

Abstract
A growing subset of metalloenzymes activates dioxygen with nonheme diiron active sites to effect substrate oxidations that range from the hydroxylation of methane and the desaturation of fatty acids to the deformylation of fatty aldehydes to produce alkanes and the six-electron oxidation of aminoarenes to nitroarenes in the biosynthesis of antibiotics. A common feature of their reaction mechanisms is the formation of O2 adducts that evolve into more reactive derivatives such as diiron(II,III)-superoxo, diiron(III)-peroxo, diiron(III,IV)-oxo, and diiron(IV)-oxo species, which carry out particular substrate oxidation tasks. In this review, we survey the various enzymes belonging to this unique subset and the mechanisms by which substrate oxidation is carried out. We examine the nature of the reactive intermediates, as revealed by X-ray crystallography and the application of various spectroscopic methods and their associated reactivity. We also discuss the structural and electronic properties of the model complexes that have been found to mimic salient aspects of these enzyme active sites. Much has been learned in the past 25 years, but key questions remain to be answered.
Graphical abstract
1. Introduction
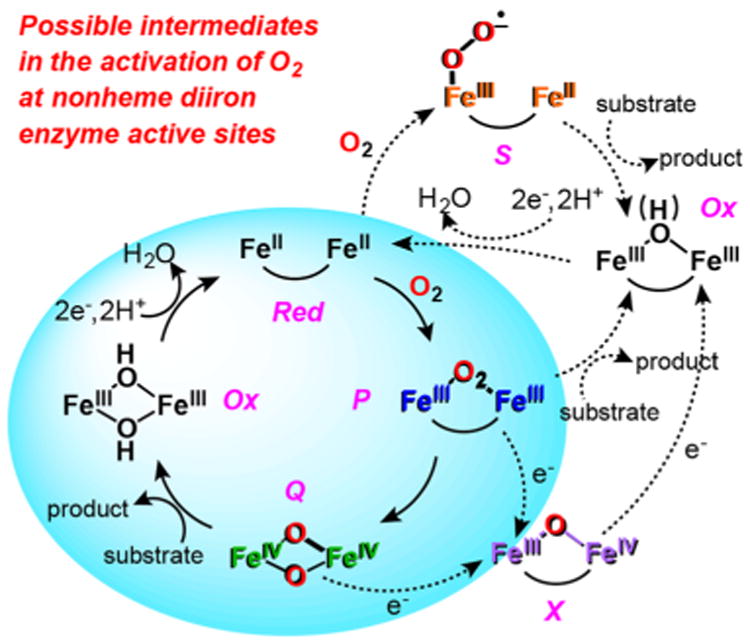
The mechanisms for dioxygen activation by metalloenzymes have fascinated chemists and biochemists ever since Mason and Hayaishi established the important role O2 plays in oxidative metabolism.1,2 One growing subset of such metalloenzymes utilizes nonheme diiron active sites with histidine and carboxylate ligands.3,4 These enzymes activate O2 at ambient temperature and pressure to catalyze reactions as simple as the oxidation of Fe2+ to Fe3+ leading to its biomineralization by ferritins in bacteria, plants, and animals5 to as challenging as the hydroxylation of the 105 kcal/mol C–H bonds of methane by soluble methane monooxygenase (sMMO) found in methanotrophs.6,7 In between these extremes are enzymes that catalyze the 6-electron oxidation of aminoarenes to nitroarenes in the biosynthesis of antibiotics (AurF8 and CmlI9), the conversion of fatty aldehydes to alkanes in algae (aldehyde deformylating oxygenase (ADO)10,11), hydroxylation of benzylic C–H bonds in chloramphenicol biosynthesis (CmlA12), arene hydroxylation (toluene/o-xylene monooxygenase (ToMO13) and toluene-4-monooxygenase (T4MO)14), fatty acid desaturation in plants (stearoyl-acyl carrier protein Δ9-desaturase (Δ9D)15), and the post-translational hydroxylation of deoxyhypusine to hypusine, the novel amino acid residue found in the eukaryotic translational initiation factor 5A required in the regulation of mammalian cell proliferation (deoxyhyupsine hydroxylase (hDOHH)16; Table 1). Enzymes with diiron sites also activate O2 to generate tyrosyl radicals that are needed to initiate the conversion of ribonucleotides to deoxyribonucleotides in DNA biosynthesis by ribonucleotide reductases (RNR).17
Table 1. Nonheme Diiron Enzymes Involved in Dioxygen Activation.
| protein | function or reaction catalyzed | protein fold | notable organisms | refs |
|---|---|---|---|---|
| hemerythrin (Hr) | reversible dioxygen binding in invertebrates | 4-helix bundle | Phascolopsis gouldii, Themiste zostericola | 18–22 |
| soluble methane monooxygenase hydroxylase (sMMOH) | conversion of methane to methanol in methanotrophs | 4-helix bundle | Methylosinus trichosporium OB3b, Methylococcus capsulatus (Bath) | 23–36 |
| ribonucleotide reductase R2 protein from E. coli (RNR-R2) | conversion of ribonucleotides to deoxyribonucleotides | 4-helix bundle | Escherichia coli | 37–44 |
| stearoyl acyl carrier protein (ACP) Δ9-desaturase (Δ9D) | conversion of stearoyl-ACP to oleyl-ACP in plants | 4-helix bundle | Ricinus communis | 45–49 |
| ferroxidase site of frog M ferritin | oxidation of Fe(II) to Fe(III) prior to biomineralization | 4-helix bundle | Rana catesbeiana | 50–53 |
| toluene/o-xylene monooxygenase hydroxylase (ToMOH) | hydroxylation of toluene and o-xylene in bacteria | 4-helix bundle | Pseudomonas stutzeri | 24,54–56 |
| toluene 4-monooxygenase hydroxylase (T4MOH) | hydroxylation of toluene to p-cresol in bacteria | 4-helix bundle | Pseudomonas mendocina | 14,28,57 |
| aldehyde deformylating oxygenase (ADO) | conversion of fatty aldehydes to formate and alkanes | 4-helix bundle | Nostoc punctiforme | 58,59 |
| AurF | p-aminobenzoate N-oxygenase in the biosynthesis of aureothin | 4-helix bundle | Streptomyces thioluteus | 8,60–62 |
| CmlI | arylamine N-oxygenase in chloramphenicol biosynthesis | 4-helix bundle | Streptomyces venezuelae | 9,63,64 |
| CmlA | β-hydroxylation of p-amino-phenylalanine in the first step of chloramphenicol biosynthesis | metallo-β-lactamase | Streptomyces venezuelae | 12,65,66 |
| human deoxyhypusine hydroxylase (hDOHH) | hydroxylation of deoxyhypusine residue on the eukaryotic initiation factor 5a that controls cell proliferation | HEAT repeat | Homo sapiens | 67,68 |
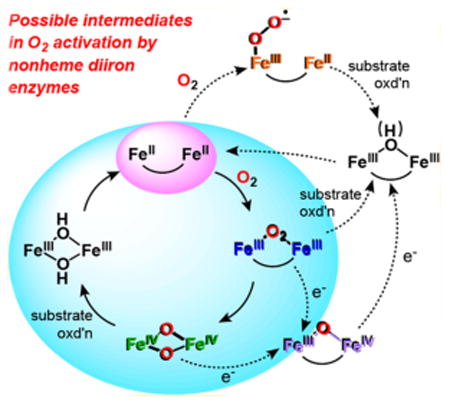
The reduced diiron sites in these enzymes react with O2 to generate various dioxygen-derived intermediates (Scheme 1) that effect the broad range of substrate transformations (Scheme 2). Some of these intermediates have been characterized and found to differ in their iron oxidation states and dioxygen binding modes. These differences can at least partially account for the catalytic versatility of these nonheme diiron enzymes. Based on the accumulated results, dioxygen activation can thus be envisioned as a series of electron transfer steps that lead to the stepwise reduction of the dioxygen unit and concomitant oxidation of the diiron site. In the first step, dioxygen binds to one of the irons in the diferrous cofactor to form an FeIIFeIII-superoxo species S. Subsequent electron transfer from the other Fe then generates a peroxodiferric intermediate P, which has been characterized for a number of enzymes.24,31,35,41,48,49,58,61,63,64,68,69 For the well-studied canonical examples of sMMO and RNR R2, the peroxodiferric intermediate undergoes O–O bond cleavage to generate high-valent (FeIV2 or FeIIIFeIV) intermediates that have been trapped and characterized. All four intermediates, S,70 P,9,54,58,71,72 Q,31,35,36,73–76 and X,6,77,78 have either been postulated or demonstrated to react with substrate in one of the enzymes in this class, and the evidence supporting these notions will be discussed in this review.
Scheme 1. Possible Intermediates in the O2 Activation Chemistry of Nonheme Diiron Enzymes Primarily Based on the Canonical Mechanism for sMMOHa.
aRed is the diferrous starting point and Ox represents the diferric end point of the cycle. P is a peroxodiferric species; Q is the bis(μ-oxo)diiron(IV) oxidant associated with sMMOH; X is the (μ-oxo)diiron(III,IV) species that generates the catalytically essential Y122• radical in RNR R2; S is the diiron(II,III)-superoxo species proposed for T4MOH.
Scheme 2. Array of Transformations Catalyzed by Soluble Diiron Enzymes.
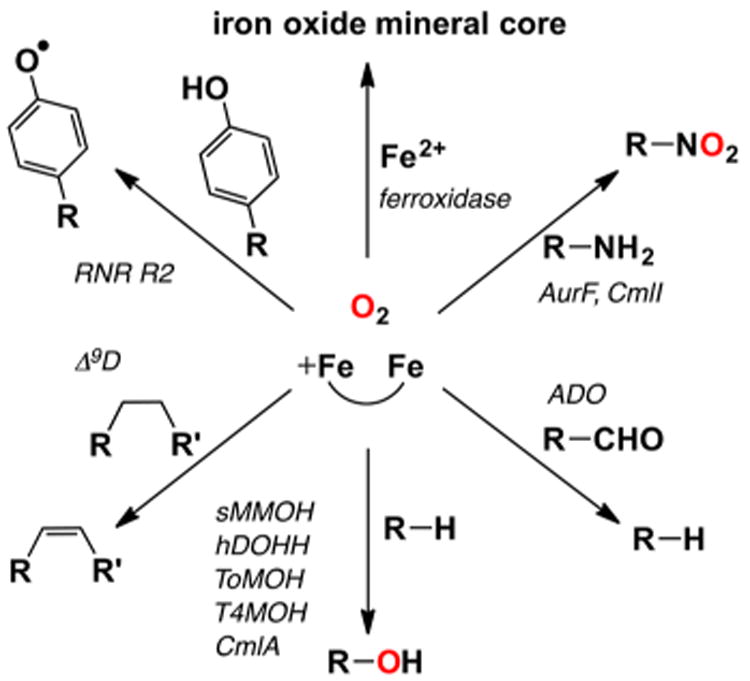
Our discussion begins with the dioxygen carrier protein hemerythrin (Hr), which can be considered as the prototype for this diiron subset of enzymes, even though it does not catalyze a reaction. Due to its relatively small size, Hr was in fact the first of this class to be structurally characterized.79 Hr has a diiron active site with five histidines and two bidentate bridging carboxylates ensconced within a 4-helix-bundle structural motif. In deoxyHr (PDB ID 1HMD), the two iron(II) centers are also bridged by a hydroxo ligand, which mediates an antiferromagnetic coupling interaction (J = 28 cm−1, Ĥ = JS1S2) observed between them.80 In this coordination environment, there is only one vacant site available, and this site has indeed been demonstrated to be utilized for dioxygen binding. Scheme 3 shows the mechanism for dioxygen binding proposed by Solomon81 based on the accumulated spectroscopic and structural evidence, in which O2 coordinates to Fe2, resulting in the transfer of an electron from Fe2 to the bound O2 to form an iron(II)iron(III)-superoxo adduct. The incipient negative charge generated on the dioxygen ligand is then stabilized by hydrogen bonding with the proton on the hydroxo bridge. Transfer of a second electron to the bound O2 this time from Fe1 results in the formation of an iron(III)iron(III)-peroxo species that is then protonated by the hydroxo bridge to form oxyhemerythrin (oxyHr; PDB ID 1HMO). OxyHr is thus best described as a (μ-oxo)diiron(III) species with terminally bound hydroperoxo ligand that is hydrogen bonded to the oxo bridge. The antiferromagnetic coupling of the two irons via the (hydr)oxo bridge and the hydrogen bonding interaction between the dioxygen ligand and the hydroxo bridge provide pathways for facile proton and electron transfer that give rise to reversible O2 binding within this protein active site. These notions serve as the framework for understanding dioxygen activation at non-heme diiron sites for this subset of enzymes.
Scheme 3. Reversible Conversion of Deoxyhemerythrin (deoxyHr, PDB ID 1HMD) to Oxyhemerythrin (oxyHr, 1HMO) via an Unobserved Iron(II)Iron(III)-Superoxo Species Postulated by Solomon81.
Crystal structures of nonheme diiron enzymes started to become available in the 1990s. The first examples were the R2 subunit of the ribonucleotide reductase from E. coli,37,44 the hydroxylase component of soluble methane monooxygenase (sMMOH) from M. capsulatus Bath34 and M. trichosporium Ob3b,29 and the stearoyl-acyl carrier protein Δ9-desaturase (Δ9D) from castor seed.45 These enzymes were all found to utilize 4-helix bundle motifs to house diiron active sites, similar to that found for Hr. However, unlike the His-rich environment of Hr, RNR R2, sMMOH, and Δ9D have carboxylate-rich ligand sets, consisting of two histidines and four carboxylates that presumably play a role in facilitating the activation of dioxygen required for these enzymes to carry out their respective functions.
Other diiron enzymes have been crystallized in the past decade. ToMOH13 and T4MOH,14 enzymes functionally related to sMMOH and belonging to the same bacterial multicomponent monooxygenase (BMM) subset,82 also have their diiron active sites within a 4-helix bundle motif. This is the case as well for other diiron enzymes that carry out more distinct functions, such as the ferroxidase site for the biomineralization of iron in frog ferritin M,51 the cyanobacterial aldehyde deformylating oxygenase (ADO),10,11 and the N-oxygenases AurF8 and CmlI9 that carry out the six-electron oxidation of an aminoaryl moiety to a nitroaryl derivative in the biosynthesis of the antibiotics aureothin and chloramphenicol, respectively.
Two other members of this class of dioxygen activating diiron enzymes deviate from the structural pattern described above. The enzyme CmlA from the biosynthetic pathway for the antibiotic chloramphenicol has a 3-His-4-carboxylate active site found in a metallo-β-lactamase fold and catalyzes the β-hydroxylation of a p-aminophenylalanine moiety on the substrate.12 On the other hand, hDOHH has a 4-His-2-carboxylate active site within a HEAT repeat protein fold16 and carries out the post-translational hydroxylation of the novel amino acid deoxyhypusine found in the eukaryotic translational initiation factor 5A, which is involved in regulating cell proliferation.
These examples demonstrate different strategies employed by Nature to achieve the activation of dioxygen at nonheme diiron sites for its metabolic functions. This review will focus on the structural and spectroscopic properties of diferrous active sites and the peroxodiferric intermediates formed upon introduction of O2. The latter species are important crossroads during enzymatic reaction cycles. In some enzymes, the peroxo species can react directly with substrates, whereas in other enzymes, O–O bond cleavage must first occur to form high-valent iron-oxo intermediates is required for substrate oxidation to occur. Spectroscopic characterization and structural analysis make it possible to better understand the O2 activation process and how Nature chooses to use peroxodiferric intermediates.
2. Structures and Properties of The Diferrous Active Sites
With very few exceptions, the catalytic cycles for dioxygen activating nonheme diiron enzymes start with the diferrous oxidation state, to which O2 binds almost invariably. As precursors to peroxodiferric intermediates, the structures of diferrous precursors can dictate what types of peroxo ligand binding modes are possible. Consequently, these geometric restrictions influence the reactivity of the diiron enzyme. Enzymatic diferrous species have been characterized by X-ray diffraction (XRD) as well as a number of spectroscopic methods, including X-ray absorption spectroscopy (XAS), magnetic circular dichroism (MCD), and Mössbauer spectroscopy. The findings from these studies are summarized in this section.
2.1. Structural Characterization by XRD and EXAFS
Three types of active site structures have been observed in crystals of diferrous enzymes by XRD, categorized into subsets A, B, and C in this review. Subset A consists of diferrous sites that exclusively use two 1,3-carboxylate bridges (Figure 1A), with two protein-derived carboxylate ligands holding the metal centers in the active site. This motif is observed in crystals of Ec RNR R2, Δ9D (Figure 2), MnII-substituted AurF, CmlI and ADO (see Table 2 for references and PDB IDs). Subset B consists of diferrous enzymes with 1,1- and 1,3-bound carboxylate bridges (Figure 1B), a motif observed in crystal structures of sMMOH, MnII-substituted ToMOH, T4MOH, and the diiron form of the RNR R2-like ligand-binding oxidase (R2lox; see Table 2 for references and PDB IDs). The 1,1-carboxylate bridge binds to one metal center in a bidentate mode and to the other metal center via one of the carboxylate O atoms. In addition, sMMOH and ToMOH each have a weakly bound μ-aqua ligand. Subset C active sites have (μ-hydroxo)diferrous cores (Figure 1C), with an additional 1,3-carboxylate bridge in the case of the ferroxidase site in frog M ferritin and with an additional 1,1-carboxylate bridge for CmlA (Table 2). hDOHH would also fall into this category. The Fe···Fe distances found for these diferrous enzymes by XRD range from 2.7 to 4.1 Å and average about 3.5 Å. The values at the extremes are provided by members of Subset A, likely reflecting the flexibility a carboxylate ligand has in adopting syn/syn, syn/anti, and anti/anti binding modes to bridge between two metal centers86,87 to accommodate the protein superstructure.88
Figure 1.
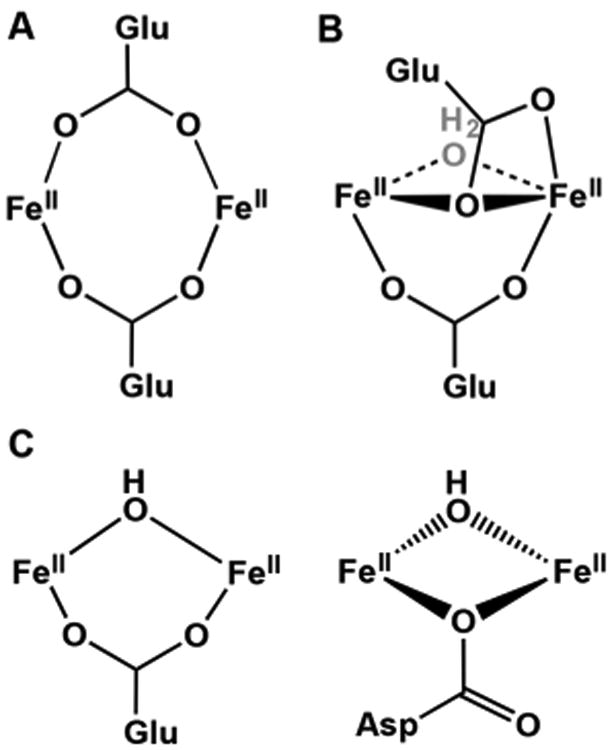
Gallery of diferrous active sites. Subset A uses exclusively two μ-1,3-carboxylate ligands, while subset B employs both a μ-1,1- and a μ-1,3-carboxylate ligand. Subset C has a hydroxo bridge and may also include either a μ-1,1- or a μ-1,3-carboxylate bridge.
Figure 2.
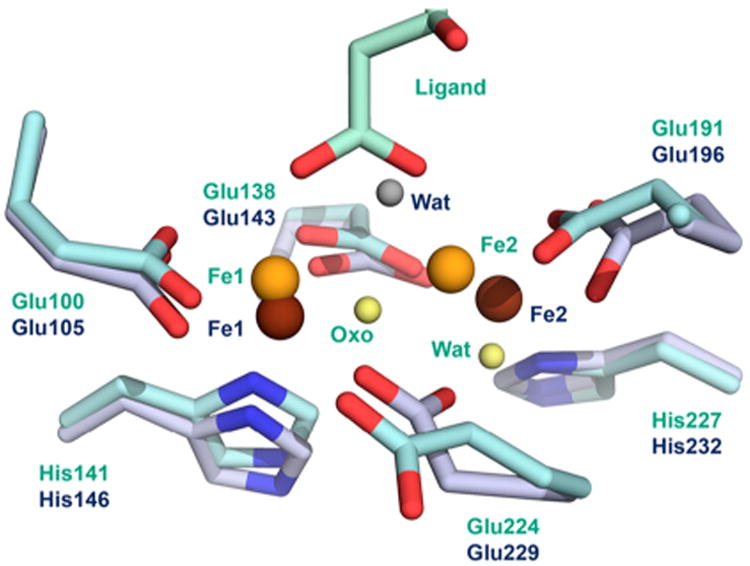
Superimposition of the diferrous active site of the castor desaturase (PDB ID 1AFR)45 and the oxidized active site of the ivy desaturase (PDB ID 2UW1).88 The reduced active site is shown in blue gray, with brown irons and gray water (Wat), while the oxidized active site is shown in cyan, with orange iron ions, and yellow oxygen atoms for the oxo bridge and the water ligand. Reproduced from ref 89 with permission. Copyright 2008 The American Society for Biochemistry and Molecular Biology. All rights reserved.
Table 2. Properties of Diferrous Active Sites of Nonheme Diiron Enzymes.
| protein | PDB ID (resolution, Å) | ligand set [Fe C.N.]a | bridge type | K-edge (eV) [pre-edge area (units)] | d(Fe···Fe) by XRD [XAS] | δ [ΔEQ] (mm/s) couplingb | refs |
|---|---|---|---|---|---|---|---|
| Hr | 1HMD (2.0) | 5H2C [5SP, 6] |
μ-OH 2 μ-1,3-O2CR |
3.3 Å [3.57] | 1.20 [2.89] antiferro | 18–20 | |
| Ec RNR-R2 | 1PFR (2.2) | 2H4C [4, 5SP]* | 2 μ-1,3-O2CR | 3.9 Å [3.41] | 1.26 [3.13] antiferro | 37–40 | |
| Δ9D | 1AFR (2.4) | 2H4C [5SP, 5SP]* | 2 μ-1,3-O2CR | 4.1 Å | 1.30 [3.04, 3.36] antiferro | 45–47 | |
| sMMOH | 1FYZ (2.15) | 2H4C [5TBP,5TBP]* |
μ-1,1-O2CR μ-1,3-O2CR μ-OH2c |
− [10, 9.6] | 3.4 Å [3.43, 3.29] | 1.30 [3.01] (g∼16 EPR) ferro | 23–28 |
| ToMOH | 2IND (2.2) | 2H4C [5, 5] |
μ-1,1-O2CR μ-1,3-O2CR μ-OH2c |
− [10.5] | 3.3 Å (MnII2) [3.37] | 1.32, 1.39 [3.06, 2.13] | 24,54–56 |
| T4MOH with bound T4MOD | 3DHI (1.68) | 2H4C [5TBP, 5TBP]* |
μ-1,1-O2CR μ-1,3-O2CR |
3.4 Å | 1.31 [3.21, 2.68] (g∼16 EPR) ferro | 14,28,57 | |
| ADO | 4RC6 (2.9) | 2H4C | 2 μ-1,3-O2CR | 2.7, 3.1 Å | 1.30 [3.10] (g∼16 EPR) ferro | 58,59,260 | |
| Ferroxidase of frog M ferritin | 4LQN (1.59) | 2H4C [5SP, 5TBP]* |
μ-OH μ-1,3-O2CR |
7122.2 [13] | 3.5 Å [3.43] | 1.30 [3.00] antiferro | 50–53 |
| R2lox | 4XBV (1.8) | 2H4C |
μ-1,1-O2CR μ-1,3-O2CR |
7121.4 | 3.6 Å [3.64] | 1.27 [2.97] | 83–85 |
| AurF | 2JCD (2.11) | 3H4C [5TBP, 5SP]* | 2 μ-1,3-O2CR | 3.6 Å (MnII2) | 1.24 [3.06] (g∼16 EPR) ferro | 8,60–62 | |
| CmlI | 5HYH (2.03) | 3H4C [5, 5] | 2 μ-1,3-O2CR | 7122.1 [8.4] | 3.6 Å [3.35] | 1.25, 1.23 [3.13, 2.80] (g∼16 EPR)d ferro | 9,63,64 |
| CmlA | 5KIK (2.2) | 3H4C [6, 6] |
μ-OH μ-1,1-O2CR |
7121.5 [8.4] | 3.3 Å [3.26] | 1.30, 1.21 [2.75, 2.90] antiferro | 65,66 |
| hDOHH | 4H2C | μ-OH | 7122.7 [8.6] | [3.47] | 1.29 [3.26, 2.90] | 67,68 |
Iron coordination number based on XRD or MCD data, the latter being distinguished by an asterisk.
Designates the nature of the coupling between the ferrous centers, whether ferro- or antiferromagnetic, based on MCD or Mössbauer experiments.
Weakly bound.
John Lipscomb, personal communication.
Extended X-ray absorption fine structure (EXAFS) analysis has also been employed to acquire frozen solution state structural metrics of the diferrous forms of nonheme diiron proteins. Of the eight systems that have parallel XRD and XAS studies, five (sMMOH, ToMOH, frog M ferritin, R2lox, and CmlA) yield structural parameters, including the Fe···Fe distance, that agree between the techniques (Table 2). In these systems XRD provides a three-dimensional picture of the diiron sites, while EXAFS can provide more precise bonding metrics, which together provide a more accurate structural picture of the active site. EXAFS can also be used to identify differences between the crystalline and frozen solution state, if they exist. Having a solution state structure is beneficial for understanding the mechanism of O2 activation of a particular system.
Three diferrous enzymes have Fe···Fe distances from XRD and EXAFS that do not agree, namely Ec RNR R2 (3.9 Å vs 3.41 Å, respectively), Hr (3.3 Å vs 3.57 Å, respectively), and CmlI (3.6 Å vs 3.35 Å, respectively). In the case of Ec RNR R2, the wild type (WT) enzyme was used for XRD experiments,37 whereas the W48A/D84E variant was used in the XAS experiments.38 The D84E mutation changes the nature of a carboxylate ligand in the primary coordination sphere, so the variance in the Fe···Fe distance may be due, at least in part, to a difference in the coordination geometry of the WT enzyme and the W48A/D84E variant.
Similarly, for CmlI the discrepancy may be due to differences in the nature of the samples used. The XRD studies were carried out with CmlIΔ33, a truncated variant of CmlI that is missing 33 amino acids on the N-terminal end, while the sample for XAS analysis used the WT enzyme. Furthermore, the crystal of the diferrous CmlIΔ33 was obtained by soaking a crystal of the μ-1,2-peroxodiferric derivative in dithionite,63 resulting in a subset A-type active site (Figure 1) with an Fe···Fe distance of 3.6 Å. The sample for EXAFS analysis was prepared by reduction of a solution of diferric CmlI to give rise to the EXAFS-derived distance of 3.35 Å,64 classifying it as a subset C active site having a single-atom bridge (Figure 1). Therefore, the differences in the XRD and XAS results may arise from the inability of the active site structure to reorganize in crystallo upon chemical reduction or photoreduction of the crystal during data collection.
On the other hand, the discrepancy in the Hr structures has no clear source. In 1988, Stern and co-workers collected EXAFS data for deoxyHr (FeII2) and oxyHr (FeIII2-OOH) and found respective Fe···Fe distances of 3.57 and 3.24 Å.19 Higher resolution (2.0 Å) crystal structures that became available in 1991 however revealed Fe···Fe distances at 3.3 Å for deoxy- and oxyHr.18 The two studies agreed on the nature of the bridging ligands, μ-hydroxo for deoxyHr and μ-oxo for oxyHr, as well as the Fe···Fe distance for oxyHr, but disagreed on the Fe···Fe distance for deoxyHr. Given that the XRD results showing comparable Fe···Fe distances provide a good basis for understanding the reversibility of dioxygen binding by hemerythrin, no further XAS studies have been reported to resolve the discrepancy in the Fe···Fe distance.
Finally, in the absence of a crystal structure, XAS is often the best method to obtain a structural model. For example, there is currently no crystal structure of the diferrous form of hDOHH, so EXAFS analysis was used to develop a structural model for hDOHH, revealing a μ-hydroxo-bridged diferrous center with an Fe···Fe distance of 3.47 Å.67 The crystal structure of the peroxodiferric form of hDOHH demonstrated that the two glutamate residues in the active site both bind as terminal monodentate ligands16 and would unlikely be able to bridge in a μ-1,3-mode. Thus, these results classify hDOHH into subset C (Figure 1).
2.2. Spectroscopic Characterization
Although XRD can provide information about the iron coordination environment in crystallo, insight into the iron coordination number in solution would also be very useful, as the interaction of dioxygen with the diiron site is more often studied in solution. Dioxygen binding would be expected to occur more readily at coordinatively unsaturated centers, where unsaturated implies that the metal center is less than six-coordinate. Two spectroscopic techniques useful for obtaining such information are X-ray absorption near edge structure (XANES)90,91 and magnetic circular dichroism (MCD).92
XANES data is obtained as part of an X-ray absorption spectrum, which is collected at a synchrotron source. In XANES analysis, one typically observes two spectroscopic features, the K-edge energy of the iron center, which represents the energy required to excite a 1s core electron from the iron nucleus into the continuum, and a much less intense pre-edge peak observed to lower energy relative to the K-edge, which corresponds to the formally spin-forbidden 1s-to-3d transition(s) of the first-row transition metal center. As illustrated in Figure 3 (left panel), the K-edge is observed to shift to higher energy in the series consisting of diferrous CmlI, the peroxodiferric intermediate of CmlI called CmlI-P, oxyhemerythrin, and the oxoiron(IV) complex [FeIV(O)(MePy2TACN)](OTf)2) (MePy2TACN = N-methyl-N′,N″-bis(2-pyridylmethyl)-1,4,7-triazacyclononane).93 The progressive increase in the K-edge energy for this series reflects a general increase in the electron affinity of the metal center in the series as the oxidation state of the metal rises. A comparison of the handful of K-edge energies reported for diferrous enzymes (Table 2) shows that they fall within a range between 7121.5 and 7122.7 eV, a 1.2-V difference.50,65,67,84 That these enzymes do not have the same K-edge energy demonstrates that the K-edge energy is sensitive to other factors besides oxidation state, particularly to coordination environment.94–97 Therefore, analysis of K-edge energies alone is not sufficient to establish the oxidation state of an Fe center.
Figure 3.
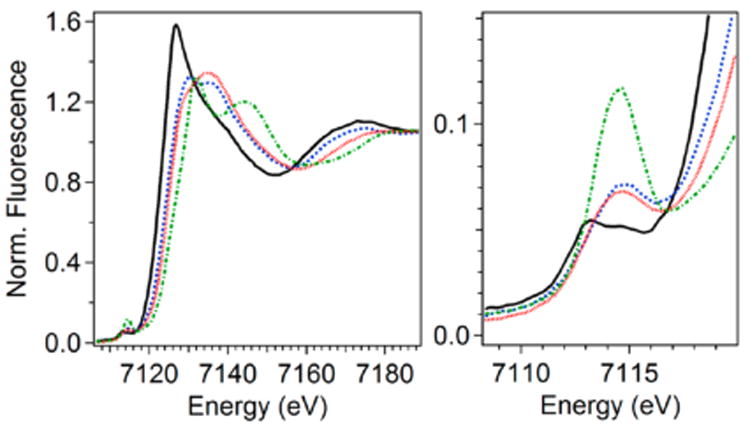
Fe K-edge XAS fluorescence spectra (normalized) of the XANES region. Representative spectra of a diferrous enzyme (black solid line, CmlI), a peroxodiferric species (red dotted line, CmlI; blue dashed line, oxyHr), and an oxoiron(IV) complex (green dashed dotted line, [FeIV(O)(MePy2TACN)](OTf)2). The left panel shows the entire XANES region, while the right panel zooms in on the pre-edge region showing peaks arising from 1s-to-3d transitions of the iron(II) centers.
The right panel of Figure 3 compares pre-edge features of four different iron centers, diferrous CmlI, the peroxodiferric intermediate of CmlI called CmlI-P, oxyhemrythrin, and the oxoiron(IV) complex [FeIV(O)(MePy2TACN)](OTf)2) (MePy2TACN = N-methyl-N′,N″-bis(2-pyridylmethyl)-1,4,7-triazacyclononane).93 As can be seen, the pre-edge transition increases in intensity in this series, reflecting the distortion of the coordination environment away from centrosymmetry due to increasingly diverging metal–ligand bond distances.98–100 Values found for the areas of the pre-edge transitions range from 3 to 25 units.
Based on a comparison of XANES data for structurally characterized synthetic complexes,98–100 the pre-edge areas can be used to gain insight into the coordination number of the metal centers. Six-coordinate centers have pre-edge areas that range from 4 to 6 units, while five-coordinate centers have pre-edge areas that range from 9 to 13 units and tetrahedral centers have average pre-edge areas of 18 units.98,100 The coordination environment is important in O2 activating systems, as open coordination sites on the metal center facilitate the binding of O2. Six of the diiron enzymes presented here have pre-edge areas reported for their diferrous forms, which range from 8.4 to 13.6 units (Table 2). Thus, CmlA (8.4 units), CmlI (8.4 units), and hDOHH (8.6 units) have all been assigned by XAS as having distorted six-coordinate iron centers, as the pre-edge values for these systems fall above the range of synthetic six-coordinate centers but below the range associated with synthetic five-coordinate centers. The crystal structure of diferrous CmlA shows six-coordinate Fe atoms and corroborates the coordination number assignment based on XANES.65
On the other hand, diferrous sMMOH and ToMOH have both been found to have pre-edge areas of about 10 units, suggesting the presence of five-coordinate iron centers.23,24 In agreement, the crystal structure of reduced sMMOH from M. capsulatus (Bath) shows each iron center to have five ligands with Fe–N/O bonds between 2.1 and 2.4 Å. However, a weakly coordinating solvent ligand also bridges the diiron site, with an Fe1–O distance of 2.5 Å and an Fe2–O distance of 2.7 Å.25 A similar picture is found in the crystal structure of the diMnII-substituted ToMOH but with the solvent bridge having an Mn1–O distance of 2.8 Å and an Mn2–O distance of 2.2 Å.55 It is conceivable that in solution this loosely bound solvent does not sufficiently perturb the Fe centers such that they can be considered five-coordinate. The diferrous ferroxidase site of frog M ferritin exhibits an even larger pre-edge area of 13.6 units50 that strongly suggests the presence of two five-coordinate iron(II) centers. This result agrees with the recently reported crystal structure of the diferrous enzyme,51 which features one five-coordinate Fe center and one six-coordinate center with a weakly bound solvent ligand at 2.5 Å.
Magnetic circular dichroism (MCD) provides another spectroscopic tool to probe the iron coordination environment. The selection rules for this technique allow the d–d transitions of the ferrous centers to be readily detected. Although the spectral analysis required is more demanding, considerably more detailed insight can be gleaned about the coordination geometry of the iron center. The spectral patterns observed have been classified in systematic studies by Solomon and co-workers into characteristic signatures that distinguish among octahedral, square pyramidal, trigonal bipyramidal, and tetrahedral geometries for the individual iron(II) ions in the active sites.92,101,102 Establishing the presence of a five-coordinate center is particularly important for understanding the binding and activation of dioxygen at these sites.
Table 2 lists the coordination numbers and geometries of the iron centers in various diferrous enzymes. Those that have been investigated by MCD are indicated by entries in the coordination number column indicated by an asterisk. In the handful of diferrous enzymes to which MCD analysis has been applied, it is clear that the individual iron sites are coordinatively unsaturated to pave the way for efficient dioxygen binding and subsequent activation.
