

Abstract
Major histocompatibility (MHC) class II molecules are strongly associated with many autoimmune disorders. In type 1 diabetes (T1D), the DQ8 molecule is common, confers significant disease risk, and is involved in disease pathogenesis. We hypothesized that blocking DQ8 antigen presentation would provide therapeutic benefit by preventing recognition of self-peptides by pathogenic T cells. We used the crystal structure of DQ8 to select drug-like small molecules predicted to bind structural pockets in the MHC antigen–binding cleft. A limited number of the predicted compounds inhibited DQ8 antigen presentation in vitro, with 1 compound preventing insulin autoantibody production and delaying diabetes onset in an animal model of spontaneous autoimmune diabetes. An existing drug with a similar structure, methyldopa, specifically blocked DQ8 in patients with recent-onset T1D and reduced inflammatory T cell responses to insulin, highlighting the relevance of blocking disease-specific MHC class II antigen presentation to treat autoimmunity.
Keywords: Autoimmunity, Endocrinology
Keywords: Diabetes, Immunotherapy, MHC class 2
Introduction
Type 1 diabetes (T1D) results from the chronic autoimmune-mediated destruction of insulin-producing β cells within pancreatic islets (1, 2). There is a wealth of knowledge regarding the disease pathogenesis and natural history of T1D, such that it can be predicted by assessment of serum islet autoantibodies years before symptom onset (3). With a predictable nature and increasing incidence (4), a number of immune therapies have been tested in well-controlled clinical trials seeking to prevent or slow the loss or decline of β cells, including therapies targeting CD2 or CD3 on T cells (5–8), CD20 on B cells (9), CD80/CD86 on antigen-presenting cells (APCs) (10), and inflammatory cytokines (IL-1 and IL-1 receptor) (11), as well as antigen-specific therapies (12–15). Unfortunately, these therapies have shown limited clinical benefit to date. To improve outcomes, therapies directed toward the molecular targets specifically involved in T1D are needed.
In many autoimmune disorders, genes within the HLA complex confer disease risk (16). Alleles of the polymorphic class II genes DQ and DR (and to a lesser extent DP) are the most important determinants of disease risk. These genes encode MHC proteins located on the B cells, DCs, and macrophages that present processed antigens to CD4 T cells. MHC class II, peptide, and the T cell receptor (TCR) form a trimolecular complex involving the presentation of self-peptides that shape autoreactive T cell responses in autoimmunity. Strong HLA disease associations exist for narcolepsy, which only develops in individuals with HLA-DQ6 (DQB1*06:02) (17); celiac disease is predominantly restricted to DQ2 and, to a lesser extent, DQ8 (18); DR3 is present in 90% of patients with neuromyelitis optica (19); multiple sclerosis is strongly associated with DR15 (20); and specific DR4 subtypes are present in rheumatoid arthritis (21, 22).
For T1D, the HLA-DQ8 allele (DQA1*03:01-DQB1*03:02) is present in 50% to 60% of all patients and provides an odds ratio for disease development of 6.5 to 11 (23–25). This is in stark contrast to HLA-DQ6 (DQB1*06:02), which confers dominant protection, with an odds ratio of 0.03 (25). The dichotomy of risk between the 2 alleles highlights the importance of MHC class II antigen presentation in T1D development. Importantly, T cells restricted to proinsulin peptides presented by DQ8 have recently been identified in inflamed pancreatic islets from recent-onset T1D organ donors (26, 27), implicating DQ8-restricted T cells in the pathogenesis of T1D. This evidence renders DQ8 an attractive therapeutic target for modifying the autoimmune response, and we hypothesized that blocking DQ8 antigen presentation would represent a pathway for treating T1D.
Previously, we used molecular docking to screen a large chemical library of drug-like small molecules to predict those capable of occupying 1 of 4 structural pockets (P1, P4, P6, and P9) in the NOD mouse MHC class II–I-Ag7–peptide–binding groove (28). The NOD mouse is a spontaneous model of autoimmune diabetes and shares similarities with human disease including MHC class II genes conferring disease risk, development of insulin autoantibodies, and T cell infiltration into pancreatic islets (29). Several compounds identified by in silico molecular docking were able to block insulin peptide binding to I-Ag7 and subsequent T cell activation in vitro (28).
In this study, we extended this approach for the purpose of inhibiting DQ8 antigen presentation. The crystal structure of DQ8 was used for molecular docking to select small molecules, which were then tested with in vitro bioassays and in preclinical animal models. By screening known drugs, we discovered that methyldopa, a clinically well-established antihypertensive drug, specifically blocked DQ8 and subsequent T cell responses. Methyldopa was used in an open-label clinical trial to treat patients with recent-onset T1D, who had the DQ8 allele and residual β cell function. Treatment resulted in the specific inhibition of DQ8 presentation by peripheral blood mononuclear cells (PBMCs) and a reduction in primary antigen–specific T cell responses to insulin. The concept of blocking specific MHC class II antigen presentation has significant relevance for T1D and other autoimmune diseases strongly associated with HLA alleles.
Results
Molecular docking to select HLA-DQ8–binding small molecules.
An in silico screening approach was used to rank small molecules predicted to bind structural pockets in the DQ8 peptide–binding groove. The sites within the antigen-binding cleft selected for molecular docking were based on the crystal structure of insulin B:11-23 bound to DQ8 (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI97739DS1), which showed that the majority of intramolecular hydrogen bond interactions between peptide and HLA occurred between the P1, P4, P6, and P9 side chains (30). We rationalized that drug binding to DQ8 in the sites critical for peptide binding would block antigen presentation and therefore used molecular docking to screen the P1, P4, P6, and P9 sites using a library consisting of 139,735 small molecules (Figure 1A). Each compound in the repository was docked in 1,000 orientations within a single pocket and scored on the basis of the sum of attractive and repulsive intermolecular forces to estimate the predicted free energy of binding (ΔG). None of the top-40 scoring compounds overlapped among P1, P4, P6, and P9 (the chemical structures, ΔG, and in silico rank are listed in Supplemental Tables 1–4 for each pocket), indicating distinct chemical composition and geometry among structural pockets in the DQ8-binding cleft.
Figure 1. In silico screening of a small-molecule library by molecular docking to structural pockets within the HLA-DQ8 peptide–binding groove and in vitro screening of the top-scoring compounds.

(A) Molecular model of the DQ8 peptide binding cleft, with pockets 1, 4, 6, and 9 depicted by colored spheres. Using a supercomputer, 139,735 repository compounds were docked into each pocket in 1,000 different orientations and scored on the basis of the free energy of binding (ΔG). (B) The top-scoring compounds were screened with an in vitro bioassay, which used an immortalized T cell that responds to an insulin epitope presented by DQ8. The TCR α/β genes from an insulin B:9-23–restricted T cell clone were retrovirally transduced into a T cell line devoid of endogenous TCR to create a TCR transductant, named clone 5. Clone 5 was cultured with insulin peptide plus a small molecule in the presence of DQ8-Tg murine splenocytes. After overnight culture, secreted IL-2 was measured in the cell culture supernatant with a highly sensitive ELISA. In the heatmap, “No antigen” represents the response without in vitro–added peptide, and “Peptide” represents the response to peptide without an in vitro–added small molecule. Numbers 1–39 depict the tested compounds, and the compounds were different for each pocket. The insulin B:9-23 peptide was used to screen pockets 1 and 9 (p1 and p9), while insulin B:13-23 was used to screen pockets 4 and 6 (p4 and p6), which resulted in a greater baseline IL-2 response. Supplemental Tables 1–4 list the corresponding chemical structures, ΔG estimates from molecular docking, and rank based on the overall free energy score.
Screening lead-candidate small molecules with a bioassay.
For each site, the top-scoring compounds were tested for their ability to alter an insulin/DQ8-specific T cell response in vitro. To establish a robust and reproducible T cell bioassay, the TCR α/β genes of an insulin/DQ8-restricted T cell clone (named clone 5) (31) were retrovirally expressed on an immortalized T cell devoid of endogenous TCR expression, thus creating a TCR transductant that secreted IL-2 when stimulated with insulin B:9-23 presented by DQ8. The use of an immortalized T cell transductant allowed us to screen hundreds of small molecules and further evaluate “hits” for specificity. Our screening of the top-scoring compounds indicated that some molecules blocked T cell responses, specifically 109 of the 142 tested compounds, and 2 of these had an increased response above the peptide alone — 1 showing an increased response without the peptide (Supplemental Table 4) and the other requiring the peptide (Supplemental Table 1). The high frequency of active compounds predicted to bind P4 and P6 (34 of 34 for P4 and 33 of 34 for P6) (Figure 1B) was in stark contrast to our previous efforts for murine I-Ag7, in which only 5 of the 160 tested compounds blocked an insulin-specific T cell response (P < 0.001, comparing DQ8 versus I-Ag7 inhibitory compounds using a Fisher’s exact test). Our evaluation of the chemical structures of compounds targeting P4 and P6 indicated that a tetraaza core chemical structure existed for the compounds selected by the P6 structural pocket (tetraazatricylclododecane [TATD]; Figure 2A) in 12 of the 34 compounds (Supplemental Table 3). We synthesized this compound at high purity and measured the concentration required to block clone 5 and another TCR transductant (TCR 489) restricted to a deamidated α-gliadin peptide presented by DQ8 (T cell clone from a patient with celiac disease) (32). TATD inhibited both TCRs, with nanomolar IC50 values and lower IC50 values for insulin compared with the posttranslationally modified α-gliadin epitope (Figure 2B), which has a high binding affinity for DQ8 upon deamidation (Q→E). To exclude off-target effects of a TATD-inhibited T cell response, TATD was incubated with recombinant α-gliadin/DQ8 protein and free α-gliadin peptide in conditions allowing for peptide exchange, washed, and then used to stimulate the 489 TCR (Figure 2C). These data demonstrated that TATD directly interacted with the peptide-DQ8 complex. Furthermore, TATD did not cause cell cytotoxicity (Supplemental Figure 2A) or inhibit an anti-CD3–stimulated T cell response (Supplemental Figure 2B) at micromolar concentrations.
