

Abstract
The formation of neurites is an important process affecting the cognitive abilities of an organism. Neurite growth requires the addition of new membranes, but the metabolic remodeling necessary to supply lipids for membrane expansion is poorly understood. Here, we show that synaptic activity, one of the most important inducers of neurite growth, transcriptionally regulates the expression of neuronal glucose transporter Glut3 and rate‐limiting enzymes of glycolysis, resulting in enhanced glucose uptake and metabolism that is partly used for lipid synthesis. Mechanistically, CREB regulates the expression of Glut3 and Siah2, the latter and LDH activity promoting the normoxic stabilization of HIF‐1α that regulates the expression of rate‐limiting genes of glycolysis. The expression of dominant‐negative HIF‐1α or Glut3 knockdown blocks activity‐dependent neurite growth in vitro while pharmacological inhibition of the glycolysis and specific ablation of HIF‐1α in early postnatal mice impairs the neurite architecture. These results suggest that the manipulation of neuronal glucose metabolism could be used to treat some brain developmental disorders.
Keywords: glycolysis, HIF‐1α, neurite growth, Siah2, synaptic activity‐mediated transcription
Subject Categories: Metabolism, Neuroscience
Introduction
Early postnatal brain development is characterized by the massive outgrowth of dendrites and axons (Wong & Ghosh, 2002; Silbereis John et al, 2016). Among the different mechanisms affecting dendritic development, signaling from afferents is particularly important for dendritic growth (Rajan & Cline, 1998; Groc et al, 2002; Wong & Ghosh, 2002; Konur & Ghosh, 2005). The most active phase of dendritic growth in the rat cerebral cortex is concurrent with the time of afferent innervations (Wong & Ghosh, 2002; Konur & Ghosh, 2005). Moreover, blocking neuronal activity in vivo alters dendritic development (Rajan & Cline, 1998; Groc et al, 2002), while increased neuronal activity by exposure to an enriched environment positively influences dendritic growth (Faherty et al, 2003).
Activity‐mediated calcium influx activates signaling events that influence dendritic architecture, modifying cytoskeleton and activating transcriptional programs (Wong & Ghosh, 2002; Puram & Bonni, 2013). Several transcription factors and co‐activators have been described to promote dendritic growth (Puram & Bonni, 2013), such as CREB, a transcription factor strongly activated by synaptic activity. In addition to its roles in cell survival and synaptic function, CREB is one of the most important transcription factors mediating activity‐dependent dendritic morphogenesis in mammalian brain neurons (Lonze & Ginty, 2002; Puram & Bonni, 2013). Activation of CREB in cortical neurons induces dendritic growth and arborization, with dominant‐negative CREB suppressing activity‐dependent neurite growth (Redmond et al, 2002). Furthermore, CREB knockout mice show impaired axonal growth and projections (Lonze et al, 2002). Despite the well‐known function of CREB in regulating dendritic development, its target genes are not yet completely known.
Neurite growth requires cytoskeletal reorganization and cell membrane extension. The latter requiring a supply of lipids. Given the impermeable nature of the blood–brain barrier (BBB), most plasma lipoproteins cannot cross the BBB and lipids need to be synthesized in the brain. Studies investigating the incorporation of radiolabeled glycerol into lipids and their transport indicate that the bulk of lipid synthesis occurs in the cell body of neurons, with the lipids then exported in vesicles to the axon and dendrites for membrane expansion (Goldberg, 2003; Pfenninger, 2009). However, little is known on where the precursors necessary for lipid synthesis come from. The intermediates of glycolysis serve as lipid precursors (Vander Heiden et al, 2009). Dihydroxyacetone phosphate (DHAP) is converted by glycerol‐3‐phosphate dehydrogenase 1 (GPD1) into glycerol‐3‐phosphate, which is then used for synthesizing glycerol‐backbone lipids. Pyruvate, the end product of glycolysis, is transported to the mitochondria where it enters the tricarboxylic acid (TCA) cycle as citrate and is further metabolized or exported to the cytoplasm to be broken down by ATP‐citrate lyase (ACLY) into oxaloacetate and acetyl‐CoA, which serves as a precursor in lipid biosynthesis. Meta‐analysis of brain and glucose consumption demonstrates that aerobic glycolysis increases in the human brain when synaptic growth rates are highest, suggesting that glycolysis provides biosynthetic support for neurite growth (Goyal Manu et al, 2014). However, it is unclear whether glycolysis is regulated by neuritogenic cues or is needed for neurite growth.
Hypoxia‐inducible factor 1 (HIF‐1) is a transcription factor regulating adaptive responses to hypoxia. It occurs as heterodimer composed of an unstable HIF‐1α and a stable HIF‐1β (also known as ARNT). The heterodimer binds hypoxia response elements (HRE) on target genes, which includes those associated with glucose transport and glycolysis genes, among others (Semenza et al, 1994; Denko, 2008). Normally, HIF‐1α is ubiquitinated and degraded in normoxic conditions. Hydroxylation of Pro‐402 and Pro‐564 by prolyl hydroxylases (PHD) in the oxygen‐dependent degradation domain (ODD) of human HIF‐1α promotes interaction with the von Hippel–Lindau ubiquitin ligase complex (VHL) which targets HIF‐1α for proteolysis (Maxwell et al, 1999; Ivan et al, 2001; Jaakkola et al, 2001). Another regulatory element of the PHD/VHL pathway is the E3 ubiquitin ligase Siah2, which contributes to HIF‐1α stabilization (Nakayama et al, 2004; Li et al, 2017). Despite its generally short half‐life in normoxia, HIF‐1α can be stabilized in normoxia by growth factors, metabolite accumulation, the expression of oncogenes, or ROS production (Denko, 2008).
Although much is known on the molecular mechanisms mediating cytoskeletal remodeling and lipid transport to induce neurite growth, little is known about the metabolic adaptations required for neuronal membrane extension. Here, we show CREB activation by synaptic activity induces the expression of the glucose transporter Glut3 and promotes Siah2‐mediated stabilization of HIF‐1α that upregulates the expression of glycolysis genes. As a consequence, glucose metabolism is enhanced and part of it is used to provide the lipid precursors necessary for neurite growth.
Results
Glucose metabolism is necessary for activity‐dependent neurite outgrowth
Neuronal activity promotes neurite growth which requires a supply of lipids to enlarge membranes. Accordingly, using an established method of network disinhibition to enhance synaptic activity by applying the GABAA receptor antagonist bicuculline (Bic) and the K+ channel antagonist 4‐aminopyridine (4AP) (Hardingham et al, 2001), we observed that active neurons have increased levels of FASN and ACLY (Appendix Fig S1A–C), two key enzymes for the novo lipid synthesis.
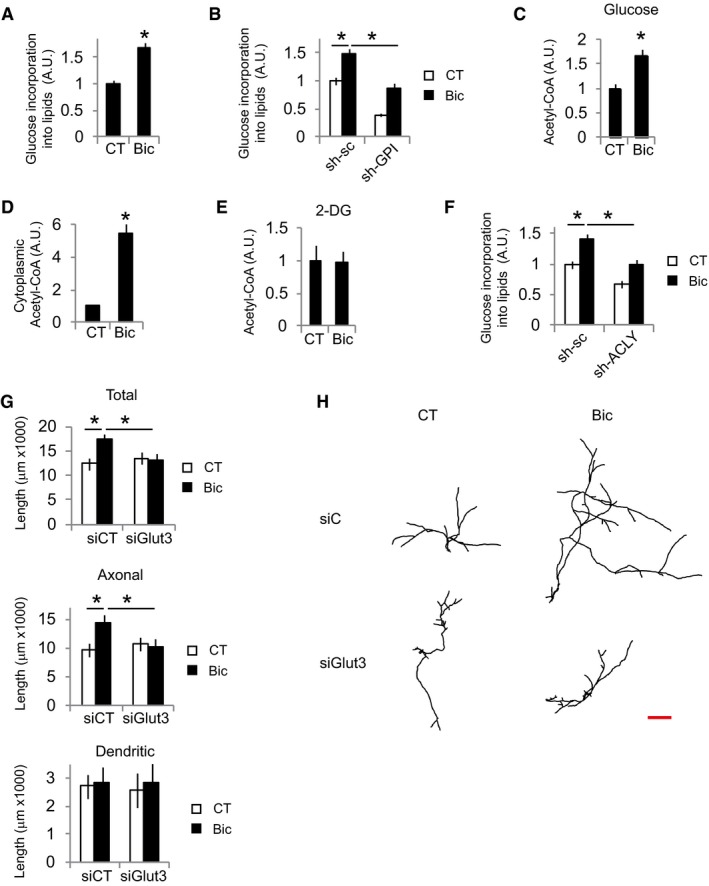
Glucose‐derived acetyl‐CoA is the main precursor for lipid biosynthesis (Pietrocola et al, 2015; Divakaruni et al, 2017). Thus, it was analyzed incorporation of radioactively labeled glucose into lipids in neurons that have experienced an episode of synaptic activity. After 48 h of stimulation, active neurons showed increased glucose incorporation into lipids (Fig 1A). This was impaired by knockdown of the glycolytic enzyme GPI (Fig 1B and Appendix Fig S1D). Glutamine metabolism is another major source of lipogenic acetyl‐CoA (Pietrocola et al, 2015); however, synaptic activity did not increase glutamine incorporation into lipids (Appendix Fig S1E).
Figure 1. Glucose uptake is necessary for activity‐dependent neurite outgrowth.
-
A, BUntransduced (A) or AAV‐shGPI or AAV‐sh‐sc (control) transduced neurons (B) were incubated with 14C‐U‐glucose and were stimulated with bicuculline plus 4‐AP (labeled Bic in this and subsequent figures) for 48 h or left unstimulated (CT). Cellular lipids were extracted and radioactive counts measured (n = 4–7 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test (A) and ANOVA one‐way ANOVA followed by Tukey's post hoc test (B).
-
CCortical neurons were stimulated for 24 h with Bic+4‐AP in the presence of glucose (n = 8 independent experiments). After 24 h, total acetyl‐CoA levels were assayed. Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
DDetermination of cytosolic acetyl‐CoA levels after 24 h of Bic+4‐AP stimulation in the presence of glucose (n = 4 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
EAcetyl‐CoA levels in cortical neurons stimulated for 24 h with Bic+4‐AP in the presence of 25 mM of the non‐metabolizable glucose analog 2‐DG (n = 4 independent experiments). Values represent mean ± s.e.m.