Another technique often used to characterize the diferrous enzymes is Mössbauer spectroscopy, which sheds light on the oxidation state of the iron center and its electronic structure. Commonly reported parameters are the isomer shift (δ), which is sensitive to the oxidation and spin state of the iron nucleus, and the quadrupole splitting (ΔEQ), which reflects the local electric field around each iron center.103 The δ values for the diferrous proteins presented here fall between 1.20 and 1.39 mm/s, while ΔEQ values range from 2.13 to 3.36 mm/s, with average values of δ = 1.28 mm/s, ΔEQ = 2.96 mm/s that are consistent with parameters for high spin (S = 2) Fe centers (refer to Table 2 for Mössbauer parameters). The relatively narrow range in isomer shifts reflects the O/N ligand environment established for these enzymes by XRD in contrast to the lower values associated with iron–sulfur proteins.
Diferrous centers can adopt three possible electronic ground states. The individual iron sites can be noninteracting and thus appear like isolated high-spin (S = 2) ferrous centers. More often than not, they interact with each other via bridging ligands, either antiferromagnetically to afford an S = 0 ground state or ferro-magnetically to afford an S = 4 ground state. These two ground states can be distinguished by analyzing Mössbauer or MCD data in externally applied magnetic fields. In addition, diiron(II) centers with ferromagnetically coupled ground states can give rise to characteristic parallel mode EPR signals at g ≈ 16, which arise from the S = 4 ground state. Such signals have been observed for the diferrous forms of sMMOH,104 T4MOH,14 the I100W variant of ToMOH,56 AurF,8 CmlI, and ADO.260 It has been a useful spectroscopic probe for monitoring changes in structural and electronic configurations of the diiron(II) site upon exposure to O2. For an overview on the electronic structures of nonheme diiron active sites, see a recent review by Solomon and Park.102
3. Peroxodiferric Intermediates
The existence of peroxodiferric species in diiron proteins has been established in the case of the invertebrate dioxygen carrier hemerythrin more than 40 years ago by Klotz and co-workers.105,106 However, much more progress has been made during the past 20 years in identifying enzymes that give rise to such intermediates, which demonstrate a fascinating diversity of structures and functions. Peroxodiferric species have been trapped as intermediates in native enzymatic cycles and serve not only as precursors to high-valent species that effect substrate oxidation but also act as the oxidants themselves. Obtaining insight into the similarities and differences of these peroxo structures will surely enhance our understanding of the mechanisms Nature uses for cleaving the O–O bond.
3.1. Generation of Peroxodiferric Species
As depicted in Scheme 1 (Red → P), the generation of peroxodiferric intermediates in nonheme diiron systems starts with the binding of O2 to a diferrous active site. In Hr, this process is fast with a first order rate constant on the order of 103 s−1 at 25 °C.107 For the diiron enzymes discussed in this review, first-order rate constants are used to describe O2 binding to form peroxo intermediates, and these rate constants are on the order of 101 to 102 s−1 (Table 3). In systems like sMMOH and R2 RNR, attempts to accumulate enzymatic peroxo intermediates were made challenging by their rapid rates of decay (Table 3) and their detection and characterization often required stopped flow and/or rapid freeze quench techniques. For CmlA, R2lox, and T4MOH, no accumulation of a transient peroxo intermediate has been reported in kinetic studies. However, longer lived peroxodiferric species have been identified for Δ9D,48 AurF,61 CmlI,9 and hDOHH,68 with lifetimes ranging from minutes to days that facilitate their spectroscopic characterization. These efforts have led to the characterization of a number of peroxodiferric species within the class of diiron proteins and enzymes, the proposed or established structures of which are depicted in Figure 4. Many of their properties are collected in Tables 3 and 4.
Table 3. Properties of Peroxodiferric Intermediates of Nonheme Diiron Proteins.
| species | kformation (s−1) [k2 (mM−1 s−1)] @ 4–5 °C | kdacay (s−1) [t1/2] @ 4–5 °C | λmax (nm) | δ [ΔEQ] (mm/s) | ν(O–O) (Δ18O2) (cm−1) | peroxo binding modee | refs |
|---|---|---|---|---|---|---|---|
| Hr | [1.2 × 104]a | 500 | 0.54 [1.92] 0.51 [1.09] | 844 (−46) [+4, Δ2H] | η1-OOH | 20,21,107,108 | |
| sMMOH | 9.1b (Mt)c 0.75b (Mc)c | 2.6 (Mt)b 0.34 (Mc)c | 725 | 0.66 [1.51] | [μ-1,2-peroxo]e | 30,31,35,109,156 | |
| Δ9D | 0.00045 [30 min] | 700 | 0.64 [1.06] 0.68 [1.90] | 898 (−54) | μ-1,2-peroxo | 48,49 | |
| E. coli W48X/D84E RNR R2 | 2.0 (X = F) | 0.26 (X = F) [2.7 s] | 700 (X = A) | 0.60 [1.47] 0.66 [1.68] (X = A) | 870 (−46) (X = F) | μ-1,2-peroxo | 38,41,110 |
| ferroxidase of frog M ferritin | 80a | 4.2a | 650 | 0.62 [1.08] | 851 (−51) | μ-1,2-peroxo | 50,52,69 |
| ToMOH | 26 | 0.045 [∼15 s] | 0.55 [0.67] | [μ-1,1-OOH]e | 54 | ||
| T201S ToMOH | 130 | 2.9 | 675 | 0.55 [0.67] 0.67 [1.51] | [μ-1,1-OOH]/[μ-1,2-peroxo]e | 111–113 | |
| ADO | 0.75 | 0.0017 | ∼450 | 0.48 [0.49] 0.55 [1.23] | [μ-η2-η2- or μ-1,1-peroxo]e | 58 | |
| AurF | 147a | [7 min]a [0.005 s]a,d | 500 (sh) | 0.54 [0.66] 0.61 [0.35] | [μ-1,2- or μ-1,1-OOH]e | 61,62 | |
| CmlI | [58, 20, 1.4] [38]d | 80d [∼3 h] | 500 (sh) | 0.54 [−0.68] 0.62 [−0.23] | 791 (−43) | μ-1,1-O–O | 9,64,259 |
| hDOHH | [>24 h]a | 630 | 0.55 [1.16] 0.58 [0.88] | 855 (−44) | μ-1,2-peroxo | 68 |
Measured at 20–25 °C.
The rate constants shown for P (or Hperoxo) formation correspond to the conversion of P* to P (or Hperoxo), where P* is an intermediate formed prior to P (or Hperoxo) in the reaction of MMOHIed with O2.
Mt and Mc correspond to M. trichosporium and M. capsulatus, respectively, the organisms from which the sMMOH's studied were purified.
In the presence of stoichiometric amino substrate.
Proposed binding modes based on limited structural characterization.
Figure 4.
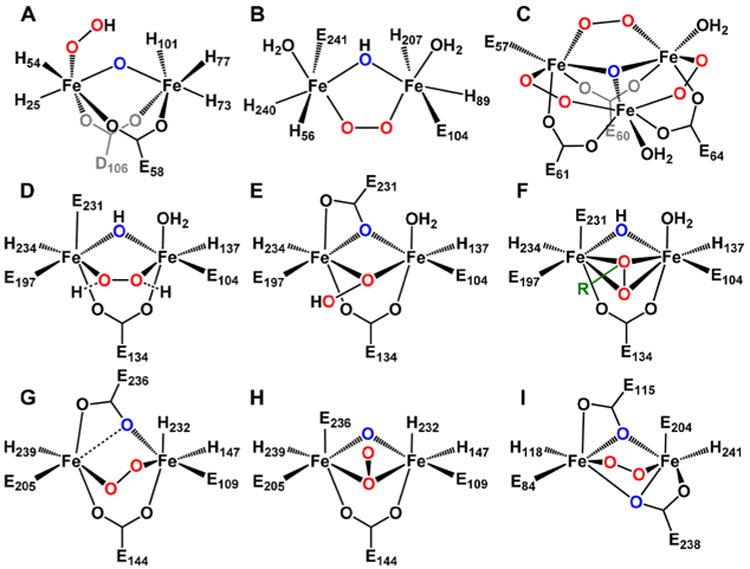
Structures of peroxodiferric intermediates from nonheme diiron proteins. Peroxo ligands are red, and single-atom bridging ligands are blue. A: OxyHr, from XRD; B: hDOHH, from XRD and XAS; C: human L ferritin, from XRD; D: T4MOH/D μ-1,2-peroxo, from XRD; E: Q228A T4MOH/D μ-1,1-(hydro)peroxo, from XRD; F: T4MOH/D μ-η2:η2-arylperoxo, R representing the substrate toluene, from XRD; G: CmlI μ-1,2-peroxo, from XRD; H: CmlI μ-1,1-peroxo based on XAS; I: proposed μ-1,2-peroxo for RNR and/or frog M ferritin based on XAS, residue numbering from W48A/D84E RNR.
Table 4. Select Structural Properties of Peroxodiferric Active Sites from XRD and XAS Studies.
| enzyme | K-edge (eV) [pre-edge area] | Fe···Fe (Å)a | bridging units (νO–O by rR) | PDB ID (res. in Å) | refs |
|---|---|---|---|---|---|
| Hr | 7124.3 [16.9]b | 3.3 (XRD) 3.24 (XAS) | μ-oxo-bis(μ-1,3-O2CR) (η1-OOH) | 1HMO (2.0) | 18 |
| W48A/D84E RNR R2 | 2.5 (XAS) | μ-1,2-peroxo- bis(μ-1,1-O2CR) | 38 | ||
| W48F/D84E RNR R2 | 3.8 (DFT based on rR data) | μ-1,2-peroxo- bis(μ-1,3-O2CR) (870 cm−1) | 38,41,110 | ||
| ferroxidase site of frog M ferritin | 7124 [13.3] | 2.53 | μ-1,2-peroxo- bis(μ-1,1-O2CR) (851 cm−1) | 50 | |
| ferroxidase site of human L ferritin | 3.2–3.5 | μ3-oxo-μ-1,2-peroxo-μ-1,3-O2CR | 5LG8 (1.98) | 114 | |
| T4MOH/D (Pμ1,2) | 3.4 | μ-1,2-peroxo- μ-1,3-O2CR μ-solvato | 3I63 (2.09) | 115 | |
| T4MOH/D (Pμ1,1) | 3.2 | μ-1,1-(hydro)peroxo- μ-1,1-O2CR μ-1,3-O2CR | 5TDV (2.0) | 70 | |
| T4MOH/D (Pη22) | 3.3 | μ–η2:η2-OOR- μ-solvato | 5TDT (1.82) | 70 | |
| hDOHH | 7125.6 [12.4] | 3.8 (3.7) 3.41 | μ-hydroxo-μ-1,2-peroxo (855 cm−1) | 4D50 (1.7) | 16,67 |
| CmlI | 7124.9 [19.2] | 3.35 | μ-oxo-μ-1,1-peroxo-μ-1,3-O2CR (791 cm−1) | 64 | |
| CmlI | 3.3 | μ-1,2-peroxo-μ-1,3-O2CR | 5HYG (2.03) | 63 |
Information on Fe···Fe distance derived from XAS indicated in italics.
See Figure S1
3.2. Spectroscopic and Structural Characterization
Spectroscopic tools available to characterize the peroxodiferric intermediates in this class of enzymes include Mössbauer spectroscopy, X-ray absorption spectroscopy, and resonance Raman spectroscopy. Mössbauer spectroscopy provides insight into the oxidation and spin states of the iron centers in these peroxo intermediates, and the parameters obtained are listed in Table 3. In general, quadrupole doublets are observed with isomer shifts ranging from 0.48 to 0.68 mm/s, consistent with high spin S = 5/2 FeIII centers. These diferric centers are antiferromagnetically coupled to afford an S = 0 ground state, as indicated by the absence of magnetic hyperfine interactions. The quadrupole splittings range from 0.23 to 1.92 mm/s, revealing variations in the extent of asymmetry in the electronic environment of the metal center, as in only a few instances do both iron centers in a diferric active site exhibit the same set of Mössbauer parameters.
Fe K-edge energies have been reported for four of the biological peroxodiferric species, namely, for the ferroxidase site of frog M ferritin (7124.0),50 oxyHr (7124.3) (Figure S1), CmlI (7124.9 eV),64 and hDOHH (7125.6),67 spanning a 1.6 eV range of values. The pre-edge areas associated with these enzyme species also vary, with hDOHH at 12.4 units, the ferroxidase site in frog M ferrtin at 13.6 units, oxyHr at 16.9 units, and CmlI at 19.2 units. These pre-edge areas are relatively large, based on available data from synthetic complexes,98,99 and suggest that the iron centers in these intermediates are significantly distorted from centrosymmetry. This outcome likely arises from the shorter Fe–O bonds formed by oxo, hydroxo, and peroxo ligands postulated for these intermediates compared to the Fe–NHis and Fe–Ocarboxylate bonds in the active sites.
The most easily recognizable characteristic of peroxodiferric complexes is the signature absorption band observed in the visible region that gives rise to the range of color, from orange to blue. This color arises from a peroxo-to–FeIII ligand-to-metal charge transfer (LMCT) transition with a maximum between 450 and 725 nm. This range presumably reflects differences in the binding mode of the peroxo ligand as well as the nature of the other ligands on the iron centers (Table 3).9,20,30,41,48,52,58,61,68,109 As an example, the UV–vis absorption spectrum of oxyHr, shown in Figure 5, has an absorption maximum at 500 nm that accounts for its magenta color. An exception to this generalization is ToMOH-P, which exhibits no visible chromophore. Its designation as a peroxo intermediate is based on its Mössbauer properties and kinetic behavior.54
Figure 5.
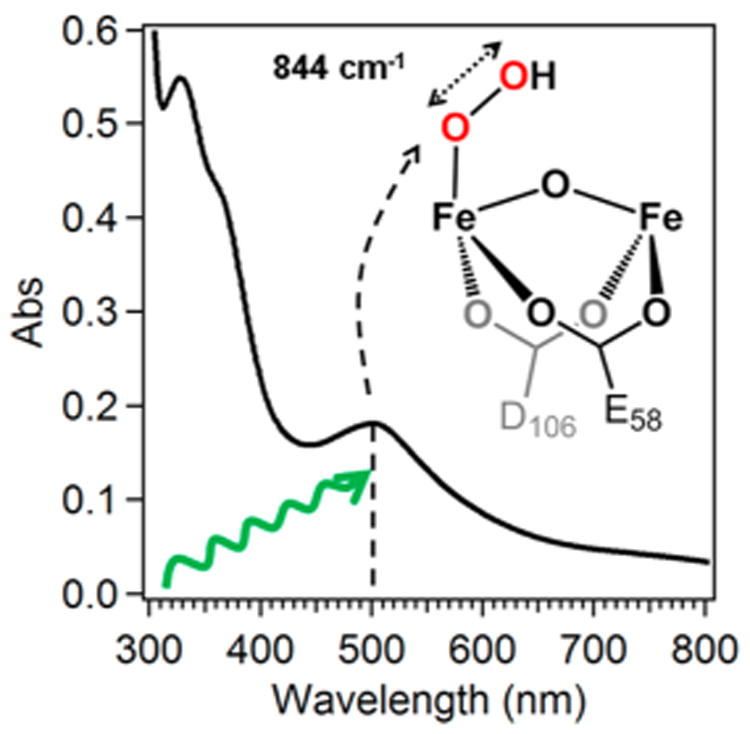
Simplified diagram for resonance Raman data collection of peroxodiferric intermediates. The absorption spectrum is of oxyHr (prepared by Dr. Anna Komor) with a peroxo-to–FeIII LMCT transition at ∼500 nm, marked by the vertical dashed line. Excitation by a laser with similar wavelength (green curved line) into the LMCT band results in the observation of resonance enhanced vibrations, such as the O–O stretch at 844 cm−1.
The peroxo-to–FeIII LMCT band is most commonly probed by resonance Raman (rR) spectroscopy using a laser of an appropriate wavelength to elicit vibrations arising from the peroxodiferric chromophore (Figure 5).116 Most enlightening is the O–O stretching vibration (ν(O–O)), which should shed light on the particular binding mode of a peroxo ligand by comparison with the Raman properties of well characterized synthetic peroxo complexes (see section 4). As listed in Table 3, the ν(O–O) values observed for the enzymatic peroxo intermediates range from 791 to 898 cm−1 and compare well with the ν(O–O) value for H2O2 (872 cm–1).
Generally, the assignment of the O–O stretching frequency must be confirmed with isotope labeling experiments, as the application of Hooke's law to a putative ν(O–O) value of 850 cm−1 would predict a downshift of 49 cm−1 with the use of 18O2 to make the sample, assuming that this feature arises from a pure diatomic stretching mode. Indeed the resonance Raman spectrum of oxyHr exhibited a peak at 844 cm−1 that shifted to 798 cm−1 for the 18O2 isotopomer, the observed 46 cm−1 downshift approaching the predicted 49 cm−1 value.18,108 In addition, the use of mixed-isotope-labeled O2 can shed further light on the symmetry of the peroxo binding mode. This aspect has been beautifully demonstrated by the experiments on oxyHr, for which distinct peaks were resolved for the four possible Fe–16O16OH, Fe–16O18OH, Fe–18O16OH, and Fe–18O18OH isotopomers obtained by exposing deoxyHr to statistically random mixed-labeled O2.106 This result led to the conclusion that dioxygen must be bound in an end-on fashion, anticipating a result that was later confirmed by X-ray crystallography.18,108
The most common peroxo ligand binding mode associated with biological peroxodiferric species is the cis-μ-1,2-peroxo mode found for the peroxodiferric intermediates of RNR R2, Δ9D, the ferroxidase site of frog M ferritin and hDOHH. These intermediates are characterized by visible absorption features between 600 and 700 nm and ν(O–O) modes in the range of 850–900 cm−1 (Table 3). Of these intermediates, subset A active sites have ν(O–O) on the higher end of the range, whereas those from the other subsets have values closer to 850 cm−1. Significantly different from these is CmlI-P, the peroxo intermediate of CmlI, with a much lower ν(O–O) frequency at 791 cm−1. Based on this unusual O–O stretch, the peroxo binding mode was initially postulated to be μ–η1:η2,9 but has subsequently been reassigned as μ-1,1-peroxo based on EXAFS data that recently became available.64 The remaining peroxo intermediates of sMMOH, ToMOH, ADO, and AurF have not yet been characterized by resonance Raman spectroscopy, so a definitive assignment of the peroxo binding mode is not possible. Various structures have been proposed based on Mössbauer and UV–vis data, in conjunction with mechanistic studies, which are listed in Tables 3 and 4.54,58,61,111
Structural parameters of peroxodiferric active sites have been determined from a combination of XRD and EXAFS data (Table 4), but only a few enzymes have afforded samples of peroxo intermediates amenable to these approaches. The active site structure of oxyHr, as discussed above, was first pieced together from a combination of spectroscopic analyses, including rR and XAS studies, and was ultimately confirmed by a high resolution crystal structure (PDB ID 1HMO).18 As found in the diferrous form, the diiron site of oxyHr is bridged by an oxo atom derived from solvent and two bidentate carboxylates, affording Fe–μ-O bonds of 1.8 Å and an Fe···Fe distance of 3.3 Å from both XRD and XAS experiments.18,19 The dioxygen moiety is bound terminally to one iron, and the distal peroxo O atom is 3 Å away from the oxo bridge (Scheme 3 and Figure 4A). The peroxo coordination observed arises from a terminally bound hydroperoxo moiety that is hydrogen bonded to the μ-oxo bridge, as evidenced by H/D dependent shifts seen in the resonance Raman spectra of oxyHr.22 It has been proposed by Solomon that this hydrogen bonding interaction is an important component of the mechanism that facilitates the reversible transfer of electrons between the diiron site and O2.81
The first peroxodiferric intermediates were trapped from diiron enzymes that were investigated in detail in the 1990s, namely sMMOH,30 Δ9D,48,49 and RNR R2.38,41 These three enzymes share a recurring 2-His-4-carboxylate ligand combination housed in a common 4-helix-bundle motif. All three peroxodiferric intermediates exhibit absorption maxima near 700 nm that likely arise from peroxo-to-iron(III) LMCT transitions. In addition, they have similar Mössbauer parameters that are associated with high-spin ferric centers, with isomer shifts greater than or equal to 0.60 mm/s and quadrupole splittings greater than 1 mm/s (Table 3), suggesting that they may share a common active site. Notably, the isomer shifts found for these intermediates fall on the high end of the high-spin ferric range, which is centered at 0.5 mm/s.103 Unfortunately, these intermediates have all proven challenging to study, and no structural information from XRD has been obtained.
The spectroscopic characterization of sMMOH-P beyond its electronic and Mössbauer spectra has been minimal. There are no XAS studies on sMMOH-P in the literature, although other sMMOH states have been studied.23,24,32,117 Resonance Raman data was reported for sMMOH-P but has since been retracted.118,119 In the absence of spectroscopic results, extensive computational efforts have been carried out and favor a (μ-1,2-peroxo)diferric structure.120–122 sMMOH-P has been shown to be the precursor to sMMOH-Q, the high-valent oxidant responsible for cleaving the 105 kcal/mol bond of methane. More spectroscopic data has been collected for sMMOH-Q, which will be discussed in the next section.
Δ9D is mechanistically related to sMMOH, as both are diiron enzymes that generate oxidizing species capable of cleaving strong (BDE > 98 kcal/mol) C–H bonds. For this enzyme, slightly more insight has been obtained on the nature of Δ9D-P, which can be formed from the fully reduced enzyme in the presence of O2 and its substrate stearoyl acyl carrier protein.48,49 Δ9D-P exhibits a visible absorption band at 700 nm and resonance Raman spectra showing a ν(O–O) of 898 cm–1, the highest frequency observed thus far for any diiron-enzyme-derived peroxo species (Table 3). 18O-labeling experiments show an isotopic distribution pattern most consistent with a μ-1,2-peroxo bridge. However, when Δ9D-P is generated and substrate-bound ACP is added to the intermediate, no reaction is observed.48 Fox and co-workers report that the addition of the appropriate reductase to Δ9D-P restores desaturase activity, emphasizing that allosteric interactions from effector proteins like the reductase can be critical for observing biological function.48
For RNR R2, peroxo intermediates can be trapped only from variants where Asp84, a carboxylate ligand of the diiron site, is replaced by Glu to make the R2 diiron site resemble sMMOH38,41,42 Peroxo samples usable for spectroscopic analysis by resonance Raman and XAS were obtained by further mutating the redox-active W48 residue into either an alanine or phenylalanine residue, as W48 lies near the diiron site and likely donates an electron to the diiron active site during the activation of the enzyme for catalysis.38,41,42 Spectroscopic studies on these intermediates reached conflicting conclusions about the nature of the diiron site, which have not been reconciled to date. Based on EXAFS analysis, W48A/D84E RNR R2-P was found to have a 2.5 Å Fe···Fe distance, a rather short distance that led the authors to postulate a diiron site with a μ-1,2-peroxo bridge and supported with two μ-1,1-carboxylates (Figure 4I).38 In contrast, a much longer Fe···Fe distance of 3.8 Å was deduced for the cis-(μ-1,2-peroxo)diferric center of W48F/D84E RNR R2-P, based on an analysis of the resonance Raman data that showed a ν(Fe–O) of 457 cm–1 and a ν(O–O) of 868 cm–1 and DFT geometry optimizations that included constraints imposed by the protein structure.110
A fourth peroxodiferric intermediate with a λmax at 650 nm was obtained upon exposure of the reduced ferroxidase site of frog M ferritin to O2.52 Like sMMOH, Δ9D, and RNR R2, the diiron active site of the ferroxidase has two His and four carboxylate ligands housed within a 4-helix bundle. The peroxodiferric intermediate from frog M ferritin exhibited a resonance Raman spectrum with a ν(O–O) of 851 cm−1,69 which is at the low end of the frequency range associated with these intermediates; a pair of frequencies was also observed at 485 and 499 cm−1, which were attributed to the νsym and νasym modes of the Fe–O2–Fe unit, respectively. EXAFS studies of this intermediate revealed a short 2.5 Å Fe···e distance,50 the same as reported for W48A/D84E RNR R2-P38 and presumably also suggestive of a (μ-1,2-peroxo)bis(μ-1,1-carboxylato)diiron(III) core (Figure 4I). However, the question of the short Fe···Fe distance remains unresolved to date.
Since the turn of the millennium, there has been increasing success in obtaining crystals of reaction intermediates generated in crystallo by exposing crystals of the reduced enzyme or the reduced enzyme–substrate complex to O2. By this method, crystallographic information on peroxo intermediates and related species along a reaction pathway have been obtained.63,70,123 One example relevant for this review is the structure of a unique peroxo species obtained from the human form of L ferritin. Crystals of apo-L-ferritin soaked in a solution containing ferrous ion in the presence of O2114 gave rise to a triiron structure that resembles the structure of a basic ferric acetate,124,125 which consists of a (μ3-oxo)triferric center with each pair of irons held together by two 1,3-carboxylate bridges. In the protein, one 1,3-bridging carboxylate of each iron pair is replaced by a 1,2-peroxo bridge to generate an analogous structure shown in Figure 4C (PDB ID 5LG8; Figure 6). Interestingly, there are no histidine ligands in this structure, unlike what is found in the structure of the diferrous form of the frog M ferritin.50,51
Figure 6.
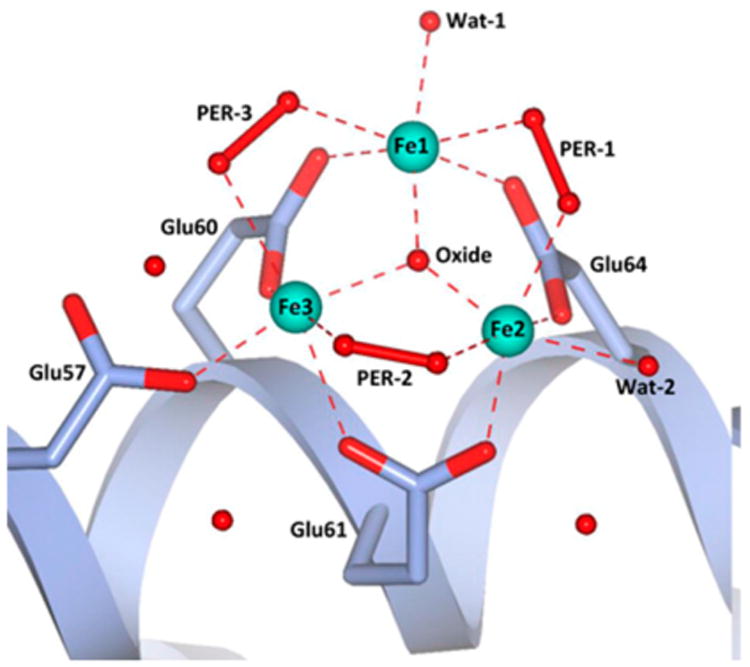
FeIII3(μ3-O)(μ-1,2-peroxo)3(μ-1,3-O2CR)3 core structure observed by Turano and co-workers in the crystal structure of human L ferritin (PDB ID 5LG8). Reproduced from ref 114 with permission. Copyright 2017 National Academy of Sciences.
The bond metrics for the protein structure can be compared to those of the basic ferric acetates as well as synthetic peroxodiferric complexes. The Fe–μ3-Ooxo bond lengths from human L ferritin are 1.8, 2.0, and 2.0 Å, typical for those found in basic ferric acetate structures that feature a μ3-O atom with Fe–O bonds averaging ∼1.9 Å.124,125 The 1.5 Å O–O bond lengths of the μ-1,2-peroxo bridges are all consistent with the peroxo designation, but the Fe–Operoxo distances are between 2.3 and 2.6 Å, which are quite a bit longer than the 1.9–2.0 Å distances typically found in synthetic models (see section 4). These elongated distances raise the possibility of photoreduction of the iron centers or possible protonation of the peroxo ligands. Lastly, the Fe···Fe distances for the three pentagonal subunits of the structure are 3.2, 3.3, and 3.5 Å, which are much longer than the 2.5 Å value reported for the peroxo intermediate in the ferroxidase site of frog M ferritin based on EXAFS analysis.50 These distances in fact correspond well to values for synthetic (μ-oxo)(μ-1,2-peroxo)diferric complexes (see section 4). A key unresolved question is whether this trinuclear assembly is relevant to the action of ferritin in the biomineralization of iron.
Crystals of a truncated hDOHH-P (Δ300) (PDB ID 4D50) have also been obtained recently (Figure 7),16 shedding light on an active site unlike those of any other diiron protein belonging to this class (Figure 4). The two iron atoms are found in a pseudo–octahedral environment, each bound by protein ligands (2 His and 1 Glu) in a meridional motif. As the two carboxylates are both bound as terminal monodentate ligands, the diiron site is held together only by a single O atom bridge assigned to a μ-hydroxo ligand and a peroxo ligand bound in a μ-1,2 mode, in agreement with rR studies.68 Terminal solvent-derived ligands serve to cap off the iron coordination spheres.
Figure 7.
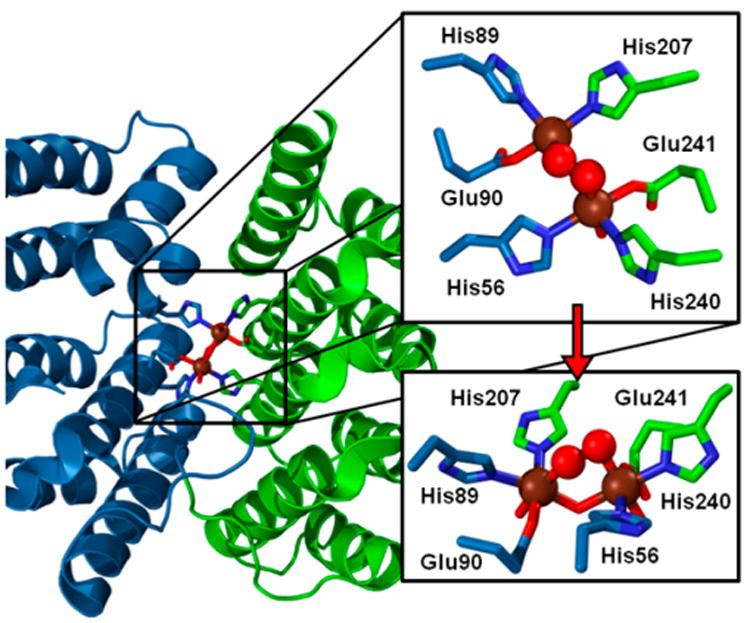
Diiron site of hDOHH-P (PDB ID 4D50). Color scheme: Fe (brown spheres), peroxo O atoms (red spheres), residues from the N-terminal domain (blue), residues from the C-terminal domain (green), amino acid ligands represented as sticks. Adapted from ref 67 with permission. Copyright 2016 Springer.
Interestingly, the peroxo ligand is bound in a hydrophobic pocket, on the opposite side of the cluster from the proposed substrate binding channel, which perhaps accounts for the unusual stability of this peroxo intermediate.16 The Fe···Fe distance was determined to be 3.7 and 3.8 Å in the two different active sites, with Fe–Operoxo distances at ∼2.2 Å. However, the metrics did not agree with those determined by XAS, which were 3.41 and 1.98 Å, respectively.67 The longer distances from the XRD study suggest the likelihood of photoreduction upon exposure of the hDOHH crystals to the X-ray beam, a well-known issue with crystallographic studies of metalloenzymes.126–128 Photoreduction can be mitigated in XAS studies, by monitoring the K-edge energy during data collection and shifting the physical spot being exposed to the X-ray beam.
Crystal structures of T4MOH have been particularly informative in revealing new modes of O2 binding to the diiron active site. Crystals of a peroxo complex were obtained by soaking crystals of as-isolated T4MOH complexed with its effector protein T4MOD in H2O2-containing buffer (T4MOH-Pμ1,2, PDB ID 3I63), as a means of bypassing the O2 binding step and go directly to the peroxide intermediate.115 T4MOH can in fact use H2O2 for catalysis via a “peroxide-shunt” pathway.115,129 This crystal structure reveals two six-coordinate iron centers with a 1,3-carboxylate bridge, a solvent-derived single-atom bridge syn to the two His ligands and a μ-1,2-peroxo ligand anti to the His ligands (Figure 4D). One Fe center has a terminal solvent-derived ligand, and the three remaining coordination sites are filled by monodentate Glu ligands. The Fe–μ-Osol distances of 2.1 and 1.9 Å and the Fe–Operoxo distances are 2.2 and 2.4 Å, leading to an Fe···Fe distance of 3.4 Å. The Fe–Operoxo bond lengths are also longer than typically observed for Fe–Operoxo bonds in synthetic peroxodiferric models (see section 4), which Bailey and Fox suggest may be indicative of the protonation of the peroxo ligand that would weaken the Fe–O bonds.115 As H2O2 was used to grow these crystals, this possibility is plausible, though there are no other examples of diiron-H2O2 adducts against which to make comparisons. Alternatively, the crystal structure could represent a peroxodiferric species that has been photoreduced during the XRD experiment. Single-crystal electronic absorption or resonance Raman spectroscopic data would be useful to obtain for the further characterization of this intermediate, as there is no observed accumulation of this species in solution.
Recently, two more crystal structures of T4MOH-peroxo intermediates have been obtained.70 One is of a μ-1,1-peroxo species obtained by bubbling O2 into an anaerobic solution of the Q228A variant of the reduced T4MOH/D complex (Pμ1,1, PDB ID 5TDV). This structure shows two six-coordinate iron centers bridged by a μ-1,1-peroxo ligand, with an Fe–O–O···Fe angle of 121° (Figure 4E). The two Fe–O–O angles (168° and 102°) differ by 66°, so the O–O bond vector tilts toward Feb (Figure 8). Also bridging the two iron centers are two glutamates, one in a 1,3-fashion and the other in a 1,1-fashion, as found in the diferrous structure of WT T4MOH/D.57 These bridges together maintain an Fe···Fe distance of 3.2 Å (Figure 4E), 0.2 Å shorter than that found in diferrous T4MOH/D. The Fe–O distances from the μ-1,1-glutamate ligand are at 2.1 Å, similar to the single-atom bridge from the T4MOH-Pμ1,2 structure. The O–O distance is 1.5 Å and the proximal Fe–Operoxo distances are also rather long at 2.4 and 2.5 Å. This may indicate some degree of photoreduction and/or protonation of the peroxo ligand. DFT calculations suggest that protonation is possible, as a hydroperoxo species yields bond lengths that are congruent with those observed in the crystal structure.70 The distal Fe–Operoxo distances are 3.2 and 3.7 Å and the O atom is pointing into the substrate binding pocket. DFT calculations based on the structural coordinates suggest that it is likely that the μ-1,1-peroxo ligand is protonated. Without protonation, one peroxide electron prefers to shift back to the diiron site to form a (μ-1,1-superoxo)diiron(II,III) species, but protonation converts it back to the (μ-1,1-hydroperoxo)diiron(III) derivative. This complex appears to be unreactive toward substrate.
Figure 8.
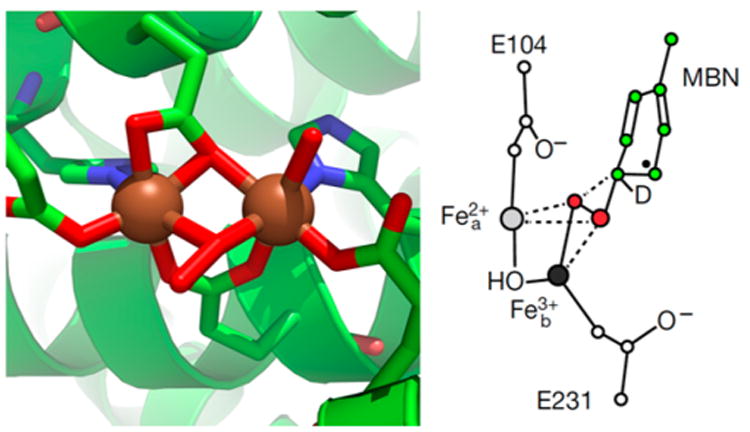
Active site structures of T4MOH/D peroxo intermediates from crystallographic studies of Fox and co-workers.70 Left: T4MOH/D μ-1,1-(hydro)peroxo species (Pμ1,1, PDB ID 5TDV). Fe, O, N, and C atoms are shown in brown, red, blue, and green, respectively. Right: T4MOH/D μ–η2:η2-OOR species (Pη22, PDB ID 5TDT). The substrate molecule is labeled MBN for methylbenzene. Reproduced from ref 70 with permission. Copyright 2017 Nature Publishing Group.