Figure 2. TATD blocks DQ8-restricted T cell responses and prevents diabetes in NOD mice.

(A) Chemical structure of TATD. (B) TATD blocked an in vitro DQ8–restricted T cell response to insulin B:13-23 (Clone 5) and deamidated α-gliadin228-240 (489 TCR). Data represent the mean ± SEM and are representative of 3 independent experiments. No antigen addition to the culture resulted in IL-2 levels below 3 pg/ml. (C) TATD was cultured with recombinant α-gliadin/DQ8 protein and free peptide in conditions to allow peptide exchange. After washing, the recombinant protein was used to stimulate 489, and IL-2 secretion was measured. Data are from triplicate wells and are representative of 3 independent experiments. 489 in culture without protein resulted in IL-2 levels below 3 pg/ml. (D) Female NOD mice were treated from 4 to 12 weeks of age with 20 mg/kg TATD (n = 10) or PBS (n = 13) by intraperitoneal injection daily for 5 days each week. P = 0.006, by log-rank test. (E) Peak serum insulin autoantibody (IAA) levels were measured using a fluid-phase RIA during the 40-week prevention study. **P = 0.003, by Mann-Whitney U test. The dotted line at 0.01 indicates a positive value. (F) Blood glucose levels during the late prevention study, in which female NOD mice were treated with 30 mg/kg TATD orally each day (n = 9), 50 μg anti-CD3 monoclonal antibody intraperitoneally for 5 consecutive days (n = 10), or PBS (n = 10) beginning at 12 weeks of age and ending at 25 weeks. Data represent the mean ± SEM. *P < 0.05, by ANOVA for comparison of TATD versus PBS. (G) Intraperitoneal GTT following cessation of the study treatments; each dot represents an individual mouse. ***P < 0.01, by 2-tailed, unpaired t test. (H) Representative H&E-stained pancreatic sections from PBS- and TATD-treated mice; insulin staining is shown in brown. Original magnification, ×15. (I) Insulitis scoring from at least 100 separate islets from TATD-treated (n = 3) and PBS-treated (n = 5) mice.
Preclinical studies in the NOD mouse.
To evaluate whether blocking MHC class II antigen presentation prevents autoimmune diabetes, TATD was administered to NOD mice. TATD cross-reacted with NOD I-Ag7, as an isomer was previously identified to block peptide binding and T cell activation (28). In vivo administration early in the NOD disease course delayed the onset of diabetes and prevented disease in half of the treated mice (Figure 2D). Since CD4+ T cells undergo cognate interactions with B cells, which can result in antibody production, we measured insulin autoantibodies in these mice. TATD treatment prevented insulin autoantibody production throughout the lives of the treated mice (Figure 2E). Treatment of the NOD mice later in the disease course resulted in the maintenance of normal blood glucose levels compared with PBS treatment and had effects similar to those seen in NOD mice treated with an anti-CD3 monoclonal antibody, which was used as a positive control (Figure 2F and Supplemental Figure 3). Additionally, TATD treatment resulted in preserved glucose tolerance (Figure 2G) and less invasive islet immune cell infiltration compared with that seen in PBS-treated mice (Figure 2, H and I).
Overall, these data indicate that therapeutic blocking of MHC class II antigen presentation in a spontaneous model of autoimmune diabetes prevents disease onset, blocks critical interactions between T and B cells, lessens tissue-specific destruction, and maintains glucose tolerance when administered in later stages of diabetes development.
Screening FDA-approved drugs that bind HLA-DQ8.
Since in vitro and preclinical NOD studies suggested that small-molecule binding pockets within the DQ8 antigen–binding cleft could be identified by molecular docking, we screened 1,207 FDA-approved small-molecule drugs predicted to occupy the P6 pocket in an attempt to repurpose an existing drug. Of the top-39 scoring drugs (Supplemental Table 5), 10 of 39 (25.6%) blocked clone 5 (Supplemental Figure 4). One of these, methyldopa (Aldomet; Figure 3A) inhibited T cell responsiveness in vitro. Methyldopa is a clinically well-established oral drug used for over 50 years to treat hypertension in both children and adults (33), which provides an opportunity to repurpose methyldopa for immune-mediated treatment of T1D.
Figure 3. Methyldopa blocks DQ8 antigen presentation in vitro.

(A) Methyldopa chemical structure. Methyldopa blocked an in vitro DQ8–restricted T cell response by (B) clone 5 to insulin B:13-23 and (C) TCR 489 to deamidated α-gliadin presented by DQ8. The negative controls (no antigen added to culture) resulted in IL-2 levels below 3 pg/ml. (D) Methyldopa specifically inhibited 489, a DQ8-restricted T cell response, while not altering C7CH17, a DR4-restricted T cell response to the HA peptide HA306-318. Cells from an EBV-transformed B cell line homozygous for DQ8 and DR4 were used as APCs in this experiment, such that each APC had DQ8 and DR4 on the cell surface. (E) Methyldopa did not inhibit a DQ8-restricted T cell response to the influenza peptide HA102-118. (B–E) Data represent the mean ± SEM and are representative of 3 independent experiments. (F) Representative isothermal titration calorimetric data for titration of methyldopa into DQ8 protein. Top: Heat released as a function of time from 2-μl injections of 4 mM methyldopa titrated into the sample cell containing 4 μM DQ8 protein at 25oC. Bottom: Fitted binding curve along with the measured thermodynamic parameters and the calculated KD from 3 independent experiments, reported as the mean ± SEM. (G) Molecular docking model of methyldopa in the DQ8 antigen–binding cleft shows potential H bonds between a methyldopa hydroxyl group and DQα62 asparagine (DQα N62) on the DQα-helix and between the methyldopa carboxylic acid and DQβ30 tyrosine (DQβ Y30) on the floor of the cleft.
Methyldopa directly binds HLA-DQ8 and blocks antigen presentation in vitro.
Methyldopa blocked in vitro DQ8–restricted T cell responses to both clone 5 (Figure 3B) and 489 (Figure 3C) in a dose-dependent manner, with IC50 values in the low micromolar range. We observed no in vitro cell cytotoxicity, and methyldopa was specific for inhibiting DQ8-restricted T cell responses, while not changing a DR4-restricted T cell response (Figure 3D). DQ8 and DR4 are in close linkage disequilibrium on chromosome 6, and both class II molecules were present on the APCs used for these studies. We next tested methyldopa against a panel of 4 TCRs restricted to the same deamidated α-gliadin peptide presented by DQ8 (34); only the TCR sequence was different between these transductants. Despite different levels of stimulation in response to peptide alone, methyldopa inhibited all 4 of the TCRs to varying degrees (Supplemental Figure 5). Methyldopa did not block a DQ8-restricted T cell response to a viral peptide (HA102-118), up to concentrations as high as 200 μM (Figure 3E). These data demonstrate that methyldopa inhibits antigen presentation by DQ8 in a peptide sequence–specific manner that may be related to the peptide affinity for DQ8 and/or the peptide sequence.
To confirm that methyldopa directly interacts with DQ8 and to assess drug-target biochemical interactions in vitro, we used isothermal titration calorimetry (ITC). ITC directly measures changes in temperature that occur during fluid-phase protein-ligand interaction (forming or breaking noncovalent interactions) and measures binding affinity as well as enthalpy and entropy. Figure 3F shows the heat released as methyldopa was titrated into DQ8 protein over time and the fitted binding curve. Given that methyldopa is a small molecule (MW <500 kDa), there was very low but favorable enthalpy (negative ΔH), which was derived from changes in interatomic interactions such as hydrogen bonding and van der Waals and electrostatic interactions. However, entropy of the reaction (removal of solvent molecules from the binding interface) was very favorable (positive ΔS), leading to a ΔG and KD of 26.2 ± 4.2 μM, similar to those reported for TCRs interacting with peptide-MHC complexes (35).
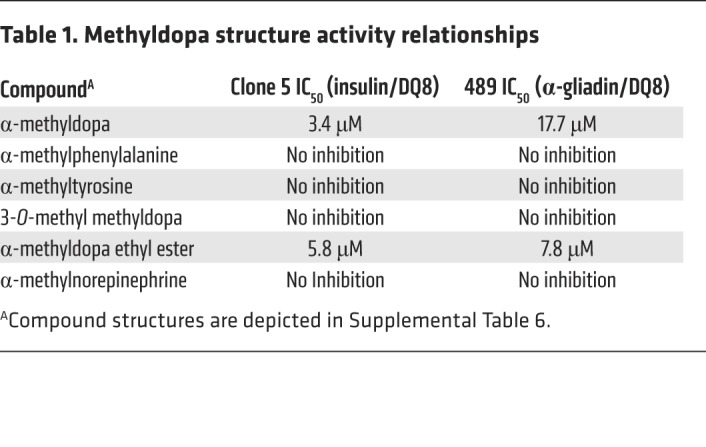
Using the crystal structure of DQ8, we modeled methyldopa interaction with amino acid residues of the DQ8 α and β chains within the antigen-binding cleft (Figure 3G). Two hydrogen bonds are predicted between methyldopa and DQ8 amino acid side chains. We tested the structurally related compounds and found that the modeled hydrogen bonds were indeed important for inhibiting a TCR response, as removal of a hydroxyl group on the benzene ring or removal of the carboxylic acid abrogated the response (Table 1 and Supplemental Table 6). Further insights from the structure activity relationships indicate that a hydroxyl group on the benzene ring is a hydrogen bond donor to DQα62 asparagine and that the carboxylic acid is a hydrogen bond acceptor for DQβ30 tyrosine.
