

Abstract
We examined whether the loss of transient receptor potential ankyrin 1 (TRPA1), an irritant-sensing ion channel, or TRPA1 antagonist treatment affects the severity inflammation and scarring during tissue wound healing in a mouse cornea injury model. In addition, the effects of the absence of TRPA1 on transforming growth factor β1 (TGF-β1)-signaling activation were studied in cell culture. The lack of TRPA1 in cultured ocular fibroblasts attenuated expression of TGF-β1, interleukin-6, and α-smooth muscle actin, a myofibroblast the marker, but suppressed the activation of Smad3, p38 MAPK, ERK, and JNK. Stroma of the healing corneas of TRPA1 −/− knockout (KO) mice appeared more transparent compared with those of wild-type mice post-alkali burn. Eye globe diameters were measured from photographs. An examination of the corneal surface and eye globes suggested the loss of TRPA1 suppressed post-alkali burn inflammation and fibrosis/scarring, which was confirmed by histology, immunohistochemistry, and gene expression analysis. Reciprocal bone marrow transplantation between mice showed that KO corneal tissue resident cells, but not KO bone marrow-derived cells, are responsible for KO mouse wound healing with reduced inflammation and fibrosis. Systemic TRPA1 antagonists reproduced the KO phenotype of healing. In conclusion, a loss or blocking of TRPA1 in mice reduces inflammation and fibrosis/scarring in the corneal stroma during wound healing following an alkali burn. The responsible mechanism may include the inhibition of TGF-β1-signaling cascades in fibroblasts by attenuated TRPA1 signaling. Inflammatory cells are considered to have a minimum involvement in the exhibition of the KO phenotype after injury.
Tissue damage evokes a local inflammatory response that promotes tissue repair. However, excess inflammation and subsequent tissue fibrosis/scarring impairs the physiological function of tissues and organs. Although growth factors and cytokines are involved in inflammatory tissue fibrosis, transforming growth factor β (TGF-β) is one of the primary determinants. TGF-β activates mitogen-activated protein kinase (MAPK) and other kinase cascades, ie, c-Jun N-terminal kinase (JNK) and p38 MAPK. Like other growth factors, Smad signaling is the major signaling stream.1,2 Smad2 or Smad3 is phosphorylated upon TGF-β binding to the cell surface receptor, and subsequently forms a complex with common Smad (Smad4) and translocates to the nuclei to regulate gene expression.3
The cornea is an avascular, transparent tissue located at the outermost part of the eye. Its surface is covered with non-keratinizing stratified epithelium, underlying connective tissue stroma with a mesenchymal cell linage, a keratocyte, and sensory nerve fibers. Ocular trauma resulting from a corneal alkali burn is a serious clinical problem, and may cause severe and permanent vision impairment by inducing tissue inflammation and fibrosis/scarring that can disrupt the transparency of the cornea and alter its regular curvature.4 Invasion of inflammatory cells (eg, macrophages and poly-morphonuclear neutrophilis (PMNs)), keratocyte activation, myofibroblast formation, and subsequent tissue fibrosis/scarring are all involved in the wound healing response or accumulation of fibrotic/scarring tissue in an alkali-burned cornea.5,6 Despite aggressive treatment with anti-inflammatory drugs and surgery, attempts to restore vision fail.4,7,8 This limitation is the basis for efforts to develop new and more effective, prevention and treatment strategies. Our previous studies showed that the gene ablation of Smad3 exhibits strong anti-inflammatory and anti-fibrotic effects in mice with alkali burns of the corneal stroma.9–11 This favorable effect was reproduced by topical adenoviral gene transfer of Smad7 (the inhibitory Smad),9 Id2/3, bone morphogenic protein-7,12 or PPARg (all of which reportedly attenuates TGF-β/Smad signaling) mainly to corneal stromal (mesenchymal) cells in the early phases of healing, before the robust invasion of inflammatory cells.13
Transient receptor potential (TRP) channels are polymodal receptors that mediate sensory transduction and are activated by a host of stimuli. The TRP superfamily includes 28 genes that are subdivided into seven subfamilies.14 Among them, TRPA1 is highly expressed in several neural tissues and is activated by many pungent or irritant compounds, eg, chemicals with alkaline pHs, which can induce acute pain, hyperalgesia, and neurogenic inflammation.15–26 Although TRPA1 expression also occurs in other cells types, its function in neurons should be investigated. In addition to its functions in the nervous system, TRPA1 participates in the control of local tissue inflammation in animal models. For example, blocking TRPA1 with an inhibitor suppresses acute airway responses to chemical exposures.15,18,27 Experimental chemical colitis was reduced in TRPA1-deficient mice and using a pharmacological inhibitor of TRPA1.28
Carrageenan-induced paw edema was also suppressed by TRPA1. We previously reported in mice that TRP vanilloid 1 (TRPV1), a TRP family member, co-expressed with TRPA1, is upregulated in the keratocytes of an alkali-burned cornea and that TRPV1 gene ablation markedly suppresses tissue inflammatory fibrosis and improved the final healing outcome in the cornea following an alkali burn.29
The present study was undertaken to elucidate the role of TRPA1 activation by corneal alkali burn in promoting inflammation and fibrosis/scarring during wound healing. TRPA1 signaling reportedly modulates tissue inflammation via the expression of substance P or calcitonin gene-related peptide. Nevertheless, the extent to which TRPA1 gene ablation affects TGF-β expression and activates TGF-β-induced signaling cascades should be investigated, because TGF-β/Smad signaling has a critical role in regulating the inflammatory fibrotic consequences in a mouse corneal stroma. It is also important to clarify whether TRPA1 and TRPV1 expression affect each other in order to better understand the inflammatory and fibrogenic effects of TRPV1 that we previously reported to be mediated by TRPA1. The present results show that TRPA1 is required for a complete activation of Smad3, p38 MAPK, ERK, and JNK in response to TGF-β1exposure in vitro and that loss of TRPA1 expression or blocking its activation suppressed severe and persistent corneal inflammation and fibrosis/scarring independently from TRPV1, resulting in a marked improvement in the restoration of tissue transparency. Therefore, we hypothesized that TRPA1 gene ablation in mice may mimic the Smad3-null phenotype in the process of inflammation and tissue fibrosis in corneal stroma during post-alkali burn healing. In contrast, the loss of TRPA1 may suppress excess inflammation and tissue fibrosis in an alkali-burned cornea. To explore this hypothesis, we examined corneal alkali burns in KO mice. The present finding adds new knowledge regarding the mechanisms of TRPA1 involvement in inflammation, tissue repair, and fibrosis/scarring.
