

Summary
Developing effective therapeutics for complex diseases such as late-onset, sporadic Alzheimer’s disease (SAD) is difficult due to genetic and environmental heterogeneity in the human population and the limitations of existing animal models. Here, we used hiPSC-derived neurons to test a compound that stabilizes the retromer, a highly conserved multiprotein assembly that plays a pivotal role in trafficking molecules through the endosomal network. Using this human-specific system, we have confirmed previous data generated in murine models and show that retromer stabilization has a potentially beneficial effect on amyloid beta generation from human stem cell-derived neurons. We further demonstrate that manipulation of retromer complex levels within neurons affects pathogenic TAU phosphorylation in an amyloid-independent manner. Taken together, our work demonstrates that retromer stabilization is a promising candidate for therapeutic development in AD and highlights the advantages of testing novel compounds in a human-specific, neuronal system.
Keywords: human induced pluripotent stem cells, Alzheimer’s disease, retromer complex stabilization
Graphical Abstract
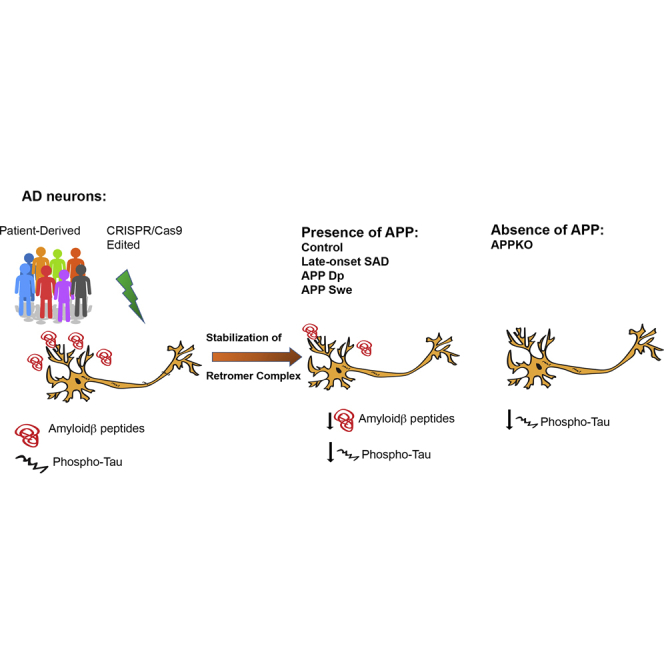
Highlights
-
•
A retromer stabilizing molecule reduces Aβ and phospho-TAU levels in human neurons
-
•
The molecule reduces Aβ and pTau in both SAD and FAD cell lines
-
•
Retromer stabilization reduces tau phosphorylation in an APP-independent manner
-
•
Confirms studies in mice and highlights hiPSCs as a preclinical model
In this work, Young and colleagues test how stabilization of a large endocytic trafficking complex, the retromer assembly, reduces cellular AD phenotypes in a human neuronal model. Using both patient-derived and genome-edited hiPSCs, they show that enhancement of retromer function affects both APP processing and tau phosphorylation independently.
Introduction
Alzheimer’s disease (AD) is a devastating disorder of the brain and a rapidly growing public health problem. Patients with rare mutations in the amyloid precursor protein (APP) and presenilin 1 and 2 genes (PSEN 1 and PSEN 2) make up about 5% of familial AD cases (FAD) while the vast majority of AD is late-onset, sporadic AD (SAD) (Avramopoulos, 2009). SAD is a complex and heterogeneous disorder with a clear heritable component that defines genetic risk (Gatz et al., 2006) but with unknown contributions from the environment. The lack of effective therapies for AD is, in large part, due to our lack of understanding of the cellular and molecular mechanisms that lead to the neuropathological outcomes of the disease. Reasons for this are several-fold. In particular, AD takes decades to manifest clinically, and irreversible cellular damage likely occurs before the overt clinical symptoms are detected. In addition, the human brain is an inaccessible organ, making it difficult to sample tissue from living patients. Human induced pluripotent stem cells (hiPSCs) have facilitated the development of human neuronal models for AD, as they can be differentiated into disease-appropriate cell types and maintain the unique patient genetic background. To date, AD has been successfully modeled using patient and isogenic cell lines, and disease-relevant phenotypes have been well-documented from stem cell-derived neurons for both FAD and SAD (Israel et al., 2012, Kondo et al., 2013, Muratore et al., 2014, Raja et al., 2016, Woodruff et al., 2013, Yagi et al., 2011, Young et al., 2015).
We and others have demonstrated a role for endocytic genes in AD risk (Karch and Goate, 2015, Rogaeva et al., 2007, Young et al., 2015), and elegant microscopic analysis has documented enlarged endosomes and suggested endocytic dysfunction as an early phenotype in AD pathogenesis (Cataldo et al., 2000, Cataldo et al., 2008, Nixon et al., 2005). This work has been recapitulated in stem cell-derived neurons (Israel et al., 2012, Raja et al., 2016); however, to date, no AD therapy targets the endosomal network, likely due to the complexity of its structure and regulation.
Retromer is a multiprotein assembly with a primary role in the sorting and trafficking of plasma membrane proteins from the endosomes, to the trans-Golgi network, or directly back to the cell surface (Hu et al., 2015). APP is trafficked through the endocytic network via retromer through its interaction with SORLA, a member of the vacuolar protein-sorting VPS10 family of sorting receptors and a well-defined AD risk gene (Fjorback et al., 2012, Reitz et al., 2011, Reitz et al., 2013, Rogaeva et al., 2007). The retromer assembly has two major components, including the cargo-recognition complex (CRC), a trimeric core composed of VPS35, VPS29, and VPS26 (Li et al., 2016). The second component of retromer comprises the membrane targeting specific nexins, chiefly SNX1, SNX3, SNX5, SNX6, and SNX27 (Burd and Cullen, 2014). Retromer deficiency, primarily loss of VPS35 and VPS26, has been described in SAD patients and increases Aβ peptides in cell culture and cognitive decline in the mouse brain (Bhalla et al., 2012, Muhammad et al., 2008, Small et al., 2005, Wen et al., 2011).
