

Abstract
Mutant RAS has remained recalcitrant to targeted therapy efforts. Here we demonstrate that combined treatment with poly ADP ribose polymerase (PARP) inhibitors and MEK inhibitors evokes unanticipated, synergistic cytotoxic effects in vitro and in vivo in multiple RAS mutant tumor models across tumor lineages where RAS mutations are prevalent. The effects of PARP and MEK inhibitor combinations are independent of BRCA1/2 and p53 mutation status suggesting that the synergistic activity is likely to be generalizable. Synergistic activity of PARP and MEK inhibitor combinations in RAS mutant tumors is associated with: 1) induction of BIM-mediated apoptosis, 2) decrease in expression of components of the homologous recombination DNA repair pathway, 3) decrease in homologous recombination DNA damage repair capacity, 4) decrease in DNA damage checkpoint activity, 5) increase in PARP inhibitor-induced DNA damage, 6) decrease in vascularity which could increase PARP inhibitor efficacy by inducing hypoxia, and 7) elevated PARP1 protein, which increases trapping activity of PARP inhibitors. Mechanistically, enforced expression of FOXO3a, which is a target of the RAS/MAPK pathway, was sufficient to recapitulate the functional consequences of MEK inhibitors including synergy with PARP inhibitors. Thus the ability of mutant RAS to suppress FOXO3a and its reversal by MEK inhibitors accounts, at least in part, for the synergy of PARP and MEK inhibitors in RAS mutant tumors. The rational combination of PARP and MEK inhibitors warrants clinical investigation in patients with RAS mutant tumors where there are few effective therapeutic options.
Introduction
Although some patients with cancer initially respond to targeted therapy, clinical responses are usually short-lived, thus warranting development and implementation of effective combination therapies to increase patient benefit. The ability of tumor cells to adapt to stress engendered by targeted therapies represents a key mechanism of resistance that, if effectively targeted, could lead to tumor cell death and improved patient outcomes. Indeed, blocking adaptive responses to targeted therapies represents an attractive means toward development of rational combination therapies (1–3).
Nowhere is the need for targeted therapies greater than for cancers driven by oncogenic RAS, which represents one of the mutations in cancer. KRAS is mutationally activated in >90% of pancreatic ductal adenocarcinomas, half of colorectal cancers, and approximately 30% of lung cancers (4). Similarly, approximately 30% of melanomas are driven by oncogenic NRAS, whereas HRAS is commonly mutated in squamous cell carcinomas (4). Despite the dominant oncogenic role of mutant RAS in these and other cancer types, activated RAS isoforms remain undruggable by current therapeutic modalities. This has led to wide research interest including establishment of the National Cancer Institute RAS Initiative (http://www.cancer.gov/research/key-initiatives/ras).
Half of Type I ovarian cancers are driven by oncogenic KRAS. This subtype consists primarily of low-grade tumors that are notoriously insensitive to chemotherapy and are almost uniformly lethal once spread to the peritoneal cavity (5). In contrast, more common Type II ovarian cancers, which predominantly consist of high-grade serous ovarian cancers (HGSOC), rarely contain RAS mutations but still exhibit RAS pathway activation in ~25% of tumors thus demonstrating the importance of the RAS pathway in both ovarian cancer subtypes (6–8).
Approximately half of all HGSOC tumors exhibit aberrations in components of the homologous recombination (HR) DNA repair pathway (6) that likely contribute to efficacy of platinum drugs and of poly-ADP ribose polymerase (PARP) inhibitors (PARPi). PARP, a critical component of the single strand break (SSB) repair (SSBR) pathway, came into focus as a target when SSBR was identified as a synthetic lethal partner with defects in the homologous recombination (HR) pathway induced by BRCA1/2 mutations(9, 10). Mechanistically, blocking PARP1 enzymatic activity compromises base excision repair (BER), resulting in conversion of SSBs to double strand breaks (DSB) during DNA replication thus inducing synthetic lethality in cancer cells with HR defects. Normal cells, in contrast, retain ability to repair DSBs through HR and are therefore resistant to PARPi (11). Because PARP1 participates in additional DNA repair processes including inhibition of nonhomologous end joining (NHEJ) and alternative-NHEJ and in recruitment of DNA repair proteins (12), patients whose tumors are HR-proficient may also benefit from PARPi. Furthermore, several PARPi “trap” PARP proteins at sites of DNA damage, with trapped PARP being more toxic than SSBs or DSBs (13, 14).
Recently a number of potent trapping PARPi including olaparib, niraparib and rucaparib have been approved for ovarian cancer therapy. However, like most other targeted therapies, responses to PARPi are all too frequently transient. A number of combination therapies have been implemented with PARPi to attempt to induce HR defects in tumors with intact HR and thus engender PARP sensitivity or to increase efficacy of PARPi by blocking DNA repair either by inducing hypoxia or by blocking DNA damage cell cycle checkpoints. These include inhibitors of signaling through the PI3K pathway (NCT01623349, NCT02208375), VEGFR (NCT02345265), and cell cycle checkpoints including WEE1 (NCT02576444, NCT02511795)(clnicaltrials.gov).
In this manuscript, we evaluated adaptive responses that could mediate resistance to PARPi through reverse phase protein arrays (RPPA), which quantitate hundreds of critical signaling molecules in terms of both total protein and post-translational modifications, analysis. Transient treatment with PARPi induced a marked increase in RAS/MAPK pathway activation including down-regulation of the key RAS/MAPK targets: FOXO3a and BIM1, which was recapitulated in PARPi-resistant cell lines. Based on this observation, we assessed activity of PARPi in combination with MEK or ERK inhibition and demonstrated that the combination was synergistic in a subset of ovarian cancer cell lines. These studies led to the serendipitous observation that RAS mutant cell lines across multiple lineages are resistant to PARPi and that this resistance could be reversed by MEK or ERK inhibition. We subsequently demonstrated that MEKi sensitize RAS mutant models to PARPi primarily through the induction of FOXO3a, at least in part through increasing apoptotic sensitivity, altering the expression of PARP1, decreasing DNA damage sensing, and decreasing HR DNA repair capacity. Consistent with these observations, PARP and MEK/ERK inhibitors demonstrated synergistic activity in vitro and in vivo in tumors with mutant RAS.
Results
PARP and MEK inhibitors induce inverse adaptive responses
To identify adaptive responses that mediate resistance to PARPi, we used RPPA to assess signaling pathway perturbations in response to treatment with BMN673 (talazoparib), a potent trapping PARPi in ten breast, ovarian, and endometrial cancer cell lines (fig. S1A). Replicates for each treatment condition (monolayer 2D, spheroid 3D, and two time points) were averaged for each line (Fig. 1A). We next rank ordered changes by summing median-centered protein amount normalized to control (DMSO-treated cells at the same time point and culture condition), and visualized data to provide an assessment of decreases or increases (represented by green and red, respectively) in expression or phosphorylation of proteins (Fig. 1 and fig. S1). Proteins exhibiting increased expression or phosphorylation (indicated by “p” before the protein name) in response to PARPi treatment include those involved in DNA damage repair or DNA damage checkpoints, such as FOXM1, CHK1/pCHK1, CHK2/pCHK2, ATM/pATM, pWEE1, RAD51, and RAD50. In addition, we observed marked changes in cell cycle regulators including pRB, CDK1, and cyclin B1, as well as activation of multiple components of the RAS/MAPK signaling pathway including pMEK, pMAPK, pPKC, pYB1, pBAD, and pS6 (15–19). PARPi treatment decreased FOXO3a, P27, and BIM, which is consistent with PARPi-induced activation of the RAS/MAPK pathway, because FOXO3a is down-regulated by RAS/MAPK pathway activation, P27 and BIM are downstream of FOXO3a (20), and BIM is targeted for proteolysis by direct ERK phosphorylation (21). This observation is also consistent with RPPA analysis of TCGA samples (Pan-Can set in TCPAportal.org v4.0) indicating that pMEK and BIM were inversely correlated (p=8.7× 10−53) across multiple tumor lineages. PARPi-induced decreases in BIM expression and increased phosphorylation of BAD would be expected to decrease propensity of cells to undergo apoptosis and thus contribute to adaptation of cells to stress induced by treatment with PARPi. Finally, we observed that PARPi treatment decreased PARP1, consistent with changes in PARP1 expression contributing to adaptation to PARPi given that PARP1 expression correlates with sensitivity to PARPi (22).
Figure 1. PARP and MEK inhibitors induce inverse adaptive responses.
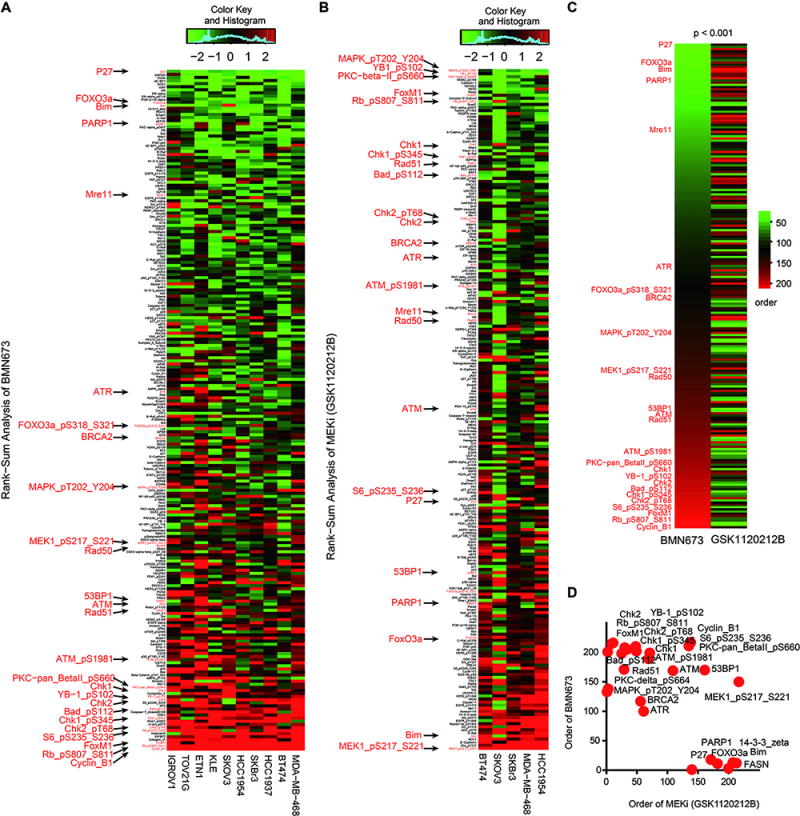
(A) 10 breast ovarian and endometrial cell lines (BT474 (PIK3CA_Mut, HER2_Amp), HCC1954 (PIK3CA_Mut, HER2_Amp), HCC1937 (BRCA1_Mut), MDA-MB-468 (EGFR_Overexpression, PTEN_Mut), IGROV-1 (BRCA1_Mut), TOV21G (PIK3CA_Mut, KRAS_Mut), KLE (TP53_Mut), ETN-1 (PTEN_Mut), SKBr3 (HER2_Amp), SKOV3 (PIK3CA_Mut, HER2_Amp)) were cultured in Matrigel (3D) or monolayer (2D), and treated for 7 or 3 days respectively with DMSO or with BMN673 at an IC50 concentration determined experimentally for each line for 2D and 3D conditions. Protein lysates were analyzed for 220 total and phosphoproteins by RPPA. For visualization, 2D and 3D conditions and time were averaged (see fig. S1 for all samples). Heatmap represents “rank ordered” changes induced by BMN673 treatment, calculated by summing median centered protein amount normalized to control. Proteins with consistent decreases are at the top (green) and increases are at the bottom (red) of the heat map.
(B) 5 cell lines from A (MDA-MB-468, HCC1954, BT474, SKOV3, and SKBr3) were cultured in Matrigel (3D) or monolayer (2D) and treated with MEK inhibitor (GSK1120212B) at 2 concentrations (2 nM and 20 nM) or DMSO for 24 or 48 hours, after which protein lysates were analyzed for 220 total and phosphoproteins by RPPA.