Table 1. Methyldopa structure activity relationships.
Next, to determine whether methyldopa acts on the cell surface of APCs to displace peptides or intracellularly block peptide loading onto DQ8, we glutaraldehyde fixed DQ8 APCs to inhibit intracellular antigen processing. Methyldopa inhibited T cell activation with fixed APCs to a degree similar to that seen with unfixed cells, indicating that the main mechanism involves the displacement of cell-surface peptides (Supplemental Figure 6, A and B). It is hypothesized that self-antigens have a lower affinity for MHC molecules than do foreign antigens, and we therefore designed experiments to test whether methyldopa more effectively blocks lower-affinity peptides. First, we tested the 489 TCR with the WT α-gliadin peptide and the peptide with deamidation (Q→E) at P1 (p1E), P9 (p9E), or both (p1E/p9E). Deamidation facilitates anchoring of the peptide to DQ8, thereby increasing the T cell response to the α-gliadin–DQ8 complex. As expected, there was a hierarchy of responses: p1E/p9E > p9E > p1E > WT (Supplemental Figure 6C). We then performed the same peptide titration experiment with methyldopa at a fixed concentration. Methyldopa blocked peptide presentation of lower-affinity peptide-DQ8 complexes to a greater extent than it did for higher-affinity peptide-DQ8 complexes (Supplemental Figure 6D). These data are consistent with our observation that methyldopa was not able to block the T cell response to a high-affinity influenza peptide presented by DQ8 (Figure 3E).
Together, these data identify a mechanism by which methyldopa binds directly within the DQ8 antigen–binding cleft on the cell surface to inhibit the presentation of peptides to T cells. Methyldopa was able to block peptides that bound DQ8 with lower affinity (i.e., insulin B:9-23) to a greater extent than it did peptides with a higher DQ8 affinity.
Methyldopa blocks DQ8 antigen presentation in vivo.
We evaluated the in vivo effects of methyldopa using DQ8-Tg mice, as methyldopa does not block NOD I-Ag7–restricted T cell responses. Using these Tg mice, we developed a biomarker assay to monitor the effect of methyldopa on DQ8 presentation (Figure 4A). Tg DQ8 mice were treated orally with vehicle or methyldopa at human-equivalent doses, and then their ex vivo splenocytes were used as APCs to stimulate clone 5 and 489 with in vitro–added peptide. The effects of methyldopa on blocking DQ8 antigen presentation in Tg mice were dose dependent, (Supplemental Figure 7), and the drug had to be administered multiple times each day, as the plasma t1/2 is 2 hours. When methyldopa was administered multiple times a day, we observed a greater than 50% reduction in T cell responses (Figure 4B), while no changes in splenic CD11b+ or CD11c+ cell percentages or numbers were detected (Figure 4, C and D). Additionally, there were no changes in cell-surface DQ8 expression on these APCs (Figure 4E), indicating that methyldopa remains bound to DQ8 and inhibits TCR recognition of a peptide-DQ8 complex ex vivo.
Figure 4. Methyldopa blocks DQ8 antigen presentation in vivo.

(A) Diagram of the assay used to monitor the potency of methyldopa to block DQ8 in Tg mice. (B) Adult DQ8-Tg mice were gavaged with vehicle or 200 mg/kg methyldopa 3 times per day for 4 days (n = 4/group). Ex vivo splenocytes were used as APCs to present different concentrations of the insulin B:13-23 peptide or a deamidated α-gliadin peptide to clone 5 or 489, respectively. No methyldopa was added to the in vitro culture. A dose of 200 mg/kg is equivalent to 1,000 mg 3 times per day in a human weighing 60 kg. Data represent the mean ± SEM and are representative of 3 independent experiments. (C) Percentages and (D) numbers of CD11b+ and CD11c+ cells in the spleens of treated mice. (E) MFI of DQ8 cell-surface staining on each cell population. Each dot represents an individual mouse.
Methyldopa is metabolized in the liver to α-methylnorepinephrine, which is responsible for lowering blood pressure by agonizing central α2 adrenergic receptors (36). The metabolite did not inhibit DQ8 antigen presentation in vitro or in vivo (Supplemental Figure 8), demonstrating that intact methyldopa is needed to interact with DQ8.
Methyldopa specifically blocks DQ8 antigen presentation in patients with recent-onset T1D.
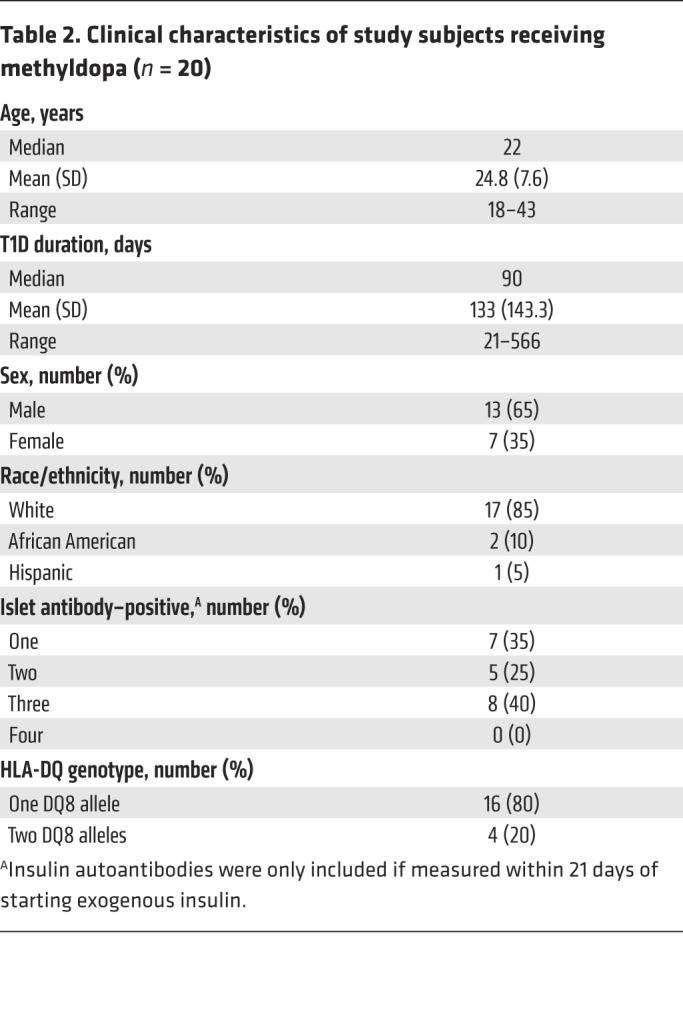
We translated these preclinical findings to human T1D in a single-arm, open-label, phase Ib dose-escalation study. We evaluated methyldopa treatment in 20 DQ8-positive T1D participants, aged 18–46 years, who had had diabetes for less than 2 years and who continued to produce endogenous insulin (Table 2 and Supplemental Table 7). All participants received methyldopa tablets orally at 3 different dosages titrated over a 6-week period to prevent hypotension and allow for assessment of the ability of the different doses to block antigen presentation (Supplemental Figure 9). Methyldopa was well tolerated, and no serious adverse events were noted.
Table 2. Clinical characteristics of study subjects receiving methyldopa (n = 20).
The primary endpoint was the change from baseline DQ8 antigen presentation by PBMCs after 6 weeks of methyldopa treatment. As with the assay used to measure DQ8 antigen presentation in Tg mice, we used primary PBMCs as APCs to present peptide to a cognate TCR transductant and measured secreted IL-2 after overnight culture (Figure 5, A–C). We found that DQ8 T cell responses from 489 were inhibited during the treatment period and returned to baseline 6 weeks after stopping therapy (Figure 5D). We noted a statistically significant inhibition of these T cell responses with low, moderate, and high doses of methyldopa. Clone 5 responses were also blocked out to 1 week after stopping therapy (Figure 5E). DQ8 presentation was inhibited by 40% compared with baseline levels for 17 of the 20 patients who responded to treatment (see Supplemental Figure 10 for individual responses); 1 nonresponder did not have detectable plasma methyldopa levels (Supplemental Figure 11), and 2 nonresponders were DQ8 homozygotes. There appeared to be a DQ8 dose effect, given that higher drug doses were required to block a response in homozygotes (n = 4) compared with that observed in heterozygotes (Supplemental Figure 12).
Figure 5. Methyldopa treatment specifically blocks DQ8 antigen presentation in recent-onset T1D patients with the DQ8 allele.