Recently, novel pharmacological chaperones were developed that stabilize the trimeric core complex of VPS35, VPS29, and VPS26. These molecules, R33 and R55, reduced Aβ in mouse hippocampal neurons in a retromer-dependent manner (Mecozzi et al., 2014). Similarly, the retromer stabilization molecule R55 reduced Aβ in in a murine neuronal cell line harboring the APP Swedish mutation (Chu and Pratico, 2017). These studies support the idea that increasing the levels and/or functions of endocytic trafficking components may be viable options for therapeutic development in AD. However, to move forward in investigation of therapeutic strategies for humans, it is critical to test candidates in a human-specific system. In addition, while Aβ is a critical pathogenic readout for AD, how retromer stabilization affects other components of the disease process, such as TAU hyperphosphorylation, and if these two events are interdependent, is unknown. To address these questions, we used hiPSC-derived neurons from SAD and FAD patients and from genome-edited hiPSC lines. This allowed us to modulate the amounts of Aβ generation and APP expression in neurons and test how retromer stabilization by the small molecule chaperone R33 affects endogenous levels of Aβ and phospho-TAU (pTAU) in a human-specific system.
Results
Retromer Pharmacological Chaperones Reduce APP Processing and Aβ Peptides in hiPSC-Derived Neurons from Non-demented Controls, SAD Patients, and FAD Mutant Cells
The pharmacological chaperone R55 was previously shown to stabilize the retromer core complex, reduce Aβ peptides, and decrease APP in endosomes in mouse primary hippocampal neurons, suggesting that retromer stabilization is beneficial in terms of AD pathogenesis (Mecozzi et al., 2014). We extended these studies to test the effect of retromer stabilization in human neurons using an hiPSC approach. We tested both R55 and its analog R33 in FACS-purified neurons derived from non-demented control (NDC) hiPSCs. Our differentiation protocol has consistently been shown to recapitulate key cellular AD phenotypes in a purified system, allowing us to accurately quantitate analytes coming from a particular cell type (i.e., human neurons) (Israel et al., 2012, Young et al., 2015). We found that, in contrast to mouse neurons, R33 had a greater effect on Aβ peptide reduction than R55 in human neurons differentiated from hiPSCs (Figure S1).
We then tested a cohort of cell lines derived from participants in the University of California, San Diego (UCSD) Alzheimer's Disease Research Center (ADRC), which included six NDCs and seven SAD patients (n = 13 patient cell lines) (Figure 1A). We found that in cell lines derived from either NDC or SAD subjects, R33 treatment for 72 hr significantly reduced both Aβ 1–40 and 1–42 peptide species (Figures 1B–1E). Because this reduction occurred equally for both Aβ species, we did not observe significant changes in the Aβ 42:40 ratio (Figures 1F and 1G). This is similar to what was previously observed in mouse hippocampal neurons, where retromer stabilization decreased Aβ 40 and 42 peptides equally (Mecozzi et al., 2014). We observed variability among neuronal cultures in the amount of increase in VPS35 stability, which could be due to inherent variability between hiPSC lines, individual patient genomes, or the limited amount of material obtained from our purified neuron protocol (Figure S2A). However, in representative experiments, we documented detectable increases in VPS35 stability from purified neuronal cultures (Figure S2B).
Figure 1.

Retromer Stabilizing Chaperone R33 Lowers Aβ Peptides in hiPSC-Derived Neurons
(A) Schematic diagram of experimental design. hiPSCs were generated from fibroblast biopsies of six NDCs and seven probable SAD patients from the UCSD ADRC. These lines have been previously published and characterized in Israel et al. (2012) and Young et al. (2015). FACS purified neural stem cells and purified neurons were generated following previously published methods (Israel et al., 2012, Yuan et al., 2011).
(B and C) Aβ1-40 (B) and Aβ1-42 (C) peptides in purified neurons derived from NDC individuals treated with vehicle (dots) or R33 (squares) for 72 hr.
(D and E) Aβ1-40 (D) and Aβ1-42 (E) peptides in purified neurons derived from SAD patients treated with vehicle (dots) or R33 (squares) for 72 hr.
(F and G) Aβ 42:40 ratios in purified neurons derived from NDC (F) or SAD (G) patients treated with vehicle (dots) or R33 (squares) for 72 hr. n = 6 NDC individuals (represented by dots/squares); two to four independent experiments/individual/treatment. n = 7 SAD individuals (represented by dots/squares); two to four independent experiments/treatment.
Non-normally distributed data (A–E) were analyzed by Wilcoxon test. ∗p < 0.05. Normally distributed data (F and G) were analyzed by two-tailed t test. NS, nonsignificant. All error bars represent SD. See also Figures S1 and S2.
We next asked if retromer stabilization by R33 had an effect in genetic backgrounds of penetrant mutations leading to early-onset, FAD, either by increasing the levels of APP (APP duplication, APPDp) or by enhancing beta cleavage of APP (APP Swedish, APPSwe). In either patient neurons from FAD APPDp (Israel et al., 2012) or in isogenic neurons in which the Swedish mutation was introduced via CRISPR/Cas9 genome editing (Figure S2D) (Woodruff et al., 2016), we observed a similar fold-decrease in Aβ peptides from APPDp patients and the APPSwe isogenic neurons as with the NDC and SAD patient neurons (Figures 2A and 2B), suggesting that this small molecule chaperone is a potent reducer of Aβ, despite patient or cell line genetic background. Indeed, we observed a significant decrease in all forms of APP processing (Figure 2C) and observed a reduction in APP β-CTF in purified neurons treated with R33 (Figure 2D). Interestingly, we did not observe a significant increase in FL APP (Figure S2C), although this may be due to the variability of APP expression across all our different patient genetic backgrounds and the dynamic process between APP cleavage and recycling, where changes in APP cleavage may affect the overall expression of APP. Future experiments are needed to determine how retromer stabilization affects these processes. Taken together, these data corroborate previous work in the mouse and suggest that retromer stabilization keeps APP out of intracellular compartments that generate β-cleavage of APP and, subsequently, Aβ.
Figure 2.

Retromer Stabilization Reduces Aβ Peptides in FAD Mutant Genetic Backgrounds and Reduces Overall APP Processing in Human Neurons
(A) R33 treatment significantly reduces Aβ peptides in hiPSC-derived neurons generated from a patient with an APP duplication mutation (APPDp). n = 1 FAD individual, three independent experiments/treatment.