(C) The left column of the heatmap shows protein changes ordered by the effects of BMN673 treatment from 1A, sorted in increasing order from top to bottom (green indicates the most consistently decreased proteins and red indicates the most consistently increased proteins after treatment). The right column in the heatmap shows protein order after GSK1120212B treatment from 1B.
(D) 2×2 comparison of selected proteins (marked in A and B) after BMN673 and GSK1120212B treatment.
Because PARPi altered expression of multiple proteins in the RAS/MAPK pathway, we also assessed adaptive responses to the GSK1120212B (MEKi) by treating five breast and ovarian cancer (Fig. 1B, fig. S1B). This analysis revealed marked GSK1120212B-mediated decreases in phosphorylation of proteins downstream of MEK (pERK, pYB1, pPKC, pBAD, pS6; Fig. 1B), though pMEK was increased likely due to a previously described positive feedback loop (23). GSK1120212B treatment also decreased the amounts or phosphorylation of proteins involved in DNA damage repair and DNA damage checkpoints including pATM, ATR, BRCA2, CHK1/pCHK1, CHK2/pCHK2, FOXM1, MRE11, RAD50, RAD51, and WEE1 (Fig 1B). As expected, MEKi increased P27, FOXO3a, and BIM, and modestly increased 53BP1 and PARP1. PARPi and MEKi induced reciprocal effects on key proteins involved in DNA damage repair, DNA damage checkpoints, and cell viability (Fig. 1C, 1D, fig. S1C, S1D, p<0.001). Together the data raised the possibility that PARPi and MEKi could each block adaptive responses induced by the other drug and thus induce a synthetic lethal interaction.
PARP and MEK inhibitors demonstrate synergy in a subset of ovarian cancer cell lines
To explore potential synergy between PARPi and MEKi, we assessed effects of mono and combination therapy on 24 well-characterized ovarian cancer cell lines selected to represent different subtypes of ovarian cancer (24) (Fig. 2A). For eight of the 24 cell lines, the PARPi/MEKi combination was synergistic with a combination index (CI) less than 0.5 (Fig. 2A, red). Some of the other cell lines exhibited marked sensitivity to BMN673 without any significant interaction with MEKi (Fig. 2A, blue), and some cell lines exhibited modest to no response to either mono or combination treatment (Fig. 2A, black). Given that the effects of PARPi most clearly manifest after multiple cell divisions, we also assessed the effects of the inhibitors in long-term assays. PARPi and MEKi at low concentrations resulted in synergistic inhibition of cell viability (fig. S2A) and colony formation in 3D (fig. S2B) and 2D (fig. S2C) of OVCAR8 compared to treatment with either inhibitor alone.
Figure 2. Mutant KRAS mediates resistance to PARP inhibitors and sensitivity to the PARP and MEK inhibitors.
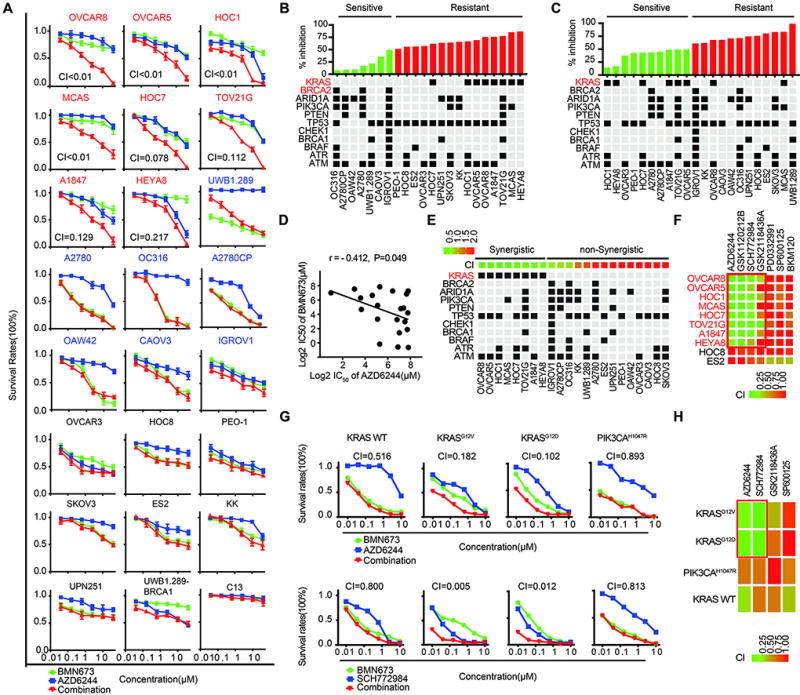
(A) Drug response curves for BMN673 combined with AZD6244 in 24 well-characterized ovarian cancer cell lines treated with varying concentrations of the two compounds for 96 hr. CI was calculated using CalcuSyn software with the Chou-Talalay equation. CI values reflect the sign and magnitude of drug-drug interaction: < 0.5 synergy, 0.5–1 additivity, and >1 antagonism. Red cell line name: synergy (CI < 0.5); blue cell line name: cells intrinsically sensitive to PARPi; black cell line name: cells exhibited modest to no response to either mono or combination treatment.
(B) Upper panel: Percentage cell growth inhibition at 10 µM BMN673 from Figure 2A, Lower panel: Selected mutations in cell lines. KRAS and BRCA2 correlations with BMN673 response were significant (p=4.7×10−5 and p=0.023, respectively).
(C) Upper panel: Percentage of cell growth inhibition by AZD6244 from Figure 2A. Lower panel: Selected mutations in cell lines. KRAS correlation with AZD6244 response was significant (p =0.026).
(D) Correlation between IC50 of BMN673 and AZD6244 (Pearson’s r = − 0.412, p=0.049).
(E) Upper panel: CI values, cells were arranged by CI into synergistic (green) and non-synergistic (red) (cutoff = 0.5) based on Figure 2A. Lower panel: Selected mutations in cell lines. KRAS mutation is correlated with synergistic effect between PARPi and MEKi (p =3×10−6).
(F) Interactions between BMN673 and AZD6244 (MEKi), GSK1120212B (MEKi), SCH772984 (ERKi), GSK2118436A (BRAFi), SP600125 (JNK inhibitor), PD0332991 (CDK4/6 inhibitor), and BKM20 (PI3Ki) were examined across a panel 10 cell lines including 8 RAS mutant lines and 2 RAS wild type lines (cell line data in fig. S3A, 3B).
(G) Response of Ba/F3 cells rendered IL3-independent with KRAS_WT, KRASG12D, KRASG12V, or PIK3CAH1047R to BMN673 and AZD6244/SCH772984.
(H) Heatmap of CI of combinations of BMN673 with AZD6244 and SCH772984 from 2G. Combinations with BMN673 and GSK2118436A (BRAFi) and SP600125 (JNK inhibitor) are also presented.
Mutant KRAS mediates resistance to PARPi and sensitivity to PARP and MEK inhibitors
We next sought to identify molecular underpinnings responsible for synergy by the PARPi and MEKi combination. In ovarian cancer cell lines examined, KRAS mutations were unexpectedly associated with resistance to BMN673 (p=4.7×10−5) (Fig. 2B). Although KRAS mutations are rare in high-grade serous ovarian cancers, the indicated ovarian cancer cell lines have classical activating KRAS mutations with the exception of OVCAR8 that has a rare KRASP121H mutation of unknown significance. OVCAR8 has a RAS/MAPK activation score (24) similar to that of the ovarian cancer cell lines with classical KRAS mutations. As expected, KRAS mutations were associated with increased sensitivity to MEKi (p=0.026; Fig. 2C). Consistent with our observation of reciprocal effects of MEKi and PARPi on adaptive responses (Fig. 1), we detected a significant inverse correlation between sensitivity of cell lines to PARPi and MEKi (Fig. 2D, p=0.049). Strikingly, KRAS mutations emerged as the most significant predictor of synergistic effects of PARPi and MEKi (p<0.001) (Fig. 2E). This synergy between PARPi and MEKi did not associate with mutation status of BRCA1 or BRCA2 (Fig. 2E), suggesting that the observed drug synergy is dependent on a mechanism(s) independent of HR deficiency induced by BRCA1/2 mutations. Furthermore, the synergistic response to the MEKi and PARPi combination was independent of p53 mutation status (Fig, 2E), indicating its potential efficacy on both p53 wild-type and mutant tumors.
To determine which targets within the RAS/MAPK pathway mediate synergy with PARPi, assessed the efficacy of targeting different nodes in the RAS/MAPK pathway. The eight KRAS mutant ovarian cancer cell lines that demonstrated synergy between BMN673 and AZD6244 were included along with two RAS wild type lines, HOC8 and ES2, which were unresponsive to the MEKi/PARPi combination (Fig. 2A). CI in the re-screening study was highly correlated with the results from our initial screen (r = 0.93; fig. S3A). Two MEKi (GSK1120212B and AZD6244) and an ERK inhibitor (SCH772984) demonstrated similar patterns of synergy with PARPi (Fig. 2F, fig. S3B). In contrast, the BRAF inhibitor (GSK2118436A) only demonstrated synergy in TOV21G and demonstrated modest additivity with PARPi in other lines. The failure of BRAF inhibitors to demonstrate consistent synergy with PARPi is likely due to the ability of other RAF homologs to bypass the effect of BRAF inhibition (25). Three unrelated inhibitors targeting JNK (SP600125), CDK4/6 (PD0332991), or PI3K (BKM120) did not demonstrate synergy with PARPi in any of the lines, which is consistent with selectivity of interactions between MEKi/ERKi and PARPi in RAS mutant cell lines (Fig. 2F). Thus, both MEK and ERK inhibitors show marked synergy with PARPi in KRAS-mutant ovarian cancer cell lines.
In order to directly demonstrate that mutant KRAS mediates resistance to PARPi and sensitizes to PARPi and MEKi combinations, we developed Ba/F3 lines (26, 27) stably expressing wild-type or activated forms of KRAS (G12V and G12C). Cells expressing wild-type KRAS or expressing activated PIK3CAH1047R were sensitive to PARPi and moderately sensitive to MEKi but did not demonstrate synergy with the PARPi and MEKi combination. This was in part due to the marked sensitivity to PARPi (Fig. 2G, H). Strikingly, both KRAS mutant expressing cell lines exhibited marked resistance to PARPi but were sensitive to combinations of PARPi and MEKi or ERKi (Fig 2G). Similar to effects in naturally occurring lines, synergistic interactions were observed with PARPi and MEK or ERK inhibitors but not with BRAF and JNK inhibitors (Fig. 2H).
We further explored the role of mutant KRAS in resistance to PARPi and in synergy between MEKi and PARPi using doxycycline (Dox)-inducible HPDE-iKRASG12D pancreatic cancer cells (28) and cell lines from a KRAS inducible mouse pancreatic cancer model (p48 Cre_tetO_LKrasG12D ROSA_rtTAL+ p53L+ mice (LKrasG12D) (29)). In HPDE-iKRASG12D, induction of mutant KRAS induced RAS/MAPK pathway activation and decreased BIM (fig. S3C) as expected. Induction of mutant KRAS decreased a second BH3-only protein, BID (BID was not on the arrays in Fig. 1), which could also contribute to decreased sensitivity to cell death. Dox-induced mutant KRAS expression decreased sensitivity to BMN673 in both inducible KRAS models (fig. S3D–E). The MEKi and PARPi combination was not synergistic in non-induced HPDE, but was synergistic in HPDE where mutant KRAS was induced by Dox (fig. S3D). Further, in two different lines from the LKrasG12D model, the combination of BMN673 with MEK or ERK inhibitors was synergistic (fig. S3F).
