


Abstract
Spinal muscular atrophy (SMA) is the leading genetic cause of infant death. We previously developed a high-throughput assay that employs an SMN2-luciferase reporter allowing identification of compounds that act transcriptionally, enhance exon recognition, or stabilize the SMN protein. We describe optimization and characterization of an analog suitable for in vivo testing. Initially, we identified analog 4m that had good in vitro properties but low plasma and brain exposure in a mouse PK experiment due to short plasma stability; this was overcome by reversing the amide bond and changing the heterocycle. Thiazole 27 showed excellent in vitro properties and a promising mouse PK profile, making it suitable for in vivo testing. This series post-translationally stabilizes the SMN protein, unrelated to global proteasome or autophagy inhibition, revealing a novel therapeutic mechanism that should complement other modalities for treatment of SMA.
Graphical abstract
INTRODUCTION
Spinal muscular atrophy (SMA) is one of the most frequent heritable causes of infant mortality that afflict all populations throughout the world.1, 2 Approximately 1 in 40 asymptomatic adults is a carrier of this autosomal recessive disease.3, 4 Infants with the most severe form of SMA (type I) typically show normal strength at birth but exhibit weakness within a few weeks or months. There is progressive motor insufficiency as the infant gains weight, and death due to respiratory failure often ensues within 2 years. Patients with types II and III SMA present weakness in early childhood, and type IV SMA is a milder, adult-onset form. These observations imply that motor neuron dysfunction and clinical symptoms develop and progress after a variable latent period. This is important as it signifies a temporal window for therapeutic intervention and stabilization or reversal of motor dysfunction.5–7
SMA was mapped to chromosome 5q13.6, 8, 9 This region contains a 500 kilobase inverted repeat and is a hotspot for chromosomal rearrangement. Within this locus are two nearly identical SMN genes: telomeric SMN1 and centromeric SMN2,6, 10, 11 which encode the identical survival motor neuron (SMN) protein. SMA carriers with one copy of SMN1 are unaffected; rather, SMA is manifested when both copies of SMN1 are deleted, disrupted, or converted to SMN2 by homologous recombination.12–15 A large amount of human mapping and correlative studies has unequivocally documented that SMA results from reduced levels of SMN protein [18–21].16–19 The SMN2 mRNA undergoes alternative splicing such that the majority (~90%) of its transcripts skip exon 7 to produce an unstable truncated SMN protein, while the remaining ~10% encode full-length SMN protein.20, 21 Although the level of SMN needed to maintain motor neurons is not known, doubling the amount of full-length SMN protein from the SMN2 gene should be clinically significant.22, 23 Several repurposed drugs, such as riluzole, phenylbutyrate, valproic acid, albuterol, and hydroxyurea, have advanced into clinical trials, but none of them have elicited convincing improvement in muscle function or survival in SMA.24, 25
There are five small molecules currently in phase I to phase III clinical trials for SMA.26 Olesoxime27 is a neuroprotective compound that retards mitochondrial permeability transition pore opening in neurons under cellular stress. After successful in vitro and extensive in vivo studies, olesoxime advanced into clinical trials for evaluation of its effects on laboratory values, vital signs, and electrocardiogram parameters in SMA patients. CK-212710728 is a fast skeletal muscle troponin activator developed by Cytokinetics in collaboration with Astellas Pharma that is currently being tested in a phase II clinical trial with SMA types II–IV patients. The other three small molecules in clinical trials upregulate SMN2-derived SMN protein through different SMA-specific pathways (Figure 1). A high-throughput screen for compounds that stimulate SMN2 transcription and subsequent structure–activity relationship (SAR) studies led to the identification of a series of 2,4-diaminoquinazolines that increased total SMN2 transcripts. After further medicinal chemistry optimization, this screen was developed into a lead compound, RG3039 (1).29 Repligen, Inc., and then Pfizer, Inc., acquired the rights to 1. While efficacy in a mouse model of SMA was observed and a phase I clinical trial suggested an acceptable safety profile, development of 1 is on hold because of unclear activity and mechanism, as elevated SMN protein levels were not detected.30, 31 PTC Therapeutics, in collaboration with Roche, developed a series of compounds that induce SMN2 exon 7 inclusion.32, 33 Clinical trials involving the lead compound, RG780032 (2), were suspended over concerns regarding an ophthalmologic side effect that developed in long-term animal studies, and it has been replaced with an alternative candidate, RG7916,34 which is currently enrolling for a phase II trial. Novartis Inc. identified a series of small molecules that increased exon 7 inclusion of SMN2, notably LMI07034 (3), through stabilization of the U1 snRNP complex. Although clinical trials were initiated to evaluate the safety of compound 3 in SMA type I patients, it presently is on hold for enrolling new patients due to toxicities in chronically exposed animals.35 Beyond small molecules, human clinical trials are evaluating the use of antisense oligonucleotides and adenovirus associated (AAV) SMN expression vectors.36–38 Nusinersen (Spinraza),39 an antisense oligonucleotide, recently became the first FDA approved therapy for SMA.
Figure 1.

Representative small molecules that have reached clinical trials for SMA treatment.
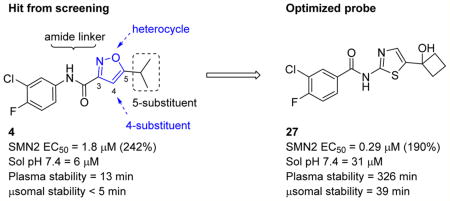
There is a clear need for multiple drug candidates with novel and distinct mechanisms of action.24, 40 We pioneered a cell-based SMN2 reporter assay to identify inducers of SMN expression, and we discovered two hit compounds: LDN-75654 (4) and LDN-76070 (5).41, 42 We demonstrated that these two series have different mechanisms of action. Compound 5 acts in a transcriptional manner, while compound 4 increases the SMN protein independent of SMN2 transcription. In vivo studies established that compound 5 increases SMN protein and the survival of severe SMA mice (SMNΔ7). Although compound 4 promoted increases in SMN expression in SMA-derived fibroblast, is compliant with Lipinski’s rules43 for drug-like molecules, and has physicochemical properties considered suitable for blood–brain barrier (BBB) penetration (MW = 283, cLogP = 3.4, PSA = 51 A2, one H-bond donor and two H-bond acceptors44–47), we found it to be inactive in vivo. Compound 4 had poor aqueous solubility (6 µM at pH 7.4) and a short half-life in mouse liver microsomes (T1/2 = 5 min). Presumably, this lack of metabolic stability and poor solubility account for its poor performance in vivo. Nevertheless, we considered 4 to be a good lead for further optimization (Figure 2). In this publication, we describe the optimization and preliminary characterization of compounds derived from isoxazole 4. The optimization of compounds derived from the 3,4-dihydroquinolinone 5 will be described elsewhere.
Figure 2.

Structures of hit molecules 4 and 5 and areas of 4 that are suitable for SAR exploration.
CHEMISTRY
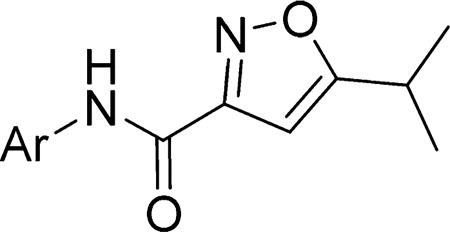
To investigate the SAR of the aryl ring, commercially available 5-isopropylisoxazole-3-carboxylic acid (6) was coupled with the appropriate aniline or amino-heterocycle in the presence of the coupling agent 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) and produced the desired amides (4–4x) in moderate to good yields (Scheme 1). Similarly, variations to the 5-position of the isoxazole were examined (e.g., 8a–i), following HATU coupling of the appropriately 5-substituted isoxazole (7a–i) with 3-chloro-4-fluoroaniline.
Scheme 1. Synthesis of 5-Isopropylisoxazole-3-carboxamides (4–4x) and 5-Substituted N-(3-Chloro-4-fluorophenyl)isoxazole-3-carboxamides (8a–i)a.

aReaction conditions and reagents: (i) ArNH2, HATU, Hunig’s base, CH2Cl2 (32–93%); (ii) 3-chloro-4-fluoroaniline, HATU, Hunig’s base, CH2Cl2 (64–89%).
The preparations of compounds with modified linkers between the aryl and isoxazole ring are depicted in Scheme 2. Reaction of the hit 4 with sodium hydride and iodomethane in THF gave the N-methylated amide (9) in 82% yield. Treatment of the amide (4) with Lawesson’s reagent proceeded smoothly to give the thioamide (10) in 75% yield. Reduction of the amide (4) to the amine (11) was accomplished in 57% yield, following lithium aluminum hydride treatment in refluxing THF. The ester (13) was prepared from the succinate ester derivative of acid (6) and reaction with 3-chloro-4-fluorophenol (12). The reverse amide analog (16) was prepared by a standard amide coupling reaction between commercially available 5-isopropylisoxazol-3-amine (14) and 3-chloro-4-fluorobenzoic acid (15) in moderate yield. The urea (19) was prepared from the commercially available isocyanate (17) and 3-chloro-4-fluoroaniline (18) in 63% yield.
Scheme 2. Syntheses of Analogs with Modified Linkersa.

aReaction conditions and reagents: (i) NaH, MeI, THF (82%); (ii) Lawesson’s reagent, toluene (75%); (iii) LiAlH4, THF, reflux (57%); (iv) N-hydroxysuccinimide, EDCI, DMAP, CH2Cl2, 0 °C (60%); (v) NaOH, 12, CH2Cl2 (93%); (vi) HATU, Hunig’s base, 15, CH2Cl2 (32%); (vii) 18, THF, reflux (63%).
The synthesis of the 4-methyl substituted isoxazole (23) is outlined in Scheme 3. Treatment of commercially available methyl 5-isopropylisoxazole-3-carboxylate (20) with NBS in DMF gave the 4-bromoisoxazole (21) in 23% yield. Palladium-catalyzed coupling of 21 with trimethylboroxine and PdCl2dppf introduced the methyl group to the 4-position, although the yield was low because of competing debromination. Hydrolysis of the ester with aqueous sodium hydroxide gave the corresponding carboxylic acid, and coupling with 3-chloro-4-fluoroaniline (18), using bromotripyrrolidinophosphonium hexafluorophosphate (PyBroP), gave the 4-methylisoxazole 23.
Scheme 3. Preparation of the 4-Methylisoxazole Analog (23)a.

aReaction conditions and reagents: (i) NBS, DMF, 60 °C (23%); (ii) C3H9B3O3, DMF, PdCl2dppf, 80 °C, (22%); (iii) 1 M NaOH (62%); (iv) PyBrop, Hunig’s base, CH2Cl2, rt (43%).
RESULTS AND DISCUSSION
Each compound was first evaluated for activity in a luciferase (luc) reporter assay that combines the SMN2 promoter, SMN exons 1–6 cDNA, followed by a splicing cassette containing introns 6, exon 7, intron 7, and exon 8.41 The terminal codon of exon 7 was changed such that when spliced onto exon 8, SMN-firefly luciferase, engineered into the latter exon, is in frame. If exon 7 is skipped, no SMN-luciferase is produced. This assay can identify compounds that increase SMN protein levels by three mechanisms: modulating alternative splicing of SMN2 exon 7; increasing transcription from the SMN2 promoter; stabilizing the full-length SMN fusion protein or mRNA. Six-point dose–response curves of each compound were generated by 3-fold serial dilutions with a final maximal concentration of 10 or 30 µM to determine EC50 values and percent maximum increases in SMN2 expression for each compound. Unless otherwise stated, all values are the mean ± SEM of at least three separate experiments. Compounds with less than 150% SMN reporter induction and >1 µM EC50 and inactive (IA) compounds defined as less than 50% SMN reporter induction were screened on two separate occasions in duplicate.