The other recent peroxo structure was generated by bubbling O2 into an anaerobic solution of diferrous WT T4MOH/D in the presence of the toluene substrate (PDB ID 5TDT). In one subunit, the iron centers are bridged by a solvent-derived ligand, a μ-1,3-glutamate, and a peroxo ligand bound in a μ–η2:η2 fashion in one of the two subunits (Figure 4F). Additionally, one O atom of the peroxo ligand (called O2peroxo) appears to be connected to the C4 atom of the toluene molecule present in the active site. The O–O bond distance is 1.4 Å, but the Fe–O1peroxo distances are both at 2.7 Å and the Fe–O2peroxo distances are at 2.5 and 2.8 Å, clearly showing that the peroxo ligand is not tightly bound to the diiron cluster. DFT geometry optimization suggests that this intermediate may be a mixed-valent FeIIFeIII-(cyclohexadienylperoxyl radical) species (Figure 8, right); however, the oxidation states of the Fe centers cannot be determined from the XRD structures.70 This series of T4MOH structures is intriguing, as it demonstrates that the 2-His-4-carboxylate-supported active site is quite versatile and can accommodate three distinct peroxo binding geometries, two of which (μ-1,1-(hydro)peroxo and μ–η2:η2-peroxo) have not been structurally characterized in a diiron complex. Interestingly, these three peroxo species have not yet been observed in the solution state, despite the fact that T4MOH shares a very similar active site structure with sMMOH and other enzymes for which peroxo species have been observed.
CmlI can also accommodate two different peroxo binding geometries based on two different studies.9,63 A crystal of the peroxodiferric intermediate of CmlIΔ33, a 33-amino-acid-truncated variant of CmlI, was obtained (CmlIΔ33-Pμ1,2, PDB ID 5HYG) from a solution of the as-isolated enzyme (Figure 9 top).63 Although neither H2O2 nor reductant was added into the crystallization media, it is conjectured that the polyethylene glycol in this solution may have generated H2O2 in the presence of air to afford the resulting structure. The diiron site observed is bridged by 1,2-peroxo and two carboxylate ligands (Figure 4G). E236 is bound in a distorted 1,3-carboxylate mode, with Fe–O1E236 distances of 2.2 and 2.6 Å, compared to the more typical 1,3-mode of E144 with Fe–O1E144 distances of 2.2 and 2.9 Å. E236 has been described as a flexible residue, as it can take on two different conformations in the active site and may be involved with substrate access to the active site.63 The binding of E236 in CmlI appears similar to the 1,1-carboxylate bridge of E231 in Q228A T4MOH/D (Figure 4E), but the Fe–OE326 distances in CmlI are longer than the Fe–OE321 distances of 2.1 Å found in the T4MOH structure. Despite the apparent flexibility of the active site ligand, the diiron cluster still maintains an Fe···Fe distance of 3.3 Å. In comparison to the other peroxo structures presented, the Fe–Operoxo distances found for CmlIΔ33-Pμ1,2 are 1.8 and 2.0 Å, consistent with those from synthetically derived peroxodiferric structures. The O–O distance is 1.5 Å, which compares well to the other peroxodiferric species described above, and the Fe–Odistal distances are at 2.8 and 2.9 Å, which are shorter than the 3.2-Å Fe–Odistal distances measured for the T4MOH-Pμ1,2 structure.
Figure 9.
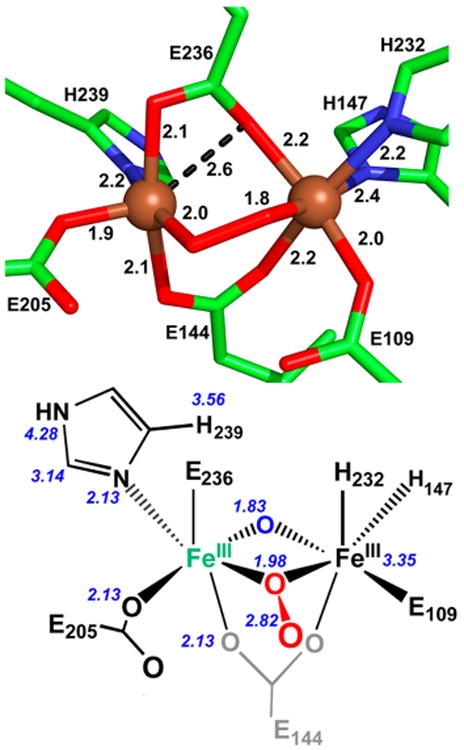
Active sites of CmlIΔ33-Pμ1,2 (top) determined by XRD (PDB ID 5HYG)63 and of WT-CmlI-P (bottom) generated in solution and analyzed by EXAFS.64 All distances are listed in angstroms. Top: the Fe, O, N, and C atoms are shown in brown, red, blue, and green, respectively. Bottom: all distances displayed are measured from the green Fe atom.
When a solution of diferric WT-CmlI or CmlIΔ33 is treated with excess H2O2, an intermediate with a broad visible absorption band at 600 nm is generated,63 reminiscent of a μ-1,2-peroxo species. This proposed structure has not been corroborated by either resonance Raman spectroscopy or EXAFS analysis. However, this intermediate is quite stable, with no observed decay over several hours at 4.5 °C. Furthermore, exposure of the Pμ1,2 species to the native substrate yields no product, showing definitively that Pμ1,2 does not facilitate N-oxygenation of arylamine substrates.63
On the other hand, a different peroxo intermediate is generated when diferrous WT or Δ33 enzyme is exposed to O2. This intermediate, called Pμ1,1, exhibits a visible absorption band at 500 nm, a blue shift observed relative to Pμ1,2 hinting at a different peroxo binding mode.9 Pμ1,1 has a half-life of ∼3 h at 4 °C9 and rapidly decays upon addition of the native substrate to yield the nitroaryl product.130,259 Based on a combination of XAS and rR data, the 500 nm chromophore from WT-CmlI is deduced to have a (μ-oxo)(μ-1,1-peroxo) core (Figure 9, bottom).64
The presence of an oxo bridge is indicated by EXAFS fits that an Fe–O distance of 1.83 Å, which is typically found for (μ-oxo)diferric units.131 This notion can be corroborated by resonance Raman spectroscopy, which reveals a feature at 487 cm−1 that is downshifted by 18 cm−1 to 469 cm−1 in H218O buffer, assigning it to be to the symmetric stretch of an Fe–O–Fe unit. The corresponding asymmetric stretch can be found at 780 cm−1 and is downshifted by 31 cm−1 to 749 cm−1 in H218O buffer. Taken together, the νsym and the νasym values correspond to an Fe–O–Fe angle of 138° based on a correlation first developed by Sanders-Loehr (Figure 10).132
Figure 10.
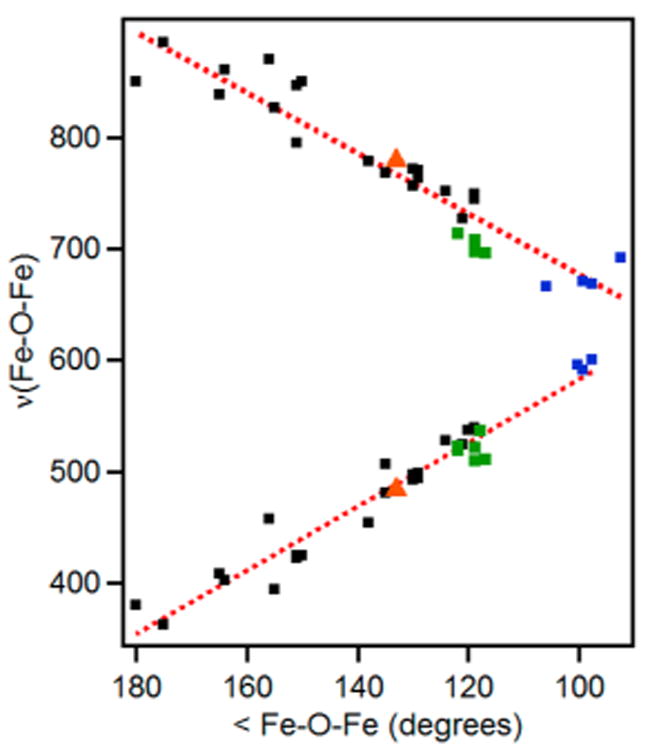
Expanded correlation between νs(Fe–O–Fe) (bottom) and νas(Fe–O–Fe) values (top) of oxo-bridged diiron complexes with their Fe–O–Fe angles: data from the original Sanders-Loehr study (black),132 augmented by data for species with (μ-oxo)(μ-1,2-peroxo)-diiron cores (green)133–137 and species with bis(μ-oxo)diiron diamond cores (blue).138,139 Red dotted lines represent the best linear fits of the data. Raman data for CmlI-P indicated by red triangles. Reproduced from ref 64 with permission. Copyright 2017 American Chemical Society.
Evidence for the μ-1,1-peroxo bridge derives from EXAFS analysis. The proximal peroxo O atom is associated with an Fe–O distance of 1.98 Å, while the distal O atom has an Fe–O distance of 2.82 Å (Figure 4H). Comparison to the crystal structure of product bound AurF (PDB ID 3CHT)8 supports the assignment of the 2.82 Å scatterer. The product of the AurF-catalyzed reaction is para-nitrobenzoic acid, and the oxygen atom from the nitro group is found in the same position in the crystal structure as the 2.82 Å scatterer from CmlI is proposed to occupy. Additional comparisons to the crystal structures of Q228A-T4MOH as well as a synthetic (μ-1,1-hydroperoxo)dicopper complex140 help establish that 2.8 Å is a plausible distance for a distal oxygen atom of a μ-1,1-peroxo ligand, requiring the distal oxygen atom to tilt out of the Fe–O–Fe plane.64
The structure proposed for WT-CmlI-Pμ1,1 based on Raman and EXAFS data differs from that proposed for AurF-P, which derives from an analysis of MCD and nuclear resonance vibrational (NRVS) data.62 Both CmlI and AurF catalyze the 6-electron oxidation of aminoarenes to nitroarenes in antibiotic biosynthesis and form similar intermediates upon exposure of reduced enzyme to O2 with broad absorption bands centered at 500 nm and similar Mössbauer parameters. However, these two species differ in thermal stability, with WT-CmlI-Pμ1,1 having a half-life of ∼3 h at 4 °C, which is 30-fold longer than that for AurF-P (7 min).
The AurF-P model derives from an MCD analysis of the diferrous form of AurF.62 The crystal structure of the enzyme-4-nitrobenzoate complex8 showing the product nitro group in close proximity to (but not ligated to) the (μ-oxo)(μ-1,3-carboxylato)diferric center was used as a starting point to construct a 3-D model for AurF-P, which was subsequently geometry optimized using DFT methods. Many models were tested, but the model that could reproduce the characteristic features measured from NRVS was a cis-μ-1,2-hydroperoxo species with two μ-1,3-carboxylate bridges (Figure 11). This calculated structure predicts an Fe···Fe distance of 3.82 Å and respective Fe–Ohydroperoxo and Fe′-OHhydroperoxo distances of 1.89 and 2.17 Å. These calculated bonding metrics are unlike any of the structurally characterized examples from above, but there are no precedents in the diiron literature against which to compare this model. Interestingly, the proposed AurF-P structure differs significantly from that of WT-CmlI-P, despite having comparable UV–vis and Mössbauer properties (Table 3), but the AurF-P model was developed without the benefit of the insights from resonance Raman and EXAFS data collected subsequently on WT-CmlI-P.
Figure 11.
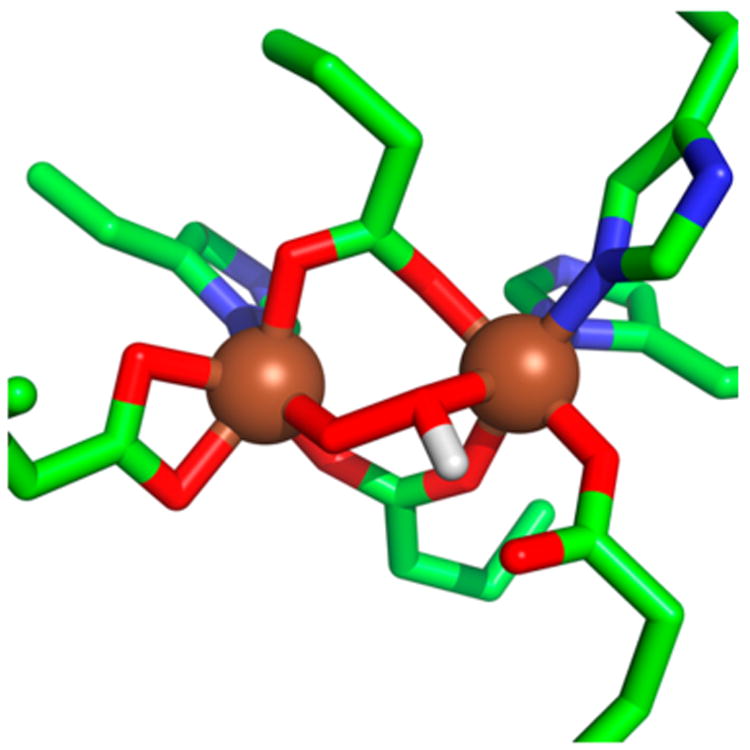
Model of AurF-P proposed by Solomon and co-workers based on a combined NRVS and DFT analysis.62 The Fe, O, N, C, and H atoms are shown in brown, red, blue, green, and white, respectively. Bond distances from DFT: Fe···Fe at 3.82 Å, Fe–Ohydroperoxo at 1.89 Å and Fe′-OHhydroperoxo at 2.17 Å, Fe-NHis ≈ 2.2 Å, and Fe–OGlu ≈ 2.0 Å.
3.3. Reactivity
Scheme 1 provides a good mechanistic overview of the dioxygen activation chemistry of nonheme diiron enzymes and emphasizes the central role played by peroxodiferric intermediates in their action. As previously demonstrated in the chemistry of heme enzymes such as aromatase and NO synthase,141 the rather basic peroxo anion is quite versatile and can carry out oxidations by nucleophilic attack of the substrate. Alternatively, the heme-bound peroxide can undergo O–O bond heterolysis with the help of a proton to generate a highly electrophilic oxidant that can attack more oxidatively resistant substrates such as alkanes. As more nonheme diiron enzymes are identified and become characterized, it is clear that they exhibit at least comparable mechanistic versatility as heme enzymes but perhaps even more.
The mechanisms for dioxygen activation at diiron sites have been most thoroughly investigated for sMMOH and E. coli RNR R2. Many intermediate species have been identified along the reaction pathway,6,7 including the peroxodiferric intermediate, P (also referred to as Hperoxo in the case of Mc sMMOH)30,31,109 and high-valent oxidants Q23,33 and X,43,77,78,142 which are derived from its decay. As sMMOH and RNR R2 share a common protein fold and similar ligand set with many other diiron enzymes,25,37,45,55,57,60,63 corresponding P intermediates have been identified for some of the latter. The working hypothesis that has thus evolved is that dioxygen activation by diiron enzymes follows the mechanistic paradigms established for sMMOH (Scheme 1, Red → P → Q) or for RNR R2 (Scheme 1, Red → P → X).
More recently, a growing number of P intermediates has been found to be capable of carrying out oxidation reactions directly. P from sMMOH has been shown to oxidize electron-rich substrates like ethyl vinyl ether, diethyl ether, and propylene.71,72,143 The corresponding ToMOH intermediate is found to decay more rapidly in the presence of the substrate analog phenol, producing catechol exclusively as the oxidized product.144 On the other hand, ADO-P is proposed to form a peroxyhemiacetal intermediate by nucleophilic attack of the aldehyde functionality of the substrate,58 and N-oxygenation of aryl-amine substrates is facilitated through P intermediates of AurF and CmlI.9,61 However, the P intermediate from Δ9D is unreactive toward the native substrate under single turnover conditions and thus may not be a catalytically competent species.49 The reason for the differences in the reactivity of P intermediates is unclear, but analysis of structural features, such as the peroxo binding geometry, may shed light on the issue.
3.3.1. Nucleophilic Oxidant in ADO
The diiron enzyme that very likely catalyzes the oxidation of its substrate via a nucleophilic peroxo mechanism is ADO. Oxidation of its fatty aldehyde substrate truncates it by one carbon atom to form an alkane, with the aldehyde functionality being converted to formate (Figure 12A).145 Isotope labeling studies of the enzyme from Nostoc punctiforme show that one O atom in the formate product derives from O2.146 This result is most readily rationalized by the attack of the nucleophilic peroxo intermediate on the electrophilic carbon atom of the aldehyde substrate to initially form a peroxyhemiacetal (structure C in Figure 12B). This mechanism is akin to those proposed for the oxidative deformylation of cyclohexane carboxaldehyde by cytochrome P4502B4 to form cyclohexene and for the last step in the aromatization of the A-ring of androst-4-ene-3,17-dione to estrone, which is catalyzed by human placental aromatase.141 However, the alkane product formed in the case of ADO (Figure 12A) differs from the dehydrogenated products found in the two heme enzyme examples mentioned above and requires the input of two more electrons. This switches the mechanism from a peroxo decomposition mechanism involving a series of two-electron steps to one entailing reductive homolysis of the O–O bond to form formate and an alkyl radical and the subsequent one-electron reduction of the radical to the alkane product (Figure 12B). Isotope labeling experiments show that the source of the H atom incorporated into the alkyl radical is a solvent-exchangeable proton that is proposed to be a water bound to the diiron site (see structure E in Figure 12B).
Figure 12.
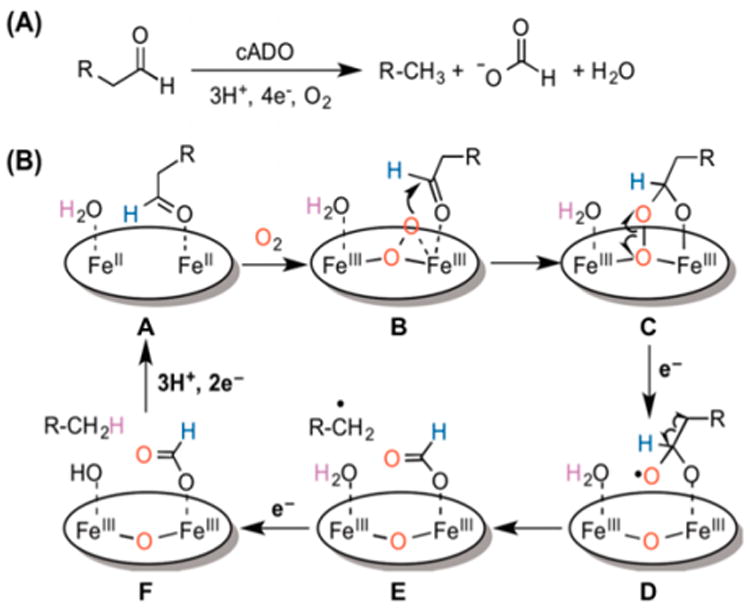
(A) Stoichiometry of reaction catalyzed by ADO. (B) ADO mechanism first proposed by Pandelia et al.58 Reproduced from ref 147 with permission. Copyright 2015 American Chemical Society.
In support of this mechanism, Pandelia et al. have trapped an intermediate with a λmax at 450 nm, presumably arising from a peroxo LMCT band, with Mössbauer parameters associated with an antiferromagnetically coupled pair of high-spin ferric ions in different coordination environments.58 A perusal of Table 3 shows that this diferric pair exhibits the most blue-shifted of the LMCT bands observed in the visible region for these intermediates. ADO-P also has individual iron centers that are most distinct from each other with respect to their Mössbauer parameters of any peroxodiferric center in this table, suggesting that the ligand environments of the two ferric ions in this intermediate differ quite significantly, perhaps as shown in structure C in Figure 12B. However, structural insight into the basis for this spectroscopic difference is not yet available. For example, it may be possible for resonance Raman measurements to distinguish between proposed peroxo and peroxohemiacetal moieties in structures B and C in Figure 12B. In support of mechanistic notions put forth by Figure 12B, a functional synthetic model for ADO has been developed to convert an aldehyde to an alkane.147 Reaction of 2-phenylpropanal with the crystallographically characterized [FeIII(TMC)(η2-O2)]− complex,148 which has been shown to be a nucleophilic oxidant, results in the oxidative deformylation of the aldehyde to form acetophenone. However, the addition into this reaction of Bu3SnH to serve as an H atom donor generates ethylbenzene instead, demonstrating the interception of the intermediate alkyl radical by an H atom from the Sn–H bond of Bu3SnH. Very likely then, ADO follows a reaction sequence of Red → P + Sub → product (Scheme 1).
Studies of the ADO from Prochlorococcus marinus with C8–10 aldehydes as substrate instead of octadecanal show formation of the expected C7–9 alkane as well as the corresponding primary alcohol and aldehyde in comparable yields.260 When carried out under 18O2, the alcohol becomes 18O-labeled. Furthermore, the use of starting aldehyde 13C-labeled at C-1 and C-2 gives rise to final alcohol and aldehyde products both labeled at C1, so the hydrocarbon product of the initial deformylation step does not necessarily escape from the active site before undergoing further oxidation. These observations have led Aukema et al. to suggest the possibility of forming an electrophilic FeIV=O oxidant upon O–O bond cleavage of the peroxohemiacetal moiety C in Figure 12B that is responsible for the observed alcohol and aldehyde byproducts.
3.3.2. Ambiphilic Oxidant in CmlI
CmlI catalyzes the oxidation of an aminoarene substrate to a nitroarene product in the biosynthesis of chloramphenicol in three successive 2-e− steps at the same active site (Figure 13, top). Evidence has accumulated for the formation of a hydroxylaminoarene as the first 2-e−-oxidized product followed by a nitrosoarene as the second 2-e−-oxidized product en route to the ultimate nitroarene product.130,259 Interestingly, the overall reaction requires two different molecules of O2, as demonstrated by the elegant labeling studies of Komor et al.130 The first O2 provides the O atom for the initially formed hydroxylamine product, while the second O2 is the source of the second O atom of the nitro group in the ultimate product. Interestingly, the second 2-e−-oxidation involving conversion of the hydroxylaminoarene to the nitrosoarene is simply a transfer of two electrons from the hydroxylamine to the diferric site, regenerating the diferrous center for a second round of O2 activation. We thus favor a reaction sequence of Red → P + Sub → product (Scheme 1) for both the arylamine and the nitrosoarene oxidation steps.
Figure 13.
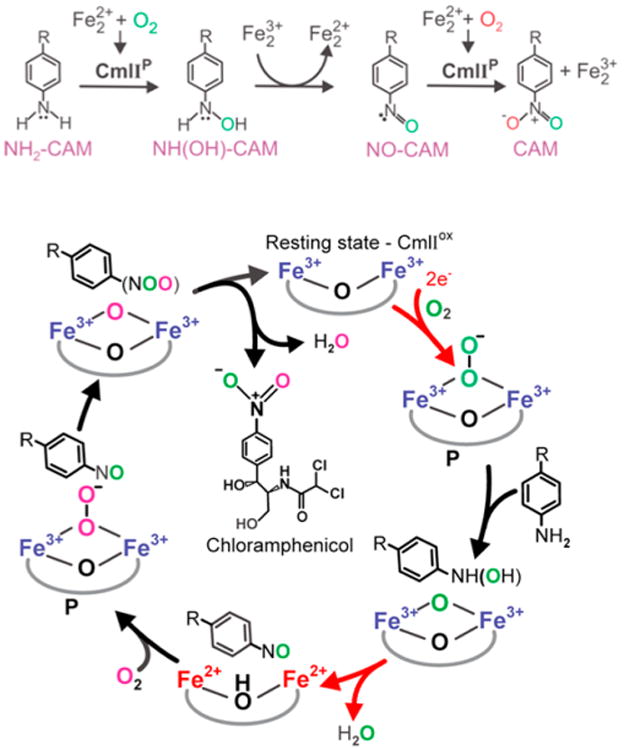
Top: Conversion of the NH2–CAM to CAM via hydroxylamino and nitroso derivatives. Bottom: Steps carried out by CmlI-P in the 6-e− oxidation of an arylamine to its nitroaryl product catalyzed by CmlI in chloramphenicol biosynthesis. The top and bottom panels were repsectively reproduced from refs 64 and 259 with permission. Copyright 2017 American Chemical Society.
The enzyme AurF carries out an analogous N-oxygenation reaction that converts 4-aminobenzoic acid to 4-nitrobenzoic acid. In 2010, the Bollinger group established that AurF requires two molecules of O2 and two exogenous electrons in order to facilitate the native reaction. Similar to the CmlI reaction, the first step generates the hydroxylaminoarene species and a diferric cluster, but unlike the CmlI mechanism, the hydroxylamine does not act as an internal reductant for the iron centers. Instead, the exogenous electrons reduce the iron cluster and the second molecule of O2 reacts to form a peroxodiferric species. This AurF-P intermediate reacts with the hydroxylaminoarene to form a putative dihydroxylaminoarene species that is proposed to serve as an internal reductant for the system. This reduction forms the nitroarene product and a diferrous iron cluster.
The chemistry of the 6-e− oxidation catalyzed by CmlI requires an ambiphilic peroxide oxidant (Figure 13, bottom). The first step entails the hydroxylation of the N–H bond of an amine (akin to C–H hydroxylation), requiring an electrophilic oxidant, but the third step involves attack of an electrophilic nitroso group (akin to the peroxo attack of the fatty aldehyde carbonyl proposed for ADO, Figure 12), which must involve a nucleophilic oxidant. The need for an ambiphilic oxidant in this 6-e− oxidation can easily be fulfilled by the recently characterized WT CmlI-Pμ1,1 intermediate (see section 3.2), which is inherently nucleophilic.64 In the first 2-e−-oxidation step, it is quite plausible for the Pμ1,1 intermediate to be protonated by the arylamine substrate, which would be in its ammonium conjugate acid form even at pH 9. Protonation of Pμ1,1 would generate an electrophilic hydroperoxo intermediate to carry out hydroxylation of the substrate amine. On the other hand, the third 2-e−-oxidation step would simply require Pμ1,1 to attack the nitroso substrate to generate the nitroaryl product. How the structural and mechanistic picture recently published for CmlI-Pμ1,164 described above can be reconciled with the somewhat different insights on the closely related AurF-P obtained by Solomon based on MCD and NRVS data62 remains to be seen.
Related to the above discussion is a study on the activation of the O–O bond in Δ9D-P using large-scale multireference ab initio calculations.149,150 It proposes a mechanism that involves protonation of the spectroscopically observed cis-(μ-1,2-peroxo)-diferric center, enlisting the assistance of an adjacent carboxylate ligand to convert it into a μ-1,1-hydroperoxo species (Figure 14). The latter is then proposed to carry out the cleavage of the first substrate C–H bond concomitant with O–O bond homolysis, generating a substrate radical and an FeIV=O that abstracts the second substrate C–H to produce the desaturated product. These mechanistic ideas should assist in addressing the corresponding mechanisms for CmlI and AurF.
Figure 14.
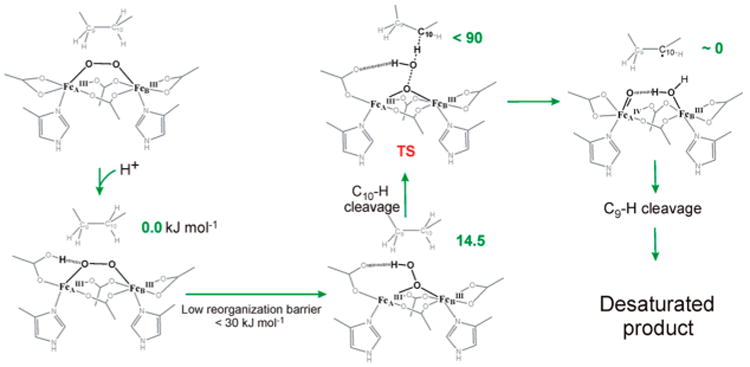
Proposed mechanism by Solomon and co-workers based on ab initio calculations on the action of Δ9D-P in fatty acid desaturation. Reproduced from ref 149 with permission. Copyright 2014 American Chemical Society.
3.3.3. Electrophilic Oxidants of sMMOH and Related Enzymes
P intermediates with a μ-1,2-peroxo binding mode (Pμ1,2) have been implicated in many reaction pathways involving electrophilic substrate oxidation, including those of sMMOH, RNR, Δ9D, T4MOH, and hDOHH. In sMMOH, Pμ1,2 is on the pathway of the native cycle and must convert to intermediate Q in order to be able to oxidize methane.30,31,73–75,109,151,152 However, sMMOH-Pμ1,2 (also called Hperoxo) has been shown to be able to react directly with electron-rich substrates.71,72,143 Thus, sMMOH may follow a sequence entailing Red → P + Sub → product for electron-rich substrates and a different sequence entailing Red → P → Q + Sub → product for harder to oxidize substrates like methane (Scheme 1).
In the case of hDOHH, the decay of its Pμ1,2 intermediate correlates with the appearance of the hydroxylated deoxy-hypusine residue on its eIF5a substrate.68 However, hypusine formation by hDOHH is very slow (∼24 h to completion at room temperature), suggesting that there may be some factor missing under the in vitro conditions. In addition, the Δ9D-Pμ1,2 intermediate is unreactive toward the native substrate under single turnover conditions,48,49 suggesting that the Pμ1,2 species of Δ9D may not be on the native pathway.
For RNR R2, hardly any Pμ1,2 intermediate accumulates in the WT enzyme but does so to a much greater extent in the carboxylate-ligand-substituted D84E variants, allowing the RNR D84E-R2-Pμ1,2 intermediate to be characterized by a variety of spectroscopic methods.38,40,41,110 Kinetic studies demonstrate that Pμ1,2 decay correlates with tyrosyl radical formation,42 supporting the notion that Pμ1,2 is on the native reaction pathway. Interestingly, when electron transfer to the peroxodiferric intermediate is precluded by replacement of the key surface Trp48 residue with Phe, the W48F/D84E-R2-Pμ1,2 species forms and instead carries out the meta-hydroxylation of the nearby F208 residue, albeit quite slowly, to form a purple product due to the coordination of the m-Tyr to Fe2. Thus, by replacing a key residue for electron transfer to the diiron site, R2 can be reprogrammed from being a 1-e−-oxidizing oxidase into a single-turnover 2-e−-oxidizing arene hydroxylase.153 Figure 15 shows a superposition of the diiron active sites of reduced D84E-R2 and the oxygenated product of W48F/D84E R2, emphasizing the similarity of the two active sites. The biggest difference is the shift of the F208 residue upon its hydroxylation to a position that makes the nascent m-tyrosine residue a ligand to Fe2, displacing the water ligand in the reduced protein. meta-Hydroxylation of residue 208 has also been reported for the F208Y-R2 variant to form a green iron-catecholate chromophore that forms within 1 min of adding 4 equiv. Fe(II) to the apoprotein.154,155 Hydroxylation of residue 208 is presumably carried out by the Pμ1,2 intermediate in a Red → P + Sub → product sequence, instead of the Red → P → X → Y• sequence associated with the R2 activation step (Scheme 1).
Figure 15.
Superposition of the diiron sites in reduced D84E-R2 (red) and the purple product obtained from the oxygenation of D84E/W48F R2 (blue) based on protein structures determined by Rosenzweig and co-workers.153 Reproduced from ref 153 with permission. Copyright 2001 American Chemical Society.
The kinetics of the conversion of sMMOH-P to Q has been investigated in great detail. Lee and Lipscomb working on the enzyme from M. trichosporium demonstrated that the decay rate of P matched the formation rate of Q and that both rates decreased sharply with increasing pH.109 The pH dependence of the rates corresponded to a single deprotonation event having a pKa of 7.6 and a solvent kinetic isotope effect (SKIE) of 1.4(1). A similar SKIE value of 1.8 was obtained by Tinberg and Lippard for the enzyme from M. capsulatus,156 emphasizing the requirement for a proton to cleave the O–O bond in P.
P decay leads to the formation of intermediate Q, which has a yellow color arising from UV–vis absorption features at 330 and 430 nm. Mössbauer studies establish the high-valent nature of the intermediate, as revealed by the low isomer shifts (δ = 0.14–0.21 mm/s) and small quadrupole splittings (ΔEQ = 0.53–0.68 mm/s). These parameters, as well as the appearance of quadrupole doublets in zero applied field and their behavior at higher applied magnetic fields, have led Münck to assign Q as having an antiferromagnetically coupled pair of S = 2 iron(IV) centers. In 1993, when Q was first characterized, not much was known about what Mössbauer parameters are associated with a high-spin FeIV center, but more recent work on synthetic oxoiron(IV) complexes157 proved Munck's intuition to be correct. The spectroscopic properties of Q are compared with those of high-valent intermediates of other diiron enzymes in Table 5.
Table 5. Properties of High-Valent Intermediates of Diiron Enzymes.
| enzyme intermediate [dimetal core structure] | λmax (nm) ε (M−1 cm−1) | δ [ΔEQ] (mm s−1) | Raman (cm−1) or EPR data | d(Fe–O) [d(Fe···Fe)] (Å) | refs |
|---|---|---|---|---|---|
| sMMOH-Qa [FeIV2(μ-O)2] | Mt: 330 (7500), 430 (7500) Mc: 420 (8400) | Mt: 0.17 [0.53] Mc: 0.14 [0.55] 0.21 [0.68] | Mt: 690 (−36) 673 | Mt: 1.78 [2.47] | 7,23,31,33,35,36,156 |
| Ct RNR R2-Q [MnIVS=3/2 FeIVS=2(μ-O)2] | ∼390 | 0.17 [−0.75] | 2.017, 2.030, 2.027 | 1.81 [2.75] | 173,181 |
| QX (or sMMOH-Q + e−) | Mc: 0.48 [−0.9] 0.14 [−0.6] | 2 | 182 | ||
| Ec RNR WT R2-X [FeIIIS=5/2–O–FeIVS=2] | 0.56 [−0.9] 0.26 [−0.6] | 2 | 1.75 [2.78] (1.8 [2.49]) | 39,43,167,168,176,177 | |
| Ct RNR R2-X [MnIVS=2 FeIIIS=5/2− (μ-O)(μ–OH)] | 1.74 [2.92] | 173,174,183,184 | |||
| Ec RNR R2-X W48•+ [FeIIIFeIV/W•] | 310 560 (3000) | 2 | 179,180 | ||
| ToMOH-X• I100W• [FeIIIFeIV/W•] | 500 (1500) | 2 | 54,56 |
In this row, Mt and Mc respectively refer to the sMMOHs from M. trichosporium and M. capsulatus
Insights into the structure of the diiron core of Q have been obtained from X-ray absorption spectroscopy.23 Examination of the XANES region shows that Q has a pre-edge area of 28 units, much larger than those observed for decayed Q (14 units) and reduced sMMOH (10 units), suggesting that the iron(IV) centers of Q are in a highly distorted geometry. Fitting the EXAFS region of Q reveals an Fe–O bond distance of 1.78 Å and an Fe···Fe separation of 2.46 Å. The short Fe–O bond is longer than the 1.71 and 1.73 Å Fe–O bonds found respectively for the (μ-oxo)diiron(IV) complexes [FeIV2(μ-O)(BPAE)2]222 and [FeIV2(μ-O)(TAML)2]2−158 but comparable to that found for the diamond core complex [FeIV2(μ-O)2(TPA*)2]4+.165 The Fe···Fe distance of 2.46 Å associated with intermediate Q has led to the postulation of an Fe2(μ-O)2 diamond core. Two crystallographically characterized synthetic complexes with Fe2(μ-O)2 diamond cores, namely [FeIII2(μ-O)2(6-Me3TPA)2]2+ and [FeIIIFeIV(μ-O)2(5-Et3TPA)2]3+,159,160 have Fe···Fe separations of ∼2.7 Å, which is a common feature of synthetic M2(μ-O)2 complexes.161 An even shorter distance of 2.58 Å has recently been found in the crystal structure of the corresponding [FeIIIFeIV(μ-O)2(TPA*)2]3+ complex,162 suggesting that there may be enough flexibility in the diamond core to accommodate the shorter separation associated with Q.