MATERIALS AND METHODS
Experimental protocols and the use of experimental mice were approved by the DNA Recombination Experiment Committee and the Animal Care and Use Committee of Wakayama Medical University and conducted in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. Wild-type (WT) C57BL/6 mice and TRPA1 −/− knockout (KO) mice (Jackson Laboratory, Bar Harbor, ME, USA) of the same strain were used in this study.
Western Blotting Analysis of Smad3, p38 MAPK, ERK, and JNK Signaling upon Exposure to TGF-β1
The eye shells (including the cornea and sclera) of WT and KO mice postnatal day 1 were minced and explanted in a 60-mm culture dish (Falcon, USA) to promote the outgrowth of ocular fibroblasts. Primary cultured cells without passage were used for the experiments. Cells were seeded on to a 60-mm culture dish, grown to confluence, and treated with recombinant human TGF-β1 (1.0 ng/ml; R&D Systems, Minneapolis, MN, USA) or vehicle control in the medium. To semi-quantify the expression levels of phospho-Smad3/Smad3, phospho-p38 MAPK/p38 MAPK, phospho-extracellular signal-regulated kinase (ERK)/ERK and phosho-JNK/JNK, we performed western blotting as previously reported.12,30 In brief, the cells were harvested in Sigma mammalian cell lysis buffer (100 μl/dish) and processed for SDS-polyacrylamide gel electrophoresis and western blotting for Smad3 (1:2000; Rockland, Gilbertsville, PA, USA), phospho-Smad3 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), p38 MAPK (1:1000; Cell Signaling, Boston, MA, USA), Phospho-p38 MAPK (1:1000; Cell Signaling, Boston, MA, USA), ERK (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho-ERK 1:2000; Cell Signaling, Boston, MA, USA), JNK (1:1000; Cell Signaling, Boston, MA, USA), and phospho-JNK(1:2000; Cell Signaling, Boston, MA, USA), as previously reported.12,30 The peak height or area was measured for quantitation (ATTO’s Densitometry Software), and the membrane was then stripped and re-stained for β-actin.
Gene Expression in Ocular Fibroblasts
Total RNA was extracted from the WT or KO cells cultured with or without 24-hr exposure to TGF-β1 (1.0 ng/ml) using a Sigma RNA extraction kit (St Louis, USA) according to the manufacturer’s protocol. The total RNA was subjected to real-time reverse transcription-PCR (RT-PCR) to determine the expression levels of TRPA1, TGF-β1, IL-6, and α-SMA following previously reported procedures. Real-time RT-PCR was performed with the TaqMan one-step RT-PCR master mix reagent kit and the Applied Biosystems Prism7300 (PE Applied Biosystems, Foster City, CA, USA). The primers and oligonucleotide probes used in this study, which are listed in Table 1, were designed according to the cDNA sequences in the GenBank database, using Primers Express software (PE Applied Biosystems, Foster City, CA, USA). Five dishes were prepared for each condition. Data were then statistically analyzed using the Mann–Whitney U test.
Table 1.
Primers and oligonucleotide probes used in this study
| α-SMA | Mm01204962_gh |
| F4/80 | Mm00802524-ml |
| MPO | Mm01298422_gl |
| Collagen 1a1 | Mm00801666_gl |
| TGF-β1 | Mm03024053_ml |
| TRPV1 | Mm01246282_ml |
| TRPA1 | Mm01227437_ml |
| VEGF | Mm01281447_ml |
| SP | Mm01166996-ml |
| IL-6 | Mm01210732_gl |
| GAPDH | Mm03302249_gl |
Expression of TRPA1 and TRPV1 in Corneas of TRPA1 −/− and TRPV1 −/− Mice
TRPV1 and TRPA1 expression in the mouse cornea was evaluated with immunohistochemistry and real-time RT-PCR, because these two cation channel receptors are co-expressed in many tissues. Intact eyes from WT, KO, and TRPV1-null mice of C57BL/6 background were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 24 h, embedded in paraffin, and then processed for histology. Paraffin sections (5 μm thick) were subjected to immunohistochemistry for TRPA1 or TRPV1 as previously reported. Rabbit polyclonal anti-TRPA1 antibody (1:200; Osenses, SA, Australia) and polyclonal anti-TRPV1 antibody (1:500; Neuromics, Edina, MN, USA) were diluted in phosphate-buffered saline (PBS). Real-time RT-PCR for TRPV1 and TRPA1 mRNAs was performed as previously reported. Total RNA was extracted from the corneal tissue excised from eight mice eyes using a Sigma RNA extraction kit (St. Louis, USA) and then processed using real-time RT-PCR. The primers and oligonucleotide probes used are listed in Table 1. Data at each time point were analyzed for significance by using the Mann–Whitney U test.
Alkali Burn in Mice Eyes
In total, 3 μl of 1 N sodium hydroxide solution was applied o the right eye of 6- to 8-week-old KO (n = 98) or WT (n = 98) mice under general anesthesia to produce an ocular surface alkali burn. A detailed description of this procedure is reported previously.31,32 Ofloxacin ointment was topically administered twice a week to reduce the risk of bacterial infection. Eyes with obvious bacterial infection were excluded from the study. Eye globe diameters were measured for each eye from photographs obtained under a microscope. The corneal tissue was then processed for histology, immunohistochemistry, or real-time RT-PCR on days 5, 10, and 20 after alkali burn using previously reported procedures.31,32
Bone Marrow Transplantation (BMT) and Ocular Alkali Burn
Reciprocal BMT was performed using a previously reported technique.29 Briefly, bone marrow cells (BM cells) were obtained by flushing the tibia and femur of KO and WT mice with PBS. A total of 2 × 106 WT BM cells were transplanted via tail vein infusion into recipient mice that had received whole-body irradiation of 12 Gy prior to BMT (from WT mice to KO mice or vice versa). Each mouse was subjected an alkali burn on the right eyes 3 weeks after BMT. A total of 10 and 20 days following BMT, mice were killed and the excised corneas were subjected to histology and immunohistochemistry examination. The repopulation of the transplanted BM cells was confirmed by RT-PCR detection of TRPA1 mRNA in the spleens of transplanted mice (data not shown).
