

Abstract
Transient receptor potential mucolipin 1 (TRPML1) is a Ca2+-releasing cation channel that mediates the calcium signalling and homeostasis of lysosomes. Mutations in TRPML1 lead to mucolipidosis type IV, a severe lysosomal storage disorder. Here we report two electron cryo-microscopy structures of full-length human TRPML1: a 3.72-Å apo structure at pH 7.0 in the closed state, and a 3.49-Å agonist-bound structure at pH 6.0 in an open state. Several aromatic and hydrophobic residues in pore helix 1, helices S5 and S6, and helix S6 of a neighbouring subunit, form a hydrophobic cavity to house the agonist, suggesting a distinct agonist-binding site from that found in TRPV1, a TRP channel from a different subfamily. The opening of TRPML1 is associated with distinct dilations of its lower gate together with a slight structural movement of pore helix 1. Our work reveals the regulatory mechanism of TRPML channels, facilitates better understanding of TRP channel activation, and provides insights into the molecular basis of mucolipidosis type IV pathogenesis.
TRPML1, a member of the mucolipin subfamily of transient receptor potential (TRPML) channels, is a mammalian endolysosomal cation-permeable channel1–3 that is an important regulator of trafficking4–6 and autophagy-related events4,7–9. The activity of TRPML1 is regulated by the concentrations of Ca2+ and H+ in the lysosomal lumen and cytosolic space3,10. TRPML1 also serves as a proton leak, responsible for preventing the excessive acidification of lysosomes11. It is activated by various small-molecule synthetic compounds12,13 (for example, mucolipin synthetic agonist 1 (ML-SA1) and MK6-83) and phosphatidylinositol-3,5-bisphosphate (PtdIns(3,5)P2)14,15, and is inhibited by sphingomyelins and PtdIns(4,5)P216. The transfer of Ca2+ from the lysosomal lumen to the cytoplasm by TRPML1 is indispensable in many endolysosome-dependent cellular processes2,3 (such as cholesterol accumulation, lipid trafficking, exocytosis and autophagy). Mutations in TRPML1 cause the neurodegenerative lysosomal storage disease mucolipidosis type IV17–19, which leads to cognitive, linguistic, and motor defects, and retinal degeneration20,21.
TRPML channels are widespread in metazoans16,22. Specifically, in mammalian cells, two TRPML proteins, TRPML2 and TRPML3, share high sequence homology with TRPML1 (Extended Data Fig. 1). TRPML2 expression is highly restricted to lymphocytes and other types of immune cells23, whereas mice with gain-of-function mutations in Trpml3 present with deafness, pigmentation defects, and circling behaviour24,25. Therefore, the structure of TRPML1 may further our understanding of Ca2+-mediated lysosomal activities, lysosome ion homeostasis, mucolipidosis type IV pathogenesis, and the functions of TRPML2 and TRPML3.
Breakthroughs in electron cryo-microscopy (cryo-EM) have accelerated structural determinations of TRP channels26–34, and provided valuable insights into the functional mechanisms and molecular dynamics of this family. Recently, structures of the human TRPML1 luminal domain have been reported35. This domain has a similar architecture to the polycystin domain of PKD232, a member of the polycystin TRP family that, along with the TRPML channels, belongs to the group 2 TRP channel family. However, the full-length structure of TRPML1 and the mechanism of its regulation are yet to be elucidated.
Overall structure of human TRPML1
We expressed full-length human TRPML1 (580 residues) by baculovirus-mediated gene transduction of mammalian cells36. The protein eluted as a monodisperse peak under 0.04% (w/v) digitonin during gel filtration, with a small fraction of an N-terminally degraded fragment (Extended Data Fig. 2a), and was subjected to structure determination by cryo-EM.
The apo TRPML1 at pH 7.0 was determined at 3.7 Å, representing a closed conformation; the ML-SA1-bound TRPML1 at pH 6.0 was determined at 3.5 Å, representing an open conformation (Extended Data Figs 2b, 3 and Extended Data Table 1). TRPML1 has a similar topology to other TRP channels, and forms the canonical homotetrameric assembly in which the ion channel is formed at its centre. Each subunit contains six transmembrane helices (S1–S6), two pore helices (PH1 and PH2), and a luminal domain of around 25 kDa (Fig. 1a–c). The electron density is clearly resolved for residues 60–526 in the apo TRPML1 and residues 38–526 in the ML-SA1-bound TRPML1 (Extended Data Fig. 4), with the exception of a short linker in the luminal domain (amino acids 206–215). The local resolution of S4–S6 is about 3.0 Å, which allowed us to build confidently the side chains of the pore region with high accuracy (Extended Data Figs 2b, 4). Whereas the overall structure of TRPML1 is similar to that of PKD232 (PDB code: 5T4D; root mean square deviation = 3.8 Å), TRPML1 contains unique features: the S2 helix of TRPML1 is longer than that of PKD2 and protrudes from the membrane bilayer-embedded part into the cytosol, and the N-terminal S1 helix has an extension (pre-S1) including two small α-helices (α1 and α2) enriched in many basic amino acids (Extended Data Figs 1, 5).
Figure 1. Overall structure of TRPML1.
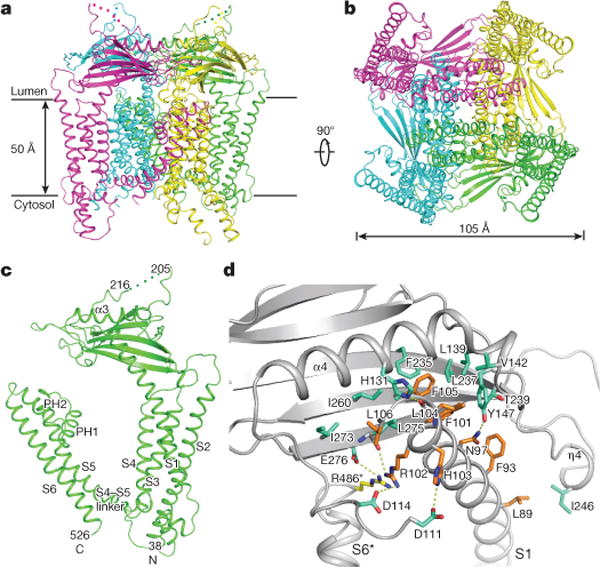
a, Ribbon representation of the structure in perspective horizontal to the plane of the membrane, with four subunits coloured differently. The flexible linker in the lumenal domain is indicated by dots. b, Structure rotated 90° around a horizontal axis. c, Ribbon representation of a TRPML1 subunit with different domains denoted. d, The interface between a transmembrane region and the luminal domain. Luminal domain residues are coloured green, S1 residues are orange, and the arginine residue from the neighbouring unit is yellow. All hydrophilic interactions are indicated by dotted lines. S6* denotes S6 of the neighbouring subunit.
