

Abstract
Cell-penetrating peptides (CPPs) can facilitate the intracellular delivery of large therapeutically relevant molecules, including proteins and oligonucleotides. Although hundreds of CPP sequences are described in the literature, predicting efficacious sequences remains difficult. Here, we focus specifically on predicting CPPs for the delivery of phosphorodiamidate morpholino oligonucleotides (PMOs), a compelling type of antisense therapeutic that has recently been FDA approved for the treatment of Duchenne muscular dystrophy. Using literature CPP sequences, 64 covalent PMO–CPP conjugates were synthesized and evaluated in a fluorescence-based reporter assay for PMO activity. Significant discrepancies were observed between the sequences that performed well in this assay and the sequences that performed well when conjugated to only a small-molecule fluorophore. As a result, we envisioned that our PMO–CPP library would be a useful training set for a computational model to predict CPPs for PMO delivery. We used the PMO activity data to fit a random decision forest classifier to predict whether or not covalent attachment of a given peptide would enhance PMO activity at least 3-fold. To validate the model experimentally, seven novel sequences were generated, synthesized, and tested in the fluorescence reporter assay. All computationally predicted positive sequences were positive in the assay, and one sequence performed better than 80% of the tested literature CPPs. These results demonstrate the power of machine learning algorithms to identify peptide sequences with particular functions and illustrate the importance of tailoring a CPP sequence to the cargo of interest.
Short abstract
A library of PMO-peptide conjugates was tested for cellular activity. The results enabled the development of a computational model to predict novel peptide sequences that improve PMO delivery.
Introduction
Although small molecules can generally diffuse through the plasma membrane, many large molecules have limited uptake into cells.1,2 These macromolecules are unable to diffuse across the plasma membrane and, if endocytosed, often remain trapped in endosomes. For example, gene-editing proteins, antisense oligonucleotides, and peptide-based proteolysis targeting chimeras (PROTACS) all mediate their effects on intracellular targets, and poor delivery limits their therapeutic potential.3−5 One promising solution to improve the intracellular delivery of these macromolecules is the covalent conjugation of cell-penetrating peptides (CPPs).6
Over the past few decades, hundreds of CPPs have been documented in the literature, and yet predicting which peptide sequences improve cytosolic delivery remains difficult. Due in part to the diverse nature of CPPs, the properties and characteristics that are necessary for cell penetration are not well understood. CPPs range from 5 to 40 residues in length, and the sequences can be highly cationic, amphipathic, or hydrophobic.6−8 Many CPPs are derived from fragments of natural proteins, such as viral proteins, DNA- or RNA-binding proteins, heparin-binding proteins, or antimicrobial peptides.9 Some sequences were rationally designed after recognizing that cationic residues or amphipathicity can improve cell penetration, while others were discovered using DNA-encoded peptide libraries.10−13 Taking advantage of machine learning techniques, one recent strategy to predict new CPPs combines experimental data sets of known CPPs with computational models, such as support vector machines or neural networks.14−17
Unfortunately, it is generally acknowledged that the existing computational models to predict CPPs are intrinsically limited.14,15,18 These models were all trained on a similar heterogeneous data set compiled from multiple experimental papers on CPPs.14−17 Since the original papers investigated CPPs for different applications, different experimental parameters were employed. For example, CPP treatment concentrations ranged from 10 to 400 μM, some included serum in the media and others did not, and different cell types were utilized including HeLa cells and primary rat cortex cells.10,19−21 All of these variables affect cellular uptake, and therefore standardized treatment conditions should be used to improve model accuracy.
Additionally, there is a need for computational models that predict CPPs specifically for macromolecule delivery. Experiments to determine putative CPP sequences generally involve the conjugation of a small-molecule fluorophore to the CPP, and the uptake of the fluorophore-CPP is then analyzed by flow cytometry or live-cell confocal imaging.22,23 However, experiments with fluorophore-labeled CPPs do not assess whether or not the CPP is suitable for the delivery of macromolecular cargo. Further, it is likely that the optimal CPP for the delivery of one type of macromolecule is different from the optimal CPP for a different type of macromolecule. One approach to manage this cargo dependence is to evaluate CPPs in the context of a functional readout for a specific macromolecule. For example, several activity-based assays have been developed to evaluate successful delivery of peptides, proteins, and antisense oligonucleotides.24−27
Phosphorodiamidate morpholino oligonucleotides (PMOs) are one particular type of macromolecule that benefits from conjugation to CPPs. PMOs are a charge-neutral antisense oligonucleotide therapeutic in which the ribose sugar is replaced with a methylenemorpholine ring and the phosphodiester backbone is replaced with a phosphorodiamidate backbone (Figure 1A).28 PMOs can be designed to bind to pre-mRNA and can alter gene splicing, resulting in the exclusion or inclusion of particular genetic fragments in the mature mRNA. To improve PMO delivery, arginine-rich CPPs and their derivatives have been covalently conjugated to PMO and investigated using gene-splicing assays, in which cellular fluorescence increases in the presence of PMO.29−32 Other types of neutral antisense oligonucleotides, such as peptide nucleic acids, have also been investigated after conjugation to CPPs.33,34
Figure 1.