-
F14C‐U‐glucose incorporation into lipids in neurons transduced with AAV expressing shRNA‐sc or targeting ACLY (sh‐ACLY) after 48 h stimulation with Bic+4‐AP (n = 7 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
-
G, HNeurite length and representative tracings of the neuritic tree of control and Bic+4‐AP‐stimulated neurons (for 48 h) after transfection with non‐targeting (siC) or Glut3‐targeting (siGlut3) siRNAs (n = 45–49 neurons from nine independent experiments). Scale bar, 500 μm. Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey's post hoc test.
One molecule of glucose is converted to two molecules of pyruvate during glycolysis. Pyruvate enters the mitochondria, where it is decarboxylated to produce acetyl‐CoA that then conjugates with oxaloacetate to produce citrate. Citrate can be further metabolized in the TCA cycle or exported to the cytoplasm, where it is converted into acetyl‐CoA by ACLY to be used for fatty acid synthesis. Total and cytosolic acetyl‐CoA levels were also increased in active neurons (Fig 1C and D), but not CoA levels (Appendix Fig S1F), which was not observed when glycolysis was inhibited with 2‐deoxy‐d‐glucose (Fig 1E), indicating a major role of glucose as a source of the increased amounts of acetyl‐CoA. In agreement, knockdown of ACLY blocked activity‐dependent enhanced glucose incorporation into lipids (Fig 1F, and Appendix Fig S1G and H).
In accordance with the need of glucose‐derived lipid production to enlarge membranes for neurite growth, knockdown of Glut3 (Fig 1G and H), the main glucose transporter in neurons, inhibition of glucose metabolism with 2‐DG (Appendix Fig S1I), or knockdown of ACLY (Appendix Fig S1J) abolished activity‐mediated neurite growth which at this developmental stage represents axonal growth.
Synaptic activity stimulates neuronal glucose uptake and metabolism at the transcriptional level
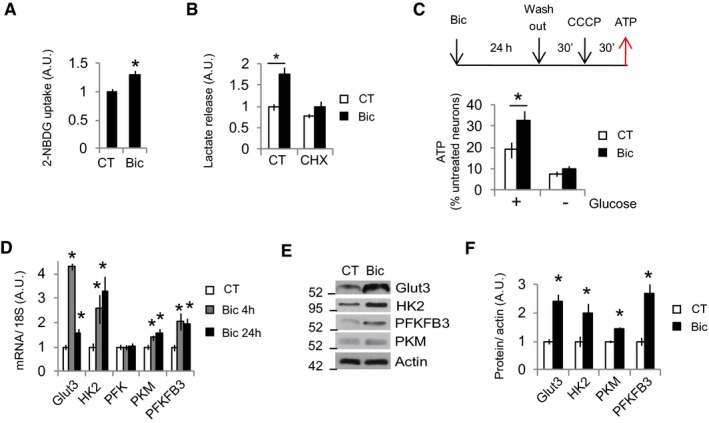
To investigate whether increased acetyl‐CoA levels correlated with increased glucose uptake and metabolism, neurons were stimulated for 24 h with Bic+4AP and washed for 30 min to allow restoration of ion gradients before analyzing glucose uptake. Bic+4AP washing effectively blocked burst firing (Appendix Fig S2A and B). It was found that neurons that had experienced synaptic activity increased their glucose uptake (Fig 2A and Appendix Fig S2C). Glycolysis‐produced pyruvate is partially metabolized into lactate in a reaction that regenerates NAD+ that is necessary for glycolysis to continue. Glycolysis is the main source of lactate, accounting for 82–90% on different types of cells (Zhang et al, 2017). Thus, lactate release into the medium is a commonly used surrogate of glycolytic flux (TeSlaa & Teitell, 2014). Active neurons released 75% more lactate than resting ones (Fig 2B). This was not due to the hyperglycemic medium used in this study, since culturing the neurons with physiological glucose concentration in rat brain (2.4 mM; Silver & Erecinska, 1994) produced similar lactate release (Appendix Fig S2D). In another approach to demonstrate increased glycolysis in active neurons, neurons were stimulated for 24 h and washed for 30 min, to allow restoration of ionic gradients, before uncoupling the mitochondria with CCCP for 30 min. This drastically reduced ATP levels, which were less reduced in the stimulated neurons than unstimulated ones, but only when glucose was present in the medium (Fig 2C), indicating that the increased glycolysis in active neurons can supply more ATP than in resting ones when the mitochondria are not functional.
Figure 2. Synaptic activity stimulates neuronal glucose uptake and metabolism at the transcriptional level.
-
A2‐NBDG uptake over 15 min in control or Bic+4‐AP‐stimulated (for 24 h) neurons after washing and medium replacement (n = 4 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
BNeurons treated or not with cycloheximide (10 μM) were stimulated with Bic+4‐AP (for 24 h) before measuring the amount of lactate released into the medium (n = 4–7 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
CATP levels in unstimulated neurons that were treated with CCCP (3 nM) following the indicated chronogram. + or − glucose indicates whether the fresh medium added after the wash step contained glucose or not (n = 3–6 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
DCortical neurons were stimulated with Bic+4‐AP for 4 or 24 h, and mRNA expression of the indicated genes was determined by real‐time qPCR (n = 3–6 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
E, F(E) Representative Western blot and (F) densitometric analysis of protein samples from control and Bic+4‐AP‐stimulated (24 h; n = 3–4 independent experiments). *P < 0.05, two‐tailed Student's t‐test.
Source data are available online for this figure.
Synaptic activity has been shown to mediate Glut3 translocation to the membrane (Ferreira et al, 2011). Moreover, synaptic activity is a potent regulator of gene expression programs (Greer & Greenberg, 2008; Bading, 2013). Thus, we aimed to determine whether activity‐mediated glycolysis induction was mediated at the transcriptional or posttranslational level. Cycloheximide was used to inhibit mRNA translation. If glucose metabolism induction was mainly due to posttranslational modifications, the increase in glucose metabolism would not be affected by cycloheximide treatment; it would be affected by cycloheximide if de novo protein synthesis was involved. We found that cycloheximide abolished the glucose metabolism induced by synaptic activity, which was established by measuring lactate release (Fig 2B). This was not the consequence of reduced neuronal viability since cycloheximide treatment did not affect viability (Appendix Fig S2E). Next, we used qPCR to analyze the expression of Glut3, the main glucose transporter in neurons; HK2, PFK, and PKM, the three regulating enzymes of the glycolysis; and PFKFB3, which produces fructose‐2,6‐bisphosphate, a potent allosteric activator of PFK. There was an increase in the expression of all these genes except PFK (Fig 2D). Strikingly, Glut3 showed a different expression pattern to these associated with glycolysis. Along with the increased mRNA levels, synaptic activity induced the protein expression of these genes (Fig 2E and F). Interestingly, treatment of neurons with BDNF, a neurotrophic factor that induces neurite growth (Park & Poo, 2013), also induced the expression of glucose metabolism genes, as well as increased glucose uptake and lactate release (Appendix Fig S2F–H), although it cannot be excluded an indirect effect due to BDNF‐induced enhanced neuronal activity (Li et al, 1998).
Glial and neuronal metabolism is coupled (Bélanger et al, 2011). Around 2% of our primary cortical neuron cultures were glial cells (Appendix Fig S2I and J). Neuronal glucose uptake experiments were analyzed by cell imaging, but the lactate release and ATP production experiments were performed using the mixed cultures. Thus, we could not rule out the possibility that activity‐dependent enhanced glycolysis occurred in the glial cells rather than neurons. To clarify this, we used two approaches. The mitosis inhibitor AraC is used in our cortical cultures to block glial proliferation. In a first approach, we prepared parallel cortical cultures that were treated with AraC or not to obtain cultures with different proportions of glial cells (Appendix Fig S2I and J). If the stimulation‐mediated increase in lactate release was due to changes in gene expression in glial cells rather than neurons, modifying the abundance of glial cells in the co‐culture would elicit a corresponding increase in lactate release after stimulation. Although cultures not treated with AraC had around tenfold more astrocytes than AraC‐treated ones, these two types of cultures showed the same degree of lactate release when stimulated, confirming that the enhanced glucose metabolism and, consequently, the expression of glucose metabolism genes when stimulated occurred in neurons rather than astrocytes (Appendix Fig S2K). In the second approach, we used pure astrocyte cultures. Astrocytes express functional BDNF receptors (Rose et al, 2003; Ohira et al, 2005); however, BDNF did not increase lactate release in pure astrocyte cultures like it did in neuronal cultures (Appendix Fig S2H and L). Thus, enhancement of glucose metabolism takes place in neurons when stimulated with neurite growth inducers.
Activity‐dependent induction of glycolysis genes depends on HIF‐1α stabilization
Next, we investigated the transcription factor responsible for the induction of activity‐mediated glucose metabolism genes. HIF‐1α is a transcription factor that stimulates glycolysis by transactivating the genes involved in intracellular glucose transport and glycolysis (Semenza et al, 1994; Denko, 2008). Although HIF‐1α is typically unstable in normoxia, certain conditions enable its stabilization in normoxic conditions (Denko, 2008). Thus, HIF‐1α could be responsible for inducing glucose metabolism genes. We observed HIF‐1α stabilization in active and BDNF‐treated neurons (Figs 3A and B, and EV1A–C), but not HIF‐2α (Fig EV1E and F), that correlated with increased HIF‐1α transcriptional activation (Fig 3C). Overexpression of HIF‐DN, a dominant‐negative form of HIF‐1α (HIF‐DN) formed by the DNA binding domain of HIF‐1α and lacking the prolyl hydroxylase and transactivation regions, completely blocked activity‐mediated transcriptional activation of HIF‐1α (Fig 3C). Transduction with AAV‐HIF‐DN blocked activity‐dependent induction of glycolysis genes, but had no effect on Glut3 gene expression (Fig 3D). This was not completely surprising because the pattern of Glut3 expression was different to that of the glycolysis genes (Fig 2D). All together, these results indicate that the activity‐dependent induction of glycolysis genes depends on HIF‐1α. Consequently, the activity‐mediated lactate release and glucose incorporation into lipids were reduced in HIF‐DN expressing neurons (Fig 3E and F).
Figure 3. Activity‐dependent induction of glycolysis genes depends on HIF‐1α stabilization.