Treatment of Corneal Alkali Burn in WT Mice with Systemic TRPA1 Antagonists
We determined whether the KO phenotype is reproduced in WT mice (n = 24) with an intraperitoneal injection of a TRPA1 antagonist after a corneal alkali burn. The antagonist, HC-030031 (100 mg/kg in PBS, daily) or its vehicle was administered daily until euthanasia. Ofloxacin ointment was topically administered twice a week to reduce the risk of bacterial infection. Eyes that developed an infection were excluded from the study. The eyes were then processed for histology or immunohistochemistry at days 5, 10, and 20 after alkali burning.
Histology and Immunohistochemistry for Analysis of Tissue Inflammation and Fibrosis
Paraffin sections (5 μm) were processed for hematoxylin and eosin (HE) staining and immunohistochemistry after measuring each eyeball as previously reported.32 The following antibodies were diluted in PBS: rabbit polyclonal anti-TRPA1 antibody, rabbit polyclonal anti-TRPV1 antibody, and mouse monoclonal anti-α smooth muscle actin (α-SMA) antibody (1:200; Neomarker, Fremont, CA, USA). The presence of monocytes and macrophages was examined using rat monoclonal F4/80 anti-macrophage antigen–antibody (1:200; BMA Biomedicals, August, Switzerland). The presence of PMN was examined using rabbit polyclonal myeloperoxidase (MPO) antibody (1:200; Neomarker, Fremont, CA, USA). We used Vevtastain ABC system (Vector Laboratories, Burlingame, CA, USA). Immunohistochemistry for TGF-β1 was performed as previously described.31,33 The antibody used to detect TGF-β1 only detects the active form of TGF-β1, but does not react with the latent form. Negative control staining was performed by omission of each primary antibody, which did not yield specific staining (data not shown).
Real-Time RT-PCR for Analysis of Tissue Inflammation and Fibrosis
Total RNA was extracted from corneal tissue excised from four burned mice eyes and then processed for real-time RT-PCR for MPO, F4/80, α-SMA, TGF-β1, IL-6, vascular endothelial growth factor (VEGF), collagen Ia1, and substance P (SP). Data collected at each time point were analyzed for significance using the Mann–Whitney U test.
RESULTS
Western Blot Analysis of TGF-β-Induced Activation of Smad3, p38 MAPK, ERK, and JNK in WT and KO Ocular Fibroblasts
Adding TGF-β1 to the culture medium upregulated Smad3 phosphorylation in 15 min, and even at 300 min phospho-Smad3 was detected in cultured ocular fibroblasts. Similarly, adding TGF-β1 to the culture medium upregulated the phosphorylation of p38 MAPK and ERK in 30 min, and even at 300 min phospho-p38 MAPK and phosphor-ERK were detected in the cultures ocular fibroblasts. Moreover, phosphorylation of JNK in 15 min, and even at 300 min phospho-JNK was detected in the cultured ocular fibroblasts. The loss of TRPA1 in the cells overall attenuated TGF-β1-induced phosphorylation of Smad3, p38 MAPK, ERK, and JNK (Figure 1).
Figure 1.

Western blot analysis of Smad3, p38 MAPK, ERK, and JNK signaling upon exposure to TGF-β1. Adding transforming growth factor-β1 (TGF-β1) to the culture medium upregulated the phosphorylation of Smad3 in 15 min, and even at 300 min phospho-Smad3 was detected in cultured ocular fibroblasts. Similarly, adding TGF-β1 to the culture medium upregulated the phosphorylation of p38 MAPK and extracellular signal-regulated kinase (ERK) in 30 min, and phosphor-p38 MAPK and phospho-ERK were maintained at 300 min. Phosphorylation of c-Jun N-terminal kinase (JNK) was seen in 15 min, and even at 300 min phosphor-JNK was detected in cultured ocular fibroblasts. A loss of TRPA1 in the cells blocked the TGF-β1-induced phosphorylation of Smad3, p38 MAPK, ERK, and JNK.
Gene Expression Analysis in Cultured WT and KO Ocular Fibroblasts
Adding exogenous TGF-β1 upregulated TRPA1 mRNA expression in WT ocular fibroblasts. mRNA expression of TGF-β1, IL-6, and α-SMA was upregulated in both WT and KO cells by adding exogenous TGF-β1. However, the expression levels of these components in the presence or absence of exogenous TGF-β1 were suppressed by the loss of the TRPA1 gene as compared with WT fibroblasts (Figure 2).
Figure 2.

Fibrogenic gene expression in cultured ocular fibroblasts. (a) Adding exogenous transforming growth factor-β1 (TGF-β1) upregulated transient receptor potential ankyrin 1 (TRPA1) mRNA expression in wild-type (WT) mice ocular fibroblasts. (b and c) Loss of TRPA1 reduced the basal mRNA expression level of TGF-β1 and interleukin (IL-6) in ocular fibroblasts, and the degree of upregulation of these genes by exogenous TGF-β1 was also attenuated by the loss of TRPA1. (d) There was no difference in the basal expression level of α-smooth muscle actin (α-SMA) mRNA in between cultured WT and TRPA1-null (KO) ocular fibroblasts. In cultured WT ocular fibroblasts, the expression of α-SMA was markedly upregulated by adding exogenous TGF-β1, which was abolished with the loss of the TRPA1 gene in the fibroblasts. *P<0.05, bar, mean±s.e.m.
Expression of TRPA1 and TRPV1 in Corneas of KO and TRPV1-Null Mice
We wondered whether expression of TRPV1 and TRPA1 were dependent or independent of the other molecule. Therefore, we examined the expression of mRNA and protein of TRPV1 or TRPA1 in the cornea of WT and KO mice. TRPV1 was detected in the basal cell of the corneal epithelium of a WT and TRPA1 KO mice (Figure 3a). mRNA expression levels of TRPV1 in the cornea was not affected by the loss of TRPA1 (Figure 3b). Similarly, TRPA1 was detected in the basal cell of the corneal epithelium in WT and TRPV1 KO mice. However, the mRNA expression levels of TRPA1 in the cornea were not affected by the loss of TRPV1 (data not shown).