The existence of a diamond core geometry for Q has recently been supported by time-resolved resonance Raman (TR3) spectroscopy.33 351 nm laser excitation of flowing solution samples of Q elicits an 18O-sensitive resonantly enhanced band at 690 cm−1. The observed 690 cm−1 frequency is too low to be associated with a terminal FeIV=O unit in Q (800–860 cm−1)163,164 but nearly matches the 674 cm−1 vibration reported for [FeIV2(μ-O)2(TPA*)2]4+. Moreover, the 690 cm−1 feature observed for Q is downshifted by 17 and 36 cm−1 with the respective use of 16O18O and 18O2 isotopes for the generation of Q. These results indicate that Q incorporates both oxygen atoms of one O2 molecule into the high-valent diiron core that is formed. Thus, TR3 spectroscopy supports the notion that intermediate Q can be best described as having an FeIV2(μ-O)2 diamond core.
The core structure of sMMOH-Q has also recently been probed by Kα high-energy-resolution fluorescence-detected X-ray absorption spectroscopy (HERFD XAS) by DeBeer and co-workers as a more sensitive alternative to conventional XAS approaches to gain insight into its electronic structure and coordination environment.166 HERFD XAS data for sMMOH-Q have been obtained and compared with data for appropriate high-valent diiron model complexes (discussed in greater detail in section 4.2). Significantly higher pre-edge energies and peak intensities are observed for synthetic complexes with open cores than those with closed diamond cores. Comparison of the results for sMMOH-Q with those of the synthetic complexes leads DeBeer to favor an open core for the structure of the diiron site of Q. Clearly, this conclusion disagrees with deductions derived from the 1997 EXAFS analysis23 and the more recent resonance Raman data,33 requiring further investigation to clarify the situation.
The trapping and characterization of Q opened the door to the possibility of assessing its role in the methane oxidation mechanism. Lipscomb and co-workers seized this opportunity to monitor the kinetics of substrate hydroxylation and measure directly the kinetic isotope effect (KIE) for cleaving the substrate C–H bond.73–75 Interestingly, the reaction of Q with CD4 was found to be 50-fold slower than the reaction with CH4, corresponding to a nonclassical KIE of 50 that implicates a reaction with a substantial quantum tunneling component. Surprisingly, no other alkane substrate including ethane gave rise to an H/D KIE in their reactions with Q, indicating that only for methane is the cleavage of its C–H bond rate determining. More detailed studies, including an investigation of the radical clock substrate norcarane,151 led to the proposal of a two-step mechanism for Q decay involving an initial binding step followed by the actual cleavage step. Only in the case of methane would the binding step be rapid. These observations suggest that sMMOH in combination of the regulatory protein sMMOB acts as a molecular sieve that provides methane with more facile access to the active site than the other substrates, so methane can react more rapidly with Q than any other hydrocarbon substrate.
3.3.4. High-Valent Oxidant of RNR R2
A parallel investigation took place in the laboratory of Stubbe on the reaction of O2 with reduced RNR R2, and evidence for a high-valent diiron intermediate called X was obtained. In contrast to the EPR silence of Q, X exhibits an S = 1/2 EPR signal with a nearly isotropic signal at g = 2, suggesting that Q and X differ by one electron.167 Furthermore, it was determined that the function of X was to oxidize the Y122 residue found near the diiron site to generate a relatively stable tyrosyl radical required for initiating ribonucleotide reduction. Reaction conditions were found to accumulate X, allowing a variety of spectroscopic characterization experiments to be carried out. Mössbauer analysis168 revealed an antiferromagnetically coupled pair consisting of an S = 5/2 iron(III) (δ = 0.56 mm/s) and an S = 2 iron(IV) (δ = 0.26 mm/s) center that is well set up to carry out the one-electron oxidation of the nearby Y122 residue, which in turn initiates ribonucleotide reduction by effecting a 1-e−-oxidation of a Cys residue 35 Å away in the R1 subunit.169
EXAFS analysis of RNR R2-X reported in 1998 found an Fe–O distance of 1.8 Å, consistent with the presence of an oxo bridge, and an Fe···Fe distance of 2.49 Å, which was essentially identical to that found for sMMOH-Q.23,43 In the nearly 20 years since this work, this short distance has been difficult to rationalize in light of computational predictions170–172 and synthetic models159,165 that favor an Fe···Fe distance of at least 2.7 Å. This experiment has recently been repeated, taking advantage of new methodologies that have allowed the generation of more concentrated samples of X, which improve the signal-to-noise ratio of the data.39 The more recent studies reveal an Fe–O distance of 1.75 Å and an Fe···Fe distance of 2.78 Å, in agreement with computational results on an FeIIIFeIV(μ-O)2 core. The resolution of the apparent discrepancy between the EXAFS-derived distance and that predicted by DFT for RNR R2-X suggests that similar efforts to resolve this issue in the case of sMMOH-Q would be highly desirable.
This new interpretation is supported by parallel studies on the Class 1c RNR from Chlamydia trachomatis (Ct),173,174 which employs an Fe/Mn center instead of the Fe/Fe center in Class 1a RNRs. Indeed, evidence has been obtained that this enzyme employs a Red → P → Q → X sequence (Scheme 1), generating Fe/Mn-Q and X analogs that are longer lived than their Fe/Fe counterparts. The greater stability of these intermediates has given rise to more concentrated samples that afford higher quality EXAFS data and unequivocally show the Fe···Mn distances of Ct RNR R2-Q173 and X174 to be 2.75 and 2.92 Å, respectively. Based on precedents in the synthetic literature,161 these distances likely derive from an MM′(μ-O)2 core and its conjugate acid (Figure 16).
Figure 16.
Proposed structures of high-valent intermediates of soluble methane monooxygenase and ribonucleotide reductases from Escherichia coli (Ec) or Chlamydia trachomatis (Ct).
Additional insight into the FeIV center of Ec RNR R2-X was obtained from MCD studies of Solomon and co-workers.175 The d–d transitions observed are inconsistent with the presence of an FeIIIFeIV(μ-O)2 core but more compatible with an [FeIIIFeIV(μ-O)(μ-OH)] core. On the other hand, 17O-ENDOR studies176,177 find that X has two nonprotein-derived ligands, an oxo bridge derived from O2 and a terminal (and not bridging) hydroxo ligand on the FeIII (Figure 16). To constrain the Fe···Fe distance to 2.8 Å, a μ-1,1-carboxylate bridge has been proposed, similar to that found in the crystal structures of diferrous CmlA66 and an FeIIIZnII purple acid phosphatase.178 A μ-1,3-carboxylate bridge commonly found in the active sites of this group of diiron enzymes completes the core. Despite the wealth of information, it is still unclear whether RNR R2-X has a site that is more consistent with a [Fe2(μ-O)(μ-OH)] or a [Fe2(μ-O)(μ-1,1-carboxylate) core. There is also the possibility that the two core structures can interconvert, but additional experiments will be required to make that determination.
An electromer of Q can also be observed in the reaction of diferrous RNR R2 with O2. Early in the reaction that forms RNR R2-X, a transient chromophore is observed with a broad absorption maximizing at around 560 nm, which resembles that of a tryptophan cation radical. This 560 nm chromophore reacts rapidly with reductants and perturbs the unique EPR and Mössbauer signatures of RNR R2-X. This species is thus best described as RNR R2-X interacting through space with a nearby Trp cation radical.179,180 This residue has been identified to be W48. It is proposed that this species forms upon electron transfer from W48 to RNR R2-P.
A closely related species is observed in the case of the I100W variant of ToMOH.54,56 ToMOH is distinct from other dioxygen activating diiron enzymes in not generating a peroxo intermediate with a visible chromophore. Upon reaction of the chemically reduced diferrous enzyme with O2, a 500 nm chromophore is observed to form rapidly and then decay, a kinetic behavior comparable to that of diferrous RNR R2 just described above. This chromophore is associated with a transient g = 2 EPR signal that is broadened when the enzyme is 57Fe-enriched, suggesting a strong resemblance between this intermediate and the corresponding [RNR R2-X W48+•] species found in RNR R2. Despite the lack of a visible chromophore, the ToMOH-P intermediate can clearly carry out the oxidative functions it is required to perform.
3.3.5. Intriguing Pair of Toluene Monooxygenases
The ToMOH and T4MOH enzymes catalyze selective hydroxylation of toluene but with different regiospecificities. As discussed above, these enzymes are structurally very similar to each other and to diiron enzymes such as sMMOH and RNR R2, but diverge in reactivity from those of the canonical diiron family. For ToMOH, a peroxodiferric intermediate was identified by Lippard and co-workers by rapid-freeze-quench Mössbauer experiments, but no visible chromophore could be associated with it.54 This surprising observation suggested that the P intermediate formed must differ from the Pμ1,2 intermediates characterized for sMMOH and RNR R2. Furthermore, in the presence of phenol, ToMOH-P decayed 40-fold faster than its self-decay, and catechol was generated as the product. Based on these observations, a Pμ1,1 hydroperoxo intermediate was proposed for ToMOH.
Further studies with the T201S variant revealed another interesting twist. Exposure of the reduced T201S-ToMOH to O2 in the presence of the regulatory protein, ToMOD, led to the formation of both the colorless ToMOH-P as well as another P-type intermediate we designate as ToMOH-P′. The latter exhibits a broad band with a maximum absorbance at ∼675 nm and a Mössbauer quadrupole doublet with δ = 0.67 mm/s and ΔEQ = 1.51 mm/s, parameters resembling those found for sMMOH-P and D84E-R2-P (Table 3). Kinetic analysis shows that ToMOH-P and ToMOH-P′ are in equilibrium, with ToMOH-P being the reactive species in arene hydroxylation.111–113 It is speculated that the T201 residue may provide a critical hydrogen bonding interaction with the peroxodiferric unit to assist in the enzyme catalysis.
In contrast, no clear spectroscopic indications for the accumulation of P intermediates for T4MOH in solution have been reported to date. However, the T4MOH system has yielded three distinct peroxodiferric intermediates in crystallo: a μ-1,2-peroxo species (Pμ1,2, PDB ID 3I63),115 a μ-1,1-(hydro)peroxo species (Pμ1,1 PDB ID STDV),70 and a μ–η2:η2-peroxo species (Pη2,2,, PDB ID STDT; Table 4, Figure 8).70 These crystal-lographic results demonstrates the versatility of the T4MOH diiron active site in accommodating multiple peroxo binding modes.
Crystals of the Pμ1,2 species of T4MOH were obtained by exposing crystals of the diferric form of the enzyme to H2O2, but there is no direct evidence that the Pμ1,2 species is reactive.115 However, toluene is hydroxylated in the peroxide shunt reaction for T4MOH in solution, albeit at a ∼600-fold slower rate than the reaction with O2. This comparison indicates that a Pμ1,2 species in T4MOH could be reactive but is not the native active oxidant for the system.
Crystals of the isomeric Pμ1,1 intermediate were obtained from the Q288A variant of T4MOH, providing the first example of a diiron site with a 1,1-peroxo bridge.70 DFT calculations based on this structure show that the μ-1,1-peroxo is likely a hydroperoxo species, as removing the proton favors electron transfer from the peroxo anion to the diferric center to generate an FeIIFeIII-superoxo species. No toluene was present in the crystallization media, so there is no direct evidence for the reactivity of the Pμ1,1 species. However, the position of the distal peroxo oxygen overlaps with the position of the C3 carbon of toluene from the Pη2,2 structure. This likely rules out the Pμ1,1 intermediate from the native reaction cycle, as substrate positioning appears to be important to achieve proper reactivity in T4MOH.70 Interestingly, Lippard proposed a reactive Pμ1,1 hydroperoxo intermediate in the ToMOH system as the active oxidant.54,111 In light o the Pμ1,1 hydroperoxo from T4MOH being off pathway, the putative peroxo intermediate structure of ToMOH may need to be reassessed.
The Pμ2,2 intermediate of T4MOH was obtained from a crystal of the diferrous form of the enzyme that was soaked with toluene substrate and exposed to O2. In the crystal structure, the O2 is bound to the diiron cluster in a μ–η2:η2 mode, which is unprecedented in iron chemistry. Furthermore, one of the oxygen atoms is connected to the C4 position of the toluene substrate present in the active site, providing evidence for the attack of the bound O2 on substrate. With longer incubation time in O2, crystals of the enzyme–product complex were obtained, with the product p-cresol bound in a μ-1,1 mode.70 This subsequent enzyme–product complex demonstrates that the Pη2,2 intermediate is on the reaction pathway and capable of attacking the C=C double bond of toluene.70 DFT calculations favor formation of an initial FeIIFeIII-superoxo intermediate (S in Scheme 1) that is responsible for the initial attack on toluene and assign the Pη2,2 species as an FeIIFeIII-arylperoxo intermediate with significant unpaired electron density on the aromatic ring.70 T4MOH thus joins an increasing number of other iron enzyme systems that invoke FeIII-superoxide species as the active oxidant.123,185–190
The T4MOH results demonstrate that Pμ1,2 and Pη2,2 may both be reactive but Pη2,2 is on the native reaction pathway. This system provides the first crystallographic evidence to support that Pη2,2 intermediates are catalytically competent in diiron enzymes. The proposed ferric superoxo intermediate as the active oxidant also departs from the mechanisms that invoke high-valent intermediates, like that in sMMOH. This alternative mechanism for the aromatic ring hydroxylation also finds support in studies of Rivard et al. on the mononuclear nonheme enzyme benzoate dioxygenase, for which an initially formed iron(III)-superoxo species is proposed to attack the aromatic ring of the substrate as the first step in the cis-dihydroxylation of the substrate.261
4. Models for Reaction Intermediates of Diiron Enzymes
Over the years, biomimetic complexes have proven helpful in enhancing our understanding of the coordination chemistry of metal centers at metalloenzyme active sites and gaining insight into mechanistic questions. The synthetic models are useful as spectroscopic and structural surrogates for enzyme active sites before protein structural metrics become available. They are even more useful in modeling reaction intermediates, as these enzymatic species may have only a fleeting existence, whereas synthetic analogs have the advantage of often being prepared in organic solvents, providing the possibility of extending the lifetimes to allow a more detailed investigation. Model complexes can also be systematically altered through synthesis and can be exposed to a wider range of conditions (pH and temperature) than can typically be achieved with biological systems. The development of a variety of synthetic complexes exhibiting a range of spectroscopic parameters has been critical to assigning metalloenzyme structural and spectroscopic features. However, most of the characterized synthetic peroxodiferric complexes have μ-1,2-peroxo binding geometries and use primarily neutral N-donor supporting ligands. Therefore, these complexes provide useful but limited points of comparison to enzymatic peroxodiferric intermediates with alternative peroxo binding geometry.
The synthetic models generally employ polydentate supporting ligands with a combination of N-donors such as tertiary amines and pyridines and O-donors like carboxylate and alkoxide ligands to mimic, respectively, ligating histidine and aspartate/glutamate residues in the active site. Some of the ligands, like the tetradentate TPA (TPA = tris(2-pyridylmethyl)amine) occupy four coordination sites on the iron center, leaving two cis labile sites to bind solvent and/or O2 (Scheme 4A).191 These monoiron species can dimerize via an oxo or hydroxo bridge to yield a diiron cluster. Alternatively, a dinucleating ligand can be used like 6-HPA (6-HPA = 1,2-bis[2-{bis(2-pyridylmethyl)amino-methyl}-6-pyridyl]ethane), which has an ethylene unit linking two TPA monomers to force the two iron centers to generate a dimeric unit (Scheme 4B).192 Both of these approaches have been successfully employed to trap dioxygen-derived diiron intermediates that exhibit functionally relevant behavior, which is the focus of this section.
Scheme 4. Examples of Synthetic Mononuclear and Dinuclear Nonheme Iron Complexesa.
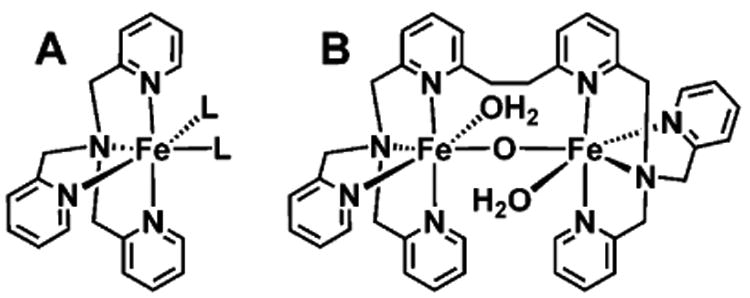
aA: mononuclear iron complex supported by the TPA ligand; B: a dinuclear iron complex supported by the 6-HPA ligand. L is derived from solvent.
4.1. Models for Peroxodiferric Intermediates
Two simple strategies have been employed to generate synthetic peroxodiferric complexes, namely by bubbling O2 into a solution of a diferrous precursor193–204 or reacting a diferric complex with H2O2.134,136,137,192,205–208 Often, these reactions are carried out at lower temperatures in order to extend the lifetime of the adduct and allow a more thorough characterization of the complex. Thus far, over 30 such peroxo complexes have been characterized, which are listed in Table 6. Crystal structures have been solved for five of these complexes,137,194,197,209 while XAS analysis has been useful for obtaining structural insight into those that have not been characterized by XRD.133–135,199,203,210
Table 6. Properties of Synthetic Peroxodiferric Complexesa.
| complex no., polydentate ligand (ancillary ligands) | λmax (εM) | ν(O–O) (Δ18O2) (cm−1) | δ [ΔEQ] (mms−1) | d(Fe···Fe) (Å)b J (cm−1)c | refs |
|---|---|---|---|---|---|
| (μ-Oxo)(μ-1,2-Peroxo)diferric Complexes | |||||
| 1, 6Me2BPP | 450 (1000) 577 (1500) | 847 (−33) | 0.50 [1.46] | 3.171 | 137 |
| 2, 6Me3TPA | 494 (1100) 648 (1200) | 847 (−44) | 0.54 [1.68] | 3.14 | 136,135 |
| 3, BQPA | 480 (1000) 620 (1000) | 844 (−44) | 3.13 | 135 | |
| 4, 6MeBQPA | 495 (1200) 640 (1300) | 853 (−45) | 3.15 | 135 | |
| 5, BnBQA | 505 (1250) 650 (1300) | 854 (−47) | 0.55 [1.43] | 3.16 J > 120 cm−1 | 133 |
| 6, indH | 690 (1500) | 874 (−38) | 3.13 | 134 | |
| 7, (PB)2 | 680 (2000) | 868 (−48) | 0.49 [-0.62] | 208,216 | |
| 8, 6HPA | 500 (1100) 610 (820) | 826 (−51) | 0.35 [1.64] | 192,219 | |
| 9, BPG2E | 452 (1420) 546 (1300) | 835 (−51) | 0.48 [1.66] | 220 | |
| 10, BPPE | 505 (1500) 595 (1400) | 816 (−45) 830 | 0.53 [1.67] | 3.04 | 135,205 |
| 11, BPG2DEV | 490 (1500) | 845 (−48) 819 (−47) | 0.58 [0.58] | 221 | |
| (μ–OH or μ-OR)(μ-1,2-Peroxo)diferric Complexes | |||||
| 1′, 6Me2BPP (μ-OH) | 644 (3000) | 919, 896 (−47) [νO–O0 = 908] | 0.50 [1.31] | 3.395 | 137 |
| 5′, BnBQA (μ-OH) | 730 (2400) | 928 (−53) | 0.57 [1.35] 0.56 [0.96] | 3.46 J = 80(15) cm−1 | 133 |
| 12, Ph-bimp (μ-OR)(μ-1,3-O2CR) [Fe2(Ph-bimp)(O2)(O2CPh)] | 500–700 (∼1700) | 0.58 [0.74] 0.65 [1.70] | 3.328 | 197 | |
| 13, N-EtHPTB (μ-OR) (OPPh3)2 | 3.463 | 209 | |||
| 13a, N-EtHPTB (μ-OR) (η1-O2CR) [Fe2(N-EtHPTB)(O2)(O2CPh)] 2+ | 500 (1500) | 900 (−50) | J ≈ 140(20) cm−1 | 198,210 | |
| 13b, N-EtHPTB (μ-OR) (η1-O2PPh2) | 621 (1800) | 897 (−49) | 0.53 [-1.03] | 3.47 | 210 |
| 13c, N-EtHPTB (μ-OR) (μ-1,3-O2PPh2) | 678 (2100) | 845, 853 (−42) [νO–O0 = 849] | 0.56 [-1.26] | 3.25 | 210 |
| 13d, N-EtHPTB (μ-OR) (μ-1,3-O2AsMe2) | 632 (2100) | 845 (−49) | 3.27 | 210 | |
| 14a, HPTP (μ-OR) (O2CPh) | 572 (2060) | 877, 893 (−51) [νO–O0 = 885] | 198 | ||
| 14b, Me4-tpdp (μ-OR) (μ-1,3-O2CPh) | 616 (2000) | 891, 918 (−47) [νO–O0 = 905] | 200 | ||
| 14c, Htppdo (μ-OR) (μ-1,3-O2CPh) | 610 (1700) | 887 (−48), 873 (−48) | 201 | ||
| (μ-1,2-Peroxo)diferric Complexes without an Additional Single-Atom Bridge | |||||
| 15a, TpiPr2 (μ-1,3-O2CCH2Ph)2 | 694 (2650) | 888 (−46) | 0.66 [1.40] | 4.004 | 194 |
| 15b, TpiPr2 (μ-1,3-O2CPh)2 | 682 (3450) | 876 (−48) | J = 66 cm−1 | 193,211 | |
| 16, (Bzim-Py)2 | 685 (1400) | 876 (−50) | 207 | ||
| Carboxylate-Rich Peroxodiferric Complexes | |||||
| 17, 5Me-HXTA (μ-OR) (μ-1,3-O2CR) | 470 (1700) | 884 | 206 | ||
| 18, PXDK/(O2CR)2 | 580 (1200) | 861 (−50) | 0.47 [0.88] 0.63 [1.20] | 202 | |
| 19, (dxlCO2)4/py2 | ∼500 (∼1000) | 822 (−43) | 0.65 [1,27] 0.52 [0.71] | μ–η1:η2(?) J ≈ 30(5) cm−1 | 196 |
| (μ-Oxo)(η1-OOH) | |||||
| 20, (DPE)2/Ph4DBA | 484 (2350) | 841 (−43) | 0.53 [1.76] 0.49 [1.15] | 3.16 | 199,215 |
| 7′, (PB)4 | 680 (2000) | 806 (−44) | 0.23 [-1.64] 0.43 [0.49] | 208,216 | |
| 21-HP, 6Me3TPA | 831 (−47) | 204 | |||
1 = [FeIII2(μ-O)(O2)(6-Me2BPP)2]2+, 6-Me2BPP = bis(6-methyl-2-pyridylmethyl)-3-aminopropionate; 2 = [FeIII2(O)(O2)(6-Me3TPA)2]2+, 6-Me3TPA = tris(6-methyl-2-pyridylmethyl)amine; 3 = [FeIII2(μ-O)(O2)(BQPA)2]2+, BQPA = bis(2-quinolylmethyl)-N-2-pyridylmethylamine; 4 = [FeIII2(μ-O)(O2)(6-Me-BQPA)2]2+, 6-Me-BQPA = bis(2-quinolylmethyl)(6-methylpyridyl-2-methyl)amine]; 5 = [FeIII2(O2)(O)(BnBQA)2]2+, BnBQA = N-benzyl-N,N-bis(2-quinolinylmethyl)amine; 6 = [FeIII2(O2)(μ-O)(IndH)2]2+, IndH = 1,3-bis(2′-pyridylimino)isoindoline; 7 = [FeIII2(O2)(μ-O)(PB)4]2+, PB = (−)- 4,5-pinenebipyridine; 8 = [FeIII2(6-HPA)(O2)(O)]2+, 6-HPA = 1,2-bis[2-{bis(2-pyridylmethyl)amino-methyl}-6-pyridyl]ethane; 9 = [FeIII2(O2)(μ-O)(BPG2E)], BPG2E = 1,2-bis[2-(N-2-pyrid-ylmethyl-N-glycinylmethyl)-6-pyridyl]ethane; 10 = [FeIII2(μ-O)(O2)(OAc)(BPPE)]+, BPPE = 1,2-bis[2-(bis(2-pyridyl)methyl)-6-pyridyl]ethane; 11 = [FeIII2(μ-O)(O2)(BPG2DEV)], BPG2DEVH2 = 2,2′-(5,5′-(4,5-dimethoxy-1,2-phenylene)bis-(ethyne-2,1-diyl)bis(pyridine-5,2-diyl))bis(methylene)bis((pyridin-2-ylmethyl)azanediyl)diacetic acid; 1′ = [FeIII2(μ–OH)(O2)(6-Me2BPP)2]1+; 5′ = [FeIII2(O2)(OH)(BnBQA)2]3+; 12 = [FeIII2(Ph-bimp)(O2)(OBz)]2+, Ph-bimp = 2,6-Bis[bish2-(1-methyl-4,5-diphenylimidazolyl)methyljaminomethyl]-4-methylphenolate; 13 = [FeIII2(O2)(N-Et-HPTB)(OPPh3)2]3+, N-Et-HPTB-H = tetrakis(2-benzimidazolylmethyl)-2-hydroxy-1,3-diaminopropane; 13a = [Fe2(N-EtHPTB)(O2)(O2CPh)]2+; 13b = [FeIII2(O2)(N-Et-HPTB)(η1–O2PPh2)(MeCN)]2+; 13c = [FeIII2(O2)(O2PPh2)(N-Et-HPTB)]2+; 13d = [FeIII2(O2)(μ-1,3-O2AsMe2)(N-Et-HPTB)]2+; 14a = [FeIII2(HPTP)(O2)(OBz)]2+, HPTP = N,N,N′,N′-Tetrakis(2-pyridylmethyl)-1,3-diaminopropan-2-olate; 14b = [FeIII2(Me4-tpdp)(O2)(OBz)]2+, Me4-tpdp = N,N,N′,N′-Tetrakis(6-methyl-2-pyridylmethyl)-1,3-diaminopropan-2-olate; 14c = [FeIII2(HTTPDO)(O2)(OBz)]2+, HTPPDO = N,N,N′,N′-Tetrakis(6-pivalamido-2-pyridylmethyl)-1,3-diaminopropan-2-ol; 15a = [FeIII2(O2)(TpiPr2)2(PPA)2], TpiPr2 = tris(3,5-diisopropyl-1-pyrazolyl)borate, PPA = phenylacetic acid; 15b = [FeIII2(O2)(TpiPr2)2(OBz)2]; 16 = [FeIII2(O2)(Bzim-Py)4(MeCN)2]4+, Bzim-Py = 2–(2′–pyridyl)–N–methylbenzimidazole; 17 = [FeIII2(O2)(5-Me-HXTA)(OAc)]2+, 5-Me-HXTAH5 = N,N′-(2-hydroxy-5-methyl-1,3-xylylene)bis(N-carboxymethylglycine); 18 = [FeIII2(O2)(PXDK)(O2CPhCy)2(py)2], HO2CPhCy = 1-phenylcyclohexanecarboxylic acid; 19 = [FeIII2(dxlCO2)4(O2)(Py)2], dxlCO2− = 2,6-bis[(2,6-dimethylphenyl)methyl]-4-tert-butylbenzoate; 20 = [FeIII2(μ-O)(OOH)(Ph4DBA)(DPE)2]+, H2Ph4DBA = Dibenzofuran-4,6-bis(diphenylacetic acid), DPE = dipyrrolidinoethane; 7′ = [FeIII2(OOH)(μ-O)(PB)4]3+; 21-HP = [(HO)(6-Me3TPA)FeIII(μ-O)FeIII(6-Me3TPA)(OOH)]2+.
Fe···Fe distance determined from EXAFS or XRD (value in italics).
J value based on Ĥ = JS1S2)
The most distinct of the crystallized complexes is [(TpiPr2)2FeIII2(O2)(μ-1,3-O2CR)2] (15), which was first characterized by Kitajima211 and subsequently crystallized by Kim and Lippard (Figure 17B).194 The average O–O distance is 1.408(9) Å, and the average Fe–Operoxo bond length is 1.885(12) Å. The dioxygen ligand binds in a cis mode, with an Fe–O–O–Fe dihedral angle of 52.9°. The Fe–O–O–Fe unit is supported by two 1,3-carboxylate bridges to afford an Fe···Fe distance of 4.004(4) Å. Interestingly, 15 exhibits Mössbauer parameters (δ = 0.66 mm/s, ΔEQ = 1.40 mm/s)194 that resemble values for the P intermediates of sMMOH, Ec RNR R2,37–40 Δ9D,45–47 and the ferroxidase site of ferritin50–53 and thus could serve as a reasonable model for the O2 adducts of these enzymes, all of which have bis(μ-carboxylato)diiron(II) active sites.
Figure 17.
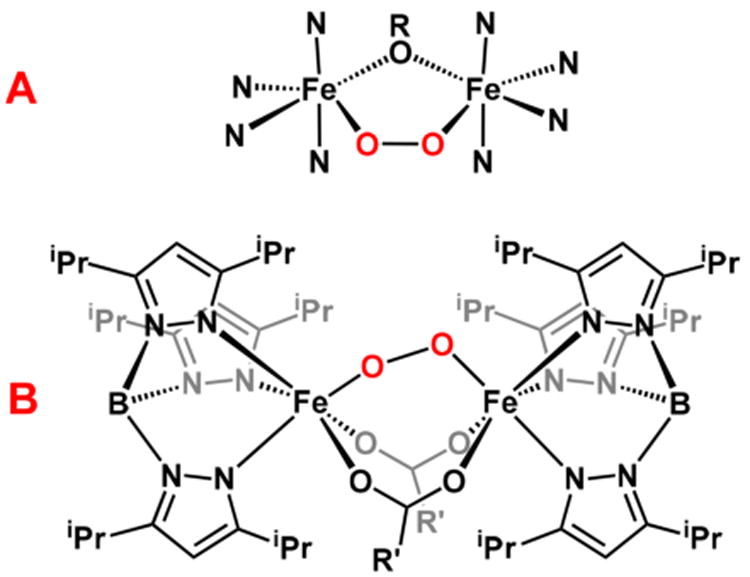
Structures of synthetic peroxodiferric intermediates. A shows the (μ-OR)(μ-1,2-peroxo)diferric core common to four of the crystallographically characterized complexes, where R can be nothing (1), a proton (1′), or an alkyl or aryl group (12 and 13). B represents the crystal structure of 15a.
On the other hand, the other four crystallized examples represent complexes with a cis-(μ-1,2-peroxo)diferric unit that has a single-atom bridge as well, either an oxo, a hydroxo, or an alkoxo ligand (Figure 17A). This subset consists of three structures reported by Suzuki137,197 and one by Que,209 for which comparable O–O distances of 1.40–.43 Å are observed. However, the nature of the single-atom bridge affects the Fe–Operoxo distances, which range from 1.86 to 1.90 Å for hydroxo- or alkoxo-bridged 1′, 12, and 13 to 2.07 to 2.11 Å for the oxo-bridged 1, demonstrating the effect of the basicity of the single atom bridge (for definitions of the complexes, see Table 6). Furthermore, the Fe···Fe distances found for this subset are much shorter than found for 15. Oxo-bridged 1 has an Fe···Fe distance of 3.17 Å, while its conjugate acid, hydroxo-bridged 1′, has a longer Fe···Fe distance of 3.395 Å due to the lengthening of the Fe–μ-O bonds from an average of 1.73 Å in 1 to an average of 1.97 Å in 1′.137 The Fe···Fe distances for the two alkoxo-bridged complexes 12 and 13 are respectively 3.328 and 3.463 Å,197,209 nicely flanking that for 1′. The resulting five-membered rings for these complexes appear relatively flat; the Fe–O–O–Fe dihedral angles of 1 and 13 are equal to 0°, while those of 1′ and 12 are −14.5° and +9.9°, which are relatively small deviations from planarity compared to the 52.9° angle found for 15.194
Extended X-ray absorption fine structure (EXAFS) analysis can be used to obtain structural information in the absence of diffraction quality crystals for XRD analysis. In Figure 18, the Fourier transformed (FT) EXAFS data of the (μ-oxo)(μ-1,2-peroxo)diferric species 4 (black trace)135 is compared to the data for 5′ (blue trace) with a (μ-hydroxo) (μ-1,2-peroxo) core133 and to [FeIV2(μ-O)2(TPA*)2]4+ (24, see Chart 1 for ligand definition; red trace) with a bis(μ-oxo)diiron(IV) “diamond” core.165 The intense peak in the FT at R + Δ ≈ 1.7 Å is comprised of contributions from the Fe–N/O bonds of the primary coordination sphere. Complex 4 has an intense feature ∼1.5 Å mainly from contributions from the short Fe–μ-O bonds (1.83 Å),135 and in 5′ this feature is much less prominent, as the Fe–μ-OH bonds (1.91 Å)133 contribute less intensely to the FT peak. The corresponding FT peak in 24 has shifted to shorter distances and increased in intensity due to the two μ-oxo ligands (1.77 Å). The other main feature in the spectrum is the FT peak associated with the Fe···Fe distance, which is designated for 4 in Figure 18 with the dotted line. The (μ-oxo/hydroxo)(μ-1,2-peroxo) cores of 4 and 5′ result in intense FT peaks at ∼3 Å that are shifted to reflect the Fe···Fe distances for each species (3.15 and 3.41 Å, respectively). In contrast, the bis(μ-oxo)diiron(IV) core of 24 results in a much more intense peak that is shifted to R + Δ ≈ 2.5 Å, reflecting a difference in the Fe···Fe distance and overall diiron cluster structure. EXAFS is thus sensitive to the active site configuration, which makes it a powerful tool for the structural characterization of intermediates.
Figure 18.
Examples of Fourier transformed unfiltered EXAFS data: 4 (black trace),135 5′ (blue trace),133 and 24 (red trace).165 The vertical dotted line represents the position of the main Fe···Fe contribution to the spectrum of 4.
Chart 1. Structures of Ligands Used for Biomimetic Complexes.
The key structural metrics for 11 peroxodiferric complexes have been determined by EXAFS analysis (see Table 6). As found for crystallographically characterized 1, complexes with (μ-oxo)(μ-1,2-peroxo)diferric cores have Fe···Fe distances between 3.13 and 3.16 Å with Fe–μ-Ooxo distances of ∼1.8 Å.133–136 The Fe···Fe distances increase to 3.46 and 3.47 Å, respectively, for complexes with (μ-hydroxo or alkoxo)(μ-1,2-peroxo)diferric cores such as 5′ and 13b133,210 but decrease significantly to 3.25–3.27 Å when an additional μ-1,3-O2X bridge is introduced, as in the cases of 13c and 13d.
Several synthetic peroxodiferric complexes have also been characterized by Mössbauer spectroscopy, affording isomer shifts and quadrupole splittings (Table 6) for comparison with those of the enzyme intermediates (Table 3). The iron(III) ions in these complexes are typically high spin (S = 5/2) centers and have isomer shifts that fall within the range of 0.47–0.66 mm/s. Two complexes stand out from this general trend, namely 8 with a δ value of 0.35 mm/s, which is at the very low end of the range for high-spin ferric centers, and 7′ with a δ value of 0.23 mm/s, which corresponds to a low-spin (S = 1/2) FeIII–OOH moiety.212 The data in Table 6 suggest that isomer shift increases with a decrease in the basicity of the auxiliary bridging ligands. Following this trend, the peroxodiferric enzyme species have an average δ of 0.62 mm/s, as they generally employ hydroxo and carboxylate bridges.