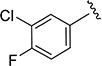
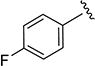
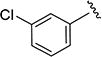
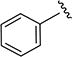
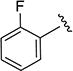
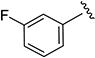
In the first series of compounds, both the 5-isopropyl group and isoxazole core were kept constant and the aryl group was modified (Table 1). Compared to compound 4, deletion of the 3-chloro had little effect on activity (4a), whereas deletion of the 4-fluoro substituent gave an increase in potency (4b), and deletion of both the 3-chloro and 4-fluoro substituents gave a significant boost in potency (4c). A fluoro substituent was well-tolerated at either the 2- or 3-position (4d or 4e), and these analogs were of comparable activity to the unsubstituted analog (4c). Conversely, substitution with other electron withdrawing or donating substituents (e.g., chloro, methyl, or methoxy 4f–l) at each of the available ring positions, in general, led to a loss in activity. On the basis of the analogs studied, unsubstituted phenyl and 2- or 3-fluorophenyl were the optimal aryl groups in terms of increasing SMN2 expression.
Table 1.
Effects of Monosubstitution on the Aryl Ring on Activity in the SMN-luc Reporter Gene Assaya
| |||||||
|---|---|---|---|---|---|---|---|
| Compd # | Ar | EC50 (µM) | % Activation | Compd # | Ar | EC50 (µM) | % Activation |
| 4 |
|
1.8 | 242 | 4a |
|
1.8 ± 0.12 | 182 ± 26 |
| 4b |
|
0.86 ± 0.13 | 160 ± 20 | 4c |
|
0.28 ± 0.03 | 190 ± 32 |
| 4d |
|
0.26 ± 0.04 | 175 ± 13 | 4e |
|
0.34 ± 0.03 | 180 ± 32 |
| 4f |
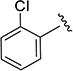
|
6.2 ± 0.3 | 114 ± 2 | 4g |
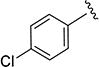
|
2.8 ± 0.61 | 168 ± 23 |
| 4h |
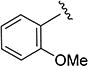
|
7.3 ± 2.4 | 80 ± 15 | 4i |
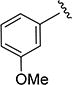
|
3.6 ± 1.0 | 147 ± 7 |
| 4j |
|
5.4 ± 0.7 | 125 ± 25 | 4k |
|
1.8 ± 0.9 | 150 ± 12 |
| 4l |
|
1.4 ± 0.3 | 200 ± 10 | ||||
Data are expressed as SEM. Compounds were analyzed in duplicate on two separate occasions, and compounds with >150% induction and EC50 < 1 µM were repeated (n = 3).
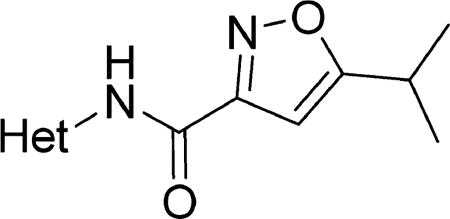
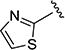
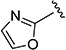
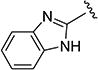
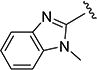
The effect of replacing the phenyl ring, by a heterocyclic ring on SMN2 expression, was next investigated (Table 2). The 5-isopropylisoxazole core and amide linker were unchanged to allow comparison between new compounds and unsubstituted phenyl analog (4c). The 2-pyridyl analog (4m) was of comparable activity as the unsubstituted phenyl analog (4c); however, the 3-pyridy analog 4n was considerably less active, and the 4-pyridyl analog (4o) was devoid of activity. The 2-thiazole analog (4q) was ~2-fold less potent than the 2-pyridyl analog (4m), although the 2-oxazole (4r) was inactive. The benzimidazole (4r) and benzothiazole (4t) analogs were of comparable activity as the 2-pyridyl analog (4m); however, the N-methylbenzimidazole (4s) and the indane (4u) were significantly less active. In general, analogs with a heteroatom at the 2-position maintained activity, but analogs with a heteroatom at the 3- or 4-position (e.g., 4n and 4m) were less active in the SMN-luc reporter gene assay.
Table 2.
Effects of Different Heterocyclic Rings on Activity in the SMN-luc Reporter Gene Assaya
| |||||||
|---|---|---|---|---|---|---|---|
| Compd # | Het | EC50 | % Activation | Compd # | Het | EC50 | % Activation |
| 4c |
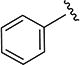
|
0.28 ± 0.03 | 190 ± 32 | 4m |
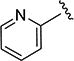
|
0.36 ± 0.09 | 195 ± 15 |
| 4n |
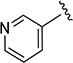
|
9.9 ± 4.2 | 94 ± 2 | 4o |
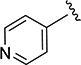
|
IA | IA |
| 4p |
|
0.78 ± 0.1 | 166 ± 23 | 4q |
|
IA | IA |
| 4r |
|
0.26 ± 0.0 | 211 ± 2 | 4s |
|
2.6 ± 0.25 | 219 ± 11 |
| 4t |
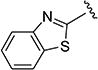
|
0.17 ± 0.01 | 124 ± 0.2 | 4u |
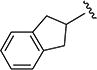
|
IA | IA |
All data are expressed as SEM. Compounds were analyzed in duplicate on two separate occasions, and compounds with >150% induction and EC50 < 1 µM were repeated (n = 3).
IA, inactive.
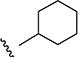
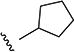
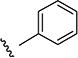
Variations to the 5-position of the isoxazole were next examined, and the effect on SMN2 expression is summarized in Table 3. Both the 3-chloro-4-fluorophenyl and the isoxazole core were retained to allow comparison between compounds. First, unsubstituted (8a), methyl (8b), and ethyl (8c) groups at the 5-position were investigated and resulted in a significant loss in activity. The more bulky sec-butyl (8d) and tert-butyl (8e) analogs were of comparable activity to 4, but the two cycloalkyl analogs (8f and 8g), the phenyl analog (8h), and the benzyl analog (8i) were not active. On the basis of the compounds studied, the original isopropyl group was the optimal 5-substituent in terms of increasing SMN2 expression.
Table 3.
Effects of the Substituent at the 5-Position of the Isoxazole on Activity in the SMN-luc Reporter Gene Assaya
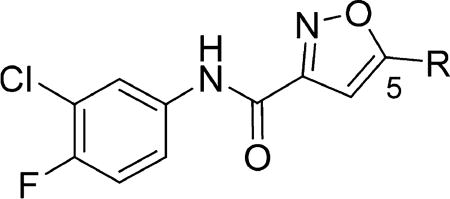
| |||||||
|---|---|---|---|---|---|---|---|
| Compd # | R | EC50 (µM) | % Activation | Compd # | R | EC50 (µM) | % Activation |
| 4 |
|
1.8 | 242 | 8a | H | IA | IA |
| 8b |
|
IA | IA | 8c |
|
8.8 ± 1.1 | 165 ± 8.4 |
| 8d |
|
1.5 ± 0.6 | 180 ± 16 | 8e |
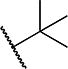
|
1.93 ± 0.7 | 140 ± 21 |
| 8f |
|
IA | IA | 8g |
|
IA | IA |
| 8h |
|
IA | IA | 8i |
|
3.56 ± 1.1 | 116 ± 15 |
Data are expressed as SEM. Compounds were analyzed in duplicate on two separate occasions, and compounds with >150% induction and EC50 < 1 µM were repeated (n = 3).
IA, inactive.
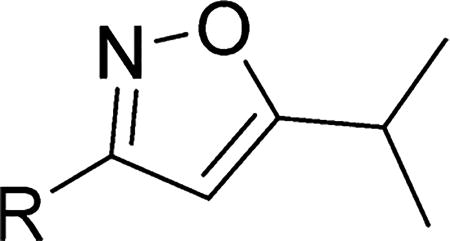
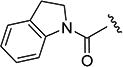
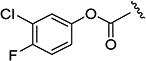
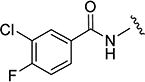
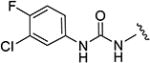
The effects of modifications to the linker between the isoxazole and the aryl group are summarized in Table 4. The 3-chloro-4-fluorophenyl and the 5-isopropylisoxazole core were kept unchanged (except for the unsubstituted indane 4v) to allow comparison between compounds in the table. N-Methylation (e.g., 9) and replacement of the amide by an ester (13) abolished activity, whereas cyclic constraint of the amide NH to the ortho-position of the aryl ring gave the indane (4v) and an improvement in activity. The activity of the indoline indicates that H-bonding is not required for activity. The thioamide (10), amine (11), reverse amide (16), and urea (19) were considerably less active than compound 4.
Table 4.
Effects of the Linker between the 5-Isopropylisoxazole and 3-Chloro-4-fluorophenyl Groups on Activity SMN-luc Reporter Gene Assaya
| |||||||
|---|---|---|---|---|---|---|---|
| Compd # |
R | EC50 (µM) | % Activation | Compd # |
R | EC50 (µM) | % Activation |
| 4 |
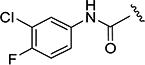
|
1.8 | 242 | 9 |
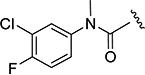
|
IA | IA |
| 10 |
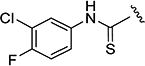
|
13.9 ± 2.4 | 120 ± 14 | 11 |
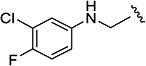
|
11.2 ± 1.2 | 137 ± 2 |
| 4v |
|
0.72 ± 0.14 | 164 ± 3.5 | 13 |
|
IA | IA |
| 16 |
|
9.9 ± 0.8 | 188 ± 13 | 19 |
|
IA | IA |
Data are expressed as SEM. Compounds were analyzed in duplicate on two separate occasions, and compounds with >150% induction and EC50 < 1 µM were repeated (n = 3).
IA, inactive.
In the final set of compounds, the effects of both reversal of the amide group and different N-linked heterocycles on activity in the SMN-luc reporter gene assay were investigated (Table 5). The reverse amide 16 (EC50 = 9.9 µM) was significantly less active than the original hit 4 (EC50 = 1.8 µM). Replacing the isoxazole core by a thiazole restored activity and the 2-amino-4-cyclobutyl analogs 26 and 27 had significantly improved activities in the SMN-luc reporter gene assay.
Table 5.
Effects of Different N-Linked Heterocycles on Activity in the SMN-luc Reporter Gene Assaya
| |||
|---|---|---|---|
| Compd # | Het | EC50 | % Activation |
| 16 |
|
9.9 ± 0.8 | 188 ± 13 |
| 24 |
|
5.5 ± 2.3 | 137 ± 13 |
| 25 |
|
1.6 ± 0.4 | 131 ± 11 |
| 26 |
|
0.5 ± 0.15 | 100 ± 16 |
| 27 |
|
0.29 ± 0.09 | 190 ± 11 |
Data are expressed as SEM. Compounds were analyzed in duplicate on two separate occasions, and compounds with >150% induction and EC50 < 1 µM were repeated (n = 3).
In order to prioritize compounds for mouse PK studies, a number of promising analogs were evaluated for stability in mouse liver microsomes and for kinetic solubility at pH 7.4 (Table 6). In comparison to lead compound 4, these analogs all displayed greater aqueous solubility; however, in the isoxazole series, only the 2-pyridyl analog (4m) had a favorable stability in mouse liver microsomes. In addition, the 5-cyclobutyl alcohol substituted thiazole (27) had good solubility and microsomal stability.
Table 6.
Physicochemical Properties, Solubility at pH 7.4, and Microsomal Stability in Mouse Liver Microsomes of Selected Compoundsa
| compd | EC50 (µM) | % activation | MW | PSA | cLogP | solubility at pH 7.4 (µM) | mouse microsome T1/2 (min) | human microsome T1/2 (min) |
|---|---|---|---|---|---|---|---|---|
| 4 | 1.8 | 242 | 282 | 51 | 3.4 | 6 | <5 | NT |
| 4c | 0.28 ± 0.03 | 190 ± 32 | 230 | 51 | 2.7 | 59 | <5 | NT |
| 23 | 0.5 ± 0.08 | 182 ± 23 | 297 | 51 | 3.9 | NT | <5 | NT |
| 4r | 0.26 ± 0.0 | 211 ± 2 | 270 | 75 | 2.4 | 12 | <5 | NT |
| 4e | 0.34 ± 0.03 | 180 ± 32 | 248 | 51 | 2.8 | 49 | <5 | NT |
| 4m | 0.36 ± 0.09 | 195 ± 15 | 231 | 63 | 2.0 | 77 | >120 | >120 |
| 26 | 0.5 ± 0.15 | 100 ± 16 | 311 | 42 | 4.8 | NT | <5 | NT |
| 27 | 0.29 ± 0.09 | 190 ± 11 | 327 | 62 | 3.4 | 31 | 39 | >120 |
NT, not tested.
