

Abstract
Centrosomes are the main microtubule‐organizing centers of animal cells. Although centrosome aberrations are common in tumors, their consequences remain subject to debate. Here, we studied the impact of structural centrosome aberrations, induced by deregulated expression of ninein‐like protein (NLP), on epithelial spheres grown in Matrigel matrices. We demonstrate that NLP‐induced structural centrosome aberrations trigger the escape (“budding”) of living cells from epithelia. Remarkably, all cells disseminating into the matrix were undergoing mitosis. This invasive behavior reflects a novel mechanism that depends on the acquisition of two distinct properties. First, NLP‐induced centrosome aberrations trigger a re‐organization of the cytoskeleton, which stabilizes microtubules and weakens E‐cadherin junctions during mitosis. Second, atomic force microscopy reveals that cells harboring these centrosome aberrations display increased stiffness. As a consequence, mitotic cells are pushed out of mosaic epithelia, particularly if they lack centrosome aberrations. We conclude that centrosome aberrations can trigger cell dissemination through a novel, non‐cell‐autonomous mechanism, raising the prospect that centrosome aberrations contribute to the dissemination of metastatic cells harboring normal centrosomes.
Keywords: biomechanical properties, invasiveness, multicellular cooperation, non‐cell‐autonomous process, structural centrosome aberrations
Subject Categories: Cancer; Cell Adhesion, Polarity & Cytoskeleton; Cell Cycle
Introduction
Centrosomes function in the organization of microtubules and in ciliogenesis (Bornens, 2012; Conduit et al, 2015; Sanchez & Dynlacht, 2016; Prosser & Pelletier, 2017; Nigg & Holland 2018), and dysfunctions of these organelles have been linked to several human diseases, notably ciliopathies and microcephaly or dwarfism (Nigg & Raff, 2009; Bettencourt‐Dias et al, 2011; Braun & Hildebrandt, 2017). Centrosome aberrations are also prominent in cancers, including pre‐invasive in situ carcinomas (Lingle et al, 2002; Pihan et al, 2003; Guo et al, 2007), suggesting that they contribute actively to carcinogenesis (Lingle et al, 1998; Nigg, 2002; Pujana et al, 2007; Zyss & Gergely, 2009; Godinho & Pellman, 2014; Gonczy, 2015). Interestingly, cancer cells adapt to centrosome aberrations in multiple ways, notably through clustering of supernumerary centrosomes (Nigg, 2002; Quintyne et al, 2005; Rhys et al, 2018). Best documented is the influence of centrosome aberrations on chromosomal instability, a hallmark of cancer (Ganem et al, 2009; Silkworth et al, 2009), and increasing evidence also suggests an impact on tissue architecture (Nigg, 2002; Godinho & Pellman, 2014; Kazazian et al, 2017; Raff & Basto, 2017). However, centrosome aberrations are generally present in only a fraction of all tumor cells and, moreover, expected to impair cell viability. Hence, the question persists of whether and how centrosome aberrations contribute to cancer in humans (Chan, 2011).
Centrosome abnormalities are subdivided into numerical and structural aberrations, but in tumors, the two defects are similarly prominent and often occur together (Lingle et al, 1998; Lingle & Salisbury, 1999; Kronenwett et al, 2005; Guo et al, 2007). The consequences of numerical aberrations have been studied extensively, culminating in the demonstration that centrosome amplification is sufficient to trigger tumorigenesis in both flies (Basto et al, 2008) and mice (Coelho et al, 2015; Sercin et al, 2016; Levine et al, 2017). In contrast, structural centrosome aberrations have received little attention. They are presumed to reflect deregulated gene expression (Lingle et al, 1998; Guo et al, 2007), as illustrated best by NLP (ninein‐like protein), a centrosomal component implicated in the nucleation and anchoring of microtubules (Casenghi et al, 2003). NLP is commonly overexpressed in human tumors, notably in breast cancers and lung carcinomas, and overexpression of NLP promotes tumorigenesis in mice (Qu et al, 2008; Yu et al, 2009; Shao et al, 2010). When overexpressed to comparable levels in 2 (2D)‐ or 3‐dimensional (3D) cultures, NLP triggers structural centrosome aberrations that closely resemble those seen in tumor sections (Salisbury et al, 1999; Casenghi et al, 2003; Kronenwett et al, 2005; Schnerch & Nigg, 2016).
Recently, we have shown that prolonged overexpression of NLP disturbs epithelial polarity and severely disrupts the architecture of MCF10A acini (Schnerch & Nigg, 2016). Here, we have used similar 3D models to explore the short‐term impact of structural centrosome aberrations, with particular focus on the ability of epithelial cells to develop invasive behavior. We found that epithelia harboring NLP‐induced centrosome aberrations trigger the selective dissemination of mitotic cells into the surrounding matrix and demonstrate that this novel type of invasive behavior is triggered by a non‐cell‐autonomous mechanism involving two concomitant processes: first, an impairment of E‐cadherin junctions and, second, an increase in cellular stiffness, which introduces heterogeneity into the biomechanical properties of the epithelium. These observations demonstrate that centrosome aberrations can trigger the dissemination of dividing cells from epithelia, and they suggest a novel mechanism for the inception of a centrosome‐related metastatic process through multicellular cooperativity.
Results
Structural centrosome aberrations trigger budding of living cells from epithelia
To determine whether centrosome aberrations influence the ability of cells to escape from an intact epithelium, we examined 3D acini grown in Matrigel, a natural hydrogel that mimics a basement membrane‐like matrix (Artym, 2016). Remarkably, MCF10A‐derived acini harboring NLP‐induced structural centrosome aberrations frequently showed dissemination of individual cells into the surrounding matrix, a phenotype hereafter termed “budding” (Fig 1A). Cell budding was observed in nearly 30% of MCF10A acini overexpressing NLP, whereas it was only seen sporadically (< 5%) in absence of transgene induction, or in acini overexpressing the control protein CEP68 (Fig 1B). Also, no budding was seen in response to overexpression of PLK4, suggesting that this phenotype is triggered primarily by structural rather than numerical centrosome aberrations. The same phenotype was seen with cysts prepared from Madine–Darby canine kidney (MDCK) cells, attesting to its robustness (Figs 1A and EV1). As judged from staining for cleaved caspase 3, a marker for apoptosis, more than 65% of cells budding from GFP‐NLP‐expressing acini were alive, while more than 80% of the rare cells disseminating from control acini were undergoing apoptosis (Fig 1C and D). Collectively, these results show that structural centrosome aberrations induce the budding of living cells from the basal surface of polarized epithelia toward the surrounding matrix, potentially triggering an invasive process.
Figure 1. Structural centrosomal aberrations cause single cell budding from 3D acini.
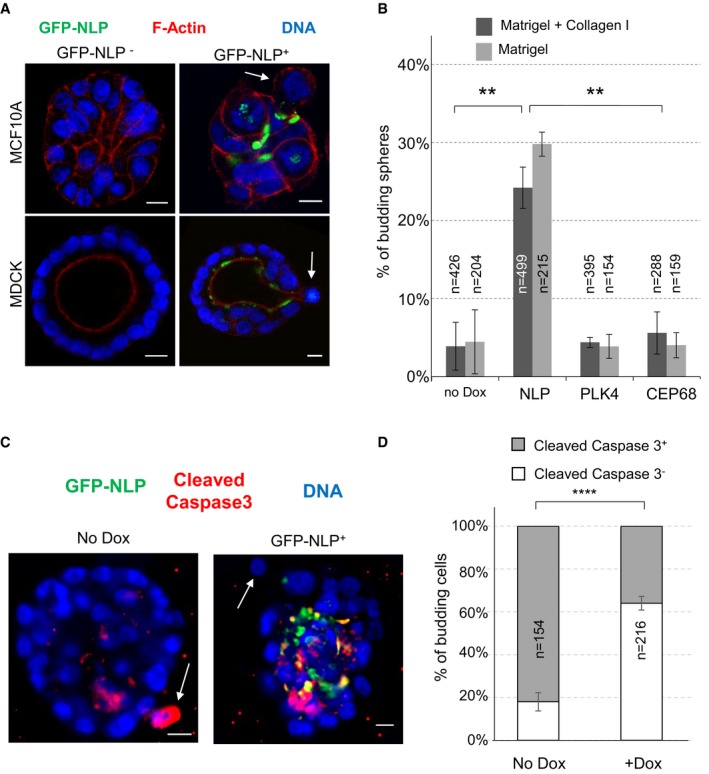
- Representative images show acini derived from MCF10A (upper panels) or MDCK cells (lower panels), with or without induction of GFP‐NLP expression. GFP‐NLP+ and GFP‐NLP− acini were stained for F‐actin (red) and DNA (blue). Arrows point to budding cells. Scale bars = 10 μm.
- Fraction of MCF10A acini that show budding in response to the indicated transgene products (GFP‐NLP, GFP‐PLK4 or GFP‐CEP68), depending on whether acini were cultured in collagen I‐enriched Matrigel (dark gray) or pure Matrigel (light gray); n indicates sample size and error bars indicate ± standard deviation (s.d.) of the mean from three independent experiments. **P < 0.01, as derived from unpaired, two‐tailed Student's t‐test.
- To determine the proportion of apoptotic cells, MCF10A acini expressing GFP‐NLP (+Dox) or not (No Dox) were fixed and stained for cleaved caspase 3. Arrows point to budding cells. Scale bars = 10 μm.
- Fraction of cells budding from MCF10A acini that are positive (gray bars) or negative (white bars) for cleaved caspase 3 staining; data are shown for acini with (+Dox) or without (No Dox) induction of GFP‐NLP expression. n indicates number of budding cells analyzed; error bars indicate ± s.d. of the mean from three independent experiments. ****P < 0.0001, as derived from unpaired, two‐tailed Student's t‐test.
Figure EV1. NLP overexpression induces budding in MDCK cysts.
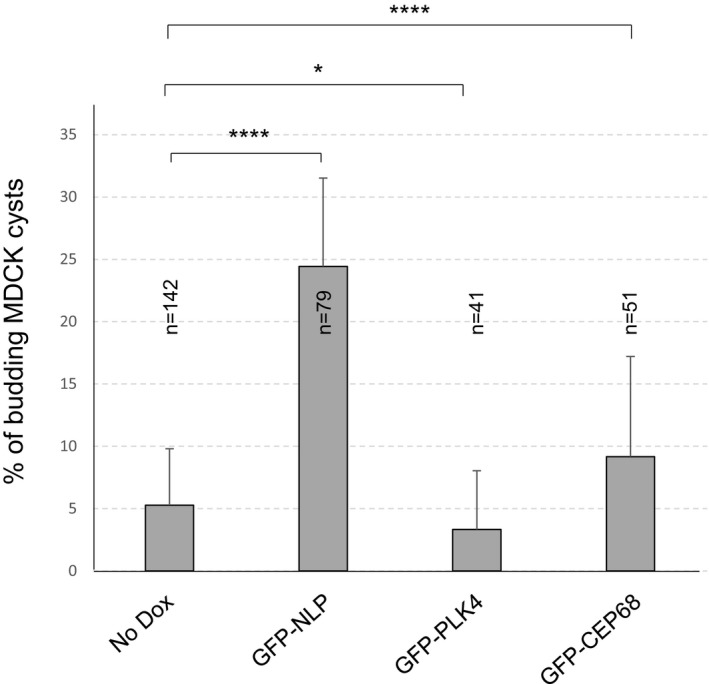
Histogram shows the proportions of MDCK cysts, cultured in pure Matrigel (without addition of collagen I), that exhibit budding after overexpression of GFP‐NLP, GFP‐PLK4, or GFP‐CEP68, in comparison to non‐induced acini (No Dox); error bars indicate + s.d. of the mean from three independent experiments, and n indicates the number of cysts analyzed from three independent experiments, P‐values were derived from unpaired, two‐tailed Student's t‐test. ****P < 0.0001 and *P < 0.05, respectively.
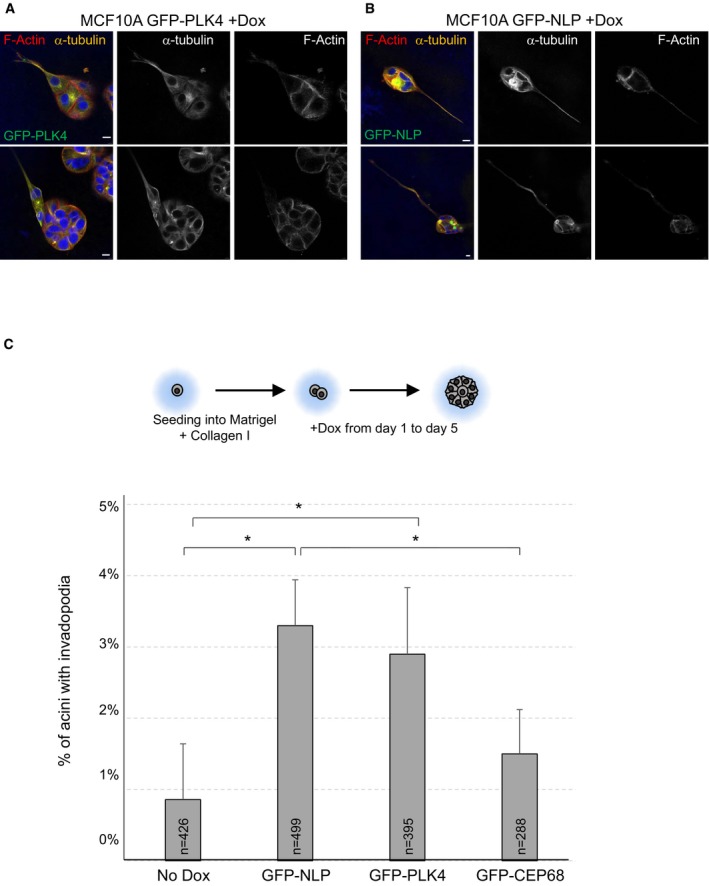
We emphasize that the budding phenotype is distinct from the formation of invasive protrusions (invadopodia) that occurs in MCF10A acini in response to PLK4‐induced centrosome amplification (Godinho et al, 2014). In agreement with Godinho et al, we also observed invadopodia formation upon overexpression of PLK4 (Fig EV2), provided that the invasion assay was sensitized by addition of type I collagen to Matrigel (Godinho et al, 2014; Artym, 2016; Schnerch & Nigg, 2016). However, we also observed invadopodia formation with acini harboring NLP‐induced structural centrosome aberrations (Fig EV2), suggesting that this phenotype may represent a more general response to centrosome aberrations than hitherto assumed.