PMOs alter gene splicing. (A) The backbone structure of phosphorodiamidate morpholino oligonucleotides, a type of antisense oligonucleotide. (B) The exon-skipping assay used in this study. HeLa-654 cells are stably transfected with a split eGFP construct that contains a mutant intron. In the absence of PMO, a nonfluorescent truncated protein is expressed. If PMO IVS2-654 is present, it hybridizes to the mutant intron, alters pre-mRNA splicing, and produces functional mRNA that is translated into fluorescent eGFP.
Here, we seek to predict CPPs specifically for PMO delivery. We hypothesized that PMO–CPP conjugates evaluated in a functional assay with standardized conditions would provide a data set for training a computational model. We synthesized a library of 64 PMO–CPP conjugates, utilizing previously reported CPPs. To benchmark each CPP, we measured the amount of PMO activity in an exon-skipping assay. We tested all PMO–CPP conjugates using the same concentration, cell-line, amount of serum in the media, and treatment time. Using select CPP sequences from our library, we directly compared the relative effectiveness of a given CPP for the delivery of small-molecule fluorophore to the delivery of PMO. We then developed a random forest classifier that can discriminate whether or not a given peptide sequence can improve PMO activity more than 3-fold. Lastly, we predicted custom peptides for PMO delivery and experimentally validated the sequences for successful delivery.
Results and Discussion
We began by measuring the effectiveness of literature-reported CPPs specifically for PMO delivery. We synthesized a library of CPPs consisting of the sequences listed in the comprehensive review by Milleti in 2012.9 All the CPPs were capped at the N-terminus with an alkyne for further conjugation. Peptides that synthesized poorly or exhibited limited solubility were discarded from the library. After purification using reversed-phase high-performance liquid chromatography (RP-HPLC), we used copper-catalyzed click chemistry to conjugate the peptides to a 6212 Da, 18-mer PMO. The PMO we chose can trigger functional eGFP expression in a modified HeLa cell line (Figure 1B).27 After another round of purification, we obtained a library of 64 PMO–CPP conjugates (Table 1). The library included all of the canonical CPPs, including TAT, pVEC, TP10, penetratin, and polyarginine. Other less commonly reported peptides, such as the heparin binding proteins (DPV3-15) and proline-rich CPPs such as Bac7, were also included. With regard to classes of CPPs, our library contained 25 sequences generally classified as cationic sequences, 8 classified as hydrophobic, and 23 classified as amphipathic.
Table 1. List of CPPs that were Conjugated to PMO and Tested in the eGFP Assaya.
| CPP name | CPP class | amino acid sequence | theoretical net charge | activity relative to PMO |
|---|---|---|---|---|
| arginine-12 | cationic | RRRRRRRRRRRR | 12 | 10.4 |
| MPG | amphipathic | GLAFLGFLGAAGSTMGAWSQPKKKRKV | 5 | 7.5 |
| Bac7 | proline rich | RRIRPRPPRLPRPRPRPLPFPRPG | 9 | 6.1 |
| TAT | cationic | RKKRRQRRR | 8 | 6.0 |
| arginine-10 | cationic | RRRRRRRRRR | 10 | 5.8 |
| DPV6 | cationic | GRPRESGKKRKRKRLKP | 9 | 5.4 |