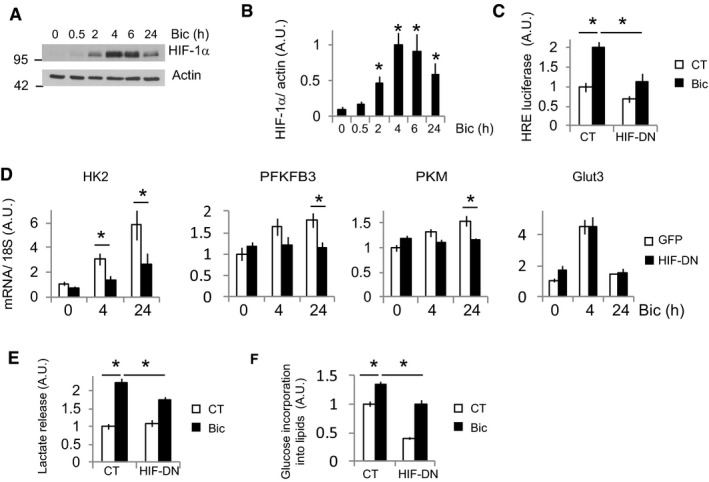
- Cortical neurons were stimulated with Bic+4‐AP over various time points, and the HIF‐1α protein was analyzed by Western blotting (n = 5 independent experiments).
- Densitometric analysis of HIF‐1α expression in (A). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
- Luciferase‐based HIF‐1α activity in neurons expressing a control plasmid (globin) or a dominant‐negative HIF‐1α (HIF‐DN) and stimulated with Bic+4‐AP for 8 h (n = 4 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
- Cortical neurons were transduced with AAV expressing HIF‐DN or control (GFP), stimulated for 4 or 24 h with Bic+4‐AP, and the mRNA expression of the indicated genes was analyzed by qPCR (n = 5 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
- Lactate released into the medium by neurons transduced with AAV expressing HIF‐DN or control (GFP) after 24 h stimulation with Bic+4‐AP (n = 3 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
- 14C‐U‐glucose incorporation into lipids in neurons transduced with AAV expressing HIF‐DN or control (GFP) and stimulated for 48 h with Bic+4‐AP (n = 4 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
Source data are available online for this figure.
Figure EV1. Synaptic activity and BDNF stabilize HIF‐1α.
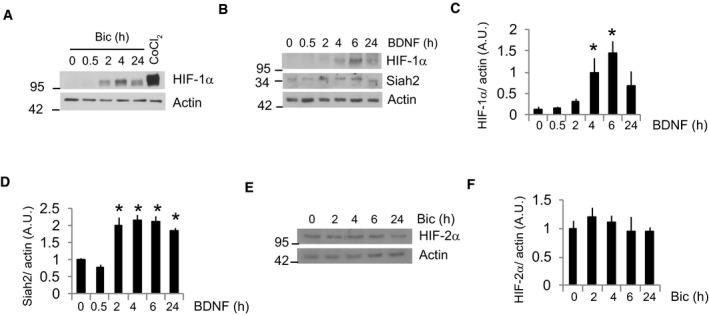
-
A–DRepresentative Western blots of cortical neurons stimulated with (A) Bic+4‐AP at the indicated time points or the hypoxia‐mimetic CoCl2 (200 μM) for 4 h and (B) BDNF (25 ng/ml) for the indicated time points (n = 4–5 independent experiments) and (C and D) densitometric analysis. Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
ENeurons were stimulated with Bic+4‐AP for the indicated times and HIF‐2α was analyzed by Western blot.
-
FDensitometric analysis (n = 4 independent experiments). Values represent mean ± s.e.m.
Source data are available online for this figure.
HIF‐1α is stabilized by Siah2 and LDH activity
HIF‐1α regulation is mainly mediated by proteasomal degradation in normoxia. Hydroxylation of prolynes in the ODD domain of HIF‐1α by PHDs promotes HIF‐1α proteolysis (Maxwell et al, 1999; Ivan et al, 2001; Jaakkola et al, 2001). We studied whether synaptic activity decreased PHD activity. Neurons were transfected with a plasmid expressing firefly luciferase fused to the oxygen‐dependent degradation domain of HIF‐1α (ODD‐Luc). Synaptic activity resulted in increased luciferase activity in ODD‐Luc, but not wild‐type firefly luciferase (Luc)‐transfected neurons (Fig 4A and B), indicating reduction in PHD activity. Siah2 is an E3 ubiquitin ligase that promotes HIF‐1α stabilization mainly by mediating proteasomal degradation of PHDs (Nakayama et al, 2004), although other mechanisms by which Siah2 promotes HIF‐1α stabilization have been also described (Li et al, 2017). Siah2 mRNA and protein levels were induced by synaptic activity (Fig 4C–E) and BDNF (Fig EV1B and D, and Appendix Fig S3B), correlating with reduced PHD activity. However, Siah1 is downregulated by synaptic activity (Appendix Fig S3A). To confirm the role of Siah2 in activity‐dependent HIF‐1α stabilization, we knocked down (KD) Siah2 by transducing the neurons with two AAV expressing different shRNAs targeting Siah2. Siah2 KD neurons showed a strong reduction of activity‐dependent HIF‐1α accumulation (Fig 4F–H and Appendix Fig S3C–G). Next, we checked the expression levels of PHD in active neurons and surprisingly we observed no changes in PHD1, PHD3, and FIH, and increased expression of PHD2 (Appendix Fig S3H and I). One alternative mechanism by which Siah2 could stabilize HIF‐1α is mediating degradation of the enzymes of the OGDHC, OGDH, and DLST (Sun Ramon & Denko Nicholas, 2014; Burr Stephen et al, 2016; Nadtochiy et al, 2016; Intlekofer et al, 2017), but we could not observe changes in protein levels of these enzymes (Appendix Fig S3H and I). However, a central role of Siah2 upregulation in the activity‐mediated enhanced glucose metabolism was further confirmed by showing that Siah2 KD blocked activity‐mediated lactate release (Fig 4I) and glucose incorporation into lipids (Fig 4J).
Figure 4. HIF‐1α is stabilized by Siah2 and LDH activities.
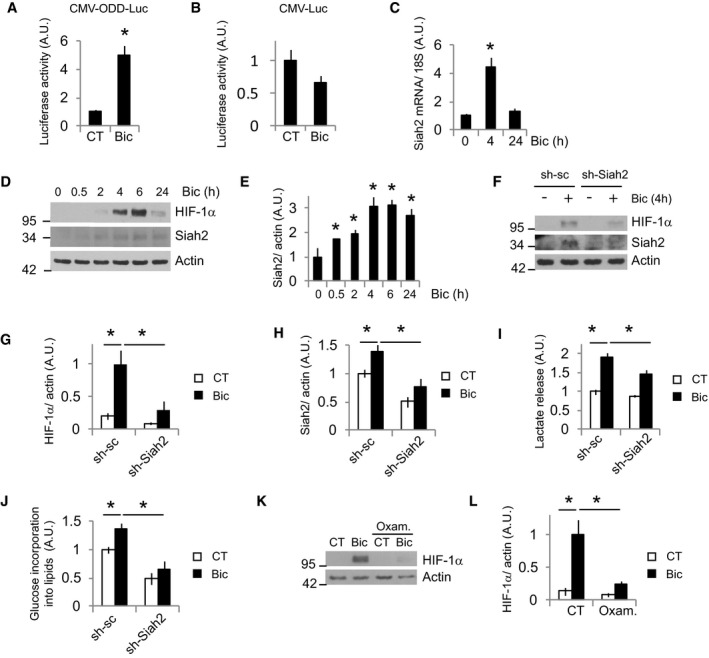
-
A, BPHD activity after Bic+4‐AP stimulation was assayed measuring luciferase activity in neurons transfected with plasmids expressing luciferase fused to the ODD domain of HIF‐1α. Non‐fused luciferase was used as control (n = 4 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
CCortical neurons were stimulated with Bic+4‐AP for 4 or 24 h, and Siah2 mRNA expression was determined by real‐time qPCR (n = 4 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
DCortical neurons were stimulated with Bic+4‐AP over various time points, and the indicated proteins were analyzed by Western blotting (n = 3–5 independent experiments).
-
EDensitometric analysis of Siah2 protein levels in (D). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
FAAV‐sh‐sc (control) or sh‐Siah2 transduced neurons were stimulated with Bic+4‐AP for 4 h before analyzing the expression of the indicated proteins (n = 5 independent experiments).
-
G, HDensitometric analysis of the indicated proteins in (F). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
-
ILactate released into the medium by neurons transduced with AAV expressing shRNA‐sc or targeting Siah2 (sh‐Siah2) after 24‐h stimulation with Bic+4‐AP (n = 4 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
-
J14C‐U‐glucose incorporation into lipids in neurons transduced with AAV expressing shRNA‐sc or targeting Siah2 (sh‐Siah2) after 48 h stimulation with Bic+4‐AP (n = 7 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
-
K, L(K) Representative Western blot and (L) densitometric analysis of neurons stimulated for 4 h with Bic+4‐AP in absence or presence of 40 mM oxamate (Oxam.; n = 5 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
Source data are available online for this figure.
HIF‐1α regulation is complex, with several regulators and mechanisms being involved. We tested whether other mechanisms different from the one mediated by Siah2 induction could participate as well in activity‐mediated HIF‐1α stabilization. HIF‐1α is an O2‐sensitive transcription factor that mediates the primary response to hypoxic stress. Excessive O2 consumption in active neurons could generate transitory hypoxia that mediates HIF‐1α stabilization. However, the use of a hypoxia sensor probe did not show hypoxia generation by synaptic activity (Appendix Fig S3J). Synaptic activity regulates a transcriptional program of defense against oxidative stress (Papadia et al, 2008). However, acute synaptic activity could boost mitochondrial metabolism and enhance ROS production which is involved in HIF‐1α stabilization in normoxia; therefore, we tested whether the use of antioxidants could block HIF‐1α activation by synaptic activity, but it did not have any effect on activity‐mediated HIF‐1α stabilization (Appendix Fig S3K and L). Increased HIF‐1α levels in active neurons were not due to the transcriptional regulation of HIF‐1α either (Appendix Fig S3M). Both rise in pyruvate level and rise in lactate level stabilize HIF‐1α (Lu et al, 2002). We inhibited LDH with oxamate, which produces accumulation of pyruvate and reduces lactate release (Lu et al, 2002). Oxamate treatment blocked activity‐mediated HIF‐1α stabilization (Fig 4K and L). Since, both, Siah2 KD and LDH inhibition almost completely blocked activity‐mediated HIF‐1α stabilization, they may be part of the same regulatory pathway.