Figure EV2. Centrosome aberrations induce invadopodia formation in collagen I‐enriched Matrigel.
-
A, BRepresentative images showing invadopodia formation in response to overexpression of either GFP‐PLK4 (A) or GFP‐NLP (B) in MCF10A acini cultured in collagen I‐enriched Matrigel. Acini were fixed and stained for α‐tubulin (orange), F‐actin (red), and DNA (blue); GFP‐tagged transgene products are shown in green. Scale bars = 10 μm.
-
CSchematic description of experiments. Histogram shows quantification of the proportions of acini showing invadopodia formation in collagen I‐enriched Matrigel; error bars indicate + s.d. of the mean from three independent experiments, and n indicates the number of acini analyzed. P‐values were derived from unpaired, two‐tailed Student's t‐test. *P < 0.05.
Budding involves mitotic cells
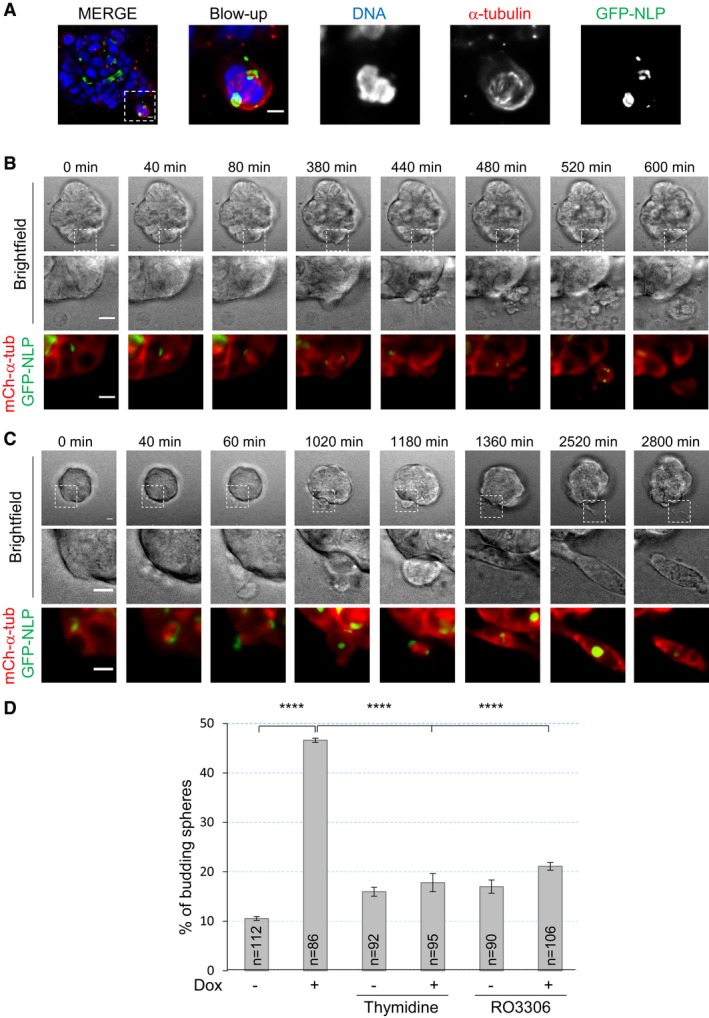
Immunofluorescence staining suggested that most, if not all, budding cells were going through mitosis (Fig 2A). To corroborate this conclusion, we performed time‐lapse microscopy on MCF10A acini and MDCK cysts derived from cells that stably expressed mCherry‐α‐tubulin. NLP overexpression was induced in acini and cysts after 5 days, and these were then imaged every 20 min for 3 days, resulting in the recording of 35 independent budding events. Remarkably, all these budding events concerned mitotic cells (Fig 2B and C): In 31 cases, the entire mitotic cell left the acinus (Fig 2B and Movie EV1), whereas in four cases, only one of two daughter cells was budding (Fig 2C and Movie EV2). Remarkably, some disseminating cells clearly continued dividing after evading from the acinus (Fig 2C and Movie EV2) and immunofluorescence performed at the end of time‐lapse microscopy confirmed that budding mitotic cells were not generally engaged in apoptosis, as judged by absence of staining for cleaved caspase 3 (Fig EV3 and Movie EV3). When cell cycle progression was arrested at the G1/S transition or in late G2, by addition of thymidine or the CDK1 inhibitor RO3386, respectively, budding of cells from GFP‐NLP‐expressing acini was markedly suppressed (Fig 2D), confirming the dependency of budding on mitosis. We conclude that NLP overexpression specifically triggers the dissemination of mitotic cells from epithelia toward the surrounding matrix.
Figure 2. NLP overexpression induces basal budding of living mitotic cells.
-
AMCF10A acini stained for α‐tubulin (red) and DNA (blue) by immunofluorescence. Images show cell that is in mitosis and apparently budding from the parental acini. Scale bars = 10 μm.
-
B, CStill series from time‐lapse experiments showing budding of entire mitotic cell (B) or one of two mitosis‐derived daughter cells (C) from MCF10A acini expressing GFP‐NLP. Upper panels show bright field microscopy, middle panels show details of the mitotic budding cells, and lower panels show the corresponding fluorescence images (GFP‐NLP in green and mCherry‐α‐tubulin in red). Scale bars = 10 μm. Images were acquired every 20 min, and time stamps are indicated.
-
DMCF10A acini were treated (+Dox) or not (−Dox) to induce GFP‐NLP expression; where indicated, thymidine or RO3306 where concomitantly added to block cells at the G1/S transition or to inhibit CDK1 activity and induce a block in G2 phase, respectively. Fraction of budding acini, with n indicating the number of acini analyzed; error bars indicate ± s.d. of the mean from three independent experiments. ****P < 0.0001, as derived from unpaired, two‐tailed Student's t‐test.
Figure EV3. A proportion of budding mitotic cells are negative for cleaved caspase 3.
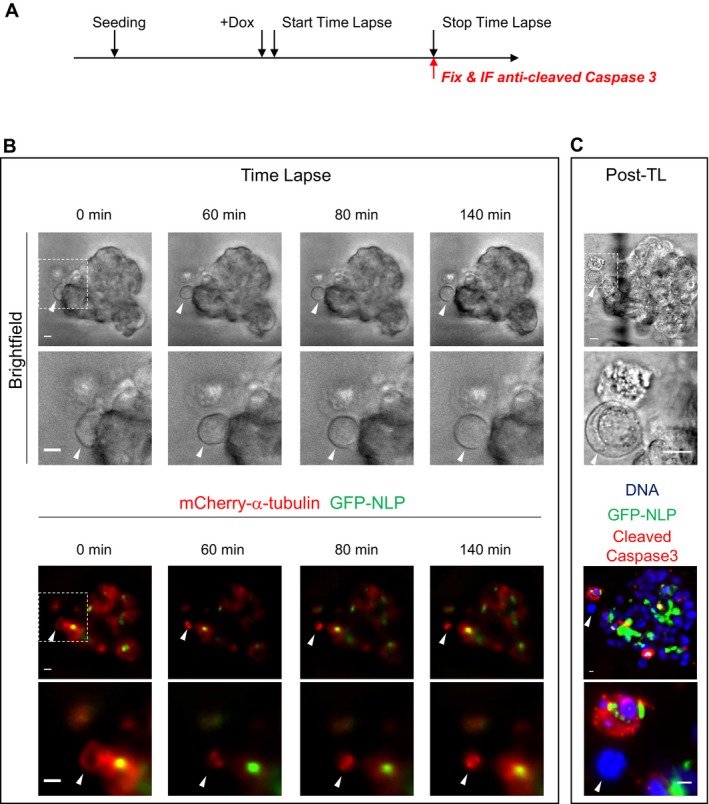
- Schematic description of experiments.
- Still series from time‐lapse experiments showing mitotic cell budding from MCF10A acini overexpressing GFP‐NLP; shown are bright field images (upper panels) and fluorescence images (lower panels) allowing detection of mCherry‐α‐tubulin (red) and GFP‐NLP (green). Dashed squares represent the regions chosen for enlargements that are shown below each bright field and fluorescence image. White arrowheads point to budding cells. Images were acquired every 20 min and time stamps are indicated. Scale bars = 10 μm.
- At the end of time‐lapse imaging, acini were fixed and processed for immunofluorescence using antibodies against cleaved caspase 3 to detect apoptotic cells. Previously recorded acini were identified through use of a gridded area and white arrowheads point to the budding cells that had been monitored. Immunofluorescence images show DNA (blue) GFP‐NLP (green) and cleaved caspase 3 (red). Scale bars = 10 μm.
Both structural and numerical centrosome aberrations destabilize E‐cadherin junctions
Overexpression of NLP caused disordering of E‐cadherin junctions in both growing MCF10A acini (Schnerch & Nigg, 2016) and MDCK cysts (Fig 3A). As shown by Western blotting, this effect cannot be attributed to altered expression of E‐cadherin (Fig 3B), suggesting that excess NLP interferes with E‐cadherin localization. To quantify the observed mislocalization phenotype, we performed immunofluorescence microscopy on 2D cultures of MDCK cells and calculated an E‐cadherin junction strength index (JSI; see Materials and Methods; Appendix Fig S1). While already established E‐cadherin junctions within confluent cell layers showed only minor alterations in response to numerical or structural centrosome aberrations (Fig 3C and D, blue bars), E‐cadherin junctions were strongly reduced when transgenes coding for either NLP or PLK4 were induced in exponentially growing cells before these reached confluence (Fig 3C and D, red bars). This suggests that centrosome aberrations interfered with the establishment of E‐cadherin junctions in proliferating cells, consistent with previous data demonstrating that E‐cadherin remodeling occurs during mitosis (Bauer et al, 1998; Ragkousi & Gibson, 2014).
Figure 3. Centrosome aberrations interfere with the establishment of E‐cadherin junctions through Rac1‐Arp2/3.
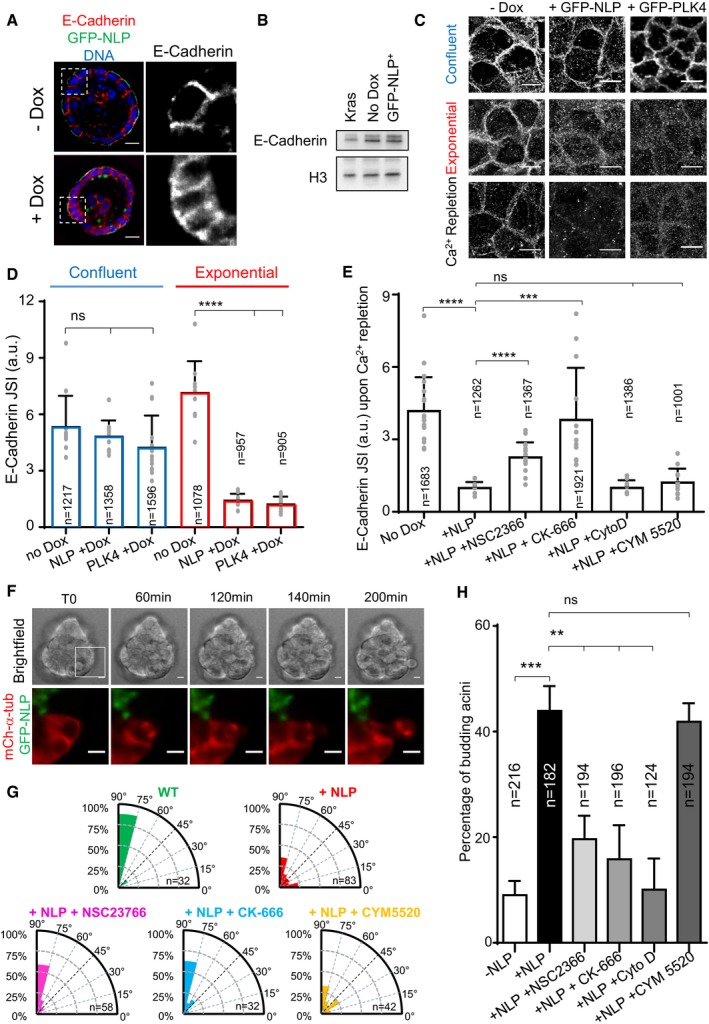
- Representative confocal images showing 8‐day‐old MDCK cysts induced (+Dox) or not (−Dox) to express GFP‐NLP during the last 3 days. Cysts were fixed and stained for E‐cadherin (red) and DNA (blue). Note that GFP‐NLP overexpression induces a broadening of the E‐cadherin signal, as compared to wild‐type cysts. Scale bars = 10 μm.
- Total extracts were prepared from MCF10A monolayer cultures overexpressing GFP‐NLP (GFP‐NLP+), K‐Ras (Kras, for control), or no transgene (No Dox) for 3 days, and analyzed by Western blotting using antibodies against E‐cadherin or histone H3 (loading control).
- Immunofluorescence images show the impact of overexpression of GFP‐NLP or GFP‐PLK4 on E‐cadherin junctions; transgenes were either induced in confluent MDCK cells (upper panels) or in proliferating cells prior to confluence (middle panels). Also shown are images illustrating the impact of GFP‐NLP or GFP‐PLK4 on E‐cadherin remodeling induced by calcium (Ca2+) repletion (lower panels) (for experimental procedure, see Appendix Fig S1). All images are z‐projected stacks of E‐cadherin staining through the entire height of the cells. Scale bars = 10 μm. Note that details on experimental procedures are provided in Appendix Fig S1A. Moreover, larger views of E‐cadherin stainings for non‐induced (No Dox) or GFP‐NLP‐expressing cells (GFP‐NLP) in confluent cultures and after Ca2+ repletion are shown in Appendix Fig S1B.
- Histogram represents the E‐cadherin junction strength index (JSI), as defined in Appendix Fig S1, for MDCK cells induced to express GFP‐NLP (NLP +Dox), GFP‐PLK4 (PLK4 +Dox), or neither transgene (No Dox) during confluence or exponential growth (corresponding to C); bars represent the means of three independent experiments + s.d. and n the numbers of cells analyzed; the values obtained for each field are plotted on the graph. P‐values were derived from unpaired, two‐tailed Student's t‐test. ns indicates not significant, and ****P‐values of < 0.0001.
