

Abstract
Autoimmune diseases, such as psoriasis and arthritis, show a patchy distribution of inflammation despite systemic dysregulation of adaptive immunity. Thus, additional tissue-derived signals, such as danger-associated molecular patterns (DAMPs), are indispensable for manifestation of local inflammation. S100A8/S100A9 complexes are the most abundant DAMPs in many autoimmune diseases. However, regulatory mechanisms locally restricting DAMP activities are barely understood. We now unravel for the first time, to our knowledge, a mechanism of autoinhibition in mice and humans restricting S100-DAMP activity to local sites of inflammation. Combining protease degradation, pull-down assays, mass spectrometry, and targeted mutations, we identified specific peptide sequences within the second calcium-binding EF-hands triggering TLR4/MD2-dependent inflammation. These binding sites are free when S100A8/S100A9 heterodimers are released at sites of inflammation. Subsequently, S100A8/S100A9 activities are locally restricted by calcium-induced (S100A8/S100A9)2 tetramer formation hiding the TLR4/MD2-binding site within the tetramer interphase, thus preventing undesirable systemic effects. Loss of this autoinhibitory mechanism in vivo results in TNF-α–driven fatal inflammation, as shown by lack of tetramer formation in crossing S100A9–/– mice with 2 independent TNF-α–transgene mouse strains. Since S100A8/S100A9 is the most abundant DAMP in many inflammatory diseases, specifically blocking the TLR4-binding site of active S100 dimers may represent a promising approach for local suppression of inflammatory diseases, avoiding systemic side effects.
Keywords: Autoimmunity, Inflammation
Keywords: Innate immunity
Introduction
The term “sterile inflammation” describes inflammatory processes not triggered by microbial agents (1, 2), but rather induced by endogenous molecules released during tissue damage or cell stress (3). For these endogenous triggers, the terms alarmins or danger-associated molecular patterns (DAMPs) have been introduced (4). Many alarmins are primarily involved in intracellular homeostasis, but also act as extracellular danger signals triggering inflammation when released by damaged or activated cells (3, 5, 6). HMGB1, heat shock proteins, and certain S100 proteins belong to this family.
Many chronic inflammatory diseases are driven by systemic autoimmune mechanisms. However, the pattern of sterile inflammation in these diseases frequently shows a local and patchy distribution, e. g., circumscribed inflammatory plaques of psoriasis beside nonaffected skin that can be explained neither by systemic dysregulation of the adaptive immune system nor by distribution of autoantigens. It is therefore evident that additional tissue-derived signals such as alarmins are indispensable for local differences in the clinical manifestation of inflammation. However, molecular mechanisms responsible for local manifestation of inflammatory activity are barely understood.
S100A8 (also known as myeloid-related protein, MRP8) and S100A9 (MRP14) constitute up to 40% of cytosolic proteins in neutrophils and 5% in monocytes (7). They are released by activated phagocytes and promote inflammation as endogenous ligands of the TLR4 complex, but molecular models for these interactions are currently missing (8–12). S100A9-deficient mice revealed a pivotal role of these proteins for many inflammatory processes (13, 14). S100A8 and S100A9 are the most abundant alarmins during infections, arthritis, allergies, autoimmune diseases, and inflammatory bowel, pulmonary, or cardiovascular diseases (6, 15). S100A8/S100A9 usually exist as heterodimeric complexes (calprotectin) that form heterotetramers in the presence of calcium (16). There are many contradictory reports about the functional activities of S100A8 and S100A9. However, most published work neglects the possibility of functional variability of different S100 complexes. Describing the structural model for S100-TLR4 interactions, we identified, for what we believe is the first time, an autoinhibitory feedback mechanism for S100 alarmin functions locally restricting their inflammatory properties. S100A8/S100A9 released by activated phagocytes is only temporarily active in the local extracellular microenvironment due to a mechanism of autoinhibition that is induced by calcium-dependent tetramerization of S100A8/S100A9. Oligomer formation of 2 heterodimers blocks the ability of the heterodimer to bind to TLR4/MD2, thus restricting the activity of these alarmins at local sites of inflammation. Loss of this control mechanism results in fatal TNF-α–driven inflammation in vivo. These properties define a specific TLR4-binding site on the active dimeric form of S100A8/S100A9 as a promising target for a very specific and locally restricted antiinflammatory therapy minimizing systemic side effects.
Results
S100A8/S100A9 tetramerization abolishes inflammatory activity.
S100A8/S100A9 heterodimers associate to (S100A8/S100A9)2 tetramers in the presence of calcium (16). Surprisingly, both isolated human and murine S100A8 and S100A9 homodimers induced a strong TNF-α release in human monocytes, whereas the S100A8/S100A9 heterocomplexes were inactive (Figure 1, A and B, and Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI89867DS1).
Figure 1. S100A8/S100A9 tetramerization blocks inflammatory activity of the alarmin.

(A and B) Effects of hS100A9 and hS100A8 homodimers or hS100A8/S100A9 (A), mS100A8 homodimer (B), or hS100A8/S100A9, hS100A8/S100A9N69A, and hS100A8/S100A9E78A (C) on TNF-α production by monocytes quantified by ELISA after 4 hours of incubation (>1 mM calcium in culture medium). (D) Representative SEC runs of human (grhS100A8/S100A9, granulocytic prepared human S100A8/S100A9 [red and green curves]; rechS100A8/S100A9, recombinant prepared human S100A8/S100A9; rechS100A8/S100A9E78A, recombinant prepared human S100A8/S100A9E78A; rechS100A8, recombinant prepared human S100A8; rechS100A9, recombinant prepared human S100A9) and murine (recmS100A8/S100A9, recombinant prepared murine S100A8/S100A9;recmS100A8, recombinant prepared murine S100A8) S100 proteins in the absence (black and red curves) and presence of 100 μM Ca2+ ions (blue and green curves) (3 independent experiments). (E) Induction of TNF-α and IL-8 mRNA in HEK293 cells expressing TLR4-MD2-CD14 and treated with either 5 or 10 μg/ml of rhS100A8/S100A9 (left), 1 or 5 μg/ml rhS100A8/S100A9N69A (middle), or 1 or 5 μg/ml hS100A8/S100A9E78A (right) for 4 hours, as detected by qRT-PCR. Results are shown as relative to baseline expression in unstimulated cells. RPL was used as a housekeeping control gene. (F) Human monocytes were left untreated or stimulated with 5 μg/ml of either S100A8/S100A9, S100A9, or S100A8 or 1 ng/ml LPS for 4 hours. RNA was isolated and used for transcriptome analyses. The 1,000 most variable genes (FDR corrected, P < 0.05) within the data set were used for HC. Data were Z score–normalized and ranked according to changes in expression upon stimulus. Data represent mean ± SD of 5 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001, 2 tailed t test.
The aa exchange of N69A or E78A in the calcium-binding EF-hand II of S100A9 prevents tetramer formation (17). In contrast with WT (S100A8/S100A9)2 tetramers, mutated S100A8/S100A9(N69A) and S100A8/S100A9(E78A) heterodimers significantly induced TNF-α release in human monocytes (Figure 1C and Supplemental Figure 1B). Size-exclusion chromatography (SEC) revealed that, in contrast with WT S100A8/S100A9 complexes, mutated heterodimers and S100A8 and S100A9 homodimers did not form tetramers in the presence of calcium (Figure 1D). Thus, loss of biological activity is strictly linked to calcium-induced tetramer formation, representing a mechanism of autoinhibition of S100 activity (Figure 1E). Finally, global gene-expression analysis confirmed that S100A8 or S100A9 homodimers, but not S100 tetramers, induced a global transcriptional response that was basically identical to that of LPS-mediated TLR4 stimulation (Figure 1F and Supplemental Table 1), confirming that activity of S100A8/S100A9 is restricted to the dimeric state. In the following experiments, we used S100A9 homodimers as a model for the in-depth analysis of dimer-mediated effects in vitro. Using transfected HEK293 cells, we confirmed that S100A9 dimer–induced activation was dependent on a functional TLR4-MD2-CD14 complex (Supplemental Figure 1C). Accordingly, blocking TLR4 with the monoclonal antibody HTA125 or TLR4 antagonist RS-LPS efficiently inhibited S100A9-induced TNF-α expression (Supplemental Figure 1D). Calcium-induced tetramers of S100A8/S100A9 were unable to block LPS or S100A8 homodimer-dependent TLR4/MD2 activation of human monocytes (Supplemental Figure 1E). Furthermore, S100A9 dimers induced TLR4-MD2 downstream signaling, activating MyD88, IRAK, PKC, ERK1/2, p38 kinases, and NF-κB (Supplemental Figure 2, A–E).
S100A8 dimers amplify TNF-α–mediated inflammation.