Glut3 and Siah2 expressions are regulated by CREB
Since Glut3 expression in active neurons did not depend on HIF‐1α activation, we explored which transcription factor could regulate it. CREB is a powerful regulator of synaptic activity‐mediated gene expression (Greer & Greenberg, 2008; Bading, 2013), controls neurite outgrowth (Redmond et al, 2002), and is involved in metabolic regulation in different cell types (Altarejos & Montminy, 2011). CREB has been shown to regulate Glut3 expression (Rajakumar et al, 2004); thus, we tested whether CREB was involved in activity‐mediated Glut3 expression. Transduction of neurons with AAV expressing a dominant‐negative CREB (A‐CREB; Ahn et al, 1998) blocked activity‐dependent Glut3 expression (Fig 5A).
Figure 5. Glut3 and Siah2 expressions are regulated by CREB .
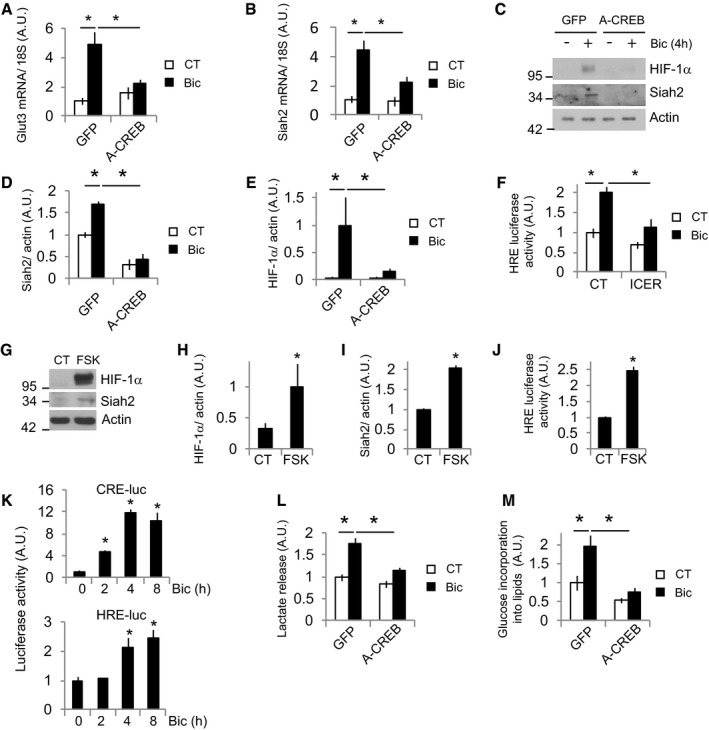
-
A, BCortical neurons transduced with AAV expressing GFP (control) or dominant‐negative A‐CREB were stimulated with Bic+4‐AP for 4 h before analyzing the mRNA expression of (A) Glut3 and (B) Siah2 by qPCR (n = 5 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey's post hoc test.
-
C–E(C) Representative Western blot and (D and E) densitometric analyses of the indicated proteins of neurons transduced with AAV expressing GFP or A‐CREB and stimulated with Bic+4‐AP for 4 h (n = 3 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
-
FLuciferase‐based HIF‐1α activity in neurons expressing a control plasmid (globin) or a CREB inhibitor ICER and stimulated with Bic+4‐AP for 8 h (n = 5 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey's post hoc test.
-
G–ICREB activator forskolin (FSK) was sufficient to induce Siah2 expression and stabilize HIF‐1α. (G) Representative Western blot and (H and I) densitometric analysis of neurons treated with 10 μM forskolin for 4 h (n = 3 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
JLuciferase‐based HIF‐1α activity in neurons treated with forskolin for 8 h (n = 4 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
KCREB activation temporally precedes HIF‐1α activation. Luciferase‐based CREB and HIF‐1α activity at different time points after Bic+4‐AP stimulation (n = 4 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
LLactate released into the medium by neurons transduced with AAV expressing A‐CREB or control (GFP) after 24 h stimulation with Bic+4‐AP (n = 5 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
-
M14C‐U‐glucose incorporation into lipids in neurons transduced with AAV expressing A‐CREB or control (GFP) and stimulated for 48 h with Bic+4‐AP (n = 5 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey post hoc test.
Source data are available online for this figure.
Given that Glut3 and Siah2 showed a similar pattern of mRNA expression following synaptic activity, a high activation after 4 h with the decay at 24 h (Figs 2D and 4C), we assessed whether Siah2 was also regulated by CREB. A‐CREB blocked Siah2 induction (Fig 5B–D) and, consequently, blocked the HIF‐1α stabilization induced by synaptic activity (Fig 5C and E). Furthermore, overexpression of the endogenous CREB inhibitor ICER (Molina et al, 1993) also blocked HIF‐1α activity (Fig 5F) but did not disturb the activity of MEF2 (Fig EV2A), an activity‐dependent transcription factor (Greer & Greenberg, 2008). Forskolin, an adenylyl cyclase activator and hence CREB activator, was sufficient to induce Siah2 expression and HIF‐1α stabilization and activation (Fig 5G–J). In agreement with an indirect CREB‐dependent activation of HIF‐1α via increased Siah2 expression, we observed that CREB activation preceded HIF‐1α activation (Fig 5K).
Figure EV2. Glut3 and Siah2 expression is regulated by CREB.
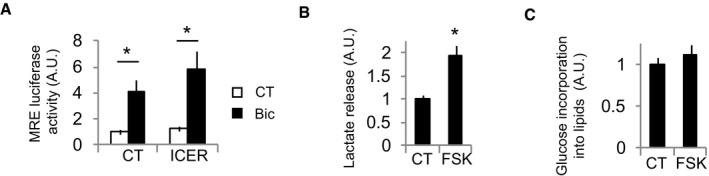
- Luciferase‐based MEF2 activity in neurons expressing a control plasmid (globin) or the CREB inhibitor ICER and stimulated with Bic+4‐AP for 8 h (n = 7 independent experiments). Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey's post hoc test.
- Lactate released into the medium by neurons stimulated with 10 μM forskolin (for 24 h; n = 4 independent experiments). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
- 14C‐U‐glucose incorporation into lipids in neurons treated with 10 μM forskolin for 48 h (n = 4 independent experiments). Values represent mean ± s.e.m.
As expected, A‐CREB expression blocked activity‐dependent lactate release (Fig 5L) and glucose incorporation into lipids (Fig 5M). Forskolin also stimulated lactate release (Fig EV2B) but had no effect on glucose incorporation into lipids (Fig EV2C), in agreement with the fact that cAMP greatly potentiates neuritic growth in response to neurotrophic factors but does not promote growth by its own (Goldberg et al, 2002).
Defective glycolysis impairs neurite growth
This and the previous results suggest that Siah2 and HIF‐1α may be necessary for neurite growth through regulating glycolysis. Indeed, HIF‐1α or Siah2 overexpression in DIV 3 immature cortical neurons increased neurite length (Fig 6A–C) that at this stage represents axonal growth (Dotti et al, 1988). However, HIF‐DN expression or Siah2 knockdown blocked activity‐dependent neurite growth in DIV 10 cortical neurons (Fig 6D–G).
Figure 6. Defective glycolysis impairs neurite growth.
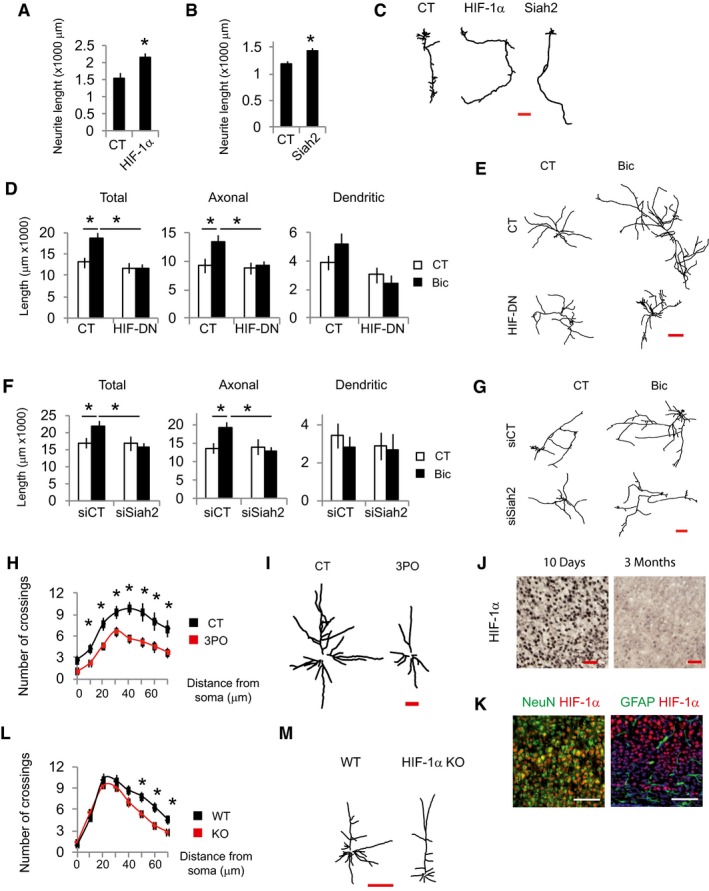
-
A–CAnalysis of neurite length of immature neurons transfected at DIV1 with GFP‐expressing plasmids plus control plasmids (globin) or expressing HIF‐1α (A), Siah2 (B) and analyzed 48 h after transfection and (C) representative tracings (n = 27–40 neurons from six independent experiments). Scale bar, 100 μm. Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
D–GAnalysis of neurite length and representative tracings of neurons transfected at DIV8 with GFP expressing plus plasmids control (globin) or expressing HIF‐DN (D and E) or a non‐targeting siRNA (siCT) or a pool of four siRNAs targeting Siah2 (F and G) and stimulated the day after with Bic+4‐AP for 48 h (n = 27–36 neurons from six independent experiments). Scale bar, 500 μm. Values represent mean ± s.e.m. *P < 0.05, one‐way ANOVA followed by Tukey's post hoc test.