(B) R33 treatment significantly reduces Aβ peptides in CRISPR/Cas9 gene-edited isogenic neurons where the APP Swedish (APPSwe) mutation was introduced into wild-type (WT) cells by homologous recombination n = 1–2 isogenic clones/genotype, three independent experiments/treatment.
(C and D) R33 treatment reduces all APP processing in human neurons, including Aβ sAPPα, sAPPβ (C), and APP β CTF (D). n = 3 independent experiments/treatment.
For each comparison, a two-tailed t test was performed. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Error bars represent SD. See also Figure S2.
Retromer Pharmacological Chaperones Reduce the Phospho-TAU/Total TAU Ratio in hiPSC-Derived Neurons by Decreasing the Level of Phosphorylated TAU on Thr231
In addition to amyloid plaques, another neuropathological hallmark of AD is the presence of neurofibrillary tangles comprising hyperphosphorylated TAU protein. The connection between Aβ and TAU is complex, with the dominant theory that Aβ pathology precedes and drives TAU pathology (Hardy and Higgins, 1992), while other evidence suggests that these pathways can occur independently (Small and Duff, 2008). Neurons purified from hiPSCs have detectable levels of phosphorylated TAU on Thr231 and this correlates with increases in Aβ from APPDp patients (Israel et al., 2012). We tested the effect of retromer stabilization by R33 on the pTAU/total TAU (tTAU) ratio on our cohort of patient neurons and found that R33 treatment significantly decreased the pTAU/tTAU ratio in all cell lines, whether they were derived from NDC or SAD individuals (Figures 3A and 3B). Interestingly, this decrease in the ratio was due to an effect on pTAU (Figure 3C), while the levels of tTAU in the cultures remained unchanged (Figure 3D). R33 treatment had a similar effect on neurons derived from an APPDp patient, with a reduction of pTAU (Figure 3E), but not tTAU (Figure 3F), leading to a lowered pTAU/tTAU ratio in these neurons (Figure 3G).
Figure 3.

Retromer Stabilization Reduces the pTAU/tTAU Ratio in hiPSC-Derived Neurons
(A) pTAU/tTAU ratio in purified neurons derived from NDC individuals treated with vehicle (dots) or R33 (squares) for 72 hr. n = 6 NDC individuals (represented by dots/squares); two to four independent experiments/individual/treatment.
(B) pTAU/tTAU ratio in purified neurons derived from NDC (SAD) individuals treated with vehicle (dots) or R33 (squares) for 72 hr. n = 7 SAD individuals (represented by dots/squares); two to four independent experiments/treatment.
(C) R33 reduces the TAU ratio by lowering phosphorylated TAU on Thr 231 in all samples (NDC and SAD neurons). n = 13 individuals (SAD + NDC) represented by dots/squares; two to four independent experiments/treatment.
(D) Levels of tTAU in all samples (NDC and SAD neurons) are unaffected by R33 treatment. n = 13 individuals (SAD + NDC) represented by dots/squares; two to four independent experiments/treatment.
(E) R33 reduces pTAU (Thr 231) in hiPSC neurons derived from an APP duplication patient.
(F) R33 does not affect tTAU levels in APP duplication neurons.
(G) R33 decreases the pTAU/tTAU ratio in APP duplication neurons.
Non-normally distributed data (A–D) were analyzed by Wilcoxon test. ∗p < 0.05, ∗∗∗p < 0.001. NS, nonsignificant. Error bars represent SD. (E–G) For APP duplication: n = 1 FAD individual, three independent experiments/treatment. For each comparison, a two-tailed t test was performed. ∗∗p < 0.01. NS, nonsignificant. Error bars represent SD. See also Figure S3.
We probed a second pTAU epitope, paired helical fragment (PHF) TAU, and observed that in the presence of compound E, a gamma-secretase inhibitor, levels of PHF TAU increased, but when neurons were concomitantly treated with both compound E and R33, the PHF signal decreased (Figure S3A). We tested whether R33 affected the activation of the TAU kinase GSK3β (Figure S3B); however, we did not observe a significant effect of R33 treatment on the activity of GSK3β, suggesting that the decrease in pTAU by R33 is mediated through a different mechanism.
Finally, we tested whether other molecules hypothesized to be neurotrophic or protective against AD phenotypes had a similar effect to R33 on the pTAU/tTAU ratio. We have previously shown that the neurotrophin brain-derived neurotrophic factor (BDNF) reduced Aβ peptides in stem cell-derived neurons and that this reduction was correlated with protective variants in the SORL1 gene, whose protein product, SORLA, is a receptor of the retromer complex (Young et al., 2015). BDNF has been shown to reduce TAU phosphorylation, although at a different epitope than is analyzed in this study, in retinoic acid-differentiated SH-SY5Y cells (Chen et al., 2014). We treated hiPSC-derived neurons from three cell lines harboring SORL1 protective variants with BDNF and measured the pTAU/tTAU ratio. Under these conditions, however, we did not observe a significant change in the TAU ratio (Figure S3C). Taken together, these data suggest that the decrease in the pTAU/tTAU ratio by R33 has a more specific effect on the stabilization of the trimeric core cargo-recognition complex than on the various retromer receptor or interacting proteins.
Genetic Knockdown of VPS35 Increases Aβ and Phospho-TAU in Stem Cell-Derived Neurons
We next tested whether destabilization of the retromer complex by knockdown of VPS35 had the opposite effect of R33 treatment in hiPSC-derived neurons. Using lentiviral transduction, we expressed two independent short hairpin RNAs (shRNAs) against VPS35 mRNA (determined from a pool of four shRNAs, Figure S4A) in purified neurons and documented significant increases in Aβ peptides similar to what has been previously reported in mice (Figures 4A and 4B) (Bhalla et al., 2012). We next measured phosphorylated TAU protein (Thr 231) from purified neurons transduced with two VPS35 shRNA and observed a small but significant increase in pTAU at Thr 231 (Figure 4C). The magnitude of the increase in pTAU after VPS35 knockdown is similar to that of the decrease in pTAU we observe after R33 treatment. Because shRNA VPS35-c gave the strongest effect, we confirmed the knockdown of VPS35 protein and VPS35 mRNA using that shRNA in multiple experiments (Figures 4D and 4E). We consistently observed an approximately 50% knockdown of VPS35 protein in our neuronal cultures (Figure 4D). Although we are only able to reduce the levels of VPS35 protein by half, it should be noted that germline deletion of VPS35 is results in embryonic lethality in mice and haploinsufficiency of VPS35 is sufficient to increase AD neuropathology in a transgenic mouse model (Wen et al., 2011). These data suggest that even modest changes in retromer subunit levels may have a large impact on cellular phenotypes. In terms of AD, we note that duplication of APP resulting in one extra copy of the gene and 50% more APP expression is sufficient to cause severe and early-onset AD (Rovelet-Lecrux et al., 2006). Thus, for factors that may increase or decrease risk but not the deterministic probability of AD, changes on the order of 20% are likely relevant in human disease.