KO of S100A9, which is generally associated with a loss of S100A8 at the protein level (13, 14), protects against overwhelming inflammation in endotoxin-induced shock, at least partly due to the lower expression of TNF-α (8). To answer the question of whether S100 proteins act up- or downstream of TNF-α–driven inflammation, we analyzed tristetraprolin-KO mice (TTP–/–) constitutively expressing high levels of murine TNF-α. TTP–/– mice appear normal at birth, but develop erosive arthritis and dermatitis during the first month of life (18, 19). Unexpectedly, TTP–/– S100A9–/– mice did not improve in their symptoms, but on the contrary, started to develop severe psoriasis-like skin inflammation, characterized by keratinocyte hyperproliferation and widespread scaling within the first days after birth (Figure 2, A and B), as reflected by an increase of an adapted Psoriasis Area and Severity Index (PASI) (20) (Figure 2A). Compared with TTP–/– mice, TTP–/– S100A9–/– animals showed a markedly thickened epidermis (acanthosis), loss of the granular layer, extension of the cornified envelope as a result of incomplete keratinization (parakeratosis), elongation of rete ridges, and a typical hyperpigmentation of the basal layer (Figure 2, B and C). Ki67 staining revealed a hyperproliferation of keratinocytes in all skin layers of TTP–/– S100A9–/– mice, which is known as a hallmark of psoriatic lesions. In contrast, the expression of keratin 10 (K10) and the terminal differentiation marker loricrin (Lor) was markedly reduced in TTP–/– S100A9–/– mice versus controls, indicating incomplete keratinocyte differentiation. As typical for psoriasiform dermatitis, TTP–/– S100A9–/– mice showed dermal invasion of macrophages, granulocytes, and T cells as well as subcorneal accumulation of invading granulocytes, i.e., Munro microabscesses (Figure 2B).
Figure 2. Phenotype of TTP–/– S100A9–/– mice.

(A) Disease progression of TTP–/– S100A9–/– mice versus controls (10 mice per group) is shown by the adapted PASI scores from P1–P9. *P < 0.05; **P < 0.01; ***P < 0.001, Mann-Whitney U test. (B) Photographs and skin sections of TTP–/– S100A9–/–, TTP–/–, S100A9–/–, and WT mice at day 9 after birth. Sections were stained with H&E or by immunohistochemistry for S100A8 or immunofluorescence for the indicated epidermal differentiation markers or invading T cells (CD3), granulocytes (Gr1), and macrophages (F4/80) (fluorescence, green signal) and Ki67 (fluorescence, red signal). Blue and red counterstaining shows nuclei. Scale bars: 200 μm (H&E); 50 μm (immunostaining). (C) Epidermis thickness was determined for at least 3 sections per mouse and for each section at 3 different sites and at least 5 mice for each group. (D and E) Expression of IFN-γ, IL-6, IL-17, IL-22, IL-23, and IL-36 in the skin of TTP–/– S100A9–/– mice at day 9 after birth compared with TTP–/– and S100A9–/– mice. The mRNA levels in the skin were measured by qRT-PCR (D) and quantified at the protein level by FACS-based analysis (E) of 3 independent experiments. Data represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001, 1-way ANOVA (C–E).
In accordance with this psoriasis-like phenotype, we detected significantly increased levels of IFN-γ, IL-6, IL-17, IL-22, IL-23, and IL-36 (21–23) in the skin of TTP–/– S100A9–/– mice versus controls (Figure 2, D and E). Although S100A9–/– mice have been shown to constitute a functional double-KO for both S100 proteins under steady-state as well as under acute inflammatory conditions (13, 14), we detected, for what we believe is the first time, a strong recovery of S100A8 protein expression under chronic TNF-α expression in the hyperproliferative epidermis of TTP–/– S100A9–/– mice compared with the skin of TTP–/–, S100A9–/–, or WT mice (Figure 2B).
Blocking TNF-α improves psoriasis-like skin disease in TTP–/– S100A9–/– mice.
Depletion of TNF-α by treatment of newborn TTP–/– S100A9–/– mice with anti–TNF-α antibodies (certolizumab) led to a significant amelioration of disease activity, as evidenced by decreased PASI scores, keratinocyte hyperproliferation, and reduced scaling of the skin (Figure 3, A and B). In parallel, S100A8 as well as cytokine expression was markedly reduced in the epidermis of anti–TNF-α-treated TTP–/– S100A9–/– mice (Figure 3, B–D). In accordance with the TNF-α–blocking studies, TTP–/– S100A9–/– mice on a TNF-α–deficient background showed a complete absence of psoriasis-like skin inflammation. Moreover, the lack of TNF-α reduced S100A8 protein expression in the skin to the level of that in healthy WT mice (Supplemental Figure 3). Finally, isolated keratinocytes from S100A9–/– mice showed a clear upregulation of S100A8 protein expression after TNF-α stimulation by Western blot analysis (Supplemental Figure 4A).
Figure 3. Improvement of psoriasis-like skin disease in TTP–/– S100A9–/– mice after treatment with anti–TNF-α.

(A) Adapted PASI score of anti–TNF-α–treated mice (40 mg certolizumab/kg mouse on days 4, 6, and 8) shows amelioration of disease progression compared with IgG control antibody–treated mice (10 mice for each group). *P < 0.05; ***P < 0.001, Mann-Whitney U test. (B) Photographs of mice and immunostainings of skin sections from anti–TNF-α–treated (right panel) and IgG-treated (left panel) TTP–/– S100A9–/– mice at day 9 after birth. Sections were stained with H&E or with antibodies against the indicated epidermal differentiation markers or with antibodies against the immune cell markers F4/80, Gr1, and CD3 (green signal). Red counterstaining shows nuclei. Scale bars: 200 μm (H&E); 50 μm (immunostaining). (C) mRNA levels of IFN-γ, IL-6, IL-17, IL-22, IL-23, and IL-36 in the skin of anti–TNF-α and IgG-treated TTP–/– S100A9–/– mice were measured by qRT-PCR. (D) Expression of IL-17 protein was analyzed in lesional skin by FACS-based protein quantification from anti–TNF-α– and IgG-treated TTP–/– S100A9–/– mice. Data represent mean ± SEM from 3 independent experiments. *P < 0.05; ***P < 0.001, 1-way ANOVA (C and D).
S100A8 protein recovery in ihTNF-tg S100A9–/– mice promotes joint and skin inflammation.
To exclude nonspecific effects of TTP KO (24, 25), we crossed S100A9–/– mice with the ihTNF-tg mouse strain, which displays features of psoriatic arthritis 3 to 6 weeks after TNF-α induction (26). Expression of human TNF-α was induced in adult mice by doxycycline (dox) for up to 8 weeks. Disease progression in ihTNF-tg S100A9–/– mice was significantly aggravated compared with that in ihTNF-tg mice, as shown by loss of body weight and grip strength as well as increased paw swelling. None of the control mice developed any abnormalities after dox application (Figure 4, A–C). Employing optical imaging (OI) (fluorescence reflectance imaging [FRI]) using Cy5.5-labeled anti-S100A8 antibodies (27), we confirmed the recovery of S100A8 expression in vivo after TNF-α induction. Significant local tracer accumulation was observed in dox-treated ihTNF-tg S100A9–/– mice in paws and knee joints as well as in the skin (Figure 4D). The recovery of the S100A8 protein in ihTNF-tg S100A9–/– mice after dox stimulation was also confirmed by immunohistochemistry in the skin (Figure 4E), nail matrix (Figure 4F), and bone marrow as well as in bone marrow–containing metacarpal sections (Figure 4G and Supplemental Figure 5) and blood cells (Figure 4H). Histological analyses of distal interphalangeal (DIP) joints revealed a stronger hyperplasia of synovial tissue and cartilage destruction in ihTNF-tg S100A9–/– mice compared with that in ihTNF-tg mice (Supplemental Figure 6). Prolonged stimulation of isolated S100A9–/– bone marrow cells (BMCs) with TNF-α induced a recovery of S100A8 homodimers on the protein level, as observed before for S100A9–/– keratinocytes (Supplemental Figure 4B). In addition, S100A8 was released into the supernatant (Supplemental Figure 4B). In the absence of prolonged TNF-α stimulation, S100A8 homodimers were rapidly degraded in the proteasome (Supplemental Figure 4C). Taken together, these data demonstrate a strong induction of the S100A8 protein after prolonged TNF-α expression in the absence of S100A9 in vivo. As shown above, S100A8 cannot form tetramers in the absence of S100A9. Lack of this autoinhibitory mechanism results in expression of dominant active S100A8 dimers, which drive uncontrolled and, in the case of TTP KO, even fatal inflammation. Healthy S100A9–/– mice are protected from fatal inflammatory reactions due to appropriate proteasome degradation over time, while this mechanism is inadequate during chronic TNF-α conditions.
Figure 4. S100A8 recovery aggravates disease progression in ihTNF-tg S100A9–/– mice.

(A–C) Changes in body weight (A), grip strength (B), and swelling of forepaws (C) of dox-treated ihTNF-tg S100A9–/– mice compared with ihTNF-tg, S100A9–/–, or control mice. Data are shown as mean ± SEM of 3 independent experiments. n = 6 mice per group in each experiment. *P < 0.05; **P < 0.01; ***P < 0.001, 1-way ANOVA. (D) Control (upper images) or dox-treated (lower images) ihTNF-tg S100A9–/– mice (n = 5 for each group) were injected with Cy5.5-labeled anti-S100A8, and whole-body OI was recorded by FRI 24 hours later. Strong fluorescence intensities were detected in all paws and skin, indicating S100A8 protein recovery during chronic TNF-α stimulation. Nonspecific background signals in the liver and bladder indicate tracer degradation. (E and F) S100A8 and S100A9 immunostainings of skin (E) and forepaw toes (F) of ihTNF-tg S100A9–/– mice in the presence (6 weeks) or absence of dox confirm S100A8 recovery in the nail matrix, skin keratinocytes, and phagocytes. The lower 2 images in F are magnifications of boxes in middle images. Scale bars: 100 μm. (G, H) Cytospin preparations of bone marrow (G) and blood cells (H) of ihTNF-tg S100A9–/– or S100A9–/– mice treated with dox for 6 (G) or 3 weeks (H). Cells were stained for S100A8/S100A9 expression. Scale bars: 50 μm. In contrast with S100A9–/– mice, ihTNF-tg S100A9–/– animals showed a pronounced recovery of S100A8 protein in BMCs after dox stimulation (middle panel), which was abolished after removal of dox for 7 days (right panel). (H) Cytospin preparations of blood cells from dox-treated ihTNF-tg S100A9–/– mice compared with S100A9–/– and WT mice. Staining of blood cells for S100A8 showed a recovery of S100A8 protein in phagocytes, but not lymphocytes, after TNF-α induction.