Figure 3.

Histology and expression of transient receptor potential vanilloid 1 (TRPV1) in corneas of wild-type (WT) and TRPA1 mice. (a) TRPV1 was detected in the basal cells of the corneal epithelium of a WT or TRPA1 KO mouse. Arrowheads indicate TRPV1-positive cells. (b) Similarly, mRNA expression level of TRPV1 in the cornea was not affected by the loss of TRPA1. Scale bar, 100 μm.
Alkali Burn in Mouse Eyes
TRPA1 expression in the intact mouse cornea is restricted to the basal epithelial cell layer. Histology of a KO cornea showed no obvious abnormalities as compared with that of a WT cornea (data not shown). TRPA1 protein was detected in both regenerated epithelium and the stroma of alkali-burned corneas 10 days after the alkali burn (Figure 4), suggesting that the alkali burn upregulated stromal cell TRPA1 expression.
Figure 4.
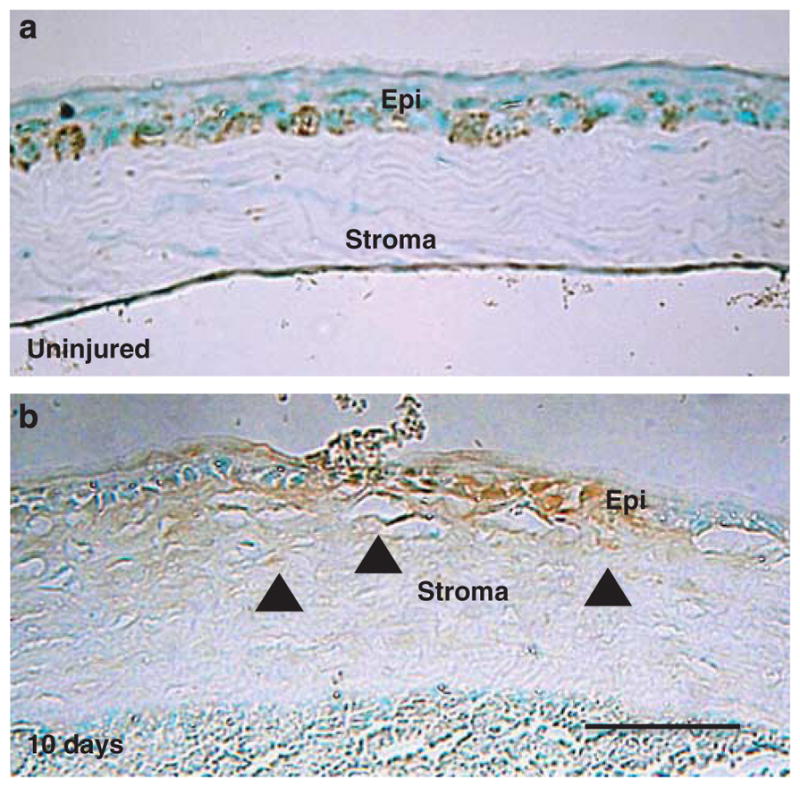
Transient receptor potential ankyrin 1 (TRPAV1) expression in a mouse cornea. (a) TRPA1 is expressed in the basal cells of corneal epithelium. (b) This was also detected in stromal cells as well as in the epithelial cells of alkali-burned corneas after 10 days of healing, suggesting that alkali burn activates stromal cell TRPA1 expression. Scale bar, 100 μm.
To examine the role that TRPA1 has in modulating wound healing of alkali-burned corneas, we first compared the corneal haze that developed in the injured corneas of both KO and WT mice. At each time point, the extent of opacification in the burned corneas appeared to be more severe in WT mice than that in TRPA1 KO mice (Figure 5a). The eye globe diameters of alkali-burned eyes were measured 5, 10, and 20 days of healing. At 20 days, the globe diameter was significantly smaller in WT than that in KO mice, indicating a more marked eye shell tissue shrinkage in WT eyes compared with that in KO eyes (Figure 5b). This finding suggested that myofibroblast transdifferentiation was greater in the WT corneas than that in KO corneas. To further characterize the tissue reaction to an alkali burn, we conducted immunohistochemistry and real-time RT-PCR to assess the variations in inflammation between WT and TRPA1 KO mice, as described below.
Figure 5.

Healing of alkali-burned corneas in wild-type (WT) and TPA1-null (KO) mice. (a) The corneas of WT and KO mice at 5, 10, and 20 days. At each time point, the incidence and degree of opacification and surface irregularity in the healing cornea was more prominent in WT mice than TRPA1 KO mice. (b) Evaluation of the changes in eyeball diameter during healing from the alkali burn shows that WT globes had a smaller diameter than KO globes at 20 days. (c) Hematoxylin and eosin (HE) histology and immunohistochemical findings of burned corneas at day 10. HE staining showed that the burned cornea exhibits a larger cell population (presumed inflammatory cells) and more severely disorganized tissue in WT corneas than KO tissues. The stroma was thicker in WT corneas than in KO corneas throughout the healing period examined. Immunohistochemistry findings suggested that the density of the myeloperoxidase (MPO)-labeled polymorphonuclear leukocytes (PMNs) and F4/80-labeled macrophages was greater in the WT cornea than in the KO cornea. Healing burned corneas in WT mice seemed to contain many α-smooth muscle actin-positive myofibroblasts. However, the majority of corneal fibroblasts were not labeled with anti-αSMA antibody in KO mice. Expression of active TGF-β1 protein was greater in the WT stroma than in the KO stroma. Scale bar, 100 μm. (d) Real-time RT-PCR also showed that lacking TRPA1 suppressed mRNA expression levels of MPO, F4/80, α-SMA, and TGF-β1 in a healing post-alkali burn cornea. Data represent mean±s.e.m. from five specimens in each condition (bar). *P<0.05.
Histology and Immunohistochemistry of Healing WT and KO Corneas
Persistent and severe inflammation and marked tissue fibrosis were observed in WT corneas following the alkali burn. For example, there seemed to be a higher level of MPO and F4/80 immunostaining, marker for PMN and macrophages, respectively, in WT mice than in KO mice (Figure 5c). Immunostaining with anti-α-SMA antibody, a marker of tissue fibrosis-related myofibroblasts,30 also suggested an immense increase in the stromal myofibroblasts of alkali-burned corneas in WT mice at 10 days. In contrast, the majority of the stromal cells in alkali-burned corneas of TRPA1 KO mice were negative for α-SMA(Figure 5c). Immunoreactivity for the active form of TGF-β1 at day 10 was also more marked in the WT stroma than in the KO stroma (Figure 5c).