| S413-PVrev | amphipathic | ALWKTLLKKVLKAPKKKRKV | 9 | 5.2 |
| HRSV | cationic | RRIPNRRPRR | 6 | 4.9 |
| HTLV-II Rex | cationic | TRRQRTRRARRNR | 8 | 4.7 |
| L-2 | amphipathic | HARIKPTFRRLKWKYKGKFW | 9 | 4.6 |
| melittin | amphipathic | GIGAVLKVLTTGLPALISWIKRKRQQ | 5 | 4.5 |
| DPV15 | cationic | LRRERQSRLRRERQSR | 6 | 4.3 |
| arginine-9 | cationic | RRRRRRRRR | 9 | 4.2 |
| penetratin | cationic | RQIKIWFQNRRMKWKK | 7 | 4.2 |
| yeast GCN4 | cationic | KRARNTEAARRSRARKLQRMKQ | 9 | 4.2 |
| PDX-1 | cationic | RHIKIWFQN RRM KWKK | 8 | 4.1 |
| arginine-8 | cationic | RRRRRRRR | 8 | 4.0 |
| BMV Gag | cationic | KMTRAQRRAAARRNRWTAR | 8 | 3.9 |
| SynB1 | amphipathic | RGGRLSYSRRRFSTSTGR | 6 | 3.9 |
| knotted-1 | cationic | KQINNWFINQRKRHWK | 6 | 3.8 |
| IVV-14 | hydrophobic | KLWMRWYSPTTRRYG | 4 | 3.7 |
| W/R | amphipathic | RRWWRRWRR | 6 | 3.5 |
| engrailed-2 | cationic | SQIKIWFQN KRAKIKK | 6 | 3.4 |
| DPV15b | cationic | GAYDLRRRERQSRLRRRERQSR | 7 | 3.3 |
| yeast PrP6 | cationic | TRRNKRNRIQEQLNRK | 6 | 3.3 |
| DPV7 | cationic | GKRKKKGKLGKKRDP | 8 | 3.2 |
| HoxA-13 | cationic | RQVTIWFQNRRVKEKK | 5 | 3.1 |
| AIP6 | amphipathic | RLRWR | 3 | 2.9 |
| (PPR)5 | cationic | PPRPPRPPRPPRPPR | 5 | 2.7 |
| CAYH | CAYHRLRRC | 4 | 2.6 | |
| DPV10 | cationic | SRRARRSPRHLGSG | 6 | 2.5 |
| (PPR)4 | amphipathic | PPRPPRPPRPPR | 4 | 2.4 |
| P22 N | cationic | NAKTRRHERRRKLAIER | 7 | 2.4 |
| DPV1047 | cationic | VKRGLKLRHVRPRVTRMDV | 7 | 2.4 |
| SVM4 | amphipathic | LYKKGPAKKGRPPLRGWFH | 7 | 2.2 |
| cp21N(12–29) | cationic | TAKTRYKARRAELIAERR | 5 | 2.1 |
| SVM3 | amphipathic | KGTYKKKLMRIPLKGT | 6 | 2.1 |
| (PPR)3 | amphipathic | PPRPPRPPR | 3 | 1.9 |
| SVM2 | RASKRDGSWVKKLHRILE | 5 | 1.9 | |
| buforin 2 | amphipathic | TRSSRAGLQWPVGRVHRLLRK | 7 | 1.9 |
| SVM1 | FKIYDKKVRTRVVKH | 6 | 1.7 | |
| SAP | amphipathic | VRLPPPVRLPPPVRLPPP | 3 | 1.7 |
| 435b | hydrophobic | GPFHFYQFLFPPV | 1 | 1.7 |
| Pept1 | hydrophobic | PLILLRLLRGQF | 2 | 1.7 |
| YTA2 | YTAIAWVKAFIRKLRK | 5 | 1.5 | |
| Pep-1 | Amphipathic | KETWWETWWTEWSQ PKKRKV | 2 | 1.4 |
| EB-1 | amphipathic | LIRLWSHLIHIWFQNRRLKWKKK | 9 | 1.4 |
| pyrrhocoricin | proline rich | VDKGSYLPRPTPPRPIYNRN | 3 | 1.4 |
| AN(1–22) | cationic | MDAQTRRRERRAEKQAQWKAAN | 4 | 1.4 |
| 439a | hydrophobic | GSPWGLQHHPPRT | 3 | 1.3 |
| MAP | amphipathic | KLALKALKALKAALKLA | 5 | 1.3 |
| Bip | hydrophobic | IPALK | 1 | 1.3 |
| Bip | hydrophobic | VPALR | 1 | 1.3 |
| pVEC | amphipathic | LLIILRRRIRKQAHAHSK | 8 | 1.2 |
| YTA4 | IAWVKAFIRKLRKGPLG | 5 | 1.2 | |
| K-FGF + NLS | amphipathic | AAVLLPVLLAAPVQRKRQKLP | 4 | 1.2 |
| HN-1 | hydrophobic | TSPLNIHNGQKL | 2 | 1.2 |
| Bip | hydrophobic | VPTLK | 1 | 1.2 |
| Bip | hydrophobic | VSALK | 1 | 1.1 |
| VT5 | amphipathic | DPKGDPKGVTVTVTVTVTGKGDPKPD | 0 | 0.8 |
| transportan 10 | amphipathic | AGYLLGKINLKALAALAKKIL | 4 | 0.8 |
| SAP(E) | amphipathic | VELPPPVELPPPVELPPP | –3 | 0.8 |
| CADY | amphipathic | GLWRALWRLLRSLWRLLWRA | 5 | 0.6 |
| PreS2-TLM | amphipathic | PLSSIFSRIGDP | 0 | 0.6 |
Each previously reported CPP was synthesized, purified, and conjugated to PMO IVS2-654. The conjugates were tested for functional PMO activity in the HeLa-654 cell assay. Individual CPPs are ranked by their activity relative to unconjugated PMO.