Identification of the S100A9-binding site on TLR4/MD2.
Structural analysis revealed several aa hidden in the (S100A8/S100A9)2 tetramer interface (Figure 5A), but freely accessible in the heterodimer (Figure 5B) (28), most of which were associated with the second calcium-binding EF-hands of both S100 subunits. We changed selected aa of S100A9 (E64, D65, K72, Q73, E77, and R85) to alanine as a model of active S100 dimers (29). These aa were not involved in the coordination of the calcium ion, showed little contact with neighboring aa, and were freely accessible in the dimer (Figure 5C). Four of the six exchanges in S100A9 (E64A, D65A, Q73A, and E77A) already showed a trend in binding reduction of about 20% compared with WT S100A9 homodimers in a quantitative TLR4/MD2-binding assay (Figure 5D). However, by changing 2 of the selected aa, S100A9 dimers lost their affinity to the TLR4/MD2 complex almost completely (Figure 5E). Changes in the overall structure of the mutated proteins were excluded by fluorescence emission spectrometry (Supplemental Figure 7A).
Figure 5. Identification of the S100A9-binding site on TLR4/MD2.

(A–C) The PDB files of the S100A8/S100A9 tetramer (1XK4, A) and S100A9 dimer (1IRJ, C) were retrieved from the RSCB PDB website. (B) 1XK4 was modified containing only the E (S100A8, gray) and G (S100A9, green) chains resembling 1 heterodimer. Masked aa in the tetramer were analyzed by Cluspro. (B, yellow- or cyan-labeled aa for S100A8 or S100A9). Labeled S100A9 aa were chosen for mutation studies (C). (D) Single mutated S100A9 was analyzed for TLR4/MD2 binding compared with WT S100A9. All S100A9 mutants showed reduced binding to TLR4/MD2. (E) Double-mutated S100A9 was analyzed for binding to TLR4/MD2 as shown in D and showed significantly reduced binding properties compared with WT S100A9. Data represent mean ± SD of 5 independent experiments. *P < 0.05; **P < 0.01, 1-way ANOVA. (F) Monocytes were stimulated for 4 hours with intact S100A9 (0 h = no trypsinization) or the fragment mixture, and TNF-α levels were quantified by ELISA. Western blot shows remaining intact S100A9 (insert). One representative of 3 independent experiments is shown. (G) Tryptic fragments of hS100A9 (F) were analyzed for TLR4/MD2 binding, and peptide 15 corresponding to aa 73–85 showed specific interaction with TLR4/MD2, as detected by nanoUPLC/ESI-Q-TOF MS/MS (m/z 1614). Peptide 6 (NIETIINTFHQYSVK) was also detected in the control setting without TLR4/MD2 and reflects unspecific binding. (H) Peptides comprising aa 63–79 of S100A9 (MEDLDTNADKQLSFEEF, MW: 2032 g/mol), control peptides 63–79 5A (MAALDTNADAALSFAEF, MW: 1758 g/mol) or a scrambled peptide (DSLEMTEENLADQFKDF, MW: 2032 g/mol) were investigated for TLR4/MD2 binding as shown in G. Unspecific binding was analyzed by using peptides 63–79 without TLR4/MD2. Only S100A9 peptides 63–79 showed binding to TLR4/MD2 (3 independent experiments). Asterisks indicate polyethylene glycol impurities.
After almost complete trypsin digestion of S100A9 (Figure 5F, insert), half of the inflammatory activity on monocytes was still present, indicating that a peptidic fragment or fragments of S100A9 are sufficient to activate monocytes (Figure 5F). Using this preparation, 1 specific peptide (peptide no. 15 of 17 possible peptides without missed cleavages) was detected by a TLR4/MD2 pull-down assay and identified by mass spectrometry analysis, revealing the sequence of S100A9 from aa 73 to 85 (Figure 5G), which is almost identical to that of the peptide identified by structural analysis and site-directed mutagenesis described above. We next synthesized a peptide comprising the whole EF-hand II of S100A9 (peptides 63–79, MEDLDTNADKQLSFEEF) and confirmed its specific binding to TLR4/MD2, while a mutated control peptide (63–79 5A, MAALDTNADAALSFAEF; bold letters indicate introduced mutations) or a scrambled peptide sequence (DSLEMTEENLADQFKDF) failed to bind to the receptor complex completely (Figure 5H). S100A9 peptides with only 2 mutations in the sequence (MEALDTNADKALSFEEF or MADLDTNADKQLSFAEF) showed reductions in binding properties similar to those observed for homologues mutations in the S100A9 full-length protein (Supplemental Figure 7B).
Model of the S100A9 homodimer interaction with TLR4/MD2.
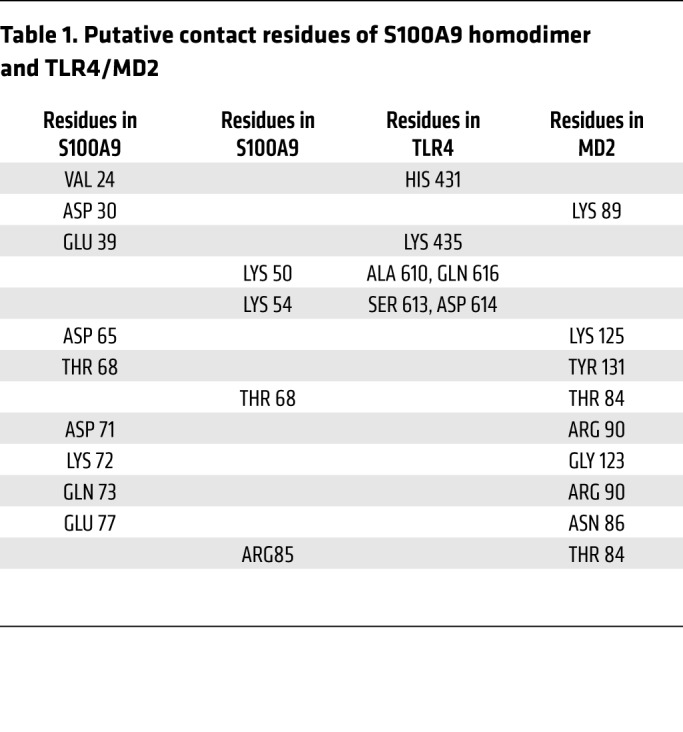
Using the known coordinates of the solved 3D structures for the human S100A9 dimer (Research Collaboratory for Structural Bioinformatics [RSCB] PDB ID: 1IRJ; https://www.rcsb.org/) and TLR4/MD2 (PDB ID: 3FXI), we simulated the binding of the synthesized peptide corresponding to EF-hand II of the S100A9 subunit with MD2 (Figure 6A). Unbiased studies of the docking of the S100A9 homodimer to TLR4/MD2 (Figure 6, B and C) confirmed this peptide-MD2 interaction for the intact S100A9 homodimer as well and identified additional aa potentially involved in binding to TLR4/MD2 (Table 1). With the exception of amino acid 64 (Glu), we were able to assign specific interaction residues for all aa mutated in S100A9 (D65, K72, Q73, E77, and R85) in MD2, including arginine 85 (second S100A9 subunit). Structural modeling of the S100A8/S100A9 heterodimer (PDB ID 1XK4, chains E and G) with TLR4/MD2 resulted in a model close to the homodimeric S100A9/TLR4/MD2 model (Figure 6, B and D). In a granuloma mouse model, we finally confirmed the presence of active dimers in vivo by crosslinking noncovalently associated S100A8/S100A9 complexes (Supplemental Figure 8, A and B). Finally, tetramer formation of S100A8/S100A9 heterodimers was significantly diminished under acidic conditions typical for inflammatory tissues/fluids (starting at pH values below 5.8; Supplemental Figure 8, C–E).
Figure 6. Proposed structural modeling of S100A9 homodimer interaction with TLR4/MD2.

(A) Modeling of the S100A9 peptides 63–79 hTLR4/MD2 complex in ribbon representation showing a close-up view with the putative interaction aa (labeled in gray for S100A9 aa and red for MD2-specific aa) as indicated. The model is based on the crystal structure of the TLR4/MD complex (PDB ID 3FXI), with LPS being omitted and the coordinates of peptides 63–79 being taken from PDB ID: 1IRJ. (B) Model of the potential binding site of S100A9 homodimer and TLR4/MD2 showing a number of contact aa summarized in Table 1. (C) Close-up view of B focused on the S100A9 EF-hand II, MD2 binding region. (B and C) Model is based on the crystal structures of S100A9 (PDB ID: 1IRJ, chains G and H) and of the human TLR4/MD2 complex (A chain of TLR4 and C chain of MD2) with LPS being omitted. The side chains of S100A9 aa are labeled in gray or brown and those of TLR4/MD2 in red. (D) Structural modeling of S100A8/S100A9 heterodimer (PDB ID 1XK4, chains E and G) interaction with TLR4/MD2 confirms identical binding regions for the homodimer S100A9 as well as for the heterodimer. Molecular modeling and graphics preparation were performed using Cluspro and PyMOL software packages.
Table 1. Putative contact residues of S100A9 homodimer and TLR4/MD2.