Acquired PARPi resistance is associated with RAS/MAPK pathway activation and sensitization to the combination of MEKi and PARPi
To explore whether MEKi could re-sensitize PARPi-resistant cells to effects of PARPi, we developed PARPi resistant cells by culturing highly PARPi-sensitive cells (UWB1.289 and A27980CP, both RAS wild type, see Fig. 2) in the continued presence of BMN673 for 3 to 4 months, at which time drug-resistant clones emerged. A2780CP PARPi resistant (A2780CP_R) and UWB1.289 PARPi resistant (UWB1.289_R) clones were highly resistant to BMN673 and cross-resistant to olaparib (Fig. 3A–B). RPPA analysis demonstrated that RAS/MAPK pathway activity (increased pMEK, pBAD, and pFOXO3a (inactive form)) was upregulated in PARPi resistant clones (Fig. 3C). Moreover, resistant clones showed lower total FOXO3a and BIM, as expected from increased RAS/MAPK pathway activity. The decreased PAR and PARP1 expression in the resistant cells could also contribute to PARPi resistance, as PARP1 expression is associated with PARPi sensitivity (22). Western blotting confirmed increased RAS/MEK pathway activity with concomitant decreases in FOXO3a and BIM in resistant cells (Fig. 3D). Overall, the signaling changes in long-term PARPi resistant cells exhibited many similarities to adaptive responses to short-term PARPi treatment (see Fig. 1). Despite increased RAS/MEK pathway activity, KRAS sequencing demonstrated that the resistant lines did not acquire classical activating KRAS mutations. However, deep NGS sequencing as well as Sanger sequencing of individual PARPi-resistant clones from A2780CP_R demonstrated the presence of KRASA146T, KRASA59T and MAP2K1A283T in 19, 11 and 6% of cells respectively but not in A2780CP parental cells. Importantly, prolonged culture of the lines without PARPi resulted in loss of the mutant KRAS and MAP2K1 clones. The KRASA146T mutant has been demonstrated to be modestly activating (30). The selection of KRAS mutations in a PARPi resistant line supports the concept that RAS mutations and RAS/MAPK pathway activation is a key mediator of PARP resistance. As expected by increases in RAS/MAPK activity in PARPi resistant cell lines and KRAS and MAPK1 mutations, A2780CP_R were markedly more sensitive and UWB1.289_R were modestly more sensitive to MEKi (Fig. 3E–F). MEKi re-sensitized both PARPi-resistant clones to PARPi (Fig. 3E–F). Thus MEKi have the potential to re-sensitize PARPi resistant human tumors to PARPi.
Figure 3. Acquired PARPi resistance is associated with RAS/MAPK pathway activation and sensitization to the combination of MEKi and PARPi.
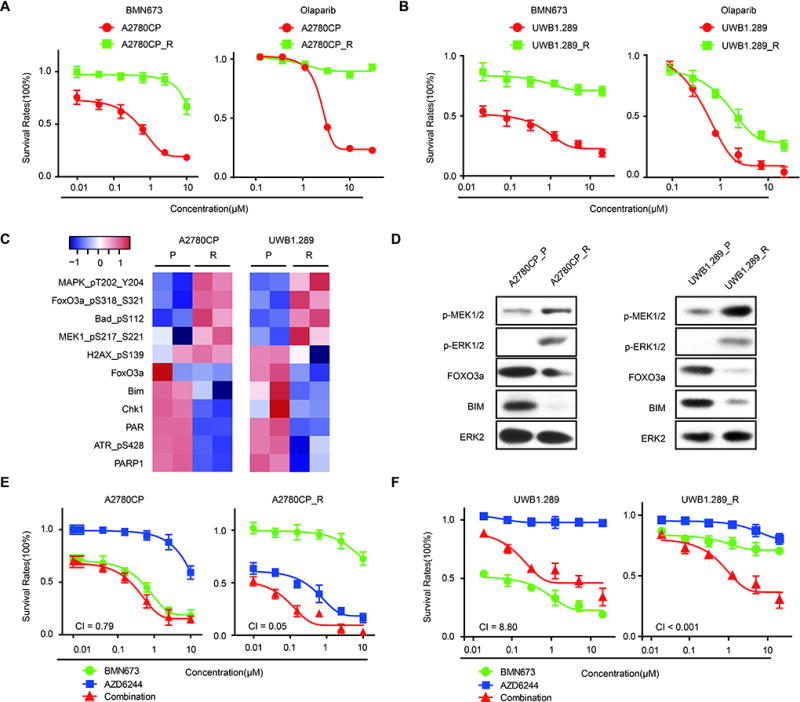
(A) Drug response curves of BMN673 (left) or olaparib (right) in parental or PARP inhibitor-resistant A2780CP incubated with indicated doses for 96 hours.
(B) Drug response curves of BMN673 (left) or olaparib (right) in parental or PARP inhibitor-resistant UWB1.289.
(C) Heatmap of differentially expressed proteins from RPPA of A2780CP _R and UWB1.289_R compared to parent cells, indicating consistent RAS/MAPK pathway activation and decreased FOXO3a, BIM, and PARP1 in both PARPi-resistant cell types.
(D) Western blot validation of RAS/ERK signaling (pMEK, pERK), FOXO3a, and BIM in both PARPi resistant and sensitive cell lines.
(E–F) Drug response curves of parental or PARP inhibitor-resistant A2780CP (E) or UWB1.289 (F) cells treated for 96 hrs with various concentrations of PARPi (BMN673) combined with MEKi (AZD6624).
Synergistic effects of PARP and MEK/ERK inhibition are lineage-independent and observed with KRAS/NRAS/BRAF mutations
To determine whether synergistic effects of combined PARPi and MEKi/ERKi can be generalized across different lineages and different RAS/ERK pathway aberrations, we next performed drug response assays using a panel of 17 KRAS mutant cancer lines (12 pancreatic, 2 lung, and 3 endometrial), 8 NRAS mutant melanoma lines, and 12 BRAF mutant melanoma lines (Fig. 4A–B, fig. S4A–B). All RAS mutant cell lines were resistant to BMN673 monotherapy, including Capan1, which has a BRCA2 mutation. However, striking synergism (CI <0.5) between BMN673 and MEKi/ERKi was observed in 25/37 of KRAS/NRAS and BRAF-mutant cell lines. In a subset of cell lines, interactions between PARPi and MEKi/ERKi were modest despite a CI indicating synergism. Most (10/12) lines that did not demonstrate synergy were highly sensitive to monotherapy with MEKi/ERKi (fig. S4A–B; WM3854, SKMEL28, and WM1779 are provided as examples of this class in Fig. 4B). Only two cell lines, HEC1B and MLE624, which contained activating mutations in KRAS and BRAF, respectively, were resistant to PARPi and MEKi/ERKi combinations. In contrast, PARPi and MEKi did not demonstrate synergy in MCF10A derived from normal mammary epithelium or in immortalized fallopian tube epithelium cells (FT33-shp53-R24C) (fig. S4C). Thus KRAS and, to a lesser degree, BRAF mutations are robust predictors of synergistic interactions between BMN673 and MEKi/ERKi inhibitors.
Figure 4. Synergistic effects of PARP and MEK/ERK inhibition are lineage-independent and observed with KRAS/NRAS/BRAF mutations.

(A) Heat map of CI values for 37 cancer cell lines with KRAS/NRAS/BRAF mutations from different cancer lineages treated for 96 hours with BMN673 and MEKi (AZD6244)/ERKi (SCH772984). Red: cells have synergy between PARPi and MEKi/ERKi; blue: highly sensitive to single drug; black: cells exhibited modest to no response to either mono or combination treatment,
(B) Representative drug response curves of combination therapy with BMN673 and AZD6244 for 96 h.
Mutant KRAS and BRAF increase HR capacity, and MEKi decreases HR capacity in RAS mutant cells, resulting in increased DNA damage
PARPi exhibit synthetic lethality with defects in HR-mediated DNA repair (31). This finding suggested that RAS-mediated resistance to PARPi and synergy of PARPi with MEKi might be explained, in part, by effects of RAS-mediated signaling on HR. Induction of mutant KRAS by doxycycline in HPDE-iKRASG12D (Fig 5A) and a KRAS-driven murine pancreatic model (fig. S5A) was associated with a decreased transcription-based HR defective (HRD) score (32). Furthermore, withdrawal of expression of mutant NRAS in a murine melanoma model (fig. S5B), BRAF and MEK inhibitors in BRAF-mutant melanoma lines (fig. S5C), knockdown of KRAS in HCT116 (fig. S5D), and inhibition of MEK in BRAF mutant but not wild type melanoma lines increased HRD scores (fig. S5E). Analysis of the NCI60 cell set demonstrated that HRAS, KRAS, and NRAS mutations were associated with resistance to the PARPi olaparib (fig. S5F). Moreover, RAS mutations are associated with HR competence (fig. S5G). Consistent with the effects of MEKi in Figure 1, induction of mutant RAS increased MRE11, RAD50, NBS1 (NBN), and BRCA1/2 transcripts, whereas mutant RAS withdrawal, RAS knockdown, or inhibition of BRAF or MEK decreased RNA expression of these genes (Fig. 5A, fig. S5A–D). qPCR confirmed that MEKi induced a marked time-dependent decrease in RAD51, BRCA1, and BRCA2 and a more modest decrease in MRE11 RNA (fig. S6A). Induction of mutant RAS also decreased BIM (BCL2L11), FOXO3a, and to a lesser degree PARP1 transcripts, whereas mutant RAS withdrawal, RAS knockdown, or inhibition of BRAF or MEK increased mRNA for these genes (Fig. 5A, fig. S5A–D). Western blot analysis confirmed that MEKi increased BIM, FOXO3a, and γ-H2AX while downregulating pERK as well as RAD51 and BRCA1 (Fig. 5B, fig S6B). Effects of MEKi on MRE11 protein at early time points were modest consistent with the limited decrease in MRE11 RNA (Fig. 5B, fig. S6B). However, at later time points, MEKi were sufficient to induce marked decreases in BRCA1, Rad51, and MRE11 (fig. S6B). MEKi failed to alter 53BP1 in OVCAR8 (Fig. 5B), consistent with the major effects of MEKi being on HR competence rather than NHEJ. Immunofluorescence analysis demonstrated low FOXO3a, BRCA1, and γ-H2AX with readily detectable diffuse RAD51 cytosolic and nuclear expression in resting OVCAR8 and HOC1 (Fig. 5C and fig. S6C). Consistent with the western blotting data, RAD51 was markedly decreased by MEKi. Strikingly, the PARPi BMN673 induced a marked increase in BRCA1 nuclear staining, which was abrogated by the addition of MEKi (Fig. 5C). The decrease in BRCA1 protein induced by MEKi and maintained in the combination of MEKi and PARPi (Fig. 5B, fig. S6B, D) may contribute, in part, to the decrease in BRCA1 nuclear localization. However, the more marked decrease in nuclear BRCA1 staining compared to total protein raises the possibility that decreased expression of multiple components of the HR pathway such as RAD51 combined to result in the marked decrease in BRCA1 nuclear localization. γ-H2AX was increased by the combination of PARPi and MEKi in multiple cell lines studied (fig. S6D). Taken together, this argues that inhibition of the RAS/ERK pathway in RAS mutant cells decreases HR capacity, rendering cells HR-defective, and increases accumulation of DNA damage in the presence of PARPi.
Figure 5. Mutant KRAS increases HR capacity and MEKi decreases HR capacity in RAS mutant cells, causing increased DNA damage.
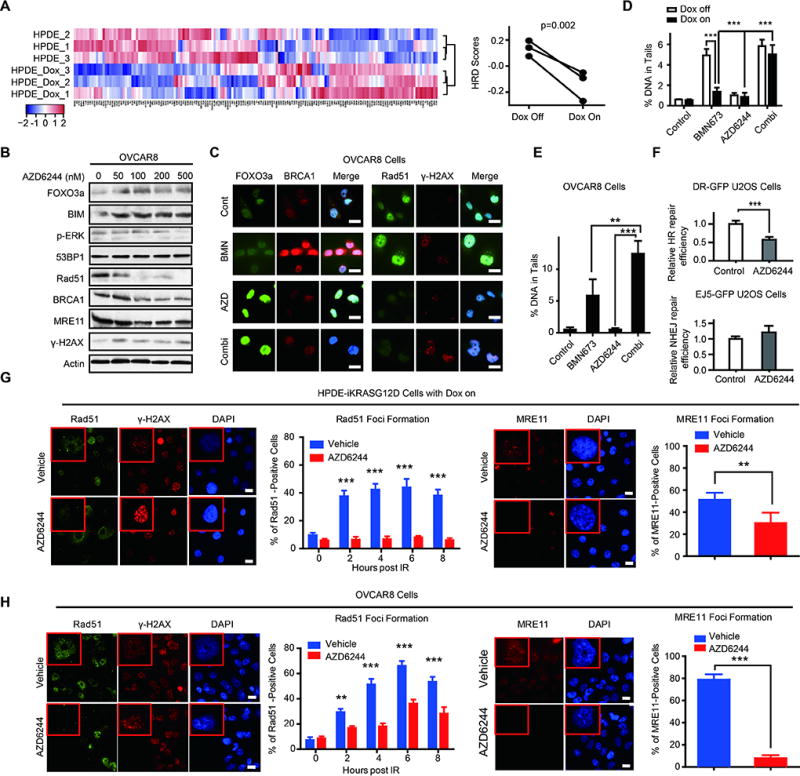
(A) Microarray data from HPDE-iKRASG12D cell lines with or without Dox induction were analyzed by unsupervised clustering for HRD gene signatures. Heatmaps of clusters indicate that cells with KRASG12D induction are more likely HR intact (left panel). Quantification of HRD scores of HPDE-iKRASG12D with or without Dox induction were calculated based on correlation to HRD gene signatures (right panel, higher scores are more likely to have HR defects).