On the basis of their activity in the SMN-luc reporter gene assay and promising stability in mouse liver microsomes and aqueous solubility, compounds 4m and 27 were selected for evaluation of their effects on the human SMN protein expression using fibroblasts from SMA patients. SMN genes are ubiquitously expressed, and even one copy of the human SMN1 gene will mask the effects of compounds on expression from the SMN2 gene. SMN null cells are not viable. The current standard is to use a human cell line derived from a severe SMA patient. The fibroblast strain 3813 (Coriell GM03813) is SMN1 null with three copies of SMN2. The 3814 cells from the carrier parent (SMN1 ± with five copies of SMN2) express more full-length SMN protein than 3813 cells.26 We used hTERT immortalized 3813T and 3814T cell clones that circumvent the problem of cell senescence.48
Patient 3813T fibroblasts were exposed to the indicated doses of 4m and 27. Analysis of SMN protein levels revealed a dose-dependent increase in SMN protein levels (Figure 3). Both compounds gave approximately a 2-fold increase in SMN protein levels at a dose of 2 µM.
Figure 3.

Effect of 4m and 27 on SMN protein levels in hTERT-immortalized spinal muscular atrophy patient fibroblast (GM0003813). The 3813T cells were treated with the indicated doses of 4m or 27 and DMSO (0.1% v/v). The 3814T cells were treated with DMSO. Cells were lysed after 48 h, proteins were separated by SDS–PAGE and immunoblotted with the indicated antibodies. (A, C) Representative immunoblot blotted with anti-SMN (Cell Signaling Technology; 2F1), anti-actin (Sigma-Aldrich; AC74), and anti-tubulin (Sigma-Aldrich; DM1α). (B, D) Densitometric analysis of SMN protein expression expressed as fold change DMSO control. Data are expressed as SEM (n ≥ 3): (*) P < 0.05.
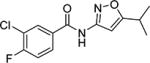
Upon these encouraging results, the synthesis of 4m was scaled up for preliminary mouse PK and efficacy experiments. Compound 4m was soluble in 40% PEG 400 + 60% water and was dosed ip to mice at 20 mg/kg. Surprisingly, 4m was detected in only very low levels in the plasma (Cmax = 2 ng/mL) and brains (Cmax = 5 ng/mL) of the mice and was below the levels of detection after 30 min (Table 7). To investigate the unexpected plasma and brain exposure, we evaluated 4m for stability in mouse plasma. Compound 4m was incubated in heparin sodium treated mouse plasma at 37 °C for 60 min. It became apparent that 4m was unstable in mouse plasma (T1/2 < 5 min). Presumably, the amide bond of compound 4m was susceptible to hydrolysis in mouse plasma. We, therefore, studied the effects of substituents located on either side of the amide bond on plasma stability (Table 8). Replacing the 2-pyridyl of 4m by phenyl (e.g., 4 or 4c) gave a very modest improvement in plasma stability, while locating a substituent on the amide nitrogen (e.g., 9 or 4v) had no effect. Introduction of a methyl substituent at the 4-position of the isoxazole (e.g., 23) resulted in a significant increase in plasma stability (T1/2 = 76 min). Presumably, the methyl group adjacent to the amide bond sterically hindered and retarded the hydrolysis. This modification resulted in a boost in activity in the SMN2 luciferase reporter gene assay, although its stability in mouse liver microsomes was poor (Table 6). Reversal of the orientation of the amide group was investigated next and resulted in a significant increase in plasma stability for both isoxazole (16, T1/2 = 76 min) and thiazole (27, T1/2 = 326 min) analogs.
Table 7.
Mouse Pharmacokinetic Data for 4m and 27
| compd | dose (mg/kg) |
plasma Cmax (ng/mL) |
plasma AUC0–24h (ng·h/mL) |
plasma Tmax (h) |
plasma T1/2 (h) |
brain Cmax (ng/mL) |
brain AUC0–24h (ng·h/mL) |
brain Tmax (h) |
brain T1/2 (h) |
B/P |
|---|---|---|---|---|---|---|---|---|---|---|
| 4m | 20 mpk ip | 3 | - | 6 | ||||||
| 27 | 20 mpk ip | 7660 | 34291 | 0.5 | 2.2 | 13500 | 51310 | 0.5 | 1.9 | 1.5/1 |
| 27 | 20 mpk po | 286 | 1641 | 0.5 | 3.4 | 446 | 2223 | 0.5 | 2.8 | 1.4/1 |
Table 8.
Effects of Modifications on Plasma Stability
| Cmpd # |
Structure | Plasma stability (T1/2) |
Cmpd # |
Structure | Plasma stability (T1/2) |
|---|---|---|---|---|---|
| 4m |
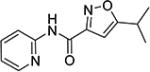
|
<5 min | 4 |
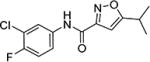
|
13 min |
| 4c |
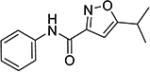
|
8 min | 9 |
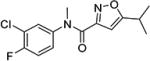
|
5 min |
| 4v |
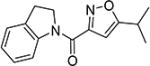
|
<5 min | 4d |
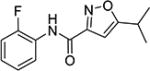
|
9 min |
| 23 |
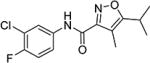
|
76 min | 16 |
|
183 min |
| 27 |
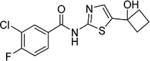
|
326 min |
Compound 27 had minimal effects on luc levels expressed from a minimal SV40 promoter and in vitro on recombinant luciferase protein (Figure 4A). It showed good activity in the SMN-luc reporter gene assay (Figure 4A) and for increasing human SMN protein expression in fibroblasts derived from SMA patients. Compound 27 had promising solubility, microsomal stability, and plasma stability and was advanced to further testing. Measurement of the plasma protein binding in mouse and human plasma indicated free fractions for 27 of 4.7% free in mouse plasma and 3.5% free in human plasma. Adequate permeability (Papp(A to B) = 15 × 10−6 cm/s) was established in the human intestinal epithelial cell line Caco-2 model. There was no effect of 27 on the viability of SH5Y5Y cells up to the maximal tested dose of 100× its EC50 after 48 h of treatment (Figure 4A). On the basis of its in vitro potency, microsomal, and plasma stability, 27 was selected for preliminary mouse PK experiments. Compound 27 demonstrated good plasma (Cmax = 23 µM) and brain (Cmax = 41 µM) levels following ip dosing at 20 mg/kg with a plasma half-life (T1/2) of 2.2 h. However, when administered orally at 20 mg/kg in adult mice, moderate plasma (Cmax = 0.9 µM) and brain (Cmax = 1.4 µM) levels were achieved (Table 7). The poor oral exposure could be due to the acid instability of 27. To evaluate this possibility, we tested the stability of 27 in 0.1 and 1 N HCL at 37 °C for 24 h. Aliquots were removed (1, 2, 4, 6, and 24 h) and analyzed by LCMS. There was less than 5% degradation of 27 at each time point. The poor oral exposure of compound 27 is likely the result of its low solubility (~10 µg/mL at pH 7.4).
Figure 4.

Exposure of SMN reporter cells to constant ratios of 4, 27, and 5. (A) SMN2, SV40 reporter cells, and SH5Y5Y were analyzed in duplicate or triplicate, with indicated concentrations of 27 for 24 or 48 h, respectively. SMN-luc (■; n = 3), Renilla luc (◆; n = 3), and SV40 luc activities (▲, n = 2) and cell viability (●; n = 3) were measured. Compound 27 was incubated with recombinant luciferase (▼, n = 2) at indicated doses, and luc activity was measured. (B) SMN reporter cells were treated with the indicated concentrations of 4 (blue) and 27 (red), individually or in combination (purple) for 24 h, and analyzed for SMN-luc activity. (C) SMN reporter cells were treated with the indicated concentrations of 5 (green) and 27 (red), individually or in combination (purple) for 24 h, and analyzed for SMN-luc activity. The theoretical additive was calculated by addition of the observed experimental activities of the individual drug exposures. Data are expressed as SEM (n ≥ 3).
To investigate whether the mechanism of 27 was similar to the parent analog 4, the effects of 27, in combination with 4 (Figure 4B) or 5 (Figure 4C), were investigated in a constant ratio experiment using the SMN2 reporter cells. We observed that combined exposure of 27 and 4 did not result in additive or above additive levels of SMN2-luc compared to their individual effects (Figure 4B). However, when cells were exposed to 27 in combination with the transcriptional activator 5, an activation above the theoretical additive was observed, indicating that these compounds act via different mechanisms (Figure 4C). We used quantitative reverse transcriptase PCR (qRT-PCR) to analyze SMN-luciferase mRNA expression from control and compound treated SMN2-luciferase cells. We previously found that 4 did not alter SMN mRNA expression or exon 7 inclusion,42 an effect that also was observed for 27 (Figure 5A). To investigate if these molecules stabilize SMN protein, we assessed whether 4 alters the SMN protein half-life. Blinded compounds (10 µM) and the proteasome inhibitor MG-132 (10 µM) were investigated for their effect on SMN protein half-life in a pulse-chase experiment. It was observed that SMN protein half-life increased by treatment with presence of 4 and MG-132 but not by the transcriptional activator 5 (Figure 5B). In a subsequent experiment to examine the effect of 4 and 27 on SMN protein stability, reporter cells were challenged simultaneously with cycloheximide (10 µM) to inhibit protein synthesis in the presence of compounds or DMSO. SMN-luc and Renilla-luc activities were measured at the indicated time points (Figure 5C, Figure 5D). Compounds 4 and 27 stabilized the SMN-luc fusion but not the renilla luciferase protein. Neither DMSO, SAHA (suberoylanilide hydroxamic acid, 10 µM), nor 5 altered stability of SMN-luc or renilla proteins. SAHA, an HDAC inhibitor, and compound 5 previously were found to increase SMN protein by increasing its transcription42, 49 and were used here as positive controls. Given the minimal activities on SV40 firefly luciferase and renilla luciferase and given that the “chase” experiments in the reporter cells, we conclude that compound 27 stabilization occurs via the SMN protein portion of the fusion and is not a result of effects on the luciferase moiety.
Figure 5.

Effect of compounds on mRNA and protein expression. (A) SMN reporter cells were analyzed for SMN-luc mRNA expression after exposure to SAHA (10 µM), 5 (10 µM), 4 (10 µM), and 27 (1, 10 µM) for 24 h. Expression of total SMN transcript and SMN exon 7-included transcript was expressed as fold change over GAPDH mRNA expression. (B) Pulse-chase analysis of endogenous SMN protein half-life in HEK293 cells. Cells were exposed with DMSO, 4 (10 µM), 5 (10 µM), or MG-132 (10 µM) for the times indicated. (C, D) SMN2 reporter cells were exposed to the indicated concentrations of SAHA, 5, 4, and 27 in combination with cycloheximide (10 µM). Cells were lysed at the indicated time points and analyzed for SMN2-luc activity (C) and Renilla-luc (D) activity. Data are expressed as SEM of n ≥ 3 independent experiments.
One possible mechanism to explain these observations is that these compounds nonspecifically block protein degradation by the proteasome. To address proteasome inhibition, we studied the effects of 27 in comparison to the proteasome inhibitor MG-132 on polyubiquitinated proteins in HEK293 cells (Figure 6). As expected, both 1 and 10 µM MG-132 increased the amount of polyubiquitinated proteins, while 27 had no effect on the basal level of polyubiquitinated proteins. To further evaluate potential changes in the amount of polyubiquitinated proteins, we pretreated HEK293 cells with 27 or MG-132 for 2 h followed by exposure to cycloheximide (10 µM) to inhibit new protein synthesis. Cycloheximide reduced basal level of polyubiquitinated proteins, an effect that was inhibited by MG-132 but not 27. Consistent with this interpretation, 27 did not alter cellular levels of short-lived proteins, including p53 and p21-Cip1, alone or after inhibition of protein synthesis with cycloheximide (Figure 7A,B). We also took advantage of the p53 in vitro degradation assay, which is a well-characterized reaction mediated by human papillomavirus (HPV) type 16 E6 and which is dependent on ubiquitination of p53 and its destruction by the proteasome.50 There was no inhibition of MBP-HPV-16 E6 mediated degradation of p53 after preincubation of the rabbit reticulocyte lysates with 27 (Figure 7C). The second classical pathway to destroy proteins is autophagy. The protein levels of microtubule-associated protein 1A/1B-light chain 3 (LC3-I and LC-3 II) and p62 levels are considered to be suitable markers of autophagic activity.51 Chloroquine was used as a positive control for autophagy inhibition and compared to the effects of 27 on the protein levels of LC3-I/II and p62 in HEK293 cells. Chloroquine led to increased p62 and LC-3 II protein levels, while 27 did not alter their levels (Figure 7D). Compound 27 also was screened in a 58 kinase panel (Nanosyn) and showed <50% inhibition at 10 µM. A similar result was found when 27 was screened in a 64 target broad panel screen (PerkinElmer). In addition, safety profiling of 27 at 10 µM showed <50% inhibition of six CYP isozymes (CYP1A2, CYP2A6, CYP2C19, CYP2C9, CYP2D6, and CYP 3A4) or hERG activity.