- Histogram shows the E‐cadherin JSI for calcium repletion experiments performed on confluent MDCK cells expressing GFP‐NLP (+NLP) or not (No Dox), and treated with or without the indicated compounds: NSC2366, inhibitor of Rac1; CK‐666, inhibitor of Arp2/3; cytochalasin D (CytoD), inhibitor of actin polymerization; CYM5520, an agonist of S1PR2. Bars represent the means of three independent experiments + s.d. and n the numbers of cells analyzed; the values obtained for each field are plotted on the graph. P‐values were derived from unpaired, two‐tailed Student's t‐test. ns indicates not significant; ***P < 0.0005 and ****P < 0.0001, respectively.
- Still series from time‐lapse experiment showing extensive rotation of the mitotic spindle during budding from an MCF10A acinus stably expressing mCherry α‐tubulin (red) and induced to express GFP‐NLP (green). Upper panels show bright field images and lower panels the corresponding enlarged fluorescence images as defined by the white square drawn on the bright field image T0 (see Movie EV6). Scale bars = 10 μm.
- Radial histograms illustrate the distributions of the acute angle between the mitotic spindle axis and the radius of the cyst for the different conditions on fixed samples. n represent the number of mitoses analyzed. Note that data for NLP+ cysts include both GFP‐NLP− and GFP‐NLP+ mitotic cells, as results for these two subclasses were virtually indistinguishable. This confirms that spindles rotate in both GFP‐NLP+ and GFP‐NLP− mitotic cells budding from NLP+ cysts (as illustrated in panel F).
- Fraction of budding acini in response to the indicated treatments. Bars represent means + s.d. and n the number of acini from three independent experiments. P‐values were derived from unpaired, two‐tailed Student's t‐test. ns indicates not significant; **P < 0.005 and ***P < 0.0005, respectively.
To obtain information about the E‐cadherin regulatory pathway perturbed by NLP overexpression, we next applied an established calcium repletion procedure (Le et al, 1999) to confluent MDCK cells harboring normal or aberrant centrosomes (Appendix Fig S1). While control cells readily re‐established E‐cadherin junctions upon calcium repletion, cells harboring NLP‐induced structural centrosome aberrations were unable to do so (Fig 3C, bottom row). Numerical centrosome aberrations triggered by PLK4 overexpression also interfered with junction formation (Fig 3C, bottom row), confirming previous observations (Godinho et al, 2014). An excess of growing microtubules was previously shown to destabilize E‐cadherin junctions by altering the cortical actin network via over‐activation of the Rac1‐Arp2/3 effectors (Waterman‐Storer et al, 1999; Akhtar & Hotchin, 2001; Chu et al, 2004; Xue et al, 2013; Godinho et al, 2014). Thus, we asked whether NLP overexpression interfered with this pathway. Indeed, partial reduction of Rac1 or Arp2/3 activity, using the respective inhibitors NSC‐23766 and CK‐666, substantially prevented the NLP‐induced inhibition of E‐cadherin junction formation (Fig 3E). In contrast, no rescue was seen with cytochalasin D, an inhibitor of actin polymerization, or with CYM5520, an agonist of the sphingosine 1 receptor 2 (S1PR2) pathway implicated in basal cell extrusion (Slattum et al, 2014; Hendley et al, 2016; Fig 3E; see Discussion). This argues that NLP overexpression impairs E‐cadherin junctions via increased stabilization of microtubules, which then results in excessive activation of the Rac1‐Arp2/3 pathway. To confirm that mitotic cell–cell contacts are impaired in response to NLP overexpression, we also used time‐lapse microscopy to monitor junction strength in 2D MCF10A cultures (Movies EV4 and EV5). As summarized in Appendix Fig S2, NLP overexpression reduced the time between metaphase onset and the loss of contact between a dividing cell and adjacent cells. This contributes to explain the selective budding of mitotic cells from 3D acini.
E‐cadherin junctions are crucial for the preservation of the orientation of cell divisions within epithelial tissues (den Elzen et al, 2009; Ragkousi & Gibson, 2014; Gloerich et al, 2017). In line with this notion, we observed marked spindle rotation in budding mitotic cells (Fig 3F, Movie EV6). To quantify this rotation phenotype, we also measured spindle orientation on fixed samples. In wild‐type MDCK cysts, the mitotic spindles were preferentially oriented orthogonally to the apico‐basal cell axis, as expected (Fig 3G). In contrast, spindle orientation was randomized in cysts harboring NLP‐induced centrosome aberrations (Fig 3G), corroborating the weakening of interactions between these latter mitotic cells and their neighbors. Partial inhibition of Rac1 (NSC23766) or Arp2/3 (CK‐666) in these cysts restored wild‐type spindle orientation (Fig 3G), confirming that the Rac1‐Arp2/3 pathway regulates E‐cadherin junctions. In contrast, CYM5520, the compound inhibiting basal cell extrusion (Slattum & Rosenblatt, 2014; Hendley et al, 2016), did not rescue the phenotype (Fig 3E–G; see Discussion). We emphasize that the ability of these three inhibitors to restore E‐cadherin junctions and spindle orientation was correlated with their ability to prevent NLP‐induced budding (Fig 3E and H).
Taken together, the above results show that centrosome aberrations interfere with E‐cadherin junctions. Importantly, however, this E‐cadherin phenotype was elicited by both structural and numerical centrosome aberrations (Fig 3A; see also Godinho et al, 2014), yet only NLP overexpression induced budding, while PLK4 overexpression did not (Fig 1B). Conversely, even though cytochalasin D prevented NLP‐induced budding, it did not restore the establishment of E‐cadherin junctions (Fig 3E–H). Taken together, these data demonstrate that weakening of mitotic E‐cadherin junctions is necessary but not sufficient to explain the budding of mitotic cells in response to NLP‐induced centrosome aberrations, implying the involvement of at least one additional mechanism.
NLP‐induced structural centrosome aberrations stabilizes microtubules
As shown by microtubule regrowth experiments, NLP overexpression results in the accumulation of microtubules at enlarged centrosomes (Casenghi et al, 2003; Schnerch & Nigg, 2016). To characterize the properties of the microtubules anchored at these structurally aberrant centrosomes, we explored their orientation and stability. Staining of MDCK cells with antibodies against γ‐tubulin and the minus‐end marker CAMSAP‐2 (Jiang et al, 2014; Akhmanova & Hoogenraad, 2015) revealed that microtubules are trapped via their minus‐ends (Fig 4A and B). Moreover, these microtubules were strongly stained by antibodies against detyrosinated α‐tubulin (Fig 4C and D), indicating that they display enhanced stability (Schulze et al, 1987; Webster et al, 1987; Kerr et al, 2015). In striking contrast, overexpression of PLK4 or CEP68 did not trigger a significant increase in detyrosinated α‐tubulin (Fig 4C and D). This falls in line with previous observations indicating that although PLK4 overexpression stimulates microtubule nucleation, these microtubules remain dynamic (Godinho et al, 2014). These results suggest that differences in microtubule stability might contribute to explain the ability of NLP‐ but not PLK4‐induced centrosome aberrations to cause budding.
Figure 4. NLP overexpression sequesters microtubule minus‐ends and stabilizes microtubules.
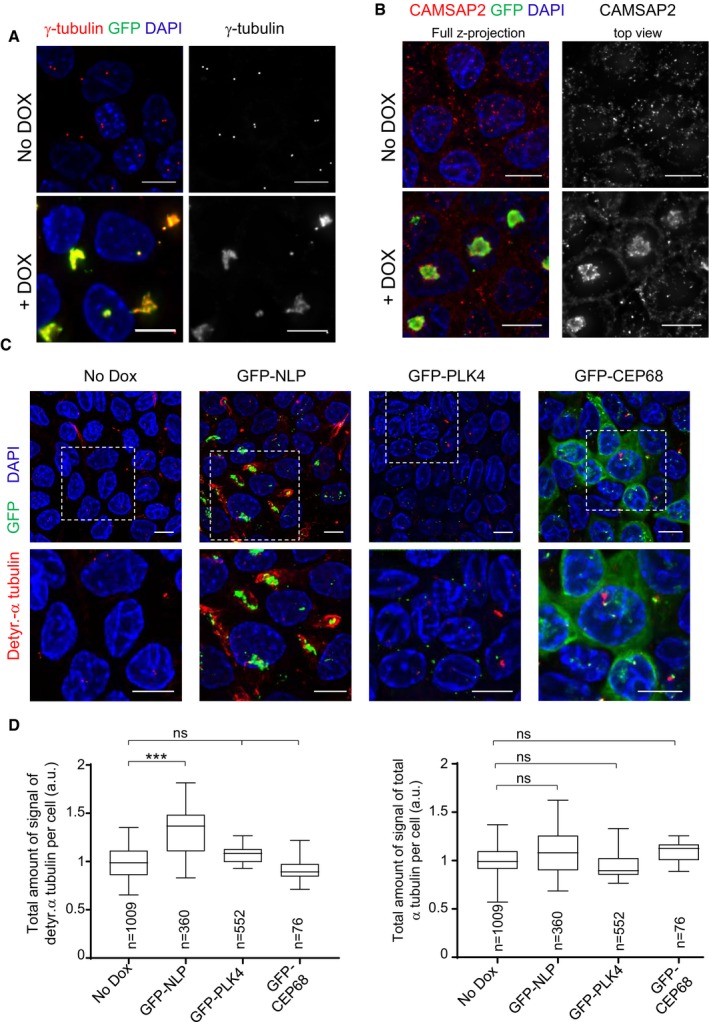
- Representative immunofluorescence images of MDCK cells show that aberrant centrosomes triggered by doxycycline‐induced GFP‐NLP overexpression (green; lower panels) over‐recruit γ‐tubulin (red), as compared to uninduced (No Dox) cells (upper panels). DNA was stained with DAPI. Scale bars = 10 μm.
- Representative immunofluorescence images show of MDCK cells that aberrant centrosomes triggered by doxycycline‐induced GFP‐NLP overexpression (green; lower panels) sequester the microtubule minus‐end‐binding protein CAMSAP2 (red), as compared to uninduced (No Dox) cells (upper panels). DNA was stained with DAPI. Scale bars = 10 μm.
- Representative immunofluorescence images of MDCK cells show that structural centrosome aberrations induced by GFP‐NLP overexpression (green), but not numerical aberrations induced by GFP‐PLK4 overexpression (green), trigger microtubule stabilization, as revealed by increased staining for detyrosinated α‐tubulin (red). Non‐induced cells (No Dox) or cells induced to express GFP‐CEP68 (which triggers no detectable centrosome aberrations) are shown for control. DNA was stained with DAPI. Dashed white squares mark the regions chosen for blow ups shown below each image. Scale bars = 10 μm.
- Histograms show the quantification of detyrosinated‐α‐tubulin (left) and total α‐tubulin (right) per cell, as deduced from analysis of immunofluorescence signals. Data are compiled from MDCK cells induced to express GFP‐NLP, GFP‐PLK4, GFP‐CEP68, or the corresponding non‐induced cells (No Dox). n indicates the number of cells analyzed in two independent experiments. Box plots show the mean (square) and median (line); whiskers are s.d. and the box is s.e.m. Statistical significance was tested using a Mann–Whitney test. ***P < 0.005; ns indicates not significant.
Budding represents a non‐cell‐autonomous process
Because not all cells express the GFP‐NLP transgene product upon induction (Appendix Figs S3 and S4), we asked whether the extent of budding could be correlated to the percentage of GFP‐NLP+ cells within mosaic acini. After estimating the proportion of GFP‐NLP+ cells per acinus (for details see Materials and Methods), these were classified into four classes differing in the proportion of GFP‐NLP+ cells, and the frequency of budding was determined for each class (Fig 5A). This analysis revealed that the frequency of budding is not proportional to the percentage of GFP‐NLP+ cells. Instead, extensive budding is triggered once the proportion of GFP‐NLP+ cells exceeds a threshold (> 50%), strongly suggesting that budding requires multicellular cooperation within the epithelium. Furthermore, no correlation was observed between the expression of GFP‐NLP within budding cells and the percentage of GFP‐NLP+ cells within the corresponding acini. Analysis of budding cells for the presence or absence of GFP‐NLP revealed that cells lacking GFP‐NLP were overrepresented, when compared to theoretical predictions (Fig 5B; for illustration see Fig EV3). This demonstrates that budding reflects a non‐cell‐autonomous mechanism and indicates that GFP‐NLP− cells are preferentially squeezed out over GFP‐NLP+ cells. This may establish a positive feed‐back loop that progressively enriches the epithelium with GFP‐NLP+ cells and favors the dissemination of mitotic cells that do not themselves harbor centrosome aberrations.
Figure 5. Budding of mitotic cells requires multicellular cooperation and reflects a non‐cell‐autonomous process.