Our structure provides details of the interface between the transmembrane helices and the luminal domain (Fig. 1d). Residues N97, R102, H103 and L104 of the S1 helix form hydrogen bonds or salt bridges with Y147, D114, D111 and H131 of the luminal domain. There is an intermolecular interaction between E276 in β-strand β7 and R486 of the PH2-S6 linker in the adjacent subunits. The S1 helical residues F93, F101, L104, F105 and L106 are buttressed against a hydrophobic area formed by the three β-strands of the luminal domain.
Structure of the TRPML1–ML–SA1 complex
ML-SA1 specifically stimulates TRPML channels without activating those of other TRP subfamilies, making it a useful molecule for studying the function of TRPML channels12. To investigate the activation mechanism of TRPML1, we incubated the protein with ML-SA1 at pH 6.0 before grid preparation for cryo-EM. Compared with the apo structure, we observed extra density between S5 and S6 (Extended Data Fig. 2b). Notably, the approximately 3.0-Å local resolution in this region supports the interpretation of this density as ML-SA1 (Extended Data Figs 2b, 4). A hydrophobic cavity created by I468 and F465 of PH1; F428, C429, V432 and Y436 of S5; F505 and F513 of S6; and Y499 and Y507 of S6 in the neighbouring subunit, tightly accommodates the agonist (Fig. 2b and Extended Data Fig. 1).
Figure 2. The structure of ML-SAl-bound TRPML1 compared with other ligand-bound channels.
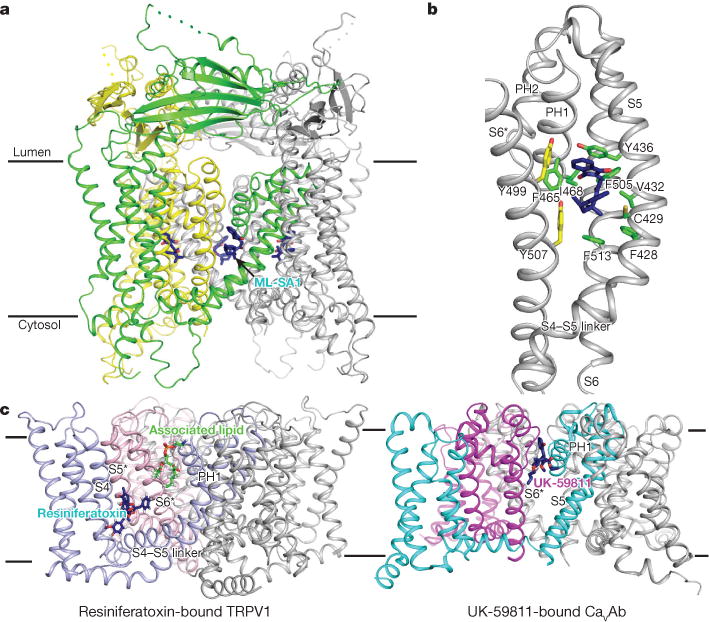
a, Overall structure. ML-SA1 is presented as sticks in dark blue. b, Detail of the interaction of TRPML1 with ML-SA1. Residues are presented as sticks in green (from the same subunit) and yellow (from the neighbouring subunit). c, Comparisons with resiniferatoxinbound TRPV1 (PDB code: 5IRX) and UK- 59811-bound CaVAb (PDB code: 5KLG). Each monomer of TRPV1 and CaVAb is shown in a different colour. Resiniferatoxin (agonist) and UK-59811 (antagonist) are shown as sticks in dark blue. The associated lipid in TRPV1 is shown as sticks in green.
The agonist-binding site in TRPML1 is different to that previously reported in TRPV1, although both binding sites require two adjacent subunits of one tetramer (Fig. 2b, c). S5, S6 and PH1 of one subunit, and S6 of the neighbouring subunit, accommodate ML-SA1 binding to TRPML1, without the involvement of the S4–S5 linker as found in TRPV1 (Fig. 2c). This binding induces a dilation of the lower selectivity filter and lower gate in TRPML1 (as discussed later). Resiniferatoxin, an agonist of TRPV1, directly binds to an S4–S5 linker of TRPV1, the S4 helix, and the S5 and S6 helices of the neighbouring subunit without binding to the pore helices (Fig. 2c). Interestingly, the comparable ML-SA1-binding site in TRPV1 and the voltage-gated calcium channel CaVAb could accommodate associated lipids29 or the CaVAb antagonist UK-5981137 (Fig. 2c), suggesting the possibility of this site for hosting other ligands in related ion channels.
To verify our findings, we performed whole-cell electrophysiology on cells expressing TRPML1 with mutations in the proposed agonist-binding hydrophobic cavity. To express TRPML1 at the cell surface, we used a leucine-to-alanine mutant that has been demonstrated to enrich functional plasmalemmal expression of TRPML1 (L15L and L577L to alanine residues, abbreviated as L/A; refs 12, 38). On the basis of our structural findings, we generated five individual mutants on the agonist-binding sites: C429G and Y436A in S5, F465A in PH1, and Y499A and F513A in S6. HEK293T cells were transiently transfected with TRPML1(L/A) constructs with an N-terminal enhanced GFP (eGFP) tag for selection. As reported previously12, application of ML-SA1 at pH 4.6 considerably activates whole-cell currents at negative potentials for TRPML1(L/A), with modest increases in current density also detected at pH 7.4 (Fig. 3a). Intracellular application of the lipid agonist PtdIns(3,5)P2 also generates a significant current at pH 4.6 without ML-SA1, and notably, with application of both PtdIns(3,5)P2 and ML-SA1, the increase in current is pronounced (Fig. 3d).
Figure 3. Electrophysiological characterization of TRPML1 and its agonist-binding pocket.