Figure 6.

Effect of 27 and MG-132 on the levels of polyubiquitinated proteins. HEK293 cells were pretreated with 27 (1 and 10 µM) and MG-132 (1 and 10 µM) for 2 h followed by addition of DMSO or cycloheximide (10 µM) and an additional incubation for 6 h. Final DMSO concentration was 0.2% (v/v). Cells were lysed, and proteins were separated on a gradient SDS–PAGE (4–15%). Polyubiquitinated proteins were identified using an anti-ubiquitin antibody. Tubulin was detected using an anti-tubulin antibody.
Figure 7.

Effect of 27 on targets of proteasomal or autophagic degradation. (A, B) HEK293 cells treated as indicated in corresponding schemes. Cycloheximide (CHX) was used to inhibit protein synthesis. After the indicated times, cells were lysed, and p21 and p53 proteins were identified by Western blot and normalized to tubulin. (C) p53 in vitro degradation assay using purified MBP-E6 protein. Lysates were incubated with DMSO or 27 for 30 min prior to the addition of MBP-E6 to initiate degradation of p53 protein. Western blot was used to determine p53 protein levels. (D) HEK293 cells were treated for 8 h with DMSO, 27, or chloroquine (CQ). Protein levels of the autophagy markers p62 and LC3-I/II were evaluated by immunoblot. Blots are representative results of three independent experiments.
CONCLUSION
We optimized the isoxazole series of compounds for potency and physicochemical properties that predicted good PK properties. However, the most promising in vitro compound 4m had unfavorable PK characteristics. We determined that plasma stability was the key contributor to poor brain and plasma exposure, and we attributed this outcome to hydrolysis of its amide bond. Compounds with the reverse amide group showed improved plasma stability. One of these compounds is 27, which had excellent plasma stability. This compound showed good brain and plasma exposure upon a 20 mg/kg dose by ip, while the oral availability of this compound was not favorable. This compound series stabilized SMN protein independent of changes to SMN mRNA levels and was not due to general inhibition of the proteasome, as 27 did not enrich polyubiquitinated proteins or stabilize polyubiquitinated proteins after inhibition of protein synthesis. In addition, 27 did not enrich p62 or LC-3 II levels, indicating that inhibition of autophagy is unlikely to contribute to the effects on protein stabilization. Compound 27 did not interact significantly (<50% inhibition at 10 µM) with 58 kinases, 64 other targets (e.g., GPCRs and ion channels), or six CYP isozymes. In summary, we have identified a suitably selective small molecule probe to investigate the post-translational stabilization of the SMN protein for the treatment of SMA. The in vivo efficacy of 27 in two mouse models of SMA will be reported elsewhere in due course.
EXPERIMENTAL SECTION
Materials and Antibodies
MG-132, cycloheximide, chloroquine, and DMSO were purchased from Sigma-Aldrich. Only chloroquine was solubilized in Milli-Q water. The following antibodies were used: anti-SMN (1:2000, 2F1, Cell Signaling Technologies); anti-p53 (PAb1801, Ref); p21 (F-5, Santa Cruz); anti-ubiquitin (FL-76, Santa Cruz); p62 (P0067, Sigma-Aldrich); LC3 (L7543, Sigma-Aldrich); anti-tubulin (DM1α, 1:4000, Sigma-Aldrich); and anti-β-actin (AC-74, 1:4000, Sigma-Aldrich).
Cell Culture
Human telomerase-immortalized SMA patient fibroblasts (GM003813) and parent fibroblasts (GM003814), HEK293, SH5Y-5Y cells, and SMN2 reporter cell line were cultured in high-glucose DMEM supplemented with 10% (v/v) fetal bovine serum (FBS) and 1× Pen/Strep.41, 52 Reporter cells medium was supplemented with hygromycin during maintenance. All cells were maintained at 37 and 5 °C CO2.
Reporter Assay
The SMN2-luc reporter cell line was seeded in 96-well white tissue culture plates at 25 000 cells per well and incubated overnight.41 Cells were exposed to six different doses of each compound by 3-fold serial dilutions, with a final maximal concentration of 10 or 30 µM or DMSO for 24 h. The final DMSO concentration was kept constant at 0.1% (v/v). Firefly and renilla luciferase expression was assayed with DualGlo (Promega E2920) and was measured on a PHERAstar FS microplate reader (BMG Labtech). Relative light units were normalized to DMSO control and expressed as a percentage. Unless otherwise stated, all values are the mean ± SEM of at least three separate trials. Synergy experiments were conducted as described previously with a constant DMSO of 0.2% (v/v).42 SMN-luc protein stability assays were assessed after simultaneous treatment with cycloheximide (10 µM) and DMSO, SAHA, 5, 4, or 27 (10 µM). Cells were lysed at the indicated time points and analyzed for SMN2-luc activity and Renilla-luc activity. Counterscreens were performed as described before.41, 42 In addition, recombinant luciferase activity was measured using QuantiLum recombinant luciferase according to the manufacturer’s instructions. In brief, an amount of 2 µL of compound was mixed with 1 mL of complete DMEM, and 15 µL of this mix was incubated with 15 µL of recombinant luciferase diluted at 1:10 000 in 1× passive lysis buffer with 1 mg/mL BSA. Activity was assayed with 30 µL of SteadyLite PE.
Cell Viability
Lead compounds were analyzed for their effect on cell viability of SH5Y-5Y cells. SH5Y-5Y cells were seeded at a density of 10 000 cells per well in a NuncEdge 96-well plate. On the following day, cells were treated with different doses of the compounds in duplicate and incubated for 48 h. At 4 h prior to the end of the experiment, CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega) was added and analyzed as per manufacturer’s instructions. Positive control for cell toxicity was cycloheximide (200 ng/mL), and the DMSO concentration was held constant at 0.2% (v/v).
Quantitative Reverse Transcriptase PCR (qRT-PCR)
Total mRNA was isolated using Trizol, treated with DNase I, and reversed transcribed as previously described. mRNA levels of SMN exon 7 included transcript, SMN full-length transcripts, and GAPDH were analyzed by qPCR using Sybr Green with primers previously described.41, 42, 53
SMN Protein Detection
SMN protein levels were analyzed after exposure to different doses of compounds in semiquantitative immunoblots to assess activity. Cells were seeded in 96-well plates at a density of 6000 cells per well. The next day, cells were exposed to the indicated concentrations of compounds and a final DMSO concentration, which was kept constant at 0.1% (v/v) for 48 h.
Cells Lysis and Immunoblotting
At the end of the indicated incubation periods, medium was aspirated, and cells were washed twice with cold PBS. Remaining PBS was aspirated, and cells were lysed in boiling lysis buffer (150 mM NaCl, 10 mM Tris-HCl, pH 8.0, 2% SDS, and 1× SigmaFAST EDTA free protease inhibitor cocktail). Lysates were boiled for 10 min, and insoluble particles were removed by centrifugation. Total protein concentration was measured, and proteins were separated by SDS–PAGE (10%). Protein levels were normalized to housekeeping proteins tubulin or actin as indicated and expressed as fold changes over DMSO control levels. Data are expressed as SEM and were analyzed by one-way ANOVA with post hoc analysis (Dunnett).
SMN Protein Half-Life
SMN protein half-life was determined by pulse-chase experiment in HEK293 cells with compounds 4 and 5 (10 µM) and the proteasome inhibitor MG-132 (10 µM) as previously described.54, 55
HPV16 E6 Mediated p53 Degradation Assay
MBP-16E6 and p53 proteins were purified as described.56 In brief, compound 27 or DMSO was incubated in assay buffer (25 mM Tris-HCl, pH 7.5, 100 mM NaCl, 2 mM DTT, 2.5 mM ATP) containing 1 µL of rabbit reticulocyte lysate (RRL) and p53 for 30 min. E6 proteins (0.1 µM) or E6 buffer was added, incubated for 2 h at 25 °C, and stopped by incubation at 75 °C. Proteins were separated on 10% SDS–PAGE, transferred onto PVDF membrane, and blotted for p53 using Pab1801 antibody and for 16E6 using anti-HPV-16 E6 antibody (ArborVita Inc.).
Chemistry, General
Melting points were determined using a capillary melting apparatus and were uncorrected. Elemental analyses were obtained for all new compounds and were within 0.4% of theoretical C, H, and N. 1H NMR spectra were recorded in deuteriochloroform (unless otherwise noted), with tetramethylsilane as the internal standard at 400 MHz. Coupling constants (J values) are quoted to the nearest 0.5 Hz. 13C NMR spectra were recorded in deuterated DMSO or deuteriochloroform (unless otherwise noted) at 100 MHz. Mass spectra were recorded on a VG 70SE magnetic sector mass spectrometer. Organic solutions were dried using anhydrous magnesium sulfate and concentrated by rotary evaporation. Analytical thin layer chromatography (TLC) was carried out on Camlab Polygram SIL G/UV254 plates. Unless otherwise stated, preparative column chromatography was carried out on 60H silica gel (Merck 9385). Compositions of solvent mixtures are quoted as ratios of volume. Known compounds gave spectral and analytical data consistent with literature values. Analytical HPLC, used to determine the purity of target compounds, was conducted using two different methods. Method 1: Analyses were performed using a 2790 HT-Alliance HPLC system (Waters Corporation, Milford, MA) and a Waters 996 diode array detector interfaced to a Waters Quattro Micro mass spectrometer. HPLC conditions: column, 4.6 mm × 150 mm, Waters XBridge C18, 5 µm; column temperature, 30 °C; UV, 254 nm; scan rate, 10 points/s; flow rate, 1.0 mL/min; injection volume, 20 µL; mobile phase A, water with 0.1% formic acid; mobile phase B, methanol; gradient, 10% B (0–1 min), 10–100% B (1–12 min), 100% B (12–19 min), 10% B (19.1 min); run time, 22 min. Method 2: Analyses were performed using an Acquity ultraperformance liquid chromatography (UPLC) system (Waters Corporation, Milford, MA) and a Waters Acquity PDA detector interfaced to a Waters ZQ mass spectrometer. HPLC conditions: column, 2.0 mm × 50 mm, Phenomenex Luna C18(2), 2.5 µm; column temperature, 30 °C; UV, 254 nm; scan rate, 10 points/s; flow rate, 0.6 mL/min; injection volume, 5 µL; mobile phase A, 95% water, 5% acetonitrile with 0.1% ammonium hydroxide; mobile phase B, 5% water, 95% acetonitrile with 0.1% ammonium hydroxide; gradient, 5% B (0–2 min), 5–100% B (2–7 min), 100% B (7–9.5 min), 5% B (9.51 min); run time, 10 min. All final compounds had purity of >95% by both methods.