Figure 4.

Knockdown of Retromer Assembly Subunit VPS35 by shRNA Increases Aβ and pTAU/tTAU Ratio in hiPSC-Derived Neurons
(A and B) Aβ peptides (A, Aβ1-40; B, Aβ1-42) measured from the cell culture media of hiPSC-derived neurons are increased when VPS35 levels are reduced by VPS35 shRNA (two shRNA, VPS35-c and VPS35-d) compared with a scrambled shRNA (SCR shRNA).
(C) pTAU levels increase in hiPSC-derived neurons when VPS35 levels are reduced by VPS35 shRNA (two shRNA, c and d) compared with scrambled shRNA (SCR shRNA).
(D) VPS35 shRNA-c reduces VPS35 protein by 50%.
(E) VPS 35 shRNA-c reduces VPS35 mRNA by 50%. n = 4 independent experiments per treatment/condition.
(A–C) For each comparison, a one-way ANOVA with a Tukey multiple comparisons posttest was performed. (D and E) For each comparison, a two-tailed t test was performed. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p<0.001. Error bars represent SD. See also Figure S4.
Phospho-TAU Reduction Correlates with, but Is Not Dependent on, Aβ Peptide Levels
Because the magnitude of decrease in pTAU after R33 treatment was similar to the decreases observed in APP processing, we performed a linear regression analysis on absolute levels of pTAU and absolute levels of Aβ in the cultures across all 13 patient cell lines and found a positive correlation between the reduction of Aβ and the reduction in pTAU (Figure 5A). Previous work in hiPSC-derived neurons has suggested that beta cleavage of APP, rather than Aβ, is more highly associated with TAU phosphorylation (Israel et al., 2012), so we also examined the levels of sAPPβ compared with pTAU and observed a similar, but less robust, correlation between the reduction in sAPPβ with pTAU (Figure 5B). Taken together, however, these data suggest that the reduction in pTAU by R33 is highly correlative with a reduction in amyloidogenic processing of APP.
Figure 5.

The Decrease of Phosphorylated TAU Mediated by Retromer Stabilization Is Correlative with, but Not Dependent on, Decreases in Aβ and sAPPβ
(A) Absolute levels of Aβ peptides from 13 patient cell lines were graphed against absolute values of pTAU from hiPSC-derived neurons and show a positive correlation between decreases in Aβ and decreases in pTAU.
(B) Absolute levels of sAPPβ peptides from 13 patient cell lines were graphed against absolute values of pTAU from hiPSC-derived neurons and show a positive correlation between decreases in sAPPβ and decreases in pTAU.
(C) hiPSC neurons with the APP Swedish (Swe) mutation introduced by genome editing show substantial increases in Aβ peptides in either heterozygous (Swe/wild-type [WT]) or homozygous (Swe/Swe) genotypes compared with the WT isogenic controls.
(D) Increased levels of Aβ peptides do not affect the observed decrease in the pTAU/tTAU ratio mediated by R33 treatment.
(E) Compound E treatment substantially decreases Aβ peptides in hiPSC-derived neurons and treatment with R33 further decreases Aβ peptides in this condition, ∗∗∗ represents statistical difference from vehicle; ∗ represents statistical difference between compound E alone and compound E + R33.
(F) Aβ reduction does not change the fold-decrease in the pTAU/tTAU ratio mediated by R33.
(A and B) A linear regression analysis was performed between Aβ and pTAU (A) and sAPPβ and pTAU (B). R2 values reported on graphs. (C) For each comparison, a one-way ANOVA with a Tukey multiple comparisons posttest was performed. (D and F) For each comparison, a two-tailed t test was performed. n = 3 independent experiments per treatment/condition.
Error bars represent SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
We next asked if modulation of Aβ levels affected the decrease in pTAU by R33. We measured pTAU from isogenic neurons harboring the APP Swedish mutation, which leads to a 2- to 3-fold increase in endogenous Aβ in heterozygous and homozygous carriers, respectively (Figure 5C). Interestingly, we did not observe increased levels of pTAU/tTAU ratio in cells harboring these mutations alone. Previous work using isogenic cell lines of another penetrant AD mutation, PSEN1ΔE9, also failed to show increased pTAU/tTAU ratios at baseline conditions (Woodruff et al., 2013). However, treatment with R33 reduced the pTAU/tTAU ratio in the isogenic neurons to the same extent across this allelic series, with no significant difference in R33-mediated TAU ratio reduction between the wild-type cells and the Swedish genotypes (Figure 5D), suggesting that retromer stabilization reduces TAU phosphorylation regardless of genetic condition. To test whether modulating level of the APP β-CTF in our cell cultures affected the R33-mediated reduction of the TAU ratio, we treated wild-type purified neurons with the gamma-secretase inhibitor compound E, which inhibits the generation of Aβ but increases the amount of the β-CTF of APP. Interestingly, R33 treatment further reduced the amount of Aβ remaining after compound E treatment (Figure 5E). The presence of compound E had no effect on the decrease pTAU/tTAU ratio after treatment with R33 (Figure 5F). Taken together, these data suggest that while the pTAU/tTAU reduction by R33 is highly correlative with Aβ peptide levels, modulation of APP processing, either up or down, does not change the R33-mediated reduction of pTAU.