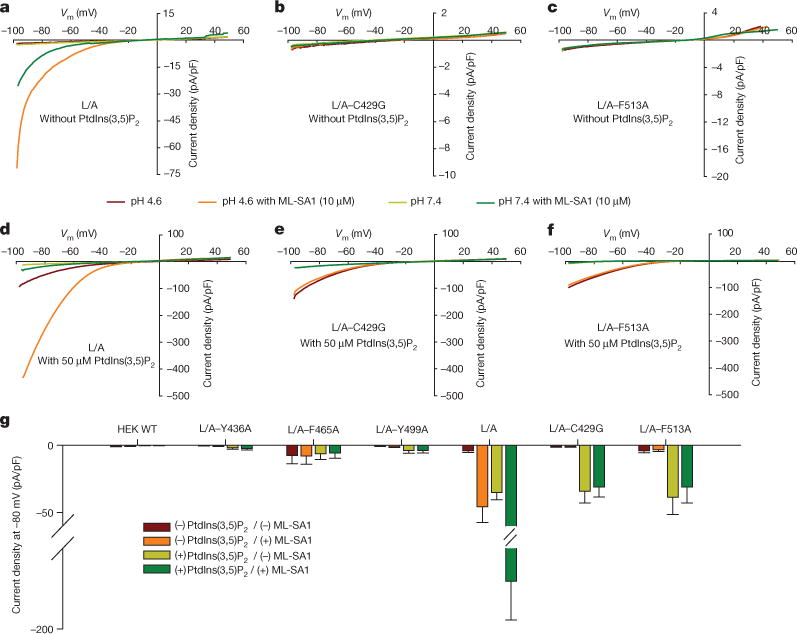
a, Whole-cell currents of HEK293T cells transfected with surface-expressing eGFP-TRPML1 with leucine-to-alanine mutants (L15L and L577L to alanine; abbreviated L/A), and L/A with binding pocket mutations. b, c, C429G (b) and F513A (c) with or without 10 μM ML-SA1 at pH 4.6 or pH 7.4 without PtdIns(3,5)P2. d–f, Whole-cell currents for constructs from panels a–c with 50 μM PtdIns(3,5)P2 substituted in the cytoplasmic solution. g, Whole-cell current density at −80 mV recorded at pH 4.6 for cells transfected with empty vector (wild type, WT), L/A, and L/A plus Y436A, F465A, Y499, C429G or F513A individually. Values are mean ±s.e.m.
Binding pocket mutations Y436A, F465A and Y499A abrogated the ML-SA1 agonist-induced currents but failed to respond significantly to PtdIns(3,5)P2 stimulation, suggesting loss of function of the channels (Fig. 3g). We hypothesize that the compact structural arrangement of the pore helices was disrupted and ion efflux stopped because these mutated sites are either in, or close to, the pore helices. Notably, mutated sites C429G and F513A, away from the pore helices, demonstrated similar expression patterns to L/A (Extended Data Fig. 6c). Like the other mutations, C429G and F513A failed to respond to ML-SA1 application (Fig. 3b, c, g); however, a positive response to PtdIns(3,5)P2 was observed, similar to wild-type TRPML1(L/A) (Fig. 3e, f). We observed a significant synergistic increase in current densities for TRPML1(L/A) when PtdIns(3,5)P2 and ML-SA1 were co-applied. For mutants C429G and F513A, this increase in current density was completely blocked (Fig. 3e–g). This indicates functional activation of the channel by the lipid agonist PtdIns(3,5)P2 and lack of functional binding by ML-SA1. Electrophysiological analysis functionally confirms this agonist-binding site in vitro.
Opening of the lower gate and selectivity filter
Superimposition of the apo and ML-SA1-bound TRPML1 structures does not reveal any notable changes in the luminal domains, consistent with previous research that did not identify luminal domain structural changes with varying pH35. The apo structure presents a closed solvent-accessible pathway (Fig. 4a) and the ML-SA1-bound structure presents an open solvent-accessible pathway (Fig. 4b). Importantly, a marked difference between the two structures is observed: a shift in the relative positions of PH1 and S6 (Fig. 4c). The selectivity filters of TRPML channels are highly conserved, including a three-residue motif (GDD472) (Extended Data Fig. 1). Upon binding to ML-SA1, the distance between the D471 Cα atoms and the distance between the G470 carbonyl oxygen atoms show no substantial change (Fig. 4c, d); notably, the carbonyl oxygens of N469 are 1.8 Å further apart than in the apo state (Fig. 4c), to open the lower selectivity filter. There is no marked conformational change in M644 of TRPV1 (PDB code: 5IRZ) after the binding of capsaicin (PDB code: 3J5R) at the equivalent position in TRPML1 (that is, at D471). By contrast, the distance between carbonyl oxygens of I642 in capsaicin-bound TRPV1 increases from 8.6 Å to 10.2 Å at the equivalent position of TPRML1 (that is, at N469).
Figure 4. Structural comparisons of apo and ML-SA1-bound TRPML1 structures.
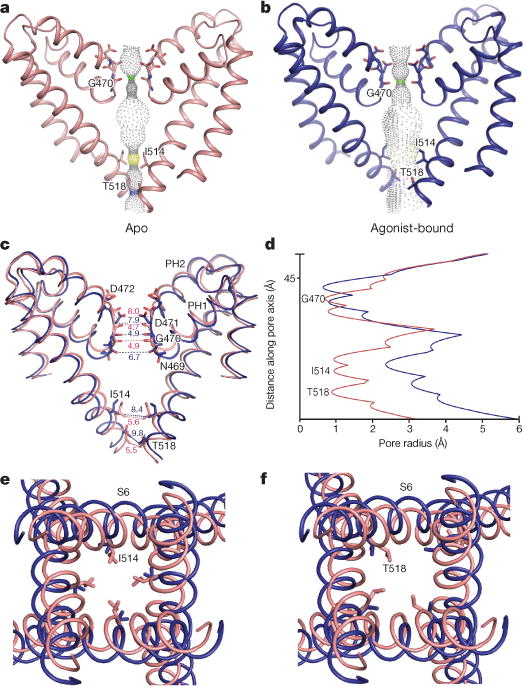
a, b, Solvent-accessible pathway along the ion permeation pore of apo (pink) and ML-SA1-bound (blue) TRPML1 generated by the program HOLE. c, Superimposition of PH1, PH2 and S2 from apo versus ML-SA1-bound TRPML1 structures. Distances from the text are numbered and indicated by dotted lines. Residues in the selectivity filter and lower gate are rendered as sticks. d, Comparison of pore radius (calculated by HOLE) for ML-SA1-bound and apo TRPML1 structures. e, f, Superimposed inner pore region from apo and ML-SA1-bound TRPML1 structures. Residues I514 and T518 are shown as sticks.
The amino acids F505IYMV509 form a π-helix in the middle of S6, a highly conserved structural feature among TRP channels. We speculate that this π-helix may control the dynamics of the bottom half of S6 and open the lower gate. I514 and T518 in S6 form two constricted sites close to the cytoplasmic side that are sufficiently narrow (approximately 5.5 Å) to restrict hydrated ion flow. This observation is consistent with the lower gate of the channel PKD232–34, which uses two residues, L677 and N681, to block the conduction of hydrated ions across the gate. In the presence of ML-SA1, both constricted sites of the lower gate expand from 5.6 Å and 5.5 Å to 8.4 Å and 9.8 Å (Fig. 4c–f), respectively. These changes in the ion permeation pathway are coupled with a slight shift of S6 away from the central channel, resulting in a wider opening of the lower gate (Fig. 4c–f). The lower gate of TRPML1 is similar to that of TRPV1, which opens from 5.3 Å to 7.6 Å when it binds to capsaicin, or to 9.3 Å when it binds to vanillotoxin or resiniferatoxin26,29.