(B) Western blot showing dose-dependent protein changes in OVCAR8 after 24 hours of treatment with the indicated concentrations of AZD6244.
(C) Immunofluorescence staining of FOXO3a/BRCA1 and RAD51/ γ-H2AX after treatment with BMN673 (1 µM)/AZD6244 (5 µM)/combination therapy in OVCAR8 for 48 hours. Scale bars: 20 µm.
(D) Comet assay in HPDE-iKRASG12D cell lines after treatment with BMN673 (200 nM)/AZD6244 (200 nM)/combination therapy for 72 h with or without Dox induction. DNA damage was quantified via % DNA in tails. Each data point represents at least 50 cells.
(E) Comet assay in OVCAR8 cell lines after treatment with BMN673 (1 µM)/ AZD6244 (5 µM)/combination therapy for 72 h. DNA damage was quantified via % DNA in tails. Each data point represents at least 50 cells.
(F) DR-GFP assay to measure HR-mediated DNA DSB repair in DR-GFP U2OS. Frequency of GFP+ cells by flow cytometry after infection with the I-Sce1 endonuclease and incubation for 48 hours with or without 100 nM AZD6244 (upper panel). NHEJ-mediated DNA DSB repair in EJ5-GFP U2OS. Frequency of GFP+ cells by flow cytometry after infection with the I-Sce1 endonuclease and incubation for 48 hours with or without 100 nM AZD6244 (lower panel).
(G–H) Immunofluorescence staining for RAD51, γ-H2AX, and MRE11 before and after IR in inducible HPDE-iKRASG12D (G) and OVCAR8 (H). Images for MRE11 foci staining and quantification of MRE11 foci-positive cells at 2 hours after IR are presented (right panels). Due to relatively late recruitment of RAD51, quantification of RAD51 staining was performed at different times (0, 2, 4, 6, 8 hours after IR), and representative images are shown (left panels). Scale bars: 20 µm. Error bars represent SEM of three independent experiments. Student’s t test: *p<0.05, **p<0.01, and ***p<0.001.
RAS mutants can activate both the MAPK pathway and the PI3K pathway. Furthermore, PI3K pathway inhibitors have demonstrated synergy with PARP inhibitors. However, as indicated in Figure 2F, PI3K inhibitors did not demonstrate synergy with PARPi in RAS mutant cells. As indicated in fig. S6E, F, although MEKi induced marked nuclear translocation of FOXO3a in the presence and absence of PARPi, the pan-PI3K inhibitor BKM120 had modest effects on FOXO3a nuclear translocation. Consistent with this observation, PI3K inhibitors had modest effects on BIM expression in RAS mutant cells (fig. S6E).
We used a comet DNA damage detection assay to directly examine whether activation of KRAS would result in decreased DNA damage that could be reversed by MEKi. In non-induced HPDE-iKRASG12D, BMN673 treatment increased the amount of DNA in tails, consistent with accumulation of DNA damage (Fig. 5D). However induction of mutant KRAS rendered cells resistant to BMN673-induced DNA damage, as indicated by decreased accumulation of DNA in tails. Combined treatment of with MEKi and PARPi reversed the effects of KRAS, as evidenced by an increase in comet formation (Fig. 5D). We observed similar effects in OVCAR8, where BMN673 but not MEKi increased DNA damage, and the combination increased comet formation further (Fig. 5E). To directly assess effects of MEKi on HR capacity, we used U2OS and a HR-GFP reporter assay. MEKi markedly decreased ability of cells to reconstitute GFP in the test plasmid (Fig 5E). In contrast, MEKi had modest effects on NHEJ (Fig. 5F), consistent with modest effects on 53BP1 (Fig. 5B).
Double strand DNA breaks (DSBs) recruit the MRN complex including MRE11, NBS1, and RAD50, which is required for resection of DNA ends at DSBs as well as for the initiation of DNA repair and DNA damage checkpoint pathways. Increased DNA damage concurrent with decreased recruitment of BRCA1 to the nucleus suggested that MRN function might be defective in cells treated with the PARPi and MEKi combination. Indeed, in both OVCAR8 and HPDE-iKRASG12D, MEKi decreased IR-induced MRE11 and RAD51 foci (Fig. 5G, H). Since efficient recruitment of RAD51 to DSBs requires MRN function, this provides independent support for MEKi compromising MRN function. The marked decrease in both MRE11 and RAD51 foci as well as the decrease in BRCA1 nuclear localization and increased accumulation of DNA damage suggests that MEKi interferes with the DNA damage repair pathway at multiple sites.
A FOXO3a-BIM cascade mediates sensitivity to PARP and MEK inhibition
As indicated in Figure 1, PARPi and MEKi demonstrated opposite effects on BIM, P27, and FOXO3a. Indeed, the amounts of basal BIM, a BH3 only protein that is a key mediator of apoptotic balance (33), in cell lines in Fig. 2 were strongly correlated with MEKi and PARPi synergism (p=0.001). Decreased FOXO3a and BIM in PARPi-resistant cells (Fig. 3C–D) and KRAS-induced cells (fig. S5) is consistent with FOXO3a and BIM contributing to resistance of RAS mutant cells to PARPi. Thus low basal BIM or treatment-induced increases in BIM could potentially identify tumors likely to respond to PARPi and MEKi combinations.
Low basal expression of BIM in KRAS-mutant cell lines or KRAS-induced lines and the ability of PARPi to decrease BIM suggest that FOXO3a- and BIM-induced apoptosis may contribute to synergy between MEKi and PARPi. Down-regulation of BIM or FOXO3a with siRNA rendered OVCAR8 more resistant to BMN673 and abrogated synergistic effects of PARPi and MEKi (Fig. 6A, see 6B for BIM and FOXO3a protein content). In contrast, enforced expression of FOXO3a or BIM was sufficient to sensitize OVCAR8 to BMN673 monotherapy (Fig. 6C left panel, see 6D for BIM and FOXO3a protein content) and to PARPi and MEKi combinations (Fig. 6C right panel). These data indicate that expression of FOXO3a or BIM is sufficient to partially mimic effects of MEKi, suggesting that BIM is both necessary and sufficient to sensitize cells to PARPi.
Figure 6. A FOXO3a-BIM cascade mediates sensitivity to PARP and MEK inhibition.
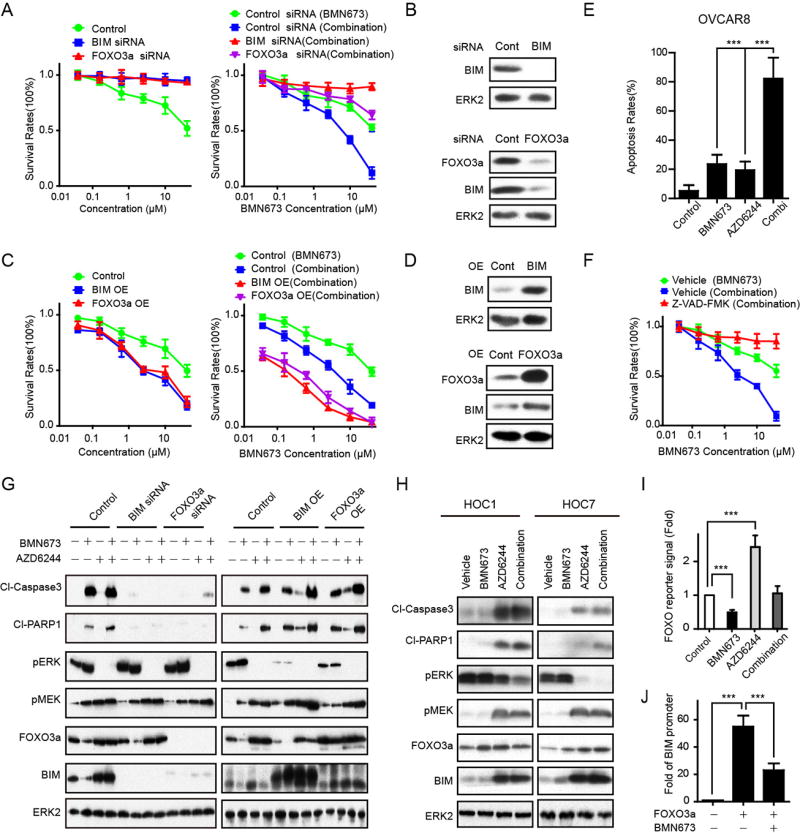
(A) OVCAR8 were treated with 10 nM control siRNA, BIM siRNA, or FOXO3a siRNA. The next day, cells were treated with increasing doses of BMN673 (left panel) or combination with increasing doses of BMN673 and 5 µM MEKi (AZD6244) (right panel). Cell viability was assessed with Prestoblue after 96 h.
(B) OVCAR8 were transfected with BIM siRNA or control siRNA. Western blotting for BIM demonstrated effective BIM knockdown by siRNA (upper panel). OVCAR8 were transfected with FOXO3a siRNA or control siRNA. Western blotting for FOXO3a demonstrated effective FOXO3a knockdown by siRNA. BIM is downregulated by FOXO3a knockdown (lower panel). ERK2 was used as loading control.
(C) BIM and FOXO3a were expressed in OVCAR8 and then treated as indicated. Cell viability was assessed with Prestoblue after 96 h. In the combination, cells were treated with increasing doses of BMN673 and a constant 5 µM MEKi (AZD6244).
(D) OVCAR8 were transfected with BIM or control plasmid. Western blotting for BIM demonstrated effective BIM overexpression (upper panel). OVCAR8 were transfected with FOXO3a or control plasmid. Western blotting for FOXO3a demonstrated effective FOXO3a overexpression. BIM is upregulated by FOXO3a overexpression (lower panel). ERK2 was used as loading control.
(E) OVCAR8 were treated with BMN673 (1 µM), AZD6244 (5 µM), or combination therapy with BMN673 (1 µM), and AZD6244 (5 µM) for 96 hours and subjected to Annexin V-FITC/PI apoptosis analysis. Each value represents mean±SEM from three independent experiments.
(F) A pan-caspase inhibitor (Z-VAD-FMK) (50 µM) was added to OVCAR8. Cells were treated as indicated. Prestoblue assay was done after 96 h of combination therapy with BMN673 dosed as indicated and a constant dose of 5 µM MEKi (AZD6244).
(G) On the left, BIM/FOXO3a were knocked down by siRNA in OVCAR8. Forty hours later, cells were treated with BMN673 (1 µM), AZD6244 (5 µM), or combination therapy with BMN673 (1 µM) and AZD6244 (5 µM) for 96 h. Western blot detection of cleaved-PARP and cleaved-Caspase-3 was used to measure apoptotic cell death (left panel). On the right, BIM/FOXO3a were overexpressed in OVCAR8 for 48 hours. Cells were then treated with BMN673 (1 µM), AZD6244 (5 µM), or combination therapy with BMN673 (1 µM) and AZD6244 (5 µM) for 96 h. Western blot detection of cleaved-PARP and cleaved-Caspase-3 was used to measure apoptotic cell death (right panel). ERK2 was used as loading control.