Discussion
The concept of sterile inflammation driven by endogenous danger signals has markedly increased our understanding of numerous inflammatory diseases. S100A8/S100A9 are the most abundant alarmins in many clinically relevant disorders, such as rheumatoid arthritis or other autoimmune diseases, inflammatory lung or bowel diseases, allergies, or cardiovascular complications (3, 6). Although the alarmin concept is supported by different gene KO models in mice, clear mechanistic models of their local regulation are lacking. We now demonstrate an autoinhibitory mechanism in which calcium-induced tetramer formation abolishes the proinflammatory activity of S100A8/S100A9 dimers, which has several implications for the field of alarmin-driven inflammation in general and S100 biology in particular. Our data unravel a model of a local self-control mechanism of S100 alarmin–driven inflammation. In the intracellular low-calcium milieu, S100A8 and S100A9 are present as heterodimers that are secreted by activated phagocytes at the site of inflammation (30–32). These dimers interact with and activate TLR4/MD2-expressing cells via a specific binding site mainly located in the second EF-hand, thus promoting inflammatory processes. Due to high extracellular calcium concentrations, heterodimers subsequently associate to (S100A8/S100A9)2 tetramers. Tetramers are unable to bind to TLR4/MD2, thereby restricting the biological activity of S100 proteins locally in order to avoid undesirable systemic inflammatory side effects. The binding of other TLR4/MD2 agonists, including LPS and S100 homodimers, is unaffected by tetramer formation. Our findings explain many contradictory reports regarding inflammatory S100 effects, since most studies did not consider the fact that different complex forms may show different inflammatory activities (33). The biological relevance of our hypothesis was confirmed in 2 independent TNF-α–driven mouse models of inflammation. In both of these models, S100A9 deficiency promoted uncontrolled inflammation due to S100A8 dimer recovery. The presence of dominant active S100A8 dimers prevented calcium-induced tetramer formation and controlled the inflammatory effects by autoinhibition due to the absence of the S100A9 subunit. Analysis of protein samples directly obtained from phagocytes, serum, or other biological samples indicated that S100A8/S100A9 heterocomplexes are the predominant form of these molecules in vivo (33). This assumption is supported by experimental evidence analyzing S100A8/S100A9 interactions in vivo (e. g., yeast 2-hybrid system) (34) and comparative biophysical analysis of homo- and heterocomplexes (35) as well as structural data (28, 29, 36), indicating that homodimers are a useful experimental tool for mimicking S100A8/S100A9 dimer effects due to the fact that homodimers cannot tetramerize, but are of minor relevance in vivo (35, 37).
The identification of a specific molecular structure responsible for S100 TLR4/MD2 interaction presented here markedly increases the chances of addressing S100A8/S100A9 as a highly relevant target for therapeutic approaches. The heterocomplex, also known as calprotectin, is widely used as a biomarker for detection and monitoring the disease activity of inflammatory bowel disease, psoriasis, or rheumatoid arthritis (15). S100A8/S100A9 expression predicts therapeutic efficiency and disease flares of arthritis, further emphasizing the functional significance of these proteins in clinically relevant diseases (38). The extraordinarily high expression of S100A8/S100A9 at local sites of active inflammation compared with clinically noninvolved tissue or systemic levels further highlights the relevance of the molecular mechanism described here for local inflammatory activity (30, 39). The frequently local and discontinuous patterns of inflammatory symptoms in many autoimmune diseases may be explained by the fact that local manifestation of clinical inflammation due to underlying systemic autoimmunity needs an additional tissue-derived stress trigger such as locally active S100 alarmins. The high local expression of these molecules during inflammation is also demonstrated in in vivo imaging studies of different inflammatory disease models in mice (27). General KO of the S100A8/S100A9 system significantly blocks inflammation, and biological relevance of this system has been confirmed in various prelinical models of inflammatory processes, such as LPS-induced septic shock, arthritis, psoriasis, and SLE-like disease, all of which reflect the expression pattern of S100A8/S1000A9 in human diseases very well (8, 11, 12, 40). Finally, several sophisticated models of metastasis development during tumor progression indicate a role of S100 TLR4–driven inflammation in the establishment of premetastatic niches and the potential use of targeted blockade of S100A8/S100A9 to prevent dissemination of tumor cells (41, 42).
TLR4 has already been shown to play a role in sterile inflammation in the absence of any microbial ligands (1–4), and S100A8 and S100A9 are the major endogenous TLR4 ligands in many inflammatory diseases, especially rheumatoid arthritis, psoriasis, and inflammatory bowel disease (38, 40, 43). TLR4-targeted therapies are currently under development for acute and chronic inflammation. In terms of therapy, targeting S100 proteins instead of TLR4 has an obvious advantage, as specific blocking of active S100A8/S100A9 heterodimers would not primarily affect the TLR4-dependent antimicrobial defense mechanisms. Although concentrations of S100A8/S100A9 are relatively high during inflammatory diseases, our data indicate that the pharmacologically relevant target is only the minor fraction of active heterodimers. There are already 2 lines of evidence that blocking of S100 proteins may be effective. Molecules of the so-called Q-compound family have been shown to inhibit the interaction of S100A8/S100A9 with TLR4, show an antiinflammatory effect, and have been successfully used in phase III trials for treatment of multiple sclerosis (44, 45). In addition, recombinant peptide bodies have been shown to block S100-dependent effects in a murine model of tumor metastasis. However, it is not clear whether Q-compounds or these peptide bodies specifically address the TLR4-binding region of S100 proteins described here (46). Along with the mechanism of locally restricted activity of S100A8/S100A9-driven inflammation shown here, these molecules are promising candidates for a very specific and local antiinflammatory therapy avoiding systemic side effects. Taking our data together, we describe a molecular mechanism by which alarmins drive sterile inflammation restricted to local sites of injury by autoinhibitory regulation.
Methods
Animal studies.
The WT C57BL/6, S100A9–/–, TTP–/–, TNF-α–/–, hTNF-tg (strain Tg197), and ihTNF-tg mouse strains have been described elsewhere (13, 18, 26). All mouse lines had a C57BL/6 genetic background except the ihTNF-tg mice, which consisted of C57BL/6 × FVB-N hybrids. For analysis of the role of S100A8/S100A9 proteins in the development of TNF-α–mediated disorders, further hybrid mice with the S100A9 deficiency in combination with other KO genes and transgenes were generated and are described in Results. Genotyping was performed by PCR using primers and conditions as described elsewhere (13, 18, 26).
Scoring criteria of the psoriasis-like skin disease of TTP–/– S100A9–/– mice.
To evaluate the severity of the psoriasis-like phenotype of TTP–/– S100A9–/– mice, an adapted PASI score was used, as follows: grade 0, 0% involved area; grade 1, less than 10% involved area; grade 2, 10%–29% involved area; grade 3, 30%–49% involved area; grade 4, 50%–69% involved area; grade 5, 70%–89% involved area; and grade 6, 90%–100% involved area.
Scoring criteria of ihTNF-tg/S100A9–/– and ihTNF-tg mice.
To induce expression of the human TNF-α transgene, 6-week-old mice were kept on drinking water containing dox (1 mg/ml, Sigma-Aldrich) and 5% sucrose for the indicated times. The water was protected from light and exchanged every 3 days. Clinical assessments, such as body weight, paw swelling, and grip strength, were performed twice a week during the dox stimulation of the mice. Paw swelling was graded from 0 to 3, and grip strength was graded from 0 to –3 as previously described (47).
Anti–TNF-α treatment.
To neutralize the excess of TNF-α in TTP–/– S100A9–/– mice, the animals received either 40 mg/kg certolizumab (UCB), a PEGylated Fab′ fragment of a humanized TNF-α inhibitor monoclonal antibody, or a monoclonal antibody of irrelevant specificity as control antibody into the back skin on days 4, 6, and 8 after birth. The antibody is already in clinical use as arthritis and psoriasis therapy.
Reagents.
LPS from E. coli (055:B5) was purchased from Sigma-Aldrich. SB202190 and PD98059, specific inhibitors for p38 MAPK and MEK were from Calbiochem. Staurosporine and GF109203X (inhibitors of protein kinase C) were from Alexis Biochemicals and Molecular Probes (via MoBiTec). The TLR4 antagonist RS-LPS (LPS from Rhodobacter sphaeroides) was from Invivogen, and the blocking mouse monoclonal antibody to human TLR4 (HTA125) was from Abcam.
Determination of cytokine concentrations.
TNF-α and IL-1β protein levels were analyzed in culture supernatants using commercial ELISA kits (BD Bioscience). Cytokine concentrations in tissues were assayed using the FACS-based FlowCytomix Multiplex Kit (eBioscience).
Cells and cell-culture conditions.
Methods of cell purification and culture conditions have been described in detail elsewhere (8). Briefly, for stimulation experiments, human monocytes (106 cells/ml) were treated with varying concentrations (100 ng/ml to 5 μg/ml) of purified S100A8, S100A9, or S100A8/S100A9 proteins or mutants thereof, LPS (1–10 ng/ml), and/or IFN-γ (500 U/ml) for the times indicated in figure legends.
Human monocytes were isolated from blood samples after leukapheresis via Pancoll (Pan Biotech) and subsequent Percoll (GE Healthcare) gradient centrifugation (48). Cells were cultivated overnight in Teflon bags in McCoy’s 5a Medium (Biochrome) supplemented with 15% FCS (Gibco Life Technologies, Thermo Fisher Scientific) prior to use for stimulation or in functional assays.