Zero-field Mössbauer spectra of these peroxodiferric complexes show quadrupole doublets, indicating that the ferric ions in these molecules are coupled to each other antiferromagnetically to afford an S = 0 ground spin state (except in the case of 7′). The strength of the coupling interaction is typically quantified with J values for the Hamiltonian Ĥ = JS1S2 and could be probed, which can be determined by magnetic susceptibility, MCD, NMR, or Mössbauer spectroscopy. Only a few J values have been determined for this group of complexes. The temperature dependence of the magnetic susceptibility of a microcrystalline sample of 15b indicates moderately strong antiferromagnetic coupling and a J value of 66 cm−1.211 For comparison, J values for related diferric complexes are 244 cm−1 for the oxo-bridged [(Tp)2FeIII2(μ-O)(O2CR)2] complex213 and 34 cm−1 for the corresponding hydroxo-bridged complex,214 which suggest that the μ-1,2-peroxo bridge is a better mediator of antiferromagnetic coupling than a hydroxo bridge but not as effective as an oxo bridge. This pattern is confirmed by variable field Mössbauer experiments on (μ-oxo)(μ-1,2-peroxo)diferric complex 5 and (μ-hydroxo)(μ-1,2-peroxo)diferric complex 5′, for which respective J values of >120 and 80 cm−1 were obtained.133 NMR experiments on (μ-alkoxo)(μ-1,2-peroxo)-diferric complex 13a estimated a J value of −140(20) cm−1.198 The strength of the antiferromagnetic interaction is likely to be modulated by the Fe–Operoxo bond distances as well as the dihedral angle of the Fe–O–O–Fe unit, but more systematic studies are needed to establish this angular dependence. Such a correlation may be helpful for gaining insight into the geometry of Fe–O–O–Fe units in peroxodiferric enzyme intermediates.
A spectroscopic signature that is typically associated with the synthetic peroxodiferric complexes is the peroxo-to-iron(III) LMCT transition in the visible region that corresponds to a visible absorption maximum ranging from 450 to 730 nm. As can be seen in Table 6, most of the synthetic complexes characterized thus far adopt a μ-1,2-peroxo binding mode with a significant subset consisting of those with an additional μ-oxo bridge. The presence of these two bridging units is manifested in the visible spectra of complexes 1–5 and 8–10 by a double-humped absorption band at ∼500 and ∼650 nm, as exemplified by those of 2 and 8 shown in Figure 19 (black solid line and blue dotted line, respectively). These two features in the cases of 2 and 5 have been assigned respectively to oxo-to-iron(III) and peroxo-to-iron(III) LMCT transitions based on excitation profiles of their resonance Raman bands.135,133 Excitation into the lower energy LMCT band enhances vibrations arising from the peroxo bridge, which are found at ∼850 cm−1 for the ν(O–O) and at ∼460 and ∼520 cm−1 for the νsym(Fe–O2–Fe) and νasym(Fe–O2–Fe) modes, respectively. On the other hand, excitation into the higher energy LMCT band gives rise to the νsym(Fe–O–Fe) and νasym(Fe–O–Fe) modes of the oxo bridge usually observed at 520 and 700 cm−1.135,133 It is assumed that the visible absorption features of the other complexes in this subset can be assigned analogously. Two complexes in this subset, namely 6 and 7, exhibit only a single broad absorption band in the visible region (Figure 19, green dashed-dotted line),134,208 perhaps because of a blue shift of the oxo LMCT band.
Figure 19.
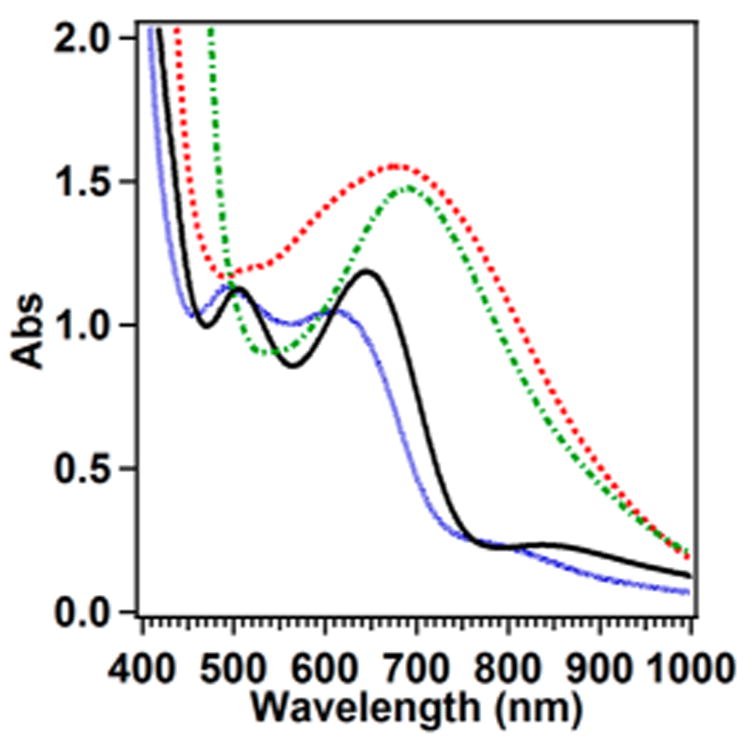
UV–vis absorption spectra of select synthetic peroxodiferric intermediates: 2a (black solid line), 6 (green dashed-dotted line), 8 (blue dotted line), and 13c (red dashed line), all generated at −40 °C in MeCN. See Table 6 and Chart 1 for identification of the complexes.
Corresponding complexes with μ-OR ligands can also be generated, where OR can be a μ-hydroxo, obtained by protonation of the μ-oxo precursor, such as in the case of 1′ and 5′, or a μ-alkoxo, with the R group built into the dinucleating ligand framework. Treatment of complexes 1 and 5 with acid generates the corresponding μ-hydroxo species (1′ and 5′), which exhibit more intense and red-shifted visible absorption features, and the oxo LMCT band disappears.133,137 Protonation typically results in an increase in the ν(O–O) frequency. Comparable features are observed in the μ-alkoxo-bridged complexes (12, 13a–c, 14a, 14b, and 14c).197,198,200,201,206,210 As an example, the visible spectrum for 13c is shown in Figure 19 (red dashed line).
Complexes 15a, 15b, and 16 lack a single-atom bridge. The absence of the highly basic single-atom bridge results in a significant red shift of the peroxo LMCT transition to near 700 nm.193,194,202,211,207 For 15a and 15b, the single-atom bridge is replaced by two 1,3-carboxylate bridges, but the (μ-1,2-peroxo)diferric unit of 16 is completely unsupported. These complexes have visible absorption features resembling those of enzymatic peroxo species in sMMOH-P, Δ9D-P, and W48A/D84E RNR R2-P (Table 3).
The three subsets of complexes from Table 6 discussed above represent the best characterized of the peroxodiferric complexes. Figure 20 plots ν(O–O) values versus Fe···Fe distance for all species in this table for which both values have been obtained and shows an empirical correlation between increasing frequency and increasing distance. The one glaring outlier is 15b (red dot), which lacks a single-atom bridge and has an Fe–O–O–Fe dihedral angle of 52.9°194 that is significantly distorted from those of the four other crystallized complexes. Because the latter four all have an additional single-atom bridge to assemble a (μ-oxo/hydroxo/alkoxo)(μ-1,2-peroxo)diferric core, the five-membered rings thus formed are relatively flat, as found in the crystal structures of 1, 1′, 12, and 13.137,197,209
Figure 20.
Empirical correlation between ν(O–O) and d(Fe···Fe) for peroxodiferric complexes listed in Table 6. Blue dots = (μ-oxo)(μ-1,2-peroxo)diferric complexes, and purple dots = (μ-hydroxo or alkoxo)(μ-1,2-peroxo)diferric complexes. The outlier represented by the red dot is 15a, which has a bis(μ-1,3-carboxylato)(μ-1,2-peroxo)diferric core with no single-atom bridge.
The utility of this correlation can be tested using some of the data in Table 3. For example, if we take ν(O–O) of 851 cm−1 observed for the peroxodiferric intermediate of the ferroxidase site of frog M ferritin and plot it against the Fe···Fe distances found for the peroxo-bridged (μ3-oxo)triferric core in the crystal structure of the ferroxidase site of human L ferritin (3.2–3.5 Å), this point would fall within the correlation shown in Figure 20. Plotting the ν(O–O) of 855 cm−1 reported for hDOHH-P against its Fe···Fe distance of 3.4 Å determined by EXAFS analysis puts hDOHH-P close to the established correlation. On the other hand, plotting the ν(O–O) of 791 cm−1 for CmlI-P9,64 against its Fe···Fe distance of 3.35 Å from EXAFS analysis64 produces a point that is quite far off from the line, suggesting that it does not have the (μ-oxo)(μ-1,2-peroxo)-diferric core structure observed in the crystal structure of CmlIΔ33-P.9,63,64
There are two subsets remaining in Table 6 to discuss. One consists of three carboxylate-rich peroxodiferric complexes. It may be surprising that there are so few of such complexes listed, given that the diiron enzyme active sites often have more carboxylate ligands than histidines, but many more complexes with nitrogen-rich polydentate ligands are reported, perhaps due to their greater ease of preparation and handling. Complex 17 uses a dinucleating ligand framework like that of 12, except for the substitution of the benzimidazoles with carboxylates. By treating [Fe2(5Me-HXTA)(O2CMe)2]− with excess H2O2 at ambient temperature, a peroxodiferric complex has been obtained with sufficient stability to be characterized by UV–vis, resonance Raman, and NMR spectroscopy. The blue shift of the absorption maximum to 484 nm (vs a broad band from 500 to 700 nm for 12) is consistent with the dinucleating ligand having more anionic donors. However, the observed ν(O–O) of 884 cm−1 is not unusual for this type of complex and no information on the Fe···Fe distance is available.
Perhaps more interesting is the following pair of complexes, 18 and 19 (Figure 21), which employ sterically bulky carboxylate ligands. The diiron unit in 18 is supported by an XDK dicarboxylate ligand (Chart 1), which is designed to serve as a stable platform with which to provide the two carboxylates that can bridge a diiron site. LeCloux et al. were successful in assembling diiron(II) complexes having a ligand arrangement approaching that found for the reduced sMMOH active site, including incorporation of a μ-1,1-carboxylate bridge.196 Exposure to O2 afforded an adduct with a broad absorption feature at ∼580 nm and a ν(O–O) of ∼860 cm−1. Mössbauer data revealed an unsymmetric diferric center, with quite different isomer shifts of 0.47 and 0.63 mm/s. Unfortunately, the binding mode of the peroxo ligand could not be established.
Figure 21.
Carboxylate-rich diiron(II) precursors to O2 adducts 18 (left) and 19 (right).
Complex 19 employed a different design strategy for introducing steric bulk on the carboxylate to obtain a diiron(II) complex with four bidentate carboxylates bridging the two metal centers to form a paddlewheel-like complex.196 Upon exposure to O2, the pale yellow precursor turned red-brown in color, corresponding to a broad band centered at ∼500 nm. Like 18, this O2 adduct also has a rather unsymmetric diferric center with Mössbauer isomer shifts of 0.52 and 0.65 mm/s. High-field Mössbauer experiments were carried out to assess the strength of the antiferromagnetic interaction and determined a J value of 30 cm−1, which is smaller than the others listed in Table 6 (≥66 cm−1). This and the observation of a relatively low ν(O–O) frequency of 822 cm−1 suggest a peroxo binding geometry different from the usual μ-1,2-peroxo binding mode and one perhaps favoring O–O bond activation. Indeed, this complex can only be prepared at −80 °C, hinting at its more reactive nature. This complex deserves further study.
The last subset consists of three complexes with a Fe–O–Fe–OOH motif that can serve as models for oxyHr. Complex 20 is the most extensively characterized of the three in this group. Like 18, 20 uses a sterically bulky dicarboxylate ligand Ph4DBA (Chart 1) to provide a template for the assembly of a diiron site.199,215 The diferrous precursor to 20 is [FeII2(μ–OH)(Ph4DBA)(DPE)2](OTf), a (μ-hydroxo)diferrous complex that has been characterized by XRD. This complex serves as a model for deoxyHr and reacts with O2 to form an adduct formulated as [FeIII2(μ-O)(OOH)(Ph4DBA)(DPE)2]+ and has a UV–vis absorption spectrum remarkably similar to that of oxyHr (λmax 334 nm (εM 6400) and 484 nm (εM 2350)) for 20 versus 330 nm (εM 6800) and 500 nm (εM 2200) for oxyHr. Like oxyHr, this adduct exhibits a Mössbauer spectrum that is fit with two different quadrupole doublets with parameters closely resembling those of oxyHr (see Table 6). Its resonance Raman spectrum shows a ν(O–O) at 841 cm−1 with a 43 cm−1 downshift with the 18O2 isotopomer. EXAFS analysis reveals an O scatterer at ∼1.8 Å and an Fe···Fe distance at 3.16 Å, consistent with the presence of a (μ-oxo)bis(μ-1,3-carboxylato)diferric core.
The two remaining complexes, 7′ and 21-HP, were not discovered by design but through serendipity. Species 7′ was found in an effort to understand the reaction mechanism of enantioselective sulfoxidation by H2O2 catalyzed by [FeIII2O-(PB)4(OH2)2]4+.208,216 At −40 °C, a broad absorption band with a maximum near 650 nm could be observed, which is reminiscent of a peroxo-to-iron(III) transition. Mössbauer characterization of this sample showed a mixture of iron centers in this solution, corresponding to three diferric components. One quadrupole doublet corresponded to the starting complex representing 38% of the Fe in the sample. A second doublet representing 36% was assigned to the (μ-oxo)(μ-1,2-peroxo)diferric complex 7 discussed earlier. The third component is 7′, which exhibits two doublets of equal intensity with δ1 = 0.43 mm/s, ΔEQ1 = 0.49 mm/s; δ2 = 0.23 mm/s, ΔEQ1 = 1.64 mm/s. While the parameters of the first doublet are similar to those of the high-spin ferric centers of 7, those of the latter resemble parameters for a low-spin ferric ion. To account for the EPR silence of the sample, these two centers must be coupled to give rise to a diferric center with an integer spin. High-field multifrequency EPR data confirmed the presence of an unprecedented S = 2 peroxodiferric species derived from the antiferromagnetic coupling of S = 5/2 and S = 1/2 iron fragments. Resonance Raman evidence confirms this assignment, with frequencies of 483 and 868 cm−1 corresponding to an Fe–O–Fe unit and frequencies of 618 and 806 cm−1 that are typical of an S = 1/2 FeIII–OOH unit and are downshifted with the use of H218O2.212,217
The last complex of interest in this section is 21-HP, which is observed in the reaction of [FeII2(OH)2(6Me3TPA)2]2+ with O2 (Figure 22).204,208 At −80 °C, an initial intermediate (designated 21-S) is observed with visible maxima at 500 nm (εM 1400) and 620 nm (εM 1200), which is the first example of a synthetic iron(II)iron(III)-superoxo intermediate (see section 4.3). This assignment is based on its resonance Raman spectrum showing a vibration at 1310 cm−1 that downshifts 71 cm−1 when 18O2 is used instead to generate the intermediate (Figure 22). This species is quite stable at −80 °C, but it decays upon warming to −60 °C. Although the (μ-oxo)(μ-1,2-peroxo)diferric product 2 is formed eventually, evidence for an intervening species 21 can be found by monitoring the time course of the visible and Raman spectra. 21 has been assigned as a diiron(III) species with an end-on bound hydroperoxide based on the observation of its ν(O–O) at 831 cm−1 and its ν(Fe–O) at 483 cm−1. This system provides an unprecedented opportunity to monitor the stepwise conversion (Figure 22) of a diferrous precursor to its (μ-oxo)(μ-1,2-peroxo)diferric product 2 one electron- or proton-transfer step at a time via two novel intermediates. Given the increasing evidence for superoxo intermediates in nonheme iron enzymes,70,186,187,190 more efforts should be focused on characterizing the nature and reactivity of such nonheme iron(III)-superoxo species.203,218
Figure 22.
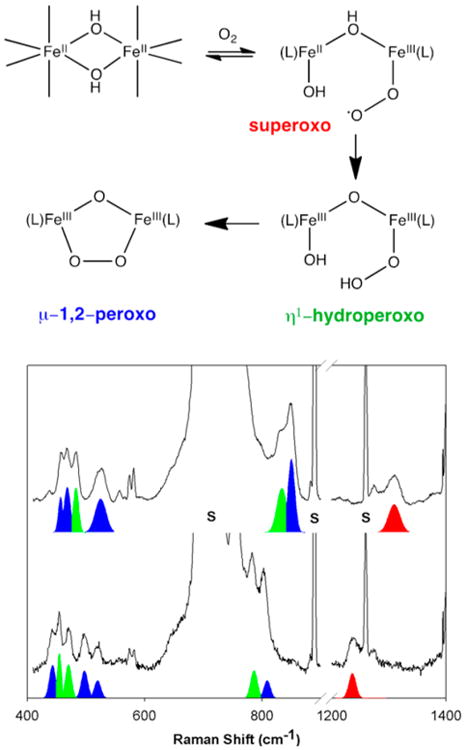
Top: Steps in the conversion of [FeII2(OH)2(6Me3TPA)2]2+ to 2. Bottom: Resonance Raman spectra of the dark olive green solution generated from the reaction of 1 with 16O2 (top) or 18O2 (bottom) in CH2Cl2 at −80 °C. Color-coded features associated with the superoxo (21-S, red), hydroperoxo (21-HP, green), and 1,2-peroxo (2, red) species. Bottom panel reproduced from ref 204 with permission. Copyright 2005 National Academy of Sciences.
4.2. Models for High-Valent Diiron Intermediates
For a number of diiron enzymes, peroxodiferric intermediates may not be powerful enough to carry out the desired substrate oxidation and therefore must undergo O–O cleavage to generate the high-valent oxidants required to cleave the relatively strong C–H bonds of the substrates for such enzymes. This mechanistic scenario is best epitomized by sMMOH, although sMMOH-P (or Hperoxo) has been shown to be capable of oxidizing electron-rich olefins like ethyl vinyl ether and propene as well as diethyl ether,71,72,143 but it must first convert to sMMOH-Q in order to oxidize methane, its natural target.
Unlike the large number of synthetic peroxodiferric complexes discussed in the preceding section that can serve as possible models for P, synthetic models for Q are fewer in number, with only seven (μ-oxo)diiron(IV) complexes that have been described thus far (Table 7).158,162,222 Among these seven complexes, only for [FeIV2O(TAML)2]2− (22) has a crystal structure been solved.158 Complex 22 exhibits an Fe–μ-O distance of 1.7284(8) Å, which is shorter than that typically found for (μ-oxo)diiron(III) complexes131 but consistent with the higher-valent nature of the iron(IV) centers.158 A similar Fe–μ-O distance (1.71 Å) is associated with [FeIV2O(BPAE)2]4+ (23), which was generated by the electrochemical oxidation of its diferric precursor 23a via the one-electron oxidized FeIIIFeIV complex 23b.222 Perhaps the most relevant of these complexes as a model for sMMOH-Q is [FeIV2(μ-O)2(TPA*)2]4+ (24), which was found by EXAFS analysis to have an Fe-μ-O distance of 1.77 Å and an Fe···Fe distance of 2.73 Å,165 core dimensions that approach those associated with sMMOH-Q.23
Table 7. Properties of Synthetic High-Valent Diiron Complexes.
| complex no. and formula UV–vis absorption data | δ [ΔEQ] (mms−1) spin state EPR g values | ν(Fe–O) (Δ18O) (cm−4) | XRD or EXAFS results (Å) | refs |
|---|---|---|---|---|
| 22, [FeIV2O(TAML)2]2− | −0.07 [3.3] S = 0 (S1 = 1/S2 = 1) | 1.7284(8) (Fe–μ-O) 3.3497(9) (Fe···Fe) 151.4(2)° (∠Fe–O–Fe) | 158 | |
| 23, [FeIV2O(BPAE)2]4+ | −0.05 [2.14] S = 0 (S1 = 1/S2 = 1) | 1.71 Å (Fe–μ-O) 3.08 Å (Fe···Fe) | 222 | |
| 23a, [FeIII2O(BPAE)2]2+ | 0.45 [1.30] S = 0 (S1 = 5/2/S2 = 5/2) | 1.795(16) Å (Fe–μ-O) 3.007(3) Å (Fe···Fe) 113.68(8)° (∠Fe–O–Fe) | 222 | |
| 23b, [FeIIIFeIV0(BPAE)2]3+ | 0.40 [1.57] S = 1/2 (S1 = 1/2/S2 = l) | 1.70 Å (Fe–μ-O) 3.07 Å (Fe···Fe) | 222 | |
| 24, [FeIV2(μ-O)2(TPA*)2]4+ λmax 485 (εM 9800) 875 (εM 2200) | −0.04 [2.09] S = 0 (S1 = 1, S2 = 1) | 674 (−30) (Fe–μ-O) | 1.77 Å (Fe–μ-O) 2.73 Å (Fe···Fe) | 165 |
| 25a, [FeIIIFeIV(μ-O)2- (5Me3TPA)2]3+ λmax 615 (εM 5500) | 0.12 [0.49] S = 3/2 (S1 = 1/2, S2 = 1) g = 4.45, 3.90, 2.01 | 676, 656 (−32) (Fe–μ-O) | 159,223,224 | |
| 25b, [FeIIIFeIV(μ-O)2- (5Et3TPA)2]3+ | 1.805, 1.860 Å (Fe–μ-O) 2.683 Å (Fe···Fe) 94.1° (∠Fe–O–Fe) | 159,223,224 | ||
| 25c, [FeIIIFeIV(μ-O)2(TPA*)2]3+ | 0.11 [0.44] S = 3/2 (S1 = 1/2, S2 = 1) | 165 | ||
| 26, [(HO)(L)FeIV–O–FeIV(O)(L)]3+ L = TPA* | 0.00 [1.96] 0.03 [0.92] S1 = 1/S2 = 1 | 1.65 (Fe=O) 1.80 Å (Fe–μ-O) 3.32 Å (Fe···Fe) ∼ 130° (∠Fe–O–Fe) | 225–228 | |
| 27, [(HO)(L)FeIII–O– FeIV(O)(L)]3+ L = TPA* | 0.40 [-0.60], 0.09 [-0.40] S = 1/2 (S1 = 5/2, S2 = 2) g = 2.008, 2.003, 1.992 (Q-band EPR) A(1H) ∼ 35 MHz(D2O exchangeable) J = 90(20) cm−1 (by EPR) | ∼130° (∠Fe–O–Fe) | 225–228,231 | |
| 28, [(F)(L)FeIII–O– FeIV(O)(L)]3+ L = TPA* | 0.45 [1.50], 0.10 [0.60] S = 1/2 (S1 = 5/2, S2 = 2) g = 1.99, 2.01, 2.01 A(19F) = (92, 113, 253) MHz J = 90(20) cm−1 (by EPR) | 1.66 (Fe=O) 1.83 (Fe–F, Fe–μ-O) 3.56 (Fe···Fe) ∼ 180° (∠Fe–O–Fe) | 225–228 | |
| [(O)(L)FeIV–O–FeIV(O)(L)]2+ L = TPA* 29a 29b | S = 0 (S1 = 2/S2 = 2) 0.14 [0.52] S = 3 (S1 = 1/S2 = 2) −0.02 [−1.17], 0.14 [0.82] | 229 | ||
| 30, [(FeIVO)2(μ-O)(6-HPA)]2+ | 0.13 [0.44] S = 0 (S1 = 2/S2 = 2) | 820 (−43) (Fe=0) | 192,219 | |
| 31, [(FeIVO)2(μ-O)(BPG2E)] | 0.20 [0.40] S = 0 (S1 = 2/S2 = 2) | 220 | ||
| 32, [(L)FeIII–O–FeIV(O)(L)]3+ (L = 6Me3TPA) | 0.50 [1.30], 0.10 [1.14] S = 1/2 (S1 = 5/2, S2 = 2) g = 1.999 | 840 (16O,16O) 835 (18O,16O) 797 (16O,18O) 794 (18O,18O) | 233 | |
| 32a, [FeIII2(μ-O)2(L)2]2+ (L = 6Me3TPA) λmax 470 (εM 560) 760 (εM 80) | 0.50 [1.93] | ∼700 (Fe–μ-O) | 1.841, 1.917 Å (Fe–μ-O) 2.95 Å (Fe···Fe) 92.5° (∠Fe–O–Fe) | 138,160 |
| 32b, [FeIII2(μ-O)(μ–OH)(L)2]3+ L = 6Me3TPA λmax ≈ 550 (800) ∼ 800 (70) | 1.82 (Fe–μ-O) 1.99 Å (Fe–μ–OH) 2.714 Å (Fe···Fe) 98.7° (∠Fe–O–Fe) | 138,234 | ||
| 33, FeIIIFeIV species from the reaction of O2 with [FeII2(O2CR)4(pyridine)2] (R = 2,6-di(p-tolyl)phenyl) | 0.56 [0.9], 0.19 [0.8] S = 1/2 (S1 = 5/2, S2 = 2) g = 1.99, 2.01, 2.01 | 235 |
Complex 24 belongs to a collection of high-valent diiron complexes supported by the TPA* ligand within a reaction landscape that has been explored by Que and co-workers over the past ten years (Scheme 5). This investigation in fact began in 1991 when Leising et al. reported that the reaction of a (μ-oxo)diiron(III) TPA precursor with H2O2 generated an intense emerald-colored chromophore with an S = 3/2 EPR signal.223 Subsequent characterization of this species by a variety of spectroscopic methods led to its identifiction as the valence-delocalized [FeIIIFeIV(μ-O)2(R-TPA)2]3+ complex,224,253,254 which was eventually confirmed by the crystal structure of its 5-Et3-TPA derivative 25b.159
Scheme 5. Landscape of High-Valent Diiron Complexes Supported by the TPA* Liganda.
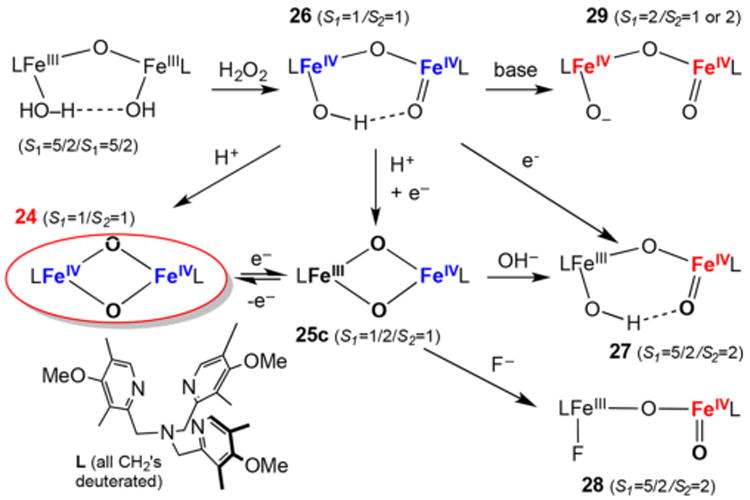
a Blue indicates an S = 1 FeIV center, while red indicates an S = 2 FeIV center.
It took another decade to obtain the diiron(IV) analog, which was achieved by the introduction of TPA*, an analog of TPA with electron donating substituents on the 3-, 4-, and 5-positions of each pyridine, which enhanced the thermal stability of the FeIV2(μ-O)2 core. In the initial experiments, 24 was prepared by one-electron electrochemical oxidation of [Fe2(μ-O)2(TPA*)2]3+ (25c), which was shown by Mössbauer spectroscopy to have S = 1 FeIV centers.165 In subsequent experiments, it was found that 24 could also be prepared by the addition of an equivalent of strong acid to 26, the product of the reaction of H2O2 with a diferric precursor [(H2O)(TPA*)FeIII–O–FeIII (TPA*)-(OH)]3+. Complex 26 was found to have an O=FeIV–O–FeIV–OH core, with both iron(IV) sites having an S = 1 spin state. This open core closed up to form the FeIV2(μ-O)2 diamond core of 24 upon acidification (Scheme 5).225–228
The discovery of such open-core complexes expanded the landscape of high-valent diiron complexes and led to the identification of three other related complexes, 27–29, shown in Scheme 5,225–229 which were all found to have S = 2 FeIV=O centers. Complex 27 can be generated either by one-electron reduction of 26 or by treating closed-core 25c with an equivalent of OH− in an acid–base reaction. This complex exhibits a nearly isotropic S = 1/2 EPR signal at g = 1.99 (Figure 23A) that resembles that observed for RNR R2-X.167 On the other hand, 28 is obtained by adding an equivalent of F− to 25c. This complex also has an isotropic S = 1/2 EPR signal, but the signal exhibits a significant 19F superhyperfine splitting, indicating 19F coordination to the diiron unit (Figure 23B). It would thus appear that the binding of a hard anion such as hydroxide or fluoride to the iron(III) site causes the iron(IV) site to switch from S = 1 in the diamond core to S = 2 in the open-core complex.
Figure 23.
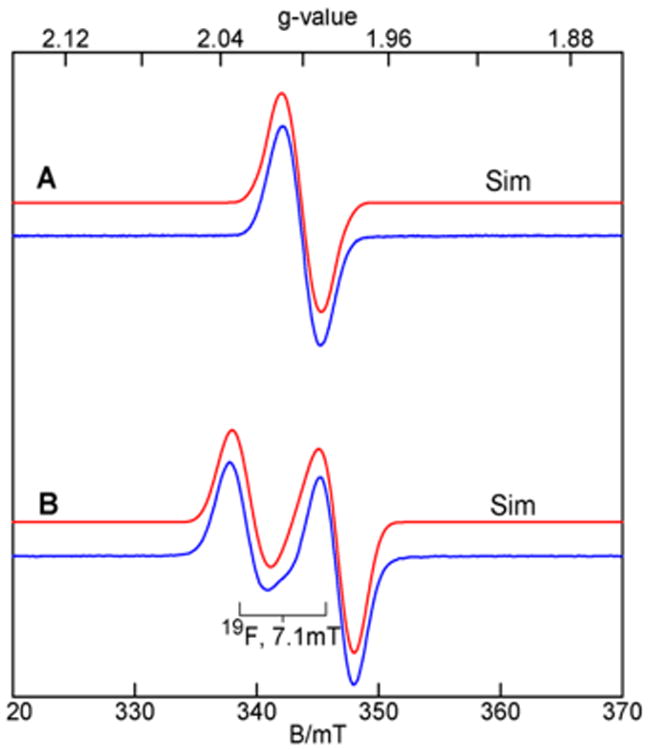
X-band (9.63 GHz) EPR spectra of (A) 27 and (B) 28 in 3:1 CH2Cl2-MeCN (blue lines) and corresponding simulations (red lines) with g = 2.002 for A and g = (1.99, 2.01, 2.01) for B. The fit in (B) includes one 19F nucleus with A = (92, 113, 253) MHz.230 Reprodued from ref 230 with permission. Copyright 2010 American Chemical Society.
EXAFS studies show that 27 and 28 differ in their Fe···Fe distances, 3.32 Å for the former and 3.56 Å for the latter (Table 7), reflecting differences in the Fe–O–Fe angle.225–228 The deviation of the Fe–O–Fe unit from linearity in 27 is proposed to arise from the presence of hydrogen bonding between the proton on the FeIII–OH half to the FeIV=O unit on the other half of the complex, resulting in the decrease in the ∠Fe–O–Fe from 180° to 130°. Support for this H-bonding interaction has been obtained experimentally from ENDOR experiments that verify the presence of an H atom at the correct distance predicted for the six-membered ring that is postulated to form (see the proposed structure of 27 in Scheme 5).231 This result demonstrates that the electrophilic FeIV=O unit also has some nucleophilic character.
Complex 29 is the last of the three open-core complexes to be discussed with S = 2 FeIV=O units. Unlike 27 and 28, 29 is a diiron(IV) complex that is generated by treating 26 with base, turning the green color of 26 to orange.229 As shown in Figure 24, the conversion of 26 to 29 is nearly quantitative and demonstrates that 26 has an acidity slightly lower than that of the tetramethylimidazolium cation in 3:1 CH2Cl2-MeCN at −85 °C. Mössbauer and parallel mode EPR studies of 29 indicate that the conjugate base is present in two forms. One isomer called 29a consists of an antiferromagnetically coupled pair of equivalent S = 2 FeIV=O sites with Mössbauer parameters (δ = 0.14 mm/s; ΔEQ = 0.52 mm/s) that match those of sMMOH-Q. The other isomer called 29b is a ferromagnetically coupled diiron(IV) complex with local S1 = 1 and S2 = 2 sites. The two isomers have different O=Fe–Fe=O dihedral angles, ∼180° for 29a and ∼90° for 29b, which rationalize the observed difference in the nature of the coupling interactions between the two ferryl units.
Figure 24.
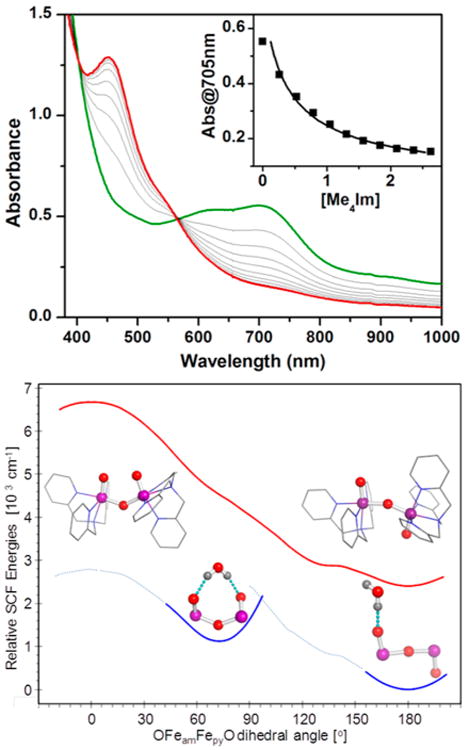
UV–vis spectroscopic changes upon titration of 0.2 mM 26 (green line) with Me4Im to generate 29 in 3:1 CH2Cl2-MeCN at −85 °C (red line).229 Inset: plot showing the change in absorbance at 705 nm vs concentration of added Me4Im. The pKa of 26 is estimated to be 0.3 units higher than that of the conjugate acid of Me4Im. Reproduced from ref 229 with permission. Copyright 2014 American Chemical Society.
The variety of high-valent diiron complexes stabilized by the same supporting ligand depicted in Scheme 5 raises the intriguing question of how the differences in geometry and electronic structure may translate into reactivity, particularly toward the cleavage of C–H bonds. This question has in fact been addressed in studies of Que and co-workers, who have compared the rates of 9,10-dihydroanthracene oxidation for the various complexes at −80 °C (the very low temperature being necessary to be able to measure the rates for the most reactive of the complexes). These results are summarized in Figure 25, which show a difference of 7.5 orders of magnitude between the least reactive complex (25c) and the most reactive one (28). Based on these observations, two main factors control the C–H bond cleavage rates, whether the oxo is bridging or terminal and whether the spin state of the Fe=O unit is 1 or 2. The message from this graph is clear: (a) a terminal oxo is at least 100-fold faster than a bridging oxo in cleaving C–H bonds and (b) an S = 2 FeIV=O is 103–104 more reactive than an S = 1 FeIV=O.
Figure 25.