(A) Diagram of the assay used to monitor specific MHC class II antigen presentation. PBMCs from participants were isolated and frozen after each study visit. These primary PBMCs were then thawed and used as APCs to stimulate engineered T cells (TCR transductants) that respond to a specific peptide presented by a given MHC class II molecule (DQ8, DR4, or DQ2). Secreted IL-2 from each TCR transductant was measured using a highly sensitive ELISA. (B) Individual study subject response for 489 (DQ8-restricted) and (C) C7CH17 (DR4-restricted). Data represent the mean ± SEM from triplicate wells at each time point during the study. Colors represent the durations on and off the drug. Participants underwent dose titration, with a low dose (500 mg 2 times/day), moderate dose (500 mg 3 times/day), and high dose (2–3 grams over the course of the day), and then went off the drug. “No antigen” indicates that there was no antigen added in vitro in the assay. (D) Summative data on study participants (n = 20) showing their responses to 489 and (E) clone 5, both of which were DQ8 restricted. *P = 0.001, **P < 0.001, ***P = 0.02, and #P < 0.01, using a longitudinal mixed-effects model that compared the least-squares mean at each time point with baseline. (F) Data from study participants with DR4 subtypes (n = 18) able to present and stimulate the HA306-318–restricted TCR transductant C7CH17. (G) Response to a DQ2 TCR transductant (233 responding to α-gliadin62-73) for the study duration in subjects with a DQ2 allele (n = 7). The negative control (no antigen added to culture) resulted in IL-2 levels below 2 pg/ml for each subject and for individual TCR transductants. Data in D–G are depicted as the least-squares mean ± SEM for the study cohort; individual responses are shown in Supplemental Figure 10.
As a control, we measured DR4 antigen presentation, as it is present on APCs with DQ8. DR4 presentation was not significantly affected in the study participants (P = NS; Figure 5F). In addition, we found that DQ2 (DQB1*02:01) antigen presentation was not altered in participants with this MHC class II molecule (Figure 5G). Flow cytometric analysis of PBMCs indicated similar numbers of APCs (B cells, myeloid DCs, and plasmacytoid DCs), with no change in the mean fluorescence intensity (MFI) of DQ or DR on the cell surface throughout the study (Supplemental Figure 13).
Together, these results indicate that methyldopa treatment specifically inhibits DQ8 antigen presentation throughout treatment with the drug and that function returns to normal upon withdrawal of therapy.
Methyldopa reduces the insulin-specific CD4+ T cell response in the peripheral circulation.
To measure primary T cell responses, as opposed to the antigen presentation described above, we performed indirect enzyme-linked immunospot (ELISPOT) assays on cryopreserved samples using an insulin B:9-23 (B22E) mimotope, which is presented by DQ8 (37, 38). Seven study subjects showed a response to the mimotope at baseline, and their responses decreased upon completion of the study (Figure 6A). This was not the case for whole tetanus toxin (Figure 6B) or DR4 epitopes within the protein (Figure 6, C and D) (39). Additionally, we have shown that the tested insulin epitope activates human CD4+ T cells isolated from the inflamed islets of T1D organ donors (26), highlighting the importance of blocking this reactivity in T1D patients.
Figure 6. Methyldopa treatment reduces primary antigen–specific T cells restricted to HLA-DQ8 but not those presented by DR4.

Cryopreserved PBMCs were thawed, cultured in the presence or absence of protein/peptide for 48 hours, washed, and then transferred to an IFN-γ monoclonal antibody–coated plate for overnight culture, followed by development and enumeration of ELISPOTs. IFN-γ ELISPOT results from study subjects for (A) an insulin B chain mimotope, B:9-23 (B22E), known to be presented by DQ8, (B) whole tetanus toxin (TT) protein, (C) an epitope of TT consisting of amino acids 506-525 known to be presented by DR4, and (D) another DR4-restricted TT epitope, amino acids 922-941. Seven study subjects responded to the insulin B chain mimotope at baseline and were further evaluated for responses to TT protein and epitopes. Each data point represents the total number of spots for a given condition from triplicate wells, minus the total number of spots without antigen (background) for an individual. Symbols represent the same individual tested for each condition. *P = 0.016, by Wilcoxon matched-pairs, signed-rank test; P = NS for TT, TT506-525, and TT922-941. Supplemental Table 8 provides ELISPOT counts at baseline and 3 months for each individual and condition, including no antigen as a negative control and whole TT as a positive control.
Methyldopa treatment results in good glycemic control and maintains β cell function over a 3-month period.
Participants had good glycemic control at the study’s conclusion, with hemoglobin A1c levels averaging 6.5% (Figure 7A), with no change in total insulin doses (Figure 7B). At 3 months, the C-peptide 2-hour AUC measurement following a mixed-meal tolerance test (MMTT), a measure of residual endogenous insulin production, was similar to baseline levels (Figure 7C). From natural history studies, it is appreciated that there is a steady decline in the C-peptide AUC after diagnosis, and age-matched historical controls would be expected to have approximately 30% loss 1 year after diagnosis (40). Although our study was a short term one, these results suggest that methyldopa treatment may limit β cell destruction and preserve function; however, trials with a longer duration and placebo arm are needed to fully evaluate metabolic efficacy.
Figure 7. Measures of glycemic control and β cell function among study participants.

Individual responses showed (A) an improvement in hemoglobin A1c, a measure of average blood glucose over the preceding 3 months, from baseline to study completion. *P = 0.008, by Wilcoxon matched-pairs, signed-rank test. (B) The amounts of exogenous insulin used per kilogram of body weight were similar from the beginning to the end of the study. (C) Maintenance of β cell function, as measured by the C-peptide 2-hour AUC following a MMTT compared with baseline levels. C-peptide is a measure of endogenous insulin secretion, as both are secreted at a 1:1 molar ratio. Individuals ingested a liquid meal (Boost) with a fixed amount of protein, fat, and carbohydrate in the fasting state, followed by the timed measurements of serum C-peptide at 0, 15, 30, 60, 90, and 120 minutes to compute the AUC. Each data point represents a single individual, with a line connecting the same study participant; n = 20 subjects in A–C, with the mean ± SD reported for the entire cohort at the start of study and upon study completion.
Discussion
Preventing pathogenic TCR activation by self-antigen–MHC class II complexes may provide a novel means for preventing and potentially reversing T1D and other autoimmune conditions. In this report, we used a rational structure-based approach to evaluate the druggability of pockets in the antigen-binding cleft of the T1D risk HLA-DQ8 molecule. Our goal was to identify drug candidates that occupy specific structural pockets and inhibit antigen presentation to T cells. Using the crystal structure of DQ8 as the basis for selection of candidate small molecules from a large chemical library, we show that DQ8 is remarkably amenable to drugs targeting pockets along the antigen-binding cleft, especially the P4 and P6 pockets. We then screened FDA-approved drugs predicted to bind the P6 pocket, in an attempt to repurpose an existing drug. We tested drugs in vitro as well as in vivo in humans to show that methyldopa specifically and durably blocked DQ8 antigen presentation. Moreover, methyldopa reduced inflammatory insulin-specific T cell responses when administered to patients with recent-onset T1D in a proof-of-concept trial. Longer-term placebo-controlled trials are warranted to evaluate methyldopa for potentially preserving residual β cell function in individuals at risk and in patents with new-onset T1D.
In our clinical trial involving the treatment of patients with recent-onset T1D with methyldopa, we observed a 40% reduction in DQ8 antigen presentation when using PBMCs as APCs (Figure 5D). Presumably, methyldopa is sequestered within DQ8 on DCs, B cells, and monocytes that are capable of presenting in vitro–added peptide to our engineered TCR transductants. Our data indicate that this is plausible, as methyldopa directly bound isolated DQ8 protein (Figure 3F) and interacted with cell-surface DQ8 as opposed to intracellular peptide loading. In fact, the processes of PBMC isolation and freezing, thawing, and washing cells did reduce the amount of inhibition measured with this assay (Figure 5A) and probably resulted in a loss of DQ8-sequestered methyldopa, indicating that a 40% reduction may be an underrepresentation of what occurs directly in vivo. Despite this limitation, DQ8 was blocked during the methyldopa treatment period and returned to baseline levels upon withdrawal of therapy. This pattern of drug treatment effect was not present for DR4, which was present on APCs with DQ8. Additionally, DQ2 presentation was not blocked with methyldopa treatment. These data demonstrate the specificity of methyldopa between MHC class II molecules and within DQ types. It is important to note that other MHC class II molecules, i.e., DR4, DR3, DQ2, and DQ8-trans (composed of the DQ2 α and DQ8 β chains), can present diabetes-relevant peptides (41). As there is epitope spreading that occurs during T1D pathogenesis, the timing of the therapeutic intervention using methyldopa to block DQ8 in the disease course is an important consideration, because those at earlier stages of the disease could potentially have greater clinical benefit.
Another observation from our data is that the methyldopa IC50 values for inhibition of T cell responses to insulin and α-gliadin were in the low micromolar range (3–50 μM). We used a separate technique to assess biochemical interactions (ITC) and obtained very similar results, with an average KD of 26 μM. Of particular interest is the fact that these values were in the reported range of affinities for TCRs binding to peptide-MHC complexes, as measured by surface plasmon resonance and ITC (35, 42, 43). Despite these somewhat low-affinity interactions of methyldopa with DQ8, primary autoantigen–specific T cell responses decreased in a subset of patients responding to an insulin B chain mimotope (Figure 6A). Using fluorescence tetramers, Fife and colleagues recently determined the phenotype of these T cells directly ex vivo in peripheral blood from patients with recent-onset T1D. These insulin-specific T cells were antigen experienced and predominantly an effector memory or central memory phenotype (44). Future studies will be needed to determine whether these insulin-specific T cells become anergic or are deleted with methyldopa treatment.