Mouse BMCs were flushed from femurs and tibias with Dulbecco’s medium. Erythrocytes were lysed with lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) for 5 minutes at room temperature, and the remaining cells were washed once with PBS. Murine whole-blood cells were collected from heparin blood after lysis of erythrocytes with lysis buffer. Collected cells were either lysed for Western blot analysis or centrifuged on glass slides for S100A8 and S100A9 staining.
A549 human alveolar epithelial cells (ATCC, CCL-185) were grown in HAM′s F-12 medium supplemented with 2 mM l-glutamine and 10% heat-inactivated FCS. The experiments reported here were performed using confluent cells in serum-rich medium.
HEK293 cells stably transfected with human TLR4, CD14, and MD2 genes or TLR4 only as well as control cells transfected with empty control vectors were purchased from Invivogen. HEK293 cells stably transfected with human receptor for advanced glycation end products (RAGE) were provided by Marco E. Bianchi (San Raffaele Scientific Institute, San Raffaele University, Milan, Italy). HEK293 cells were grown in DMEM with 4.5 g/l glucose, 10% FBS at 37°C in 5% CO2.
Primary epidermal keratinocytes were isolated from the skin of 2- to 3-day-old newborn mice as described (49) and cultured in FAD medium containing 50 μM calcium ions. All cell culture media (Merck Millipore) and all other reagents were entirely free of endotoxin, as checked by limulus amebocyte lysate (LAL) assay (BioWhittaker).
Antibody labeling.
Rabbit-derived antibodies addressing S100A8 were purified via protein G–sepharose and labeled with the fluorochrome Cy5.5 according to the manufacturer’s instructions (GE Healthcare). Briefly, 5 mg of the antibody was dialyzed toward 100 mM Na2CO3 buffer, pH 8.0, and a 20-fold excess of the fluorochrome was added for 90 minutes at room temperature. The resulting tracer was purified from unbound dye using SEC (PD10 column). The labeling efficacy (dye/antibody ratio) was determined on the basis of UV-spectra of the purified dye–antibody compound using PBS as a reference buffer. Typically, the labeling resulted in 2.5–3.0 fluorochrome molecules per antibody.
FRI.
FRI was performed using the In-Vivo FX PRO Imaging Station (Bruker Biospin MRI GmbH). For imaging of Cy5.5-labeled S100A8 antibody, the excitation light was set to 630 nm using an appropriate bandpass filter. Emission at 700 nm was recorded using a filter-equipped high-sensitivity (4-million-pixel) cooled charge-coupled device (CCD) camera. Acquisition time was 5 seconds for each image, followed by a photography-style white light image for the biodistribution, respectively. Mice were held under isoflurane inhalation anesthesia (2.0% isoflurane in air) for the duration of the scan. As the total scan time was usually less than 1 minute, physiological effects due to a significant decrease of body temperature were not to be expected. Mice were intravenously injected with Cy5.5-labeled S100A8 antibody (α-S100A8–Cy5.5, 2 nmol of Cy5.5 ~100 μg antibody in total) 24 hours prior to OI.
Protein expression and purification of S100 WT and mutant proteins.
Recombinant S100 proteins were expressed in E. coli BL21 (DE3) cells and purified as previously described (17, 35). Briefly, bacteria cells were transformed with pET11/20 expression vectors containing the cDNAs for murine or human WT S100A8 or WT S100A9 or the human S100A9 mutants N69A, E78A, E64A, D65A, K72A, Q73A, E77A, R85A, E64A+D65A, Q73A+E77A, E64A+E77A, and D65A+Q73A; after growing at 37°C in 2× yeast tryptone medium for 24 hours, cells were harvested and lysed. The inclusion bodies (IB) were pelleted and dissolved in 8 M urea buffer. To establish proper refolding, samples were first adjusted to pH 2.0–2.5 by adding hydrochloric acid, and after 60 minutes incubation at room temperature, proteins were stepwise dialyzed against Tris buffers in the presence of 2 mM DTT to become adapted to pH 7.4. After centrifugation (10 minutes, 60,000 g, 4°C) to pellet the aggregated material, samples were further dialyzed against appropriate buffers for anion exchange column and gel filtration chromatography. The protein concentrations were determined by UV absorption at 280 nm using a specific absorption coefficient of 1.02 (humS100A8), 0.53 (humS100A9 and mutants), 0.76 (humS100A8/S100A9 and mutants) 0.40 (murS100A8), 0.11 (murS100A9), or 0.25 (murS100A8/S100A9) ml/mg-1/cm-1, respectively. Proper complex formation of S100A8/S100A9 and mutants thereof was enabled by including a denaturation/renaturation step. Both S100 subunits were mixed in an equimolar ratio. Samples were adjusted to pH 2.0–2.5 by adding hydrochloric acid in 0.1 M glycine buffer. After 60 minutes incubation at room temperature, samples were dialyzed stepwise to become adapted to 20 mM Tris (pH 8.5), 1 mM EDTA, 1 mM EGTA, and 2 mM DTT, pH 8.5, for proper refolding and complex formation. After centrifugation (10 minutes, 60,000 g, 4°C) to pellet the aggregated material, samples were applied to anion exchange column chromatography. Only fractions that contained equal amounts of both proteins, as detected by SDS-PAGE, were collected. Samples were stored as stock solutions at –20°C. Correct refolding and complex formation were assessed by SDS-PAGE, MALDI–mass spectrometry (MALDI-MS), and electrospray ionization–MS (ESI-MS) (16, 50). To confirm the structural integrity of double-mutated S100A9 homodimers, fluorescence spectra were recorded at 20°C with a spectrofluorometer (Spex, Model FluoroMax II, Instruments SA) using protein concentrations of 5 μg/ml. Changes in the conformation of S100A9 due to the mutation or mutations would be easily detectable by altered intrinsic Trp and Tyr fluorescence properties. All fluorescence spectra were buffer corrected (35). Possible endotoxin contaminations were excluded by LAL assay (BioWhitaker). LPS could not be detected or was lower than 1 pg/μg S100 protein in the different batches. As an additional control, S100A9 protein samples were completely heat inactivated at temperatures at which LPS was still fully active, but no biological response was registered. Moreover, LPS contaminations were further excluded by blocking experiments using polymyxinB sulfate (Sigma-Aldrich) for all preparations.
DNA microarray hybridization.
Total cellular RNA (Macherey-Nagel) was isolated from human monocytes that were exposed to 5 μg/ml of S100A8, S100A9, or S100A8/S100A9 or 1 ng/ml LPS or to medium for 4 hours (3 independent experiments) and used for analysis by Illumina human HT-12 Expression BeadChips. The protocol was performed according to the manufacturer′s instructions. Data were imported into Partek Genomics Suite V6.6 (PGS) (Partek) using robust multiarray average (RMA), which is an algorithm used for background correction, log2 transformation, and quantile normalization of expression data prior to batch correction. A 1-way ANOVA model was performed to calculate the 1,000 most variable and differentially expressed genes between the different stimulating conditions. Differentially expressed genes were defined by a fold change (FC) greater than 2.5 or less than –2.5 and a false discovery rate–corrected (FDR-corrected) P value of less than 0.05. To visualize the structure within the data, we performed hierarchical clustering (HC) on the 1,000 most variable genes, with default settings in PGS, on FDR-corrected P values according to the expression values of the samples across the conditions as described (9).
Quantitative real-time PCR.
RNA from HEK cells, human monocytes, or snap-frozen mouse tissue was isolated using the NucleoSpin RNA II Kit (Macherey-Nagel), and 1 μg of total RNA was reverse-transcribed to cDNA using RevertAid H Minus Transcriptase (Fermentas). Expression levels of selected genes were confirmed by quantitative reverse-transcriptase PCR (RT-PCR), which was performed using Sybr Green (PeqLab). Data were acquired with the CFX 384 Real-Time System and CFX-manager software, version 3.0 (Bio-Rad). Each measurement was set up in duplicate, and the relative expression was calculated as 2–ΔΔCt using ribosomal protein L (RPL), GAPDH, or β-actin as the housekeeping control genes. Sample data are presented as fold induction compared with naive cells.
The following primers were used for human PCR analysis: IL-6 forward, 5′-AGAGGCACTGGCAGAAAACAAC-3′; IL-6 reverse, 5′-AGGCAAGTCTCCTCATTAATCC-3′; SOCS1 forward, 5′-AGCTCCTTCCCCTTCCAGATTT-3′; SOCS1 reverse, 5′-CCACATGGTTCCAGGCAAGTAATA-3′; SOCS3 forward, 5′-CCTCAAGACCTTCAGCTCCAA-3′; SOCS3 reverse, 5′-ACTGCGCTCCAGTAGAAGCC-3′; TNF-AIP3 forward, 5′-CGTCAGAAGCTGGCTGATTTG-3′; TNF-AIP3 reverse, 5′-CCTTGTCGGTCTCCTCCATTTC-3′; IL-10 forward, 5′-GCTGAGAACCAAGACCCAGACA-3′; IL-10 reverse, 5′-CGGCCTTGCTCTTGTTTTCA-3′; IL-1α forward, 5`-AGACCTACGCCTGGTTTTCC-3′; IL-1α reverse, 5`-GCTGAAGGAGATGCCTGAGAT-3′; CD80 forward; 5′-CTGCTTTGCCCCAAGATGC-3′; CD80 reverse, 5′-CAGATCTTTTCAGCCCCTTGC-3′; SOCS2 forward, 5′-CTGACCAAACCGCTCTACACGT-3′; SOCS2 reverse, 5′-TTGTTGGTAAAGGCAGTCCCC-3′; NF-κB forward, 5′-TTATGTATGTGAAGGCCCATCC-3′; NF-κB reverse, 5′-TGGTCCCACATAGTTGCAGATT-3′; TNF-α forward, 5′-CTTCTCGAACCCCGAGTGAC-3′; TNF-α reverse, 5′-TGAGGTACAGGCCCTCTGATG-3′; RAGE forward; 5′-CTACCGAGTCCGTGTCTACCA-3′; RAGE reverse, 5′-CATCCAAGTGCCAGCTAAGAG-3′; TLR-4 forward, 5′-CCAGAACCAAACGATGGACT-3′; TLR-4 reverse, 5′-CCTGTCCCTGAACCCTATGA-3′; CXCL10/IP-10 forward, 5′-AGACATCTCTTCTCACCCTTC-3′; CXCL10/IP-10 reverse, 5′-GGAACCTCCAGTCTCAGCACCA-3′; CCL5 forward, 5′-CAGTGGCAAGTGCTCCAACC-3′; CCL5 reverse, 5′-CCATCCTAGCTCATCTCCAAAGAGT-3′; RPL forward, 5′-AGGTATGCTGCCCCACAAAAC-3′; RPL reverse, 5′-TGTAGGCTTCAGACGCACGAC-3′; GAPDH forward, 5′-CAAATTCCATGGCACCGTCA-3′; and GAPDH reverse, 5′-TCGCCCCACTTGATTTTGG-3′.