(H) Two RAS-mutant cell lines (HOC1 and HOC7) were treated with BMN673 (1 µM), AZD6244 (1 µM), or combination therapy with BMN673 (1 µM) and AZD6244 (1 µM) for 96 h. WB detection of cleaved-PARP and cleaved-Caspase-3 was used to measure apoptotic cell death. ERK2 was used as loading control.
(I) OVCAR8 were transfected with FOXO reporter for 24 h, then treated as indicated. FOXO activity was assayed 48 h after treatments (n = 3). In all reporter assays, the luciferase-based reporter signal was normalized to the expression of a co-transfected renilla luciferase control plasmid.
(J) OVCAR8 were treated with BMN673 for 48 h. CHIP-PCR was used to determine percentage input of the BIM promoter precipitated with endogenous FOXO3a.
Results are shown as means±SEM of three independent experiments. Student’s t test: *P<0.05, **P<0.01, and ***P<0.001.
PARPi and MEKi monotherapy each induced a modest increase in apoptosis as assessed by an Annexin V apoptosis assay (Fig. 6E). However, treatment with both PARPi and MEKi led to a robust increase in apoptosis (Fig. 6E). Consistent with a role for apoptosis in effects of PARPi and MEKi combinations, Z-VAD-FMK (a pan caspase inhibitor) markedly inhibited the effects of PARPi and MEKi combinations (Fig. 6F).
FOXO3a knockdown decreased BIM as expected, yet depletion of BIM had no effect on FOXO3a (Fig. 6G). MEKi efficiently decreased pERK and increased pMEK, likely due to a feedback loop. Changes in cleaved caspase 3 and cleaved PARP were consistent with Annexin V results (Fig. 6F) and demonstrated that PARPi, MEKi, and particularly the combination induced apoptosis that was decreased in cells with knockdown of BIM or FOXO3a (Fig. 6G). Similarly, over-expression of BIM or FOXO3a increased apoptosis induced by PARPi, MEKi, and the combination of both agents (Fig. 6G). The effects of BIM and FOXO3a on pERK likely reflect a feedback loop from these downstream mediators. Both MEKi and the combination of MEKi and PARPi increased apoptosis, as indicated by increased cleaved PARP and cleaved caspase in HOC1 and HOC7 KRAS mutant cells (Fig. 6H) suggesting generalizability.
Consistent with effects of PARPi and MEKi on FOXO3a protein (Fig. 1), PARPi and MEKI decreased and increased, respectively, FOXO3a activity as assessed by a FOXO reporter assay (34) in OVCAR8 (Fig. 6I). The addition of MEKi to PARPi reversed effects of PARPi on FOXO3a transcription in OVCAR8 (Fig. 6I). In a chromatin immunoprecipitation (CHIP) assay, PARPi decreased association of FOXO with the BIM promoter (Fig. 6J), consistent with decreases in BIM in response to PARPi (Fig. 1).
The ability of FOXO3a expression to mimic the effects of MEKi in combination with PARPi (Fig. 6A, C) and of siRNA to FOXO3a to reverse the effects of the MEKi and PARPi combination on cell viability prompted us to explore whether FOXO3a contributed to the effects of mutant RAS and MEKi on DNA damage repair. Based on published transcriptional profiles, induction of FOXO3a in RAS-mutant DL23 colon cancer cells induced a marked decrease in hallmarks associated with DNA damage repair based on GSEA and IPA analysis (35) (Fig. 7A,B). Furthermore, induction of FOXO3a markedly increased HRD transcriptional scores (33) (Fig. 7C). In addition, expression of FOXO3a in U20S decreased the ability of cells to repair a defective plasmid by HR (Fig. 7D). Consistent with the alterations in HR, FOXO3a expression decreased RAD51, BRCA1, and BRCA2 RNA as well as BRCA1 and RAD51 protein, including those induced by either IR or BMN673 (Fig. 7E,F). This was associated with increased DNA damage as reflected in γ-H2AX accumulation. As expected, expression of FOXO3a increased BIM and P21 RNA as well as BIM protein and induced apoptosis as indicated by accumulation of cleaved PARP. While FOXO3a induction had modest effects at early time points, prolonged induction of FOXO3a was sufficient to decrease MRE11 protein (Fig. 7F). This was similar to the effects of MEKi on MRE11 that were only observed at late time points (fig. S6B).
Figure 7. FOXO3a regulates HR function and DNA damage.

(A) GSEA demonstrating a negative enrichment of DNA damage geneset after 4-OHT (4-Hydroxytestosterone) induced FOXO3a expression in DL23.
(B) Genes statistically downregulated after 4-OHT induced FOXO3a expression were analyzed by Ingenuity Pathway Analysis (IPA) software, demonstrating downregulation of DNA damage response and DNA repair pathways.
(C) Microarray data from DL23 cell lines with or without 4-OHT induction were analyzed by unsupervised clustering for HRD gene signatures. Heatmap of clusters indicates that cells with FOXO3a induction are more likely HR defective (left panel). Quantification of HRD scores based on correlation to HRD gene signatures (right panel, higher scores are more likely to have HR defects).
(D) DR-GFP assay to measure HR-mediated DNA DSB repair in U2OS-DR-GFP after overexpression of FOXO3a.
(E) Upregulation of FOXO3a, BIM, and P21 and downregulation of RAD51, BRCA1, and BRCA2 mRNAs in DL23 treated with 4-OHT were confirmed by RT-PCR.
(F) Western blot showing indicated protein changes in DLD1 (parent cell) and DL23 cells after 4 and 24 hours (left panel) and 72 hours (right panel) induction with 4-OHT.
(G) Immunofluorescence staining of FOXO3a/BRCA1 and RAD51/γ-H2AX after treatment with 10 Gy IR for 5 hours (upper panel) or BMN673 (500 nM) for 48 hours (lower panel) in DL23 cells with/without 4-OHT induction. Scale bars: 20 µm.
(H–I) Western blot showing indicated protein changes after treatment with 10 Gy IR for 5 hours (H) or BMN673 for 48 hours (I) in DL23 cells with/without 4-OHT induction.
(J–K) Comet assay after treatment with 10 Gy IR for 5 hours (J) or BMN673 for 48 hours (K) in DL23 cells with/without 4-OHT induction.
(L) Drug response curves of BMN673 in DL23 cells with/without 4-OHT induction.
(M) Clone formation assay for BMN673 treatment in DL23 cells with/without 4-OHT induction. Clone formation rates are presented as percentage relative to control.
As assessed by immunofluorescence, induction of FOXO3a markedly decreased nuclear localization of BRCA1 induced by IR (Fig. 7G). Similarly, induction of FOXO3a markedly decreased RAD51 and increased γ-H2AX foci in cells treated with IR and PARPi (Fig. 7G). Further, induction of FOXO3a was sufficient to decrease BRCA1 protein (Fig. 7H,I) and to inhibit the increase in RAD51 induced by IR and the increase in BRCA1 and RAD51 induced by PARPi (Fig. 7H,I). In addition, expression of FOXO3a was sufficient to modestly increase DNA damage as indicated by γ-H2AX accumulation (Fig. 7F, G, H, I) and comet formation (Fig. 7J, K) and to markedly increase DNA damage induced by either IR or PARPi. Expression of FOXO3a was sufficient to markedly decrease cell number as assessed dye conversion (Fig. 7L) and colony formation (Fig. 7M). Together, these data indicate that FOXO3a induction is sufficient to mimic many of the effects of MEKi in RAS mutant cells and to explain, at least in part, the synergy between PARPi and MEKi.
PARP1 expression contributes to sensitivity to PARPi and MEKi combinations
PARP1 expression is decreased by PARPi, increased by MEKi (Fig. 1) and decreased in PARPi-resistant cells (Fig. 3), suggesting that PARP1 could contribute to synergistic activity of PARPi and MEKi, since PARP1 is required for the optimal activity of PARPi. Indeed PARP1 is inversely correlated with RAS/MAPK pathway activation, including a direct correlation with BIM across more than 700 cell lines ((http://tcpaportal.org/mclp/#/), fig. S7A). Furthermore, PARP1 is directly correlated with expression of multiple members of DNA damage and DNA checkpoint pathways, consistent with the role of PARP1 in DNA damage repair (fig. S7A). We thus determined effects of knockdown of PARP1 on sensitivity of OVCAR8 to PARPi and MEKi. Indeed, knockdown of PARP1 markedly decreased sensitivity to PARPi and PARPi and MEKi combinations (fig. S7B) consistent with effects of PARPi and MEKi combinations being due, at least in part, to PARP trapping.
MRE11 contributes to sensitivity to PARPi and MEKi combination
The MRN complex that includes MRE11, RAD50, and NBS1 is required for DNA damage sensing and for excision of overhangs in double strand DNA breaks as well as for activity of PARPi (36, 37). Based on RPPA analysis of over 700 cell lines (http://tcpaportal.org/mclp/#/), MRE11 protein correlates with RAS/MAPK pathway activity, as evidenced by a direct correlation between MRE11 and pMEK as well as an inverse correlation between MRE11 and FOXO3a and BIM protein (fig. S7C). Furthermore, MRE11 protein correlated with multiple DNA damage repair and DNA damage checkpoint mediators (fig. S7C). Similarly, MRE11A RNA was significantly elevated in KRAS mutant tumors within the Cancer Cell Line Encyclopedia ((cBioportal.org) p=4.3×10−4).
Our data indicate that MEKi leads to decreased MRE11 and RAD50 protein (NBS1 is not in the protein array) (Fig. 1) and RNA (fig. S5) concomitant with decreased phosphorylation of multiple DNA damage repair and DNA damage checkpoint mediators,. MRE11 foci were also decreased by MEKi (Fig. 5G, H). These data suggest that knockdown of MRE11 might mimic effects of MEKi and increase activity of PARPi in RAS-mutant tumors. Indeed, MRE11 siRNA increased sensitivity of OVCAR8 to PARPi (fig. S7D). Knockdown of MRE11 did not further increase activity of PARPi and MEKi combinations consistent with the marked decrease in MRE11 induced by MEKi.
FOXM1 does not mediate sensitivity to the PARPi and MEKi combination
PARPi and MEKi treatment also led to increases and decreases, respectfully, in another forkhead factor, FOXM1, which has been implicated in tumorigenesis (38). However, FOXM1 siRNA did not alter activity of PARPi in RAS-mutant OVCAR8 or MCAS (fig. S7E). Furthermore, the FOXM1 inhibitors sinomycin or FDI-6 did not demonstrate synergism with BMN673 (fig. S7F). Thus, FOXM1 does not appear to be a rate-limiting mediator of synergy between PARPi and MEKi.
MEKi and PARPi are synergistic in KRAS-mutant tumors in vivo
Based on synergy of MEKi and PARPi in RAS-mutant cell lines in vitro, we explored effects of MEKi and PARPi in KRAS-mutant tumors in vivo. In established OVCAR8 tumors, combinations of MEKi and PARPi induced tumor regression followed by prolonged decreases in tumor volume (Fig. 8A). In contrast, OVCAR8 tumors were completely resistant to BMN673 at the dose used and demonstrated only a modest response to MEKi.
Figure 8. PARPi and MEKi are synergistic in OVCAR8 xenograft, HPDE-iKRASG12D xenograft, and LPA1-T127 syngeneic breast cancer models.