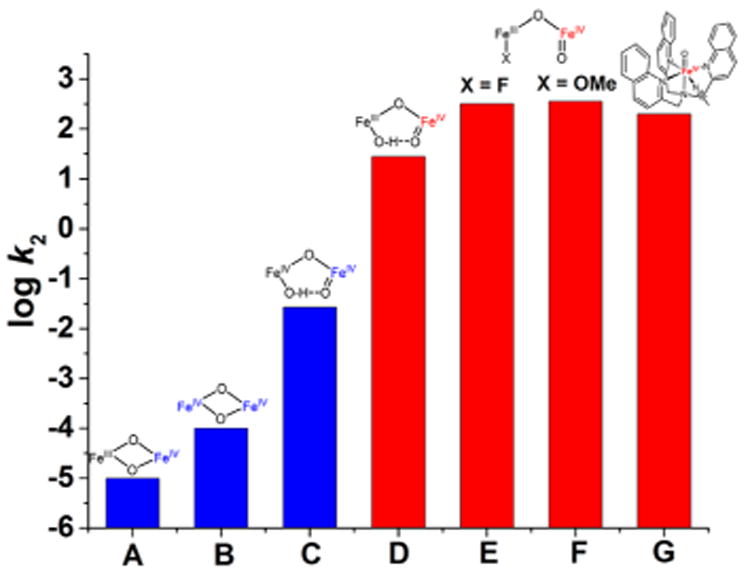
Comparison of DHA oxidation rates by high-valent diiron complexes supported by the TPA* ligand at −80 °C in a 3:1 CH2Cl2/MeCN solvent mixture. Blue bars indicate complexes with S = 1 FeIV centers, while red bars correspond to complexes with S = 2 FeIV centers.157 Legend: A = 25c, B = 24, C = 26, D = 27, E = 28, F = [(MeO)(TPA*)FeIV–O–FeIV(TPA*)]3+, and G = [FeIV(O)(TQA)]2+, a highly reactive monoiron-oxo complex which is included for comparison. Adapted from ref 157 with permission. Copyright 2015 American Chemical Society.
In parallel to the efforts in the Que laboratory, Kodera has reported some truly provocative results on synthetic high-valent diiron complexes by pursuing a strategy focusing on complexes of dinucleating variants of the tripodal tetradentate ligands, exemplified by the 6-HPA and BPG2E ligands (Chart 1). These ligands are essentially dimeric versions of the tripodal TPA and BPG ligands tethered by an ethylene linker that connects the 6-positions of one pyridine on each half of the dinucleating ligand. These studies start with the (μ-oxo)diferric complexes with one coordination site on each iron occupied by solvent, as illustrated in Scheme 4. Addition of H2O2 leads to the efficient catalytic oxidation of alkanes and alkenes at 25 °C.220,232 At lower temperatures, however, it is possible to see the formation of visible chromophores suggestive of the formation of (μ-1,2-peroxo)diferric species, as discussed in section 4.1.192,219,220
Kodera has accumulated intriguing evidence for showing an equilibrium between the (μ-oxo)(μ-1,2-peroxo)diferric species and the corresponding (μ-oxo)diferryl species by reversible O–O bond cleavage (Scheme 6). Most convincing are Mössbauer data on solids precipitated from solution demonstrating the reversible interconversion of the two isomers as the temperature is changed between 23 and 295 K. Two quadrupole doublets are observed in these spectra; one doublet has parameters typical of the (μ-oxo)(μ-1,2-peroxo)diferric species (compare data for 8 and 9 with other complexes in Table 6), while the other doublet has parameters associated with S = 2 FeIV=O complexes (compare data for 30 and 31 with other complexes in Table 7). In fact, the parameters for the latter resemble those of 28a (Table 7) as well as those of sMMOH-Q (Table 5). In the case of the 6-HPA complex, the ratio between the two isomers is 60:40 in favor of the peroxodiferric isomer at 23 K but becomes 15:85 at 295 K, clearly favoring the diferryl isomer. The situation is similar for the BPG2E complex, with the peroxodiferric isomer favored 90:10 at 23 K and the ratio becoming 45:55 at 295 K.
Scheme 6. Proposed Conversion of 8 to 30 and 9 to 31192,219,220.

Support for this equilibrium can in principle be obtained from resonance Raman studies by monitoring the ν(O–O) of the peroxo isomer and the ν(Fe=O) of the diferryl isomer. However, these features have similar frequencies and the data obtained are somewhat noisy, so the Raman results are not as persuasive.192,219,220 The possibility of the equilibrium shown in Scheme 6 was initially suggested by isotope labeling results in the catalytic oxidation experiments in which the 18O-label on the oxo bridge of the diferric catalyst was partially incorporated into the epoxide product of trans-β-methylstyene epoxidation.232 This result is most easily rationalized by label scrambling among O atoms in the (μ-oxo)diferryl isomer.
4.3. Models for Iron-Superoxo Intermediates
There are only four nonheme iron-superoxo complexes described with sufficient characterization to be listed in Table 8. The small number is likely due mainly to the lack of relevant enzyme intermediates to model. However, increased activity in this biomimetic endeavor may be anticipated, given the recent exciting crystallographic results on T4MOH implicating a catalytically relevant iron(II)iron(III)-superoxo species in the hydroxylation of toluene.70 These developments can be coupled with emergent findings about the key roles played by iron(III)-superoxo intermediates in the catalytic cycles of mononuclear nonheme iron enzymes such as the extradiol cleaving catechol dioxygenases,123,236–238 Rieske dioxygenases,187 and isopenicillin N synthase.190 In addition, a superoxo intermediate has been trapped and characterized spectroscopically by Bollinger and Krebs for the diiron enzyme myo-inositol oxidase,10,186,239–241 which is not discussed in this review.
Table 8. Properties of Synthetic Iron-Superoxo Complexes.
| complex no. and structural details when available | λmax (εM) | δ [ΔEQ] (mms−1) spin state | ν(O–O) (Δ18O2) (cm−1) | refs |
|---|---|---|---|---|
| 21-S, [(HO)(6-Me3TPA)FeII–OH–FeIII(6-Me3TPA)-(O2)]2+, the first intermediate formed in the reaction of O2 with [FeII2(μ–OH)2(6Me3TPA)2]2+ | 325 (10 300) 500 (1400) 620 (1200) | 1310 (−71) | 204 | |
| 34, FeIIFeIII-superoxo species trapped upon O2 exposure of [FeII2([G-3]COO)4(L)2]ad(Fe–O)ave = 2.02–2.05 Å d(Fe···Fe) = 3.00–3.05 | 442 (6000) | 1.20 [3.10] 0.48 [0.71] | 203 | |
| 35, [Fe(BDPP)O2] | 330 (9400) | 0.58 [−1.65] S = 3 (S1 = 5/2/S2 = 1/2) | 1125 (−63) | 218,243 |
| 36, [FeIII(η2-O2)(TAML)]3− d(Fe–O)ave = 1.92 Å d(O–O) = 1.323, 1.306 Å ∠O–Fe–O = 40.4°, 39.9° | 490 (2600) | 0.10 [2.66] EPR silent S = 1 (3.26 μB by Evans method) (S1 = 3/2/S2 = 1/2) | 1260 (−77) | 242 |
| MlOXb intermediate G (FeIIIFeIII-superoxo) | S = 1/2 g = 2.05, 1.98, 1.90 | 186 |
[G-3]COO− = 3rd generation dendrimer-appended terphenyl carboxylate ligand; L = 4-pyrrolidinopyridine.
MIOX = myo-inositol oxygenase.
The first complex listed is a species designated 21-S that is trapped in the oxygenation of [FeII2(μ-OH)2(6Me3TPA)2]2+ at −80 °C in CH2Cl2 en route to its conversion to the (μ-oxo) (μ-1,2-peroxo)diferric derivative 2 discussed in Section 4.1.204 It exhibits a UV–vis absorption spectrum with three relatively intense absorption bands at 325, 500, and 620 nm, and excitation into the lowest energy band elicits an enhanced vibration at 1310 cm−1 that downshifts 71 cm−1 with 18O2 (Figure 22). These observations clearly assign this vibration to a superoxo ligand, which must derive from the one-electron transfer from the diferrous center to O2 upon binding. In turn, the diiron(II) center is oxidized to the iron(II)iron(III) oxidation state. Unfortunately, the need for CH2Cl2 as solvent precludes the use of Mössbauer spectroscopy to ascertain the iron oxidation states in the intermediate. The use of mixed-labeled O2 to form the adduct affords an isotope pattern strongly suggestive of an end-on bound dioxygen unit. Upon warming to −60 °C, 21-S decays and converts to 21-HP, which has been tentatively assigned to be an FeIII–O–FeIII–OOH species on the basis of its resonance Raman spectrum (Table 6). In the final step, 21-HP converts to 2 (Figure 22, top).
The second example in Table 8 (34) is an O2 adduct derived from a carboxylate-rich diiron(II) center (related to complexes 18 and 19 in Table 6) reported by Lippard and co-workers.203 This diiron(II) center is embedded in a dendrimer that serves to slow down the O2 chemistry and allows the initial O2 adduct to be trapped. This adduct exhibits an intense visible absorption band at 442 nm, which is blue-shifted relative to those observed for the carboxylate-rich peroxo complexes 17–19. Mössbauer data is available for this adduct, showing two quadrupole doublets in a 1:1 ratio corresponding to a coupled pair of high-spin FeII and high-spin FeIII centers. The demonstrated one-electron oxidation of the diiron(II) center would suggest that O2 has been reduced to the superoxide level in the adduct, but there is no vibrational data reported to shed light on the O2-derived ligand. EXAFS studies suggest the possibility of a relatively short Fe···Fe distance of 3–3.05 Å, which led the authors to propose a μ–η1:η2-O2 superoxo ligand.
Due to the paucity of biomimetic superoxo complexes, two recent mononuclear examples have also been included. Complex 35, reported by Lee and co-workers,218 is the O2 adduct of [FeII(BDPP)], where BDPP is a dianionic pentadentate N3O2 ligand that provides a square pyramidal coordination environment about the metal center. The two alkoxide ligands donate sufficient electron density to the iron(II) center to enable it to bind O2 reversibly at −80 °C. The adduct exhibits a rather intense UV–vis absorption band at 330 nm that when probed by a 413.1 nm laser gives rise to an enhanced resonance Raman feature at 1125 cm−1, which downshifts by 71 cm−1 with the use of 18O2. The mixed-isotope experiment was not carried out because the vibrational feature was considered not sharp enough to make this experiment worthwhile. The Mössbauer spectrum showed a single quadrupole doublet with an isomer shift indicative of a high-spin ferric center. The observation of a quadrupole doublet instead of a more complex pattern shows that the S = 5/2 iron center must be exchange coupled to S = 1/2 superoxo ligand. The use of the glass forming 2-MeTHF solvent significantly improved the resolution of the high-field Mössbauer spectra and allowed the exchange coupling to be determined as ferromagnetic, that is, 35 has a S = 3 ground spin state.243
The second example of a mononuclear iron(III)-superoxo complex is 36, reported by Nam and co-workers.242 Complex 36 is formed from the reaction of KO2 with Na[FeIII(TAML)] in the presence of excess 2.2.2-cryptand and persists for days at −20 °C. Because of its stability, crystals have been obtained and a crystal structure has been solved to reveal an iron(III) adduct with a side-on bound dioxygen unit (Figure 26). Consistent with the superoxo assignment, the O–O bond distance is found to be 1.323 Å and a vibration at 1260 cm−1 is observed in MeCN solution that downshifts by 77 cm−1 when K18O2 is used in place of K16O2. The Mössbauer spectrum of 36 shows a quadrupole doublet with an isomer shift and quadrupole splitting quite different from those of 35 and more consistent with an S = 3/2 spin state. This notion is corroborated by an Evans susceptibility measurement showing an S = 1 spin state for the complex, which would result from the antiferromagnetic coupling of the S = 3/2 iron(III) center and the S = 1/2 superoxo ligand. Even with only four examples, the iron(III)-superoxo units found in these complexes exhibit some diversity in their properties, and our understanding of the factors that determine these characteristics would be enhanced by the synthesis of more examples.
Figure 26.
Crystal structure of [FeIII(TAML)(O2)]2− (36) reported by Nam and co-workers, with an O1–O2 bond distance of 1.323 Å, consistent with a superoxo assignment for the O2 unit.242
5. Summary and Perspectives
Dioxygen activating nonheme diiron enzymes have active sites consisting of a combination of histidine and carboxylate ligands that can form distinct O2 adducts to carry out a broad array of physiological functions. Nature has been able to execute these various functions using diiron sites mainly with the same 2-His-4-carboxylate active site assembled within a 4-helix bundle protein fold. This variety of function has led to the discovery of a diverse landscape of activated O2 intermediates that span the range from diiron(II,III)-superoxo (S), through diiron(III)-peroxo (P), to oxo-bridged diiron(III,IV) (X) or diiron(IV) species (Q), all of which can in one enzyme or another, be responsible for substrate oxidation (Scheme 1).
In the canonical diiron systems like sMMOH and RNR R2, P intermediates were initially regarded as a means to obtain the high-valent intermediates Q and X, respectively, but were not considered for their potential as active oxidants in a catalytic cycle. As these two enzymes were the earliest investigated and the best characterized, an assumption was made that other diiron systems would function analogously. However, advances in crystallography, spectroscopy, and computation, as well as in methods for trapping intermediates over the past two decades have contributed to the identification a growing number of catalytically competent S and P intermediates. This boom in biologically relevant oxidants has stimulated chemists to explore the model chemistry of reactive S and P species, in addition to the coveted high-valent oxidants that are employed to cleave the strongest C–H bonds in substrates.
Still, many questions remain to be answered about intermediates from O2 activation. Finding a rationale for the ability of CmlI and T4MOH active sites to support multiple P intermediates is ripe for exploration. In the case of CmlI, the choice of oxidant generates either a Pμ1,2 (from H2O2) or Pμ1,1 (from O2) species, but only the Pμ1,1 intermediate participates in the catalytic cycle.63,64 Crystallographic results show that T4MOH can support Pμ1,2, Pμ1,1, and Pη2,2 intermediates, but follow-up studies in solution should be carried out to assess which of these peroxo intermediates lead to productive enzyme function.70 Finally, further work into the roles of accessory proteins such as sMMOB258 and T4MOD70 in steering the chemistry toward a particular pathway and away from off-pathway intermediates should be undertaken.
What is clear is that not all activated-O2 intermediates in catalytic pathways are alike, and Nature has been clever in her selection of the various types of intermediates used by diiron enzymes. For example, the Pμ1,1 species is sufficient for N-oxygenation in CmlI but may not be a potent enough oxidant for the hydroxylation of aromatic rings or methane. Alternatively, generation of a diiron(II,III)-superoxo or higher-valent species could lead to deleterious off-pathway reactions. Thus, as illustrated in Scheme 1, there is not just one O2 activation mechanism for all diiron enzymes but many related pathways that employ differing diiron oxidants in response to the requirements for the oxidation of the particular substrate. Unraveling the connections among these different pathways and the O2-derived intermediates poses some of the next challenges ahead for bioinorganic chemists and metallobiochemists alike.
Another frontier is represented by the much less well characterized class of integral-membrane-bound nonheme diiron enzymes.89,245,246 Examples of this class include AlkB from Pseudomonas putida (formerly Ps. oleovorans), which is an ω-hydroxylase that catalyzes the hydroxylation of terminal methyl groups on long-chain alkanes, xylene monoooxygenases, and mammalian fatty acid desaturases. An enzyme in the last group of particular interest is stearoyl-CoA desaturase (SCD), which introduces a cis-double bond at C9 and C10 of stearoyl- and palmitoyl-CoA Such monounsaturated derivatives are important precursors of membrane phospholipids, cholesterol esters, and triglycerides, making SCD pivotal in fatty acid metabolism. Thus, SCD is a pharmocological target in the treatment of obesity and diabetes.244
Sequence comparisons among enzymes in this class reveal the presence of eight to nine highly conserved His residues, suggesting a different diiron active site from the soluble enzymes that are the focus of this review.89,245,246 Mössbauer studies on as-isolated AlkB reveal a pair of quadrupole doublets with δ = 0.55 mm/s and ΔEQ = 1.70 mm/s for Fe(1) and δ = 0.51 mm/s and ΔEQ = 1.13 mm/s for Fe(2),247 features that point to an antiferromagnetically coupled oxo-bridged pair of high-spin Fe(III) centers. These values closely resemble those found for oxyhemerythrin (δ = 0.54 mm/s and ΔEQ = 1.92 mm/s for Fe(1) and δ = 0.51 mm/s and ΔEQ = 1.09 mm/s for Fe(2))18 and for toluene-4-monooxygense (δ; = 0.56 mm/s and ΔEQ = 1.55 mm/s for Fe(1) and δ = 0.51 mm/s and ΔEQ = 0.93 mm/s for Fe(2)).14 High-field studies estimate J to be >80 cm−1. This form can be reduced by sodium dithionite to form an air-stable diferrous center with isomer shifts of 1.05–1.15 mm/s. As these values are lower than those found for deoxyhemerythrin (1.20 mm/s) and for the diferrous forms of the soluble diiron enzymes (1.24–1.39 mm/s; Table 2), a His-rich diiron(II) active site has been proposed for AlkB versus the carboxylate-rich sites of the soluble diiron enzymes, in agreement with the sequence homology data.
A recently published X-ray absorption spectroscopic study of oxidized AlkB finds a K-edge at 7125.6 eV and a pre-edge area of 11.2 units,248 which support the Mössbauer assignment for an oxo-bridged high-spin diferric site. The observed pre-edge area is comparable in magnitude to those reported for diiron proteins with oxo-bridged diferric sites such as methemerythrin azide (10.4 units), RNR R2 (10.1 units), and Δ9D (10.8–11.5 units)249,250 but higher than those of diferric enzymes with no oxo bridges such as MMOH32 and phosphate and arsenate complexes of oxidized uteroferrin (6.8 and 7.4 units, respectively).249 However, the presence of an Fe–O–Fe unit is not corroborated by EXAFS analysis, as the best fit of the first coordination sphere shows only a shell of 5 N/O scatterers at 2.02 Å with no evidence found for an O scatterer at ca. 1.8 Å corresponding to the oxo bridge. The best fit also provides some evidence for an Fe scatterer that is found at 3.08 Å, resembling those reported for oxidized sMMOH.32,251 In the latter case, X-ray crystallography has shown an active site with an FeIII2(μ-OH)2 core.29,34 So the structural evidence for oxidized AlkB from XAS needs to be reconciled with somewhat conflicting insight from the Mössbauer analysis.
In strong support of the postulated His-rich active site for this class of enzymes is the crystal structure of the mouse stearoyl-CoA desaturase (SCD) at 2.6 Å resolution (Figure 27),252 which reveals two metal centers with a combined total of nine His ligands and no bound carboxylates. The metal sites in this structure are occupied by zinc ions with a metal–metal separation of 6.4 Å. The fatty acid substrate is positioned nearby, with the C9 and C10 atoms of the acyl side chain in closest proximity to the Zn ions in the presumed active site, in a conformation that rationalizes the observed regioselectivity and stereospecificity of fatty acid desaturation.
Figure 27.
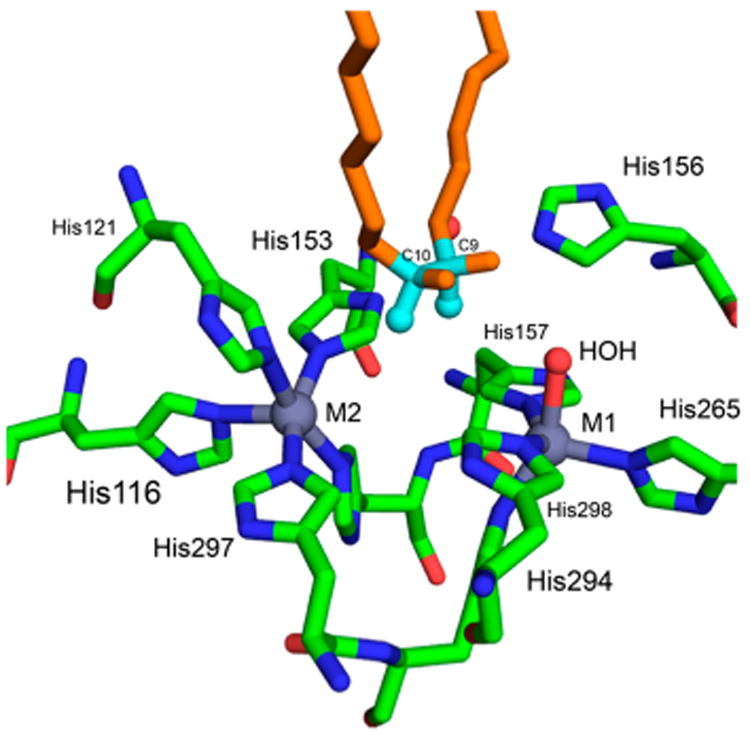
Dimetal active site structure of mouse stearoyl-CoA desaturase (PDB ID 4YMK), showing the C9–H and C10–H bonds of the bound substrate in position for cleavage to afford the monounsaturated fatty acid product by the high-valent dimetal center.252 In this structure, the metal sites are occupied by Zn2+ ions. This image was generously provided by Prof. Brian Fox.
These intriguing results support the notion that the integral-membrane-bound nonheme diiron enzymes have diiron active sites different from their soluble counterparts. Presumably, intermediates parallel to those of soluble diiron enzymes depicted in Scheme 1 participate in their reaction mechanisms, but this notion must be tested in future investigations. The His-rich diiron active sites associated with this class of enzymes represent yet another bioinorganic strategy that has evolved in nature to activate dioxygen for the cleavage of strong C–H bonds, joining the ranks of the heme-containing cytochromes P450,255 the copper-requiring particulate methane monooxygenases,256,257 mononuclear nonheme iron enzymes that have a 2-His-1-carboxylate facial triad in their active sites,262 and the soluble diiron enzymes that are the focus of this review. Collectively these oxygenases demonstrate Nature's ingenuity in dealing the challenge of functionalizing inert C–H bonds.
Supplementary Material
Figure S1 showing the pre-edge region of the Fe K-edge X-ray absorption spectrum of oxyhemerythrin. (PDF)
Acknowledgments
Support for this effort was provided by the National Institutes of Health (Grant GM38767 to L.Q.). We thank Apparao Draksharapu, Johannes E. M. N. Klein, Anna J. Komor, and Waqas Rasheed for their assistance in the assembly of this manuscript. We very much appreciate the critical reading of our manuscript provided by our long-term collaborator and my colleague Professor John Lipscomb.
Biographies
Andrew J. Jasniewski is from West Allis, WI and received his B.S. degree (2011) from the University of Wisconsin—Madison working for Thomas Brunold on functional models of the Mn-dependent superoxide dismutase. He then moved to the University of Minnesota to study the structures and spectroscopy of nonheme diiron enzymes and related model complexes with Larry Que, receiving his PhD. degree in 2017. He currently works at the University of California, Irvine with Markus Ribbe on the biochemistry and spectroscopy of nitrogenase.
Lawrence Que, Jr. received his B.S. degree (1969) from Ateneo de Manila University and Ph.D. degree (1973) from the University of Minnesota working under Louis H. Pignolet. He then took postdoctoral positions with Richard H. Holm at MIT (1973–74) and Eckard Munck at the Gray Freshwater Biological Institute of the University of Minnesota (1975–77). He began his independent career at Cornell University (1977–83) and then returned to the University of Mnnesota in 1983 where he is now Regents Professor. His research has focused on understanding the activation of dioxygen by nonheme iron enzymes and developing model complexes that shed light on the spectroscopic and reactivity properties of the reaction intermediates in these reactions, particularly those in high-valent iron oxidation states. His research accomplishments have been recognized by the American Chemical Society with the 2008 Alfred Bader Award in Bioorganic or Chemistry and the 2017 ACS Award in Inorganic Chemistry, by the Royal Society of Chemistry with the 2011 Inorganic Reaction Mechanisms Award, and by the Max-Planck-Institut fur Bioanorganische Chemie with the 2005 Frontiers in Biological Chemistry Award. He is most proud of what his former students have achieved in their own independent careers.
Footnotes
Associated Content: Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrev.7b00457.
Special Issue: Oxygen Reduction and Activation in Catalysis
Notes: The authors declare no competing financial interest.
References
- 1.Mason HS, Fowlks WL, Peterson E. Oxygen Transfer and Electron Transport by the Phenolate Complex. J Am Chem Soc. 1955;77:2914–2915. [Google Scholar]
- 2.Hayaishi O, Katagiri M, Rothberg S. Mechanism of the Pyrocatechase Reaction. J Am Chem Soc. 1955;77:5450–5451. [Google Scholar]
- 3.Lundin D, Poole AM, Sjöberg BM, Hoögbom M. Use of Structural Phylogenetic Networks for Classification of the Ferritin-Like Superfamily. J Biol Chem. 2012;287:20565–20575. doi: 10.1074/jbc.M112.367458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nordlund P, Eklund H. Di-Iron-Carboxylate Proteins. Curr Opin Struct Biol. 1995;5:758–766. doi: 10.1016/0959-440x(95)80008-5. [DOI] [PubMed] [Google Scholar]
- 5.Liu X, Theil EC. Ferritins: Dynamic Management of Biological Iron and Oxygen Chemistry. Acc Chem Res. 2005;38:167–175. doi: 10.1021/ar0302336. [DOI] [PubMed] [Google Scholar]
- 6.Wallar BJ, Lipscomb JD. Dioxygen Activation by Enzymes Containing Binuclear Non-Heme Iron Clusters. Chem Rev. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 7.Tinberg CE, Lippard SJ. Dioxygen Activation in Soluble Methane Monooxygenase. Acc Chem Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. In Vitro Reconstitution and Crystal Structure of P-Aminobenzoate N-Oxygenase (AurF) Involved in Aureothin Biosynthesis. Proc Natl Acad Sci U S A. 2008;105:6858–6863. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makris TM, Vu VV, Meier KK, Komor AJ, Rivard BS, Munck E, Que L, Jr, Lipscomb JD. An Unusual Peroxo Intermediate of the Arylamine Oxygenase of the Chloramphenicol Biosynthetic Pathway. J Am Chem Soc. 2015;137:1608–1617. doi: 10.1021/ja511649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krebs C, Bollinger JM, Jr, Booker SJ. Cyanobacterial Alkane Biosynthesis Further Expands the Catalytic Repertoire of the Ferritin-Like Di-Iron-Carboxylate' Proteins. Curr Opin Chem Biol. 2011;15:291–303. doi: 10.1016/j.cbpa.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das D, Eser BE, Han J, Sciore A, Marsh ENG. Oxygen-Independent Decarbonylation of Aldehydes by Cyanobacterial Aldehyde Decarbonylase: A New Reaction of Diiron Enzymes. Angew Chem, Int Ed. 2011;50:7148–7152. doi: 10.1002/anie.201101552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makris TM, Knoot CJ, Wilmot CM, Lipscomb JD. Structure of a Dinuclear Iron Cluster-Containing β-Hydroxylase Active in Antibiotic Biosynthesis. Biochemistry. 2013;52:6662–6671. doi: 10.1021/bi400845b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bochevarov AD, Li J, Song WJ, Friesner RA, Lippard SJ. Insights into the Different Dioxygen Activation Pathways of Methane and Toluene Monooxygenase Hydroxylases. J Am Chem Soc. 2011;133:7384–7397. doi: 10.1021/ja110287y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pikus JD, Studts JM, Achim C, Kauffmann KE, Münck E, Steffan RJ, McClay K, Fox BG. Recombinant Toluene-4-Monooxygenase: Catalytic and Mössbauer Studies of the Purified Diiron and Rieske Components of a Four-Protein Complex. Biochemistry. 1996;35:9106–9119. doi: 10.1021/bi960456m. [DOI] [PubMed] [Google Scholar]
- 15.Fox BG, Lyle KS, Rogge CE. Reactions of the Diiron Enzyme Stearoyl-Acyl Carrier Protein Desaturase. Acc Chem Res. 2004;37:421–429. doi: 10.1021/ar030186h. [DOI] [PubMed] [Google Scholar]
- 16.Han Z, Sakai N, Boettger LH, Klinke S, Hauber J, Trautwein AX, Hilgenfeld R. Crystal Structure of the Peroxo-Diiron(III) Intermediate of Deoxyhypusine Hydroxylase, an Oxygenase Involved in Hypusination. Structure. 2015;23:882–892. doi: 10.1016/j.str.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Nordlund P, Reichard P. Ribonucleotide Reductases. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 18.Holmes MA, Le Trong I, Turley S, Sieker LC, Stenkamp RE. Structures of Deoxy and Oxy Hemerythrin at 2.0 Å Resolution. J Mol Biol. 1991;218:583–593. doi: 10.1016/0022-2836(91)90703-9. [DOI] [PubMed] [Google Scholar]
- 19.Zhang K, Stern EA, Ellis F, Sanders-Loehr J, Shiemke AK. The Active Site of Hemerythrin as Determined by X-Ray Absorption Fine Structure. Biochemistry. 1988;27:7470–7479. doi: 10.1021/bi00419a045. [DOI] [PubMed] [Google Scholar]
- 20.Okamura MY, Klotz IM, Johnson CE, Winter MR, Williams RJ. The State of Iron in Hemerythrin. A Mössbauer Study. Biochemistry. 1969;8:1951–1958. doi: 10.1021/bi00833a027. [DOI] [PubMed] [Google Scholar]
- 21.Shiemke AK, Loehr TM, Sanders-Loehr J. Resonance Raman Study of the μ-Oxo-Bridged Binuclear Iron Center in Oxyhemerythrin. J Am Chem Soc. 1984;106:4951–4956. doi: 10.1021/ja00269a050. [DOI] [PubMed] [Google Scholar]
- 22.Shiemke AK, Loehr TM, Sanders-Loehr J. Resonance Raman Study of Oxyhemerythrin and Hydroxhemerythrin: Evidence for Hydrogen Bonding of Ligands to the Fe-O-Fe Center. J Am Chem Soc. 1986;108:2437–2443. doi: 10.1021/ja00269a050. [DOI] [PubMed] [Google Scholar]
- 23.Shu L, Nesheim JC, Kauffmann K, Munck E, Lipscomb JD, Que L., Jr An FeIV2O2 Diamond Core Structure for the Key Intermediate Q of Methane Monooxygenase. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 24.Rudd DJ, Sazinsky MH, Lippard SJ, Hedman B, Hodgson KO. X-Ray Absorption Spectroscopic Study of the Reduced Hydroxylases of Methane Monooxygenase and Toluene/O-Xylene Monooxygenase: Differences in Active Site Structure and Effects of the Coupling Proteins MMOB and TOMOD. Inorg Chem. 2005;44:4546–4554. doi: 10.1021/ic048794w. [DOI] [PubMed] [Google Scholar]
- 25.Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Geometry of the Soluble Methane Monooxygenase Catalytic Diiron Center in Two Oxidation States. Chem Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- 26.Pulver SC, Froland WA, Lipscomb JD, Solomon EI. Ligand Field Circular Dichroism and Magnetic Circular Dichroism Studies of Component B and Substrate Binding to the Hydroxylase Component of Methane Monooxygenase. J Am Chem Soc. 1997;119:387–395. [Google Scholar]
- 27.DeWitt JG, Bentsen JG, Rosenzweig AC, Hedman B, Green J, Pilkington S, Papaefthymiou GC, Dalton H, Hodgson KO, Lippard SJ. X-Ray Absorption, Mössbauer, and EPR Studies of the Dinuclear Iron Center in the Hydroxylase Component of Methane Monooxygenase. J Am Chem Soc. 1991;113:9219–9235. [Google Scholar]
- 28.Schwartz JK, Wei Pp, Mitchell KH, Fox BG, Solomon EI. Geometric and Electronic Structure Studies of the Binuclear Nonheme Ferrous Active Site of Toluene-4-Monooxygenase: Parallels with Methane Monooxygenase and Insight into the Role of the Effector Proteins in O2 Activation. J Am Chem Soc. 2008;130:7098–7109. doi: 10.1021/ja800654d. [DOI] [PubMed] [Google Scholar]
- 29.Elango NA, Radhakrishnan R, Froland WA, Wallar BJ, Earhart CA, Lipscomb JD, Ohlendorf DH. Crystal Structure of the Hydroxylase Component of Methane Monooxygenase from Methylosinus Trichosporium Ob3b. Protein Sci. 1997;6:556–568. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu KE, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. Spectroscopic Detection of Intermediates in the Reaction of Dioxygen with the Reduced Methane Monooxygenase Hydroxylase from Methylococcus Capsulatus (Bath) J Am Chem Soc. 1994;116:7465–7466. [Google Scholar]
- 31.Liu KE, Valentine AM, Wang DL, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. Kinetic and Spectroscopic Characterization of Intermediates and Component Interactions in Reactions of Methane Monooxygenase from Methylococcus Capsulatus (Bath) J Am Chem Soc. 1995;117:10174–10185. [Google Scholar]
- 32.Shu L, Liu Y, Lipscomb JD, Que L., Jr X-Ray Absorption Spectroscopic Studies of the Methane Monooxygenase Hydroxylase Component from Methylosinus Trichosporium Ob3b. JBIC, J Biol Inorg Chem. 1996;1:297–304. [Google Scholar]
- 33.Banerjee R, Proshlyakov Y, Lipscomb JD, Proshlyakov DA. Structure of the Key Species in the Enzymatic Oxidation of Methane to Methanol. Nature. 2015;518:431–434. doi: 10.1038/nature14160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Crystal Structure of a Bacterial Non-Haem Iron Hydroxylase That Catalyses the Biological Oxidation of Methane. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 35.Lee SK, Nesheim JC, Lipscomb JD. Transient Intermediates of the Methane Monooxygenase Catalytic Cycle. J Biol Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- 36.Lee SK, Fox BG, Froland WA, Lipscomb JD, Munck E. A Transient Intermediate of the Methane Monooxygenase Catalytic Cycle Containing an FeIV FeIV Cluster. J Am Chem Soc. 1993;115:6450–6451. [Google Scholar]