The following primers were used for murine PCR analysis: TNF-α forward, 5′-AGAAACACAAGATGCTGGGACAGT-3′; TNF-α reverse, 5′-CCTTTGCAGAACTCAGGAATGG-3′; IL-1β forward, 5′-TGGGCTGGACTGTTTCTAATGC-3′; IL-1β reverse, 5′-TGTCTTGGCCGAGGACTAAGG-3′; IL-6 forward, 5′-TGAGATCTACTCGGCAAACCTAGTG-3′; IL-6 reverse, 5′-CTTCGTAGAGAACAACATAAGTCAGATACC-3′; IL-17A forward, 5′-CAGAAGGCCCTCAGACTACC-3′; IL-17A reverse, 5′-TCTCGACCCTGAAAGTGAAG-3′; IL-22 forward, 5′-ACCTTTCCTGACCAAACTCA-3′; IL-22 reverse, 5′-AGCTTCTTCTCGCTCAGACG-3′; IL-23 forward, 5′-ATCCAGTGTGAAGATGGTTGTGA-3′; IL-23 reverse, 5′-CTGGAGGAGTTGGCTGAGTC-3′; IL-36 forward 5′-TACATGGGAGTGCAAAGGCC-3′; IL-36 reverse 5′-AGTGGGCAGCTCCCTTTAGAGC-3′; IFN-γ forward 5′-TGCTGATGGGAGGAGATGTCTAC-3′; IFN-γ reverse 5′-TTTCTTTCAGGGACAGCCTGTTAC-3′; RPL forward, 5′-TGGTCCCTGCTGCTCTCAAG-3′; RPL reverse, 5′-GGCCTTTTCCTTCCGTTTCTC-3′; GAPDH forward, 5′-CCCACTCTTCCACCTTCGATG-3′; GAPDH reverse, 5′-GTCCACCACCCTGTTGCTGTAG-3′; β-actin forward, 5′-AAGGAGAAGCTGTGCTACGTC-3′; and β-actin reverse, 5′-AACCGCTCATTGCCAATGGTG-3′.
Histology and immunohistochemistry.
The paws were fixed overnight in 4% paraformaldehyde, decalcified with 0.5 M EDTA solution, and embedded in paraffin. Sections measuring 4 μm in thickness were analyzed. The extent of inflammation and cartilage damage in the paws was determined by morphometry as described elsewhere (47). Usually, 4 different sections per mouse sample were quantified from 5 to 6 mice for each mouse line. Proteoglycan loss was determined as the percentage of destained cartilage area. For skin samples, mice were sacrificed at day 9, and skin from the back was fixed in 4% paraformaldehyde or embedded in Tissue-Tek (Sakura) for cryosectioning. Fixed skin samples were embedded in paraffin according to procedures used for H&E staining and immunohistochemistry. For immunohistochemistry, paraffin skin sections (4 μm) were dewaxed, blocked 30 minutes with 10% normal goat serum (Invitrogen), and incubated with rabbit anti-S100A9 or anti-S100A8 antibodies for 1 hour at room temperature. Vectastain ABC-AP (BA-1000) and Vector Red Substance Kits (SK-5100) (Vector Laboratories) were used for visualization of the stained proteins. For immunofluorescence analyses, sections (5 μm) of frozen tissue were fixed in ice-cold acetone. After blocking with 10% normal goat serum in PBS for 30 minutes, the sections were washed in PBS (3 × 5 minutes). Primary antibodies dissolved in PBS were applied for 1 hour at room temperature or overnight at 4°C. After washing in PBS, sections were incubated with anti-rabbit or anti-rat Alexa Fluor 488–coupled IgG. Sections were then counterstained with propidium iodide or DAPI. Finally, after washing in PBS, sections were fixed with 4% paraformaldehyde and mounted in Mowiol (Sigma-Aldrich). The following primary polyclonal antibodies were used: anti-keratin 14 (1:100; clone poly 19053, BioLegend), anti-K10 (1:100; clone poly 19054, BioLegend), anti-Lor (1:2000; clone poly 19051, BioLegend) (Covance). Anti-Ki67 (1:100, ab15580, Abcam) rat monoclonal antibodies were used on frozen sections as follows: F4/80 (1:100; clone Cl:A3-1, Bio-Rad), anti–Gr-1 (1:20; clone RB6-8C5, BD Bioscience), and anti-CD3 (1:1000; clone 17A2, Thermo Fisher). Sections were counterstained with propidium iodide to visualize the nuclei. Fluorescence stainings were analyzed using a Zeiss Axio Vision Microscope.
Detection of MyD88 translocation by immunofluorescence microscopy.
A549 cells were cultured on fibronectin-coated LabTec chamber slides (Nunc, Thermo Scientific) until confluence. After stimulation, as indicated in the figure legends, cells were fixed with formaldehyde (3.7%, 20°C, 10 minutes) and permeabilized with Triton-X 100 (0.5%, 20°C, 10 minutes). After blocking of unspecific binding sites with FCS (10%, 4°C, overnight), samples were processed for single-labeling immunofluorescence using a polyclonal rabbit antibody to human MyD88 (ALX-210-900, Alexis Biochemicals) (51).
Immunoprecipitation of IRAK-1.
Human monocytes (20 × 106 cells per sample) were preincubated overnight with human recombinant IFN-γ (500 U/ml, Bender MedSystems) or with medium alone and subsequently stimulated with 10 ng/ml LPS or S100A9 (5 μg/ml) for 45 minutes. Cells were then lysed in 1 ml lysis buffer (0.5% Nonidet P-40, 10% glycerol, 50 mM HEPES, pH 7.9, 250 mM NaCl, 20 mM glycerophosphate, 5 mM p-nitrophenyl phosphate, 1 mM EDTA, 1 mM sodium orthovanadate, 5 mM dithioerythritol, 1× complete protease inhibitors; Roche). IRAK-1 was immunoprecipitated using 2 μg per sample IRAK-1–specific monoclonal antibody (catalog sc-5288, Santa Cruz Biotechnology Inc.) and a mixture of protein G–sepharose and protein A–agarose (Roche). In vitro kinase assays were performed as described previously (52).
Western blot analysis.
Nuclear cell extracts were prepared using the extraction kit (Epigentek). For each stimulus, equal cytosolic amounts of monocytes (p–IRAK-1, catalog sc-130197, Santa Cruz Biotechnology Inc.), keratinocytes (murS100A8, homemade), BMCs (murS100A8), or nuclear extracts of monocytes (P50 [ab7971], P65 [ab7970] Abcam) were separated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Schleicher & Schuell BioScience). Peptidic fragments of S100A9 generated by trypsin digestion were separated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes, which were then blocked with 5% skim milk powder and subsequently probed with primary antibodies overnight at 4°C. Afterwards, bound primary antibody was detected with HRP-conjugated secondary antibodies and developed with enhanced chemiluminescence system (ECL).
Proteasome and ubiquitin enzymes E1 and E2 inhibition assays.
BMCs were isolated from S100A9–/– mice. Cell pellet was resuspended in BMC growth medium (DMEM, 10% FCS, 20% L929 cell supernatant, 1% penicillin/streptomycin, 2% l-glutamine), and cells were seeded at 1 × 106 cells/well in a 24-well plate at 1 ml per well. Cells were then treated with inhibitors of the proteasome (MG132, UBPBio), ubiquitin-activating enzyme E1 (PYR-41, UBPBio), or ubiquitin-conjugating enzyme E2 (BAY-11-7821, Tocris Bioscience) at the indicated time points and concentrations and incubated at 37°C, 5% CO2. The control cells were treated with DMSO.
Tryptic digestion of human S100A9 homodimers.
Immobilized TPCK trypsin (25 μl of settled gel, Pierce) was used to digest 30 μg human S100A9 at 37°C for different time points, as indicated in Figure 5F. Subsequently, samples were centrifuged (5 minutes, 400 g) using a resin separator to remove trypsin-loaded beads. Aliquots of the supernatants were taken either for analyses by SDS-PAGE/Western blotting or for stimulation of human monocytes for 4 hours. TNF-α concentrations in supernatants of stimulated monocytes were then determined by ELISA (OptEIA, BD Biosciences). The fragments generated by tryptic digestion of hS100A9 were incubated with TLR4/MD2-coupled protein A/G agarose beads in the presence of 100 μM Ca2+ ions. Bound fragments were eluted at pH 2.0 and analyzed via MALDI-TOF and LC-coupled tandem mass spectrometry.