Representative Synthesis of N-(3-Chloro-4-fluorophenyl)-3-isopropylisoxazole-5-carboxamide Using HATU (4)
5-Isopropyl-isoxazole-3-carboxylic acid (6) (50 mg, 0.32 mmol) was added to a solution of 3-chloro-4-fluoroaniline (46 mg, 0.32 mmol), N,N-diisopropylethylamine (DIPEA) (113 µL, 0.65 mmol), HATU (133 mg, 0.35 mmol) in dichloromethane (4.0 mL). The resulting mixture was stirred at room temperature for 12 h. The mixture was concentrated and purified by silica gel column chromatography (elution with a 40:60% cyclohexane/ethyl acetate gradient) and gave the title compound (4) (80 mg, 88%). MS 283 (M + 1). 1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 3.11–3.20 (m, 1H), 6.51 (s, 1H), 7.14 (t, J = 9.0 Hz, 1H), 7.45 (ddd, J = 3.0, 4.0, 9.0 Hz, 1H), 7.86 (dd, J = 3.0, 7.0 Hz, 1H), 8.48 (s, 1H). Mp 98–100.5 °C. HRMS calcd for C13H12ClFN2O2 283.0650 [M + H+], found 283.0646 [M + H+].
The following compounds were prepared using the same general procedure.
N-(4-Fluorophenyl)-5-isopropylisoxazole-3-carboxamide (4a)
1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 3.10–3.20 (m, 1H), 6.51 (s, 1H), 7.06 (t, J = 8.5 Hz, 2H), 7.62 (dd, J = 5.0, 8.5 Hz, 2H), 8.49 (s, 1H). HRMS calcd for C13H13FN2O2 249.1039 [M + H+], found 249.1034 [M + H+].
N-(3-Chlorophenyl)-5-isopropylisoxazole-3-carboxamide (4b)
1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 3.10–3.20 (m, 1H), 6.51 (s, 1H), 7.14 (ddd, J = 1.0, 2.0, 8.0 Hz, 1H), 7.29 (t, J = 8.0 Hz, 1H), 7.48 (ddd, J = 1.0, 2.0, 8.0 Hz, 1H), 7.79 (t, J = 2.0 Hz, 1H), 8.52 (s, 1H). HRMS calcd for C13H13ClN2O2 265.0744 [M + H+], found 265.0750 [M + H+].
5-Isopropyl-N-phenylisoxazole-3-carboxamide (4c)
1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 3.10–3.20 (m, 1H), 6.52 (s, 1H), 7.17 (t, J = 7.5 Hz, 1H), 7.38 (t, J = 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 8.51 (s, 1H). Mp 83.1–84.1 °C. HRMS calcd for C13H14N2O2 231.1134 [M + H+], found 231.1126 [M + H+].
N-(2-Fluorophenyl)-5-isopropylisoxazole-3-carboxamide (4d)
1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 3.10–3.20 (m, 1H), 6.51 (s, 1H), 6.84–6.89 (m, 1H), 7.26–7.34 (m, 2H), 7.63 (dt, J = 2.0, 10.5 Hz, 1H), 8.57 (s, 1H). HRMS calcd for C13H13FN2O2 249.1039 [M + H+], found 249.1041 [M + H+].
N-(3-Fluorophenyl)-5-isopropylisoxazole-3-carboxamide (4e)
1H NMR (CDCl3) δ: 1.33 (d, J = 7.0 Hz, 6H), 3.07–3.17 (m, 1H), 6.49 (s, 1H), 6.81–6.86 (m, 1H), 7.24–7.31 (m, 2H), 7.60 (dt, J = 2.0, 10.5 Hz, 1H), 8.57 (s, 1H). Mp 55.5–57.8 °C. HRMS calcd for C13H13FN2O2 249.1039 [M + H+], found 249.1034 [M + H+].
N-(2-Chlorophenyl)-5-isopropylisoxazole-3-carboxamide (4f)
1H NMR (CDCl3) δ: 1.37 (d, J = 7.0 Hz, 6H), 3.11–3.21 (m, 1H), 6.52 (s, 1H), 7.10 (td, J = 1.5, 7.5 Hz, 1H), 7.32 (td, J = 1.5, 7.5 Hz, 1H), 7.42 (dd, J = 1.5, 8.0 Hz, 1H), 8.48 (dd, J = 1.5, 8.0 Hz, 1H), 9.13 (s, 1H). HRMS calcd for C13H13ClN2O2 265.0744 [M + H+], found 265.0748 [M + H+].
N-(4-Chlorophenyl)-5-isopropylisoxazole-3-carboxamide (4g)
1H NMR (CDCl3) δ: 1.35 (d, J = 7.0 Hz, 6H), 3.09–3.20 (m, 1H), 6.50 (s, 1H), 7.33 (d, J = 9.0 Hz, 2H), 7.61 (d, J = 9.0 Hz, 2H), 8.52 (s, 1H). HRMS calcd for C13H13ClN2O2 265.0744 [M + H+], found 265.0743 [M + H+].
5-Isopropyl-N-(2-methoxyphenyl)isoxazole-3-carboxamide (4h)
1H NMR (CDCl3) δ: 1.35 (d, J = 7.0 Hz, 6H), 3.09–3.19 (m, 1H), 3.91 (s, 3H), 6.50 (s, 1H), 6.91 (dd, J = 1.0, 8.0 Hz, 1H), 6.99 (td, J = 1.0, 8.0 Hz, 1H), 7.09 (td, J = 1.5, 8.0 Hz, 1H), 8.45 (dd, J = 1.5, 8.0 Hz, 1H), 9.16 (s, 1H). HRMS calcd for C14H16N2O3 261.1239 [M + H+], found 261.1246 [M + H+].
5-Isopropyl-N-(3-methoxyphenyl)isoxazole-3-carboxamide (4i)
1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 3.10–3.20 (m, 1H), 3.83 (s, 3H), 6.51 (s, 1H), 6.72 (dd, J = 2.5, 8.0 Hz, 1H), 7.13 (dd, J = 2.0, 8.0 Hz, 1H), 7.26 (t, J = 8.0 Hz, 1H), 7.40 (t, J = 2.5 Hz, 1H), 8.49 (s, 1H). HRMS calcd for C14H16N2O3 261.1239 [M + H+], found 261.1237 [M + H+].
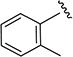
5-Isopropyl-N-(o-tolyl)isoxazole-3-carboxamide (4j)
1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 2.35 (s, 3H), 3.10–3.20 (m, 1H), 6.53 (s, 1H), 7.12 (td, J = 1.0, 7.5 Hz, 1H), 7.22–7.27 (m, 2H), 8.01 (d, J = 7.5 Hz, 1H), 8.46 (s, 1H). HRMS calcd for C14H16N2O2 245.1290 [M + H+], found 245.1296 [M + H+].
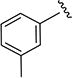
5-Isopropyl-N-(m-tolyl)isoxazole-3-carboxamide (4k)
1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 2.37 (s, 3H), 3.09–3.20 (m, 1H), 6.51 (s, 1H), 6.98 (d, J = 7.5 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 7.49 (s, 1H), 8.47 (s, 1H). HRMS calcd for C14H16N2O2 245.1290 [M + H+], found 245.1293 [M + H+].
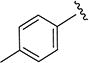
5-Isopropyl-N-(p-tolyl)isoxazole-3-carboxamide (4l)
1H NMR (CDCl3) δ: 1.35 (d, J = 7.0 Hz, 6H), 2.33 (s, 3H), 3.08–3.19 (m, 1H), 6.50 (s, 1H), 7.17 (d, J = 8.0 Hz, 2H), 7.53 (d, J = 8.5 Hz, 2H), 8.49 (s, 1H). HRMS calcd for C14H16N2O2 245.1290 [M + H+], found 245.1282 [M + H+].
5-Isopropyl-N-(pyridin-2-yl)isoxazole-3-carboxamide (4m)
1H NMR (CDCl3) δ: 1.35 (d, J = 7.0 Hz, 6H), 3.08–3.19 (m, 1H), 6.50 (s, 1H), 7.09 (ddd, J = 1.0, 5.0, 7.5 Hz, 1H), 7.75 (ddd, J = 2.0, 7.5, 8.5 Hz, 1H), 8.29 (dd, J = 1.0, 8.5 Hz, 1H), 8.33 (ddd, J = 1.0, 2.0, 5.0 Hz, 1H), 9.22 (s, 1H). HRMS calcd for C12H13N3O2 232.1086 [M + H+], found 232.1093 [M + H+].
5-Isopropyl-N-(pyridin-3-yl)isoxazole-3-carboxamide (4n)
1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 3.11–3.21 (m, 1H), 6.52 (s, 1H), 7.33 (dd, J = 4.5, 8.5 Hz, 1H), 8.23 (ddd, J = 1.5, 2.5, 8.5 Hz, 1H), 8.42 (dd, J = 1.5, 4.5 Hz, 1H), 8.58 (s, 1H), 8.75 (d, J = 2.5 Hz, 1H). HRMS calcd for C12H13N3O2 232.1086 [M + H+], found 232.1082 [M + H+].
5-Isopropyl-N-(pyridin-4-yl)isoxazole-3-carboxamide (4o)
1H NMR (CDCl3) δ: 1.34 (d, J = 7.0 Hz, 6H), 3.08–3.19 (m, 1H), 6.51 (s, 1H), 7.62 (dd, J = 1.5, 5.0 Hz, 2H), 8.55 (dd, J = 1.5, 5.0 Hz, 2H), 8.81 (s, 1H). HRMS calcd for C12H13N3O2 232.1086 [M + H+], found 232.1092 [M + H+].
5-Isopropyl-N-(thiazol-2-yl)isoxazole-3-carboxamide (4p)
1H NMR (CDCl3) δ: 1.37 (d, J = 7.0 Hz, 6H), 3.11–3.22 (m, 1H), 6.54 (s, 1H), 7.05 (d, J = 3.5 Hz, 1H), 7.69 (d, J = 3.5 Hz, 1H). HRMS calcd for C10H11N3O2S 238.0650 [M + H+], found 238.0656 [M + H+].
5-Isopropyl-N-(oxazol-2-yl)isoxazole-3-carboxamide (4q)
1H NMR (CDCl3) δ: 1.35 (d, J = 7.0 Hz, 6H), 3.09–3.20 (m, 1H), 6.53 (s, 1H), 7.21 (s, 1H), 7.51 (s, 1H). HRMS calcd for C10H11N3O3 222.0879 [M + H+], found 222.0884 [M + H+].
N-(1H-Benzo[d]imidazol-2-yl)-5-isopropylisoxazole-3-car-boxamide (4r)
1H NMR (DMSO) δ: 1.29 (d, J = 7.0 Hz, 6H), 3.09–3.20 (m, 1H), 6.58 (s, 1H), 7.19 (dd, J = 3.0, 6.0 Hz, 2H), 7.44 (dd, J = 3.0, 6.0 Hz, 2H). HRMS calcd for C14H14N4O2 271.1195 [M + H+], found 271.1201 [M + H+].
5-Isopropyl-N-(1-methyl-1H-benzo[d]imidazol-2-yl)-isoxazole-3-carboxamide (4s)
1H NMR (CDCl3) δ: 1.33 (d, J = 7.0 Hz, 6H), 3.06–3.17 (m, 1H), 3.77 (s, 3H), 6.48 (s, 1H), 7.25–7.33 (m, 3H), 7.41–7.43 (m, 1H). HRMS calcd for C15H16N4O2 285.1352 [M + H+], found 285.1350 [M + H+].
N-(Benzo[d]thiazol-2-yl)-5-isopropylisoxazole-3-carboxamide (4t)
1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 3.11–3.22 (m, 1H), 6.57 (s, 1H), 7.34 (t, J = 7.5 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 7.82 (dd, J = 8.0, 17.5 Hz, 2H). HRMS calcd for C14H13N3O2S 288.0807 [M + H+], found 288.0812 [M + H+].
N-(2,3-Dihydro-1H-inden-2-yl)-5-isopropylisoxazole-3-carboxamide (4u)
1H NMR (CDCl3) δ: 1.31 (d, J = 7.0 Hz, 6H), 2.92 (dd, J = 5.0, 16.0 Hz, 2H), 3.04–3.14 (m, 1H), 3.38 (dd, J = 7.0, 16.0 Hz, 2H), 4.87–4.93 (m, 1H), 6.42 (s, 1H), 7.02 (s, 1H), 7.17–7.20 (m, 2H), 7.21–7.24 (m, 2H). HRMS calcd for C16H18N2O2 271.1447 [M + H+], found 271.1442 [M + H+].