- 37.Logan DT, Su XD, Aberg A, Regnstrom K, Hajdu J, Eklund H, Nordlund P. Crystal Structure of Reduced Protein R2 of Ribonucleotide Reductase - the Structural Basis for Oxygen Activation at a Dinuclear Iron Site. Structure. 1996;4:1053–1064. doi: 10.1016/s0969-2126(96)00112-8. [DOI] [PubMed] [Google Scholar]
- 38.Baldwin J, Krebs C, Saleh L, Stelling M, Huynh BH, Bollinger JM, Jr, Riggs-Gelasco P. Structural Characterization of the Peroxodiiron(III) Intermediate Generated During Oxygen Activation by the W48A/D84E Variant of Ribonucleotide Reductase Protein R2 from Escherichia Coli. Biochemistry. 2003;42:13269–13279. doi: 10.1021/bi035198p. [DOI] [PubMed] [Google Scholar]
- 39.Dassama LMK, Silakov A, Krest CM, Calixto JC, Krebs C, Bollinger JM, Jr, Green MT. A 2.8 Å Fe-Fe Separation in the Fe2III/IV Intermediate (X) from Escherichia Coli Ribonucleotide Reductase. J Am Chem Soc. 2013;135:16758–16761. doi: 10.1021/ja407438p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang YS, Baldwin J, Ley BA, Bollinger JM, Solomon EI. Spectroscopic and Electronic Structure Description of the Reduced Binuclear Non-Heme Iron Active Site in Ribonucleotide Reductase from E. coli: Comparison to Reduced Δ9 Desaturase and Electronic Structure Contributions to Differences in O2 Reactivity. J Am Chem Soc. 2000;122:8495–8510. [Google Scholar]
- 41.Moenne-Loccoz P, Baldwin J, Ley BA, Loehr TM, Bollinger JM., Jr O2 Activation by Non-Heme Diiron Proteins: Identification of a Symmetric μ-1,2-Peroxide in a Mutant of Ribonucleotide Reductase. Biochemistry. 1998;37:14659–14663. doi: 10.1021/bi981838q. [DOI] [PubMed] [Google Scholar]
- 42.Bollinger JM, Jr, Krebs C, Vicol A, Chen S, Ley BA, Edmondson DE, Huynh BH. Engineering the Diiron Site of Escherichia coli Ribonucleotide Reductase Protein R2 to Accumulate an Intermediate Similar to Hperoxo, the Putative Peroxodiiron(III) Complex from the Methane Monooxygenase Catalytic Cycle. J Am Chem Soc. 1998;120:1094–1095. [Google Scholar]
- 43.Riggs-Gelasco PJ, Shu L, Chen S, Burdi D, Huynh BH, Que L, Jr, Stubbe J. Exafs Characterization of the Intermediate X Generated During the Assembly of the Escherichia Coli Ribonucleotide Reductase R2 Diferric Tyrosyl Radical Cofactor. J Am Chem Soc. 1998;120:849–860. [Google Scholar]
- 44.Nordlund P, Sjoberg BM, Eklund H. Three-Dimensional Structure of the Free Radical Protein of Ribonucleotide Reductase. Nature. 1990;345:593–598. doi: 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- 45.Lindqvist Y, Huang WJ, Schneider G, Shanklin J. Crystal Structure of Δ9 Stearoyl-Acyl Carrier Protein Desaturase from Castor Seed and Its Relationship to Other Diiron Proteins. EMBO J. 1996;15:4081–4092. [PMC free article] [PubMed] [Google Scholar]
- 46.Fox BG, Shanklin J, Somerville C, Münck E. Stearoyl-Acyl Carrier Protein Δ9 Desaturase from Ricinus communis Is a Diiron-Oxo Protein. Proc Natl Acad Sci U S A. 1993;90:2486–2490. doi: 10.1073/pnas.90.6.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang YS, Broadwater JA, Pulver SC, Fox BG, Solomon EI. Circular Dichroism and Magnetic Circular Dichroism Studies of the Reduced Binuclear Non-Heme Iron Site of Stearoyl-ACP Δ9-Desaturase: Substrate Binding and Comparison to Ribonucleotide Reductase. J Am Chem Soc. 1999;121:2770–2783. [Google Scholar]
- 48.Broadwater JA, Achim C, Münck E, Fox BG. Mössbauer Studies of the Formation and Reactivity of a Quasi-Stable Peroxo Intermediate of Stearoyl-Acyl Carrier Protein Δ9-Desaturase. Biochemistry. 1999;38:12197–12204. doi: 10.1021/bi9914199. [DOI] [PubMed] [Google Scholar]
- 49.Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Peroxodiferric Intermediate of Stearoyl-Acyl Carrier Protein Δ9 Desaturase: Oxidase Reactivity During Single Turnover and Implications for the Mechanism of Desaturation. Biochemistry. 1998;37:14664–14671. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- 50.Hwang J, Krebs C, Huynh BH, Edmondson DE, Theil EC, Penner-Hahn JE. A Short Fe-Fe Distance in Peroxodiferric Ferritin: Control of Fe Substrate Versus Cofactor Decay? Science. 2000;287:122–125. doi: 10.1126/science.287.5450.122. [DOI] [PubMed] [Google Scholar]
- 51.Pozzi C, Di Pisa F, Lalli D, Rosa C, Theil E, Turano P, Mangani S. Time-Lapse Anomalous X-Ray Diffraction Shows How Fe2+ Substrate Ions Move through Ferritin Protein Nanocages to Oxidoreductase Sites. Acta Crystallogr, Sect D: Biol Crystallogr. 2015;71:941–953. doi: 10.1107/S1399004715002333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pereira AS, Small W, Krebs C, Tavares P, Edmondson DE, Theil EC, Huynh BH. Direct Spectroscopic and Kinetic Evidence for the Involvement of a Peroxodiferric Intermediate During the Ferroxidase Reaction in Fast Ferritin Mineralization. Biochemistry. 1998;37:9871–9876. doi: 10.1021/bi980847w. [DOI] [PubMed] [Google Scholar]
- 53.Schwartz JK, Liu XS, Tosha T, Theil EC, Solomon EI. Spectroscopic Definition of the Ferroxidase Site in M Ferritin: Comparison of Binuclear Substrate vs Cofactor Active Sites. J Am Chem Soc. 2008;130:9441–9450. doi: 10.1021/ja801251q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murray LJ, Naik SG, Ortillo DO, Garcia-Serres R, Lee JK, Huynh BH, Lippard SJ. Characterization of the Arene-Oxidizing Intermediate in TOMOH as a Diiron(III) Species. J Am Chem Soc. 2007;129:14500–14510. doi: 10.1021/ja076121h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCormick MS, Sazinsky MH, Condon KL, Lippard SJ. X-Ray Crystal Structures of Manganese(II)-Reconstituted and Native Toluene/O-Xylene Monooxygenase Hydroxylase Reveal Rotamer Shifts in Conserved Residues and an Enhanced View of the Protein Interior. J Am Chem Soc. 2006;128:15108–15110. doi: 10.1021/ja064837r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murray LJ, García-Serres R, Naik S, Huynh BH, Lippard SJ. Dioxygen Activation at Non-Heme Diiron Centers: Characterization of Intermediates in a Mutant Form of Toluene/O-Xylene Monooxygenase Hydroxylase. J Am Chem Soc. 2006;128:7458–7459. doi: 10.1021/ja062762l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bailey LJ, Mc Coy JG, Phillips GN, Jr, Fox BG. Structural Consequences of Effector Protein Complex Formation in a Diiron Hydroxylase. Proc Natl Acad Sci U S A. 2008;105:19194–19198. doi: 10.1073/pnas.0807948105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pandelia ME, Li N, Noergaard H, Warui DM, Rajakovich LJ, Chang WC, Booker SJ, Krebs C, Bollinger JM., Jr Substrate-Triggered Addition of Dioxygen to the Diferrous Cofactor of Aldehyde-Deformylating Oxygenase to Form a Diferric-Peroxide Intermediate. J Am Chem Soc. 2013;135:15801–15812. doi: 10.1021/ja405047b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jia C, Li M, Li J, Zhang J, Zhang H, Cao P, Pan X, Lu X, Chang W. Structural Insights into the Catalytic Mechanism of Aldehyde-Deformylating Oxygenases. Protein Cell. 2015;6:55–67. doi: 10.1007/s13238-014-0108-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zocher G, Winkler R, Hertweck C, Schulz GE. Structure and Action of the N-Oxygenase Aurf from Streptomyces thioluteus. J Mol Biol. 2007;373:65–74. doi: 10.1016/j.jmb.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 61.Korboukh VK, Li N, Barr EW, Bollinger JM, Jr, Krebs C. A Long-Lived, Substrate-Hydroxylating Peroxodiiron(III/III) Intermediate in the Amine Oxygenase, Aurf, from Streptomyces thioluteus. J Am Chem Soc. 2009;131:13608–13609. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park K, Li N, Kwak Y, Srnec M, Bell CB, Liu LV, Wong SD, Yoda Y, Kitao S, Seto M, Hu M, Zhao J, Krebs C, Bollinger JM, Solomon EI. Peroxide Activation for Electrophilic Reactivity by the Binuclear Non-Heme Iron Enzyme AurF. J Am Chem Soc. 2017;139:7062–7070. doi: 10.1021/jacs.7b02997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knoot CJ, Kovaleva EG, Lipscomb JD. Crystal Structure of Cmli, the Arylamine Oxygenase from the Chloramphenicol Biosynthetic Pathway. JBIC, J Biol Inorg Chem. 2016;21:589–603. doi: 10.1007/s00775-016-1363-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jasniewski AJ, Komor AJ, Lipscomb JD, Que L. An Unprecedented (μ-1,1-Peroxo)Diferric Structure for the Ambiphilic Orange Peroxo Intermediate of the Nonheme N-Oxygenase Cmli. J Am Chem Soc. 2017;139:10472–10485. doi: 10.1021/jacs.7b05389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jasniewski AJ, Knoot CJ, Lipscomb JD, Que L., Jr A Carboxylate Shift Regulates Dioxygen Activation by the Diiron Nonheme β-Hydroxylase Cmla Upon Binding of a Substrate-Loaded Nonribosomal Peptide Synthetase. Biochemistry. 2016;55:5818–5831. doi: 10.1021/acs.biochem.6b00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Makris TM, Chakrabarti M, Munck E, Lipscomb JD. A Family of Diiron Monooxygenases Catalyzing Amino Acid Beta-Hydroxylation in Antibiotic Biosynthesis. Proc Natl Acad Sci U S A. 2010;107:15391–15396. doi: 10.1073/pnas.1007953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jasniewski AJ, Engstrom LM, Vu VV, Park MH, Que L. X-Ray Absorption Spectroscopic Characterization of the Diferric-Peroxo Intermediate of Human Deoxyhypusine Hydroxylase in the Presence of Its Substrate EIF5A. JBIC, J Biol Inorg Chem. 2016;21:605–618. doi: 10.1007/s00775-016-1373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vu VV, Emerson JP, Martinho M, Kim YS, Munck E, Park MH, Que L., Jr Human Deoxyhypusine Hydroxylase, an Enzyme Involved in Regulating Cell Growth, Activates O2 with a Nonheme Diiron Center. Proc Natl Acad Sci U S A. 2009;106:14814–14819. doi: 10.1073/pnas.0904553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moenne-Loccoz P, Krebs C, Herlihy K, Edmondson DE, Theil EC, Huynh BH, Loehr T. The Ferroxidase Reaction of Ferritin Reveals a Diferric μ-1,2 Bridging Peroxide Intermediate in Common with Other O2-Activating Non-Heme Diiron Proteins. Biochemistry. 1999;38:5290–5295. doi: 10.1021/bi990095l. [DOI] [PubMed] [Google Scholar]
- 70.Acheson JF, Bailey LJ, Brunold TC, Fox BG. In-Crystal Reaction Cycle of a Toluene-Bound Diiron Hydroxylase. Nature. 2017;544:191–195. doi: 10.1038/nature21681. [DOI] [PubMed] [Google Scholar]
- 71.Beauvais LG, Lippard SJ. Reactions of the Peroxo Intermediate of Soluble Methane Monooxygenase Hydroxylase with Ethers. J Am Chem Soc. 2005;127:7370–7378. doi: 10.1021/ja050865i. [DOI] [PubMed] [Google Scholar]
- 72.Tinberg CE, Lippard SJ. Oxidation Reactions Performed by Soluble Methane Monooxygenase Hydroxylase Intermediates Hperoxo and Q Proceed by Distinct Mechanisms. Biochemistry. 2010;49:7902–7912. doi: 10.1021/bi1009375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nesheim JC, Lipscomb JD. Large Kinetic Isotope Effects in Methane Oxidation Catalyzed by Methane Monooxygenase: Evidence for C–H Bond Cleavage in a Reaction Cycle Intermediate. Biochemistry. 1996;35:10240–10247. doi: 10.1021/bi960596w. [DOI] [PubMed] [Google Scholar]
- 74.Brazeau BJ, Lipscomb JD. Kinetics and Activation Thermodynamics of Methane Monooxygenase Compound Q Formation and Reaction with Substrates. Biochemistry. 2000;39:13503–13515. doi: 10.1021/bi001473l. [DOI] [PubMed] [Google Scholar]
- 75.Brazeau BJ, Wallar BJ, Lipscomb JD. Unmasking of Deuterium Kinetic Isotope Effects on the Methane Monooxygenase Compound Q Reaction by Site-Directed Mutagenesis of Component B. J Am Chem Soc. 2001;123:10421–10422. doi: 10.1021/ja016632i. [DOI] [PubMed] [Google Scholar]
- 76.Jin Y, Lipscomb JD. Desaturation Reactions Catalyzed by Soluble Methane Monooxygenase. JBIC, J Biol Inorg Chem. 2001;6:717–725. doi: 10.1007/s007750100250. [DOI] [PubMed] [Google Scholar]
- 77.Bollinger JM, Jr, Edmondson DE, Huynh BH, Filley J, Norton JR, Stubbe J. Mechanism of Assembly of the Tyrosyl Radical-Dinuclear Iron Cluster Cofactor of Ribonucleotide Reductase. Science. 1991;253:292–298. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- 78.Bollinger JM, Jr, Stubbe J, Huynh BH, Edmondson DE. Novel Diferric Radical Intermediate Responsible for Tyrosyl Radical Formation in Assembly of the Cofactor of Ribonucleotide Reductase. J Am Chem Soc. 1991;113:6289–6291. [Google Scholar]
- 79.Stenkamp RE. Dioxygen and Hemerythrin. Chem Rev. 1994;94:715–726. [Google Scholar]
- 80.Brunold TC, Solomon EI. Reversible Dioxygen Binding to Hemerythrin. 1. Electronic Structures of Deoxy- and Oxyhemerythrin. J Am Chem Soc. 1999;121:8277–8287. [Google Scholar]
- 81.Brunold TC, Solomon EI. Reversible Dioxygen Binding to Hemerythrin. 2. Mechanism of the Proton-Coupled Two-Electron Transfer to O2 at a Single Iron Center. J Am Chem Soc. 1999;121:8288–8295. [Google Scholar]
- 82.Sazinsky MH, Lippard SJ. Correlating Structure withFunction in Bacterial Multicomponent Monooxygenases and RelatedDiiron Proteins. Acc Chem Res. 2006;39:558–566. doi: 10.1021/ar030204v. [DOI] [PubMed] [Google Scholar]
- 83.Griese JJ, Roos K, Cox N, Shafaat HS, Branca RMM, Lehtiö J, Graslund A, Lubitz W, Siegbahn PEM, Hogbom M. Direct Observation of Structurally Encoded Metal Discrimination and Ether Bond Formation in a Heterodinuclear Metalloprotein. Proc Natl Acad Sci U S A. 2013;110:17189–17194. doi: 10.1073/pnas.1304368110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Griese JJ, Kositzki R, Schrapers P, Branca RMM, Nordstrom A, Lehtiö J, Haumann M, Hogbom M. Structural Basis for Oxygen Activation at a Heterodinuclear Manganese/Iron Cofactor. J Biol Chem. 2015;290:25254–25272. doi: 10.1074/jbc.M115.675223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kutin Y, Srinivas V, Fritz M, Kositzki R, Shafaat HS, Birrell J, Bill E, Haumann M, Lubitz W, Högbom M, Griese JJ, Cox N. Divergent Assembly Mechanisms of the Manganese/Iron Cofactors in R2lox and R2c Proteins. J Inorg Biochem. 2016;162:164–177. doi: 10.1016/j.jinorgbio.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 86.Rardin RL, Tolman WB, Lippard SJ. Monodentate Carboxylate Complexes and the Carboxylate Shift: Implications for Polymetalloprotein Structure and Function. New J Chem. 1991;15:417–430. [Google Scholar]
- 87.Tshuva EY, Lippard SJ. Synthetic Models for Non-Heme Carboxylate-Bridged Diiron Metalloproteins: Strategies and Tactics. Chem Rev. 2004;104:987–1012. doi: 10.1021/cr020622y. [DOI] [PubMed] [Google Scholar]
- 88.Guy JE, Whittle E, Kumaran D, Lindqvist Y, Shanklin J. The Crystal Structure of the Ivy Δ4-16:0-Acp Desaturase Reveals Structural Details of the Oxidized Active Site and Potential Determinants of Regioselectivity. J Biol Chem. 2007;282:19863–19871. doi: 10.1074/jbc.M702520200. [DOI] [PubMed] [Google Scholar]
- 89.Shanklin J, Guy JE, Mishra G, Lindqvist Y. Desaturases: Emerging Models for Understanding Functional Diversification of Diiron-Containing Enzymes. J Biol Chem. 2009;284:18559–18563. doi: 10.1074/jbc.R900009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.de Groot F. High-Resolution X-Ray Emission and X-Ray Absorption Spectroscopy. Chem Rev. 2001;101:1779–1808. doi: 10.1021/cr9900681. [DOI] [PubMed] [Google Scholar]
- 91.Sarangi R. X-Ray Absorption near-Edge Spectroscopy in Bioinorganic Chemistry: Application to M-O2 Systems. Coord Chem Rev. 2013;257:459–472. doi: 10.1016/j.ccr.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 93.Company A, Sabenya G, González-Béjar M, Gómez L, Clemancey M, Blondin G, Jasniewski AJ, Puri M, Browne WR, Latour JM, Que L, Costas M, Perez-Prieto J, Lloret-Fillol J. Triggering the Generation of an Iron(IV)-Oxo Compound and Its Reactivity toward Sulfides by RuII Photocatalysis. J Am Chem Soc. 2014;136:4624–4633. doi: 10.1021/ja412059c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Colpas GJ, Maroney MJ, Bagyinka C, Kumar M, Willis WS, Suib SL, Mascharak PK, Baidya N. X-Ray Spectroscopic Studies of Nickel Complexes, with Application to the Structure of Nickel Sites in Hydrogenases. Inorg Chem. 1991;30:920–928. [Google Scholar]
- 95.Wirt MD, Sagi I, Chen E, Frisbie SM, Lee R, Chance MR. Geometric Conformations of Intermediates of B12 Catalysis by X-Ray Edge Spectroscopy: Cobalt(I) B12, Cobalt(II) B12, and Base-Off Adenosylcobalamin. J Am Chem Soc. 1991;113:5299–5304. [Google Scholar]
- 96.Penner-Hahn JE, Fronko RM, Pecoraro VL, Yocum CF, Betts SD, Bowlby NR. Structural Characterization of the Manganese Sites in the Photosynthetic Oxygen-Evolving Complex Using X-Ray Absorption Spectroscopy. J Am Chem Soc. 1990;112:2549–2557. [Google Scholar]
- 97.Rudd DJ, Goldsmith CR, Cole AP, Stack TDP, Hodgson KO, Hedman B. X-Ray Absorption Spectroscopic Investigation of the Spin-Transition Character in a Series of Single-Site Perturbed Iron(II) Complexes. Inorg Chem. 2005;44:1221–1229. doi: 10.1021/ic048765l. [DOI] [PubMed] [Google Scholar]
- 98.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. A Multiplet Analysis of Fe K-Edge 1s → 3d Pre-Edge Features of Iron Complexes. J Am Chem Soc. 1997;119:6297–6314. [Google Scholar]
- 99.Roe AL, Schneider DJ, Mayer RJ, Pyrz JW, Widom J, Que L., Jr X-Ray Absorption Spectroscopy of Iron-Tyrosinate Proteins. J Am Chem Soc. 1984;106:1676–1681. [Google Scholar]
- 100.Randall CR, Shu L, Chiou YM, Hagen KS, Ito M, Kitajima N, Lachicotte RJ, Zang Y, Que L., Jr X-Ray Absorption Pre-Edge Studies of High Spin Iron(II) Complexes. Inorg Chem. 1995;34:1036–1039. [Google Scholar]
- 101.Solomon EI, Goudarzi S, Sutherlin KD. O2 Activation by Non-Heme Iron Enzymes. Biochemistry. 2016;55:6363–6374. doi: 10.1021/acs.biochem.6b00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Solomon EI, Park K. Structure/Function Correlations over Binuclear Non-Heme Iron Active Sites. JBIC, J Biol Inorg Chem. 2016;21:575–588. doi: 10.1007/s00775-016-1372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Münck E. In: Physical Methods in Bioinorganic Chemistry. Que L Jr, editor. University Science Books; Sausalito, CA: 2000. [Google Scholar]
- 104.Hendrich MP, Munck E, Fox BG, Lipscomb JD. Integer-Spin Epr Studies of the Fully Reduced Methane Monooxygenase Hydroxylase Component. J Am Chem Soc. 1990;112:5861–5865. [Google Scholar]
- 105.Garbett K, Darnall DW, Klotz IM, Williams RJP. Spectroscopy and Structure of Hemerythrin. Arch Biochem Biophys. 1969;135:419–434. doi: 10.1016/0003-9861(69)90559-1. [DOI] [PubMed] [Google Scholar]
- 106.Kurtz DM, Shriver DF, Klotz IM. Resonance Raman Spectroscopy with Unsymmetrically Isotopic Ligands. Differentiation of Possible Structures of Hemerythrin Complexes. J Am Chem Soc. 1976;98:5033–5035. doi: 10.1021/ja00432a064. [DOI] [PubMed] [Google Scholar]
- 107.Armstrong GD, Sykes AG. Reactions of O2 with Hemerythrin, Myoglobin, and Hemocyanin: Effects of D2O on Equilibration Rate Constants and Evidence for H-Bonding. Inorg Chem. 1986;25:3135–3139. [Google Scholar]
- 108.Dunn JBR, Shriver DF, Klotz IM. Resonance Raman Studies of the Electronic State of Oxygen in Hemerythrin. Proc Natl Acad Sci U S A. 1973;70:2582–2584. doi: 10.1073/pnas.70.9.2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lee SK, Lipscomb JD. Oxygen Activation Catalyzed by Methane Monooxygenase Hydroxylase Component: Proton Delivery During the O–O Bond Cleavage Steps. Biochemistry. 1999;38:4423–4432. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- 110.Skulan AJ, Brunold TC, Baldwin J, Saleh L, Bollinger JM, Solomon EI. Nature of the Peroxo Intermediate of the W48F/D84E Ribonucleotide Reductase Variant: Implications for O2 Activation by Binuclear Non-Heme Iron Enzymes. J Am Chem Soc. 2004;126:8842–8855. doi: 10.1021/ja049106a. [DOI] [PubMed] [Google Scholar]
- 111.Song WJ, Lippard SJ. Mechanistic Studies of Reactions of Peroxodiiron(III) Intermediates in T201 Variants of Toluene/o-Xylene Monooxygenase Hydroxylase. Biochemistry. 2011;50:5391–5399. doi: 10.1021/bi200340f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Song WJ, Behan RK, Naik SG, Huynh BH, Lippard SJ. Characterization of a Peroxodiiron(III) Intermediate in the T201S Variant of Toluene/o-Xylene Monooxygenase Hydroxylase from Pseudomonas Sp Ox1. J Am Chem Soc. 2009;131:6074–6075. doi: 10.1021/ja9011782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Song WJ, McCormick MS, Behan RK, Sazinsky MH, Jiang W, Lin J, Krebs C, Lippard SJ. Active Site Threonine Facilitates Proton Transfer During Dioxygen Activation at the Diiron Center of Toluene/o-Xylene Monooxygenase Hydroxylase. J Am Chem Soc. 2010;132:13582–13585. doi: 10.1021/ja1063795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pozzi C, Ciambellotti S, Bernacchioni C, Di Pisa F, Mangani S, Turano P. Chemistry at the Protein–Mineral Interface in L-Ferritin Assists the Assembly of a Functional (μ 3-Oxo)Tris[(μ 2-Peroxo)] Triiron(III) Cluster. Proc Natl Acad Sci U S A. 2017;114:2580–2585. doi: 10.1073/pnas.1614302114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bailey LJ, Fox BG. Crystallographic and Catalytic Studies of the Peroxide-Shunt Reaction in a Diiron Hydroxylase. Biochemistry. 2009;48:8932–8939. doi: 10.1021/bi901150a. [DOI] [PubMed] [Google Scholar]
- 116.Spiro TG, Czernuszewicz RS. In: Physical Methods in Bioinorganic Chemistry. Que L Jr, editor. University Science Books; Sausalito, CA: 2003. [Google Scholar]
- 117.DeWitt JG, Rosenzweig AC, Salifoglou A, Hedman B, Lippard SJ, Hodgson KO. X-Ray Absorption Spectroscopic Studies of the Diiron Center in Methane Monooxygenase in the Presence of Substrate and the Coupling Protein of the Enzyme System. Inorg Chem. 1995;34:2505–2515. [Google Scholar]
- 118.Liu KE, Valentine AM, Qiu D, Edmondson DE, Appelman EH, Spiro TG, Lippard SJ. Characterization of a Diiron(III) Peroxide Intermediate in the Reaction Cycle of Methane Monooxygenase Hydroxylase from Methylococcus capsulatus (Bath) J Am Chem Soc. 1995;117:4997–4998. [Google Scholar]
- 119.Liu KE, Valentine AM, Qiu D, Edmondson DE, Appelman EH, Spiro TG, Lippard SJ. Characterization of a Diiron(III) Peroxo Intermediate in the Reaction Cycle of Methane Monooxygenase Hydroxylase from Methylococcus capsulatus (Bath) J Am Chem Soc. 1995;117:4997–4998. [Google Scholar]; J Am Chem Soc. 1997;119:11134–11135. [Google Scholar]
- 120.Gherman BF, Baik MH, Lippard SJ, Friesner RA. Dioxygen Activation in Methane Monooxygenase: A Theoretical Study. J Am Chem Soc. 2004;126:2978–2990. doi: 10.1021/ja036506+. [DOI] [PubMed] [Google Scholar]
- 121.Rinaldo D, Philipp DM, Lippard SJ, Friesner RA. Intermediates in Dioxygen Activation by Methane Monooxygenase: A QM/MM Study. J Am Chem Soc. 2007;129:3135–3147. doi: 10.1021/ja0654074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Han WG, Noodleman L. Structural Model Studies for the Peroxo Intermediate P and the Reaction Pathway from P → Q of Methane Monooxygenase Using Broken-Symmetry Density Functional Calculations. Inorg Chem. 2008;47:2975–2986. doi: 10.1021/ic701194b. [DOI] [PubMed] [Google Scholar]
- 123.Kovaleva EG, Lipscomb JD. Crystal Structures of Fe2+ Dioxygenase Superoxo, Alkylperoxo, and Bound Product Intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Holt EM, Holt SL, Tucker WF, Asplund RO, Watson KJ. Preparation and Properties of Iron(III)-Amino Acid Complexes. Iron(III)-Alanine, a Possible Ferritin Analog. J Am Chem Soc. 1974;96:2621–2623. doi: 10.1021/ja00815a055. [DOI] [PubMed] [Google Scholar]
- 125.Thundathil RV, Holt EM, Holt SL, Watson KJ. Preparation and Properties of Iron(III)-Amino Acid Complexes. 2. The Crystal and Molecular Structure of Monoclinic Tri-μ-Oxo-Triaquohexakis(Glycine)Triiron(III) Perchlorate. J Am Chem Soc. 1977;99:1818–1823. doi: 10.1021/ja00448a024. [DOI] [PubMed] [Google Scholar]
- 126.Sousa CM, Carpentier P, Matias PM, Testa F, Pinho F, Sarti P, Giuffrè A, Bandeiras TM, Romao CV. Superoxide Reductase from Giardia intestinalis: Structural Characterization of the First Sor from a Eukaryotic Organism Shows an Iron Centre That Is Highly Sensitive to Photoreduction. Acta Crystallogr, Sect D: Biol Crystallogr. 2015;71:2236–2247. doi: 10.1107/S1399004715015825. [DOI] [PubMed] [Google Scholar]
- 127.Gudmundsson M, Kim S, Wu M, Ishida T, Momeni MH, Vaaje-Kolstad G, Lundberg D, Royant A, Ståhlberg J, Eijsink VGH, Beckham GT, Sandgren M. Structural and Electronic Snapshots During the Transition from a Cu(II) to Cu(I) Metal Center of a Lytic Polysaccharide Monooxygenase by X-Ray Photoreduction. J Biol Chem. 2014;289:18782–18792. doi: 10.1074/jbc.M114.563494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sigfridsson KGV, Chernev P, Leidel N, Popovic-Bijelic A, Graslund A, Haumann M. Rapid X-Ray Photoreduction of Dimetal-Oxygen Cofactors in Ribonucleotide Reductase. J Biol Chem. 2013;288:9648–9661. doi: 10.1074/jbc.M112.438796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Andersson KK, Froland WA, Lee SK, Lipscomb JD. Dioxygen Independent Oxygenation of Hydrocarbons by Methane Monooxygenase Hydroxylase Component. New J Chem. 1991;15:411–415. [Google Scholar]
- 130.Komor AJ, Rivard BS, Fan R, Guo Y, Que L, Jr, Lipscomb JD. Mechanism for Six-Electron Aryl-N-Oxygenation by the Non-Heme Diiron Enzyme Cmli. J Am Chem Soc. 2016;138:7411–7421. doi: 10.1021/jacs.6b03341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kurtz DM. Oxo- and Hydroxo-Bridged Diiron Complexes: A Chemical Perspective on a Biological Unit. Chem Rev. 1990;90:585–606. [Google Scholar]
- 132.Sanders-Loehr J, Wheeler WD, Shiemke AK, Averill BA, Loehr TM. Electronic and Raman Spectroscopic Properties of Oxo-Bridged Dinuclear Iron Centers in Proteins and Model Compounds. J Am Chem Soc. 1989;111:8084–8093. [Google Scholar]
- 133.Cranswick MA, Meier KK, Shan X, Stubna A, Kaizer J, Mehn MP, Münck E, Que L., Jr Protonation of a Peroxodiiron-(III) Complex and Conversion to a Diiron(III/IV) Intermediate: Implications for Proton-Assisted O-O Bond Cleavage in Nonheme Diiron Enzymes. Inorg Chem. 2012;51:10417–10426. doi: 10.1021/ic301642w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pap JS, Cranswick MA, Balogh-Hergovich E, Barath G, Giorgi M, Rohde GT, Kaizer J, Speier G, Que L., Jr An Iron(II)[1,3-Bis(2′-Pyridylimino)Isoindoline] Complex as a Catalyst for Substrate Oxidation with H2O2 - Evidence for a Transient Peroxidodiiron(III) Species. Eur J Inorg Chem. 2013;2013:3858–3866. doi: 10.1002/ejic.201300162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fiedler AT, Shan X, Mehn MP, Kaizer J, Torelli S, Frisch JR, Kodera M, Que L., Jr Spectroscopic and Computational Studies of (μ-Oxo)(μ-1,2-Peroxo)Diiron(III) Complexes of Relevance to Nonheme Diiron Oxygenase Intermediates. J Phys Chem A. 2008;112:13037–13044. doi: 10.1021/jp8038225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dong Y, Zang Y, Kauffmann K, Shu L, Wilkinson EC, Munck E, Que L., Jr Models for Nonheme Diiron Enzymes. Assembly of a High-Valent Fe2(μ-O)2 Diamond Core from Its Peroxo Precursor. J Am Chem Soc. 1997;119:12683–12684. [Google Scholar]
- 137.Zhang X, Furutachi H, Fujinami S, Nagatomo S, Maeda Y, Watanabe Y, Kitagawa T, Suzuki M. Structural and Spectroscopic Characterization of (μ-Hydroxo or μ-Oxo)(μ-Peroxo)-Diiron(III) Complexes: Models for Peroxo Intermediates of Non-Heme Diiron Proteins. J Am Chem Soc. 2005;127:826–827. doi: 10.1021/ja045594a. [DOI] [PubMed] [Google Scholar]
- 138.Zheng H, Zang Y, Dong Y, Young VG, Jr, Que L., Jr Complexes with FeIII2(μ-O)(μ-OH), FeIII2(μ-O)2, and [FeIII3(μ2-O)3] Cores: Structures, Spectroscopy, and Core Interconversions. J Am Chem Soc. 1999;121:2226–2235. [Google Scholar]
- 139.Wilkinson EC, Dong YH, Zang Y, Fujii H, Fraczkiewicz R, Fraczkiewicz G, Czernuszewicz RS, Que L., Jr Raman Signature of the Fe2O2 “Diamond” Core. J Am Chem Soc. 1998;120:955–962. [Google Scholar]
- 140.Kindermann N, Dechert S, Demeshko S, Meyer F. Proton-Induced, Reversible Interconversion of a μ-1,2-Peroxo and a μ-1,1-Hydroperoxo Dicopper(II) Complex. J Am Chem Soc. 2015;137:8002–8005. doi: 10.1021/jacs.5b04361. [DOI] [PubMed] [Google Scholar]
- 141.Meunier B, de Visser SP, Shaik S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 142.Cotruvo JA, Stubbe J. Class I Ribonucleotide Reductases: Metallocofactor Assembly and Repair in Vitro and in Vivo. Annu Rev Biochem. 2011;80:733–767. doi: 10.1146/annurev-biochem-061408-095817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Valentine AM, Stahl SS, Lippard SJ. Mechanistic Studies of the Reaction of Reduced Methane Monooxygenase Hydroxylase with Dioxygen and Substrates. J Am Chem Soc. 1999;121:3876–3887. [Google Scholar]
- 144.Murray LJ, Lippard SJ. Substrate Trafficking and Dioxygen Activation in Bacterial Multicomponent Monooxygenases. Acc Chem Res. 2007;40:466–474. doi: 10.1021/ar600040e. [DOI] [PubMed] [Google Scholar]
- 145.Warui DM, Li N, Nørgaard H, Krebs C, Bollinger JM, Booker SJ. Detection of Formate, Rather Than Carbon Monoxide, as the Stoichiometric Coproduct in Conversion of Fatty Aldehydes to Alkanes by a Cyanobacterial Aldehyde Decarbonylase. J Am Chem Soc. 2011;133:3316–3319. doi: 10.1021/ja111607x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li N, Nørgaard H, Warui DM, Booker SJ, Krebs C, Bollinger JM. Conversion of Fatty Aldehydes to Alka(e)nes and Formate by a Cyanobacterial Aldehyde Decarbonylase: Cryptic Redox by an Unusual Dimetal Oxygenase. J Am Chem Soc. 2011;133:6158–6161. doi: 10.1021/ja2013517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shokri A, Que L. Conversion of Aldehyde to Alkane by a Peroxoiron(III) Complex: A Functional Model for the Cyanobacterial Aldehyde-Deformylating Oxygenase. J Am Chem Soc. 2015;137:7686–7691. doi: 10.1021/jacs.5b01053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cho J, Jeon S, Wilson SA, Liu LV, Kang EA, Braymer JJ, Lim MH, Hedman B, Hodgson KO, Valentine JS, Solomon EI, Nam W. Structure and Reactivity of a Mononuclear Non-Haem Iron(III)–Peroxo Complex. Nature. 2011;478:502–505. doi: 10.1038/nature10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chalupský J, Rokob TA, Kurashige Y, Yanai T, Solomon EI, Rulíšek L, Srnec M. Reactivity of the Binuclear Non-Heme Iron Active Site of Δ9 Desaturase Studied by Large-Scale Multireference Ab Initio Calculations. J Am Chem Soc. 2014;136:15977–15991. doi: 10.1021/ja506934k. [DOI] [PubMed] [Google Scholar]
- 150.Srnec M, Rokob TA, Schwartz JK, Kwak Y, Rulíšek L, Solomon EI. Structural and Spectroscopic Properties of the Peroxodiferric Intermediate of Ricinus Communis Soluble Δ9 Desaturase. Inorg Chem. 2012;51:2806–2820. doi: 10.1021/ic2018067. [DOI] [PubMed] [Google Scholar]
- 151.Brazeau BJ, Austin RN, Tarr C, Groves JT, Lipscomb JD. Intermediate Q from Soluble Methane Monooxygenase Hydroxylates the Mechanistic Substrate Probe Norcarane: Evidence for a Stepwise Reaction. J Am Chem Soc. 2001;123:11831–11837. doi: 10.1021/ja016376+. [DOI] [PubMed] [Google Scholar]