Inflammatory and Fibrogenic Gene Expression in the Healing Corneas of a WT and KO Mice
To confirm the immunohistochemical observations, we performed real-time RT-PCR for mRNA expression of MPO, F4/80, α-SMA, and TGF-β1. The data confirmed that there were significantly fewer inflammatory cells and myofibroblasts in KO tissue than in WT tissue at several time points (Figure 5d). The expression of TGF-β1 mRNA expression in a healing cornea was also significantly suppressed by the loss of TRPA1. A lacking of TRPA1 in the healing alkali-burned corneas also significantly suppressed mRNA levels of IL-6, VEGF, collagen Ia1, and substance P at certain time points throughout the wound closure interval (Figure 6).
Figure 6.

Expression pattern of wound healing-related genes in an alkali-burned cornea. (a) Expression of interleukin-6 (IL-6) is transiently increased at 5 days in the healing alkali-burned wild-type (WT) corneas, but then decreased. Lacking transient receptor potential ankyrin 1 (TRPV1) abolishes the IL-6 mRNA expression peak. (b) Expression of vascular endothelial growth factor (VEGF) was transiently increased at 10 days in the healing alkali-burned WT corneas but then decreased. Lacking TRPA1 abolished the VEGF mRNA expression peak. (c) Expression of collagen Ia1 mRNA increases at 5 days in the healing alkali-burned WT corneas, and declines due to TRPA1 loss. (d) Expression of Substance P (SP) increased markedly at 20 days in the healing alkali-burned WT corneas, but was later suppressed, which is associated with TRPA1 loss in the healing alkali-burned mouse corneas at this time point. Data represent mean±s.e.m. from five specimens in each condition (bar). *P<0.05.
BMT and Ocular Alkali Burn Healing
A recent cell culture study showed that TGF-β1-derived signaling is attenuated in mesenchymal cells by the loss of TRPA1. However, whether the KO phenotype that showed healing after alkali exposure is attributed to resident corneal cells or infiltrated inflammatory cells is unclear. To answer this question, and to determine the roles that infiltrating inflammatory cells have in eliciting the KO-healing phenotype, we used in vivo chimera mice generated with reciprocal BMT of WT and KO mice. We compared the corneal response with an alkali exposure in chimera mice that had received reciprocal BM from WT to KO mice and vice versa (KO-to-WT and WT-to-KO group) at 10 and 20 days after the alkali burn. The chimeric WT mice that received WT BM cells (WT-to-WT) and KO BM cells (KO-to-WT) showed no differences compared with other groups in their healing patterns after an alkali burn (data not shown). RT-PCR detected TRPA1 mRNA in the spleen of WT-to-KO mice, indicating that WT BM cells had successfully reconstituted in KO mice (data not shown). In contrast, 10 days after the alkali burn, the chimeric KO mice receiving WT BM cells (WT-to-KO) still displayed less opacification (scarring) than that of WT mice receiving KO BM cells (KO-to-WT) and of WT mice (compare Figure 7a with Figure 5a). HE histology in the corneas of KO-to-WT chimeras showed greater stromal cellularity and swelling than those of WT-to-KO chimeras (Figure 7b). Typically, the cornea of a WT-to-KO chimera mouse had less stromal α-SMA immunohistochemical staining and levels of MPO, F4/80, and active TGF-β1 immunoreactivity than those of KO-to-WT chimeras (Figure 7b). These findings are consistent with the notion that TRPA1 expression in corneal resident cells with a WT genetic background is necessary to elicit severe inflammation and tissue fibrosis in alkali-burned corneas.
Figure 7.

Healing an alkali-burned cornea in a bone marrow transplanted (BMT) mouse. (a) A total of 10 and 20 days after alkali burning, a wild-type (WT) mouse that had received bone marrow (BM) from a WT mouse (WT-to-WT) and a WT mouse that had received BM from a TRPA1-null (KO) mouse (KO-to-WT) exhibited greater opacification and neovascularization as compared with those by the KO mouse that had received BM from a WT mouse (WT-to-KO) group. (b) HE histology shows increases in cell density in the swollen stroma of a KO-to-WT cornea as compared with a WT-to-KO tissue. Immunohistochemistry indicated the cornea of a WT-to-KO mouse has less stromal α-SMA staining and lower levels of immunoreactivity for MPO, F4/80, and active TGF-β1 as compared with the KO-to-WT tissue. Scale bar, 100 μm.
Treatment of Corneal Alkali Burn in WT Mice with a TRPA1 Antagonist
Finally, we attempted to reproduce the favorable phenotype of a KO mouse showing decreased inflammatory fibrosis in the alkali-burned cornea using a chemical TRPA1 antagonist. Corneal transparency restoration was markedly improved in the mice treated with HC-030031 (100 mg/kg, daily). The TRPA1 antagonist treatment suppressed the shrinkage of the globe diameter at 20 days (Figures 8a and b), suggesting that tissue contraction caused by wound healing was more marked in the untreated control group than that in the antagonist group. The destruction of the stromal matrix organization was also inhibited by the treatment, according to the HE histology. The antagonist-treated mice exhibited lower levels of infiltration by MPO-labeled PMNs and F4/80-positive macrophages as well as increased α-SMA staining (Figure 8c). The expression of the active TGF-β1 protein at day 10 was greater in the untreated mouse stroma than that in the antagonist-treated stroma (Figure 8b). These results indicate that inflammatory cell infiltration and attenuated myofibroblast transdifferentiation had decreased in the antagonist-treated group compared with those in the untreated mice. Therefore, the TRPA1 antagonist treatment reproduced the KO phenotype that demonstrated wound healing of an alkali-burned cornea.
Figure 8.