R33 Reduction of Phospho-TAU in hiPSC-Derived Neurons Is Independent of APP Expression
Because hiPSCs are amenable to genome engineering, we generated an APP knockout (APP KO) hiPSC line from our parental APP duplication hiPSC line (APPDp1) using CRISPR/Cas9 genome editing. APP KO hiPSCs were generated from FAD APPDp parental line by excision of one copy of APP and the introduction of two premature stop codons via non-homologous end-joining in the remaining copies (R. Van der Kant et al., personal communication; Figures S5A and S5B). APP KO hiPSCs have no detectable APP protein by western blot analysis (Figure S5C). We differentiated and purified neurons from APP KO hiPSCs and confirmed that these neurons do not have detectable levels of secreted Aβ peptides by ELISA (Figure 6A). We next treated APP KO neurons with R33 and observed decreased pTAU from APP KO neurons (Figure 6B) with no change in tTAU (Figure 6C), again leading to a significant reduction in the pTAU/tTAU ratio in APP KO neurons (Figure 6D). When APP KO neurons were transduced with shVPS35-c, we documented a significant increase in the amount of pTAU (Figure 6E), and this effect was confirmed with a second shRNA, shVPS35-d (Figure S6A). Finally, we tested whether VPS35 knockdown would change the effect of R33 on pTAU levels in APP KO neurons. Interestingly, knockdown of VPS35 did not affect the R33-mediated reduction in pTAU in these cells (Figure S6B). However, this result is not overly surprising, as we have an incomplete loss of VPS35 (∼50%) and the R33 chaperone is still stabilizing the remaining retromer. Indeed, previous work demonstrates that retromer stabilization chaperones continued to stabilize two of the three cargo-recognition core proteins (VPS29 and VPS35) under single knockdown conditions (shVPS26) (Mecozzi et al., 2014). Taken together, these experiments suggest that retromer stability influences TAU phosphorylation and that this is mediated through an APP-independent mechanism. Furthermore, these data imply that retromer enhancement may be beneficial even under less than optimal retromer conditions, such as loss of VPS35 expression, a condition documented in AD patient tissue (Small et al., 2005).
Figure 6.

Retromer Stabilization Decreases the pTAU/tTAU Ratio and Knockdown of Retromer Assembly Subunit VPS35 Increases pTAU/tTAU Ratio Independently of APP Expression in Human Neurons
(A) APP KO neurons were generated from APPDp cells by CRISPR/Cas9 genome editing have no detectable levels of Aβ peptides.
(B) APP KO neurons show a significant reduction in pTAU (Thr 231) after treatment with R33.
(C) tTAU levels are not affected in APP KO neurons after treatment with R33.
(D) The pTAU/tTAU ratio is significantly reduced in APP KO neurons after treatment with R33.
(E) APP KO neurons transduced with VPS35 shRNA show a significant increase in pTAU/tTAU ratio compared with scrambled shRNA (SCR shRNA).
n = 3 independent experiments per treatment/condition. For each comparison, a two-tailed t test was performed. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. NS, nonsignificant. Error bars represent SD. See also Figures S5 and S6.
Discussion
In this work, we demonstrate that a small molecule stabilizer of the retromer complex reduces endogenous cellular AD phenotypes in human neurons. This small molecule, R33, has been previously shown to reduce Aβ peptides and decrease APP in early endosomes, consistent with its in vitro action as a pharmacological chaperone that stabilizes the retromer complex (Mecozzi et al., 2014) (Chu and Pratico, 2017). We extended this work in human neurons to test how retromer stabilization affects TAU phosphorylation, another critical pathologic phenotype in AD and one that correlates more highly with cognitive decline than the presence of amyloid plaques in patients (Arriagada et al., 1992). In this work, we demonstrate that retromer stabilization in human neurons reduces TAU phosphorylation at two epitopes, suggesting that this strategy may be beneficial for pathologies extending beyond amyloid. Indeed, while the focus on Aβ as the premier pathological event has undoubtedly yielded insights into the disease, particularly for FAD, it is equally conceivable to envision a “dual-pathway” hypothesis where an upstream molecular driver influences Aβ and TAU pathologies separately (Small and Duff, 2008). This may especially be the case for SAD, where immense genetic heterogeneity and complex environmental interactions have unknown influences on the course of the disease. Here we decouple the effect of retromer stabilization on both APP processing and TAU phosphorylation using gene-edited hiPSC-derived neurons. In both neurons engineered to have 2- to 3-fold higher amounts of Aβ (APPSwe) or neurons engineered to not express APP and which have no detectable Aβ (APP KO), retromer stabilization decreases TAU phosphorylation to the same extent, suggesting that manipulation of endocytic trafficking via retromer may be an upstream driver of these two pathologies independently from one another. This approach of gene editing in pluripotent stem cells allows the dissection of relevant disease biology in a human-specific system.
The link between retromer regulation of endosomal trafficking and TAU phosphorylation is currently unclear, although compelling studies are emerging to suggest that endocytic trafficking may directly affect TAU pathology. Michel et al., 2014, suggest that monomeric TAU is endocytosed from the extracellular space and its transit through endosomes facilitates TAU seeding and aggregation. Work in flies demonstrates that removing cathepsin D, a retromer cargo and integral enzyme in lysosomal degradation, exacerbates TAU-induced neurodegeneration (Khurana et al., 2010). Extracellular TAU can be taken up and propagated via exosomes in microglia, the resident immune cells of the CNS (Asai et al., 2015), and retromer deficiency impairs the recycling of TREM2 protein, a product of the TREM2 gene that is also associated with increased AD risk, which can inhibit microglial activation (Yin et al., 2016). Developing methods of improving endocytic trafficking and function in multiple CNS types will likely be protective in many neurodegenerative disorders, underscoring the importance of elucidating the role of the endocytic network in both Aβ and TAU pathology. Finally, we highlight the potential of hiPSC models to confirm and elaborate on work done in non-human systems. We have replicated work previously published in mice to show that retromer stabilization decreases pathogenic APP processing (Mecozzi et al., 2014) and extended our studies to show a beneficial effect of retromer stabilization on TAU in human neurons, which, to our knowledge, has not been documented previously. Furthermore, we demonstrate that this process correlates with, but is not dependent upon, APP processing.
Considering that the current failure rate of AD clinical trials is very high (Posner et al., 2017), new therapeutic strategies aimed at improving retromer and other endocytic network component function are likely to be promising therapeutic options. Indeed, recent genetic findings implicate endosomal and vesicular trafficking genes in both early- and late-onset AD (Bellenguez et al., 2017, Kunkle et al., 2017, Rosenthal and Kamboh, 2014). For that reason, and because defects in the endocytic pathway occur early in AD pathogenesis, targeting this pathway represents a valid cell biological target for therapeutic development (Small et al., 2017).