The comparison of apo versus ML-SA1-bound structures suggests a molecular mechanism of TRPML1 activation. F505 and F513, in contact with ML-SA1, force helix S6 away from the central axis, opening the lower gate (Fig. 5a). F465 and I468 change their conformations to create a hydrophobic cavity that facilitates ML-SA1 binding. These changes, in turn, move N469 to a lower position allowing the formation of a hydrophilic interaction (at a distance of 3.5 Å) with Y507 in S6 of the neighbouring subunit, causing a slight opening of the selectivity filter (Fig. 5a).
Figure 5. Structural rearrangements in the outer pore region and lower gate.
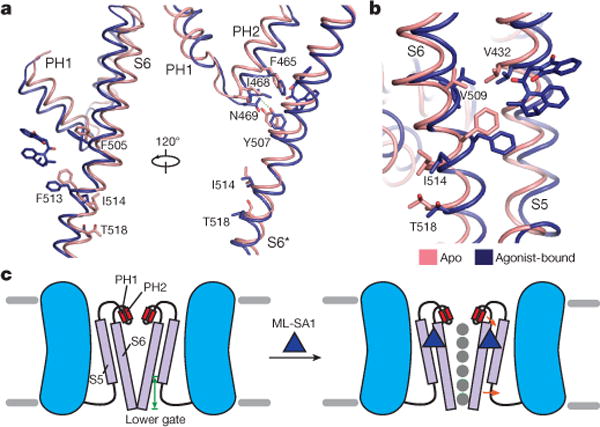
a, An allosteric coupling between the selectivity filter and lower gates. The residues in apo (pink) and ML-SA1-bound (blue) TRPML1 structures are shown as sticks. ML-SA1 is shown as sticks in dark blue. The hydrogen bond between N469 and Y507 (3.5 Å distance) is indicated by a dotted line. b, Proposed mechanism of V432-mediated lower gate regulation. c, Model of agonist-mediated TRPML1 activation. Small ligands (for example, ML-SA1 and MK6-83) bind within a hydrophobic pocket formed by S5, S6 and PH1, inducing conformational changes that expand the selectivity filter and lower gate (orange arrows), allowing ions (grey balls) across the channel.
A proline substitution at V432 (TRPML1(V432P)) causes a gain-of-function phenotype that activates TRPML1 channels at the plasma and lysosomal membranes25,39. Notably, the transfection of TRPML1(V432P) into HEK293 cells appears to enhance lysosomal exocytosis39. In light of our structure, this gain-of-function mutation can now be rationalized in molecular detail. In the apo state, the Cα distances between V432 and V509 and between V432 and F513 are 3.9 Å and 4.7 Å, respectively, facilitating strong van der Waals interactions to maintain S6 in the closed state. The V432P mutation disrupts these hydrophobic interactions and releases S6 from S5; it may allow S6 to swing open, akin to the action of ML-SA1, which interrupts the V432–F513 interaction and triggers the lower gate to open (Fig. 5b). Besides this putative mechanism, V432P mutation might also break S5, introducing more marked structural arrangements that result in channel opening. However, the rationale behind notably increased exocytosis and plasma membrane localization of TRPML1(V432P) requires additional work.
Summary
We report two structures of TRPML1 in distinct conformations by cryo-EM. These structures allow us to map the mutations that cause mucolipidosis type IV3 (Extended Data Fig. 7). Like other lysosomal storage disease-causing proteins40,41, several mutations1 (such as T232P and D362Y) in TRPML1 lead to mislocalization and block the flow of Ca2+ across the lysosomal membrane. A previous study identified luminal domain mutations35, including L106P, C166F and T232P. Other clinically defined loss-of-function mutations may be explained by our structure. For instance, aromatic–aromatic interactions between F465 in PH1 and Y499 in S6 of the neighbouring subunit could support the conformation of pore helices; the F465L mutation may disrupt this interaction and the dynamics of the selectivity filter. Y436 forms a hydrophilic interaction with S461 in PH1, potentially stabilizing the selectivity filter. Our electrophysiological experiments support the idea that Y436A, F465A and Y499A mutants could not be stimulated by agonist ML-SA1 and PtdIns(3,5)P2.
The ML-SA1-bound TRPML1 structure presents a novel agonist-binding site among TRP channels. Remarkably, ML-SA1 is buried in an aromatic pocket created by two TRPML1 subunits. Structural analysis reveals an allosteric coupling between the selectivity filter and the lower gate, which may explain the molecular mechanism of this channel (Fig. 5c). The ML-SA1 agonist releases the restriction of the lower gate and induces a structural rearrangement of PH1. Both changes create a new hydrophilic bond between Y507 (S6) and N469 (PH1) of the neighbouring subunit, leading to a slight opening of the selectivity filter. Previous studies have shown that there is a PtdIns(3,5)P2-binding site in the positively charged pre-S1 region of TRPML115, which is distant from the ML-SA1-binding site that we mapped in the membrane domain. A related Nature paper provides specific insights into TRPML1 in the lipid environment42.
Combined with previous structural observations on TRP channels, our work suggests some commonalities of TRP channels as a whole: first, the inter-subunit agonist-binding site is found in both the TRPML and TRPV families, although these two sites are in different positions of the membrane region; and second, the lower gate in the channels of group 2 TRP family proteins have two constriction sites. Moreover, owing to the topological similarities between PKD2 and TRPML1, PKD2 may have a similar agonist-binding site, and thus our study might provide insights into the activation of PKD2 and the pathogenic mechanism of PKD2-related autosomal dominant polycystic kidney disease.
METHODS
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Protein expression and purification
Human TRPML1 was cloned into pEG BacMam with an N-terminal Flag tag. The protein was expressed using baculovirus-mediated transduction of mammalian HEK293S GnTI− cells. At 48 h post infection at 37 °C, cells were disrupted by sonication in buffer A (20 mM HEPES pH 7.0, 150 mM NaCl) with 1 mM PMSF and 5 μg ml−1 each of leupeptin and aprotinin. After low-speed centrifugation, the resulting supernatant was incubated in buffer A with 1% (w/v) lauryl maltose neopentyl glycol (MNG, Anatrace) for 1 h at 4 °C. The lysate was centrifuged again and the supernatant was loaded onto an anti-Flag M2 affinity column (Sigma). After washing twice, the protein was eluted in 20 mM HEPES pH 7.0, 150 mM NaCl, 0.1 mg ml−1 Flag peptide and 0.01% MNG, and then concentrated. The concentrated protein was purified by Superdex-200 (GE Healthcare) size-exclusion chromatography in a buffer containing buffer A and 0.04% (w/v) digitonin (Sigma). The peak fractions were collected and concentrated to 7 mg ml−1 for grid preparation. The mutated DNA constructs were generated using QuikChange (Agilent).