Immunoprecipitation studies to identify TLR4/MD2-binding peptides.
Anti-His antibody (catalog 37-2900, Invitrogen) and His-tagged TLR4/MD2 (R&D Systems) were mixed and coupled to protein A/G agarose (Pierce, Thermo Scientific). Tryptic peptides of S100A9 were added in the presence of 1 mM calcium. Bound peptidic fragments were eluted at pH 2.0 and analyzed by nano-ESI-Q-TOF and MALDI-TOF mass spectrometry. Identical experiments were performed to analyze the binding of the chemically synthesized peptides of S100A9 (aa 63–79, MEDLDTNADKQLSFEEF), corresponding control peptides (aa 63–79 5A, MAALDTNADAALSFAEF, aa 63–79 2A, MEALDTNADKALSFEEF, aa 63–79 2A, MADLDTNADKQLSFAEF; bold letters indicate introduced mutations), or a scrambled peptide of S100A9 (aa DSLEMTEENLADQFKDF).
Binding of WT S100A9 and mutated S100A9 to TLR4/MD2.
Binding of S100A9 to TLR4/MD2 was analyzed by a modified S100 ELISA. Briefly, TLR4/MD2 (R&D Systems) was coupled to the wells of a plate and served as a capturing molecule. S100A9 or mutant S100A9 proteins were added at a concentration of 2 μg/ml each in the presence or absence of 100 μM calcium. Bound S100 proteins were detected by the addition of a primary anti-S100A9 antibody (1 μg/ml, polyclonal rabbit, homemade) and the secondary anti-rabbit IgG antibody coupled to HRP (1 μg/ml, Cell Signalling Technology). Tetramethylbenzidine (TMB) was used as substrate to quantify binding efficiency by absorbance readings at 450 nm (Anthos Mikrosysteme).
Computer-assisted ligand/receptor interaction studies.
PDB files of an S100A8/S100A9 tetramer (PDB ID: 1XK4, chains E,F,G,H), S100A9 tetramer (PDB ID: 1IRJ), and TLR4/MD2 dimer (PDB ID: 3FXI) were retrieved from the RSCB PDB website. The S100A8/S100A9 PDB file was modified so that it contained only the E and G chains resembling the heterodimer. The S100A9 PDB file was modified so that it contained only the G and H chains resembling 1 homodimer. The TLR4/MD2 PDB file was modified so that it contained only the A chain of TLR4 and C chain of MD2. The modified PDB files of S100 proteins were then docked against TLR4/MD2 using Cluspro (http://cluspro.bu.edu/login.php) (53). PyMOL (https://pymol.org/2/) and Swiss-Pdb viewer (https://spdbv.vital-it.ch/) software packages were used to visualize the docking interactions. We concentrated our analyses on the identification of aa that are not involved in Ca2+ binding and are sterically free for binding to TLR4. The aa in S100A9 (positions 64, 65, 72, 73, 77, and 85) were chosen for mutation studies.
SEC.
SEC experiments were performed using a Superdex 75 loaded column connected with an Äktapurifier 900 FPLC (GE Healthcare) controlled by UNICORN software. The SEC separation runs were performed on a 5.0 × 640 mm column, and UV detection was conducted at a wavelength of 280 nm by running a linear flow rate of 0.25 ml/min. The column was calibrated with 8.56 kDa ubiquitin, 12.4 kDa cytochrome c, 23.3 kDa trypsin, 43 kDa ovalbumin, and 66 kDa serum albumin, and the running buffer was either HBS, 1 mM EDTA, 1 mM DTT, pH 7.4. or HBS, 5 mM Ca2+, 1 mM DTT, pH 7.4.
Statistics.
Values are shown as mean ± SD unless otherwise stated. P values are given in the figure legends, and values of P > 0.05 were considered not to be significant. Statistical analyses were performed by parametric tests (2 tailed t test or 1-way ANOVA) and the Mann-Whitney U test.
Study approval.
Experimental procedures were approved by the local ethics committee in accordance with German regulations of the Society for Laboratory Animal Science, the European Health Law of the Federation of Laboratory Animal Science Associations, and the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen.
Data availability.
All original microarray data were deposited in the NCBI’s Gene Expression Omnibus database (GEO GSE109646). Further information about the data and about the reagents used is available upon request to the corresponding author.
Author contributions
T Vogl, AS, JR, SL, TL, and TP designed and supervised the study and the experiments, performed animal studies, and wrote the manuscript. VW, T Völler, ST, SZ, DC, KL, MF, PP, and CW performed animal studies and experiments and analyzed the data. SH, CG, and M Schäfers performed PET and OI experiments. SKJ, OP, M Stoll, and JLS performed the microarray study and analyzed the data. SK and AD performed mass spectrometry experiments. All authors edited the manuscript.
Supplementary Material
Acknowledgments
We thank H. Berheide, H. Hater, and D. Lagemann for excellent technical support. This work was supported by grants from Cluster-of-Excellence EXC 1003 Cells in Motion, Münster, Germany (FF project-2013-24 to JR), the Interdisciplinary Center of Clinical Research at the University of Muenster (Vo2/004/14, Ro2/003/15, Lo2/004/16 and core unit PIX to T Vogl, JR, and KL), the German Research Foundation (DFG) (CRC 1009 B7, B8, and B9 to KL, T Vogl, and JR), CRC Transregio-128 (A2 to JR and A10 to KL, CRC 656 A9 to T Vogl and SH), the Federal Ministry of Education and Research (BMBF), project AID-NET and E-RARE, Treat-AID (to JR), and the CRC 704 and Excellence Cluster ImmunoSensation (to JLS). T Völler was supported by a DFG postdoctoral fellowship (grant VO 2273/1-1).
Version 1. 04/03/2018
Electronic publication
Version 2. 05/01/2018
Print issue publication
Funding Statement
Funding for Thomas Vogl
Funding for Johannes Roth
Funding for Sven Hermann
Funding for Karin Loser
Funding for Thomas Vogl
Funding for Johannes Roth
Funding for Johannes Roth
Funding for Thomas Vogl and Sven Hermann
Funding for Johannes Roth
Funding for Joachim L. Schultze
Funding for Joachim L. Schultze
Funding for Tom Völler
Footnotes
T Völler’s present address is: Institute of Microbiology, Eidgenössische Technische Hochschule (ETH) Zurich, Zurich, Switzerland.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(5):1852–1866.https://doi.org/10.1172/JCI89867.
Contributor Information
Thomas Vogl, Email: vogl@uni-muenster.de.
Athanasios Stratis, Email: stratis@uni-muenster.de.
Viktor Wixler, Email: vwixler@uni-muenster.de.
Tom Völler, Email: Tom.Voeller@gmx.de.
Sumita Thurainayagam, Email: sumitathurai@yahoo.co.uk.
Stefanie Zenker, Email: zenkers@uni-muenster.de.
Alena Dreiling, Email: Alena.Dreiling@gmx.de.
Deblina Chakraborty, Email: deblina90@gmail.com.
Mareike Fröhling, Email: MareikeFroehling@gmx.de.
Peter Paruzel, Email: paruzelp@uni-muenster.de.
Corinna Wehmeyer, Email: cwehmeye@uni-muenster.de.
Sven Hermann, Email: hermann.sven@uni-muenster.de.
Olympia Papantonopoulou, Email: papanton@uni-bonn.de.
Christiane Geyer, Email: christiane.geyer@uni-muenster.de.
Karin Loser, Email: karin.loser@ukmuenster.de.
Stephan Ludwig, Email: Ludwigs@uni-muenster.de.
Monika Stoll, Email: mstoll@uni-muenster.de.
Tomas Leanderson, Email: tomas.leanderson@activebiotech.com.
Simone König, Email: simone.koenig@ifg.uni-muenster.de.
Thomas Pap, Email: thomas.pap@uni-muenster.de.
Johannes Roth, Email: rothj@uni-muenster.de.