The use of hiPSCs, which can be differentiated into all cell types of the CNS, represents an ideal model to test novel and existing drugs and other therapies in a preclinical system. In particular, the advantage of a system that does not rely on overexpression of disease proteins has already been shown: patient-derived neurons behave differently from APP-overexpressing cell lines and animal AD model-derived neurons when exposed to drug candidates, such as gamma-secretase modulators (Liu et al., 2014). Recently, cortical neurons from hiPSCs have been used to screen modulators of Aβ peptide length (Brownjohn et al., 2017), and work in scalable three-dimensional culture formats has shown detection of TAU aggregates in a quantifiable manner in 384-well plates (Medda et al., 2016), representing a growing technology involving complex cellular interactions that will likely be highly translational.
Abnormalities in the endo/lysosomal network are a common theme in multiple neurodegenerative disorders (Hu et al., 2015, Schreij et al., 2016), and retromer-related mutations or defects are implicated not only in AD, but Parkinson disease and Down syndrome, as well as other non-neurological conditions (Berman et al., 2015). Our work, for the first time, examines a small molecule that enhances the function of retromer, which is an integral component of the endosomal network, using human neurons. This pharmacologic chaperone, R33, demonstrated pronounced and potentially beneficial effects on the two main pathological molecules in AD: Aβ and pTAU. We predict that using hiPSCs, both from patients and isogenic lines with key mutations or variants, to test and screen novel therapeutic molecules and other therapeutic strategies will represent a major advance in therapeutic development for AD and other complex neurodegenerative disorders.
Experimental Procedures
Cell Lines and Cell Culture
All patient-derived hiPSC lines were generated by four-factor reprogramming and have been previously characterized and published (Israel et al., 2012, Young et al., 2015). Briefly, hiPSCs were maintained on a mouse embryonic fibroblast (MEF) feeder layer in HUES medium and were routinely tested for mycoplasma. NSCs and neurons were generated using previously described methods (Israel et al., 2012, Yuan et al., 2011) and purified by fluorescence-activated cell sorting (FACS). After FACS purification, depending on the percentage of neurons in each differentiation, 2 to 4 independent purified cultures were re-plated for each experiment. From these independent cultures and for each assay, 2 to 4 technical replicates from each well were analyzed.
CRISPR/Cas9 Gene Editing
Genome-edited lines were generated using published protocols. Briefly, guide RNAs (gRNAs) to APP were generated using the Zhang Lab Crispr design Web site at MIT (crispr.mit.edu) and cloned into vector px458 (Addgene) that co-expresses the Cas9 nuclease and GFP. hiPSCs were electroporated with the px458 plasmid containing the gRNAs to target APP. We generated the APP KO cells from our parental APPDp cell line (APPDp1), first described in Israel et al., 2012. To generate APP KO cells, no repair template was added. For generating the APP Swedish mutation, we targeted wild-type hiPSCs derived from J. Craig Venter (Gore et al., 2011) and 120-bp single-stranded oligonucleotide was designed to have the 2-bp Swedish mutation (GA to TC, corresponding to exon 16 of APP). Electroporated hiPSCs were sorted for GFP and plated a clonal density on 10-cm MEF plates. After 1.5 to 2.0 weeks, sub-cloned colonies were picked and transferred to a 96-well plate. Colonies were spit in duplicate and one set was maintained for cell line generation and the other was analyzed for the mutation of interest by Sanger Sequencing. All gene-edited lines were digitally karyotyped as euploid by the Illumina Infinium HumanCoreExomeBeadChip, as has been previously described (Young et al., 2015)
Drug Treatment and ELISA Assays
Pure R55 and R33 were obtained from MedKoo Biosciences (products TPT-260 and TPT-172) and re-suspended at designated concentrations in a solution of 50% DMSO/50% double-distilled H2O at low pH (pH 4.3). For these studies, R33 was used for every experiment. Purified neurons were treated with R33 or vehicle (DMSO/H2O) for 72 hr and then harvested for analysis. For Aβ and TAU ELISA assays, purified neurons were seeded at 200,000 cells/well of a 96-well plate. After treatment, cell culture media and cell lysate were harvested from triplicate wells and run on Aβ Triplex ELISA plates and pTAUThr231/tTAU ELISA plates (Meso Scale Discovery). Protein concentrations were determined by BCA assay (Pierce) and Aβ levels were normalized to the amount of total protein.
Western Blotting
Cell lysates were run on NuPAGE 4% to 12% Bis-Tris Gels, transferred to nitrocellulose membranes, and probed with antibodies to VPS35 (Abcam #ab57632), APP (Life Tech #512700), TAU (Thermo #MN1000 and MN#1020). Quantification of western blots was performed on the Odyssey system and using ImageJ software.
shRNA Design and Virus
shRNAs against VPS35 were ordered from Origene (#TL300553). VPS35 and scrambled control shRNAs were packed into lentivirus using HEK293FT cells. Virus was purified by PEG-it and cells were transduced with lentivirus for 72 hr. Knockdown of VPS35 was confirmed by qRT-PCR and western blot analysis.
gRNA, ssODN, and Primer Sequences
gRNA to generate APP Swedish and APP KO cells: GGAGATCTCTGAAGTGAAGA TGG.
ssODN for APP Swedish homologous recombination 2-bp change in bold: TGATGTAATACAGGTTCTGGGTTGACAAATATCAAGACGGAGGAGATCTCTGAAGTGAATCTGGATGCAGAATTCCGACATGACTCAGGATATGAAGTTCATCATCAAAAATTGGTACGT.
VPS35 primer for qRT-PCR: F primer, TGCCCACCAGACTATCAGTG; R primer, AGCTGCACTTGTAGAGAGGG.
Statistical Methods
Statistics for all datasets were performed using GraphPad Prism. Normality for each dataset was assessed using the D'Agostino-Pearson test. When data were normally distributed, a parametric two-tailed t test was used. The comparisons between multiple cells lines from different individuals usually resulted in non-normally distributed data and these data were analyzed using the Wilcoxon-signed rank test.
Author Contributions
J.E.Y., S.A.S., G.A.P., and L.S.B.G. conceived the project. J.E.Y. designed the experiments. J.E.Y., L.F., and H.F. performed the experiments. J.E.Y. wrote the manuscript. S.A.S., G.A.P., and L.S.B.G. edited and provided feedback on the manuscript. J.E.Y. and L.S.B.G. acquired funding for the project.