Whole-cell patch-clamp electrophysiology
The full-length human TRPML1(L/A) was subcloned to peGFP-C1 for electrophysiological assays. Patch-clamp recordings of cell electrical parameters were performed as described previously12,35,43, using MATLAB-based Capmeter v7.244 with a National Instruments digital acquisition board and an Axopatch 200B patch-clamp amplifier. Square-wave voltage perturbation (20 mV; 0.2–0.5 kHz) was used for capacitance measurement. Input resistances were 2–10 MΩ, and the apparent cell resistances were 0.5–2 GΩ. Cells were held at 0 mV holding potentials with a voltage ramp protocol set from −100 to +50 mV; 2 s with a 2 s minimum interval between pulses and a 60 s interval between solution changes. External solutions were adjusted to room temperature (23–25 °C) in gravity-fed parallel solution lines with outlet flow velocities of 2–5 mm s−1, allowing extracellular solution changes within 2–3 s. Borosilicate glass pipettes were fire-polished and backfilled with the cytoplasmic solutions containing: 120 mM caesium methanesulfonate, 4 mM NaCl, 10 mM EGTA, 2 mM MgCl2, 20 mM HEPES, 2 mM Tris-ATP pH 7.2 with CsOH (~25 mM). For experiments containing 50 μM cytoplasmic PtdIns(3,5)P2, a 5 mM stock of C8:0 PtdIns(3,5)P2 (Avanti Polar Lipids) in ddH2O was added at 1:100 to the pipette solution before backfilling. Dialysis of patched cells occurred typically within 2 min after membrane rupture as observed by peak differences between the pH 7.4 and 4.6 current densities when PtdIns(3,5)P2 was present in the cytoplasmic solution. Extracellular solutions were set to pH 7.4 and contained 140 mM NaCl, 5 mM KCl, 10 mM glucose, 20 mM HEPES, 1 mM MgCl2, 2 mM CaCl2 and 8 mM NaOH, or were set to pH 4.6 and contained 140 mM sodium gluconate, 5 mM KCl, 10 mM glucose, 10 mM HEPES, 10 mM MES, 1 mM MgCl2, 2 mM CaCl2 and 8 mM HCl. 10 μM ML-SA1 was added to the appropriate bath solution immediately before recording. Low passage HEK293T (ATCC CRL-11268) cells used for electrophysiology were maintained in DMEM (GIBCO) with 10% FBS, penicillin–streptomycin and L-glutamine (Sigma) and routinely monitored for Mycoplasma infection using MycoSensor PCR Kits (Agilent). Cells were transiently transfected with Lipofectamine 3000 (Invitrogen) and used within 48–72 h. Before recording, cells were detached with trypsin (0.05%) and placed in a bath solution on an inverted Nikon TE2000U inverted microscope equipped with a 60 × oil immersion, 1.45-NA objective. A Lambda DG-4 xenon epifluorescence power supply was used to detect cells expressing surface eGFP–TRPML1 mutants.
For concurrent confocal imaging, a Nikon EZ-C1confocal system containing a 40 mW 163-Argon laser (Spectra Physics, Newport Corporation) operating at 488 nm and 7.5% maximum capacity was used with a 500–540 nm emission filter (Semrock). Typical resolution was 512 × 512, set with a pixel dwell-time yielding approximately 1 s exposure times and a pinhole of 150 μm with images averaged over 3 frames.
Statistical analyses of the electrophysiology were performed in MATLAB and SigmaPlot (SigmaStat). Current–voltage relations represent the mean of 4 voltage ramps, current density changes extracted at −80 mV represent the mean and s.e.m. of at least 4 independent cells per condition.
Electron microscopy sample preparation and imaging
A freshly purified protein sample at 7 mg ml−1 was added to Quantifoil R1.2/1.3 400 mesh Au holey carbon grids (Quantifoil), blotted with Vitrobot Mark IV (FEI), and frozen in liquid ethane. For ML-SA1-bound protein, the protein in a buffer containing 20 mM acetate pH 6.0, 150 mM NaCl and 0.04% digitonin was incubated with 0.5 mM ML-SA1 (Tocris Bioscience, dissolved in DMSO as a 20 mM stock) on ice for 1 h before grid preparation and freezing. The grids were imaged with a 300 keV Titan Krios electron microscope (FEI) with a Gatan K2 Summit direct electron detector (Gatan). Data were collected at 1 Å per pixel with a dose rate of 8 electrons per physical pixel per second. Images were recorded for 10 s exposures in 50 subframes to give a total dose of 80 electrons per Å2.
Imaging processing and 3D reconstruction
Dark-subtracted images were first normalized by gain reference that resulted in a pixel size of 1 Å per pixel. Drift correction was performed using the program Unblur45. The contrast transfer function was estimated using CTFFIND446. To generate templates for automatic picking, around 2,000 particles were manually picked and classified by 2D classification in RELION47. After automatic picking and manual micrograph inspection, about 111,000 (apo) and 201,000 (ML-SA1 bound) particles were extracted for subsequent 2D and 3D classification. Motion correction of individual particles was performed using the program alignparts_lmbfgs48. Using the structure of Caenorhabditis elegans TRPML1 (EMDB code: EMD-6670) low-pass filtered to 60 Å as the initial model, 3D classification was carried out in RELION47. The best classes of both structures, containing ~60,000 particles, provided a 6–7 Å map. Refinement was performed in FREALIGN49 using this best class as the initial model, and all particles were subject to post-motion correction to generate the final map for model construction.
Model construction
To obtain better side-chain densities for model building, we sharpened one half-map using BFACTOR.EXE (author: N. Grigorieff) with a resolution limit of 3.7 Å (apo) and 3.5 Å (ML-SA1-bound), and a B-factor value of 100 Å2. The entire model was built from de novo in COOT50. The crystal structure of the TRPML1 luminal domain (residues 84–296, PDB code: 5TJA) was used to check the registers of the luminal domain of our model.
Model refinement and validation
The model was refined in real space using PHENIX51 and also in reciprocal space using REFMAC52,53. Structural factors were calculated from a half-map (working) using the program sfall54. Fourier shell correlations (FSCs) were calculated between the two half maps, the model against the working map, the other (free) half map, and full (sum) map55. Local resolutions were estimated using blocres56. MolProbity57 was used to validate the geometries of the model. Structure figures were generated using PyMOL (http://www.pymol.org), Chimera58 and HOLE59.