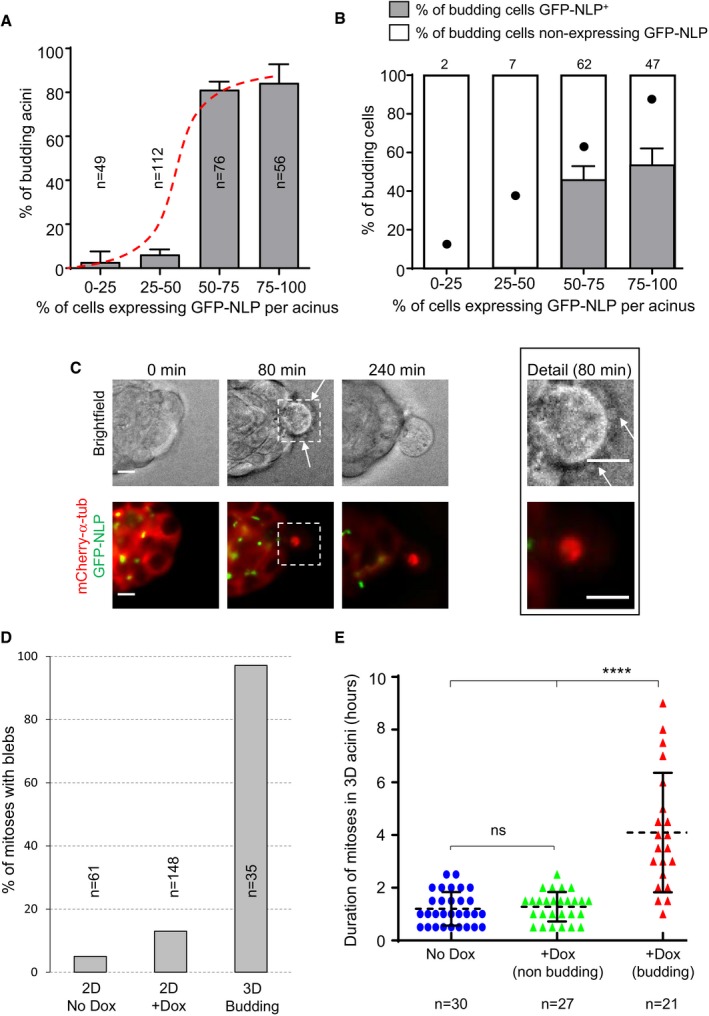
- Fraction of budding MCF10A acini as a function of the estimated percentage of GFP‐NLP+ cells. Acini were grouped into four classes, depending on the percentage of GFP‐NLP+ cells within each acinus. As illustrated by the sigmoidal curve (red dashed curve), the frequency of budding is not proportional to the percentage of GFP‐NLP+ cells within each group, but instead requires a threshold (> 50%) above which budding occurs with high frequency. Bars represent the means of four independent experiments + s.d. (totally 293 acini; the number of acini analyzed for each class is indicated in each bar).
- Histogram distinguishes budding cells for each class (as in A) according to whether or not they express GFP‐NLP. Each bar indicates the percentage of budding cells that lack a detectable GFP‐NLP signal (white) as well as the percentage of GFP‐NLP+ cells (gray). For calibration, filled dots indicate the percentages of GFP‐NLP+ budding cells that would be expected if budding occurred in proportion to the average frequency of such cells in the acini of the corresponding class (e.g., 12.5% for the class 0–25, 87.5% for the class 75–100). Bars represent the means of four independent experiments + s.d. Data were compiled from 293 MCF10A acini (as in A) displaying at least one budding event (totally 118 budding cells). Absolute numbers of budding cells for each class are indicated above each bar. Note that budding was a very rare event for the acini showing fewer than 50% of GFP‐NLP‐positive cells (see A), and therefore, the numbers of budding cells were also very small. Regarding the acini expressing more than 50% of NLP‐positive cells, two‐tailed Student's t‐tests confirm the statistical significance of the observed bias in favor of dissemination of GFP‐NLP‐negative cells, yielding P < 0.001 and < 0.005 for the acini classes 50–75% and 75–100%, respectively.
- Representative stills from time‐lapse recordings show a mitotic cell budding from an MCF10A acinus stably expressing mCherry α‐tubulin (red) and induced to express GFP‐NLP (green). Upper panels show bright field images and lower panels the corresponding fluorescence images; arrows point to membrane blebs. Images were acquired every 20 min, and time stamps are indicated; scale bars = 10 μm.
- Fraction of mitotic cells associated with extensive blebbing in different MCF10A populations observed by time‐lapse for 2 days. The graph compares MCF10A cells cultured in a 2D monolayers without (2D No Dox) or after induction of GFP‐NLP expression (2D + Dox) and budding cells from 3D acini (3D Budding). Bars are means of two independent experiments for the 2D cultures and of 16 independent experiments for the 3D acini; n represents the number of mitoses analyzed.
- Scatter plot shows the mitotic duration, determined from time‐lapse experiments, of cells dividing within MCF10A acini. The graph compares mitoses in acini without GFP‐NLP induction (No Dox, blue circles) and mitoses within acini expressing GFP‐NLP (+Dox) that either undergo budding (budding, red triangles) or not (non‐budding, green triangles). Error bars represent ± s.d. of the means, and n shows the numbers of mitoses analyzed. Statistical significance was tested using a Mann–Whitney test. ****P < 0.0001; ns indicates not significant.
Budding mitotic cells displayed two striking characteristics. First, almost all budding cells monitored by time‐lapse microscopy displayed extensive membrane blebbing (Fig 5D). This blebbing was also seen in budding cells that lacked detectable GFP‐NLP (Fig 5C, Movie EV7, indicating that it is not a cell‐autonomous consequence of NLP overexpression. Corroborating this conclusion, NLP overexpression induced only a minor increase in the blebbing of mitotic cells in 2D cultures (13% vs. 5% for control cells), in stark contrast to the blebbing seen in 97% of cells budding from acini (see Fig 5D). Second, budding cells showed markedly extended M phase durations and typically remained in metaphase for several hours (Figs 2B and 5E). In contrast, non‐budding mitoses within acini overexpressing NLP lasted approximately 1 h, similar to the duration of mitoses in non‐induced controls or 2D cultures (Fig 5E; Appendix Fig S4). This demonstrates that both blebbing and prolonged mitoses are related to budding rather than NLP overexpression per se.
Budding is governed by biomechanical properties of epithelia
Both blebbing and delayed mitotic progression have recently been observed in confined mitotic cells exposed to increasing pressure (Cattin et al, 2015). This led us to postulate that cells harboring structural centrosome aberrations might display altered biomechanical properties. If such cells were to display increased stiffness, epithelia containing a sufficient proportion of GFP‐NLP+ cells might confine mitotic cells and eventually squeeze them out. Support for this notion stems from the observation that excess NLP triggers an increase in detyrosinated α‐tubulin (Fig 4C and D), which is known to correlate with enhanced microtubule stability and increased cellular stiffness (Schulze et al, 1987; Webster et al, 1987; Kerr et al, 2015). To further explore this hypothesis, we used atomic force microscopy (AFM) to map the mechanical stiffness of various cell populations. Indeed, when compared to cells overexpressing either PLK4 or CEP68 within a confluent MDCK monolayer, interphase cells overexpressing NLP showed a significant increase in cellular stiffness (Figs 6A and EV4A). Furthermore, probing the basal surface of MDCK 3D cysts revealed a similar increase in cellular stiffness in response to NLP overexpression (Fig 6B). For comparison, we also measured cellular stiffness within confluent MDCK monolayers after application of different drugs acting on the cytoskeleton (Fig 6C). Treatment with taxol, a stabilizer of microtubules, also increased stiffness, in line with previous results (Kerr et al, 2015). In wild‐type cells, the taxol‐induced stiffness was comparable to that seen upon overexpression of NLP, but in GFP‐NLP+ cells, which already contain stabilized microtubules (Fig 4; Schnerch & Nigg, 2016), taxol produced only a minor additional effect. Conversely, inhibition of microtubule polymerization by nocodazole did not affect wild‐type cells and slightly reduced cellular stiffness in GFP‐NLP+ cells (Fig 6C). The relatively minor but significant effect falls in line with immunofluorescence experiments showing that nocodazole treatment caused only partial depolymerization of microtubules in confluent interphase cells [Fig EV4B; see also (Pepperkok et al, 1990)]. Finally, inhibition of actin polymerization by cytochalasin D reduced the stiffness of both control cells and GFP‐NLP+ cells (Fig 6C), confirming that actin exerts a major contribution to cellular stiffness (Fletcher & Mullins, 2010; Bruckner & Janshoff, 2015). Taken together, these results support the notion that NLP‐induced centrosomal aberrations increase cellular stiffness by stabilizing microtubules, which in turn influences the actin cytoskeleton.
Figure 6. Structural but not numerical centrosome aberrations increase stiffness of epithelial cells.
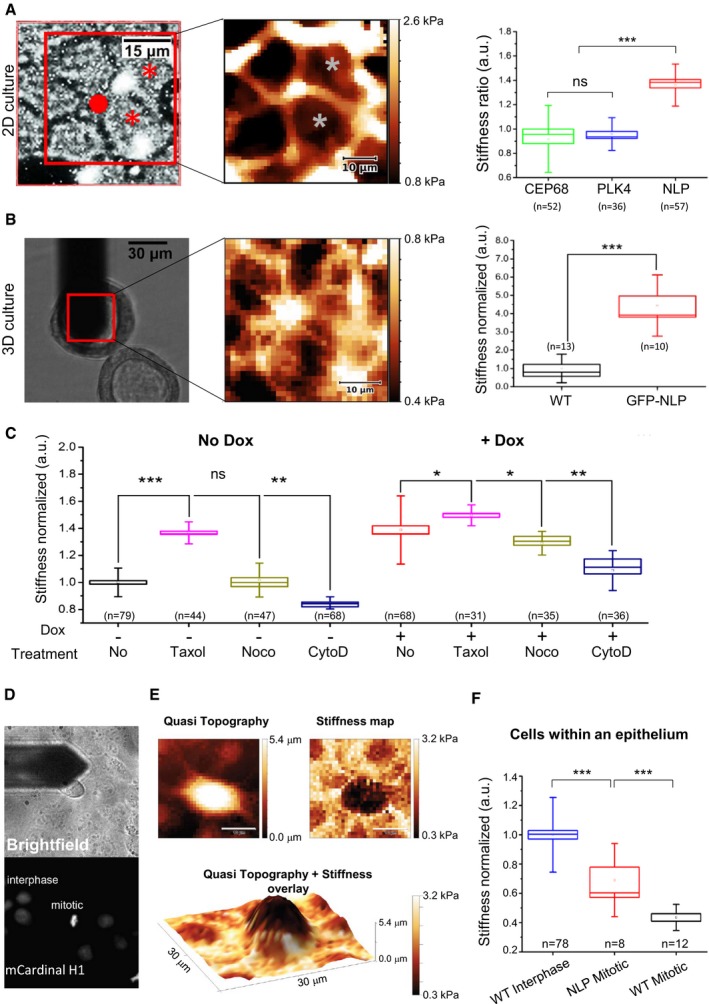
- AFM performed on MDCK cells cultured in 2D. (Left) Example of epifluorescence microscopy image; cells harboring structural centrosome aberrations (GFP‐NLP) are marked (asterisks). The scan area (red square) and position of the AFM probe (red dot) are monitored in the bright field signal for each experiment. (Middle) Stiffness map visualizes local nanomechanical heterogeneities. As seen in epifluorescence, corresponding cells that overexpress GFP‐NLP are marked by asterisks. (Right) Stiffness ratios compare cells overexpressing NLP, PLK4 or CEP68 with neighboring non‐expressing cells (WT), see Materials and Methods section; NLP overexpressing cells are stiffer than WT cells while cells overexpressing either PLK4 or CEP68 exhibit mechanical phenotypes similar to WT cells.
- AFM performed on 3D MDCK cysts. (Left) Bright field image showing the AFM probe positioned above the isolated cyst as marked by the red square. (Middle) Stiffness map recorded within the red square visualizes the basal surface of individual cells. (Right) Quantitative analysis confirms that NLP overexpression induces cell stiffening in 3D.
- Drug‐induced changes of cytoskeletal structures alter cellular stiffness of WT (No Dox) or GFP‐NLP+ (+Dox) MDCK cells in 2D.
- Cell cycle dependence of cellular stiffness. Position of the AFM probe is monitored in the bright field signal (top) while corresponding detection of condensed chromatin mCardinal‐histone H1 by epifluorescence identifies mitoses from neighboring interphase MDCK cells (bottom). Images are 90 × 90 μm.
- Differences in stiffness between mitotic and neighboring interphase MDCK cells in confluency. (Left) 2D quasi topography map reveals the elongated shape of the mitotic cell that protrudes out of the confluent layer. (Right) 2D stiffness map reveals that the mitotic cell is significantly softer than the surrounding interphase cells. (Lower panel) An overlay map of quasitopography and stiffness visualizes the same soft mitotic cell as in (D). Scale bars = 10 μm.
- Quantitative AFM analysis of confluent MDCK cells in interphase or in mitosis, in comparison with surrounding cells in interphase that express GFP‐NLP (NLP) or not (WT).
Figure EV4. Impact of cell cycle and cytoskeleton on stiffness of 2D MDCK cells.
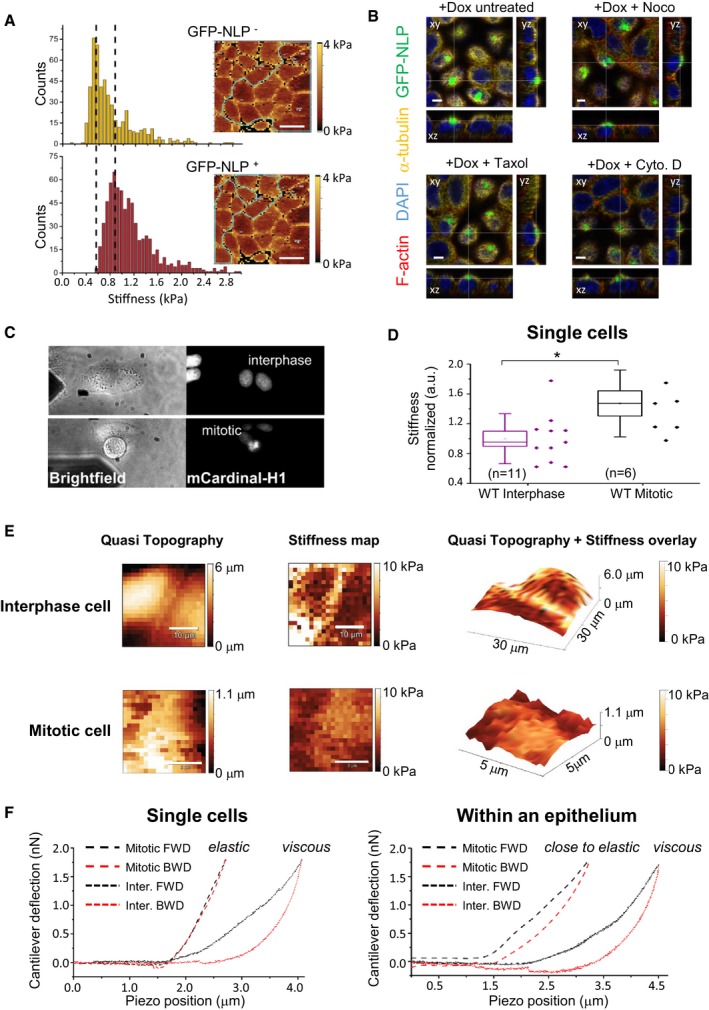
- Exemplary stiffness histograms of MDCK cells (such as in Fig 6A), comparing GFP‐NLP− cells (top) and GFP‐NLP+ MDCK cells (bottom) in the same 2D culture. Counts report the number of force curves per bin. (Insets) Corresponding stiffness maps show cells (marked by green lines) for GFP‐NLP− cells (top) and GFP‐NLP+ cells (bottom) in the same 2D culture. Note the increased stiffness of the GFP‐NLP+ cells (peak shift to the right), as illustrated by the dotted lines.