-
H, I(H) Sholl analysis and (I) representative tracings of Golgi‐stained layer 5 pyramidal neurons of the somatosensory cortex of 14‐day‐old rats administered with 3PO (50 mg/kg) for 5 days (n = 15–20 neurons from four mice per condition). Scale bar, 100 μm. Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
JImmunohistochemical detection of HIF‐1α in sections of the somatosensory cortex from 10‐day and 3‐month‐old mice (n = 3 mice per condition). Scale bar, 50 μm.
-
KImmunofluorescence detection of HIF‐1α, NeuN, and GFAP in cortical sections from 10‐day‐old mice (n = 3 mice per condition). Scale bar, 100 μm.
-
L, M(L) Sholl analysis and (M) representative tracings of Golgi‐stained layer 5 pyramidal neurons of the somatosensory cortex of WT and early postnatal deleted HIF‐1α mice (n = 16 neurons from three mice per condition). Scale bar, 100 μm. Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
The first 3 weeks after birth is when maximum neurite growth occurs in mice, being synaptic activity one of the effectors of this growth (Wong & Ghosh, 2002). In order to provide evidence about the in vivo role of glucose metabolism on neurite growth, we administered PFKFB3 inhibitor 3PO (Schoors et al, 2014) for 5 days to rat pups that were 8 days old. Golgi staining showed reduced complexity of neuritic arbors in the cortex of 3PO administered rats (Fig 6H and I).
HIF‐1α deletion in neural precursor cell progenitors leads to atrophy of the cerebral cortex, indicating an important role for HIF‐1α in neuronal development in vivo (Tomita et al, 2003). Thus, we compared HIF‐1α expression in the brains of mice that were 10 days and 3 months old. We observed a higher density of HIF‐1α‐positive cells and increased protein levels in 10‐day‐old mice compared to 3‐month‐old mice (Figs 6J, and EV3A and B). Expression of HIF‐1α in 10‐day‐old mice brain was highest in the cortex, hippocampus, and corpus callosum (Fig EV3C). Dual staining revealed colocalization of HIF1α with the neuronal marker NeuN and complete absence in astrocytes expressing GFAP (Fig 6K).
Figure EV3. HIF‐1α is highly expressed in the brain of 10‐day‐old mice.
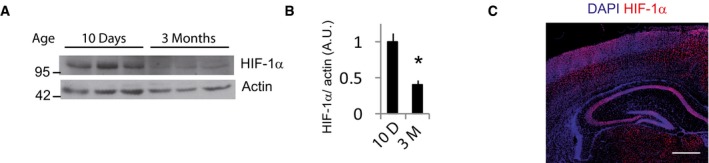
-
A, B(A) Representative Western blot of HIF‐1α expression in protein extracts of the cortex of 10‐day and 3‐month‐old mice and (B) densitometric analysis (n = 4 mice per condition). Values represent mean ± s.e.m. *P < 0.05, two‐tailed Student's t‐test.
-
CImmunofluorescence detection of HIF‐1α in coronal sections from 10‐day‐old mice (n = 3 mice). Scale bar, 100 μm.
Source data are available online for this figure.
In order to provide evidence about the in vivo role of HIF‐1α in neurite growth, we deleted HIF‐1α in tamoxifen‐inducible HIF‐1α KO mice that were 3–5 days old, when gross anatomy of the brain resembles to that of the adult but is characterized by outgrowth of dendrites and axons. Once these animals reached adulthood (4–6 months), the neuronal architecture of the cortex of WT and HIF‐1α KO mice was analyzed by Golgi staining, which showed reduced complexity of neuritic arbors in the KO mice (Fig 6L and M). Interestingly, glucose metabolism is necessary for axonal growth in vitro with no effect on dendritic growth meanwhile in vivo the effect observed accounts mainly for dendritic growth. This is possibly due to the fact that in the developmental stage when neurites were analyzed in vitro, dendrites are mostly stable and axons are growing while postnatal dendrites are still growing in vivo. Taken together, our results indicate that induction of glucose metabolism in neurons is required for proper neuronal development regardless whether the neurons are in axonal or dendritic growth state.
Discussion
Signaling from afferents plays an essential role in neurite growth (Rajan & Cline, 1998; Groc et al, 2002; Wong & Ghosh, 2002). Here, we show that activity‐dependent neurite growth requires induction of glucose metabolism. Synaptic activity enhances glucose uptake and metabolism, some of which is diverted toward lipid synthesis that is required for cell membrane enlargement during outgrowth. The induction of glucose metabolism is mediated at the transcriptional level, and CREB induces Glut3 and Siah2, the latter promoting the normoxic stabilization of HIF‐1α that regulates the expression of the rate‐limiting enzymes involved in glycolysis (Fig 7A and B).
Figure 7. Model for synaptic activity inducing glucose metabolism to supply lipids for neurite growth.
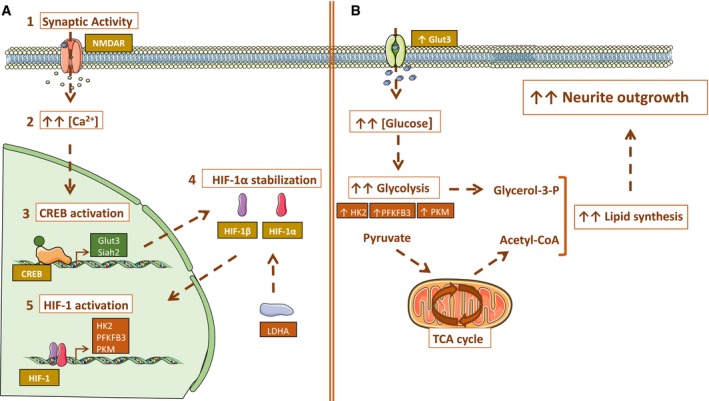
- Synaptic activity induces glucose metabolism at transcriptional level: Synaptic activity (1) induces Ca2+ transients (2) that activate the transcription factor CREB (3). CREB upregulates Glut3 expression to increase glucose uptake by neurons. CREB also induces Siah2 expression that together with LDH activity promotes the stabilization and activation of HIF‐1α (4, 5), which in turn activates the expression of genes encoding the rate‐limiting enzymes of glycolysis.
- Transcriptional changes induced by synaptic activity increase glucose uptake and enhance glycolytic flux. As a result, metabolites such as acetyl‐CoA are generated and used for lipid synthesis necessary for membrane extension during neurite growth. Some graphics in the figure were obtained and modified from Smart Servier Medical Art (https://smart.servier.com/).
Most of the glucose in the brain is oxidized to supply the large amounts of ATP required for maintaining membrane ion gradients and processes regulating to synaptic transmission (Harris Julia et al, 2012; Rangaraju et al, 2014; Magistretti Pierre & Allaman, 2015). However, measurements of human brain glucose and oxygen consumption reveal that the total amount of glucose consumed is in excess of oxygen consumption (Vaishnavi et al, 2010; Goyal Manu et al, 2014). The utilization of aerobic glycolysis in the human brain differs across the developmental stages, with 35% of glucose being consumed aerobically after birth that falls to 10–12% in adulthood (Goyal Manu et al, 2014). This increased aerobic glycolysis during infancy correlates with the period of maximal neurite growth (Goyal Manu et al, 2014; Silbereis John et al, 2016). Neuronal differentiation in vitro shows increased expression levels of Glut3 and glycolysis enzymes, with 2‐DG treatment of DIV1 cortical neurons blocking differentiation (Agostini et al, 2016). Our data show that glucose metabolism is necessary for axonal growth in vitro and dendritic growth in vivo. This may indicate that enhanced glycolysis does not trigger axonal or dendritic growth per se, but it is necessary to supply the building blocks required to enlarge the membranes regardless of whether the neurons are in axonal or dendritic growth state. Highly proliferative cancer cells rely on aerobic glycolysis, which it is known as the Warburg effect (Vander Heiden et al, 2009). Despite aerobic glycolysis being a less efficient way of generating ATP, it confers advantages to cancer cells such as the production of intermediates that support cell growth and division (Vander Heiden et al, 2009). Although postmitotic neurons do not divide, they undergo massive growth during differentiation. A neurite with a diameter of 1 μm elongating 0.55 μm/day must expand its surface at a rate of around 1 μm2/min to reach 250,000 μm2 which is the typical neuron surface area, compared with 1,256 μm2 membrane surface of a spherical cell with 20 μm diameter (Pfenninger, 2009). Despite that glucose incorporation into lipids in the growth cone was not analyzed, several evidences strongly support the view that glucose‐derived lipids are used to enlarge membranes: (i) Synaptic activity induced glucose uptake and metabolism and increased glucose incorporation into lipids but not glutamine incorporation into lipids; (ii) synaptic activity increased the total and cytosolic levels of the lipids precursor acetyl‐CoA, which was dependent of glucose metabolism; (iii) pharmacological inhibition of glycolysis affected neurite growth in vitro and in vivo; (iv) knockdown of GPI, the second enzyme in the glycolysis, blocked both glucose incorporation into lipids and activity‐mediated neurite growth; (v) knockdown of ACLY, the enzyme linking glycolysis with fatty acid metabolism, blocked both glucose incorporation into lipids and activity‐mediated neurite growth; (vi) genetic manipulation of CREB, HIF‐1α, and Siah2 block both glucose incorporation into lipids and activity‐mediated neurite growth; and (vii) HIF‐1α knockout in postnatal mice reduced dendritic architecture in vivo.
Astrocytes and neurons are metabolically coupled. The astrocyte‐neuron lactate shuttle (ANLS) hypothesis proposes that the lactate produced by astrocytes is released and taken up by neurons to produce energy (Pellerin & Magistretti, 1994). Although there is a lot of evidence supporting the ANLS, there are also data that oppose this hypothesis (Pellerin & Magistretti, 2012; Patel et al, 2014; Lundgaard et al, 2015; Dienel, 2017). Here, we clearly demonstrate that synaptic activity boosts glucose uptake and metabolism in neurons, rather than glial cells, by upregulating Glut3 and glycolysis genes. Increasing the proportion of glial cells in our primary cultures did not result in increased lactate release after stimulation, neither BDNF treatment affected lactate release in astrocytes unlike the stimulatory effect in lactate release in neuronal cultures. Moreover, glucose uptake in neurons was observed by live imaging, and HIF‐1α activation measured with luciferase reporters is based in neuronal‐specific transfection protocol which nearly the totality of transfected cells are neurons (Soriano et al, 2008). In accordance with our results, a very recent study (Bas‐Orth et al, 2017) showed synaptic activity caused an upregulation of glycolytic genes with the subsequent increase in glycolytic metabolism. Isolating neurons by FACS allowed the authors to conclude that the induction of glycolytic genes was produced in neurons. Despite strong evidence indicating that glucose metabolism is activated by synaptic activity in neurons, these results do not oppose the ANLS, but supports the notion that neurons take up and metabolize through glycolysis in certain conditions (Zala et al, 2013; Lundgaard et al, 2015; Jang et al, 2016; Ashrafi et al, 2017; Díaz‐García et al, 2017). Cell division, migration, and neurite growth require extraordinary levels of energy that would explain why the supply of energy in neurons is not limited to the astrocytic lactate during pre‐ and postnatal stages. This is also seen in the support of cholesterol synthesis in neurons by astrocytes, which can supply sufficient levels of cholesterol to adult neurons unable to synthesize cholesterol, but cannot do so during the phase of maximal membrane growth (Fünfschilling et al, 2012).