In addition to small-molecule drugs that occupy structural pockets in disease-related HLA molecules, there are alternate therapies to inhibit HLA-specific antigen presentation including antibodies targeting specific HLA molecules or peptide-HLA complexes. Although serotype-specific anti-HLA antibodies are available, their lack of specificity toward single HLA molecules would likely result in more off-target effects compared with those of an allele-specific agent. Moreover, it is difficult to generate allele-specific antibodies, as key polymorphisms that distinguish closely related molecules encode amino acid differences at sites within the antigen-binding cleft that are inaccessible to antibodies. Monoclonal antibodies against specific peptide/MHC molecules have been generated (45, 46) but are only specific for single epitopes, while immune responses to many different self-antigens are present in an autoimmune disease. Small-molecule drugs have not been widely utilized to target HLA molecules, as ligands for conventional polymorphic HLA molecules have primarily been described as peptides. Until recently, small molecules were not thought to bind in the antigen-binding cleft. The antiretroviral drug abacavir was shown by x-ray crystallography to directly interact with a structural pocket (F pocket, P9, or PΩ) in the peptide-binding cleft of HLA-B*57:01 (47, 48). These data, along with the results presented here, suggest that specific pockets within MHC class I and II antigen–binding grooves have chemical and geometric features amenable to binding small molecules.
The use of small-molecule drugs to target MHC II antigen presentation presents several concerns. First, negative effects on immune function may result from a hole in the immune repertoire created when a specific MHC class II molecule is blocked. However, detrimental effects on immune function are not expected, as other class II molecules exist to provide normal adaptive immune responses. For methyldopa, DQ8-restricted T cell responses to lower-affinity self-antigens were inhibited, while T cell responses to a higher-affinity viral pathogen remained unaltered. Additionally, DR4 and DQ2 responses remained intact, and it is also likely that DP functioned normally.
Second, drugs may trigger unwanted immune responses (hypersensitivity) by binding specific HLA molecules and creating neoantigen epitopes. Adverse drug reactions can be severe, but are rare (1–2/1,000,000 for Stevens-Johnson syndrome and 0.4–1.2/1,000,000 each year for toxic epidermal necrolysis) and have been identified only after treating large numbers of patients with the offending drug (49). Moreover, the modest affinity of methyldopa for DQ8 (KD of ~26 μM) was significantly lower than that of abacavir, a drug that caused hypersensitivity in a HLA-associated manner and bound a self-peptide and HLA-B*57:01 with an estimated KD of 0.2 nM (50). These data suggest that methyldopa is not likely to bind DQ8 with sufficient affinity to alter the repertoire of peptides or the consequent presentation of neoantigens that trigger hypersensitivity.
One advantage of “repurposing” existing drugs is that well-characterized safety profiles indicate potential complications and off-target effects. The safety profile of methyldopa, which is on the World Health Organization’s list of essential medicines, is based on over 50 years of clinical use, and the drug is indicated for the treatment of pregnancy-induced hypertension. Since HLA-DQ8 is present in Americans of European descent at a frequency of approximately 10% (51), methyldopa has been administered to large numbers of individuals expressing DQ8 and is considered safe.
In conclusion, we describe what we believe to be a novel approach to identifying immunomodulatory small molecules targeted to DQ8. As many autoimmune diseases are associated with specific HLA alleles and high-resolution crystal structures exist for many of these MHC class II molecules, strategies for the rapid selection HLA allele–specific compounds and for testing their activity in experimental systems can be implemented. The ability to identify HLA allele–specific drugs has broad applicability for treating autoimmune diseases and other HLA-associated conditions.
Methods
Molecular modeling and docking.
We used the crystal structure of HLA-DQ8 complexed to insulin B:11-23 (Protein Data Bank code 1JK8; https://www.rcsb.org/structure/1JK8) as the basis for molecular docking (52). To prepare the site for docking, all water molecules were removed, and protonation of DQ8 residues was done with SYBYL software (Tripos, Certara) (53). We explored the molecular surface of the structure using sets of spheres to describe potential binding pockets. The sites selected for molecular docking were defined using the SPHGEN program (54), which generates a grid of points that reflect the shape of the selected site, and filtered through CLUSTER (54). The CLUSTER program groups the selected spheres to define the points that are used by DOCK, version 5.1.0 (54), to match potential ligand atoms with spheres. Intermolecular AMBER energy scoring (van der Waals plus columbic), contact scoring, and bump filtering were implemented in the DOCK program algorithm. Atomic coordinates for 139,735 small molecules in the NIH Developmental Therapeutics Program repository of drug-like compounds were positioned in each structural pocket in 1,000 different orientations and scored on the basis of predicted polar (hydrogen bond) and nonpolar (van der Waals) interactions. The most favorable orientation and scores (contact and electrostatic) were calculated. PyMOL (https://pymol.org/2/) was used to generate graphic images.
Generation of TCR transductants.
TCR transductants were generated as previously described using retroviral transduction of the 5KC murine T cell hybridoma cell line lacking endogenous TCR expression (55). The sequences for the α/β TCR clones were either obtained as gifts from investigators (clone 5 from Bart Roep, Leiden University, Leiden, Netherlands; 489 from Ludvig Sollid, Oslo University, Oslo, Norway; and DQ8-flu from Bill Kwok, Benaroya Research Institute, Seattle, Washington, USA) or from published literature (clones 13, 22, and 38, which are DQ8 restricted; clone 233, which is DQ2 restricted; and C7CH17, which is DR4 restricted) (31, 32, 34, 56). Supplemental Table 9 lists each TCR transductant, its corresponding peptide, and its MHC class II restriction. Briefly, to create a TCR transductant, a single TCR sequence (α and β chains linked by the PTV1.2A sequence) was cloned into murine stem cell virus–based (MSCV-based) retroviral vectors carrying the GFP (pMIGII) or pMSCV puro retroviral vector (Clontech), followed by production of replication-incompetent retroviruses encoding TCR sequences. 5KC cells transduced with retroviruses were then selected either by flow cytometric cell sorting of GFP-positive cells or with puromycin according to the selection genes carried by the retroviral vectors; TCR expression was confirmed by staining with anti-mouse TCR β antibody (BD Biosciences; clone H57-597). The 5KC cells were also transduced to express native human CD4, which was confirmed by flow cytometric staining with anti-CD4 antibody (BioLegend; clone RPA-T4).
In vitro screening of small molecules.
Small molecules for screening were obtained from the NIH’s Developmental Therapeutics Program (DTP) repository in vialed sets, dissolved in DMSO, and diluted in PBS for a final concentration of 0.1% DMSO in each well. Each small molecule was screened at a concentration of 100 μM. The clone 5 TCR transductant was cultured with peptide, with or without a small molecule, in the presence of murine DQ8-Tg splenic cells. DQ8-Tg splenic cells were dispersed to single cells, followed by red blood cell lysis, and then 2 × 105 splenoctyes were cultured with peptide for 4 hours in a 96-well, U-bottomed plate followed by the addition of 2 × 105 clone 5 cells for overnight culture. Murine IL-2 secreted by the 5KC cells was measured using a highly sensitive ELISA (V-PLEX IL-2 Kit; Meso Scale Diagnostics), followed by detection using a MESO QuickPlex SQ120 instrument. Pocket 1 and pocket 9 small molecules were screened using clone 5 in the presence of insulin B:9-23 (100 μg/ml), while pocket 4 and pocket 6 compounds were screened using insulin B:13-23 (100 μg/ml), which provides a greater amount of IL-2 secretion than does B:9-23.
Peptides, protein, and small molecules.
The peptides used for TCR stimulation assays were obtained from Genemed Synthesis at a greater than 95% purity. Recombinant α-gliadin/DQ8 protein containing the deamidated peptide sequence QQYPSGEGSFQPSQENPQ was expressed in S2 Drosophila cells and purified as previously described (55). TATD (1,3,6,8-tetraaza-tricyclo[4.4.1.13,8]dodecane) was synthesized at greater than 95% purity and characterized by 1H NMR and 13C NMR (The Chemistry Research Solutions). Methyldopa was purchased at greater than 98% purity from Sigma-Aldrich (catalog 857416); the metabolite α-methylnorepinephrine was obtained from Toronto Research Chemicals (catalog D454000); and compounds structurally related to methyldopa were purchased from Sigma-Aldrich. DQ8 protein used for ITC was isolated and purified by affinity chromatography using a DQ monoclonal antibody (clone SPV-L3) and an EBV-transformed B cell line derived from a DQ8 homozygote (57).
Characterization of TATD in vitro.
TATD was dissolved in PBS and stored in low-binding Eppendorf tubes (Costar; catalog 3207). Dose titrations were performed in a manner similar to that for the small-molecule screening assays: 2 × 105 DQ8-Tg splenocytes were culture with cognate peptide (100 μg/ml B:13-23 for clone 5 and 1 μM deamidated α-gliadin for TATD) for 4 hours, followed by the addition of 2 × 105 TCR transductant cells for overnight culture and measurement of secreted IL-2. Cell viability was measured with the CellTiter-Glo Luminescent assay (Promega) according to the manufacturer’s instructions. Experiments with recombinant deamidated α-gliadin/DQ8 protein were performed by incubating 0.01 μg protein in 100 μl buffer (25 mM sodium phosphate at pH 8.0) with 1 μM deamidated α-gliadin peptide plus TATD concentrations for 24 hours at 50°C to allow peptide exchange. Following the initial incubation, the contents were transferred to a 96-well, high-binding plate (Costar; catalog 9018) and incubated for 2 hours at room temperature. The plate was washed twice with 0.05% Tween-20 in PBS to remove any unbound protein, peptide, and TATD. Then, 489 cells (105 cells) were incubated with the protein overnight at 37° with 5% CO2, and the supernatant was assayed for secreted IL-2.