- Confocal microscopy experiments visualize drug‐induced changes in the actin and microtubule cytoskeletons of GFP‐NLP+ cells; shown are confocal slices with top views (xy; main panels) and lateral views (xz, lower panels, and yz, right hand panels). Thin white lines refer to the positions of the optical sections. Scale bars = 5 μm.
- Single (non‐confluent) wild‐type MDCK cells are depicted in the bright field signal with the AFM probe located in proximity (left). Interphase cells (top) are differentiated from mitotic cells (bottom) that show a positive epifluorescence signal for mCardinal‐H1 (right). Bright field and epifluorescence images are 90 × 45 μm.
- Stiffness of single MDCK cells in interphase or mitosis. Note that mitotic cells are significantly stiffer than interphase cells. All box plots show the mean (square) and median (line); the whiskers are s.d., and the box is s.e.m. The statistical significance was tested using a Mann–Whitney test. *P < 0.05.
- Upper row shows representative maps of quasi topography (left), cellular stiffness (middle), and the quasitopography/stiffness overlay (right) of interphase single cells; scale bars = 10 μm. Lower row shows representative maps of quasi topography (left), stiffness (middle), and the quasitopography/stiffness overlay (right) of isolated mitotic cell; scale bars = 2 μm. Note that analyzed areas were limited to the top of the mitotic cells (ca. 25 μm2) to ensure correct measurements and avoid any bias caused by the increase of cortex curvature induced by mitotic rounding. Scale bars = 10 μm (upper panels); 2 μm (lower panels).
- Representative loading (forward/FWD, black dashed lines) and unloading (backward/BWD, red dashed lines) force curves highlight differences in the elastic and viscous regimes for single (left) versus confluent (right) cells in mitosis (Mitotic) or in interphase (Inter.). Individual mitotic MDCK cells are purely elastic whereas mitotic cells in confluency tend to be more viscous.
As budding selectively involves mitotic cells, we next compared the stiffness of interphase and mitotic cells. Specifically, we analyzed stiffness in near‐confluent monolayers of MDCK cells expressing mCardinal‐histone H1 to allow monitoring of cell cycle phases (Fig 6D). Interphase cells were generally stiffer than mitotic cells and overexpression of NLP increased the stiffness of the latter (Fig 6E). These results fall in line with previous in situ observations, suggesting that the presence of soft cells in tumor biopsies correlates with metastatic spreading (Swaminathan et al, 2011; Lekka et al, 2012; Plodinec et al, 2012), and they suggest that the higher stiffness of GFP‐NLP+ mitotic cells contributes to explain the preferential budding of wild‐type mitotic cells (Fig 5B; Cadart et al, 2014). However, these results seemed to conflict with earlier data showing that cells become stiffer when entering mitosis (Kunda et al, 2008; Stewart et al, 2011; Chugh et al, 2017). We suspected that this difference might relate to the fact that previous measurements were performed on isolated single cells lacking cell–cell contact. Indeed, we could readily confirm that isolated MDCK cells going through mitosis display a higher stiffness than interphase cells (Fig EV4). These data demonstrate that cellular stiffness is drastically influenced by physical constraints, notably the confinement of cells within an epithelium (Saw et al, 2017). Collectively, our data lead us to propose that centrosome aberrations trigger not only cytoskeletal remodeling but also heterogeneity in the biomechanical properties of epithelia, which then results in the selective budding of mitotic cells through a non‐cell‐autonomous process (see schematic models in Figs 7 and EV5).
Figure 7. Structural centrosome aberrations trigger mitotic cell budding through non‐cell‐autonomous mechanism.
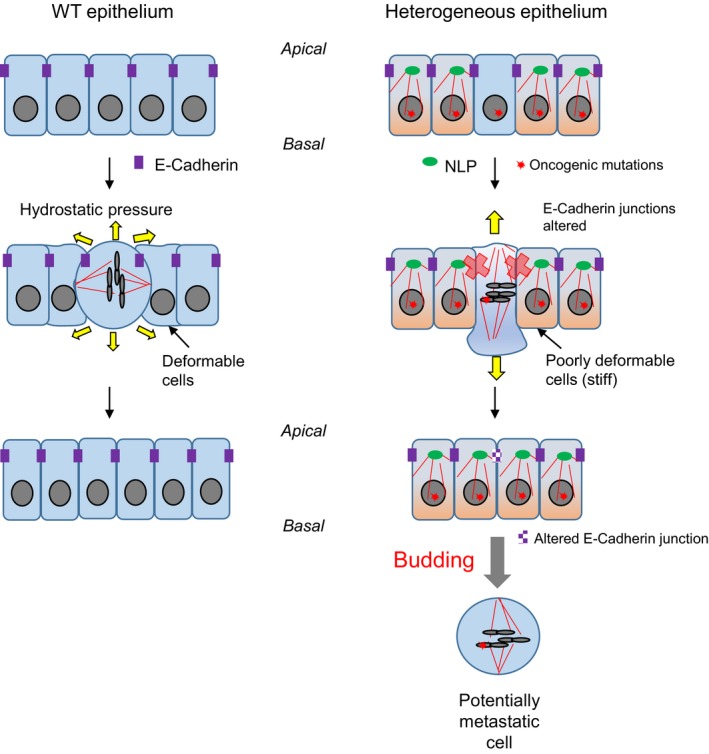
This schematic model presents the key conclusions emerging from the present study, including plainly speculative elements. In wild‐type epithelia (left), cell division does not impair epithelial integrity. Normal E‐cadherin dynamics ensures correct positioning of the mitotic spindle and maintenance of cell–cell junctions. When mitotic cells exert hydrostatic pressure against confinement, the surrounding cells are sufficiently “deformable” to accommodate the products of cell division in the plane of the epithelium. In striking contrast, in mosaic epithelia comprising cells with NLP‐induced structural centrosome aberrations, E‐cadherin junctions are impaired and, consequently, spindle orientation randomized. Furthermore, cells harboring such structural centrosome aberrations show increased stiffness, and their reduced deformability is expected to cause resistance against insertion of dividing cells. As a consequence, mitotic cells escape the epithelium through budding. We emphasize that this non‐cell‐autonomous process offers a solution to the long‐standing conundrum that many tumors comprise substantial subpopulations of cells with centrosome aberrations, even though these aberrations are expected to impair cell viability. Specifically, we propose that the non‐cell‐autonomous mechanism described here triggers the budding of potentially invasive cells in response to centrosome aberrations within the tumor. Importantly, these disseminating cells need not necessarily harbor any centrosome aberrations themselves, even though they may carry oncogenic mutations, including mutations that favor metastasis.
Figure EV5. Structural centrosome aberrations trigger budding of mitotic cells through decreasing E‐cadherin junctions and increasing cellular stiffness.
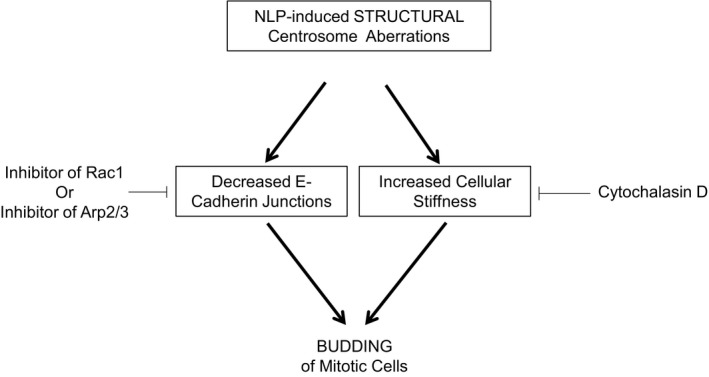
Scheme shows that NLP‐induced structural centrosome aberrations favor budding of mitotic cells through two distinct pathways that are both necessary but not sufficient. On the one side, NLP‐induced centrosome aberrations induce a decreased of E‐cadherin junctions that can be rescued by partial inhibition of the Rac1 or Arp2/3 activities, which consequently prevents budding. On the other side, budding also requires an increased of cellular stiffness that can be restored by depolymerizing the actin network by a cytochalasin D treatment. Such a treatment can also prevent budding induced by NLP overexpression.
Discussion
Centrosome aberrations are common in human tumors (Lingle et al, 1998; Guo et al, 2007; Chan, 2011), but their role in carcinogenesis remains subject to intense debate (Nigg, 2002; Godinho & Pellman, 2014; Gonczy, 2015; Raff & Basto, 2017). Studies in cells and animals support the view that centrosome amplification induces aneuploidy through chromosome mis‐segregation (Ganem et al, 2009; Levine et al, 2017), but other possible contributions to cancer development have also been proposed (Basto et al, 2008; Godinho et al, 2014; Coelho et al, 2015; Sercin et al, 2016; Kazazian et al, 2017). Yet, a conundrum persists in that centrosome aberrations are a priori expected to impair the viability of those tumor cell subpopulations that harbor these aberrations. Thus, the functional significance of centrosome aberrations in human tumors remains difficult to ascertain. Our study identifies a novel mechanism through which cells harboring centrosome aberrations may contribute to promote an invasive phenotype through a non‐cell‐autonomous process, thereby offering a solution to the above conundrum. Specifically, we propose a model with the potential to explain how centrosome aberrations could contribute to metastasis, without the disseminating cells carrying these deleterious alterations (Figs 7 and EV5).
We show that structural centrosome aberrations, induced by overexpression of NLP (Casenghi et al, 2003; Schnerch & Nigg, 2016), trigger the selective budding of mitotic cells from 3D epithelial acini. Furthermore, we identify two complementary mechanisms supporting this cell dissemination: first, increased microtubule stability in GFP‐NLP+ cells causes a weakening of E‐cadherin junctions between mitotic cells and their neighbors. Second, cytoskeletal aberrations induce heterogeneity in the biomechanical properties (stiffness) of cells, causing extensive blebbing and marked delays in mitotic progression, indicative of confinement (Cattin et al, 2015; Sorce et al, 2015). Consequently, when epithelia contain cells whose stiffness is increased by centrosome aberrations, soft mitotic cells are selectively squeezed out. Considering that mitotic cells devoid of centrosome aberrations are softer than GFP‐NLP+ mitotic cells, the former are expected to offer reduced resistance to extrusion forces (Cadart et al, 2014; Sorce et al, 2015) and hence bud preferentially, as observed.
In recent years, an epithelial delamination process known as “basal cell extrusion” has attracted increasing attention (Slattum et al, 2009; Marshall et al, 2011; Slattum & Rosenblatt, 2014). This process contributes to remove dying or unwanted cells from epithelia (Eisenhoffer et al, 2012; Slattum et al, 2014) and has also been proposed to play a role in tumor cell invasion (Slattum & Rosenblatt, 2014). However, although the budding mechanism described here can formally be considered as a form of basal extrusion, we emphasize that the underlying mechanism differs in several fundamental aspects from the basal cell extrusion phenomenon pioneered by Rosenblatt and coworkers. First, basal extrusion of dying cells is based on constriction of an actomyosin ring (Slattum et al, 2009, 2014; Marshall et al, 2011) and considering that apical activation of actomyosin contractility is incompatible with mitosis (Grosshans & Wieschaus, 2000; Mata et al, 2000; Seher & Leptin, 2000), such a mechanism is not consistent with the budding of mitotic cells. Second, basal cell extrusion is inhibited by CYM5520, an agonist of S1PR2 (Hendley et al, 2016), while we show here that this compound does not prevent budding (Fig 3). Finally, while basal cell extrusion is a cell‐autonomous phenomenon (Slattum et al, 2014), budding represents a non‐cell‐autonomous phenomenon based on multicellular cooperation. Hence, cell dissemination by budding is mechanistically distinct from basal cell extrusion.
Ninein‐like protein is frequently overexpressed in different human cancers (Qu et al, 2008; Yu et al, 2009; Shao et al, 2010) and reported to confer resistance to paclitaxel in breast cancer (Zhao et al, 2012). Moreover, NLP‐induced centrosome aberrations in 2D and 3D culture models recruit γ‐tubulin and centrin (Casenghi et al, 2003; Schnerch & Nigg, 2016), highly reminiscent of the aberrations seen in human tumors (Lingle et al, 1998; Salisbury et al, 2004; Guo et al, 2007). Accordingly, we used NLP to study the impact of structural centrosome aberrations on epithelial architecture and invasiveness. In future, it will be interesting to determine whether other centrosomal proteins affecting the dynamics of the microtubule network (Delaval & Doxsey, 2010; Fogeron et al, 2013) trigger a similar invasive phenotype. It is tempting to speculate that any cellular alteration able to exert comparable effects on both E‐cadherin junctions and the biomechanical properties of epithelia may trigger the dissemination of mitotic cells through a similar non‐cell‐autonomous mechanism.