References
- 1.Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol. 2010;28:321–342. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140(6):871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 3.Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oppenheim JJ, Tewary P, de la Rosa G, Yang D. Alarmins initiate host defense. Adv Exp Med Biol. 2007;601:185–194. doi: 10.1007/978-0-387-72005-0_19. [DOI] [PubMed] [Google Scholar]
- 5.Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol. 2012;8(4):195–202. doi: 10.1038/nrrheum.2011.222. [DOI] [PubMed] [Google Scholar]
- 6.Chan JK, et al. Alarmins: awaiting a clinical response. J Clin Invest. 2012;122(8):2711–2719. doi: 10.1172/JCI62423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem. 1991;266(12):7706–7713. [PubMed] [Google Scholar]
- 8.Vogl T, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13(9):1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 9.Fassl SK, et al. Transcriptome assessment reveals a dominant role for TLR4 in the activation of human monocytes by the alarmin MRP8. J Immunol. 2015;194(2):575–583. doi: 10.4049/jimmunol.1401085. [DOI] [PubMed] [Google Scholar]
- 10.Liu JZ, et al. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe. 2012;11(3):227–239. doi: 10.1016/j.chom.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loser K, et al. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat Med. 2010;16(6):713–717. doi: 10.1038/nm.2150. [DOI] [PubMed] [Google Scholar]
- 12.van Lent PL, et al. Myeloid-related proteins S100A8/S100A9 regulate joint inflammation and cartilage destruction during antigen-induced arthritis. Ann Rheum Dis. 2008;67(12):1750–1758. doi: 10.1136/ard.2007.077800. [DOI] [PubMed] [Google Scholar]
- 13.Manitz MP, et al. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol. 2003;23(3):1034–1043. doi: 10.1128/MCB.23.3.1034-1043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hobbs JA, et al. Myeloid cell function in MRP-14 (S100A9) null mice. Mol Cell Biol. 2003;23(7):2564–2576. doi: 10.1128/MCB.23.7.2564-2576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehrchen JM, Sunderkötter C, Foell D, Vogl T, Roth J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol. 2009;86(3):557–566. doi: 10.1189/jlb.1008647. [DOI] [PubMed] [Google Scholar]
- 16.Strupat K, Rogniaux H, Van Dorsselaer A, Roth J, Vogl T. Calcium-induced noncovalently linked tetramers of MRP8 and MRP14 are confirmed by electrospray ionization-mass analysis. J Am Soc Mass Spectrom. 2000;11(9):780–788. doi: 10.1016/S1044-0305(00)00150-1. [DOI] [PubMed] [Google Scholar]
- 17.Leukert N, Vogl T, Strupat K, Reichelt R, Sorg C, Roth J. Calcium-dependent tetramer formation of S100A8 and S100A9 is essential for biological activity. J Mol Biol. 2006;359(4):961–972. doi: 10.1016/j.jmb.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 18.Taylor GA, et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4(5):445–454. doi: 10.1016/S1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 19.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184(4):1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, et al. Activated macrophages are essential in a murine model for T cell–mediated chronic psoriasiform skin inflammation. J Clin Invest. 2006;116(8):2105–2114. doi: 10.1172/JCI27180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129(6):1339–1350. doi: 10.1038/jid.2009.59. [DOI] [PubMed] [Google Scholar]
- 22.Tortola L, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest. 2012;122(11):3965–3976. doi: 10.1172/JCI63451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Meglio P, Villanova F, Nestle FO. Psoriasis. Cold Spring Harb Perspect Med. 2014;4(8):a015354. doi: 10.1101/cshperspect.a015354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bollmann F, et al. Endothelial dysfunction in tristetraprolin-deficient mice is not caused by enhanced tumor necrosis factor-α expression. J Biol Chem. 2014;289(22):15653–15665. doi: 10.1074/jbc.M114.566984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molle C, et al. Tristetraprolin regulation of interleukin 23 mRNA stability prevents a spontaneous inflammatory disease. J Exp Med. 2013;210(9):1675–1684. doi: 10.1084/jem.20120707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Retser E, et al. Doxycycline-induced expression of transgenic human tumor necrosis factor α in adult mice results in psoriasis-like arthritis. Arthritis Rheum. 2013;65(9):2290–2300. doi: 10.1002/art.38026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogl T, et al. Alarmin S100A8/S100A9 as a biomarker for molecular imaging of local inflammatory activity. Nat Commun. 2014;5:4593. doi: 10.1038/ncomms5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korndörfer IP, Brueckner F, Skerra A. The crystal structure of the human (S100A8/S100A9)2 heterotetramer, calprotectin, illustrates how conformational changes of interacting alpha-helices can determine specific association of two EF-hand proteins. J Mol Biol. 2007;370(5):887–898. doi: 10.1016/j.jmb.2007.04.065. [DOI] [PubMed] [Google Scholar]
- 29.Itou H, et al. The crystal structure of human MRP14 (S100A9), a Ca(2+)-dependent regulator protein in inflammatory process. J Mol Biol. 2002;316(2):265–276. doi: 10.1006/jmbi.2001.5340. [DOI] [PubMed] [Google Scholar]
- 30.Frosch M, et al. Myeloid-related proteins 8 and 14 are specifically secreted during interaction of phagocytes and activated endothelium and are useful markers for monitoring disease activity in pauciarticular-onset juvenile rheumatoid arthritis. Arthritis Rheum. 2000;43(3):628–637. doi: 10.1002/1529-0131(200003)43:3<628::AID-ANR20>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 31.Rammes A, Roth J, Goebeler M, Klempt M, Hartmann M, Sorg C. Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J Biol Chem. 1997;272(14):9496–9502. doi: 10.1074/jbc.272.14.9496. [DOI] [PubMed] [Google Scholar]
- 32.Pruenster M, et al. Extracellular MRP8/14 is a regulator of β2 integrin-dependent neutrophil slow rolling and adhesion. Nat Commun. 2015;6:6915. doi: 10.1038/ncomms7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogl T, Gharibyan AL, Morozova-Roche LA. Pro-inflammatory S100A8 and S100A9 proteins: self-assembly into multifunctional native and amyloid complexes. Int J Mol Sci. 2012;13(3):2893–2917. doi: 10.3390/ijms13032893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pröpper C, Huang X, Roth J, Sorg C, Nacken W. Analysis of the MRP8-MRP14 protein-protein interaction by the two-hybrid system suggests a prominent role of the C-terminal domain of S100 proteins in dimer formation. J Biol Chem. 1999;274(1):183–188. doi: 10.1074/jbc.274.1.183. [DOI] [PubMed] [Google Scholar]
- 35.Vogl T, Leukert N, Barczyk K, Strupat K, Roth J. Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochim Biophys Acta. 2006;1763(11):1298–1306. doi: 10.1016/j.bbamcr.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 36.Ishikawa K, Nakagawa A, Tanaka I, Suzuki M, Nishihira J. The structure of human MRP8, a member of the S100 calcium-binding protein family, by MAD phasing at 1.9 A resolution. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 5):559–566. doi: 10.1107/s0907444900002833. [DOI] [PubMed] [Google Scholar]
- 37.Leukert N, Sorg C, Roth J. Molecular basis of the complex formation between the two calcium-binding proteins S100A8 (MRP8) and S100A9 (MRP14) Biol Chem. 2005;386(5):429–434. doi: 10.1515/BC.2005.051. [DOI] [PubMed] [Google Scholar]
- 38.Foell D, et al. Methotrexate withdrawal at 6 vs 12 months in juvenile idiopathic arthritis in remission: a randomized clinical trial. JAMA. 2010;303(13):1266–1273. doi: 10.1001/jama.2010.375. [DOI] [PubMed] [Google Scholar]
- 39.Broome AM, Ryan D, Eckert RL. S100 protein subcellular localization during epidermal differentiation and psoriasis. J Histochem Cytochem. 2003;51(5):675–685. doi: 10.1177/002215540305100513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schonthaler HB, et al. S100A8-S100A9 protein complex mediates psoriasis by regulating the expression of complement factor C3. Immunity. 2013;39(6):1171–1181. doi: 10.1016/j.immuni.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 41.Hiratsuka S, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10(11):1349–1355. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 42.Hiratsuka S, et al. Primary tumours modulate innate immune signalling to create pre-metastaic vascular hyperpermeability foci. Nat Commun. 2013;4:1853. doi: 10.1038/ncomms2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kostakis ID, Cholidou KG, Vaiopoulos AG, Vlachos IS, Perrea D, Vaos G. Fecal calprotectin in pediatric inflammatory bowel disease: a systematic review. Dig Dis Sci. 2013;58(2):309–319. doi: 10.1007/s10620-012-2347-5. [DOI] [PubMed] [Google Scholar]
- 44.Björk P, et al. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009;7(4):e97. doi: 10.1371/journal.pbio.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Comi G, et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;371(9630):2085–2092. doi: 10.1016/S0140-6736(08)60918-6. [DOI] [PubMed] [Google Scholar]
- 46.Qin H, et al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat Med. 2014;20(6):676–681. doi: 10.1038/nm.3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korb-Pap A, et al. Early structural changes in cartilage and bone are required for the attachment and invasion of inflamed synovial tissue during destructive inflammatory arthritis. Ann Rheum Dis. 2012;71(6):1004–1011. doi: 10.1136/annrheumdis-2011-200386. [DOI] [PubMed] [Google Scholar]
- 48.Roth J, Burwinkel F, van den Bos C, Goebeler M, Vollmer E, Sorg C. MRP8 and MRP14, S-100-like proteins associated with myeloid differentiation, are translocated to plasma membrane and intermediate filaments in a calcium-dependent manner. Blood. 1993;82(6):1875–1883. [PubMed] [Google Scholar]
- 49.Tscharntke M, et al. Impaired epidermal wound healing in vivo upon inhibition or deletion of Rac1. J Cell Sci. 2007;120(Pt 8):1480–1490. doi: 10.1242/jcs.03426. [DOI] [PubMed] [Google Scholar]
- 50.Vogl T, Roth J, Sorg C, Hillenkamp F, Strupat K. Calcium-induced noncovalently linked tetramers of MRP8 and MRP14 detected by ultraviolet matrix-assisted laser desorption/ionization mass spectrometry. J Am Soc Mass Spectrom. 1999;10(11):1124–1130. doi: 10.1016/S1044-0305(99)00085-9. [DOI] [PubMed] [Google Scholar]
- 51.Ahmad-Nejad P, Häcker H, Rutz M, Bauer S, Vabulas RM, Wagner H. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur J Immunol. 2002;32(7):1958–1968. doi: 10.1002/1521-4141(200207)32:7<1958::AID-IMMU1958>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 52.Bosisio D, et al. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-gamma: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood. 2002;99(9):3427–3431. doi: 10.1182/blood.V99.9.3427. [DOI] [PubMed] [Google Scholar]
- 53.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20(1):45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original microarray data were deposited in the NCBI’s Gene Expression Omnibus database (GEO GSE109646). Further information about the data and about the reagents used is available upon request to the corresponding author.