Characterization of methyldopa in vitro.
Methyldopa was dissolved in dilute HCl and the pH adjusted to 7.0 prior to each in vitro assay, as air oxidation was visible over the ensuing days. Dose titrations with TCR transductants were performed in a manner identical to titrations done with TATD. To assess in vitro specificity, EBV-transformed B cells from a DQ8/DR4 homozygote (DQA1*03:01-DQB1*03:02-DRB1*04:01) were used as APCs (57). For these assays, EBV-transformed B cells (105 cells) were cultured with cognate peptide for 4 hours, followed by addition of 105 TCR transductant cells for overnight culture and the measurement of secreted IL-2. For experiments done with the DQ8-flu TCR, previously produced M12C3 cells (murine B cell line) expressing HLA-DQ8 were used as APCs (5 × 104) (26), cultured with 10 μg/ml influenza peptide for 4 hours, followed by the addition of TCR transductant cells (105 cells) for overnight culture and measurement of secreted IL-2. To assess the effects of intracellular uptake of methyldopa, APC fixation experiments were conducted. M12C3 DQ8 cells were glutaraldehyde fixed. To fix cells, these APCs (5 × 106 cells/ml) were incubated with 0.05% glutaraldehyde for 30 seconds, followed by quenching the reaction with 0.2 M glycine. Fixed APCs (5 × 104) were cultured with 10 μM deamidated α-gliadin peptide and methyldopa for 24 hours, and then 489 TCR transductant cells (105 cells) were added for overnight culture, followed by the measurement of secreted IL-2.
ITC.
ITC experiments were performed using a MicroCal iTC200 Calorimeter (GE Healthcare) at 25°C. DQ8 protein was dialyzed in citrate-phosphate buffer (pH 5.0) and loaded into the calorimeter cell at a concentration of 4 μM. Methyldopa was dissolved in the same citrate-phosphate buffer at a concentration of 4 mM, and the pH was adjusted to 5.0. The injection volumes of methyldopa were 2 μl every 4 minutes over a total of 80 minutes. The first data point was removed from analysis because of dilution across the injection needle tip. Binding isotherms were generated using Origin Software (MicroCal, OriginLab) with a single-site binding model by nonlinear regression. For baseline correction, methyldopa was titrated into buffer prior to each experiment and subtracted from each data set. KD values were calculated as the reciprocal of the measured binding affinity, and free energy of binding was calculated using the following equation: ΔG = ΔH – TΔS, where H = enthalpy, T = temperature in kelvin, and S = entropy.
Animals.
Mice were bred and housed under specific pathogen–free conditions at the University of Colorado Denver Center for Comparative Medicine. Female NOD mice were purchased from The Jackson Laboratory and used for the early and late TATD intervention studies. The late prevention study (therapy beginning at 12 weeks of age) was conducted at Novartis Research Facilities in Basel, Switzerland. TATD was dissolved in PBS for intraperitoneal administration and in sterile water for oral gavage. An anti-CD3 monoclonal antibody (eBioscience; clone 2C11) was administered by intraperitoneal injection at 50 μg daily for 5 consecutive days in the late prevention study.
DQ8-Tg mice (containing a human DQ8/CD4 transgene, murine IA-null on a NOD Rag-KO background) were obtained from The Jackson Laboratory (catalog 006022) and bred at the University of Colorado Denver. Spleens from these mice were used to obtain splenocytes to present peptides to TCR transductants in in vitro T cell stimulation assays and for in vivo testing of methyldopa and metabolites. DQ8-Tg mice were used on a Rag-KO background, such that T and B cells would not be present to secrete IL-2. For the methyldopa in vivo studies, methyldopa powder was dissolved in dilute HCl prior to each administration, and control mice received dilute HCl (vehicle).
Assessment of murine diabetes, insulin antibodies, and glucose tolerance.
Blood glucose was measured weekly with a ReliOn Blood Glucose Monitoring System (Solartek), and mice were considered diabetic after having 2 consecutive blood glucose values above 250 mg/dl (13.9 mmol/l). Insulin antibodies were measured with a fluid-phase RIA at 4 weeks of age and then every 4 weeks thereafter as previously described (58). Intraperitoneal glucose tolerance tests (GTTs) were performed following an overnight fast, and D-glucose (2 g/kg body weight) in 200 μl PBS was administered by intraperitoneal injection. Blood glucose values were obtained at 0, 15, 30, 60, 90, and 120 minutes using a ReliOn meter, and the 2-hour glucose AUC was calculated.
Histological analysis.
Parafin-embedded pancreatic sections were H&E stained, followed by insulin staining. Islets were identified by glucagon staining and graded for insulitis by an individual blinded to the experimental design. A minimum of 100 islets per cohort (PBS or TATD) were scored for islet infiltration as follows: 0, no infiltration; 1, peri-insulitis; 2, islets with lymphocyte infiltration in less than 50% of the area; 3, islets with lymphocyte infiltration in more than 50% of the area.
Study patients and assessments.
Subjects for the phase Ib dose-escalation study were recruited from clinics of the Barbara Davis Center for Diabetes. Eligibility criteria included: (a) age between 18 and 45 years; (b) diagnosis of T1D, according to American Diabetes Association criteria, within the previous 2 years; (c) positive for at least one T1D-related autoantibody (insulin autoantibodies were only considered positive if measured within 3 weeks of starting exogenous insulin therapy); (d) positive for at least one DQ8 allele (DQB*03:02); and (e) a random serum C-peptide level of greater than 0.1 nmol/l, which is a measure of residual β cell function.
Visits and clinical assessments were performed at baseline and in weeks 1, 3, 6, 7, and 12, for a total of 6 visits. Blood samples were obtained at each visit for immunologic assessments. PBMCs were isolated from heparinized blood using Ficoll-Paque (GE Healthcare); freshly isolated PBMCs were used for flow cytometric panels to analyze peripheral blood B cells and DCs, while the remainder were cryopreserved in FBS with 10% DMSO. Physical examination and metabolic samples (glycated hemoglobin [HbA1c] and MMTTs) were obtained at baseline and in week 12. Pill counts were performed at the visits in weeks 1, 3, and 6 to assess drug compliance. Adverse events and insulin use (basal and bolus) were recorded at each study visit.
Measurement of serum islet autoantibodies and HLA genotyping.
Peripheral blood was obtained for islet autoantibody assays and HLA genotyping. Islet autoantibodies against insulin, GAD65, IA-2, and ZnT8 were measured from the serum by fluid-phase RIA as previously described (59). HLA-DRB1, DQA1, and DQB1 alleles were typed using oligonucleotide probes as previously described (60).
Tolerability and safety of methyldopa.
Methyldopa (Aldomet; 500-mg tablets) was obtained from Mylan Pharmaceuticals (lots 3038543 and 3062517). The study subjects self-administered doses throughout the open-label study. All participants tolerated the low dose (500 mg 2 times/day) and moderate dose (500 mg 3 times/day), while 18 of 20 tolerated the high dose (2–3 grams self-administered over the course of a day). For the 2 individuals who did not tolerate the high dose, the moderate dose was continued during the treatment phase of the study. Methyldopa has a well-known safety profile, and in our trial we observed: (a) no lowering of systolic or diastolic blood pressure; (b) no elevation of liver function tests (aspartate aminotransferase [AST] or alanine aminotransferase [ALT]); and (c) no anemia. There were no serious adverse events related to study drug administration.
Immunologic assessments.
For the measurement of specific MHC class II presentation, cryopreserved PBMCs were thawed and 2 × 105 cells cultured with peptide (200 μg/ml B:13-23 for clone 5; 10 μM deamidated α-gliadin for 489; 100 μM α-II-gliadin for 233; or 1 μg/ml HA306-318 for C7CH17) in 96-well, U-bottomed plates for 4 hours at 37° with 5% CO2. After 4 hours, the corresponding TCR transductant (105 cells) was added and incubated overnight at 37° with 5% CO2. Each condition was tested in triplicate wells, including 1 with no antigen and anti-CD3 antibody (eBioscience; clone 2C11), which was a positive control for the TCR transductant. PBMCs from each study visit for a single participant were included on the same 96-well plate for each TCR transductant (i.e., PBMCs from each of the 6 visits were on a single plate for clone 5) to avoid batch effects between study visits and participants. Following overnight culture, the plate was centrifuged at 500 g for 5 minutes, and the supernatant was removed and frozen at –80° until measurement of IL-2 in the supernatant with a highly sensitive ELISA (Meso Scale Diagnostics).
ELISPOT assays.