Treatment of corneal alkali burn in wild-type (WT) mice with a transient receptor potential ankyrin 1 (TRPV1) antagonist (HC-030031). (a) The corneas of WT mice treated with or without HC-030031 at 5, 10, or 20 days. At each time point, corneal transparency restoration was markedly improved in the mice treated with the TRPA1 antagonist HC-030031. At Day 20, was observed through the transparent cornea in an antagonist-treated mouse, but not in an untreated mouse. (b) Evaluation of changes in eyeball diameter during wound healing from an alkali burn shows that untreated globes have a smaller diameter at 20 days than antagonist-treated globes. (c) The histology of burned corneas stained with HE and immunohistochemical findings at day 10. The stromal organization is poorer in untreated mice than in antagonist-treated mice. The antagonist-treated mice have lower infiltration levels for MPO-labeled neutrophils and F4/80-positive macrophages, in addition to marked α-SMA and active TGF-β1 staining at both time points. Scale bar, 100 μm.
DISCUSSION
An injury inflicted with alkali burn induces severe inflammation and subsequent tissue fibrosis/scarring in corneal stroma of the affected eye that eventually causes opacification and surface curvature alteration of the corneal tissue, leading to visual impairment.6,34 In addition to its clinical importance, an alkali burn of the cornea in an animal model provides suitable tissue to investigate the pathobiology of tissue inflammation and fibrosis because of its relatively simple structure and avascularity without contaminating resident blood vessels cells and hair follicle cells.
In the present study, we first conducted an in vitro experiment using primarily cultured ocular fibroblasts obtained from the mutant mouse. The loss of TRPA1 in cultured fibroblasts attenuated the activation of Smad, p38 MAPK, ERK, and JNK in the presence of TGF-β1. Adding TGF-β1 to the medium resulted in a subsequent and significant upregulation TGF-β1 mRNA expression in both WT and KO fibroblasts. However, the KO fibroblasts express significantly less TGF-β1 mRNA in the absence or presence of exogenous TGF-β1, as compared with that of WT cells. Lacking this channel receptor also suppressed mRNA expression of the inflammatory cytokines, IL-6, and α-SMA are myofibroblast markers—the hallmark of tissue fibrosis. The loss of this receptor also counteracted TGF-β1-induced upregulation of these two components in cultured ocular fibroblasts. This change in gene expression patterns may be mediated by alterations of TGF-β1-dependent signaling cascades; fibrogenic gene expression and myofibroblast conversion in fibroblasts are reportedly mediated by Smad and p38 MAPK cascades activated by TGF-β1.9,33,35–37 The expression of TRPA1 is upregulated in the cells with exposure to TGF-β1, suggesting the presence of positive feedback through a TGF-β1-induced activation of signaling cascades mediated by TRPA1. A previous report indicated that the TRPA1 receptor participates in tissue inflammation. For example, TRPA1 signals modulate local tissue inflammation in animal models of acute pulmonary inflammation induced by acrolein and cigarette smoke,38 experimental colitis,28 or carrageenan-induced paw edema.39 Although TGF-β1 activates multiple signaling streams, it is well established that Smad3 signaling is most involved in in vivo inflammatory tissue fibrosis with wound healing of an alkali-burned cornea; the loss of Smad3 reportedly exhibits an anti-fibrosis or anti-inflammatory phenotype in wound healing tissues, including in an alkali-burned mouse cornea.9
Immunohistochemistry detected the upregulation of TRPA1 protein in the corneal stromal fibroblasts or keratocytes in a healing, alkali-burned cornea, suggesting that TGF-β1 upregulates TRPA1 expression in corneal stromal cells. This notion was supported by results obtained with cultured ocular fibroblasts; adding TGF-β1 directly upregulated TRPA1 expression in cultured ocular fibroblasts. In healing alkali-burned corneas of both animals and humans, many PMNs first invade the injured stroma upon tissue damage followed by macrophage invasion and upregulation of proinflamamtory and profibrogenic growth factors and cytokines. Macrophage infiltration further augments local tissue inflammation through the accumulation of proinflamamtory growth factors and cytokines in a positive feedback system.
In the present study, we show for the first time that a lack of TRPA1 signaling was beneficial for restoring corneal transparency following an alkali burn to mouse corneas. A more prominent pathogenic tissue response, ie, inflammation and subsequent tissue fibrogenic reaction, as indicated by greater myofibroblast transdifferentiation and matrix elaboration, was observed in an alkali-burned WT mouse cornea as compared with those of a KO cornea. The phenotype observed in a KO cornea coincides with the effects of the loss of TRPA1 in other tissues. The loss of TRPA1 or TRPV1 did not affect expression levels of TRPV1 and TRPA1, respectively. We previously reported the suppressive effects of tissue inflammation and subsequent fibrosis and scarring in a mouse cornea resulting from TRPV1 gene ablation or chemical TRPV1 blocking.29 The present study showed that similar favorable (suppressive) effects on the ablation of the functions of TRPA1 may be independent of those of TRPV1.
Histology, immunohistochemistry, and real-time RT-PCR showed suppression of PMN infiltration and of macrophage in an alkali-burned KO cornea as compared with that in WT mice. It should be noted that mRNA expression of IL-6 and collagen Ia1 (exclusively expressed by corneal fibroblasts) was significantly less in KO tissues than in WT tissues at day 5, whereas there was no difference in the infiltration levels of PMNs and macrophages. This finding is considered to be consistent with the notion that TRPA1 gene ablation in ocular fibroblasts attenuated TGF-β1 signaling and both proinflammatory and profibrogenic gene expression in cell culture. KO ocular fibroblast exposure to TGF-β1 did not elicit myofibroblast transdifferentiation as was indicated by the lack of α-SMA mRNA expression. In the present results, the in vivo expression of IL-6 was lower in a KO-healing cornea as compared with that in a WT tissue at day 5, when PMN and macrophages infiltration was similar between WT and KO tissues. Suppression of the initial expression of inflammatory cytokines and growth factors in KO resident corneal cells appears to interrupt the inflammatory augmentation cycle by infiltrating inflammatory cells in the healing tissue. Differences among macrophage populations between the WT and KO corneas were detected at day 20, but not at day 10. At this time point (day 10), mRNA expression of α-SMA and TGF-β1 was lower in KO corneas than that in WT corneas. This phenotype at day 10 may also be attributed to the dependency of the KO genotype on resident tissue cells, but not macrophages. Suppressing of TGF-β1 and VEGF expression through chemoattractants to macrophages may explain further the attenuation of macrophage infiltration and reduction of macrophage-derived growth factors in tissues. The suppression of fibrosis in KO ocular tissue is also consistent with the finding that a WT eye globe showed greater and more prominent shriveling than that of a KO globe during healing. A decline in active TGF-β in the healing tissues of KO mice may result in fewer contractile α-SMA-positive myofibroblasts as compared with those of WT, which may in part explain the smaller eye globe diameters observed in WT mice.