The dissemination of individual cells or cell clusters, accompanied by complete or partial epithelial–mesenchymal transition (Nieto et al, 2016), is considered a first key step in metastasis (Aceto et al, 2014; Lambert et al, 2017). Moreover, the importance of short‐range dispersal of individual tumor cells is well recognized (Waclaw et al, 2015). A complete understanding of cell dissemination will ultimately require a combination of intra‐vital imaging (Alexander et al, 2013; Paul et al, 2017) and simulation of invasive processes using defined in vitro systems (Shamir & Ewald, 2014; Discher et al, 2017). Results obtained with established 2D and 3D culture models lead us to propose a novel mechanism through which centrosome aberrations could trigger the dissemination of incipient tumor cells. Extrapolating to an in vivo situation, our findings have several implications. First, they bear on the question of when disseminating cancer cells first arise (Ghajar & Bissell, 2016). Considering that centrosome aberrations can be observed already in pre‐malignant lesions, the mechanism proposed here would allow dissemination of cells with metastatic potential from very early tumors, in line with recent proposals (Harper et al, 2016; Hosseini et al, 2016). Second, we conclude that cytoskeletal aberrations affecting tissue architecture need not necessarily occur within the same cells that harbor oncogenic mutations, offering a new explanation for how centrosome aberrations could contribute to aggressive cancer development in spite of being a priori deleterious. Third, the non‐cell‐autonomous nature of the observed process implies that aberrations conferring metastatic properties may not necessarily be detectable within the disseminating cancer cells themselves, implying that drivers of metastasis may escape detection by genetic methods comparing metastatic cells with primary tumor cells. Collectively, our data contribute to focus attention on the microenvironment surrounding tumors cells (Bissell & Hines, 2011; Tabassum & Polyak, 2015) and on the biomechanical properties of tumor tissues (Swaminathan et al, 2011; Lee et al, 2012; Plodinec et al, 2012). In particular, our data support previous observations suggesting that metastatic spreading correlates with the presence of low stiffness cells within tumor biopsies (Swaminathan et al, 2011; Plodinec et al, 2012). Finally, our findings may also have implications for normal development. In particular, it will be interesting to explore whether differences in stiffness could contribute to trigger developmentally controlled epithelial invaginations for which mitotic progression appears to be a pre‐requisite (Kondo & Hayashi, 2013).
Materials and Methods
Generation of expression constructs and cell lines
cDNA encoding mCherry‐α‐tubulin, mCardinal‐H1, and dTomato‐VE‐cadherin were PCR‐amplified from pmCherry_α_tubulin_IRES_puro2 [kindly provided by Daniel Gerlich (Steigemann et al, 2009)], mCardinal‐H1‐10 [a gift from Michael Davidson (Chu et al, 2014; Addgene plasmid # 56161)], and tdTomato‐VE‐cadherin‐N‐10 (a gift from Michael Davidson (Addgene plasmid # 58142), respectively, and subsequently ligated into pMXs‐IRES‐Blasticidin (Cell Biolabs Inc.).
MCF10A ecoR cell lines allowing doxycycline‐inducible expression of EGFP‐NLP, EGFP‐CEP68 and EGFP‐PLK4 were described previously (Schnerch & Nigg, 2016). Doxycycline‐inducible MDCK II cells were generated using the same two‐step transduction strategy and enriched by antibiotic selection using hygromycin at 400 μg ml−1 and puromycin at 2 μg ml−1. Inducible MDCK and MCF10A cells stably expressing mCherry‐α‐tubulin, dTomato‐E‐cadherin and mCardinal H1 were generated by retroviral transduction and sorted by flow cytometry using BD FACSAria IIIu cell sorter (FACS Core Facility of Biozentrum). MCF10A ecoR cells stably expressing K‐Ras were obtained by ecotropic retroviral transduction with pBabe K‐Ras 12V [Addgene plasmid #12544, gift from Channing Der (Khosravi‐Far et al, 1996)].
Cell culture
MCF10A ecoR cells (a kind gift from Tilman Brummer; University of Freiburg) were grown as described previously (Debnath et al, 2003). Briefly, MCF10A cells were grown in DMEM:F12 (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 10 μg ml−1 insulin (Sigma‐Aldrich, St. Louis, MO, USA), 0.5 μg ml−1 hydrocortisone (Sigma‐Aldrich, St. Louis, MO, USA), 100 ng ml−1 cholera toxin (Sigma‐Aldrich, St. Louis, MO, USA), and penicillin–streptomycin (Life Technologies, Carlsbad, CA, USA), 2% or 5% horse serum (Life Technologies, Carlsbad, CA, USA) and 5 ng ml−1 or 20 ng ml−1 epidermal growth factor (PeproTech, London, UK) for 3D and 2D culture, respectively. MDCK II cells (a kind gift from Inke Naethke, University of Dundee, UK) were grown in Minimum Essential Medium Eagle (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 10% fetal calf serum (GE Healthcare, Chicago, Illinois, USA) and penicillin–streptomycin (Life Technologies, Carlsbad, CA, USA). Phoenix cells and HEK293T cells (provided by Stefan Zimmermann and Ralph Wäsch; University Medical Center Freiburg) were grown in DMEM medium (Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (GE Healthcare, Chicago, Illinois, USA), sodium pyruvate (Life Technologies, Carlsbad, CA, USA), and penicillin–streptomycin (Life Technologies, Carlsbad, CA, USA). Tissue cultures were routinely tested for mycoplasma contamination by PCR using growth medium from high‐density cultures as a template. MCF10A acini and MDCK II cysts were generated by plating single cell solutions onto beds of pure Matrigel (356231, Corning) or Matrigel enriched by collagen I (1.6 mg ml−1, Life Technologies, Carlsbad, CA, USA), as described previously (Debnath et al, 2003; Godinho et al, 2014). To monitor invadopodia formation, MCF10A cells were seeded in collagen I‐enriched Matrigel and transgene expression was induced by doxycycline the day following seeding.
Compounds
Expression of transgenes coding for EGFP‐tagged centrosomal proteins was initiated by addition of 2.5 μg ml−1 of doxycycline. Inhibitors used in 3D acini experiments and for determination of E‐cadherin JSIs were used at the following concentrations: 25 μM NSC23766 (Sigma‐Aldrich MO, USA), 50 μM CK‐666 (Sigma‐Aldrich MO, USA), 2.5 μM cytochalasin D (Sigma‐Aldrich MO, USA), 5 μM RO‐3306 (Merck Millipore Darmstadt Germany), 2 mM Thymidine (Sigma‐Aldrich MO, USA), 10 μM of CYM 5520 (Sigma‐Aldrich MO, USA). For rescue experiments, doxycycline and drugs were added simultaneously to medium.
Fluorescence microscopy
2D monolayers of MDCK II or MCF10A cells were grown and processed for immunolabeling in Ibidi 8‐well microscopy slides, and 3D acini derived from MCF10A or MDCK cells were grown in eight‐chamber slides (354108, Falcon Corning) and processed for immunostaining essentially as described previously (Debnath et al, 2003; Schnerch & Nigg, 2016). Briefly, cells and cysts were fixed using 2% formalin or 4% paraformaldehyde in PBS for 15 min at room temperature. Cells were permeabilized using PBS 0.5% Triton X‐100 for 5 min and blocked in 2% BSA in PBS for 30 min and antibodies diluted in PBS‐0.5% Tween‐2%BSA. Primary antibodies used were anti‐α‐tubulin (T9026, Sigma, St. Louis, Mo, USA), anti‐cleaved caspase 3 (D175; 9661, Cell Signaling Technology) and anti‐E‐cadherin (610182, BD, Franklin Lakes, NJ, USA). F‐actin fibers were stained using Alexa Fluor 647‐linked phalloidin at 33 nM (A22287, Life Technologies, Carlsbad CA, USA) and DNA stained with 1 μg ml−1 DAPI. For stainings of γ‐tubulin (T6557, Sigma, St. Louis, Mo, USA), CAMSAP2 (17880‐1‐AP, Proteintech, Chicago, USA), detyrosinated‐α‐tubulin (ab48389, Abcam, Cambridge, U.K.), and α‐tubulin in cells cultured in 2D, fixation, and permeabilization were achieved by incubating cells in ice‐cold methanol for 5 min at −20°C. Secondary antibodies were Alexa Fluor 647 goat anti‐mouse (A21236), Alexa Fluor 568 goat anti‐mouse (A11004), and Alexa Fluor 568 donkey anti‐rabbit (A10042) (all from Life Technologies, Carlsbad CA, USA). 3D acini were finally mounted in ProLong Antifade mounting medium (Molecular Probes) and analyzed rapidly.
Confocal images were acquired using a Leica SP5‐II‐MATRIX point scanning confocal microscope equipped with a 20×/0.70 HCX Plan Apo CS air objective and a 63×/1.40–1.60 HCX Plan Apo lambda blue oil immersion objective. 405 nm diode laser light was applied for DAPI staining, 488 nm Argon laser light for visualization of GFP, 561 nm DPSS laser light for visualization of Alexa Fluor 568 stainings, and 633 nm HeNe laser light for visualization of Alexa Fluor 647 stainings. Image analyses and final adjustments of confocal images taken in the sphere equator planes were carried out in Omero 5.1.2. Image analyses, 3D reconstructions, and final adjustments of confocal z‐stacks (spacing 0.3 μm between confocal planes) were carried out in Imaris 8.1.2.
For live‐cell imaging of 3D cultured acini, single cells were seeded in Matrigel in eight‐well slides and processed as described (Debnath et al, 2003) for 5 days until the beginning of the time‐lapse. The entire acini were filmed by taking stacks of pictured spaced of 0.37 μm. Gridded bottom slides (80827 or 80826‐G500, Ibidi Germany) were used when previously recorded acini had to be identified for subsequent immunofluorescence analyses. For time‐lapse microscopy on 2D monolayers, cells were grown on collagen IV‐coated ibidi eight‐well slides (80822, Ibidi, Germany) and GFP‐NLP expression was induced 48 h prior to the onset of recording.
All live‐cell imaging experiments were carried out using a FEI MORE wide‐field system (FEI Munich, Graefelfing, Germany) equipped with a 40×/0.95 U Plan S Apo air objective. For visualization of EGFP and mCherry signals, LEDs, combined with a quad band‐pass filter, were used as a light source for trans‐illumination at 515/18 and 595/19 nm, whereas dTomato and mCardinal signals were distinguished using single‐band‐pass filters at 590 nm and 685 nm, respectively. Pictures acquisition was performed at 37°C, 5% CO2, and > 70% air humidity. Image analyses were carried out using Image J or Imaris 8.1.2 following deconvolution using Huygens Remote manager 3.3.0‐rc9.
Determination of E‐cadherin junction strength index
Madine–Darby canine kidney cells were stained for DNA (DAPI) and E‐cadherin, and z‐stacks (spacing 0.2 μm) of the whole epithelium within the field were imaged (around 70 planes). Images were then processed for deconvolution using Huygens Remote manager 3.3.0‐rc9, and z‐projections of the maximum intensities of the whole stacks were performed using Fiji software. E‐cadherin signals were processed to determine an E‐cadherin JSI using Icy software (http://icy.bioimageanalysis.org/). Briefly, E‐cadherin signals were segmented into three classes, termed background, junctions, and speckles/aggregates, by using k‐means to define two thresholds. A mask was then created to identify the junction and a median filter with half size 2 used to remove noise. This mask was used to determine the entropy and the area of the signal using Icy's ROI statistics. E‐cadherin JSI was finally defined as the ratio of area to entropy of signal per nucleus.
Mitotic spindle angle measurements
3D reconstructions from z‐stack images (spacing 0.2 μm) of MDCK cysts stained for α‐tubulin and DNA and were performed using Imaris 8.1.2 and the acute angles between the spindle axes and the radius of the cysts (hitting the center of the mitotic spindle); in all cases, cysts were rotated to ensure positioning the mitotic spindle within the equatorial plane of the cyst.
Determination of total amount of α‐tubulin and detyrosinated‐α‐tubulin
Madine–Darby canine kidney cells were stained for DNA (DAPI), total α‐tubulin and detyrosinated‐α‐tubulin, and z‐stacks (spacing 0.2 μm) of the whole epithelium within the field were imaged (around 70 planes). Images were then processed for deconvolution using Huygens Remote manager 3.3.0‐rc9 and z‐projections of the maximum intensities of the whole stacks were performed using Fiji software. Regions of interest (ROI) were manually delineated using α‐tubulin and GFP signals to determine the areas of positive cells; the same regions of interest were used to quantify total amount of α‐tubulin and detyrosinated‐α‐tubulin signals.
Estimation of the percentage of cells GFP‐NLP + within a sphere
The numbers of GFP‐NLP+ CRBs (centrosome‐related bodies; Schnerch & Nigg, 2016) and nuclei (stained with DAPI) within a given acinus were determined using IMARIS software on 3D reconstructed acini. Assuming that MCF10A cultured in 2D or in 3D react similarly to doxycycline, we determined the average number of GFP‐NLP+ CRBs per GFP‐NLP+ cell (= 2.09 ± 0.1208 s.e.m.) by analyzing 2D cultured MCF10A (n = 182 cells) treated with doxycycline for the same duration as used for 3D experiments. For each acinus, the number of GFP‐NLP+ cells was then estimated by dividing the number of CRBs within the acinus by the average number of CRBs per NLP‐GFP+ cell as determined above (= 2.09). Finally, the percentage of GFP‐NLP+ cells within the acinus was then calculated by dividing the estimated number of GFP‐NLP+ cells by the total number of cells within the acinus determined by DAPI staining.
Western blots
Cells were harvested and proteins extracted in extract buffer (50 mM Tris–HCl, pH 7.4, 0.5% IGEPAL (Sigma‐Aldrich, St. Louis, MO, USA), 150 mM NaCl, 1 mM DTT, 5% glycerol, 50 mM NaF, 1 mM PMSF, 25 mM β‐glycerophosphate, 1 mM vanadate, complete mini protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). Proteins were then separated on Bio‐Rad mini‐protean 4–15% gels (456–1,086, Bio‐Rad Hercules, CA, USA) and transferred onto nitrocellulose membranes. Primary antibodies used were anti‐E‐cadherin (1:5,000, 610182, BD, Franklin Lakes, NJ, USA) and anti‐histone H3 (1:1,000, ab1791, Abcam Cambridge, U.K), and secondary antibodies were HRP‐conjugated anti‐mouse immunoglobulin (170‐6516, 1:2,000, Bio‐Rad, Hercules, CA, USA) or anti‐rabbit immunoglobulin (170‐6515, 1:2,000, Bio‐Rad, Hercules, CA, USA).