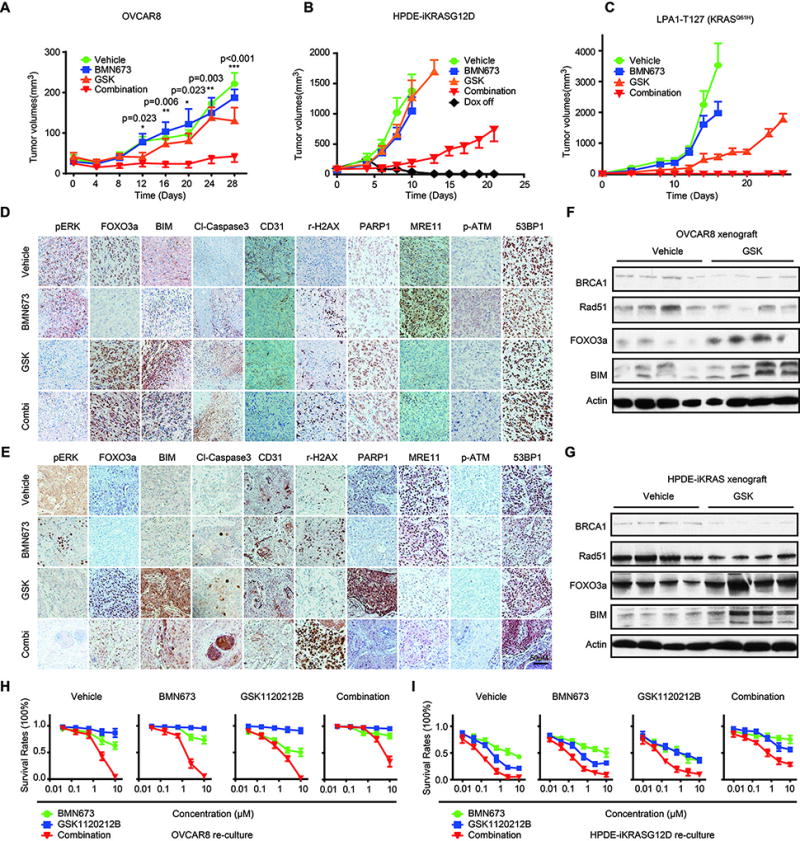
(A) Human OVCAR8 were grown as xenograft tumors in athymic nude mice. When tumors were 50–200 mm3, mice were randomized into treatment cohorts: vehicle (0.5% hydroxypropylmethylcellulose (HPMC) and 0.2% Tween-80), BMN673 (0.333 mg/kg/day), GSK1120212B (2 mg/kg/day), or the combination of BMN673 and GSK1120212B (n=8 for each group). Tumor measurements were performed 2 times per week by calipers, and average tumor volume ± SEM for each cohort is displayed. p values were determined by one-way ANOVA test.
(B) HPDE-iKRASG12D were injected into athymic nude mice subcutaneously. 7 days after tumor injection, mice were randomly assigned into Dox Off and 4 treatment groups with Dox on (via doxycycline diet (200 mg/kg; BioServ)): vehicle, BMN673 (0.333 mg/kg/day), GSK1120212B (2 mg/kg/day), or the combination of BMN673 and GSK1120212B (n = 8 per group).
(C) LPA1-T127 tumor tissues were transplanted into the mammary fat pads of FVB mice. Eight days later, mice were randomly assigned and treated accordingly. Mice were followed until death or sacrificed when tumor size reached 2 cm in diameter.
(D–E) Tumor tissues from OVCAR8 (D) and HPDE-iKRASG12D(E) xenografts were subjected to IHC analyses and probed with indicated antibodies. Representative images of IHC are shown with treatment indicated.
(F–G) Western blots showing indicated protein changes in tumor tissues after treatment with MEKi or vehicle in OVCAR8 xenografts (F) and HPDE-iKRASG12D xenografts (G).
(H–I) Cells from OVCAR8 (H) and HPDE-iKRASG12D (I) tumor-bearing mice were recultured in the absence of drug. Drug response curves are shown for individual and combination therapy with BMN673 and GSK1120212B for 96 h in recultured cells from OVCAR8 and HPDE-iKRASG12D xenografts. 3 recultured cell lines from each group were assessed, and representative results are presented as mean±SEM of three independent experiments.
We also assessed efficacy of PARPi and MEKi combinations in HPDE-iKRASG12D tumors. Mutant KRAS was induced with Dox, and tumors were allowed to establish in vivo. In this model, PARPi or MEKi alone were without activity (Fig. 8B). Once again, MEKi and PARPi combinations markedly decreased tumor growth. As demonstrated in previous studies (28), removal of Dox and thus KRAS expression resulted in complete tumor regression.
We also assessed effects of PARPi and MEKi combinations in a MMTV-LPA receptor transgene-induced transplantable tumor that had acquired a spontaneous KRASQ61H mutation (LPA1-127) (39). Like human PDX, the LPA1-127 tumor has never been cultured on plastic and thus may be more representative of human tumors. Furthermore, LPA receptor transgene induced tumors are late onset, heterogeneous, and associated with an inflammatory response similar to human tumors (38). In this syngeneic tumor model, PARPi and MEKi combinations induced tumor regressions that were maintained (Fig. 8C). Indeed, complete tumor regressions extended beyond the need to terminate mice due to tumor volume in other treatment groups. There was no evidence of toxicity as indicated by weight loss in mice with either mono or combination therapy with PARPi and MEKi (fig. S8A).
Immunohistochemical analysis of OVCAR8 and HPDE-iKRASG12D tumors at the termination of the study demonstrated that pERK was decreased in MEKi-treated tumors, consistent with MEKi fully inhibiting its target at doses used throughout the duration of the study (Fig. 8D, E). As expected, FOXO3a and BIM were decreased in BMN673-treated tumors, increased in MEKi-treated tumors, and increased compared to PARPi-treated tumors in mice that received MEKi/PARPi combinations. This was associated with induction of activated caspase 3 indicative of apoptosis and of γ-H2AX indicative of DNA damage, particularly in MEKi and PARPi treated cells (Fig. 8D–E).
CD31, which is expressed by endothelial cells, was markedly decreased in MEKi/PARPi-treated tumors (Fig. 8D–E). This was consistent with a blanched appearance of tumors excised from the mice (fig. S8B). The observations that mutant RAS induces VEGF production and that VEGFR signals through the RAS/MAPK pathway (40, 41) could contribute to decreases in vascularity observed with PARPi and MEKi combinations. Indeed, VEGF expression is dependent on RAS pathway activity in multiple RAS-mutant cell line models (fig. S5A–D). PARP1 was decreased in PARPi-treated cells but increased in MEKi-treated tumors, with PARP1 being higher in PARPi plus MEKi-treated tumors than in PARPi monotherapy treated tumors (Fig. 8D, E).
MRE11 was markedly decreased in MEKi-treated tumors and remained low in tumors treated with both PARPi and MEKi (Fig. 8D, E). In data from TCGA samples (TCPAportal.com), MRE11 protein was highly correlated with pCHK2 (r=0.5, p=3e−287) and CD31 (r=0.47 p=4.9e−255) suggesting that the relationship between MRE11 and CD31 in MEKi and PARPi treated tumors may reflect concurrent effects of RAS/MAPK activity on both pathways. Since MRE11 and the MRN complex are required for optimal activation of ATM (42), parallel effects on MRE11 and pATM supports importance of decreased MRE11 and MRN complex function in activity of PARPi and MEKi combinations. 53BP1 was not significantly altered in tumors treated with PARPi or MEKi or the combination (Fig. 8D, E). This is consistent with the major effects of the MEKi on sensitivity to PARPi being mediated by effects on HR rather than NHEJ (Fig. 5). Western blotting data of tumors confirmed that MEKi decreased expression of BRCA1and RAD51 while increasing BIM and FOXO3a (Fig. 8F, G).
Our in vitro studies provided evidence that PARPi-resistant cells would be sensitive to effects of PARPi and MEKi combinations (Fig 3). To test this further, we re-cultured cells from residual OVCAR8 and HPDE-iKRASG12D tumors after treatment and determined their sensitivity to the drugs. As noted in Fig. 8H–I, responses of re-cultured cells from vehicle-treated tumors recapitulated responses of parental cells to PARPi and MEKi. Although OVCAR8 from BMN673-treated tumors did not demonstrate differential responses to the inhibitors, HPDE-iKRASG12D from BMN673 treated tumors were resistant to PARPi but not to MEKi or PARPi and MEKi combinations. Similarly, HPDE-iKRASG12D from MEKi-treated tumors demonstrated decreased sensitivity to MEKi (OVCAR8 are constitutively resistant to MEKi, so a change could not be assessed) but were sensitive to both PARPi and the combination. Thus, both OVCAR8 and HPDE-iKRASG12D from PARPi or MEKi treated tumors retained sensitivity to PARPi and MEKi combinations, suggesting that MEKi or PARPi treated patients could still benefit from PARPi plus MEKi combinations. In contrast, OVCAR8 and HPDE-iKRASG12D from tumors treated with PARPi and MEKi combinations were modestly resistant to individual drugs and more resistant to combinations than DMSO-treated cells, suggesting that patients who fail the combination are likely to need alternative therapies.
Discussion
We demonstrated that KRAS and NRAS mutant tumors and to a lesser extent BRAF mutant tumors are highly sensitive to PARPi and MEKi/ERKi combinations in vitro and, for KRAS mutant models and the PARPi and MEKi combination, in vivo. The combination was well tolerated at active doses in mice, and further doses used in mice were similar to those used in previous studies and represent achievable monotherapy doses in humans (43, 44). Synergistic activity was observed with two structurally distinct PARPi, indicating that the activity is likely on target and a class effect. Similarly, two different MEKi and an ERKi demonstrated synergy with PARPi, again consistent with the RAS/MAPK pathway being a direct mediator. However, BRAF inhibitors were less effective in combination with PARPi, suggesting that targeting specific nodes in the RAS/MAPK pathway might demonstrate differential efficacy potentially due to feedback or bypass mechanisms activated by inhibition of different nodes (25, 45). The sensitivity of RAS mutant cells to the combination appears to be independent of intrinsic gene expression patterns as it is observed across multiple different lineages. Because synergistic responses to MEKi and PARPi combinations were independent of p53 mutation status, the approach should be effective in both p53 wild type and mutant tumors. Together, the in vitro and in vivo data argue that a MEKi and PARPi combination has the potential to induce cell death and increase the magnitude, duration, and spectrum of PARPi activity.
Many patients who initially respond to PARPi rapidly develop resistance and progress (46). Cell lines rendered resistant to PARPi have elevated RAS/MAPK signaling independent of mutations in KRAS. Resistance to PARPi in cell lines selected for resistance in vitro as well as in cells recultured from PARPi treated tumors in vivo was reversed, at least in part, by MEKi, suggesting that PARPi and MEKi combination may be active in patients who have failed PARPi. Because many breast and ovarian cancer patients are receiving PARPi and patients rapidly become resistant to PARPi, PARPi-resistant patients will soon represent a large population of breast and ovarian cancer patients who require therapeutic alternatives.
PARPi and MEKi combinations were effective in cells that did not have aberrations in BRCA1/2, suggesting that the combination could expand the spectrum of patients likely to benefit from PARPi beyond those with tumors with intrinsic HR defects. Indeed, PARPi and MEKi combinations were highly effective in cell lines with high HR competence due to RAS or BRAF mutations. Although RAS, and to a lesser degree BRAF mutations, are robust predictors of synergistic interactions between PARPi and MEK/ERK inhibitors, not all RAS or BRAF mutant lines demonstrated synergy with the combination. The combination demonstrated marked activity in 95% of the RAS and BRAF mutant lines tested based on synergy in 8/8 RAS mutant ovarian lines and 25/37 of RAS and BRAF mutant lines from other lineages combined with monotherapy activity of PARPi or MEKi in 10 of the 12 RAS or BRAF mutant lines that did not demonstrate synergism. Thus if effective biomarkers of response with greater predictive value than RAS/BRAF mutations or BIM protein cannot be identified, and if the combination has acceptable tolerability in humans as it does in mice, the combination would be expected to demonstrate activity in the majority of patients with RAS or BRAF mutant tumors (defined as response to the monotherapy or the combination therapy).
Given that only a subset of tumors with BRCA1/2 mutants respond to PARPi, it is reasonable to hypothesize that RAS mutations or pathway activation plays a role in resistance to PARPi in some BRCA1/2 mutant tumors such as those represented by Capan1, a BRCA2/KRAS mutant cell line resistant to PARPi. RAS mutations are rare in BRCA1/2 mutated ovarian and breast cancers, but physiological pathway activation (6, 7) could mediate PARPi resistance, which could potentially be reversed by MEKi. In contrast, in other lineages such as pancreas and stomach, BRCA1/2 and KRAS mutations do co-occur (cBIOportal.org).