Our previous experiments showed that decreased inflammatory fibrosis in a healing cornea following an alkali burn in a TRPV1-null mouse depended on the genotype of mesenchymal cells, but not of inflammatory cells.29 Our previous study of adenoviral transfer of anti-Smad genes to an alkali-burned mouse cornea9 also supports the notion that the gene expression and behavior of tissue mesenchymal cells play a critical role in the tissue-repair process. In those studies, the administration of adenoviral vectors that carried anti-Smad genes to an injured cornea at an early phase of the healing process with minimal infiltration of inflammatory cell populations mimicked the Smad3-null phenotype of healing and exerted a significant therapeutic effect in the suppression of inflammation and fibrosis in the local tissue.9 Although we could not exclude the possibility that adenoviral gene transfer conveyed genes to inflammatory cells in the early phase of healing, the dependency of the phenotype on resident corneal cells may be much supported. The present in vitro finding of a decreased reaction of KO ocular fibroblast to exogenous TGF-β1 suggests that the KO phenotype of healing (less inflammation and less fibrosis) may be attributed to the absence of TRPA1 in tissue resident cells rather than in inflammatory cells (either PMNs or macrophages) of BM with origins similar to those of the healing phenotype of TRPV1-null mouse. In order to clarify this point, we performed an experiment with corneal alkali burning in chimeric mice of reciprocal BMT. The results from that study showed that KO mice receiving WT BM exhibited a KO-like phenotype of healing with less inflammation and fibrosis, but the WT counterpart chimeras constituting BM of KO mice did not. The present findings obtained from BMT experiments further support the notion that the phenotype in a KO mouse depends on tissue resident cells, but not on inflammatory cells.
We demonstrated in mice that the loss of TRPA1 signaling suppresses inflammatory and fibrogenic reactions in the cornea following a chemical injury. The underlying mechanisms may include the inhibition of TGF-β1-related signaling cascades in keratocytes or corneal fibroblasts. Therefore, blocking of TRPA1 channel pharmacologically could be of a therapeutic value in treating inflammation-based corneal diseases. To explore this possibility, we examined the effects of a systemic i.p. administration of a TRPA1 antagonist, HC-0030031, on the wound healing outcome in an alkali-burned mouse cornea. The inhibition of the TRPA1 receptor reproduced the KO phenotype of healing of suppression of inflammation and less tissue fibrosis, suggesting that a novel strategy for treating a chemical corneal burn by suppressing TRPA1-related inflammatory and fibrogenic reactions.
Acknowledgments
This study was supported by the grants from the Ministry of Education, Science, Sports and Culture of Japan (C21592241 to YO, C19592036 to SS), Mitsui Life Social Welfare Foundation, Mochida Memorial Foundation, Takeda Science Foundation and Uehara Foundation (SS) and EY04795 (to PSR).
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Piek E, Ju W, Heyer J, et al. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. 2001;276:19945–19953. doi: 10.1074/jbc.M102382200. [DOI] [PubMed] [Google Scholar]
- 2.Massague J. Integration of Smad and MAPK pathways: a link and a linker revisited. Genes Dev. 2003;17:2993–2997. doi: 10.1101/gad.1167003. [DOI] [PubMed] [Google Scholar]
- 3.Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005;24:5742–5750. doi: 10.1038/sj.onc.1208928. [DOI] [PubMed] [Google Scholar]
- 4.Brodovsky SC, McCarty CA, Snibson G, et al. Management of alkali burns: an 11-year retrospective review. Ophthalmology. 2000;107:1829–1835. doi: 10.1016/s0161-6420(00)00289-x. [DOI] [PubMed] [Google Scholar]
- 5.Ishizaki M, Zhu G, Hasebe T, et al. Expression of collagen I, smooth muscle a-actin, and vimentin during the healing of alkali-burned and lacerated corneas. Invest Ophthalmol Vis Sci. 1993;34:3320–3328. [PubMed] [Google Scholar]
- 6.Saika S, Kobata S, Hashizume N, et al. Epithelial basement membrane in alkali-burned corneas in rats. Immunohistochemical study Cornea. 1993;12:383–390. doi: 10.1097/00003226-199309000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Sridhar MS, Bansal AK, Sangwan VS, et al. Amniotic membrane transplantation in acute chemical and thermal injury. Am J Ophthalmol. 2000;130:124–137. doi: 10.1016/s0002-9394(00)00500-6. [DOI] [PubMed] [Google Scholar]
- 8.Meller D, Pires RT, Mack RJ, et al. Amniotic membrane transplantation for acute chemical or thermal burns. Ophthalmology. 2000;107:980–989. doi: 10.1016/s0161-6420(00)00024-5. [DOI] [PubMed] [Google Scholar]
- 9.Saika S, Ikeda K, Yamanaka O, et al. Expression of Smad7 in mouse eyes accelerates healing of corneal tissue after exposure to alkali. Am J Pathol. 2005;166:1405–1418. doi: 10.1016/S0002-9440(10)62358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saika S, Yamanaka O, Sumioka T, et al. Fibrotic disorders in the eye: targets of gene therapy. Prog Retin Eye Res. 2008;27:177–196. doi: 10.1016/j.preteyeres.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Saika S. TGFβ pathobiology in the eye. Lab Invest. 2006;86:106–115. doi: 10.1038/labinvest.3700375. [DOI] [PubMed] [Google Scholar]
- 12.Saika S, Ikeda K, Yamanaka O, et al. Adenoviral gene transfer of BMP-7, Id2 or Id3 suppresses injury-induced epithelial-mesenchymal transition of lens epithelium in mice. Am J Physiol Cell Physiol. 2006;290:C282–C289. doi: 10.1152/ajpcell.00306.2005. [DOI] [PubMed] [Google Scholar]
- 13.Saika S, Yamanaka O, Okada Y, et al. Effect of overexpression of PPAR gamma on the healing process of corneal alkali burn in mice. Am J Physiol Cell Physiol. 2007;293:C75–C86. doi: 10.1152/ajpcell.00332.2006. [DOI] [PubMed] [Google Scholar]