Acknowledgments
This work was supported by the BrightFocus Foundation (Postdoctoral Fellowship to J.E.Y.), the Tietze Foundation (to J.E.Y.), the University of Washington (UW) ADRC (NIH AG005136) pilot award (to J.E.Y.), a generous gift from the Ellison Foundation (to UW), and the NIH: NIH-NIA 2P50AG005131-31 to L.S.B.G. S.A.S. and G.A.P. are on the advisory board of MeiraCTx. S.A.S. is also on the Scientific Advisory Board of Denali Therapeutics Janssen Pharmaceutical. G.A.P. has consulting arrangements with Amicus Therapeutics, Proclara Biosciences, and QR Pharma.
Published: March 1, 2018
Footnotes
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.01.031.
Contributor Information
Jessica E. Young, Email: jeyoung@uw.edu.
Lawrence S.B. Goldstein, Email: lgoldstein@ucsd.edu.
Supplemental Information
References
- Arriagada P.V., Growdon J.H., Hedley-Whyte E.T., Hyman B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Asai H., Ikezu S., Tsunoda S., Medalla M., Luebke J., Haydar T., Wolozin B., Butovsky O., Kugler S., Ikezu T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015;18:1584–1593. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avramopoulos D. Genetics of Alzheimer's disease: recent advances. Genome Med. 2009;1:34. doi: 10.1186/gm34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellenguez C., Charbonnier C., Grenier-Boley B., Quenez O., Le Guennec K., Nicolas G., Chauhan G., Wallon D., Rousseau S., Richard A.C. Contribution to Alzheimer's disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging. 2017;59:220.e1–220.e9. doi: 10.1016/j.neurobiolaging.2017.07.001. [DOI] [PubMed] [Google Scholar]
- Berman D.E., Ringe D., Petsko G.A., Small S.A. The use of pharmacological retromer chaperones in Alzheimer's disease and other endosomal-related disorders. Neurotherapeutics. 2015;12:12–18. doi: 10.1007/s13311-014-0321-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla A., Vetanovetz C.P., Morel E., Chamoun Z., Di Paolo G., Small S.A. The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol. Dis. 2012;47:126–134. doi: 10.1016/j.nbd.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownjohn P.W., Smith J., Portelius E., Serneels L., Kvartsberg H., De Strooper B., Blennow K., Zetterberg H., Livesey F.J. Phenotypic screening identifies modulators of amyloid precursor protein processing in human stem cell models of Alzheimer's disease. Stem Cell Rep. 2017;8:870–882. doi: 10.1016/j.stemcr.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd C., Cullen P.J. Retromer: a master conductor of endosome sorting. Cold Spring Harb. Perspect. Biol. 2014;6 doi: 10.1101/cshperspect.a016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo A.M., Mathews P.M., Boiteau A.B., Hassinger L.C., Peterhoff C.M., Jiang Y., Mullaney K., Neve R.L., Gruenberg J., Nixon R.A. Down syndrome fibroblast model of Alzheimer-related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am. J. Pathol. 2008;173:370–384. doi: 10.2353/ajpath.2008.071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo A.M., Peterhoff C.M., Troncoso J.C., Gomez-Isla T., Hyman B.T., Nixon R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Zhou Z., Zhang L., Xu S., Chen C., Yu Z. The cellular distribution and Ser262 phosphorylation of tau protein are regulated by BDNF in vitro. PLoS One. 2014;9:e91793. doi: 10.1371/journal.pone.0091793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J., Pratico D. The retromer complex system in a transgenic mouse model of AD: influence of age. Neurobiol. Aging. 2017;52:32–38. doi: 10.1016/j.neurobiolaging.2016.12.025. [DOI] [PubMed] [Google Scholar]
- Fjorback A.W., Seaman M., Gustafsen C., Mehmedbasic A., Gokool S., Wu C., Militz D., Schmidt V., Madsen P., Nyengaard J.R. Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J. Neurosci. 2012;32:1467–1480. doi: 10.1523/JNEUROSCI.2272-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatz M., Reynolds C.A., Fratiglioni L., Johansson B., Mortimer J.A., Berg S., Fiske A., Pedersen N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry. 2006;63:168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- Gore A., Li Z., Fung H.L., Young J.E., Agarwal S., Antosiewicz-Bourget J., Canto I., Giorgetti A., Israel M.A., Kiskinis E. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J.A., Higgins G.A. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hu Y.B., Dammer E.B., Ren R.J., Wang G. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl. Neurodegener. 2015;4:18. doi: 10.1186/s40035-015-0041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel M.A., Yuan S.H., Bardy C., Reyna S.M., Mu Y., Herrera C., Hefferan M.P., Van Gorp S., Nazor K.L., Boscolo F.S. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch C.M., Goate A.M. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry. 2015;77:43–51. doi: 10.1016/j.biopsych.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana V., Elson-Schwab I., Fulga T.A., Sharp K.A., Loewen C.A., Mulkearns E., Tyynela J., Scherzer C.R., Feany M.B. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 2010;6:e1001026. doi: 10.1371/journal.pgen.1001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T., Asai M., Tsukita K., Kutoku Y., Ohsawa Y., Sunada Y., Imamura K., Egawa N., Yahata N., Okita K. Modeling Alzheimer's disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell. 2013;12:487–496. doi: 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Kunkle B.W., Vardarajan B.N., Naj A.C., Whitehead P.L., Rolati S., Slifer S., Carney R.M., Cuccaro M.L., Vance J.M., Gilbert J.R. Early-onset Alzheimer disease and candidate risk genes involved in endolysosomal transport. JAMA Neurol. 2017;74:1113–1122. doi: 10.1001/jamaneurol.2017.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Shah S.Z., Zhao D., Yang L. Role of the retromer complex in neurodegenerative diseases. Front. Aging Neurosci. 2016;8:42. doi: 10.3389/fnagi.2016.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Waltz S., Woodruff G., Ouyang J., Israel M.A., Herrera C., Sarsoza F., Tanzi R.E., Koo E.H., Ringman J.M. Effect of potent gamma-secretase modulator in human neurons derived from multiple presenilin 1-induced pluripotent stem cell mutant carriers. JAMA Neurol. 2014;71:1481–1489. doi: 10.1001/jamaneurol.2014.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecozzi V.J., Berman D.E., Simoes S., Vetanovetz C., Awal M.R., Patel V.M., Schneider R.T., Petsko G.A., Ringe D., Small S.A. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014;10:443–449. doi: 10.1038/nchembio.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medda X., Mertens L., Versweyveld S., Diels A., Barnham L., Bretteville A., Buist A., Verheyen A., Royaux I., Ebneth A. Development of a scalable, high-throughput-compatible assay to detect tau aggregates using iPSC-derived cortical neurons maintained in a three-dimensional culture format. J. Biomol. Screen. 2016;21:804–815. doi: 10.1177/1087057116638029. [DOI] [PubMed] [Google Scholar]