Indolin-1-yl(5-isopropylisoxazol-3-yl)methanone (4v)
1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 3.09–3.19 (m, 1H), 3.22 (t, J = 8.5 Hz, 2H), 4.52 (t, J = 8.5 Hz, 2H), 6.45 (s, 1H), 7.10 (t, J = 7.5 Hz, 1H), 7.24–7.28 (m, 2H), 8.31 (d, J = 8.0 Hz, 1H). HRMS calcd for C15H16N2O2 257.1290 [M + H+], found 257.1292 [M + H+].
Compounds bearing different substituents at the 5-position of the isoxazole (8a–i) were prepared following HATU coupling of the appropriately 5-substituted isoxazole with 3-chloro-4-fluoroaniline.
N-(3-Chloro-4-fluorophenyl)isoxazole-3-carboxamide (8a)
1H NMR (CDCl3) δ: 6.92 (d, J = 1.5 Hz, 1H), 7.15 (t, J = 8.5 Hz, 1H), 7.47 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 7.87 (dd, J = 2.5, 6.5 Hz, 1H), 8.51–8.54 (m, 2H). HRMS calcd for C10H6ClFN2O2 241.0180 [M + H+], found 241.0177 [M + H+].
N-(3-Chloro-4-fluorophenyl)-5-methylisoxazole-3-carboxamide (8b)
1H NMR (CDCl3) δ: 2.51 (s, 3H), 6.51 (s, 1H), 7.12 (t, J = 9.0 Hz, 1H), 7.45 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 7.85 (dd, J = 2.5, 6.5 Hz, 1H), 8.53 (s, 1H). HRMS calcd for C11H8ClFN2O2 255.0337 [M + H+], found 255.0340 [M + H+].
N-(3-Chloro-4-fluorophenyl)-5-ethylisoxazole-3-carboxamide (8c)
1H NMR (CDCl3) δ: 1.35 (t, J = 7.5 Hz, 3H), 2.85 (q, J = 7.5 Hz, 2H), 6.52 (s, 1H), 7.13 (t, J = 9.0 Hz, 1H), 7.45 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 7.86 (dd, J = 2.5, 6.5 Hz, 1H), 8.51 (s, 1H). HRMS calcd for C12H10ClFN2O2 269.0493 [M + H+], found 269.0498 [M + H+].
N-(3-Chloro-4-fluorophenyl)-5-isobutylisoxazole-3-carboxamide (8d)
1H NMR (CDCl3) δ: 1.00 (d, J = 6.5 Hz, 6H), 2.03–2.14 (m, 1H), 2.71 (d, J = 7.0 Hz, 2H), 6.53 (s, 1H), 7.14 (t, J = 9.0 Hz, 1H), 7.46 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 7.86 (dd, J = 2.5, 6.5 Hz, 1H), 8.52 (s, 1H). HRMS calcd for C14H14ClFN2O2 297.0806 [M + H+], found 297.0801 [M + H+].
5-(tert-Butyl)-N-(3-chloro-4-fluorophenyl)isoxazole-3-carboxamide (8e)
1H NMR (CDCl3) δ: 1.39 (s, 9H), 6.49 (s, 1H), 7.14 (t, J = 9.0 Hz, 1H), 7.46 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 7.85 (dd, J = 2.5, 6.5 Hz, 1H), 8.50 (s, 1H). HRMS calcd for C14H14ClFN2O2 297.0806 [M + H+], found 297.0811 [M + H+].
N-(3-Chloro-4-fluorophenyl)-5-cyclohexylisoxazole-3-carboxamide (8f)
1H NMR (CDCl3) δ: 1.25–1.34 (m, 1H), 1.37–1.5 (m, 4H), 1.72–1.77 (m, 1H), 1.81–1.86 (m, 2H), 2.07–2.11 (m, 2H), 2.83–2.88 (m, 1H), 6.49 (s, 1H), 7.14 (t, J = 9.0 Hz, 1H), 7.45 (ddt, J = 2.0, 4.0, 9.0 Hz, 1H), 7.86 (dd, J = 2.5, 6.5 Hz, 1H), 8.48 (s, 1H). HRMS calcd for C16H16ClFN2O2 323.0963 [M + H+], found 323.0957 [M + H+].
N-(3-Chloro-4-fluorophenyl)-5-cyclopentylisoxazole-3-carboxamide (8g)
1H NMR (CDCl3) δ: 1.70–1.82 (m, 6H), 2.10–2.17 (m, 2H), 3.24–3.30 (m, 1H), 6.50 (s, 1H), 7.14 (t, J = 9.0 Hz, 1H), 7.45 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 7.86 (dd, J = 2.5, 6.5 Hz, 1H), 8.50 (s, 1H). HRMS calcd for C15H14ClFN2O2 309.0806 [M + H+], found 309.0811 [M + H+].
N-(3-Chloro-4-fluorophenyl)-5-phenylisoxazole-3-carboxamide (8h)
1H NMR (DMSO) δ: 7.46 (t, J = 9.0 Hz, 1H), 7.51 (s, 1H), 7.56–7.60 (m, 3H), 7.79 (ddd, J = 2.5, 4.5, 9.0 Hz, 1H), 7.97–7.99 (m, 2H), 8.09 (dd, J = 2.5, 7.0 Hz, 1H), 11.0 (s, 1H). HRMS calcd for C16H10ClFN2O2 317.0493 [M + H+], found 317.0499 [M + H+].
5-Benzyl-N-(3-chloro-4-fluorophenyl)isoxazole-3-carboxamide (8i)
1H NMR (CDCl3) δ: 4.16 (s, 2H), 6.48 (s, 1H), 7.13 (t, J = 9.0 Hz, 1H), 7.27–7.37 (m, 5H), 7.44 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 7.83 (dd, J = 2.5, 6.5 Hz, 1H), 8.46 (s, 1H). HRMS calcd for C17H12ClFN2O2 331.0650 [M + H+], found 331.0654 [M + H+].
N-(3-Chloro-4-fluorophenyl)-5-isopropyl-N-methylisoxa-zole-3-carboxamide (9)
Under an inert environment, N-(3-chloro-4-fluorophenyl)-5-isopropylisoxazole-3-carboxamide (50 mg, 0.177 mmol) was dissolved in DMF (2.2 mL). Cesium carbonate (183 mg, 0.562 mmol) then was added before the dropwise addition of iodomethane (16.9 µL, 0.271 mmol). The reaction was stirred for 24 h at room temperature and concentrated down after the product was observed. The solution was purified by silica gel column chromatography (elution with a 75:25% cyclohexane/ethyl acetate gradient) to yield N-(3-chloro-4-fluorophenyl)-5-isopropyl-N-methylisoxazole-3-carboxamide (9). 1H NMR (DMSO) δ: 1.17 (br, 6H), 2.95–3.1 (br, 1H), 3.35 (s, 3H), 6.29 (s, 1H), 7.36 (br, 2H) 7.65 (s, 1H). HRMS calcd for C14H14ClFN2O2 297.0806 [M + H+], found 297.0806 [M + H+].
N-(3-Chloro-4-fluorophenyl)-5-isopropylisoxazole-3-carbo-thioamide (10)
N-(3-Chloro-4-fluorophenyl)-5-isopropylisoxazole-3-carboxamide 4 (50 mg, 0.18 mmol) was dissolved in anhydrous toluene (2 mL) under nitrogen. 2,4-Bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (36 mg, 0.09 mmol) and sodium bicarbonate (15 mg, 0.18 mmol) were added to the solution. The reaction was refluxed for 24 h. The reaction was washed with sodium bicarbonate and extracted with ethyl acetate. The organic layer then was washed with saturated sodium chloride, dried over sodium sulfate, and concentrated. The crude product was purified via silica gel column chromatography (elution with a 0:100% cyclohexane/ethyl acetate gradient) to give N-(3-chloro-4-fluorophenyl)-5-isopropylisoxazole-3-carbothioamide (10). 1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 3.07–3.18 (m, 1H), 6.67 (s, 1H), 7.20 (t, J = 9.0 Hz, 1H), 7.66 (ddd, J = 3.0, 4.0, 9.0 Hz, 1H), 8.08 (dd, J = 2.5, 6.5 Hz, 1H), 10.03 (s, 1H). HRMS calcd for C13H12ClFN2O2S 299.0421 [M + H+], found 299.0419 [M + H+].
3-Chloro-4-fluoro-N-((5-isopropylisoxazol-3-yl)methyl)-aniline (11)
N-(3-Chloro-4-fluorophenyl)-5-isopropylisoxazole-3-carboxamide (100 mg, 0.35 mmol) was dissolved in tetrahydrofuran (4.5 mL) before the addition of lithium aluminum hydride (20.2 mg, 0.53 mmol). The reaction was refluxed for 24 h. Once complete, the reaction was quenched with water. The product was extracted with ether, concentrated down, and purified by silica gel column chromatography (elution with a 30:70% cyclohexane/ethyl acetate gradient) to give 3-chloro-4-fluoro-N-((5-isopropylisoxazol-3-yl)-methyl)aniline (11). 1H NMR (CDCl3) δ: 1.29 (d, J = 7.0 Hz, 6H), 3.0–3.10 (m, 1H), 4.31 (s, 2H), 5.91 (s, 1H), 6.51 (ddd, J = 3.0, 3.5, 9.0 Hz, 1H), 6.67 (dd, J = 3.0, 6.0 Hz, 1H), 6.96 (t, J = 9.0 Hz, 1H). HRMS calcd for C13H14ClFN2O 269.0857 [M + H+], found 269.0859 [M + H+].
3-Chloro-4-fluorophenyl-5-isopropylisoxazole-3-carboxylate (13)
5-Isopropylisoxazole-3-carboyxlic acid 6 (250 mg, 1.61 mmol), N-hydroxysuccinimide (276 mg, 2.41 mmol), and dimethylaminopyridine (15 µL) were dissolved in dichloromethane (10 mL). The solution was cooled to 0 °C, and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) (372 mg, 2.41 mmol) was added. The mixture was stirred for 12 h, warming to room temperature before washing with water (2 × 10 mL) and saturated ammonium chloride (10 mL). The solution was dried over magnesium sulfate and evaporated to give 2,5-dioxopyrrolidin-1-yl 5-isopropylisoxazole-3-carboxylate (245 mg, 60%), which was used directly in the next step. 3-Chloro-4-fluorophenol (12) (60 mg, 0.40 mmol) and sodium hydroxide (48 mg, 0.60 mmol) were dissolved in dichloromethane (2 mL) and stirred for 15 min. The resulting solution was added to 2,5-dioxopyrrolidin-1-yl-5-isopropylisoxazole-3-carboxylate (100 mg, 0.40 mmol) in dichloromethane (2 mL). The solution was stirred at room temperature for 24 h. The solution was evaporated and purified by silica gel column chromatography (elution with a 0:100% cyclohexane/ethyl acetate gradient) to give the title compound (13) (94 mg, 93%). MS 284 (M + 1). 1H NMR (CDCl3) δ: 1.36 (d, J = 7.0 Hz, 6H), 3.12–3.23 (m, 1H), 6.50 (s, 1H), 7.11–7.21 (m, 2H), 7.33 (dd, J = 2.5, 6.0 Hz, 1H). HRMS calcd for C13H11ClFNO3 284.0490 [M + H+], found 284.0488 [M + H+].
3-Chloro-4-fluoro-N-(5-isopropylisoxazol-3-yl)benzamide (16)
16 was prepared by the representative HATU procedure described above. MS 283 (M + 1). 1H NMR (CDCl3) δ: 1.31 (d, J = 7.0 Hz, 6H), 3.0–3.10 (m, 1H), 6.42 (s, 1H), 7.30 (t, J = 8.5 Hz, 1H), 7.79 (ddd, J = 2.5, 4.5, 8.5 Hz, 1H), 8.0 (dd, J = 2.5, 7.0 Hz, 1H), 8.61 (s, 1H). HRMS calcd for C13H12ClFN2O2 283.0650 [M + H+], found 283.0654 [M + H+].