- 14.Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38:233–252. doi: 10.1016/j.ceca.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 15.McNamara CR, Mandel-Brehm J, Bautista DM, et al. TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci USA. 2007;104:13525–13530. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bandell M, Story GM, Hwang SW, et al. Noxious cold ion channel TRPA1 is activated by compounds and bradykinin. Neuron. 2004;41:849–857. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- 17.Bautista DM, Jordt SE, Nakai T, et al. TRPA1 mediates the inflammatory actions of environmental irritants and oroalgesic agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 18.Bautista DM, Movahed P, Hinman A, et al. Pungent oroducts from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci USA. 2005;102:2248–12252. doi: 10.1073/pnas.0505356102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordt SE, Bautista DM, Chuang HH, et al. Mustard oils and cannabinoida excite sensory nerve fibers through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- 20.Macpherson LJ, Geierstanger BH, Viswanath V, et al. The pungency of garlic: activation of TRPA1 and TRPV1 in response to allicin. Curr Biol. 2005;15:929–934. doi: 10.1016/j.cub.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Macpherson LJ, Xiao B, Kwan KY, et al. An ion channel essential for sensing chemical damage. J Neurosci. 2007;27:11412–11415. doi: 10.1523/JNEUROSCI.3600-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diogenes A, Akopian AN, Hargreaves KM. NGF up-regulates TRPA1: implications for orfacial pain. J Dent Res. 2007;86:550–555. doi: 10.1177/154405910708600612. [DOI] [PubMed] [Google Scholar]
- 23.Ro JY, Lee JS, Zhang Y. Activation of TRPV1 and TRPA1 leads to muscle nociception and mechanical hyperalgesia. Pain. 2009;144:270–277. doi: 10.1016/j.pain.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Namer B, Seifert F, Handwaeker HO, et al. TRPA1 and TRPM8 avtivation in humans: effects of cinnamaldehyde and menthol. Neuroreport. 2005;16:955–959. doi: 10.1097/00001756-200506210-00015. [DOI] [PubMed] [Google Scholar]
- 25.Ward L, Wright E, McMahon SB. A comparison of the effects of noxious and innocuous counterstimuli on experimentally induced itch and pain. Pain. 1996;64:129–138. doi: 10.1016/0304-3959(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 26.Fujita F, Uchida K, Moriyama T, et al. Intracellular alkalization causes pain sensation through activation of TRPA1 in mice. J Clin Invest. 2008;118:4049–4057. doi: 10.1172/JCI35957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eid SR, Crown ED, Moore EL, et al. HC-030031, a TRPA1 selective antagonist, attenuates inflammatory and neuropathy-induced mechanical hypersensitivity. Mol Pain. 2008;4:48. doi: 10.1186/1744-8069-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poole DP, Pelayo JC, Cattaruzza F, et al. Transient receptor potential ankyrin 1 is expressed by inhibitory motoneurons of the mouse intestine. Gastroenterology. 2011;141:565–575. doi: 10.1053/j.gastro.2011.04.049. [DOI] [PubMed] [Google Scholar]
- 29.Okada Y, Reinach PS, Shirai K, et al. TRPV1 involvement in inflammatory tissue fibrosis in mice. Am J Pathol. 2011;178:2654–2664. doi: 10.1016/j.ajpath.2011.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saika S, Yamanaka O, Okada Y, et al. TGF beta in fibroproliferative diseases in the eye. Front Biosci (Schol ED) 2009;1:376–390. doi: 10.2741/S32. [DOI] [PubMed] [Google Scholar]
- 31.Saika S, Miyamoto T, Yamanaka O, et al. Therapeutic effect of topical administration of SN50, an inhibitor of nuclear factor-B, in treatment of corneal alkali burns in mice. Am J Pathol. 2005;166:1393–1403. doi: 10.1016/s0002-9440(10)62357-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saika S, Ikeda K, Yamanaka O, et al. Loss of tumor necrosis factor a potentiates transforming growth factor b-mediated pathogenic tissue response during wound healing. Am J Pathol. 2006;168:1848–1860. doi: 10.2353/ajpath.2006.050980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flanders KC, Ludecke G, Engels S, et al. Localization and actions of transforming growth factor-bs in the embryonic nervous system. Development. 1991;113:183–191. doi: 10.1242/dev.113.1.183. [DOI] [PubMed] [Google Scholar]
- 34.Saika S, Uenoyama K, Hiroi K, et al. Ascorbic acid phosphate ester and wound healing in rabbit corneal alkali burns: epithelial basement membrane and stroma. Graefes Arch Clin Exp Ophthalmol. 1993;231:221–227. doi: 10.1007/BF00918845. [DOI] [PubMed] [Google Scholar]
- 35.Yamanaka O, Saika S, Ohnishi Y, et al. Inhibition of p38MAP kinase suppresses fibrogenic reaction in conjunctiva in mice. Mol Vis. 2007;18:1730–1739. [PubMed] [Google Scholar]
- 36.Yan W, Wang P, Zhao CX, et al. Decorin gene delivery inhibits cardiac fibrosis in spontaneously hypertensive rats by modulation of transforming growth factor-beta/Smad and p38 mitogen-activated protein kinase signaling pathways. Hum Gene Ther. 2009;20:1190–1200. doi: 10.1089/hum.2008.204. [DOI] [PubMed] [Google Scholar]
- 37.Ratkaj I, Bujak M, Jurišić D, et al. Microarray analysis of Dupuytren’s disease cells: the profibrogenic role of the TGFβ-inducible p38 MAPK pathway. Cell Physiol Biochem. 2012;30:927–942. doi: 10.1159/000341470. [DOI] [PubMed] [Google Scholar]
- 38.Nassini R, Pedretti P, Moretto N, et al. Transient receptor potential ankyrin 1 channel localized to non-neuronal airway cells promotes non-neurogenic inflammation. PLoS One. 2012;7:e42454. doi: 10.1371/journal.pone.0042454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moilanen LJ, Laavola M, Kukkonen M, et al. TRPA1 contributes to the acute inflammatory response and mediates carrageenan-induced paw edema in the mouse. Sci Rep. 2012;2:380. doi: 10.1038/srep00380. [DOI] [PMC free article] [PubMed] [Google Scholar]