- Michel C.H., Kumar S., Pinotsi D., Tunnacliffe A., St George-Hyslop P., Mandelkow E., Mandelkow E.M., Kaminski C.F., Kaminski Schierle G.S. Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J. Biol. Chem. 2014;289:956–967. doi: 10.1074/jbc.M113.515445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammad A., Flores I., Zhang H., Yu R., Staniszewski A., Planel E., Herman M., Ho L., Kreber R., Honig L.S. Retromer deficiency observed in Alzheimer's disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc. Natl. Acad. Sci. USA. 2008;105:7327–7332. doi: 10.1073/pnas.0802545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratore C.R., Rice H.C., Srikanth P., Callahan D.G., Shin T., Benjamin L.N., Walsh D.M., Selkoe D.J., Young-Pearse T.L. The familial Alzheimer's disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014;23:3523–3536. doi: 10.1093/hmg/ddu064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon R.A., Wegiel J., Kumar A., Yu W.H., Peterhoff C., Cataldo A., Cuervo A.M. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Posner H., Curiel R., Edgar C., Hendrix S., Liu E., Loewenstein D.A., Morrison G., Shinobu L., Wesnes K., Harvey P.D. Outcomes assessment in clinical trials of Alzheimer's disease and its precursors: readying for short-term and long-term clinical trial needs. Innov. Clin. Neurosci. 2017;14:22–29. [PMC free article] [PubMed] [Google Scholar]
- Raja W.K., Mungenast A.E., Lin Y.T., Ko T., Abdurrob F., Seo J., Tsai L.H. Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer's disease phenotypes. PLoS One. 2016;11:e0161969. doi: 10.1371/journal.pone.0161969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C., Cheng R., Rogaeva E., Lee J.H., Tokuhiro S., Zou F., Bettens K., Sleegers K., Tan E.K., Kimura R. Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch. Neurol. 2011;68:99–106. doi: 10.1001/archneurol.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C., Tosto G., Vardarajan B., Rogaeva E., Ghani M., Rogers R.S., Conrad C., Haines J.L., Pericak-Vance M.A., Fallin M.D. Independent and epistatic effects of variants in VPS10-d receptors on Alzheimer disease risk and processing of the amyloid precursor protein (APP) Transl. Psychiatry. 2013;3:e256. doi: 10.1038/tp.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E., Meng Y., Lee J.H., Gu Y., Kawarai T., Zou F., Katayama T., Baldwin C.T., Cheng R., Hasegawa H. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal S.L., Kamboh M.I. Late-onset Alzheimer's disease genes and the potentially implicated pathways. Curr. Genet. Med. Rep. 2014;2:85–101. doi: 10.1007/s40142-014-0034-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovelet-Lecrux A., Hannequin D., Raux G., Le Meur N., Laquerriere A., Vital A., Dumanchin C., Feuillette S., Brice A., Vercelletto M. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Schreij A.M., Fon E.A., McPherson P.S. Endocytic membrane trafficking and neurodegenerative disease. Cell. Mol. Life Sci. 2016;73:1529–1545. doi: 10.1007/s00018-015-2105-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small S.A., Duff K. Linking Abeta and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron. 2008;60:534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small S.A., Kent K., Pierce A., Leung C., Kang M.S., Okada H., Honig L., Vonsattel J.P., Kim T.W. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann. Neurol. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- Small S.A., Simoes-Spassov S., Mayeux R., Petsko G.A. Endosomal traffic jams represent a pathogenic Hub and therapeutic target in Alzheimer's disease. Trends Neurosci. 2017;40:592–602. doi: 10.1016/j.tins.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L., Tang F.L., Hong Y., Luo S.W., Wang C.L., He W., Shen C., Jung J.U., Xiong F., Lee D.H. VPS35 haploinsufficiency increases Alzheimer's disease neuropathology. J. Cell Biol. 2011;195:765–779. doi: 10.1083/jcb.201105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff G., Reyna S.M., Dunlap M., Van Der Kant R., Callender J.A., Young J.E., Roberts E.A., Goldstein L.S. Defective transcytosis of APP and lipoproteins in human iPSC-derived neurons with familial Alzheimer's disease mutations. Cell Rep. 2016;17:759–773. doi: 10.1016/j.celrep.2016.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff G., Young J.E., Martinez F.J., Buen F., Gore A., Kinaga J., Li Z., Yuan S.H., Zhang K., Goldstein L.S. The presenilin-1 DeltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013;5:974–985. doi: 10.1016/j.celrep.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T., Ito D., Okada Y., Akamatsu W., Nihei Y., Yoshizaki T., Yamanaka S., Okano H., Suzuki N. Modeling familial Alzheimer's disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- Yin J., Liu X., He Q., Zhou L., Yuan Z., Zhao S. Vps35-dependent recycling of Trem2 regulates microglial function. Traffic. 2016;17:1286–1296. doi: 10.1111/tra.12451. [DOI] [PubMed] [Google Scholar]
- Young J.E., Boulanger-Weill J., Williams D.A., Woodruff G., Buen F., Revilla A.C., Herrera C., Israel M.A., Yuan S.H., Edland S.D. Elucidating molecular phenotypes caused by the SORL1 Alzheimer's disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell. 2015;16:373–385. doi: 10.1016/j.stem.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S.H., Martin J., Elia J., Flippin J., Paramban R.I., Hefferan M.P., Vidal J.G., Mu Y., Killian R.L., Israel M.A. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One. 2011;6:e17540. doi: 10.1371/journal.pone.0017540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.