ELISPOT assays were conducted as previously described using human IFN-γ ELISPOT kits (U-CyTech Biosciences) (61). Briefly, cryopreserved PBMCs were thawed, and 106 cells were cultured in 250 μl serum-free AIM-V Medium (Invitrogen, Thermo Fisher Scientific) with 10 μg/ml peptide. Cells were supplemented with an additional 250 μl medium after 24 hours and harvested 24 hours later. After washing, the cells were resuspended in 300 μl medium and transferred in three 100-μl aliquots onto 96-well, clear polystyrene culture plates coated with an IFN-γ capture monoclonal antibody and subsequently treated with 1× blocking solution (U-CyTech Biosciences). Seventeen hours later, the cells were removed by decanting and the wells washed. Spots were then formed by sequential incubations with the biotinylated second-site anti–IFN-γ, gold-labeled goat anti-biotin, and a precipitating silver substrate. Spots were enumerated with a BIOREADER 4000 Pro X (BIOSYS GmbH). Wells with no antigen served as a negative control, and tetanus toxoid vaccine was used as a positive control in each assay.
Flow cytometry.
Freshly isolated PBMCs were stained with cell-surface markers for the following panels: B cells (CD19, HLA-DR, HLA-DQ); myeloid DCs 1 (CD11c, CD1c [BDCA-1], HLA-DR, HLA-DQ, CD4/CD8); myeloid DCs 2 (CD11c, CD141 [BDCA-3], HLA-DQ, HLA-DQ, CD4/CD8); and plasmacytoid DCs (CD11c, CD303 [BDCA-2], CD304 [BDCA-4], CD123, HLA-DR, HLA-DQ). The specific antibody clones and suppliers are listed in Supplemental Table 10; isotype controls were used for CD11c and CD123. Analysis was done using a BD LSR-II, and data were analyzed using FlowJo Software, version 9.
Metabolic assessments.
At 0 and 3 months, HbA1c was measured using a DCA 2000 Analyzer (Siemens Healthcare Diagnostics). A MMTT (360 ml of Boost) was administered and serum C-peptide measured at 0, 15, 30, 60, 90, and 120 minutes using an ELISA assay (ALPCO), and the 2 hour C-peptide AUC was calculated.
Plasma methyldopa levels.
Methyldopa concentrations were measured in a 100-μl cryopreserved sample (1:1 plasma/PBS) collected at each study visit. Samples were thawed and underwent protein precipitation with 25 μl of 10% perchloric acid in Milli-Q water, followed by centrifugation at 13,300 g for 15 minutes. The supernatants were analyzed by liquid chromatography–tandem mass spectrometry. Positive ion electrospray ionization mass spectra were obtained with an MDS Sciex 3200 Q-TRAP triple quadrupole mass spectrometer (Applied Biosystems) with a turbo ion spray source interfaced with an Agilent 1200 Series Binary Pump SL HPLC system. Samples were chromatographed using a Sunfire C8, 5-μm, 4.6 × 50 mm column (Waters Corporation) protected by a C18 guard cartridge (4.0 × 2.0 mm; Phenomenex). Methyldopa standard curves were prepared in human plasma diluted 1:1, spiked with 5 μl of the appropriate standard concentration with PBS, and were linear over a range of 5 to 5,000 ng/ml. The linearity of the standard curves was greater than r2 = 0.99, using 1/x2 weighting.
Statistics.
Statistical analyses were performed using SAS 9.4 (SAS Institute) and GraphPad Prism 6.0 (GraphPad Software), including calculations for IC50 values from dose titrations with TATD and methyldopa. The statistical tests, including the log-rank test, Mann-Whitney U test, ANOVA, unpaired t test, Wilcoxon matched-pairs signed rank test, used for each experiment are indicated in the corresponding figure legends. P values of less than 0.05 were considered significant. For MHC class II–specific antigen presentation in study subjects, longitudinal mixed-effects models adjusted for the baseline value were used to examine changes over time. Residual plots were examined in order to evaluate the fit of the models. P values compare the differences of least-squares means between 2 time points. For safety outcomes (blood pressure, liver function tests, and hemoglobin), mixed-effects models were used to compare mean values on and off treatment, without an adjustment for baseline.
Study approval.
The animal studies were conducted under protocols approved by the University of Colorado Denver and Novartis Animal Care and Use Committees. The clinical investigation in this study was conducted in accordance with Declaration of Helsinki principles, with study approval provided by the Colorado Multiple Institutional Review Board. The clinical trial (ClinicalTrials.gov Identifier NCT01883804) was investigational new drug (IND) exempt by the FDA (PIND 118119). Written informed consent was obtained after the nature and possible consequences of the study were explained to each participant.
Author contributions
DAO, MAA, PAG, and AWM designed the studies and wrote the manuscript. AA, KAM, SC, EEB, BP, and KJS performed experiments and reviewed the manuscript. LP conducted independent statistical analyses of the study data. SE, VNS, SKG, PAG, and AWM designed, obtained regulatory approval for, and conducted the phase Ib clinical trial. AWM is the guarantor of this work, had full access to all the data in the study, and assumes responsibility for the data integrity and accuracy of analysis.
Supplementary Material
Acknowledgments
We thank the late George S. Eisenbarth (1942–2012) for helpful scientific discussions, inspiration, and encouragement. We also thank the National Cancer Institute (NCI), Division of Cancer Treatment and Diagnosis (DCTD), DTP for providing the small molecules used for screening in the study, and Lori Sussel and Edwin Liu at the University of Colorado for critical editing of the manuscript. This work was supported by NIH grants (DK095995, DK108868, DK110845, DK032083, DK104223, and AI110503); the Juvenile Diabetes Research Foundation (17-2010-744 and 2014-143-C-R); the Children’s Diabetes Foundation; the Barbara Davis Center Translational Research Unit; and the Colorado Clinical and Translational Science Institute (TR001082).
Version 1. 02/13/2018
In-Press Preview
Version 2. 04/03/2018
Electronic publication
Version 3. 05/01/2018
Print issue publication
Footnotes
Conflict of interest: AWM and DAO are inventors on a patent titled “Compounds that modulate autoimmunity and methods of using the same,” licensed to ImmunoMolecular Therapeutics (US patent number 9,629,848). AWM and PAG are scientific cofounders of ImmunoMolecular Therapeutics and own shares in the company. KJS and BP are former employees of Novartis.
Reference information: J Clin Invest. 2018;128(5):1888–1902.https://doi.org/10.1172/JCI97739.
Contributor Information
David A. Ostrov, Email: ostroda@pathology.ufl.edu.
Aimon Alkanani, Email: aimon.alkanani@ucdenver.edu.
Kristen A. McDaniel, Email: kristen.mcdaniel@ucdenver.edu.
Stephanie Case, Email: s.r.case@hotmail.com.
Erin E. Baschal, Email: erin.baschal@ucdenver.edu.
Laura Pyle, Email: laura.pyle@ucdenver.edu.
Bernadette Pöllinger, Email: bpoellinger@hotmail.com.
Katherine J. Seidl, Email: kseidl@mac.com.
Viral N. Shah, Email: viral.shah@ucdenver.edu.
Satish K. Garg, Email: satish.garg@ucdenver.edu.
Mark A. Atkinson, Email: atkinson@pathology.ufl.edu.
Peter A. Gottlieb, Email: peter.gottlieb@ucdenver.edu.
Aaron W. Michels, Email: aaron.michels@ucdenver.edu.