Activity‐mediated CREB activation plays a pivotal role in enhanced glucose metabolism. In neurons, CREB is a key transcription factor that regulates plasticity, cell survival, and neurite growth (Lonze & Ginty, 2002; Redmond et al, 2002; Greer & Greenberg, 2008; Bading, 2013). In peripheral tissues, CREB has a major metabolic role, regulating glucose and lipid metabolism in insulin‐sensitive tissues (Altarejos & Montminy, 2011). As previously described (Rajakumar et al, 2004), we observed CREB‐mediated regulation of Glut3 gene expression, the main glucose transporter in neurons. In addition, CREB promoted Siah2 gene expression and, consequently, HIF‐1α stabilization, which resulted in the increased gene expression of the rate‐limiting enzymes involved in glycolysis. HIF‐1α is best known as a transcription factor that mediates adaptation to hypoxia. Genes associated with glucose metabolism are the largest functional group regulated by HIF‐1α (Semenza et al, 1994; Denko, 2008). HIF‐1α KO leads to embryonic lethality at E11, producing cardiovascular and brain malformations (Iyer et al, 1998; Ryan et al, 1998). HIF‐1α deletion in neuronal progenitors elicits cerebral cortex atrophy, defective brain development with fewer neural cells and impaired of spatial memory (Tomita et al, 2003); however, HIF‐1α deletion in mature neurons using CaMKII‐CRE does not generate any evident major morphological defect (Helton et al, 2005). Here, we show that HIF‐1α deletion in early postnatal mice, a period of maximal afferent innervations and neurite growth, disturbs neurite architecture in the adult.
HIF‐1α activation is regulated at multiple levels. We observed that synaptic activity reduced PHD activity, while CREB upregulated the expression of the ubiquitin ligase Siah2 and that activity‐dependent induction of Siah2 is necessary for stabilizing HIF‐1α. Siah2 KO mice exhibit mild phenotypes and do not show obvious brain abnormalities (Frew et al, 2003). However, the presence of other Siah proteins (1a and 1b) may explain this lack of phenotype, given that double Siah1a/2 KO animals show a more severe phenotype regarding HIF‐1α activity than Siah2 KO mice (Nakayama et al, 2004). The first mechanism described by which Siah1 and 2 proteins stabilize HIF‐1α was by promoting proteasomal degradation of PHDs (Nakayama et al, 2004). However, we could not detect reduction in PHDs after synaptic activity. HIF‐1α is known to interact with more than one hundred proteins (Semenza, 2017), most of them regulating HIF‐1α stability. Among the interacting proteins, some of them have been shown to be targeted by Siah2, such as Polo‐like kinase 3 (PLK3) that phosphorylates and destabilizes HIF‐1α (Xu et al, 2010), and Siah2‐mediated PLK3 degradation results in HIF‐1α activation (Li et al, 2017). Sprouty2 promotes HIF‐1α ubiquitination (Hicks & Patel, 2016) and is regulated by Siah2 (Qi et al, 2008), and its downregulation promotes axonal growth (Hausott et al, 2009, 2012; Marvaldi et al, 2015); however, the link between these three observations has not been established. We observed that both Siah2 knockdown and LDH inhibition almost completely blocked activity‐dependent HIF‐1α stabilization, what indicates that they may share a common regulatory pathway. Acid pH and LDH promote the non‐canonical conversion of 2‐oxoglutarate to L‐2‐HG which functions as a potent inhibitor of PHDs (Burr Stephen et al, 2016; Nadtochiy et al, 2016; Intlekofer et al, 2017). Disruption of the OGDHC results in accumulation of oxoglutarate with the subsequent increase in L‐2‐HG and HIF‐1α stabilization (Burr Stephen et al, 2016). Siah2 has been reported to disrupt the OGDHC complex (Habelhah et al, 2004; Sun Ramon & Denko Nicholas, 2014), but acute synaptic activity did not affect the protein levels of OGDHC. Future studies will be aimed to understand the interrelationship between Siah2 and LDH to promote activity‐mediated HIF‐1α stabilization.
Disruption of dendritic development is the most consistent anatomical finding in mental retardation (Kaufmann & Moser, 2000). The role of abnormal cytoskeletal remodeling has been associated with mental retardation in some syndromes but further studies are required to determine how glucose metabolism is affected. Glut1 deficiency syndrome or chronic hypoglycemia (congenital or early infantile) causes the syndrome of glycopenia which is characterized by seizures, developmental delay, and mental retardation (Pascual et al, 2007). Here, we have shown that activity‐mediated neurite growth requires enhanced glucose metabolism to supply lipids for membrane enlargement. Further studies are required to determine how glucose metabolism is affected in intellectual development disorders. Those studies could lead to the possibility of manipulating glucose metabolism to treat of some forms of intellectual development disorders.
Material and Methods
Cell culture and stimulation
Cortical neurons from E21 Sprague Dawley rats were cultured as described previously (Martorell‐Riera et al, 2015). See Appendix Supplementary Methods.
HIF‐1α gene inactivation in vivo
To study the in vivo role of HIF‐1α in neuronal architecture, we used previously described HIF1α floxed UBC‐CRE‐ERT2 mice (Soro‐Arnaiz et al, 2016). These mice ubiquitously express a tamoxifen‐inducible CRE recombinase (cre‐ERT2) that allows global inactivation of HIF‐1α locus flanked by two LoxP sites upon 4OH‐tamoxifen treatment. For HIF‐1α gene inactivation, the newborn mice received tamoxifen via breastfeeding from the mother. Weaning mothers were injected intraperitoneally with 4OH‐tamoxifen daily for 5 days (2 mg/day) starting 3–5 days postpartum. After this period, females were returned to a standard mouse diet. Once newborns reached adulthood (4–6 months), neuronal architecture was analyzed. Mice were kept under specific pathogen‐free conditions at the animal facility at the Autonomous University of Madrid (UAM).
3PO administration
Histology
Transfection, plasmids, and virus generation
Neurons were transfected at DIV8 using Lipofectamine 2000 (Invitrogen). Transfection efficiency was approximately 5% in which nearly the totality of transfected cells is neurons (Soriano et al, 2008).
Neurons were infected with rAAV at DIV4. Infection efficiencies were determined at DIV 10–11 by analyzing GFP fluorescence or immunocytochemical analysis and were observed to range from 70 to 85% of the viable neurons. See Appendix Supplementary Methods.
Neurite length measurement
Cortical neurons were transfected with a plasmid expressing GFP, and neurons were fixed 48 h later with 4% paraformaldehyde, permeabilized, blocked, and incubated overnight at 4°C with anti‐GFP antibody (1:750, A11122, Life Technologies). Antibody binding was visualized using a biotinylated secondary antibody (1:200, Jackson Immuno Research) and Cy3‐conjugated streptavidin (1:500, Jackson Immuno Research). Preparations were mounted on VECTASHIELD Mounting Medium with DAPI (Vector Laboratories).
Images were taken blindly at 4× magnification using an Olympus BX61 microscope equipped with an Olympus DP70 camera. Neurites were manually traced and analyzed using Simple Neurite Tracer software (Longair et al, 2011).
Luciferase assays
Cells were transfected with firefly luciferase‐based reporter plasmid along with a Renilla expressing vector (pTK‐RL; Promega), together with, where relevant, an HIF‐DN or A‐CREB expression vector. Luciferase assays were performed using the Dual Glo Luciferase Assay system (Promega) with firefly luciferase‐based reporter gene activity normalized to the Renilla control (pTK‐RL plasmid), except the CMV‐ODD‐Luc and CMV‐Luc experiments that were normalized to CMV‐Renilla.
RNA isolation, RT–PCR, and qPCR
RNA was isolated using an PureLink™ RNA mini kit (Life Technologies). For qPCR, cDNA was synthesized from RNA using the SuperScript® III First‐Strand Synthesis SuperMix (Life Technologies) following the manufacturer's instructions. qPCR was performed in a StepOne Real‐Time PCR System (Applied Biosystem) using GoTaq qPCR Master Mix (Promega) according to the manufacturer's instructions. See Appendix Supplementary Methods.
Western blotting and antibodies
Acetyl‐CoA and CoA determination
Acetyl‐CoA and CoA levels were measured using the acetyl‐coenzyme A and CoA Assay Kits, respectively (Sigma). See Appendix Supplementary Methods.
Glucose uptake measurements
The uptake of 2‐[N‐(7‐nitrobenz‐2‐oxa‐1,3‐diazol‐4‐yl)amino]‐2‐deoxy‐glucose (2‐NBDG, Life Technologies), a fluorescent glucose analog, was used to measure glucose transport. Cortical neurons were rinsed three times with phenol‐red free SGG medium with reduced glucose concentration (0.5 mM) and incubated with 100 μM 2‐NBDG in reduced glucose SGG medium for 30 min at 37°C and 5% CO2. Cultures were washed three times with phenol‐red free SGG medium to remove free 2‐NBDG. Accumulation of intracellular 2‐NBDG, measured using an excitation wavelength of 488 nm, was imaged under a Leica DMIRB microscope equipped with a Leica DFC 550 camera at 40× magnification. ROIs of the same surface were drawn in the soma, and fluorescence intensity was analyzed using ImageJ (Schneider et al, 2012).
Imaging studies
Neurons were visualized using a TCS SP2 Leica confocal laser scanning microscope (Leica Lasertechnik GmbH, Mannheim, Germany) adapted to an inverted Leitz DMIRBE microscope at 37°C in a controlled 5% CO2 atmosphere (Life Imaging Services). Pictures were acquired using a 40× (1.25–0.75 NA) Leitz Plan‐Apochromatic objective. Images were analyzed using ImageJ software.