1-(3-Chloro-4-fluorophenyl)-3-(5-isopropylisoxazol-3-yl)-urea (19)
A stirred mixture of 3-chloro-4-fluoroaniline (36 mg, 0.25 mmol) and 5-isopropyl-3-isoxazolyl isocyanate (38 mg, 0.25 mmol) in THF (1 mL) was heated at 50 °C for 15 h. After cooling, the reaction was concentrated under reduced pressure and purified by silica gel flash chromatography (elution with a 0:100% cyclohexane/ethyl acetate gradient) to give the title compound (19) (47 mg, 63%). 1H NMR (CDCl3) δ: 1.33 (d, J = 7.0 Hz, 6H), 3.02–3.08 (m, 1H), 5.83 (s, 1H), 7.10 (t, J = 9.0 Hz, 1H), 7.32 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 7.74 (dd, J = 2.5, 6.5 Hz, 1H), 7.84 (s, 1H), 9.41 (s, 1H). HRMS calcd for C13H13ClFN3O2 298.0759 [M + H+], found 298.0756 [M + H+].
Methyl 4-Bromo-5-isopropylisoxazole-3-carboxylate (21)
Methyl 5-isopropyl-3-isoxazolecarboxylate (20) (625 mg, 3.7 mmol) and N-bromosuccinimide (7.92 g, 22.1 mmol) were dissolved in DMF (12 mL) and stirred at room temperature for 12 h. The mixture then was washed with sodium thiosulfate (3 × 10 mL) and ethyl acetate (10 mL) and dried over sodium sulfate. The mixture was concentrated and purified by silica gel column chromatography (elution with a 90:10% cyclohexane/ethyl acetate gradient) to give the title compound (21) (212 mg, 23%). MS 348 (M + 1). 1H NMR (CDCl3) δ: 1.34 (d, J = 7.0 Hz, 6H), 3.21–3.32 (m, 1H), 3.96 (s, 3H).
Methyl 5-Isopropyl-4-methylisoxazole-3-carboxylate (22)
Methyl 4-bromo-5-isopropylisoxazole-3-carboxylate (21) (414 mg, 1.67 mmol), trimethylboroxine (0.70 mL, 5.0 mmol), and potassium carbonate (689 mg, 5.01 mmol) were dissolved in dry DMF (6 mL) and degassed under argon. [1,1′-Bis(diphenylphosphino)ferrocene]-dichloropalladium(II) complex with dichloromethane (PdCl2dppf) (122 mg, 0.17 mmol) was added. The resulting mixture was heated to 80 °C and stirred under argon for 48 h. The mixture was washed with ethyl acetate (2 × 10 mL) and saturated sodium chloride solution (2 × 10 mL), dried over sodium sulfate, and concentrated. The mixture was purified by five (20 cm × 20 cm, 2000 µm) preparative TLC plates and run in 80:20% cyclohexane/ethyl acetate solution to give the title compound (22) (65 mg, 22%). MS 184 (M + 1). 1H NMR (CDCl3) δ: 1.31 (d, J = 7.0 Hz, 6H), 2.14 (s, 3H), 3.10–3.18 (m, 1H), 3.94 (s, 3H).
3-Chloro-4-fluorophenyl-5-isopropyl-4-methylisoxazole-3-carboxamide (23)
Methyl 5-isopropyl-4-methylisoxazole-3-carboxylate (22) (65 mg, 0.36 mmol) was dissolved in water (3.4 mL). Sodium hydroxide (15.6 mg, 0.39 mmol) was added, and the resulting mixture was stirred at room temperature for 12 h. 0.1 M hydrochloric acid was added until the reaction mixture was at pH 1. The mixture was extracted with ethyl acetate (3 × 5 mL) and concentrated to give the crude carboxylic acid (37 mg, 62%) that was used directly in the next step.
N,N-Diisopropylethylamine (DIPEA) (0.08 mL, 0.44 mmol) was added dropwise to a solution of bromotripyrrolidinophosphonium hexafluorophosphate (PyBroP) (122.6 mg, 0.26 mmol) in tetrahydrofuran (1.1 mL). Crude 5-isopropyl-4-methylisoxazole-3-carboxylic acid (37 mg, 0.22 mmol) and 3-chloro-4-fluoroaniline (31.83 mg, 0.22 mmol) were added to the resulting solution, and the mixture was stirred at 75 °C for 12 h. The mixture was washed with a saturated solution of sodium bicarbonate (2 × 5 mL), ethyl acetate (2 × 5 mL), and saturated sodium chloride solution (2 × 5 mL). The mixture was dried over sodium sulfate, concentrated, and purified via silica gel column chromatography (elution with a 90:10% cyclohexane/ethyl acetate gradient) to give the title compound (23) (28 mg, 43%). MS 297 (M + 1). 1H NMR (CDCl3) δ: 1.34 (d, J = 7.0 Hz, 6H), 2.24 (s, 3H), 3.12–3.23 (m, 1H), 7.13 (t, J = 9.0 Hz, 1H), 7.41 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 7.88 (dd, J = 2.5, 6.5 Hz, 1H), 8.50 (s, 1H). HRMS calcd for C14H14ClFN2O2 297.0806 [M + H+], found 297.0812 [M + H+].
Compounds with different core heterocycles (24–27) were prepared following HATU coupling of the appropriate heterocyclic carboxylic acid with 3-chloro-4-fluoroaniline.
3-Chloro-4-fluoro-N-(3-isopropylisoxazol-5-yl)benzamide (24)
1H NMR (CDCl3) δ: 1.31 (d, J = 7.0 Hz, 6H), 3.00–3.10 (m, 1H), 6.42 (s, 1H), 7.30 (t, J = 8.5 Hz, 1H), 7.80 (ddd, J = 2.5, 4.5, 8.5 Hz, 1H), 8.00 (dd, J = 2.5, 7.0 Hz, 1H), 8.66 (s, 1H). HRMS calcd for C13H12ClFN2O2 283.0650 [M + H+], found 283.0650 [M + H+].
3-Chloro-4-fluoro-N-(2-isopropylthiazol-4-yl)benzamide (25)
1H NMR (CDCl3) δ: 1.45 (d, J = 7.0 Hz, 6H), 3.30–3.36 (m, 1H), 7.13 (t, J = 9.0 Hz, 1H), 7.55 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 7.90 (dd, J = 2.5, 6.5 Hz, 1H), 8.08 (s, 1H), 9.20 (s, 1H). HRMS calcd for C13H12ClFN2OS 299.0421 [M + H+], found 299.0427 [M + H+].
3-Chloro-N-(5-cyclobutylthiazol-2-yl)-4-fluorobenzamide (26)
1H NMR (CDCl3) δ: 1.92–1.97 (m, 1H), 2.01–2.07 (m, 1H), 2.13–2.21 (m, 2H), 2.40–2.46 (m, 2H), 3.63–3.69 (m, 1H), 6.87 (s, 1H), 7.23–7.31 (m, 1H), 7.99 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 8.18 (dd, J = 2.5, 7.0 Hz, 1H). HRMS calcd for C14H12ClFN2OS 311.0421 [M + H+], found 311.0421 [M + H+].
3-Chloro-4-fluoro-N-(5-(1-hydroxycyclobutyl)thiazol-2-yl)-benzamide (27)
1H NMR (DMSO) δ: 1.62–1.71 (m, 1H), 1.78–1.84 (m, 1H), 2.30–2.39 (m, 4H), 5.97 (s, 1H), 7.47 (s, 1H), 7.60 (t, J = 9.0 Hz, 1H), 8.10 (ddd, J = 2.5, 4.0, 9.0 Hz, 1H), 8.33 (d, J = 7.0 Hz, 1H), 12.62 (s, 1H). HRMS calcd for C14H12ClFN2O2S 327.0370 [M + H+], found 327.0364 [M + H+].
Supplementary Material
Acknowledgments
The authors are supported by grants from the National Institutes of Health (Grants R01 HD064850, R21 NS064349, R21 HD057402), FightSMA, and Gwendolyn Strong Foundation.
ABBREVIATIONS USED
- AAV
adenovirus associated
- BBB
blood–brain barrier
- CHX
cycloheximide
- CQ
chloroquine
- DIPEA
N,N-diisopropylethylamine
- DMSO
dimethyl sulfoxide
- FBS
fetal bovine serum
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo-[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HDAC
histone deacetylase
- HPV
human papillomavirus
- IA
inactive
- Luc
luciferase
- NA
not applicable
- PyBroP
bromotripyrrolidinophosphonium hexafluorophosphate
- qRT-PCR
quantitative reverse transcriptase PCR
- RRL
rabbit reticulocyte lysate
- SMA
spinal muscular atrophy
- SMN
survival motor neuron
- SAHA
suberoylanilide hydroxamic acid
- SAR
structure–activity relationship
- T1/2
half-life
- TLC
thin layer chromatography
- UPLC
ultraperformance liquid chromatography
Footnotes
ASSOCIATED CONTENT
- Molecular formula strings and some data (CSV)
The authors declare no competing financial interest.