References
- 1.Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;383(9911):69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464(7293):1293–1300. doi: 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ziegler AG, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA. 2013;309(23):2473–2479. doi: 10.1001/jama.2013.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayer-Davis EJ, et al. Incidence Trends of Type 1 and Type 2 Diabetes among Youths, 2002-2012. N Engl J Med. 2017;376(15):1419–1429. doi: 10.1056/NEJMoa1610187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rigby MR, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest. 2015;125(8):3285–3296. doi: 10.1172/JCI81722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherry N, et al. Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378(9790):487–497. doi: 10.1016/S0140-6736(11)60931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keymeulen B, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352(25):2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 8.Herold KC, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346(22):1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 9.Pescovitz MD, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361(22):2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orban T, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011;378(9789):412–419. doi: 10.1016/S0140-6736(11)60886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moran A, et al. Interleukin-1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double-blind, placebo-controlled trials. Lancet. 2013;381(9881):1905–1915. doi: 10.1016/S0140-6736(13)60023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wherrett DK, et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet. 2011;378(9788):319–327. doi: 10.1016/S0140-6736(11)60895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Näntö-Salonen K, et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet. 2008;372(9651):1746–1755. doi: 10.1016/S0140-6736(08)61309-4. [DOI] [PubMed] [Google Scholar]
- 14.Skyler JS, et al. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial — Type 1. Diabetes Care. 2005;28(5):1068–1076. doi: 10.2337/diacare.28.5.1068. [DOI] [PubMed] [Google Scholar]
- 15.Diabetes Prevention Trial--Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med. 2002;346(22):1685–1691. doi: 10.1056/NEJMoa012350. [DOI] [PubMed] [Google Scholar]
- 16.Tsai S, Santamaria P. MHC class II polymorphisms, autoreactive T-cells, and autoimmunity. Front Immunol. 2013;4:321. doi: 10.3389/fimmu.2013.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inoko H, Ando A, Tsuji K, Matsuki K, Juji T, Honda Y. HLA-DQ beta chain DNA restriction fragments can differentiate between healthy and narcoleptic individuals with HLA-DR2. Immunogenetics. 1986;23(2):126–128. doi: 10.1007/BF00377973. [DOI] [PubMed] [Google Scholar]
- 18.Green PH, Cellier C. Celiac disease. N Engl J Med. 2007;357(17):1731–1743. doi: 10.1056/NEJMra071600. [DOI] [PubMed] [Google Scholar]
- 19.Bukhari W, Barnett MH, Prain K, Broadley SA. Molecular pathogenesis of neuromyelitis optica. Int J Mol Sci. 2012;13(10):12970–12993. doi: 10.3390/ijms131012970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moutsianas L, et al. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat Genet. 2015;47(10):1107–1113. doi: 10.1038/ng.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viatte S, Plant D, Raychaudhuri S. Genetics and epigenetics of rheumatoid arthritis. Nat Rev Rheumatol. 2013;9(3):141–153. doi: 10.1038/nrrheum.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raychaudhuri S, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44(3):291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu X, et al. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat Genet. 2015;47(8):898–905. doi: 10.1038/ng.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med. 2009;360(16):1646–1654. doi: 10.1056/NEJMra0808284. [DOI] [PubMed] [Google Scholar]
- 25.Erlich H, et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes. 2008;57(4):1084–1092. doi: 10.2337/db07-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michels AW, et al. Islet-Derived CD4 T Cells Targeting Proinsulin in Human Autoimmune Diabetes. Diabetes. 2017;66(3):722–734. doi: 10.2337/db16-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pathiraja V, et al. Proinsulin-specific, HLA-DQ8, and HLA-DQ8-transdimer-restricted CD4+ T cells infiltrate islets in type 1 diabetes. Diabetes. 2015;64(1):172–182. doi: 10.2337/db14-0858. [DOI] [PubMed] [Google Scholar]
- 28.Michels AW, et al. Structure-based selection of small molecules to alter allele-specific MHC class II antigen presentation. J Immunol. 2011;187(11):5921–5930. doi: 10.4049/jimmunol.1100746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaparro RJ, Dilorenzo TP. An update on the use of NOD mice to study autoimmune (Type 1) diabetes. Expert Rev Clin Immunol. 2010;6(6):939–955. doi: 10.1586/eci.10.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee KH, Wucherpfennig KW, Wiley DC. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat Immunol. 2001;2(6):501–507. doi: 10.1038/88694. [DOI] [PubMed] [Google Scholar]
- 31.Eerligh P, et al. Functional consequences of HLA-DQ8 homozygosity versus heterozygosity for islet autoimmunity in type 1 diabetes. Genes Immun. 2011;12(6):415–427. doi: 10.1038/gene.2011.24. [DOI] [PubMed] [Google Scholar]
- 32.Tollefsen S, et al. HLA-DQ2 and -DQ8 signatures of gluten T cell epitopes in celiac disease. J Clin Invest. 2006;116(8):2226–2236. doi: 10.1172/JCI27620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mah GT, Tejani AM, Musini VM. Methyldopa for primary hypertension. Cochrane Database Syst Rev. 2009;(4):CD003893. doi: 10.1002/14651858.CD003893.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Broughton SE, et al. Biased T cell receptor usage directed against human leukocyte antigen DQ8-restricted gliadin peptides is associated with celiac disease. Immunity. 2012;37(4):611–621. doi: 10.1016/j.immuni.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Davis MM, et al. Ligand recognition by alpha beta T cell receptors. Annu Rev Immunol. 1998;16:523–544. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 36.Sjoerdsma A, Vendsalu A, Engelman K. Studies on the metabolism and mechanism of action of methyldopa. Circulation. 1963;28:492–502. doi: 10.1161/01.CIR.28.4.492. [DOI] [PubMed] [Google Scholar]
- 37.Nakayama M, et al. Regulatory vs. inflammatory cytokine T cell responses to mutated insulin peptides in healthy and type 1 diabetic subjects. Proc Natl Acad Sci U S A. 2015;112(14):4429–4434. doi: 10.1073/pnas.1502967112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J, et al. Autoreactive T cells specific for insulin B:11-23 recognize a low-affinity peptide register in human subjects with autoimmune diabetes. Proc Natl Acad Sci U S A. 2014;111(41):14840–14845. doi: 10.1073/pnas.1416864111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.James EA, Bui J, Berger D, Huston L, Roti M, Kwok WW. Tetramer-guided epitope mapping reveals broad, individualized repertoires of tetanus toxin-specific CD4+ T cells and suggests HLA-based differences in epitope recognition. Int Immunol. 2007;19(11):1291–1301. doi: 10.1093/intimm/dxm099. [DOI] [PubMed] [Google Scholar]
- 40.Greenbaum CJ, et al. Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite Type 1 Diabetes TrialNet data. Diabetes. 2012;61(8):2066–2073. doi: 10.2337/db11-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roep BO, Peakman M. Antigen targets of type 1 diabetes autoimmunity. Cold Spring Harb Perspect Med. 2012;2(4):a007781. doi: 10.1101/cshperspect.a007781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krogsgaard M, et al. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol Cell. 2003;12(6):1367–1378. doi: 10.1016/S1097-2765(03)00474-X. [DOI] [PubMed] [Google Scholar]
- 43.Willcox BE, et al. TCR binding to peptide-MHC stabilizes a flexible recognition interface. Immunity. 1999;10(3):357–365. doi: 10.1016/S1074-7613(00)80035-7. [DOI] [PubMed] [Google Scholar]
- 44.Spanier JA, et al. Increased effector memory insulin-specific CD4+T cells correlate with insulin autoantibodies in patients with recent-onset type 1 diabetes. Diabetes. 2017;66(12):3051–3060. doi: 10.2337/db17-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang L, et al. Monoclonal antibody blocking the recognition of an insulin peptide-MHC complex modulates type 1 diabetes. Proc Natl Acad Sci U S A. 2014;111(7):2656–2661. doi: 10.1073/pnas.1323436111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dahan R, et al. Antigen-specific immunomodulation for type 1 diabetes by novel recombinant antibodies directed against diabetes-associates auto-reactive T cell epitope. J Autoimmun. 2013;47:83–93. doi: 10.1016/j.jaut.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 47.Ostrov DA, et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc Natl Acad Sci U S A. 2012;109(25):9959–9964. doi: 10.1073/pnas.1207934109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Illing PT, et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature. 2012;486(7404):554–558. doi: 10.1038/nature11147. [DOI] [PubMed] [Google Scholar]
- 49.White KD, Chung WH, Hung SI, Mallal S, Phillips EJ. Evolving models of the immunopathogenesis of T cell-mediated drug allergy: The role of host, pathogens, and drug response. J Allergy Clin Immunol. 2015;136(2):219–34; quiz 235. doi: 10.1016/j.jaci.2015.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pompeu YA, Stewart JD, Mallal S, Phillips E, Peters B, Ostrov DA. The structural basis of HLA-associated drug hypersensitivity syndromes. Immunol Rev. 2012;250(1):158–166. doi: 10.1111/j.1600-065X.2012.01163.x. [DOI] [PubMed] [Google Scholar]
- 51.Klitz W, et al. New HLA haplotype frequency reference standards: high-resolution and large sample typing of HLA DR-DQ haplotypes in a sample of European Americans. Tissue Antigens. 2003;62(4):296–307. doi: 10.1034/j.1399-0039.2003.00103.x. [DOI] [PubMed] [Google Scholar]
- 52.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 53.Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL. Assessing scoring functions for protein-ligand interactions. J Med Chem. 2004;47(12):3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 54.Ewing TJ, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des. 2001;15(5):411–428. doi: 10.1023/A:1011115820450. [DOI] [PubMed] [Google Scholar]
- 55.Liu E, et al. Exploring T cell reactivity to gliadin in young children with newly diagnosed celiac disease. Autoimmune Dis. 2014;2014:927190. doi: 10.1155/2014/927190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hennecke J, Wiley DC. Structure of a complex of the human alpha/beta T cell receptor (TCR) HA1.7, influenza hemagglutinin peptide, and major histocompatibility complex class II molecule, HLA-DR4 (DRA*0101 and DRB1*0401): insight into TCR cross-restriction and alloreactivity. J Exp Med. 2002;195(5):571–581. doi: 10.1084/jem.20011194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sidney J, et al. Measurement of MHC/peptide interactions by gel filtration or monoclonal antibody capture. Curr Protoc Immunol. 2013;Chapter 18:Unit 18.3. doi: 10.1002/0471142735.im1803s100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu L, Eisenbarth G, Bonifacio E, Thomas J, Atkinson M, Wasserfall C. The second murine autoantibody workshop: remarkable interlaboratory concordance for radiobinding assays to identify insulin autoantibodies in nonobese diabetic mice. Ann N Y Acad Sci. 2003;1005:1–12. doi: 10.1196/annals.1288.002. [DOI] [PubMed] [Google Scholar]
- 59.Yu L, et al. Antiislet autoantibodies usually develop sequentially rather than simultaneously. J Clin Endocrinol Metab. 1996;81(12):4264–4267. doi: 10.1210/jcem.81.12.8954025. [DOI] [PubMed] [Google Scholar]
- 60.Rewers A, et al. Ethnic differences in the associations between the HLA-DRB1*04 subtypes and type 1 diabetes. Ann N Y Acad Sci. 2003;1005:301–309. doi: 10.1196/annals.1288.047. [DOI] [PubMed] [Google Scholar]
- 61.Nagata M, Kotani R, Moriyama H, Yokono K, Roep BO, Peakman M. Detection of autoreactive T cells in type 1 diabetes using coded autoantigens and an immunoglobulin-free cytokine ELISPOT assay: report from the fourth immunology of diabetes society T cell workshop. Ann N Y Acad Sci. 2004;1037:10–15. doi: 10.1196/annals.1337.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.