Cytoplasmic Ca2+ was monitored with Fluo‐4 (Life Technologies). Neurons were loaded 2 μM Fluo‐4 for 45 min at room temperature in phenol‐red free SGG medium with 10 mM HEPES and 10 mM glucose. After three washes with phenol‐red free SGG medium, neurons were de‐esterified for 30 min at room temperature, excited at 488 nm, and emission captured with a 516‐nm filter.
For hypoxia analysis, neurons were loaded with 10 μM Image‐IT Hypoxia Reagent (Life Technologies) in HBSS medium and placed in an incubator chamber attached to the microscope, which was flushed with 95% N2/5% CO2 at a flow rate of 20 l/min at 37°C for 30 min. Non‐hypoxic neurons were maintained in normoxic conditions during probe incubation. Neurons were excited at 490 nm, and emission was measured using a 610‐nm filter.
Lactate measurement
The culture medium was filtered using 10K Amicon Ultra‐0.5 ml centrifugal filters (EMD Millipore). Then, 50 μl of medium was incubated with 200 μl of reaction buffer (320 mM glycine, 320 mM hydrazine, 2.4 mM NAD+, and 2 U/ml of lactate dehydrogenase (LDH). After 30 min of incubation at room temperature, the lactate‐dependent generation of NADH was measured at 340 nm using the Infinite 200 PRO multimode reader (Tecan). Lactate levels were normalized by total protein levels, quantified using Pierce BCA Protein Assay Kit (Thermo Scientific).
Glucose and glutamine incorporation into lipids
Neurons were grown on glass coverslips for 48 h in a medium containing 0.8 μCi/ml 14C‐U‐glucose (Perkin‐Elmer) or 2 μCi/ml L‐3‐4‐3H(N)‐glutamine (Perkin‐Elmer). Lipid isolation was performed as described previously by Folch et al (1957). See Appendix Supplementary Methods.
ATP measurement
ATP levels were measured using the ATPlite Luminescence Assay System (Perkin‐Elmer) on the Infinite 200 PRO multimode reader (TECAN) following the manufacturer's instructions. ATP levels were normalized by total protein levels, quantified using Pierce BCA Protein Assay Kit (Thermo Scientific).
Statistical analysis
Statistical analysis involved two‐tailed Student's t‐tests. For any multiple comparisons within data sets, we used a one‐way ANOVA followed by Tukey's post hoc test. All data are presented as the mean ± s.e.m. of at least three independent experiments (n). A P value < 0.05 was considered statistically significant.
Author contributions
MS‐M designed and performed most of the experiments and analyzed data; SC‐D, MR‐P, CM‐S designed and performed experiments and analyzed data; AM‐R and IH performed experiments and analyzed data; JA performed HIF‐1α gene inactivation in vivo and had critical input into the manuscript preparation; MR and OMM‐E analyzed data and had critical input into the manuscript preparation; FXS conceived, designed and performed experiments, analyzed data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
This work was supported by research grants from the Spanish Ministerio de Economía y Competitividad/FEDER funds (SAF2011‐30283 and SAF2014‐59872‐P to FXS; SAF2016‐76815‐R to JA; BFU2015‐68135‐P to OMM‐E) and Fundación Ramón Areces (to OMM‐E). MS‐M is the recipient of a predoctoral fellowship from Fundación Tatiana Pérez de Guzmán el Bueno.
The EMBO Journal (2018) 37: e97368
References
- Agostini M, Romeo F, Inoue S, Niklison‐Chirou MV, Elia AJ, Dinsdale D, Morone N, Knight RA, Mak TW, Melino G (2016) Metabolic reprogramming during neuronal differentiation. Cell Death Differ 23: 1502–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C (1998) A dominant‐negative inhibitor of CREB reveals that it is a general mediator of stimulus‐dependent transcription of c‐fos. Mol Cell Biol 18: 967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarejos JY, Montminy M (2011) CREB and the CRTC co‐activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol 12: 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Wu Z, Farrell RJ, Ryan TA (2017) GLUT4 mobilization supports energetic demands of active synapses. Neuron 93: 606–615.e603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H (2013) Nuclear calcium signalling in the regulation of brain function. Nat Rev Neurosci 14: 593–608 [DOI] [PubMed] [Google Scholar]
- Bas‐Orth C, Tan Y‐W, Lau D, Bading H (2017) Synaptic activity drives a genomic program that promotes a neuronal Warburg effect. J Biol Chem 292: 5183–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti Pierre J (2011) Brain energy metabolism: focus on astrocyte‐neuron metabolic cooperation. Cell Metab 14: 724–738 [DOI] [PubMed] [Google Scholar]
- Burr Stephen P, Costa Ana SH, Grice Guinevere L, Timms Richard T, Lobb Ian T, Freisinger P, Dodd Roger B, Dougan G, Lehner Paul J, Frezza C, Nathan James A (2016) Mitochondrial protein lipoylation and the 2‐oxoglutarate dehydrogenase complex controls hif1α stability in aerobic conditions. Cell Metab 24: 740–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denko NC (2008) Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8: 705–713 [DOI] [PubMed] [Google Scholar]
- Díaz‐García CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G (2017) Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab 26: 361–374.e364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA (2017) Lack of appropriate stoichiometry: strong evidence against an energetically important astrocyte‐neuron lactate shuttle in brain. J Neurosci Res 95: 2103–2125 [DOI] [PubMed] [Google Scholar]
- Divakaruni AS, Wallace M, Buren C, Martyniuk K, Andreyev AY, Li E, Fields JA, Cordes T, Reynolds IJ, Bloodgood BL, Raymond LA, Metallo CM, Murphy AN (2017) Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J Cell Biol 216: 1091–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti C, Sullivan C, Banker G (1988) The establishment of polarity by hippocampal neurons in culture. J Neurosci 8: 1454–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faherty CJ, Kerley D, Smeyne RJ (2003) A Golgi‐Cox morphological analysis of neuronal changes induced by environmental enrichment. Dev Brain Res 141: 55–61 [DOI] [PubMed] [Google Scholar]
- Ferreira JM, Burnett AL, Rameau GA (2011) Activity‐dependent regulation of surface glucose transporter‐3. J Neurosci 31: 1991–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH (1957) A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem 226: 497–509 [PubMed] [Google Scholar]
- Frew IJ, Hammond VE, Dickins RA, Quinn JMW, Walkley CR, Sims NA, Schnall R, Della NG, Holloway AJ, Digby MR, Janes PW, Tarlinton DM, Purton LE, Gillespie MT, Bowtell DDL (2003) Generation and analysis of Siah2 mutant mice. Mol Cell Biol 23: 9150–9161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U, Jockusch WJ, Sivakumar N, Möbius W, Corthals K, Li S, Quintes S, Kim Y, Schaap IAT, Rhee J‐S, Nave K‐A, Saher G (2012) Critical time window of neuronal cholesterol synthesis during neurite outgrowth. J Neurosci 32: 7632–7645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JL, Espinosa JS, Xu Y, Davidson N, Kovacs GTA, Barres BA (2002) Retinal ganglion cells do not extend axons by default. Neuron 33: 689–702 [DOI] [PubMed] [Google Scholar]
- Goldberg JL (2003) How does an axon grow? Genes Dev 17: 941–958 [DOI] [PubMed] [Google Scholar]
- Goyal Manu S, Hawrylycz M, Miller Jeremy A, Snyder Abraham Z, Raichle Marcus E (2014) Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab 19: 49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, Greenberg ME (2008) From synapse to nucleus: calcium‐dependent gene transcription in the control of synapse development and function. Neuron 59: 846–860 [DOI] [PubMed] [Google Scholar]
- Groc L, Petanjek Z, Gustafsson B, Ben‐Ari Y, Hanse E, Khazipov R (2002) In vivo blockade of neural activity alters dendritic development of neonatal CA1 pyramidal cells. Eur J Neurosci 16: 1931–1938 [DOI] [PubMed] [Google Scholar]
- Habelhah H, Laine A, Erdjument‐Bromage H, Tempst P, Gershwin ME, Bowtell DDL, Ronai Z (2004) Regulation of 2‐oxoglutarate (α‐Ketoglutarate) dehydrogenase stability by the ring finger ubiquitin ligase siah. J Biol Chem 279: 53782–53788 [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJL, Bading H (2001) Nuclear calcium signaling controls CREB‐mediated gene expression triggered by synaptic activity. Nat Neurosci 4: 261–267 [DOI] [PubMed] [Google Scholar]
- Harris Julia J, Jolivet R, Attwell D (2012) Synaptic energy use and supply. Neuron 75: 762–777 [DOI] [PubMed] [Google Scholar]
- Hausott B, Vallant N, Auer M, Yang L, Dai F, Brand‐Saberi B, Klimaschewski L (2009) Sprouty2 down‐regulation promotes axon growth by adult sensory neurons. Mol Cell Neurosci 42: 328–340 [DOI] [PubMed] [Google Scholar]
- Hausott B, Vallant N, Schlick B, Auer M, Nimmervoll B, Obermair GJ, Schwarzer C, Dai F, Brand‐Saberi B, Klimaschewski L (2012) Sprouty2 and ‐4 regulate axon outgrowth by hippocampal neurons. Hippocampus 22: 434–441 [DOI] [PubMed] [Google Scholar]
- Helton R, Cui J, Scheel JR, Ellison JA, Ames C, Gibson C, Blouw B, Ouyang L, Dragatsis I, Zeitlin S, Johnson RS, Lipton SA, Barlow C (2005) Brain‐specific knock‐out of hypoxia‐inducible factor‐1α reduces rather than increases hypoxic‐ischemic damage. J Neurosci 25: 4099–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks KC, Patel TB (2016) Sprouty2 protein regulates hypoxia‐inducible factor‐α (HIFα) protein levels and transcription of HIFα‐responsive genes. J Biol Chem 291: 16787–16801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Wang B, Liu H, Shah H, Carmona‐Fontaine C, Rustenburg AS, Salah S, Gunner MR, Chodera JD, Cross JR, Thompson CB (2017) L‐2‐Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat Chem Biol 13: 494–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr (2001) HIFα targeted for VHL‐mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292: 464–468 [DOI] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL (1998) Cellular and developmental control of O2 homeostasis by hypoxia‐inducible factor 1α. Genes Dev 12: 149–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian Y‐M, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim AV, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Targeting of HIF‐α to the von hippel‐lindau ubiquitylation complex by O2‐regulated prolyl hydroxylation. Science 292: 468–472 [DOI] [PubMed] [Google Scholar]