Data availability
The 3D cryo-EM density maps of TRPML1 have been deposited in the Electron Microscopy Data Bank (EMDB) under the accession numbers EMD-8840 (apo TRPML1) and EMD-8841 (ML-SA1-bound TRPML1). Atomic coordinates for the atomic model have been deposited in the Protein Data Bank (PDB) under the accession numbers 5WJ5 (apo TRPML1) and 5WJ9 (ML-SA1-bound TRPML1).
Extended Data
Extended Data Figure 1. Sequence alignment of human TRPML1, TRPML2 and TRPML3.

Residues discussed in the paper are annotated using symbols at their positions.
Extended Data Figure 2. Biochemical properties and cryo-EM studies of human TRPML1.
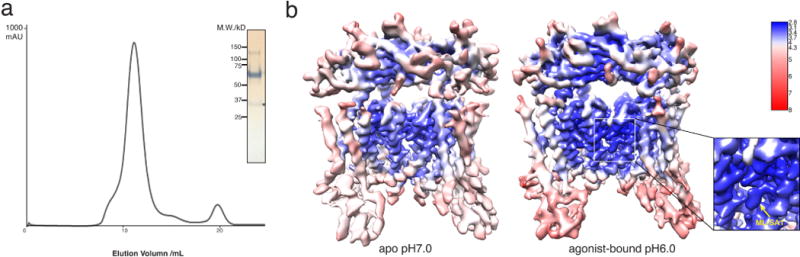
a, Size-exclusion chromatogram and SDS–PAGE gel of the purified TRPML1. A degradation fragment is presented on the gel and is indicated by an asterisk. b, Density maps of structures coloured by local resolution estimation using blocres56.
Extended Data Figure 3. Data and model quality assessment.
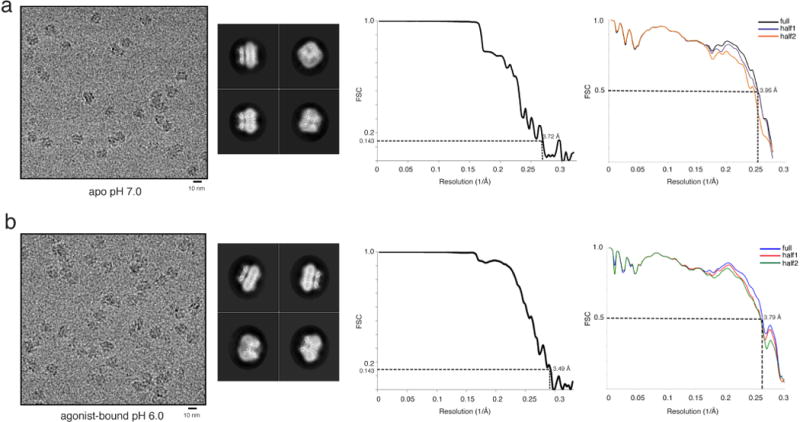
a, Left to right, a representative electron micrograph at defocus −2.0 μm; 2D classification; and FSC curves of the apo structure. The left curve shows a plot of the FSC as a function of resolution using Frealign output, the right curve shows the FSC calculated between the refined structure and the half map used for refinement, the other half map, and the full map. b, Left to right, a representative micrograph at defocus −2.0 μm; 2D classification; and FSC curves of ML-SA1-bound TRPML1 structure. The two FSC curves represent the same as for panel a.
Extended Data Figure 4. Electron microscopy density of different portions of the structures.
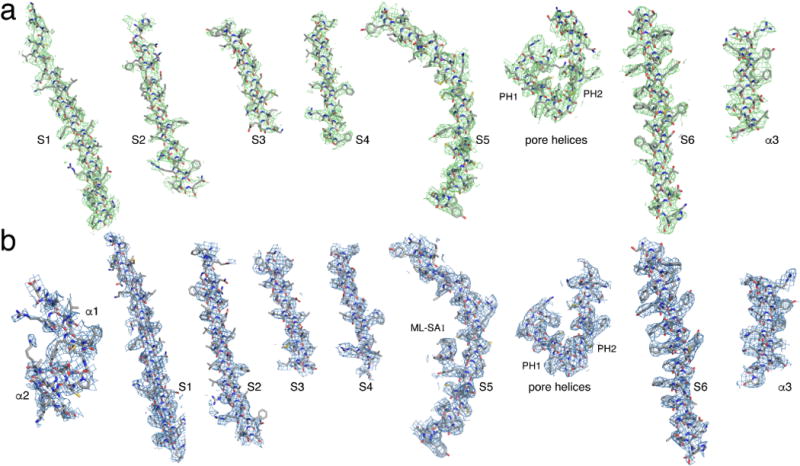
a, The apo TRPML1 structure. b, The ML-SA1-bound TRPML1 structure.
Extended Data Figure 5. Comparisons of TRPML1 and PKD2.
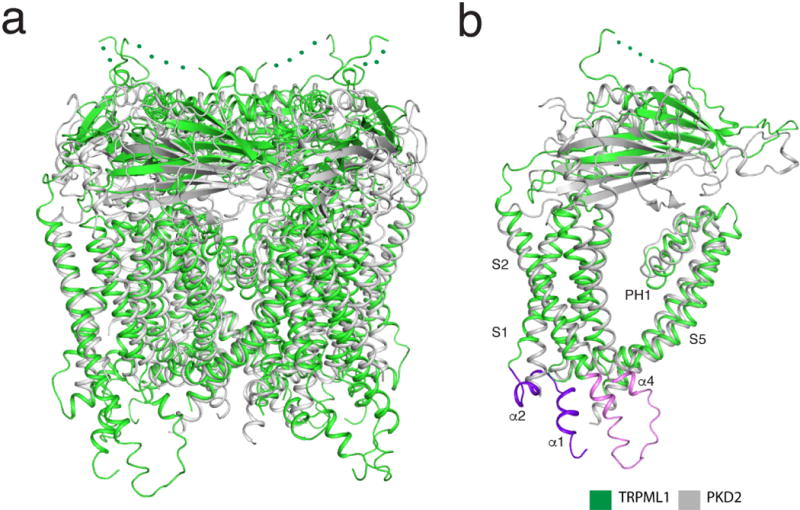
a, Superimposition of overall structures of TRPML1 and PKD2 (PDB code: 5T4D). b, Superimposition of one subunit of TRPML1 and PKD2. The extended structural elements of pre-S1 (α1 and α2), and α4 and S2 of TRPML1, are highlighted in purple and pink, respectively.
Extended Data Figure 6. Whole-cell currents of HEK wild-type cells and TRPML1(L/A) with Y436A, F465A or Y499A mutations.