The adaptive responses induced by PARPi and MEKi suggested multiple potential mechanisms underlying synergistic activity of the combination that were further validated in vitro and in vivo. The synergistic activity of PARPi and MEKi combinations is associated with: 1) altered RAS/MAPK signaling, 2) decreased HR repair capacity including decreased BRCA1/2 and DNA checkpoint activity leading to increased DNA damage, 3) decreased expression of MRN components, in particular MRE11, 4) FOXO3a and BIM mediated apoptosis, 5) increased PARP1, which is required for optimal activity of trapping PARPi, and 6) decreased vascularity, which could increase hypoxia and resultant PARPi efficacy. The majority of changes in total protein amounts could be detected at an mRNA level, suggesting that many of the effects are transcriptional. However, several were best reflected by changes in phosphorylation consistent with post-translational effects.
The ability of MEKi to increase FOXO3a appears to be central to the effects of MEKi in RAS mutant cells and to explain, to a major degree, the synergy between PARPi and MEKi. Enforced expression of FOXO3a was sufficient to mimic many of the effects of MEKi. Although the induction of apoptosis due to FOXO3a induction of BIM was expected, surprisingly, FOXO3a was sufficient to decrease MRE11, BRCA1, and RAD51 consistent with decreased ability to repair DNA damage in untreated cells or in the presence of either IR or PARPi. Although PI3K is located downstream of mutant RAS, and PI3K regulates FOXO3a (47), PI3K inhibitors were not synergistic with PARPi in RAS mutant cells. Furthermore, the effects of PI3K inhibitors on BIM and FOXO3a were modest compared to that of MEKi in RAS mutant tumors. This may be in part due to RAS/MAPK regulating BIM stability as well as transcription. PARPi and MEKi combinations were associated with decreased vascularity and CD31 expression in treated tumors. The observations that mutant RAS induces VEGF production and that VEGFR signals through the RAS/MAPK pathway (40, 41) could contribute to decreases in vascularity observed with MEKi. Indeed, our data indicate that VEGF expression are RAS-dependent in multiple systems. Furthermore, FOXO3a regulates VEGF production through FOXM1 (48). Because hypoxia increases the activity of PARPi and the combination of a PARPi and cedirainib (a VEGFR inhibitor) is highly active in patients (49), the effect of MEKi and PARPi on vascularity could contribute to activity of the combination in vivo. The correlation between MRE11 and CD31 in TCGA patient samples before treatment suggests that there may be an additional unexpected relationship between DNA damage and tumor vascularity.
MRE11 and decreased activity of the MRN complex and subsequent activation of DNA damage repair and DNA damage checkpoint activity induced by MEKi appears to contribute to the efficacy of PARPi and MEKi combinations. Indeed, down-regulation of MRE11 was sufficient to increase activity of PARPi. MEKi as well as FOXO3a induction induced a delayed decrease in MRE11 as monotherapy. However, FOXO3a and MEKi were not sufficient to markedly decrease RAD50 or NBS1 protein in vitro. In vivo, MEKi monotherapy markedly decreased MRE11 protein, likely due to the prolonged in vivo treatment. Thus downregulation of MRE11 appears to require prolonged exposure to MEKi. MRE11 inhibitors have been identified (36) and may represent an alternative approach to MEKi to increase activity of PARPi. These inhibitors only block the enzyme activity of MRE11 and not the scaffolding activity, and thus may not fully recapitulate loss of MRE11. Further, MRE11 inhibition may not be sufficient to mimic the full spectrum of activities of MEKi in RAS mutant tumors.
The ability of MEKi to increase PARP1 could not be explained by actions of FOXO3a alone. The ability of PARPi and MEKi combinations to optimally decrease cell viability is dependent on the amount of PARP1. This is compatible with the contention that PARP1 is necessary for toxicity mediated by trapping PARPi (13, 22). This suggests that non-trapping PARPi may not demonstrate the same degree of synergy with MEKi as do trapping PARPi. FOXO3a induction was insufficient to increase PARP.
The studies herein add to a number of previous studies that suggest that adaptive responses to targeted therapies provide an opportunity to rapidly and efficiently identify potential therapeutic interventions that would convert cytostatic responses observed with many targeted therapies into cytotoxicity with a greater benefit (1–3). While analysis of RNA or other omics approaches such as metabolomics or lipidomics provides an opportunity to identify adaptive responses, most targeted therapies that are being developed target proteins, and changes in protein function mediate resistance to targeted therapies. RNA and protein expression and particularly the amounts of post-translationally modified and functional proteins are frequently weakly correlated (50). Although it may be possible to impute protein function by quantifying downstream RNA master regulators (51), analysis of proteins and protein function may provide a more direct assessment. Thus, analysis of protein function may provide a more efficient approach to identify adaptive responses. Although emerging mass spectrometry approaches have the ability to identify adaptive responses, RPPA provides a cost-effective, efficient approach for sparse unbiased analysis of pathways involved in adaptive responses. In particular, RPPA has increased sensitivity for a number of phosphoproteins over mass spectrometry, and mass spectrometry is “blind” to a number of phosphoproteins due to a lack of convenient cleavage sites (52).
This study has a number of limitations. While we have demonstrated synergy between the PARP and MEK combination in KRAS and NRAS and to a lesser degree BRAF mutant lines in vitro, we have not assessed HRAS mutant models and have only assessed KRAS mutant models in vivo. Thus we do not know whether the combination would be effective when the RAS/MAPK pathway is activated by other mechanisms such as loss of NF1 or activation of cell surface receptors. While OVCAR8 used in these studies has a KRAS mutation and activation of the RAS/MAPK pathway to similar levels to cells with KRAS mutations, it is not clear whether the KRAS mutation in OVCAR8 is a driver or passenger. At most it is modestly activating. While we have demonstrated that PARPi-resistant A2780CP acquire KRASA146T or KRASA59T and MAP2K1A283T mutations and that these are modulated by incubation in PARPi, we have not demonstrated that these mutations drive resistance to PARPi and further, we have not identified a mechanism explaining the PARPi resistance in the UWB1.289 clone. This is of particular importance as the parental UWB1.289 clone has a deleterious BRCA1 mutant and is PARP sensitive. Although we are able to explain almost all of the effects of inhibition of MEKi on the sensitization of cells to PARPi by an induction of FOXO3A function, we have not elucidated the mechanism underlying increases in PARP1 and, while there is literature support for a FOXO3A pathway to CD31, we have not demonstrated this functionally in the models tested. Finally, of course, while the preclinical data is strongly supportive of both efficacy and acceptable toxicity, we have not yet demonstrated activity and tolerability of the combination in human patients.
In summary, MEK and ERK inhibitors are synergistic with PARPi in the majority of RAS-mutant lines evaluated as well as in PARPi-resistant cells. MEKi alter apoptotic balance, induce HR deficiency, and decrease DNA damage checkpoint activity in RAS-mutant cells. These act together to increase DNA damage induced by PARPi and to increase apoptosis in response to PARPi. The activity of the combination is independent of BRCA1/2 and p53 mutation status. Together the data indicate that RAS mutant tumors as well as tumors that have become resistant to PARPi are likely to benefit from the combination. These data indicate that exciting clinical activity of PARPi in BRCA1/2 mutant tumors could be extended to RAS and BRAF mutant tumors. Thus PARPi and MEKi/ERKi combination therapy warrants exploration in human trials.
Materials and Methods
Study design
The objective of this study was to evaluate adaptive resistance to PARPi through high-throughput pathway surveillance tools (RPPA), and to develop a rational combinational therapy that would lead to synthetic lethality. We demonstrate that PARP and MEK inhibitors demonstrate synergy in vitro and in vivo in a wide spectrum of RAS-mutant tumor models. Sample size in experiments was selected based on previous experience and was sufficient to detect statistically significant differences between treatments. Mice were randomized to different treatment groups, without blinding. Study groups were followed until individual tumor or entire cohort measurements reached 2 cm in diameter, at which point sacrifice was indicated in accordance with Institutional Animal Care and Use Committee (IACUC) protocols. All in vitro experiments were run in biological triplicates, except where specified otherwise
Statistical analysis
Two-sided Student’s t test was used to compare differences between two groups of cells in vitro. Data are presented as means ± SEM and p<0.05 is considered significant. Analysis of variance was used to compare differences among multiple groups. Differences in survival of mice were examined by Kaplan–Meier curves with the log-rank test. Two-sided p less than 0.05 was considered significant. All statistical analyses were done using SPSS 17.0 (SPSS Inc.).
Supplementary Material
Supplementary materials and methods
Fig S1. PARP and MEK inhibitors induce inverse adaptive responses
Fig S2. Long-term assay of combination therapy with PARPi and MEKi demonstrate low doses of drugs show synergy in OVCAR8.
Fig S3. PARPi plus MEKi /ERKi demonstrate synergy in cells with RAS mutations.
Fig S4. Synergistic effects of PARP and MEK/ERK inhibition are lineage independent and observed with KRAS/NRAS/BRAF mutations.
Fig S5. RAS/ERK pathway activity is associated with HR competence, DNA damage checkpoints and apoptotic sensitivity
Fig S6. MEKi decrease HR competence, and enhance DNA damage induced by IR or PARPi
Fig S7. PARP and MRE11 but not FoxM1 contribute to the synergistic effects of PARPi and MEKi
Fig. S8. Effect of PARPi, MEKi and combination therapy on the body weight of mice.
Summary.
Few therapeutic options are available for patients with RAS mutant tumors or for patients who previously failed treatment with PARP inhibitors. We demonstrate that the rational combination of PARP and MEK inhibitors warrants investigation in patients with RAS or RAF mutant tumors or in patients who have failed single agent PARP inhibitor therapy.
Acknowledgments
We thank Prof. Anirban Maitra (Division of Pathology/Lab Medicine, The University of Texas MD Anderson Cancer Center) for iKRAS1, iKRAS2, Pa01c, Pa02c, Pa03C, Pa04c, Pa09c, Pa16c, Pa18c, Pa21c, and Prof. Boudewijn M T Burgering (Department of Molecular Cancer Research, University Medical Centre, Utrecht, The Netherlands) for DLD1 and DL23.
Funding: Susan G Komen SAC110052 (GBM), NIH grants 5U01CA168394 (GBM and KS), 5P50CA098258 (GBM, SNW), 5P50CA083639 (GBM, SNW) and Andrew Sabin Family Fellowship Program (SNW) and CCSG grant CA016672 as well as a gift from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (GBM and JB).
Footnotes
Author Contributions: C.S., Y.F., J.Y., and J.C. conceived and performed experiments. Z.J. analyzed RPPA data. X.C., C.P., and Y.L. performed RPPA. D.Z. conducted some immunohistochemical evaluation, K. J., and A. K. performed deep NGS sequencing as well as Sanger sequencing of PARPi-resistant clones. P.K. established wild-type KRAS or mutant KRAS and PIK3CA BaF3 cell lines. N.B., S.N.W., J.R.G., S.L, K.L.S., G.P., J.B., and G.B.M. provided expertise and feedback. G.B.M. conceived and coordinated the project and wrote the manuscript.
Competing interest: GBM has licensed a HRD assay to Myriad Genetics and is a SAB member/consultant with AstraZeneca, Catena Pharmaceuticals, Critical Outcome Technologies, ImmunoMET, Ionis, Medimmune, Nuevolution, Pfizer, Precision Medicine, Signalchem Lifesciences, Symphogen, Takeda/Millennium Pharmaceuticals, and Tarveda, and has stock options with Catena Pharmaceuticals, ImmunoMet, Spindle Top Ventures, and Tarveda. SW is a consultant with AstraZeneca, Clovis Oncology, Genentech, Medivation, Casdin Capital and Vermillion. GBM receives research support from Abbvie, Adelson Medical Research Foundation, AstraZeneca, Breast Cancer Research Foundation, Critical Outcomes Technology, Horizon Diagnostics, Illumina, Ionis, Karus Therapeutics, Komen Research Foundation, Nanostring, Pfizer, Takeda/Millennium Pharmaceuticals, and Tesaro.
