL1 Genotyping Assay for Nondiabetic Chronic Kidney Disease Risk Assessment. J Mol Diagn JMD. 2016;18:260–266. doi: 10.1016/j.jmoldx.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nadkarni GN, Gottesman O, Linneman JG, et al. Development and validation of an electronic phenotyping algorithm for chronic kidney disease. AMIA Annu Symp Proc AMIA Symp. 2014;2014:907–916. [PMC free article] [PubMed] [Google Scholar]
- 34.Tangri N, Grams ME, Levey AS, et al. Multinational Assessment of Accuracy of Equations for Predicting Risk of Kidney Failure: A Meta-analysis. JAMA. 2016;315:164–174. doi: 10.1001/jama.2015.18202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Participants with and without composite outcome by number of biomarkers elevated
Association of Plasma TNFR1, TNFR2, KIM1 with the Renal Outcome in AA with APOL1 risk genotype in Subgroups A. with Available UACR/UPCR and B. with Preserved eGFR (> 60 ml/min/1.73 m2 ) C. In participants without type 2 diabetes at baseline.