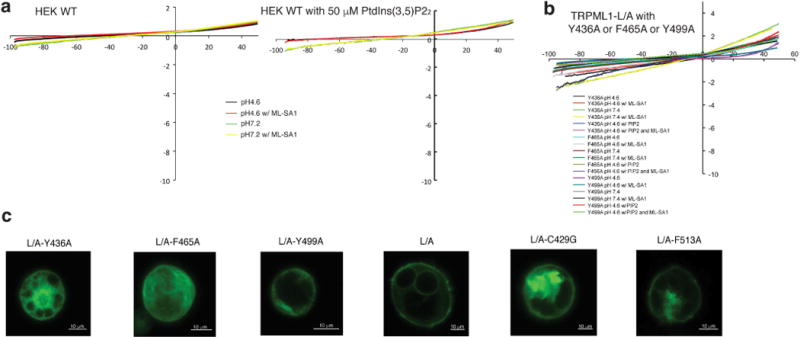
a, Whole-cell currents of HEK293T cells transfected with empty vector at pH 4.6 or pH 7.2 with or without 10 μM ML-SA1 and 50 μM PtdIns(3,5)P2. b, Whole-cell currents of HEK293T cells transfected with surface-expressing mutant eGFP–TRPML1(L/A) containing Y436A, F465A or Y499A under the same conditions as in panel a. c, Confocal images of patched cells. Scale bars, 10 μm.
Extended Data Figure 7. The distribution of mutations that cause mucolipidosis type IV in the TRPML1 structure.
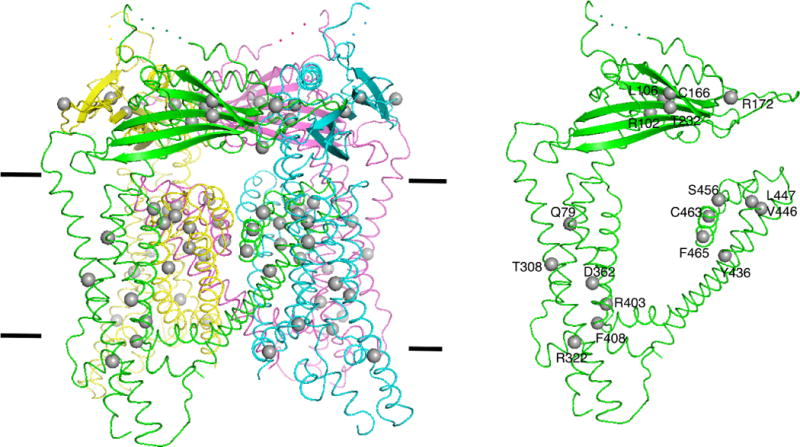
The mutations are shown as grey balls.
Extended Data Table 1.
Statistics of data collection and refinement
| Apo pH7.0 | Agonist-bound | |
|---|---|---|
| Data Collection | ||
| Microscope | Titan Krios (FEI) | Titan Krios (FEI) |
| Voltage (kV) | 300 | 300 |
| Detector | K2 Summit (Gatan) |
K2 Summit (Gatan) |
| Pixel size (Å) | 1.0 | 1.0 |
| Defocus range (μm) | −1.0 to −3.0 | −1.0 to −2.5 |
| Movies | 1172 | 1179 |
| Frames/movie | 50 | 50 |
| Dose rate (electrons/pixel/s) | 8 | 8 |
| Total dose (electrons/Å2) | 80 | 80 |
| Number of particle | 111762 | 201368 |
|
| ||
| Model composition | ||
| Non-hydrogen atoms | 14640 | 15538 |
| Protein residues | 1828 | 1920 |
| Ligand | 0 | 4 |
| Refinement | ||
| Number of particle for refinement | 56105 | 63111 |
| Resolution (Å) | 3.72 | 3.49 |
| RMS deviations | ||
| Bond lengths (Å) | 0.0085 | 0.0077 |
| Bond angles (°) | 1.2531 | 1.1373 |
| Ramachandran plot (%) | ||
| Favored | 90.73 | 90.63 |
| Allowed | 8.99 | 9.11 |
| Disallowed | 0.28 | 0.26 |
| Clashscore, all atoms | 4.42 | 3.55 |
| Molprobity score | 2.10 | 1.87 |
Supplementary Material
Acknowledgments
We thank M. Ebrahim and J. Sotiris at the Rockefeller Cryo-EM Resource Center for assistance in data collection, Z. Zhang for help in EM data processing, D. Hilgemann, E. Coutavas and E. Debler for help in manuscript preparation, and J. Fernandez and H. Molina for mass spectrometry analyses. This work was supported by funds from the Howard Hughes Medical Institute (to G.B.) and the Endowed Scholars Program in Medical Science of University of Texas Southwestern Medical Center (to X.L.). M.F was supported by the National Institutes of Health (T32DK007257 and HL119843). X.L. was the recipient of a Gordon and Betty Moore Foundation Fellow of the Life Sciences Research Foundation.
Footnotes
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Supplementary Information is available in the online version of the paper.
Author Contributions
P.S. and X.L. purified proteins and carried out cryo-EM work. M.F. carried out functional characterization by electrophysiology. All authors analysed data and contributed to manuscript preparation. X.L. conceived the project and wrote the paper.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer Information Nature thanks S. Hansen, C. Ulens and the other anonymous reviewer(s) for their contribution to the peer review of this work.