References and notes
- 1.Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, Gao S, Mills GB, Brugge JS. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer cell. 2012;21:227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kugel CH, 3rd, Hartsough EJ, Davies MA, Setiady YY, Aplin AE. Function-blocking ERBB3 antibody inhibits the adaptive response to RAF inhibitor. Cancer research. 2014;74:4122–4132. doi: 10.1158/0008-5472.CAN-14-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwong LN, Boland GM, Frederick DT, Helms TL, Akid AT, Miller JP, Jiang S, Cooper ZA, Song X, Seth S, Kamara J, Protopopov A, Mills GB, Flaherty KT, Wargo JA, Chin L. Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. The Journal of clinical investigation. 2015;125:1459–1470. doi: 10.1172/JCI78954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diaz-Padilla I, Malpica AL, Minig L, Chiva LM, Gershenson DM, Gonzalez-Martin A. Ovarian low-grade serous carcinoma: a comprehensive update. Gynecologic oncology. 2012;126:279–285. doi: 10.1016/j.ygyno.2012.04.029. [DOI] [PubMed] [Google Scholar]
- 6.N. Cancer Genome Atlas Research. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hew KE, Miller PC, El-Ashry D, Sun J, Besser AH, Ince TA, Gu M, Wei Z, Zhang G, Brafford P, Gao W, Lu Y, Mills GB, Slingerland JM, Simpkins F. MAPK Activation Predicts Poor Outcome and the MEK Inhibitor, Selumetinib, Reverses Antiestrogen Resistance in ER-Positive High-Grade Serous Ovarian Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22:935–947. doi: 10.1158/1078-0432.CCR-15-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patton SE, Martin ML, Nelsen LL, Fang X, Mills GB, Bast RC, Jr, Ostrowski MC. Activation of the ras-mitogen-activated protein kinase pathway and phosphorylation of ets-2 at position threonine 72 in human ovarian cancer cell lines. Cancer research. 1998;58:2253–2259. [PubMed] [Google Scholar]
- 9.Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–3790. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 10.Schultz N, Lopez E, Saleh-Gohari N, Helleday T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic acids research. 2003;31:4959–4964. doi: 10.1093/nar/gkg703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iglehart JD, Silver DP. Synthetic lethality--a new direction in cancer-drug development. N Engl J Med. 2009;361:189–191. doi: 10.1056/NEJMe0903044. [DOI] [PubMed] [Google Scholar]
- 12.Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic acids research. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer research. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, Pommier Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13:433–443. doi: 10.1158/1535-7163.MCT-13-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, Liu W, Yang JY, Yoshihara K, Li J, Ling S, Seviour EG, Ram PT, Minna JD, Diao L, Tong P, Heymach JV, Hill SM, Dondelinger F, Stadler N, Byers LA, Meric-Bernstam F, Weinstein JN, Broom BM, Verhaak RG, Liang H, Mukherjee S, Lu Y, Mills GB. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nature communications. 2014;5:3887. doi: 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toulany M, Schickfluss TA, Eicheler W, Kehlbach R, Schittek B, Rodemann HP. Impact of oncogenic K-RAS on YB-1 phosphorylation induced by ionizing radiation. Breast Cancer Res. 2011;13:R28. doi: 10.1186/bcr2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang X, Yu S, Eder A, Mao M, Bast RC, Jr, Boyd D, Mills GB. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene. 1999;18:6635–6640. doi: 10.1038/sj.onc.1203076. [DOI] [PubMed] [Google Scholar]
- 18.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. The Journal of biological chemistry. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panupinthu N, Yu S, Zhang D, Zhang F, Gagea M, Lu Y, Grandis JR, Dunn SE, Lee HY, Mills GB. Self-reinforcing loop of amphiregulin and Y-box binding protein-1 contributes to poor outcomes in ovarian cancer. Oncogene. 2014;33:2846–2856. doi: 10.1038/onc.2013.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. Journal of immunology. 2002;168:5024–5031. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- 21.Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 22.Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, Giri U, Peyton M, Fan YH, Diao L, Masrorpour F, Shen L, Liu W, Duchemann B, Tumula P, Bhardwaj V, Welsh J, Weber S, Glisson BS, Kalhor N, Wistuba II, Girard L, Lippman SM, Mills GB, Coombes KR, Weinstein JN, Minna JD, Heymach JV. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer discovery. 2012;2:798–811. doi: 10.1158/2159-8290.CD-12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nature reviews. Cancer. 2015;15:577–592. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- 24.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nature communications. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolch W, Halasz M, Granovskaya M, Kholodenko BN. The dynamic control of signal transduction networks in cancer cells. Nature reviews. Cancer. 2015;15:515–527. doi: 10.1038/nrc3983. [DOI] [PubMed] [Google Scholar]
- 26.Cheung LW, Yu S, Zhang D, Li J, Ng PK, Panupinthu N, Mitra S, Ju Z, Yu Q, Liang H, Hawke DH, Lu Y, Broaddus RR, Mills GB. Naturally occurring neomorphic PIK3R1 mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathway inhibitors. Cancer cell. 2014;26:479–494. doi: 10.1016/j.ccell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang H, Cheung LW, Li J, Ju Z, Yu S, Stemke-Hale K, Dogruluk T, Lu Y, Liu X, Gu C, Guo W, Scherer SE, Carter H, Westin SN, Dyer MD, Verhaak RG, Zhang F, Karchin R, Liu CG, Lu KH, Broaddus RR, Scott KL, Hennessy BT, Mills GB. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 2012;22:2120–2129. doi: 10.1101/gr.137596.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsang YH, Dogruluk T, Tedeschi PM, Wardwell-Ozgo J, Lu H, Espitia M, Nair N, Minelli R, Chong Z, Chen F, Chang QE, Dennison JB, Dogruluk A, Li M, Ying H, Bertino JR, Gingras MC, Ittmann M, Kerrigan J, Chen K, Creighton CJ, Eterovic K, Mills GB, Scott KL. Functional annotation of rare gene aberration drivers of pancreatic cancer. Nature communications. 2016;7:10500. doi: 10.1038/ncomms10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, DePinho RA. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tyner JW, Erickson H, Deininger MW, Willis SG, Eide CA, Levine RL, Heinrich MC, Gattermann N, Gilliland DG, Druker BJ, Loriaux MM. High-throughput sequencing screen reveals novel, transforming RAS mutations in myeloid leukemia patients. Blood. 2009;113:1749–1755. doi: 10.1182/blood-2008-04-152157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 32.Peng G, Chun-Jen Lin C, Mo W, Dai H, Park YY, Kim SM, Peng Y, Mo Q, Siwko S, Hu R, Lee JS, Hennessy B, Hanash S, Mills GB, Lin SY. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nature communications. 2014;5:3361. doi: 10.1038/ncomms4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. The EMBO journal. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kannike K, Sepp M, Zuccato C, Cattaneo E, Timmusk T. Forkhead transcription factor FOXO3a levels are increased in Huntington disease because of overactivated positive autofeedback loop. The Journal of biological chemistry. 2014;289:32845–32857. doi: 10.1074/jbc.M114.612424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tenbaum SP, Ordonez-Moran P, Puig I, Chicote I, Arques O, Landolfi S, Fernandez Y, Herance JR, Gispert JD, Mendizabal L, Aguilar S, Ramon y Cajal S, Schwartz S, Jr, Vivancos A, Espin E, Rojas S, Baselga J, Tabernero J, Munoz A, Palmer HG. beta-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nature medicine. 2012;18:892–901. doi: 10.1038/nm.2772. [DOI] [PubMed] [Google Scholar]
- 36.Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, Ismail A, Ismalaj E, Petricci E, Neale MJ, Bristow RG, Masson JY, Wyman C, Jeggo PA, Tainer JA. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Molecular cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vilar E, Bartnik CM, Stenzel SL, Raskin L, Ahn J, Moreno V, Mukherjee B, Iniesta MD, Morgan MA, Rennert G, Gruber SB. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer research. 2011;71:2632–2642. doi: 10.1158/0008-5472.CAN-10-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zona S, Bella L, Burton MJ, Nestal de Moraes G, Lam EW. FOXM1: an emerging master regulator of DNA damage response and genotoxic agent resistance. Biochimica et biophysica acta. 2014;1839:1316–1322. doi: 10.1016/j.bbagrm.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F, Yu S, Stephens LC, Cui X, Murrow G, Coombes K, Muller W, Hung MC, Perou CM, Lee AV, Fang X, Mills GB. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer cell. 2009;15:539–550. doi: 10.1016/j.ccr.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi O, Komaki R, Smith PD, Jurgensmeier JM, Ryan A, Bekele BN, Wistuba II, Jacoby JJ, Korshunova MV, Biernacka A, Erez B, Hosho K, Herbst RS, O’Reilly MS. Combined MEK and VEGFR inhibition in orthotopic human lung cancer models results in enhanced inhibition of tumor angiogenesis, growth, and metastasis. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:1641–1654. doi: 10.1158/1078-0432.CCR-11-2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel RS. Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer research. 1995;55:4575–4580. [PubMed] [Google Scholar]
- 42.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science (New York, N.Y.) 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 43.Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, Wang B, Lord CJ, Post LE, Ashworth A, BMN 673. a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:5003–5015. doi: 10.1158/1078-0432.CCR-13-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, Zappacosta F, Annan R, Sutton D, Laquerre SG. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 45.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, Santiago-Walker AE, Letrero R, D’Andrea K, Pushparajan A, Hayden JE, Brown KD, Laquerre S, McArthur GA, Sosman JA, Nathanson KL, Herlyn M. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Macpherson E, Watkins C, Carmichael J, Matulonis U. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. The New England journal of medicine. 2012;366:1382–1392. doi: 10.1056/NEJMoa1105535. [DOI] [PubMed] [Google Scholar]
- 47.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 48.Karadedou CT, Gomes AR, Chen J, Petkovic M, Ho KK, Zwolinska AK, Feltes A, Wong SY, Chan KY, Cheung YN, Tsang JW, Brosens JJ, Khoo US, Lam EW. FOXO3a represses VEGF expression through FOXM1-dependent and -independent mechanisms in breast cancer. Oncogene. 2012;31:1845–1858. doi: 10.1038/onc.2011.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF, Rimel B, Buss MK, Nattam S, Hurteau J, Luo W, Quy P, Whalen C, Obermayer L, Lee H, Winer EP, Kohn EC, Ivy SP, Matulonis UA. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. The Lancet. Oncology. 2014;15:1207–1214. doi: 10.1016/S1470-2045(14)70391-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cenik C, Cenik ES, Byeon GW, Grubert F, Candille SI, Spacek D, Alsallakh B, Tilgner H, Araya CL, Tang H, Ricci E, Snyder MP. Integrative analysis of RNA, translation, and protein levels reveals distinct regulatory variation across humans. Genome Res. 2015;25:1610–1621. doi: 10.1101/gr.193342.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, Califano A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet. 2016;48:838–847. doi: 10.1038/ng.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, Kawaler E, Mundt F, Krug K, Tu Z, Lei JT, Gatza ML, Wilkerson M, Perou CM, Yellapantula V, Huang KL, Lin C, McLellan MD, Yan P, Davies SR, Townsend RR, Skates SJ, Wang J, Zhang B, Kinsinger CR, Mesri M, Rodriguez H, Ding L, Paulovich AG, Fenyo D, Ellis MJ, Carr SA, Nci C. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science (New York, N.Y.) 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eijkelenboom A, Mokry M, de Wit E, Smits LM, Polderman PE, van Triest MH, van Boxtel R, Schulze A, de Laat W, Cuppen E, Burgering BM. Genome-wide analysis of FOXO3 mediated transcription regulation through RNA polymerase II profiling. Molecular systems biology. 2013;9:638. doi: 10.1038/msb.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Fig S1. PARP and MEK inhibitors induce inverse adaptive responses
Fig S2. Long-term assay of combination therapy with PARPi and MEKi demonstrate low doses of drugs show synergy in OVCAR8.
Fig S3. PARPi plus MEKi /ERKi demonstrate synergy in cells with RAS mutations.
Fig S4. Synergistic effects of PARP and MEK/ERK inhibition are lineage independent and observed with KRAS/NRAS/BRAF mutations.
Fig S5. RAS/ERK pathway activity is associated with HR competence, DNA damage checkpoints and apoptotic sensitivity
Fig S6. MEKi decrease HR competence, and enhance DNA damage induced by IR or PARPi
Fig S7. PARP and MRE11 but not FoxM1 contribute to the synergistic effects of PARPi and MEKi
Fig. S8. Effect of PARPi, MEKi and combination therapy on the body weight of mice.