Mechano‐optical microscopy
All AFM experiments were carried out using a customized mechano‐optical microscope (MOM) comprised of an AFM (JPK Instruments AG, Germany and SPECS Zurich GmbH, Switzerland) and epifluorescence/spinning disk confocal microscope (Visitron Systems GmbH, Germany). Epifluorescence microscopy was used in all experiments to confirm the GFP‐NLP status of cells to be analyzed. Bright field and fluorescence images were taken with 10 and 40× air objectives (Leica, Germany) and recorded using an ORCA Flash 4 sCMOS (Hamamatsu Photonics K.K., Japan). Triangular DNP‐S10 D (Bruker AFM Probes, USA) with a nominal spring constant of 0.06 N m−1, an average tip length of 5 μm, and a nominal tip radius of 15 nm was used in all 2D cultures. Rectangular HQ‐CSC38/CR‐AU B (MikroMasch, Nanoworld AG, Switzerland) cantilevers with a nominal spring constant of 0.03 N m−1, and average tip length of 15 μm and a nominal tip radius of 20 nm were used for 3D cultures. The spring constant of each cantilever was determined by the thermal tune method (Sader et al, 1995). For cell stiffness measurements, the AFM was operated in a “Force‐Volume Mode”. Briefly, this consisted of either 20 × 20, 32 × 32, or 64 × 64 force–displacement curves that were recorded in square arrays (i.e., stiffness maps) over selected sample areas ranging from 5 × 5 μm2 to 80 × 80 μm2, depending on the experimental setting. All force–displacement curves were sampled at 4 kHz with indentations ranging between 1 and 3 μm at a defined maximum force load (A‐Hassan et al, 1998; Wu et al, 1998) that was set to 1.8 nN at an indentation velocity of 16 μm s−1. Individual stiffness maps were completed within 5–45 min depending on the experiment.
All AFM experiments were carried out at temperatures close to 37°C. During the measurements, culture dishes were replenished with fresh cell medium saturated with 5% CO2 to maintain pH at 7.5 and compensate for evaporation. For 2D experiments, cells were plated on Ibidi μ‐Dish 35 mm, low width glass bottom or TPP 9.2‐cm2 dishes at 75,000 or 150,000 cells cm−2 and grown for 48 or 72 h, respectively, to reach confluency. Doxycycline was added at 2.5 μg ml−1 for 24 h to induce overexpression of GFP‐NLP, GFP‐PLK4, or GFP‐CEP68. Before AFM experimentation, confluent cells were rinsed with PBS and fresh doxycycline‐containing growth medium was added to the cells. Drugs were added in the medium 30 min before AFM measurements; taxol (Merck Millipore Darmstadt Germany) was added at a final concentration of 50 nM, nocodazole (Sigma‐Aldrich MO, USA) at 200 ng ml−1, and cytochalasin D at 10 μM. Epifluorescence microscopy was used to identify GFP‐CEP68, GFP‐PLK4, and GFP‐NLP overexpressing cells in 2D and GFP‐NLP+ cells in 3D cultures. Furthermore, epifluorescence was used during AFM experiments to discriminate mitotic from interphase cells by visualization of mCardinal‐histone H1 signal. Stiffness measurements of living 3D MDCK cysts were performed in culture dishes (TPP, Switzerland) that were coated with poly‐L‐lysine (P4707, Sigma‐Aldrich, USA) for 30 min followed by rinsing in PBS before cyst placement. Individual cysts were first isolated from large pieces of embedding Matrigel with a pipette and placed in the culture dish. Cellular morphology and stiffness were used as an indicator to verify that each isolated cyst was free of Matrigel.
Bright field and fluorescence images were overlaid to identify WT and NLP overexpressing cells. The optical images were analyzed using Fiji/ImageJ (http://imagej.nih.gov). AFM data were further analyzed using a custom‐made “OfflineReader” (LABWIEV, National Instruments, USA), OriginPro 2016 (OriginLab Corporation, USA), and Gwyddion (http://gwyddion.net/) software. Force–volume data were analyzed using a customized software. This includes a set of commonly used criteria (signal‐to‐noise ratio, forward and backward curve tilt, hydrodynamic drag between forward and backward curves) to select suitable maps for analysis, as described in detail previously (Plodinec & Lim, 2015). First, force vs. piezo displacement data were transformed into force vs. tip‐sample distance curves where indentation corresponds to the tip‐sample contact point and a user‐defined maximal load. Second, the slope was determined from a linear fit to the upper 50% of each unloading force curve. Third, the contact area was calculated from the tip geometry and known indentation depth. Fourth, the elastic modulus (in kPa) was computed using the contact area and the slope according to Oliver and Pharr theory (Oliver & Pharr, 1992), as described in detail previously (Plodinec et al, 2011, 2012). Finally, the elastic modulus of each individual force curve was spatially plotted in 2D to yield a color‐coded stiffness map. It should be noted that all quantitative graphical comparisons excluded data from the cell junctions that were clearly discriminated by AFM. The stiffness values of transgene‐expressing cells (NLP, PLK4, or CEP68) in 2D were divided by the stiffness of the neighboring WT cells in the same stiffness map or in close proximity. Thus, a stiffness ratio is reported (see box plot Fig 6A). For GFP‐NLP overexpressing vs. WT cysts in 3D (see box plot Fig 6B), values were normalized to the average of the stiffness of WT cysts. Similarly, in drug treatment experiments (see Fig 6C), the stiffness was normalized to the mean stiffness of cells treated with no drug. For mitotic vs. interphase cells in confluency (see boxplot Fig 6F as well as Fig EV4D), the stiffness is normalized to the mean stiffness of WT interphase cells. In general, box plots show the mean (square), median (line), the standard error of mean (box), and the standard deviation (whiskers). Statistical significance was tested using two‐sample Student's t‐test for normally distributed samples or otherwise using a Mann–Whitney test (Figs 6B and D, and EV4C).
Author contributions
OG and DS contributed equally to this work. OG designed and analyzed most budding experiments, generated stable cell lines, performed and analyzed immunofluorescence and time‐lapse experiments, designed and performed E‐cadherin JSI experiments, and determined and interpreted the estimated number of GFP‐NLP+ cells per sphere. DS initiated the project and performed preliminary work, generated stable cell lines, designed and analyzed budding experiments, and performed and analyzed invadopodia formation assays. PO and MP designed and analyzed AFM experiments. PO performed all AFM experiments. RYHL contributed to design and analysis of AFM experiments. EAN supervised the project, designed experiments, analyzed the data, and wrote the paper. All authors contributed to editing the manuscript.
Conflict of interest
The University of Basel has filed patents related to the AFM technology based on the inventions of M.P., P.O., and R.Y.H.L.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Acknowledgments
We thank Inke Naethke (University of Dundee, UK), Channing Der (Pharmacology—UNC School of Medicine, USA), Daniel Gerlich (IMBA, Austria), Michael Davidson, Tilman Brummer (University of Freiburg), Stefan Zimmermann, and Ralph Wäsch (University Medical Center Freiburg) for reagents. We also thank Elena Nigg for expert technical assistance; Kai Schleicher, Alexia Loynton‐Ferrand, Wolf Heusermann, and Frederik Grüll (imaging core facility) for assistance with imaging; and Janine Bögli (FACS core facility) for assistance with flow cytometry. Work in E.A.N.'s laboratory was supported by the Swiss National Science Foundation (310030B_149641) and the University of Basel. Work in the laboratory of R.Y.H.L was supported by the Canton of Aargau (Argovia Professorship awarded to R.Y.H.L), the Biozentrum, the Swiss Nanoscience Institute, and the Swiss National Science Foundation Nanotera Project awarded to the PATLiSci II Consortium.
The EMBO Journal (2018) 37: e98576
See also: https://doi.org/10.15252/embj.201899422 (May 2018)
References
- Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, Brannigan BW, Kapur R, Stott SL, Shioda T, Ramaswamy S, Ting DT, Lin CP, Toner M, Haber DA, Maheswaran S (2014) Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158: 1110–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- A‐Hassan E, Heinz WF, Antonik MD, D'Costa NP, Nageswaran S, Schoenenberger CA, Hoh JH (1998) Relative microelastic mapping of living cells by atomic force microscopy. Biophys J 74: 1564–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmanova A, Hoogenraad CC (2015) Microtubule minus‐end‐targeting proteins. Curr Biol 25: R162–R171 [DOI] [PubMed] [Google Scholar]
- Akhtar N, Hotchin NA (2001) RAC1 regulates adherens junctions through endocytosis of E‐cadherin. Mol Biol Cell 12: 847–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander S, Weigelin B, Winkler F, Friedl P (2013) Preclinical intravital microscopy of the tumour‐stroma interface: invasion, metastasis, and therapy response. Curr Opin Cell Biol 25: 659–671 [DOI] [PubMed] [Google Scholar]
- Artym VV (2016) Dense fibrillar collagen is a master activator of invadopodia. Mol Cell Oncol 3: e1035476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW (2008) Centrosome amplification can initiate tumorigenesis in flies. Cell 133: 1032–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A, Lickert H, Kemler R, Stappert J (1998) Modification of the E‐cadherin‐catenin complex in mitotic Madin‐Darby canine kidney epithelial cells. J Biol Chem 273: 28314–28321 [DOI] [PubMed] [Google Scholar]
- Bettencourt‐Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA (2011) Centrosomes and cilia in human disease. Trends Genet 27: 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, Hines WC (2011) Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 17: 320–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornens M (2012) The centrosome in cells and organisms. Science 335: 422–426 [DOI] [PubMed] [Google Scholar]
- Braun DA, Hildebrandt F (2017) Ciliopathies. Cold Spring Harbor Perspect Biol 9: a028191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckner BR, Janshoff A (2015) Elastic properties of epithelial cells probed by atomic force microscopy. Biochim Biophys Acta 1853: 3075–3082 [DOI] [PubMed] [Google Scholar]
- Cadart C, Zlotek‐Zlotkiewicz E, Le Berre M, Piel M, Matthews HK (2014) Exploring the function of cell shape and size during mitosis. Dev Cell 29: 159–169 [DOI] [PubMed] [Google Scholar]
- Casenghi M, Meraldi P, Weinhart U, Duncan PI, Korner R, Nigg EA (2003) Polo‐like kinase 1 regulates Nlp, a centrosome protein involved in microtubule nucleation. Dev Cell 5: 113–125 [DOI] [PubMed] [Google Scholar]
- Cattin CJ, Duggelin M, Martinez‐Martin D, Gerber C, Muller DJ, Stewart MP (2015) Mechanical control of mitotic progression in single animal cells. Proc Natl Acad Sci USA 112: 11258–11263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JY (2011) A clinical overview of centrosome amplification in human cancers. Int J Biol Sci 7: 1122–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu YS, Thomas WA, Eder O, Pincet F, Perez E, Thiery JP, Dufour S (2004) Force measurements in E‐cadherin‐mediated cell doublets reveal rapid adhesion strengthened by actin cytoskeleton remodeling through Rac and Cdc42. J Cell Biol 167: 1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Haynes RD, Corbel SY, Li P, Gonzalez‐Gonzalez E, Burg JS, Ataie NJ, Lam AJ, Cranfill PJ, Baird MA, Davidson MW, Ng HL, Garcia KC, Contag CH, Shen K, Blau HM, Lin MZ (2014) Non‐invasive intravital imaging of cellular differentiation with a bright red‐excitable fluorescent protein. Nat Methods 11: 572–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh P, Clark AG, Smith MB, Cassani DAD, Dierkes K, Ragab A, Roux PP, Charras G, Salbreux G, Paluch EK (2017) Actin cortex architecture regulates cell surface tension. Nat Cell Biol 19: 689–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho PA, Bury L, Shahbazi MN, Liakath‐Ali K, Tate PH, Wormald S, Hindley CJ, Huch M, Archer J, Skarnes WC, Zernicka‐Goetz M, Glover DM (2015) Over‐expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol 5: 150209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conduit PT, Wainman A, Raff JW (2015) Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol 16: 611–624 [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS (2003) Morphogenesis and oncogenesis of MCF‐10A mammary epithelial acini grown in three‐dimensional basement membrane cultures. Methods 30: 256–268 [DOI] [PubMed] [Google Scholar]
- Delaval B, Doxsey SJ (2010) Pericentrin in cellular function and disease. J Cell Biol 188: 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher DE, Smith L, Cho S, Colasurdo M, Garcia AJ, Safran S (2017) Matrix mechanosensing: from scaling concepts in ‘omics data to mechanisms in the nucleus, regeneration, and cancer’. Annu Rev Biophys 46: 295–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien CB, Morcos PA, Rosenblatt J (2012) Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 484: 546–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Elzen N, Buttery CV, Maddugoda MP, Ren G, Yap AS (2009) Cadherin adhesion receptors orient the mitotic spindle during symmetric cell division in mammalian epithelia. Mol Biol Cell 20: 3740–3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher DA, Mullins RD (2010) Cell mechanics and the cytoskeleton. Nature 463: 485–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogeron ML, Muller H, Schade S, Dreher F, Lehmann V, Kuhnel A, Scholz AK, Kashofer K, Zerck A, Fauler B, Lurz R, Herwig R, Zatloukal K, Lehrach H, Gobom J, Nordhoff E, Lange BM (2013) LGALS3BP regulates centriole biogenesis and centrosome hypertrophy in cancer cells. Nat Commun 4: 1531 [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature 460: 278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghajar CM, Bissell MJ (2016) Metastasis: pathways of parallel progression. Nature 540: 528–529 [DOI] [PubMed] [Google Scholar]
- Gloerich M, Bianchini JM, Siemers KA, Cohen DJ, Nelson WJ (2017) Cell division orientation is coupled to cell‐cell adhesion by the E‐cadherin/LGN complex. Nat Commun 8: 13996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho SA, Pellman D (2014) Causes and consequences of centrosome abnormalities in cancer. Philos Trans R Soc Lond B Biol Sci 369: 20130467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho SA, Picone R, Burute M, Dagher R, Su Y, Leung CT, Polyak K, Brugge JS, Thery M, Pellman D (2014) Oncogene‐like induction of cellular invasion from centrosome amplification. Nature 510: 167–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonczy P (2015) Centrosomes and cancer: revisiting a long‐standing relationship. Nat Rev Cancer 15: 639–652 [DOI] [PubMed] [Google Scholar]