References
- 1.Zeevi DA, Frumkin A, Bach G. TRPML and lysosomal function. Biochim Biophys Acta. 2007;1772:851–858. doi: 10.1016/j.bbadis.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol. 2015;77:57–80. doi: 10.1146/annurev-physiol-021014-071649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Venkatachalam K, Wong CO, Zhu MX. The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium. 2015;58:48–56. doi: 10.1016/j.ceca.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samie M, et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell. 2013;26:511–524. doi: 10.1016/j.devcel.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miedel MT, et al. Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. J Exp Med. 2008;205:1477–1490. doi: 10.1084/jem.20072194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, et al. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol. 2016;18:404–417. doi: 10.1038/ncb3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Venkatachalam K, et al. Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell. 2008;135:838–851. doi: 10.1016/j.cell.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vergarajauregui S, Connelly PS, Daniels MP, Puertollano R. Autophagic dysfunction in mucolipidosis type IV patients. Hum Mol Genet. 2008;17:2723–2737. doi: 10.1093/hmg/ddn174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109. doi: 10.1038/ncomms12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong XP, Wang X, Xu H. TRP channels of intracellular membranes. J Neurochem. 2010;113:313–328. doi: 10.1111/j.1471-4159.2010.06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soyombo AA, et al. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem. 2006;281:7294–7301. doi: 10.1074/jbc.M508211200. [DOI] [PubMed] [Google Scholar]
- 12.Shen D, et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun. 2012;3:731. doi: 10.1038/ncomms1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen CC, et al. A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat Commun. 2014;5:4681. doi: 10.1038/ncomms5681. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, Li X, Xu H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc Natl Acad Sci USA. 2012;109:11384–11389. doi: 10.1073/pnas.1202194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong XP, et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat Commun. 2010;1:38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang W, Zhang X, Gao Q, Xu H. TRPML1: an ion channel in the lysosome. Handb Exp Pharmacol. 2014;222:631–645. doi: 10.1007/978-3-642-54215-2_24. [DOI] [PubMed] [Google Scholar]
- 17.Sun M, et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum Mol Genet. 2000;9:2471–2478. doi: 10.1093/hmg/9.17.2471. [DOI] [PubMed] [Google Scholar]
- 18.Bargal R, et al. Identification of the gene causing mucolipidosis type IV. Nat Genet. 2000;26:118–123. doi: 10.1038/79095. [DOI] [PubMed] [Google Scholar]
- 19.Bassi MT, et al. Cloning of the gene encoding a novel integral membrane protein, mucolipidin—and identification of the two major founder mutations causing mucolipidosis type IV. Am J Hum Genet. 2000;67:1110–1120. doi: 10.1016/s0002-9297(07)62941-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weitz R, Kohn G. Clinical spectrum of mucolipidosis type IV. Pediatrics. 1988;81:602–603. [PubMed] [Google Scholar]
- 21.Bach G. Mucolipidosis type IV. Mol Genet Metab. 2001;73:197–203. doi: 10.1006/mgme.2001.3195. [DOI] [PubMed] [Google Scholar]
- 22.Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.García-Añoveros J, Wiwatpanit T. TRPML2 and mucolipin evolution. Handb Exp Pharmacol. 2014;222:647–658. doi: 10.1007/978-3-642-54215-2_25. [DOI] [PubMed] [Google Scholar]
- 24.Grimm C, Barthmes M, Wahl-Schott C. Trpml3. Handb Exp Pharmacol. 2014;222:659–674. doi: 10.1007/978-3-642-54215-2_26. [DOI] [PubMed] [Google Scholar]
- 25.Di Palma F, et al. Mutations in Mcoln3 associated with deafness and pigmentation defects in varitint-waddler (Va) mice. Proc Natl Acad Sci USA. 2002;99:14994–14999. doi: 10.1073/pnas.222425399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao E, Liao M, Cheng Y, Julius D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature. 2013;504:113–118. doi: 10.1038/nature12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulsen CE, Armache JP, Gao Y, Cheng Y, Julius D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature. 2015;520:511–517. doi: 10.1038/nature14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao Y, Cao E, Julius D, Cheng Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature. 2016;534:347–351. doi: 10.1038/nature17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zubcevic L, et al. Cryo-electron microscopy structure of the TRPV2 ion channel. Nat Struct Mol Biol. 2016;23:180–186. doi: 10.1038/nsmb.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huynh KW, et al. Structure of the full-length TRPV2 channel by cryo-EM. Nat Commun. 2016;7:11130. doi: 10.1038/ncomms11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen PS, et al. The structure of the polycystic kidney disease channel PKD2 in lipid nanodiscs. Cell. 2016;167:763–773.e11. doi: 10.1016/j.cell.2016.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilkes M, et al. Molecular insights into lipid-assisted Ca2+ regulation of the TRP channel Polycystin-2. Nat Struct Mol Biol. 2017;24:123–130. doi: 10.1038/nsmb.3357. [DOI] [PubMed] [Google Scholar]
- 34.Grieben M, et al. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2) Nat Struct Mol Biol. 2017;24:114–122. doi: 10.1038/nsmb.3343. [DOI] [PubMed] [Google Scholar]
- 35.Li M, et al. Structural basis of dual Ca2+/pH regulation of the endolysosomal TRPML1 channel. Nat Struct Mol Biol. 2017;24:205–213. doi: 10.1038/nsmb.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goehring A, et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat Protocols. 2014;9:2574–2585. doi: 10.1038/nprot.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang L, et al. Structural basis for inhibition of a voltage-gated Ca2+ channel by Ca2+ antagonist drugs. Nature. 2016;537:117–121. doi: 10.1038/nature19102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vergarajauregui S, Puertollano R. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic. 2006;7:337–353. doi: 10.1111/j.1600-0854.2006.00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong XP, et al. Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J Biol Chem. 2009;284:32040–32052. doi: 10.1074/jbc.M109.037184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, Saha P, Li J, Blobel G, Pfeffer SR. Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2. Proc Natl Acad Sci USA. 2016;113:10079–10084. doi: 10.1073/pnas.1611956113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pipalia NH, et al. Histone deacetylase inhibitors correct the cholesterol storage defect in most Niemann-Pick C1 mutant cells. J Lipid Res. 2017;58:695–708. doi: 10.1194/jlr.M072140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Q, et al. Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature. 2017 doi: 10.1038/nature24035. http://doi.org/10.1038/nature24035. [DOI] [PMC free article] [PubMed]
- 43.Fine M, et al. Massive endocytosis driven by lipidic forces originating in the outer plasmalemmal monolayer: a new approach to membrane recycling and lipid domains. J Gen Physiol. 2011;137:137–154. doi: 10.1085/jgp.201010469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang TM, Hilgemann DW. Ca-dependent nonsecretory vesicle fusion in a secretory cell. J Gen Physiol. 2008;132:51–65. doi: 10.1085/jgp.200709950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 2015;4:e06980. doi: 10.7554/eLife.06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohou A, Grigorieff N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rubinstein JL, Brubaker MA. Alignment of cryo-EM movies of individual particles by optimization of image translations. J Struct Biol. 2015;192:188–195. doi: 10.1016/j.jsb.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 49.Grigorieff N. Frealign: an exploratory tool for single-particle cryo-EM. Methods Enzymol. 2016;579:191–226. doi: 10.1016/bs.mie.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 53.Brown A, et al. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr D. 2015;71:136–153. doi: 10.1107/S1399004714021683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ten Eyck LF. Efficient structure-factor calculation for large molecules by the fast Fourier transform. Acta Crystallogr A. 1977;33:486–492. [Google Scholar]
- 55.Wang Z, et al. An atomic model of brome mosaic virus using direct electron detection and real-space optimization. Nat Commun. 2014;5:4808. doi: 10.1038/ncomms5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heymann JB, Belnap DM. Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol. 2007;157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 57.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 59.Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MS. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph. 1996;14:354–360. 376. doi: 10.1016/s0263-7855(97)00009-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 3D cryo-EM density maps of TRPML1 have been deposited in the Electron Microscopy Data Bank (EMDB) under the accession numbers EMD-8840 (apo TRPML1) and EMD-8841 (ML-SA1-bound TRPML1). Atomic coordinates for the atomic model have been deposited in the Protein Data Bank (PDB) under the accession numbers 5WJ5 (apo TRPML1) and 5WJ9 (ML-SA1-bound TRPML1).