References
- 1.McAndrew PE, Parsons DW, Simard LR, Rochette C, Ray PN, Mendell JR, Prior TW, Burghes AH. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997;60:1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearn J. Classification of spinal muscular atrophies. Lancet. 1980;315:919–922. doi: 10.1016/s0140-6736(80)90847-8. [DOI] [PubMed] [Google Scholar]
- 3.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frezal M, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 4.Wirth B, Schmidt T, Hahnen E, Rudnik-Schöneborn S, Krawczak M, Müller-Myhsok B, Schönling J, Zerres K. De novo rearrangements found in 2% of index patients with spinal muscular atrophy: Mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am. J. Hum. Genet. 1997;61:1102–1111. doi: 10.1086/301608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jablonka S, Wiese S, Sendtner M. Axonal defects in mouse models of motoneuron disease. J. Neurobiol. 2004;58:272–286. doi: 10.1002/neu.10313. [DOI] [PubMed] [Google Scholar]
- 6.Melki J, Lefebvre S, Burglen L, Burlet P, Clermont O, Millasseau P, Reboullet S, Benichou B, Zeviani M, Le Paslier D, Cohen D, Weissenbach J, Munnich A. De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Science. 1994;264:1474–1477. doi: 10.1126/science.7910982. [DOI] [PubMed] [Google Scholar]
- 7.Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Brzustowicz LM, Lehner T, Castilla LH, Penchaszadeh GK, Wilhelmsen KC, Daniels R, Davies KE, Leppert M, Ziter F, Wood D, Dubowitz V, Zerres K, Hausmanowa-Petrusewicz I, Ott J, Munsat TL, Gilliam TC. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2–13.3. Nature. 1990;344:540–541. doi: 10.1038/344540a0. [DOI] [PubMed] [Google Scholar]
- 9.Melki J, Abdelhak S, Sheth P, Bachelot M, Burlet P, Marcadet A, Aicardi J, Barois A, Carriere J, Fardeau M, Fontan D, Ponsot G, Billette T, Angelini C, Barbosa C, Ferriere C, Lanzi G, Ottolini A, Babron M, Cohen D, Hanauer A, Clerget-Darpoux F, Lathrop M, Munnich A, Frezal J. Gene for chronic spinal muscular atrophies maps to chromosome 5q. Nature. 1990;344:767–768. doi: 10.1038/344767a0. [DOI] [PubMed] [Google Scholar]
- 10.Echaniz-Laguna A, Guiraud-Chaumeil C, Tranchant C, Reeber A, Melki J, Warter JM. Homozygous exon 7 deletion of the SMN centromeric gene (SMN2): a potential susceptibility factor for adult-onset lower motor neuron disease. J. Neurol. 2002;249:290–293. doi: 10.1007/s004150200007. [DOI] [PubMed] [Google Scholar]
- 11.Melki J. Spinal muscular atrophy. Curr. Opin. Neurol. 1997;10:381–385. doi: 10.1097/00019052-199710000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Boda B, Mas C, Giudicelli C, Nepote V, Guimiot F, Levacher B, Zvara A, Santha M, LeGall I, Simonneau M. Survival motor neuron SMN1 and SMN2 gene promoters: identical sequences and differential expression in neurons and non-neuronal cells. Eur. J. Hum. Genet. 2004;12:729–737. doi: 10.1038/sj.ejhg.5201217. [DOI] [PubMed] [Google Scholar]
- 13.Burglen L, Lefebvre S, Clermont O, Burlet P, Viollet L, Cruaud C, Munnich A, Melki J. Structure and organization of the human survival motor neuron (SMN) gene. Genomics. 1996;32:479–482. doi: 10.1006/geno.1996.0147. [DOI] [PubMed] [Google Scholar]
- 14.Echaniz-Laguna A, Miniou P, Bartholdi D, Melki J. The promoters of the survival motor neuron gene (SMN) and its copy (SMNc) share common regulatory elements. Am. J. Hum. Genet. 1999;64:1365–1370. doi: 10.1086/302372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lefebvre S, Bürglen L, Frézal J, Munnich A, Melki J. The role of the SMN gene in proximal spinal muscular atrophy. Hum. Mol. Genet. 1998;7:1531–1536. doi: 10.1093/hmg/7.10.1531. [DOI] [PubMed] [Google Scholar]
- 16.Crawford TO, Paushkin SV, Kobayashi DT, Forrest SJ, Joyce CL, Finkel RS, Kaufmann P, Swoboda KJ, Tiziano D, Lomastro R, Li RH, Trachtenberg FL, Plasterer T, Chen KS. Pilot study of biomarkers for spinal muscular atrophy trial group. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS One. 2012;7:e33572. doi: 10.1371/journal.pone.0033572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 18.Sleigh JN, Gillingwater TH, Talbot K. The contribution of mouse models to understanding the pathogenesis of spinal muscular atrophy. Dis. Models & Mech. 2011;4:457–467. doi: 10.1242/dmm.007245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wee CD, Kong L, Sumner CJ. The genetics of spinal muscular atrophies. Curr. Opin. Neurol. 2010;23:450–458. doi: 10.1097/WCO.0b013e32833e1765. [DOI] [PubMed] [Google Scholar]
- 20.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U. S. A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AHM, McPherson JD. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 22.Meyer K, Marquis J, Trub J, Nlend Nlend R, Verp S, Ruepp MD, Imboden H, Barde I, Trono D, Schumperli D. Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum. Mol. Genet. 2009;18:546–555. doi: 10.1093/hmg/ddn382. [DOI] [PubMed] [Google Scholar]
- 23.Zanetta C, Nizzardo M, Simone C, Monguzzi E, Bresolin N, Comi GP, Corti S. Molecular therapeutic strategies for spinal muscular atrophies: current and future clinical trials. Clin. Ther. 2014;36:128–140. doi: 10.1016/j.clinthera.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 24.Cherry JJ, Androphy EJ. Therapeutic strategies for the treatment of spinal muscular atrophy. Future Med. Chem. 2012;4:1733–1750. doi: 10.4155/fmc.12.107. [DOI] [PubMed] [Google Scholar]
- 25.Calder AN, Androphy EJ, Hodgetts KJ. Small molecules in development for the treatment of spinal muscular atrophy. J. Med. Chem. 2016;59:10067–10083. doi: 10.1021/acs.jmedchem.6b00670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cherry JJ, Calder AN, Hodgetts KJ, Androphy EJ. Small Molecule Approaches to Upregulate SMN Expression from the SMN2 Locus. Academic Press; San Diego, CA: 2017. [Google Scholar]
- 27.Bordet T, Buisson B, Michaud M, Drouot C, Galea P, Delaage P, Akentieva NP, Evers AS, Covey DF, Ostuni MA, Lacapere JJ, Massaad C, Schumacher M, Steidl EM, Maux D, Delaage M, Henderson CE, Pruss RM. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007;322:709–720. doi: 10.1124/jpet.107.123000. [DOI] [PubMed] [Google Scholar]
- 28.Hwee DT, Kennedy AR, Hartman JJ, Ryans J, Durham N, Malik FI, Jasper JR. The small-molecule fast skeletal troponin activator, CK-2127107, improves exercise tolerance in a rat model of heart failure. J. Pharmacol. Exp. Ther. 2015;353:159–168. doi: 10.1124/jpet.114.222224. [DOI] [PubMed] [Google Scholar]
- 29.Jarecki J, Chen X, Bernardino A, Coovert DD, Whitney M, Burghes A, Stack J, Pollok BA. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: Early leads towards a therapeutic for spinal muscular atrophy. Hum. Mol. Genet. 2005;14:2003–2018. doi: 10.1093/hmg/ddi205. [DOI] [PubMed] [Google Scholar]
- 30.Gogliotti RG, Cardona H, Singh J, Bail S, Emery C, Kuntz N, Jorgensen M, Durens M, Xia B, Barlow C, Heier CR, Plasterer HL, Jacques V, Kiledjian M, Jarecki J, Rusche J, DiDonato CJ. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum. Mol. Genet. 2013;22:4084–4101. doi: 10.1093/hmg/ddt258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Meerbeke JP, Gibbs RM, Plasterer HL, Miao W, Feng Z, Lin MY, Rucki AA, Wee CD, Xia B, Sharma S, Jacques V, Li DK, Pellizzoni L, Rusche JR, Ko CP, Sumner CJ. The DcpS inhibitor RG3039 improves motor function in SMA mice. Hum. Mol. Genet. 2013;22:4074–4083. doi: 10.1093/hmg/ddt257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, Ling KK, Karp GM, Qi H, Woll MG, Chen G, Zhang N, Gabbeta V, Vazirani P, Bhattacharyya A, Furia B, Risher N, Sheedy J, Kong R, Ma J, Turpoff A, Lee CS, Zhang X, Moon YC, Trifillis P, Welch EM, Colacino JM, Babiak J, Almstead NG, Peltz SW, Eng LA, Chen KS, Mull JL, Lynes MS, Rubin LL, Fontoura P, Santarelli L, Haehnke D, McCarthy KD, Schmucki R, Ebeling M, Sivaramakrishnan M, Ko CP, Paushkin SV, Ratni H, Gerlach I, Ghosh A, Metzger F. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345:688–693. doi: 10.1126/science.1250127. [DOI] [PubMed] [Google Scholar]
- 33.Ratni H, Karp GM, Weetall M, Naryshkin NA, Paushkin SV, Chen KS, McCarthy KD, Qi H, Turpoff A, Woll MG, Zhang X, Zhang N, Yang T, Dakka A, Vazirani P, Zhao X, Pinard E, Green L, David-Pierson P, Tuerck D, Poirier A, Muster W, Kirchner S, Mueller L, Gerlach I, Metzger F. Specific correction of alternative survival motor neuron 2 splicing by small molecules: Discovery of a potential novel medicine to treat spinal muscular atrophy. J. Med. Chem. 2016;59:6086–6100. doi: 10.1021/acs.jmedchem.6b00459. [DOI] [PubMed] [Google Scholar]
- 34.Hoffmann-La Roche. A study to investigate the safety, tolerability, pharmacokinetics and pharmacodynamics of RO7034067 (RG7916) given by mouth in healthy volunteers. National Library of Medicine; Bethesda, MD, U.S: Dec 15, 2015. [cited March 1, 2017]. ClinicalTrials.gov, Identifier: NCT02633709. https://clinicaltrials.gov/ct2/show/NCT02633709. [Google Scholar]
- 35.Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, Van Hoosear M, Shin Y, Chin DN, Keller CG, Beibel M, Renaud NA, Smith TM, Salcius M, Shi X, Hild M, Servais R, Jain M, Deng L, Bullock C, McLellan M, Schuierer S, Murphy L, Blommers MJ, Blaustein C, Berenshteyn F, Lacoste A, Thomas JR, Roma G, Michaud GA, Tseng BS, Porter JA, Myer VE, Tallarico JA, Hamann LG, Curtis D, Fishman MC, Dietrich WF, Dales NA, Sivasankaran R. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 2015;11:511–517. doi: 10.1038/nchembio.1837. [DOI] [PubMed] [Google Scholar]
- 36.Benkhelifa-Ziyyat S, Besse A, Roda M, Duque S, Astord S, Carcenac R, Marais T, Barkats M. Intramuscular scAAV9-SMN injection mediates widespread gene delivery to the spinal cord and decreases disease severity in SMA mice. Mol. Ther. 2013;21:282–290. doi: 10.1038/mt.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glascock JJ, Osman EY, Wetz MJ, Krogman MM, Shababi M, Lorson CL. Decreasing disease severity in symptomatic, Smn(−/−);SMN2(+/+), spinal muscular atrophy mice following scAAV9-SMN delivery. Hum. Gene Ther. 2012;23:330–335. doi: 10.1089/hum.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rigo F, Hua Y, Krainer AR, Bennett CF. Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol. 2012;199:21–25. doi: 10.1083/jcb.201207087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, Yamashita M, Rigo F, Hung G, Schneider E, Norris DA, Xia S, Bennett CF, Bishop KM. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388:3017–3026. doi: 10.1016/S0140-6736(16)31408-8. [DOI] [PubMed] [Google Scholar]
- 40.d’Ydewalle C, Sumner CJ. Spinal muscular atrophy therapeutics: Where do we stand? Neurotherapeutics. 2015;12:303–316. doi: 10.1007/s13311-015-0337-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cherry JJ, Evans MC, Ni J, Cuny GD, Glicksman MA, Androphy EJ. Identification of novel compounds that increase SMN protein levels using an improved SMN2 reporter cell assay. J. Biomol. Screening. 2012;17:481–495. doi: 10.1177/1087057111431605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cherry JJ, Osman EY, Evans MC, Choi S, Xing X, Cuny GD, Glicksman MA, Lorson CL, Androphy EJ. Enhancement of SMN protein levels in a mouse model of spinal muscular atrophy using novel drug-like compounds. EMBO Mol. Med. 2013;5:1103–1118. doi: 10.1002/emmm.201202305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 44.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hitchcock SA, Pennington LD. Structure-brain exposure relationships. J. Med. Chem. 2006;49:7559–7583. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- 46.Hitchcock SA. Structural modifications that alter the P-glycoprotein efflux properties of compounds. J. Med. Chem. 2012;55:4877–4895. doi: 10.1021/jm201136z. [DOI] [PubMed] [Google Scholar]
- 47.Hodgetts K. Case Studies of CNS Drug Optimization—Medicinal Chemistry and CNS Biology Perspectives. John Wiley & Sons, Inc.; Hoboken, NJ: 2015. [Google Scholar]
- 48.Fuller HR, Man NT, Lam LT, Shamanin VA, Androphy EJ, Morris GE. Valproate and bone loss: iTRAQ proteomics show that valproate reduces collagens and osteonectin in SMA cells. J. Proteome Res. 2010;9:4228–4233. doi: 10.1021/pr1005263. [DOI] [PubMed] [Google Scholar]
- 49.Hahnen E, Eyupoglu IY, Brichta L, Haastert K, Trankle C, Siebzehnrubl FA, Riessland M, Holker I, Claus P, Romstock J, Buslei R, Wirth B, Blumcke I. In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J. Neurochem. 2006;98:193–202. doi: 10.1111/j.1471-4159.2006.03868.x. [DOI] [PubMed] [Google Scholar]
- 50.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 51.Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J. Pathol. 2010;221:117–124. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao J, Marugan JJ, Zheng W, Titus S, Southall N, Cherry JJ, Evans M, Androphy EJ, Austin CP. Discovery, synthesis, and biological evaluation of novel SMN protein modulators. J. Med. Chem. 2011;54:6215–6233. doi: 10.1021/jm200497t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Evans MC, Cherry JJ, Androphy EJ. Differential regulation of the SMN2 gene by individual HDAC proteins. Biochem. Biophys. Res. Commun. 2011;414:25–30. doi: 10.1016/j.bbrc.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burnett BG, Munoz E, Tandon A, Kwon DY, Sumner CJ, Fischbeck KH. Regulation of SMN protein stability. Mol. Cell. Biol. 2009;29:1107–1115. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burnett BG, Xiao J, Southall N, Zheng W, Ferrer M, Cherry JJ, Androphy EJ, Fischbeck K, Marugan JJ. Probe Reports from the NIH Molecular Libraries Program. National Center for Biotechnology Information; Bethesda, MD, U.S: 2010. SMN modulator ML372 increases SMN protein abundance, body weight, lifespan, and rescues motor function in SMNDelta7 SMA mice. [PubMed] [Google Scholar]
- 56.Rietz A, Petrov DP, Bartolowits M, DeSmet M, Davisson VJ, Androphy EJ. Molecular probing of the HPV-16 E6 protein alpha helix binding groove with small molecule inhibitors. PLoS One. 2016;11:e0149845. doi: 10.1371/journal.pone.0149845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.