- Jang S, Nelson Jessica C, Bend Eric G, Rodríguez‐Laureano L, Tueros Felipe G, Cartagenova L, Underwood K, Jorgensen Erik M, Colón‐Ramos Daniel A (2016) Glycolytic enzymes localize to synapses under energy stress to support synaptic function. Neuron 90: 278–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW (2000) Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex 10: 981–991 [DOI] [PubMed] [Google Scholar]
- Konur S, Ghosh A (2005) Calcium signaling and the control of dendritic development. Neuron 46: 401–405 [DOI] [PubMed] [Google Scholar]
- Li Y‐X, Zhang Y, Lester HA, Schuman EM, Davidson N (1998) Enhancement of neurotransmitter release induced by brain‐derived neurotrophic factor in cultured hippocampal neurons. J Neurosci 18: 10231–10240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Park S, Zhang X, Dai W, Xu D (2017) Mutual regulation between Polo‐like kinase 3 and SIAH2 E3 ubiquitin ligase defines a regulatory network that fine‐tunes the cellular response to hypoxia and nickel. J Biol Chem 292: 11431–11444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longair MH, Baker DA, Armstrong JD (2011) Simple neurite tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 27: 2453–2454 [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605–623 [DOI] [PubMed] [Google Scholar]
- Lonze BE, Riccio A, Cohen S, Ginty DD (2002) Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron 34: 371–385 [DOI] [PubMed] [Google Scholar]
- Lu H, Forbes RA, Verma A (2002) Hypoxia‐inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem 277: 23111–23115 [DOI] [PubMed] [Google Scholar]
- Lundgaard I, Li B, Xie L, Kang H, Sanggaard S, Haswell JDR, Sun W, Goldman S, Blekot S, Nielsen M, Takano T, Deane R, Nedergaard M (2015) Direct neuronal glucose uptake heralds activity‐dependent increases in cerebral metabolism. Nat Commun 6: 6807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti Pierre J, Allaman I (2015) A cellular perspective on brain energy metabolism and functional imaging. Neuron 86: 883–901 [DOI] [PubMed] [Google Scholar]
- Martorell‐Riera A, Segarra‐Mondejar M, Reina M, Martínez‐Estrada OM, Soriano FX (2015) Mitochondrial fragmentation in excitotoxicity requires ROCK activation. Cell Cycle 14: 1365–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvaldi L, Thongrong S, Kozłowska A, Irschick R, Pritz CO, Bäumer B, Ronchi G, Geuna S, Hausott B, Klimaschewski L (2015) Enhanced axon outgrowth and improved long‐distance axon regeneration in sprouty2 deficient mice. Dev Neurobiol 75: 217–231 [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang G‐W, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature 399: 271–275 [DOI] [PubMed] [Google Scholar]
- Molina CA, Foulkes NS, Lalli E, Sassone‐Corsi P (1993) Inducibility and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell 75: 875–886 [DOI] [PubMed] [Google Scholar]
- Nadtochiy SM, Schafer X, Fu D, Nehrke K, Munger J, Brookes PS (2016) Acidic pH is a metabolic switch for 2‐hydroxyglutarate generation and signaling. J Biol Chem 291: 20188–20197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument‐Bromage H, Tempst P, Frappell PB, Bowtell DD, Ronai Z (2004) Siah2 regulates stability of prolyl‐hydroxylases, controls HIF1α abundance, and modulates physiological responses to hypoxia. Cell 117: 941–952 [DOI] [PubMed] [Google Scholar]
- Ohira K, Kumanogoh H, Sahara Y, Homma KJ, Hirai H, Nakamura S, Hayashi M (2005) A truncated tropo‐myosine‐related kinase B receptor, T1, regulates glial cell morphology via rho GDP dissociation inhibitor 1. J Neurosci 25: 1343–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE (2008) Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci 11: 476–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Poo M‐M (2013) Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 14: 7–23 [DOI] [PubMed] [Google Scholar]
- Pascual JM, Wang D, Hinton V, Engelstad K, Saxena CM, Van Heertum RL, De Vivo DC (2007) Brain glucose supply and the syndrome of infantile neuroglycopenia. Arch Neurol 64: 507–513 [DOI] [PubMed] [Google Scholar]
- Patel AB, Lai JCK, Chowdhury GMI, Hyder F, Rothman DL, Shulman RG, Behar KL (2014) Direct evidence for activity‐dependent glucose phosphorylation in neurons with implications for the astrocyte‐to‐neuron lactate shuttle. Proc Natl Acad Sci USA 111: 5385–5390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA 91: 10625–10629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ (2012) Sweet sixteen for ANLS. J Cereb Blood Flow Metab 32: 1152–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenninger KH (2009) Plasma membrane expansion: a neuron's Herculean task. Nat Rev Neurosci 10: 251–261 [DOI] [PubMed] [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo‐San Pedro JM, Madeo F, Kroemer G (2015) Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab 21: 805–821 [DOI] [PubMed] [Google Scholar]
- Puram SV, Bonni A (2013) Cell‐intrinsic drivers of dendrite morphogenesis. Development 140: 4657–4671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Nakayama K, Gaitonde S, Goydos JS, Krajewski S, Eroshkin A, Bar‐Sagi D, Bowtell D, Ronai Z (2008) The ubiquitin ligase Siah2 regulates tumorigenesis and metastasis by HIF‐dependent and ‐independent pathways. Proc Natl Acad Sci USA 105: 16713–16718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakumar A, Thamotharan S, Raychaudhuri N, Menon RK, Devaskar SU (2004) Trans‐activators regulating neuronal glucose transporter isoform‐3 gene expression in mammalian neurons. J Biol Chem 279: 26768–26779 [DOI] [PubMed] [Google Scholar]
- Rajan I, Cline HT (1998) Glutamate receptor activity is required for normal development of tectal cell dendrites in vivo . J Neurosci 18: 7836–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, Ryan Timothy A (2014) Activity‐driven local ATP synthesis is required for synaptic function. Cell 156: 825–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, Ghosh A (2002) Calcium regulation of dendritic growth via CaM kinase IV and CREB‐mediated transcription. Neuron 34: 999–1010 [DOI] [PubMed] [Google Scholar]
- Rose CR, Blum R, Pichler B, Lepier A, Kafitz KW, Konnerth A (2003) Truncated TrkB‐T1 mediates neurotrophin‐evoked calcium signalling in glia cells. Nature 426: 74–78 [DOI] [PubMed] [Google Scholar]
- Ryan HE, Lo J, Johnson RS (1998) HIF‐1α is required for solid tumor formation and embryonic vascularization. EMBO J 17: 3005–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoors S, De Bock K, Cantelmo Anna R, Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong Brian W, Quaegebeur A, Goveia J, Bifari F, Wang X, Blanco R, Tembuyser B, Cornelissen I, Bouché A, Vinckier S, Diaz‐Moralli S, Gerhardt H, Telang S et al (2014) Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab 19: 37–48 [DOI] [PubMed] [Google Scholar]
- Semenza GL, Roth PH, Fang HM, Wang GL (1994) Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia‐inducible factor 1. J Biol Chem 269: 23757–23763 [PubMed] [Google Scholar]
- Semenza GL (2017) A compendium of proteins that interact with HIF‐1α. Exp Cell Res 356: 128–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbereis John C, Pochareddy S, Zhu Y, Li M, Sestan N (2016) The cellular and molecular landscapes of the developing human central nervous system. Neuron 89: 248–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver I, Erecinska M (1994) Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo‐, hypo‐, and hyperglycemic animals. J Neurosci 14: 5068–5076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Léveillé F, Papadia S, Higgins LG, Varley J, Baxter P, Hayes JD, Hardingham GE (2008) Induction of sulfiredoxin expression and reduction of peroxiredoxin hyperoxidation by the neuroprotective Nrf2 activator 3H‐1,2‐dithiole‐3‐thione. J Neurochem 107: 533–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soro‐Arnaiz I, Li Qilong Oscar Y, Torres‐Capelli M, Meléndez‐Rodríguez F, Veiga S, Veys K, Sebastian D, Elorza A, Tello D, Hernansanz‐Agustín P, Cogliati S, Moreno‐Navarrete Jose M, Balsa E, Fuertes E, Romanos E, Martínez‐Ruiz A, Enriquez Jose A, Fernandez‐Real Jose M, Zorzano A, De Bock K et al (2016) Role of mitochondrial complex IV in age‐dependent obesity. Cell Rep 16: 2991–3002 [DOI] [PubMed] [Google Scholar]
- Sun Ramon C, Denko Nicholas C (2014) Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab 19: 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- TeSlaa T, Teitell MA (2014) Chapter Five – Techniques to monitor glycolysis In Methods in enzymology, Galluzzi L, Kroemer G. (eds), Vol. 542, pp 91–114. San Diego, CA: Academic Press; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Ueno M, Sakamoto M, Kitahama Y, Ueki M, Maekawa N, Sakamoto H, Gassmann M, Kageyama R, Ueda N, Gonzalez FJ, Takahama Y (2003) Defective brain development in mice lacking the Hif‐1α gene in neural cells. Mol Cell Biol 23: 6739–6749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnavi SN, Vlassenko AG, Rundle MM, Snyder AZ, Mintun MA, Raichle ME (2010) Regional aerobic glycolysis in the human brain. Proc Natl Acad Sci USA 107: 17757–17762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ROL, Ghosh A (2002) Activity‐dependent regulation of dendritic growth and patterning. Nat Rev Neurosci 3: 803–812 [DOI] [PubMed] [Google Scholar]
- Xu D, Yao Y, Lu L, Costa M, Dai W (2010) Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF‐1α. J Biol Chem 285: 38944–38950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zala D, Hinckelmann M‐V, Yu H, Lyra da Cunha Marcel M, Liot G, Cordelières Fabrice P, Marco S, Saudou F (2013) Vesicular glycolysis provides on‐board energy for fast axonal transport. Cell 152: 479–491 [DOI] [PubMed] [Google Scholar]
- Zhang W, Guo C, Jiang K, Ying M, Hu X (2017) Quantification of lactate from various metabolic pathways and quantification issues of lactate isotopologues and isotopmers. Sci Rep 7: 8489 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5