- Grosshans J, Wieschaus E (2000) A genetic link between morphogenesis and cell division during formation of the ventral furrow in Drosophila . Cell 101: 523–531 [DOI] [PubMed] [Google Scholar]
- Guo HQ, Gao M, Ma J, Xiao T, Zhao LL, Gao Y, Pan QJ (2007) Analysis of the cellular centrosome in fine‐needle aspirations of the breast. Breast Cancer Res 9: R48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, Avivar‐Valderas A, Nagi C, Girnius N, Davis RJ, Farias EF, Condeelis J, Klein CA, Aguirre‐Ghiso JA (2016) Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 540: 588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendley AM, Wang YJ, Polireddy K, Alsina J, Ahmed I, Lafaro KJ, Zhang H, Roy N, Savidge SG, Cao Y, Hebrok M, Maitra A, Reynolds AB, Goggins M, Younes M, Iacobuzio‐Donahue CA, Leach SD, Bailey JM (2016) p120 catenin suppresses basal epithelial cell extrusion in invasive pancreatic neoplasia. Can Res 76: 3351–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini H, Obradovic MM, Hoffmann M, Harper KL, Sosa MS, Werner‐Klein M, Nanduri LK, Werno C, Ehrl C, Maneck M, Patwary N, Haunschild G, Guzvic M, Reimelt C, Grauvogl M, Eichner N, Weber F, Hartkopf AD, Taran FA, Brucker SY et al (2016) Early dissemination seeds metastasis in breast cancer. Nature 540: 552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Hua S, Mohan R, Grigoriev I, Yau KW, Liu Q, Katrukha EA, Altelaar AF, Heck AJ, Hoogenraad CC, Akhmanova A (2014) Microtubule minus‐end stabilization by polymerization‐driven CAMSAP deposition. Dev Cell 28: 295–309 [DOI] [PubMed] [Google Scholar]
- Kazazian K, Go C, Wu H, Brashavitskaya O, Xu R, Dennis JW, Gingras AC, Swallow CJ (2017) Plk4 promotes cancer invasion and metastasis through Arp2/3 complex regulation of the actin cytoskeleton. Can Res 77: 434–447 [DOI] [PubMed] [Google Scholar]
- Kerr JP, Robison P, Shi G, Bogush AI, Kempema AM, Hexum JK, Becerra N, Harki DA, Martin SS, Raiteri R, Prosser BL, Ward CW (2015) Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat Commun 6: 8526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi‐Far R, White MA, Westwick JK, Solski PA, Chrzanowska‐Wodnicka M, Van Aelst L, Wigler MH, Der CJ (1996) Oncogenic Ras activation of Raf/mitogen‐activated protein kinase‐independent pathways is sufficient to cause tumorigenic transformation. Mol Cell Biol 16: 3923–3933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Hayashi S (2013) Mitotic cell rounding accelerates epithelial invagination. Nature 494: 125–129 [DOI] [PubMed] [Google Scholar]
- Kronenwett U, Huwendiek S, Castro J, Ried T, Auer G (2005) Characterisation of breast fine‐needle aspiration biopsies by centrosome aberrations and genomic instability. Br J Cancer 92: 389–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunda P, Pelling AE, Liu T, Baum B (2008) Moesin controls cortical rigidity, cell rounding, and spindle morphogenesis during mitosis. Curr Biol 18: 91–101 [DOI] [PubMed] [Google Scholar]
- Lambert AW, Pattabiraman DR, Weinberg RA (2017) Emerging biological principles of metastasis. Cell 168: 670–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TL, Yap AS, Stow JL (1999) Recycling of E‐cadherin: a potential mechanism for regulating cadherin dynamics. J Cell Biol 146: 219–232 [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Wu PH, Staunton JR, Ros R, Longmore GD, Wirtz D (2012) Mismatch in mechanical and adhesive properties induces pulsating cancer cell migration in epithelial monolayer. Biophys J 102: 2731–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekka M, Gil D, Pogoda K, Dulinska‐Litewka J, Jach R, Gostek J, Klymenko O, Prauzner‐Bechcicki S, Stachura Z, Wiltowska‐Zuber J, Okon K, Laidler P (2012) Cancer cell detection in tissue sections using AFM. Arch Biochem Biophys 518: 151–156 [DOI] [PubMed] [Google Scholar]
- Levine MS, Bakker B, Boeckx B, Moyett J, Lu J, Vitre B, Spierings DC, Lansdorp PM, Cleveland DW, Lambrechts D, Foijer F, Holland AJ (2017) Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev Cell 40: 313–322.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL (1998) Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci USA 95: 2950–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Salisbury JL (1999) Altered centrosome structure is associated with abnormal mitoses in human breast tumors. Am J Pathol 155: 1941–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W, Whitehead CM, Reynolds C, Salisbury JL (2002) Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci USA 99: 1978–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall TW, Lloyd IE, Delalande JM, Nathke I, Rosenblatt J (2011) The tumor suppressor adenomatous polyposis coli controls the direction in which a cell extrudes from an epithelium. Mol Biol Cell 22: 3962–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata J, Curado S, Ephrussi A, Rorth P (2000) Tribbles coordinates mitosis and morphogenesis in Drosophila by regulating string/CDC25 proteolysis. Cell 101: 511–522 [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, Thiery JP (2016) EMT: 2016. Cell 166: 21–45 [DOI] [PubMed] [Google Scholar]
- Nigg EA (2002) Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer 2: 815–825 [DOI] [PubMed] [Google Scholar]
- Nigg EA, Holland AJ (2018) Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol https://doi.org/10.1038/nrm.2017.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Raff JW (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139: 663–678 [DOI] [PubMed] [Google Scholar]
- Oliver WC, Pharr GM (1992) An improved technique for determining hardness and elastic modulus using load and displacement sensing indentation experiments. J Mater Res 7: 1564–1583 [Google Scholar]
- Paul CD, Mistriotis P, Konstantopoulos K (2017) Cancer cell motility: lessons from migration in confined spaces. Nat Rev Cancer 17: 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepperkok R, Bre MH, Davoust J, Kreis TE (1990) Microtubules are stabilized in confluent epithelial cells but not in fibroblasts. J Cell Biol 111: 3003–3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihan GA, Wallace J, Zhou Y, Doxsey SJ (2003) Centrosome abnormalities and chromosome instability occur together in pre‐invasive carcinomas. Can Res 63: 1398–1404 [PubMed] [Google Scholar]
- Plodinec M, Loparic M, Suetterlin R, Herrmann H, Aebi U, Schoenenberger CA (2011) The nanomechanical properties of rat fibroblasts are modulated by interfering with the vimentin intermediate filament system. J Struct Biol 174: 476–484 [DOI] [PubMed] [Google Scholar]
- Plodinec M, Loparic M, Monnier CA, Obermann EC, Zanetti‐Dallenbach R, Oertle P, Hyotyla JT, Aebi U, Bentires‐Alj M, Lim RY, Schoenenberger CA (2012) The nanomechanical signature of breast cancer. Nat Nanotechnol 7: 757–765 [DOI] [PubMed] [Google Scholar]
- Plodinec M, Lim RY (2015) Nanomechanical characterization of living mammary tissues by atomic force microscopy. Methods Mol Biol 1293: 231–246. [DOI] [PubMed] [Google Scholar]
- Prosser SL, Pelletier L (2017) Mitotic spindle assembly in animal cells: a fine balancing act. Nat Rev Mol Cell Biol 18: 187–201 [DOI] [PubMed] [Google Scholar]
- Pujana MA, Han JD, Starita LM, Stevens KN, Tewari M, Ahn JS, Rennert G, Moreno V, Kirchhoff T, Gold B, Assmann V, Elshamy WM, Rual JF, Levine D, Rozek LS, Gelman RS, Gunsalus KC, Greenberg RA, Sobhian B, Bertin N et al (2007) Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet 39: 1338–1349 [DOI] [PubMed] [Google Scholar]
- Qu D, Qu H, Fu M, Zhao X, Liu R, Sui L, Zhan Q (2008) Increased expression of Nlp, a potential oncogene in ovarian cancer, and its implication in carcinogenesis. Gynecol Oncol 110: 230–236 [DOI] [PubMed] [Google Scholar]
- Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS (2005) Spindle multipolarity is prevented by centrosomal clustering. Science 307: 127–129 [DOI] [PubMed] [Google Scholar]
- Raff JW, Basto R (2017) Centrosome amplification and cancer: a question of sufficiency. Dev Cell 40: 217–218 [DOI] [PubMed] [Google Scholar]
- Ragkousi K, Gibson MC (2014) Cell division and the maintenance of epithelial order. J Cell Biol 207: 181–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhys AD, Monteiro P, Smith C, Vaghela M, Arnandis T, Kato T, Leitinger B, Sahai E, McAinsh A, Charras G, Godinho SA (2018) Loss of E‐cadherin provides tolerance to centrosome amplification in epithelial cancer cells. J Cell Biol 217: 195–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sader JE, Larson I, Mulvaney P, White LR (1995) Method for the calibration of atomic force microscope cantilevers. Rev Sci Instrum 66: 3789–3798 [Google Scholar]
- Salisbury JL, Whitehead CM, Lingle WL, Barrett SL (1999) Centrosomes and cancer. Biol Cell 91: 451–460 [PubMed] [Google Scholar]
- Salisbury JL, D'Assoro AB, Lingle WL (2004) Centrosome amplification and the origin of chromosomal instability in breast cancer. J Mammary Gland Biol Neoplasia 9: 275–283 [DOI] [PubMed] [Google Scholar]
- Sanchez I, Dynlacht BD (2016) Cilium assembly and disassembly. Nat Cell Biol 18: 711–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saw TB, Doostmohammadi A, Nier V, Kocgozlu L, Thampi S, Toyama Y, Marcq P, Lim CT, Yeomans JM, Ladoux B (2017) Topological defects in epithelia govern cell death and extrusion. Nature 544: 212–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnerch D, Nigg EA (2016) Structural centrosome aberrations favor proliferation by abrogating microtubule‐dependent tissue integrity of breast epithelial mammospheres. Oncogene 35: 2711–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze E, Asai DJ, Bulinski JC, Kirschner M (1987) Posttranslational modification and microtubule stability. J Cell Biol 105: 2167–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seher TC, Leptin M (2000) Tribbles, a cell‐cycle brake that coordinates proliferation and morphogenesis during Drosophila gastrulation. Curr Biol 10: 623–629 [DOI] [PubMed] [Google Scholar]
- Sercin O, Larsimont JC, Karambelas AE, Marthiens V, Moers V, Boeckx B, Le Mercier M, Lambrechts D, Basto R, Blanpain C (2016) Transient PLK4 overexpression accelerates tumorigenesis in p53‐deficient epidermis. Nat Cell Biol 18: 100–110 [DOI] [PubMed] [Google Scholar]
- Shamir ER, Ewald AJ (2014) Three‐dimensional organotypic culture: experimental models of mammalian biology and disease. Nat Rev Mol Cell Biol 15: 647–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Liu R, Wang Y, Song Y, Zuo L, Xue L, Lu N, Hou N, Wang M, Yang X, Zhan Q (2010) Centrosomal Nlp is an oncogenic protein that is gene‐amplified in human tumors and causes spontaneous tumorigenesis in transgenic mice. J Clin Investig 120: 498–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth WT, Nardi IK, Scholl LM, Cimini D (2009) Multipolar spindle pole coalescence is a major source of kinetochore mis‐attachment and chromosome mis‐segregation in cancer cells. PLoS One 4: e6564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattum G, McGee KM, Rosenblatt J (2009) P115 RhoGEF and microtubules decide the direction apoptotic cells extrude from an epithelium. J Cell Biol 186: 693–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattum GM, Rosenblatt J (2014) Tumour cell invasion: an emerging role for basal epithelial cell extrusion. Nat Rev Cancer 14: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattum G, Gu Y, Sabbadini R, Rosenblatt J (2014) Autophagy in oncogenic K‐Ras promotes basal extrusion of epithelial cells by degrading S1P. Curr Biol 24: 19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorce B, Escobedo C, Toyoda Y, Stewart MP, Cattin CJ, Newton R, Banerjee I, Stettler A, Roska B, Eaton S, Hyman AA, Hierlemann A, Muller DJ (2015) Mitotic cells contract actomyosin cortex and generate pressure to round against or escape epithelial confinement. Nat Commun 6: 8872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW (2009) Aurora B‐mediated abscission checkpoint protects against tetraploidization. Cell 136: 473–484 [DOI] [PubMed] [Google Scholar]
- Stewart MP, Helenius J, Toyoda Y, Ramanathan SP, Muller DJ, Hyman AA (2011) Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature 469: 226–230 [DOI] [PubMed] [Google Scholar]
- Swaminathan V, Mythreye K, O'Brien ET, Berchuck A, Blobe GC, Superfine R (2011) Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Can Res 71: 5075–5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabassum DP, Polyak K (2015) Tumorigenesis: it takes a village. Nat Rev Cancer 15: 473–483 [DOI] [PubMed] [Google Scholar]
- Waclaw B, Bozic I, Pittman ME, Hruban RH, Vogelstein B, Nowak MA (2015) A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 525: 261–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman‐Storer CM, Worthylake RA, Liu BP, Burridge K, Salmon ED (1999) Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat Cell Biol 1: 45–50 [DOI] [PubMed] [Google Scholar]
- Webster DR, Gundersen GG, Bulinski JC, Borisy GG (1987) Differential turnover of tyrosinated and detyrosinated microtubules. Proc Natl Acad Sci USA 84: 9040–9044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HW, Kuhn T, Moy VT (1998) Mechanical properties of L929 cells measured by atomic force microscopy: effects of anticytoskeletal drugs and membrane crosslinking. Scanning 20: 389–397 [DOI] [PubMed] [Google Scholar]
- Xue B, Krishnamurthy K, Allred DC, Muthuswamy SK (2013) Loss of Par3 promotes breast cancer metastasis by compromising cell‐cell cohesion. Nat Cell Biol 15: 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Song Y, Zhang Q, Zhan Q (2009) Ninein‐like protein is overexpressed in head and neck squamous cell carcinoma and contributes to cancer growth and resistance to apoptosis. Oncol Rep 22: 789–798 [DOI] [PubMed] [Google Scholar]
- Zhao W, Song Y, Xu B, Zhan Q (2012) Overexpression of centrosomal protein Nlp confers breast carcinoma resistance to paclitaxel. Cancer Biol Ther 13: 156–163 [DOI] [PubMed] [Google Scholar]
- Zyss D, Gergely F (2009) Centrosome function in cancer: guilty or innocent? Trends Cell Biol 19: 334–346 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7