- 152.Lipscomb JD, Que L., Jr MMO: P450 in Wolf's Clothing? JBIC, J Biol Inorg Chem. 1998;3:331–336. [Google Scholar]
- 153.Baldwin J, Voegtli WC, Khidekel N, Moënne-Loccoz P, Krebs C, Pereira AS, Ley BA, Huynh BH, Loehr TM, Riggs-Gelasco PJ, Rosenzweig AC, Bollinger JM. Rational Reprogramming of the R2 Subunit of Escherichia Coli Ribonucleotide Reductase into a Self-Hydroxylating Monooxygenase. J Am Chem Soc. 2001;123:7017–7030. doi: 10.1021/ja002114g. [DOI] [PubMed] [Google Scholar]
- 154.Ormö M, deMaré F, Regnström K, Aberg A, Sahlin M, Ling J, Loehr TM, Sanders-Loehr J, Sjöberg BM. Engineering of the Iron Site in Ribonucleotide Reductase to a Self-Hydroxylating Monooxygenase. J Biol Chem. 1992;267:8711–8714. [PubMed] [Google Scholar]
- 155.Aaberg A, Ormö M, Nordlund P, Sjöberg BM. Autocatalytic Generation of Dopa in the Engineered Protein R2 F208Y from Escherichia Coli Ribonucleotide Reductase and Crystal Structure of the Dopa-208 Protein. Biochemistry. 1993;32:9845–9850. doi: 10.1021/bi00088a040. [DOI] [PubMed] [Google Scholar]
- 156.Tinberg CE, Lippard SJ. Revisiting the Mechanism of Dioxygen Activation in Soluble Methane Monooxygenase from M. capsulatus (Bath): Evidence for a Multi-Step, Proton-Dependent Reaction Pathway. Biochemistry. 2009;48:12145–12158. doi: 10.1021/bi901672n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Puri M, Que L. Toward the Synthesis of More Reactive S = 2 Non-Heme Oxoiron(IV) Complexes. Acc Chem Res. 2015;48:2443–2452. doi: 10.1021/acs.accounts.5b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ghosh A, Tiago de Oliveira F, Yano T, Nishioka T, Beach ES, Kinoshita I, Munck E, Ryabov AD, Horwitz CP, Collins TJ. Catalytically Active μ-Oxodiiron(IV) Oxidants from Iron(III) and Dioxygen. J Am Chem Soc. 2005;127:2505–2513. doi: 10.1021/ja0460458. [DOI] [PubMed] [Google Scholar]
- 159.Hsu HF, Dong Y, Shu L, Young VG, Que L. Crystal Structure of a Synthetic High-Valent Complex with an Fe2(μ-O)2 Diamond Core. Implications for the Core Structures of Methane Monooxygenase Intermediate Q and Ribonucleotide Reductase Intermediate X. J Am Chem Soc. 1999;121:5230–5237. [Google Scholar]
- 160.Zang Y, Dong Y, Que L, Kauffmann K, Muenck E. The First Bis(μ-Oxo)Diiron(III) Complex. Structure and Magnetic Properties of [Fe2(μ-O)2(6TLA)2](ClO4)2. J Am Chem Soc. 1995;117:1169–1170. [Google Scholar]
- 161.Que L, Jr, Tolman WB. Bis(μ-Oxo)Dimetal “Diamond” Cores in Copper and Iron Complexes Relevant to Biocatalysis. Angew Chem, Int Ed. 2002;41:1114–1137. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 162.Rohde GT., Ph.D . Dissertation. University of Minnesota; Minneapolis, MN: 2015. [Google Scholar]
- 163.McDonald AR, Que L. High-Valent Nonheme Iron-Oxo Complexes: Synthesis, Structure, and Spectroscopy. Coord Chem Rev. 2013;257:414–428. [Google Scholar]
- 164.Klein JEMN, Que L., Jr . Biomimetic High-Valent Mononuclear Nonheme Iron-Oxo Chemistry. In: Scott RA, editor. Encyclopedia of Inorganic and Bioinorganic Chemistry. John Wiley; Chichester, U.K: 2016. 10.1002/9781119951438.eibc2344. [Google Scholar]
- 165.Xue G, Wang D, De Hont R, Fiedler AT, Shan X, Munck E, Que L. A Synthetic Precedent for the [FeIV2(μ-O)2] Diamond Core Proposed for Methane Monooxygenase Intermediate Q. Proc Natl Acad Sci U S A. 2007;104:20713–20718. doi: 10.1073/pnas.0708516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Castillo RG, Banerjee R, Allpress CJ, Rohde GT, Bill E, Que L, Lipscomb JD, DeBeer S. High-Energy Resolution Fluorescence Detected X-Ray Absorption of the Q Intermediate of Soluble Methane Monooxygenase. J Am Chem Soc. 2017;139:18024–18033. doi: 10.1021/jacs.7b09560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tong WH, Chen S, Lloyd SG, Edmondson DE, Huynh BH, Stubbe J. Mechanism of Assembly of the Diferric Cluster–Tyrosyl Radical Cofactor of Escherichia coli Ribonucleotide Reductase from the Diferrous Form of the R2 Subunit. J Am Chem Soc. 1996;118:2107–2108. [Google Scholar]
- 168.Sturgeon BE, Burdi D, Chen S, Huynh BH, Edmondson DE, Stubbe J, Hoffman BM. Reconsideration of X, the Diiron Intermediate Formed During Cofactor Assembly in E. coli Ribonucleotide Reductase. J Am Chem Soc. 1996;118:7551–7557. [Google Scholar]
- 169.Stubbe J, Nocera DG, Yee CS, Chang MCY. Radical Initiation in the Class I Ribonucleotide Reductase: Long-Range Proton-Coupled Electron Transfer? Chem Rev. 2003;103:2167–2202. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 170.Han WG, Liu T, Lovell T, Noodleman L. Seven Clues to the Origin and Structure of Class-I Ribonucleotide Reductase Intermediate X. J Inorg Biochem. 2006;100:771–779. doi: 10.1016/j.jinorgbio.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 171.Han WG, Liu T, Lovell T, Noodleman L. Density Functional Theory Study of Fe(IV) D–D Optical Transitions in Active-Site Models of Class I Ribonucleotide Reductase Intermediate X with Vertical Self-Consistent Reaction Field Methods. Inorg Chem. 2006;45:8533–8542. doi: 10.1021/ic060566+. [DOI] [PubMed] [Google Scholar]
- 172.Han WG, Noodleman L. DFT Calculations of Comparative Energetics and ENDOR/Mössbauer Properties for Two Protonation States of the Iron Dimer Cluster of Ribonucleotide Reductase Intermediate X. Dalton Trans. 2009:6045–6057. doi: 10.1039/b903847g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jiang W, Hoffart LM, Krebs C, Bollinger JM. A Manganese(IV)/Iron(IV) Intermediate in Assembly of the Manganese(IV)/Iron(III) Cofactor of Chlamydia trachomatis Ribonucleotide Reductase. Biochemistry. 2007;46:8709–8716. doi: 10.1021/bi700906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Younker JM, Krest CM, Jiang W, Krebs C, Bollinger JM, Green MT. Structural Analysis of the Mn(IV)/Fe(III) Cofactor of Chlamydia trachomatis Ribonucleotide Reductase by Extended X-Ray Absorption Fine Structure Spectroscopy and Density Functional Theory Calculations. J Am Chem Soc. 2008;130:15022–15027. doi: 10.1021/ja804365e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mitic N, Clay MD, Saleh L, Bollinger JM, Solomon EI. Spectroscopic and Electronic Structure Studies of Intermediate X in Ribonucleotide Reductase R2 and Two Variants: A Description of the FeIV-Oxo Bond in the FeIII–O–FeIV Dimer. J Am Chem Soc. 2007;129:9049–9065. doi: 10.1021/ja070909i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Burdi D, Willems JP, Riggs-Gelasco P, Antholine WE, Stubbe J, Hoffman BM. The Core Structure of X Generated in the Assembly of the Diiron Cluster of Ribonucleotide Reductase: 17O2 and H217O Endor. J Am Chem Soc. 1998;120:12910–12919. [Google Scholar]
- 177.Doan PE, Shanmugam M, Stubbe J, Hoffman BM. Composition and Structure of the Inorganic Core of Relaxed Intermediate X(Y122F) of Escherichia coli Ribonucleotide Reductase. J Am Chem Soc. 2015;137:15558–15566. doi: 10.1021/jacs.5b10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sträter N, Klabunde T, Tucker P, Witzel H, Krebs B. Crystal Structure of a Purple Acid Phosphatase Containing a Dinuclear Fe(III)-Zn(II) Active Site. Science. 1995;268:1489–1492. doi: 10.1126/science.7770774. [DOI] [PubMed] [Google Scholar]
- 179.Baldwin J, Krebs C, Ley BA, Edmondson DE, Huynh BH, Bollinger JM. Mechanism of Rapid Electron Transfer During Oxygen Activation in the R2 Subunit of Escherichia coli Ribonucleotide Reductase. 1. Evidence for a Transient Tryptophan Radical. J Am Chem Soc. 2000;122:12195–12206. [Google Scholar]
- 180.Bollinger JM, Tong WH, Ravi N, Huynh BH, Edmondson DE, Stubbe J. Mechanism of Assembly of the Tyrosyl Radical-Diiron(III) Cofactor of E. coli Ribonucleotide Reductase. 3. Kinetics of the Limiting Fe2+ Reaction by Optical, EPR, and Moessbauer Spectroscopies. J Am Chem Soc. 1994;116:8024–8032. [Google Scholar]
- 181.Martinie RJ, Blaesi EJ, Krebs C, Bollinger JM, Silakov A, Pollock CJ. Evidence for a Di-μ-Oxo Diamond Core in the Mn(IV)/Fe(IV) Activation Intermediate of Ribonucleotide Reductase from Chlamydia trachomatis. J Am Chem Soc. 2017;139:1950–1957. doi: 10.1021/jacs.6b11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Valentine AM, Tavares P, Pereira AS, Davydov R, Krebs C, Hoffman BM, Edmondson DE, Huynh BH, Lippard SJ. Generation of a Mixed-Valent Fe(III)Fe(IV) Form of Intermediate Q in the Reaction Cycle of Soluble Methane Monooxygenase, an Analog of Intermediate X in Ribonucleotide Reductase R2 Assembly. J Am Chem Soc. 1998;120:2190–2191. [Google Scholar]
- 183.Jiang W, Yun D, Saleh L, Barr EW, Xing G, Hoffart LM, Maslak MA, Krebs C, Bollinger JM. A Manganese(IV)/Iron(III) Cofactor in Chlamydia trachomatis Ribonucleotide Reductase. Science. 2007;316:1188–1191. doi: 10.1126/science.1141179. [DOI] [PubMed] [Google Scholar]
- 184.Kwak Y, Jiang W, Dassama LMK, Park K, Bell CB, Liu LV, Wong SD, Saito M, Kobayashi Y, Kitao S, Seto M, Yoda Y, Alp EE, Zhao J, Bollinger JM, Krebs C, Solomon EI. Geometric and Electronic Structure of the Mn(IV)Fe(III) Cofactor in Class Ic Ribonucleotide Reductase: Correlation to the Class Ia Binuclear Non-Heme Iron Enzyme. J Am Chem Soc. 2013;135:17573–17584. doi: 10.1021/ja409510d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Worsdorfer B, Lingaraju M, Yennawar NH, Boal AK, Krebs C, Bollinger JM, Jr, Pandelia ME. Organophosphonate-Degrading PhnZ Reveals an Emerging Family of HD Domain Mixed-Valent Diiron Oxygenases. Proc Natl Acad Sci U S A. 2013;110:18874–18879. doi: 10.1073/pnas.1315927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM., Jr Evidence for C–H Cleavage by an Iron-Superoxide Complex in the Glycol Cleavage Reaction Catalyzed by Myo-Inositol Oxygenase. Proc Natl Acad Sci U S A. 2006;103:6130–6135. doi: 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rivard BS, Rogers MS, Marell DJ, Neibergall MB, Chakrabarty S, Cramer CJ, Lipscomb JD. Rate-Determining Attack on Substrate Precedes Rieske Cluster Oxidation During Cis-Dihydroxylation by Benzoate Dioxygenase. Biochemistry. 2015;54:4652–4664. doi: 10.1021/acs.biochem.5b00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Capece L, Lewis-Ballester A, Yeh SR, Estrin DA, Marti MA. Complete Reaction Mechanism of Indoleamine 2,3-Dioxygenase as Revealed by QM/MM Simulations. J Phys Chem B. 2012;116:1401–1413. doi: 10.1021/jp2082825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.van der Donk WA, Krebs C, Bollinger JM. Substrate Activation by Iron Superoxo Intermediates. Curr Opin Struct Biol. 2010;20:673–683. doi: 10.1016/j.sbi.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tamanaha E, Zhang B, Guo Y, Chang Wc, Barr EW, Xing G, St Clair J, Ye S, Neese F, Bollinger JM, Krebs C. Spectroscopic Evidence for the Two C–H-Cleaving Intermediates of Aspergillus nidulans Isopenicillin N Synthase. J Am Chem Soc. 2016;138:8862–8874. doi: 10.1021/jacs.6b04065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zang Y, Kim J, Dong YH, Wilkinson EC, Appelman EH, Que L., Jr Models for Nonheme Iron Intermediates: Structural Basis for Tuning the Spin States of Fe(TPA) Complexes. J Am Chem Soc. 1997;119:4197–4205. [Google Scholar]
- 192.Kodera M, Kawahara Y, Hitomi Y, Nomura T, Ogura T, Kobayashi Y. Reversible O–O Bond Scission of Peroxodiiron(III) to High-Spin Oxodiiron(IV) in Dioxygen Activation of a Diiron Center with a Bis-Tpa Dinucleating Ligand as a Soluble Methane Monooxygenase Model. J Am Chem Soc. 2012;134:13236–13239. doi: 10.1021/ja306089q. [DOI] [PubMed] [Google Scholar]
- 193.Brunold TC, Tamura N, Kitajima N, Moro-Oka Y, Solomon EI. Spectroscopic Study of [Fe2(O2)(Obz)2{Hb(Pz′)3}2]: Nature of the μ-1,2 Peroxide-Fe(III) Bond and Its Possible Relevance to O2 Activation by Non-Heme Iron Enzymes. J Am Chem Soc. 1998;120:5674–5690. [Google Scholar]
- 194.Kim K, Lippard SJ. Structure and Mössbauer Spectrum of a (μ-1,2-Peroxo)Bis(μ-Carboxylato)Diiron(III) Model for the Peroxo Intermediate in the Methane Monooxygenase Hydroxylase Reaction Cycle. J Am Chem Soc. 1996;118:4914–4915. [Google Scholar]
- 195.Kryatov SV, Taktak S, Korendovych IV, Rybak-Akimova EV, Kaizer J, Torelli S, Shan X, Mandal S, MacMurdo VL, Mairata i Payeras A, Que L., Jr Dioxygen Binding to Complexes with Fe(II)2(μ-OH)2 Cores: Steric Control of Activation Barriers and O2-Adduct Formation. Inorg Chem. 2005;44:85–99. doi: 10.1021/ic0485312. [DOI] [PubMed] [Google Scholar]
- 196.Chavez FA, Ho RYN, Pink M, Young VG, Jr, Kryatov SV, Rybak-Akimova EV, Andres H, Munck E, Que L, Jr, Tolman WB. Unusual Peroxo Intermediates in the Reaction of Dioxygen with Carboxylate-Bridged Diiron(II,II) Paddlewheel Complexes. Angew Chem, Int Ed. 2002;41:149–152. doi: 10.1002/1521-3773(20020104)41:1<149::aid-anie149>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 197.Ookubo T, Sugimoto H, Nagayama T, Masuda H, Sato T, Tanaka K, Maeda Y, Okawa H, Hayashi Y, Uehara A, Suzuki M. cis- μ-1,2-Peroxo Diiron Complex: Structure and Reversible Oxygenation. J Am Chem Soc. 1996;118:701–702. [Google Scholar]
- 198.Dong Y, Menage S, Brennan BA, Elgren TE, Jang HG, Pearce LL, Que L., Jr Dioxygen Binding to Diferrous Centers. Models for Diiron-Oxo Proteins. J Am Chem Soc. 1993;115:1851–1859. [Google Scholar]
- 199.Mizoguchi TJ, Kuzelka J, Spingler B, DuBois JL, Davydov RM, Hedman B, Hodgson KO, Lippard SJ. Synthesis and Spectroscopic Studies of Non-Heme Diiron(III) Species with a Terminal Hydroperoxide Ligand: Models for Hemerythrin. Inorg Chem. 2001;40:4662–4673. doi: 10.1021/ic010076b. [DOI] [PubMed] [Google Scholar]
- 200.Hayashi Y, Kayatani T, Sugimoto H, Suzuki M, Inomata K, Uehara A, Mizutani Y, Kitagawa T, Maeda Y. Synthesis, Characterization, and Reversible Oxygenation of μ-Alkoxo Diiron(II) Complexes with the Dinucleating Ligand N,N,N′,N′-Tetrakis{(6-Methyl-2-Pyridyl)Methyl}-1,3-Diamino-Propan-2-Olate. J Am Chem Soc. 1995;117:11220–11229. [Google Scholar]
- 201.Arii H, Nagatomo S, Kitagawa T, Miwa T, Jitsukawa K, Einaga H, Masuda H. A Novel Diiron Complex as a Functional Model for Hemerythrin. J Inorg Biochem. 2000;82:153–162. doi: 10.1016/s0162-0134(00)00163-x. [DOI] [PubMed] [Google Scholar]
- 202.LeCloux DD, Barrios AM, Mizoguchi TJ, Lippard SJ. Modeling the Diiron Centers of Non-Heme Iron Enzymes. Preparation of Sterically Hindered Diiron(II) Tetracarboxylate Complexes and Their Reactions with Dioxygen. J Am Chem Soc. 1998;120:9001–9014. [Google Scholar]
- 203.Zhao M, Helms B, Slonkina E, Friedle S, Lee D, DuBois J, Hedman B, Hodgson KO, Frechet JMJ, Lippard SJ. Iron Complexes of Dendrimer-Appended Carboxylates for Activating Dioxygen and Oxidizing Hydrocarbons. J Am Chem Soc. 2008;130:4352–4363. doi: 10.1021/ja076817a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Shan X, Que L., Jr Intermediates in the Oxygenation of a Nonheme Diiron(II) Complex, Including the First Evidence for a Bound Superoxo Species. Proc Natl Acad Sci U S A. 2005;102:5340–5345. doi: 10.1073/pnas.0409640102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kodera M, Taniike Y, Itoh M, Tanahashi Y, Shimakoshi H, Kano K, Hirota S, Iijima S, Ohba M, Okawa H. Synthesis, Characterization, and Activation of Thermally Stable μ-1,2-Peroxodiiron(III) Complex. Inorg Chem. 2001;40:4821–4822. doi: 10.1021/ic0155434. [DOI] [PubMed] [Google Scholar]
- 206.Murch BP, Bradley FC, Que L., Jr A Binuclear Iron Peroxide Complex Capable of Olefin Epoxidation. J Am Chem Soc. 1986;108:5027–5028. [Google Scholar]
- 207.Pap JS, Draksharapu A, Giorgi M, Browne WR, Kaizer J, Speier G. Stabilisation of M-Peroxido-Bridged Fe(III) Intermediates with Non-Symmetric Bidentate N-Donor Ligands. Chem Commun. 2014;50:1326–1329. doi: 10.1039/c3cc48196d. [DOI] [PubMed] [Google Scholar]
- 208.Duboc-Toia C, Menage S, Ho RYN, Que L, Lambeaux C, Fontecave M. Enantioselective Sulfoxidation as a Probe for a Metal-Based Mechanism in H2O2-Dependent Oxidations Catalyzed by a Diiron Complex. Inorg Chem. 1999;38:1261–1268. doi: 10.1021/ic980958j. [DOI] [PubMed] [Google Scholar]
- 209.Dong Y, Yan S, Young VG, Jr, Que L., Jr The Crystal Structure of a Synthetic Nonheme Diiron-O2 Adduct: Insight into Oxygen Activation. Angew Chem, Int Ed Engl. 1996;35:618–620. [Google Scholar]
- 210.Frisch JR, Vu VV, Martinho M, Munck E, Que L., Jr Characterization of Two Distinct Adducts in the Reaction of a Nonheme Diiron(II) Complex with O2. Inorg Chem. 2009;48:8325–8336. doi: 10.1021/ic900961k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kitajima N, Tamura N, Amagai H, Fukui H, Moro-Oka Y, Mizutani Y, Kitagawa T, Mathur R, Heerwegh K. Monomeric Carboxylate Ferrous Complexes as Models for the Dioxygen Binding Sites in Non-Heme Iron Proteins. The Reversible Formation and Characterization Of μ-Peroxo Diferric Complexes. J Am Chem Soc. 1994;116:9071–9085. [Google Scholar]
- 212.Roelfes G, Vrajmasu V, Chen K, Ho RYN, Rohde JU, Zondervan C, la Crois RM, Schudde EP, Lutz M, Spek AL, Hage R, Feringa BL, Munck E, Que L. End-on and Side-on Peroxo Derivatives of Non-Heme Iron Complexes with Pentadentate Ligands: Models for Putative Intermediates in Biological Iron/Dioxygen Chemistry. Inorg Chem. 2003;42:2639–2653. doi: 10.1021/ic034065p. [DOI] [PubMed] [Google Scholar]
- 213.Armstrong WH, Lippard SJ. (μ-Oxo)Bis(μ-Acetato)Bis-(Tri-1-Pyrazolylborato)Diiron(III), [(HBpz3)FeO(CH3CO2)2Fe-(HBpz3)]: Model for the Binuclear Iron Center of Hemerythrin. J Am Chem Soc. 1983;105:4837–4838. [Google Scholar]
- 214.Armstrong WH, Lippard SJ. Reversible Protonation of the Oxo Bridge in a Hemerythrin Model Compound. Synthesis, Structure, and Properties of (μ-Hydroxo)Bis(μ-Acetato)Bis[Hydrotris(1-Pyrazolyl)Borato]Diiron(III) [(Hb(Pz)3)Fe(OH)(O2CCH3)2Fe(HB-(Pz)3)2]+ J Am Chem Soc. 1984;106:4632–4633. [Google Scholar]
- 215.Mizoguchi TJ, Lippard SJ. Synthetic Models of the Deoxy and Oxy Forms of the Non-Heme Dioxygen-Binding Protein Hemerythrin. J Am Chem Soc. 1998;120:11022–11023. [Google Scholar]
- 216.Hummel H, Mekmouche Y, Duboc-Toia C, Ho RYN, Que L, Jr, Schunemann V, Thomas F, Trautwein AX, Lebrun C, Fontecave M, Menage S. A Diferric Peroxo Complex with an Unprecedented Spin Configuration: An S = 2 System Arising from an S = 5/2, 1/2 Pair. Angew Chem, Int Ed. 2002;41:617–620. [Google Scholar]
- 217.Ho RYN, Roelfes G, Feringa BL, Que L. Raman Evidence for a Weakened O–O Bond in Mononuclear Low-Spin Iron(III)–Hydroperoxides. J Am Chem Soc. 1999;121:264–265. [Google Scholar]
- 218.Chiang CW, Kleespies ST, Stout HD, Meier KK, Li PY, Bominaar EL, Que L, Munck E, Lee WZ. Characterization of a Paramagnetic Mononuclear Nonheme Iron-Superoxo Complex. J Am Chem Soc. 2014;136:10846–10849. doi: 10.1021/ja504410s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kodera M, Ishiga S, Tsuji T, Sakurai K, Hitomi Y, Shiota Y, Sajith PK, Yoshizawa K, Meda K, Ogura T. Formation and High Reactivity of the Anti-Dioxo Form of High-Spin μ-Oxodioxodiiron(IV) as the Active Species That Cleaves Strong C–H Bonds. Chem - Eur J. 2016;22:5924–5936. doi: 10.1002/chem.201600048. [DOI] [PubMed] [Google Scholar]
- 220.Kodera M, Tsuji T, Yasunaga T, Kawahara Y, Hirano T, Hitomi Y, Nomura T, Ogura T, Kobayashi Y, Sajith PK, Shiota Y, Yoshizawa K. Roles of Carboxylate Donors in O-O Bond Scission of Peroxodiiron(III) to High-Spin Oxodiiron(IV) with a New Carboxylate-Containing Dinucleating Ligand. Chem Sci. 2014;5:2282–2292. [Google Scholar]
- 221.Friedle S, Kodanko JJ, Morys AJ, Hayashi T, Moenne-Loccoz P, Lippard SJ. Modeling the Syn Disposition of Nitrogen Donors in Non-Heme Diiron Enzymes. Synthesis, Characterization, and Hydrogen Peroxide Reactivity of Diiron(III) Complexes with the Syn N-Donor Ligand H2BPG2dev. J Am Chem Soc. 2009;131:14508–14520. doi: 10.1021/ja906137y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wang D, Farquhar ER, Stubna A, Münck E, Que L. A Diiron(IV) Complex That Cleaves Strong C–H and O–H Bonds. Nat Chem. 2009;1:145–150. doi: 10.1038/nchem.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Leising RA, Brennan BA, Que L, Fox BG, Munck E. Models for Non-Heme Iron Oxygenases: A High-Valent Iron-Oxo Intermediate. J Am Chem Soc. 1991;113:3988–3990. [Google Scholar]
- 224.Dong Y, Fujii H, Hendrich MP, Leising RA, Pan G, Randall CR, Wilkinson EC, Zang Y, Que L. A High-Valent Nonheme Iron Intermediate. Structure and Properties of [Fe2(μ-O)2(5-Me-TPA)2](ClO4)3. J Am Chem Soc. 1995;117:2778–2792. [Google Scholar]
- 225.Xue G, Fiedler AT, Martinho M, Munck E, Que L. Insights into the P-to-Q Conversion in the Catalytic Cycle of Methane Monooxygenase from a Synthetic Model System. Proc Natl Acad Sci U S A. 2008;105:20615–20620. [Google Scholar]
- 226.Xue G, De Hont R, Munck E, Que L. Million-Fold Activation of the [Fe2(μ-O)2] Diamond Core for C–H Bond Cleavage. Nat Chem. 2010;2:400–405. doi: 10.1038/nchem.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Xue G, Pokutsa A, Que L. Substrate-Triggered Activation of a Synthetic [Fe2(μ-O)2] Diamond Core for C–H Bond Cleavage. J Am Chem Soc. 2011;133:16657–16667. doi: 10.1021/ja207131g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Xue G, Geng C, Ye S, Fiedler AT, Neese F, Que L. Hydrogen-Bonding Effects on the Reactivity of [X–FeIII–O–FeIV=O] (X = OH, F) Complexes toward C–H Bond Cleavage. Inorg Chem. 2013;52:3976–3984. doi: 10.1021/ic3027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Stoian SA, Xue G, Bominaar EL, Que L, Münck E. Spectroscopic and Theoretical Investigation of a Complex with an [O=FeIII-O–FeIV=O] Core Related to Methane Monooxygenase Intermediate Q. J Am Chem Soc. 2014;136:1545–1558. doi: 10.1021/ja411376u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.De Hont RF, Xue G, Hendrich MP, Que L, Bominaar EL, Munck E. Mössbauer, Electron Paramagnetic Resonance, and Density Functional Theory Studies of Synthetic S = 1/2 FeIII–O–FeIV=O Complexes. Superexchange-Mediated Spin Transition at the FeIV=O Site. Inorg Chem. 2010;49:8310–8322. doi: 10.1021/ic100870v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Shanmugam M, Xue G, Que L, Hoffman BM. 1H-Endor Evidence for a Hydrogen-Bonding Interaction That Modulates the Reactivity of a Nonheme FeIV=O Unit. Inorg Chem. 2012;51:10080–10082. doi: 10.1021/ic3015783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kodera M, Itoh M, Kano K, Funabiki T, Reglier M. A Diiron Center Stabilized by a Bis-TPA Ligand as a Model of Soluble Methane Monooxygenase: Predominant Alkene Epoxidation with H2o2. Angew Chem, Int Ed. 2005;44:7104–7106. doi: 10.1002/anie.200501825. [DOI] [PubMed] [Google Scholar]
- 233.Zheng H, Yoo SJ, Munck E, Que L. The Flexible Fe2(μ-O)2 Diamond Core: A Terminal Iron(IV)–Oxo Species Generated from the Oxidation of a Bis(μ-Oxo)Diiron(III) Complex. J Am Chem Soc. 2000;122:3789–3790. [Google Scholar]
- 234.Zang Y, Pan G, Que L, Fox BG, Munck E. First Diferric Complex with an Fe2(μ-O)(μ-OH) Core. Structure and Reactivity of [Fe2(μ-O)(μ-OH)(6TLA)2](ClO4)3. J Am Chem Soc. 1994;116:3653–3654. [Google Scholar]
- 235.Lee D, Pierce B, Krebs C, Hendrich MP, Huynh BH, Lippard SJ. Functional Mimic of Dioxygen-Activating Centers in Non-Heme Diiron Enzymes: Mechanistic Implications of Paramagnetic Intermediates in the Reactions between Diiron(II) Complexes and Dioxygen. J Am Chem Soc. 2002;124:3993–4007. doi: 10.1021/ja012251t. [DOI] [PubMed] [Google Scholar]
- 236.Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Finding Intermediates in the O2 Activation Pathways of Non-Heme Iron Oxygenases. Acc Chem Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kovaleva EG, Lipscomb JD. Versatility of Biological Non-Heme Fe(II) Centers in Oxygen Activation Reactions. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Jeoung JH, Bommer M, Lin TY, Dobbek H. Visualizing the Substrate-, Superoxo-, Alkylperoxo-, and Product-Bound States at the Nonheme Fe(II) Site of Homogentisate Dioxygenase. Proc Natl Acad Sci U S A. 2013;110:12625–12630. doi: 10.1073/pnas.1302144110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Brown PM, Caradoc-Davies TT, Dickson JMJ, Cooper GJS, Loomes KM, Baker EN. Crystal Structure of a Substrate Complex of Myo-Inositol Oxygenase, a Di-Iron Oxygenase with a Key Role in Inositol Metabolism. Proc Natl Acad Sci U S A. 2006;103:15032–15037. doi: 10.1073/pnas.0605143103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Xing G, Hoffart LM, Diao Y, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM. A Coupled Dinuclear Iron Cluster That Is Perturbed by Substrate Binding in Myo-Inositol Oxygenase. Biochemistry. 2006;45:5393–5401. doi: 10.1021/bi0519607. [DOI] [PubMed] [Google Scholar]
- 241.Snyder RA, Bell CB, Diao Y, Krebs C, Bollinger JM, Solomon EI. Circular Dichroism, Magnetic Circular Dichroism, and Variable Temperature Variable Field Magnetic Circular Dichroism Studies of Biferrous and Mixed-Valent Myo-Inositol Oxygenase: Insights into Substrate Activation of O2 Reactivity. J Am Chem Soc. 2013;135:15851–15863. doi: 10.1021/ja406635k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Hong S, Sutherlin KD, Park J, Kwon E, Siegler MA, Solomon EI, Nam W. Crystallographic and Spectroscopic Characterization and Reactivities of a Mononuclear Non-Haem Iron(III)-Superoxo Complex. Nat Commun. 2014;5:5440. doi: 10.1038/ncomms6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Stout HD, Kleespies ST, Chiang CW, Lee WZ, Que L, Munck E, Bominaar EL. Spectroscopic and Theoretical Study of Spin-Dependent Electron Transfer in an Iron(III) Superoxo Complex. Inorg Chem. 2016;55:5215–5226. doi: 10.1021/acs.inorgchem.6b00134. [DOI] [PubMed] [Google Scholar]
- 244.Zhang Z, Dales NA, Winther MD. Opportunities and Challenges in Developing Stearoyl-Coenzyme a Desaturase-1 Inhibitors as Novel Therapeutics for Human Disease. J Med Chem. 2014;57:5039–5056. doi: 10.1021/jm401516c. [DOI] [PubMed] [Google Scholar]
- 245.Shanklin J, Whittle E, Fox BG. Eight Histidine Residues Are Catalytically Essential in a Membrane-Associated Iron Enzyme, Stearoyl-CoA Desaturase, and Are Conserved in Alkane Hydroxylase and Xylene Monooxygenase. Biochemistry. 1994;33:12787–12794. doi: 10.1021/bi00209a009. [DOI] [PubMed] [Google Scholar]
- 246.Shanklin J, Whittle E. Evidence Linking the Pseudomonas Oleovorans Alkane H-Hydroxylase, an Integral Membrane Diiron Enzyme, and the Fatty Acid Desaturase Family. FEBS Lett. 2003;545:188–192. doi: 10.1016/s0014-5793(03)00529-5. [DOI] [PubMed] [Google Scholar]
- 247.Shanklin J, Achim C, Schmidt H, Fox BG, Munck E. Mössbauer Studies of Alkane ω-Hydroxylase: Evidence for a Diiron Cluster in an Integral-Membrane Enzyme. Proc Natl Acad Sci U S A. 1997;94:2981–2986. doi: 10.1073/pnas.94.7.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Tsai YF, Luo WI, Chang JL, Chang CW, Chuang HC, Ramu R, Wei GT, Zen JM, Yu SSF. Electrochemical Hydroxylation of C3–C12 N-Alkanes by Recombinant Alkane Hydroxylase (AlkB) and Rubredoxin-2 (AlkG) from Pseudomonas Putida Gpo1. Sci Rep. 2017;7:8369. doi: 10.1038/s41598-017-08610-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.True AE, Scarrow RC, Randall CR, Holz RC, Que L. EXAFS Studies of Uteroferrin and Its Anion Complexes. J Am Chem Soc. 1993;115:4246–4255. [Google Scholar]
- 250.Shu L, Broadwater JA, Achim C, Fox BG, Munck E, Que L., Jr Exafs and Mössbauer Characterization of the Diiron(III) Site in Stearoyl-Acyl Carrier Protein Δ9- Desaturase. JBIC, J Biol Inorg Chem. 1998;3:392–400. [Google Scholar]
- 251.Jackson Rudd D, Sazinsky MH, Merkx M, Lippard SJ, Hedman B, Hodgson KO. Determination by X-Ray Absorption Spectroscopy of the Fe–Fe Separation in the Oxidized Form of the Hydroxylase of Methane Monooxygenase Alone and in the Presence of MMOD. Inorg Chem. 2004;43:4579–4589. doi: 10.1021/ic049716b. [DOI] [PubMed] [Google Scholar]
- 252.Bai Y, McCoy JG, Levin EJ, Sobrado P, Rajashankar KR, Fox BG, Zhou M. X-Ray Structure of a Mammalian Stearoyl-Coa Desaturase. Nature. 2015;524:252. doi: 10.1038/nature14549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Skulan AJ, Hanson MA, Hsu Hf, Que L, Jr, Solomon EI. Spectroscopic Study of [Fe2O2(5-Et3-TPA)2]3+: Nature of the Fe2O2 Diamond Core and Its Possible Relevance to High-Valent Binuclear Non-Heme Enzyme Intermediates. J Am Chem Soc. 2003;125:7344–7356. doi: 10.1021/ja021137n. [DOI] [PubMed] [Google Scholar]
- 254.Skulan AJ, Hanson MA, Hsu Hf, Dong Y, Que L, Jr, Solomon EI. EPR Spectroscopy of [Fe2O2(5-Et3-TPA)2]3+: Electronic Origin of the Unique Spin-Hamiltonian Parameters of the Fe2III,IVO2 Diamond Core. Inorg Chem. 2003;42:6489–6496. doi: 10.1021/ic034170z. [DOI] [PubMed] [Google Scholar]
- 255.Ortiz de Montellano PR, editor. Structure, Mechanism and Biochemistry. 3rd. Kluwer Academic/Plenum Publishers; New York: 2005. Cytochrome P-450. [Google Scholar]
- 256.Sirajuddin S, Rosenzweig AC. Enzymatic Oxidation of Methane. Biochemistry. 2015;54:2283–2294. doi: 10.1021/acs.biochem.5b00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ross MO, Rosenzweig AC. A tale of two methane monooxygenases. JBIC, J Biol Inorg Chem. 2017;22:307–319. doi: 10.1007/s00775-016-1419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Lee SJ, McCormick MS, Lippard SJ, Cho US. Controlof substrate access to the active site in methane monooxygenase. Nature. 2013;494:380–384. doi: 10.1038/nature11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Komor AJ, Rivard BS, Fan R, Guo Y, Que L, Jr, Lipscomb JD. CmlI N-Oxygenase Catalyzes the Final Three Steps in Chloramphenicol Biosynthesis without Dissociation of Intermediates. Biochemistry. 2017;56:4940–4950. doi: 10.1021/acs.biochem.7b00695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Aukema KG, Makris TM, Stoian SA, Richman JE, Munck E, Lipscomb JD, Wackett LP. Cyanobacterial Aldehyde Deformylase Oxygenation of Aldehydes Yields n – 1 Aldehydes and Alcohols in Addition to Alkanes. ACS Catal. 2013;3:2228–2238. doi: 10.1021/cs400484m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Rivard BS, Rogers MS, Marell DJ, Neibergall MB, Chakrabarty S, Cramer CJ, Lipscomb JD. Rate-Determining Attack on Substrate Precedes Rieske Cluster Oxidation during cis-Dihydroxylation by Benzoate Dioxygenase. Biochemistry. 2015;54:4652–4664. doi: 10.1021/acs.biochem.5b00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kal S, Que L., Jr Dioxygen Activation by Nonheme Iron Enzymes with the 2-His-1-carboxylate Facial Triad That Generate High-valent Oxoiron Oxidants. JBIC, J Biol Inorg Chem. 2017;22:339–365. doi: 10.1007/s00775-016-1431-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 showing the pre-edge region of the Fe K-edge X-ray absorption spectrum of oxyhemerythrin. (PDF)