For a functional readout, the PMO–CPP conjugates were tested in the eGFP HeLa PMO assay (Figure 1B). In this assay, HeLa-654 cells are stably transfected with an eGFP coding sequence interrupted by an intron from the human β-globin gene (IVS2-654) containing a mutation that alters the normal pre-mRNA splice site to a formerly cryptic splice site. The change in splicing leads to retention of an unnatural mRNA fragment in the spliced eGFP mRNA and the translation of a nonfluorescent form of eGFP. The PMO IVS2-654 hybridizes to the mutant β-globin exon in the stably transfected HeLa cells, altering gene splicing and leading to full-length eGFP expression. The amount of PMO delivered is therefore correlated to the amount of functional eGFP expressed. In the experiment, eGFP HeLa cells are incubated with 5 μM of each PMO–CPP conjugate in media containing 10% fetal bovine serum (FBS) for 24 h. Then the cellular fluorescence is analyzed by flow cytometry. Given that the effectiveness of CPPs can be sensitive to treatment conditions such as the amount of serum in the treatment media and the amount of time treated, we kept these variables constant, enabling us to directly compare all of the CPPs under similar conditions.
Testing the library under these unified conditions led to the observation that several literature CPPs had little effect on promoting PMO delivery (Table 1). While seven peptides increased PMO activity above 5-fold, many peptides exhibited marginal improvement for PMO delivery. Twenty-seven CPPs (42% of library) led to under a 2-fold increase in eGFP fluorescence, and five CPPs actually decreased the amount of eGFP fluorescence compared to unconjugated PMO. In particular, the commonly used CPP transportan-10 (TP10) exhibited a negative effect on eGFP fluorescence, suggesting it is ineffective for PMO delivery under these conditions.
Additionally, we observed that net positive charge was one of the strongest predictors of a successful CPP. Cationic sequences represented 70% of the CPPs with over a 3-fold improvement in PMO activity (19 out of 27 sequences). This trend is specific for the number of arginine residues, with more arginine residues leading to more observed eGFP fluorescence. In particular, attachment of the arginine-12 CPP led to the greatest enhancement of PMO activity. However, a high net charge is by no means necessary—the peptide MPG has just one arginine residue and a relatively minor theoretical net charge of +5, yet it exhibited the second highest activity of all the CPPs that we tested.
To understand if the trends in CPP effectiveness were specific for PMO delivery, we evaluated select members of our CPP library for fluorophore delivery (Figure 2A). The chosen CPPs cover the range of physiochemical properties present in our library, as well as the most commonly utilized CPP sequences. We used copper-catalyzed click chemistry to conjugate the CPPs to cyanine 5.5 (Cy5.5 – λex 684 nm, λem 710 nm). Next, eGFP HeLa cells were treated for 2 h with 5 μM of each Cy5.5–CPP conjugate in media containing 10% serum, and the cellular fluorescence was analyzed by flow cytometry. The amount of Cy5.5 fluorescence was normalized to the mean fluorescence intensity of Cy5.5–YTA-4, the conjugate with the highest fluorescence intensity. Then, the amount of Cy5.5 fluorescence measured for each Cy5.5–CPP conjugate was compared to the relative amount of eGFP fluorescence for the equivalent PMO–CPP conjugate. We observed little correlation between the relative effectiveness of PMO and Cy5.5 delivery for a given CPP (Figure 2B). For example, arginine-12 led to the highest eGFP fluorescence of the CPPs evaluated, yet only moderate Cy5.5 fluorescence. In contrast, YTA4 and TP10 both led to substantial Cy5.5 fluorescence but demonstrated no practical improvement in PMO delivery.
Figure 2.

Cargo identity alters relative CPP efficacy. (A) Each CPP sequence was analyzed for delivery of both a fluorophore (Cy5.5, MW = 665 Da) and a PMO (IVS2-654, MW = 6212 Da). For Cy5.5-CPP conjugates, HeLa-654 cells were treated with 5 μM of the conjugate in media containing 10% FBS. After 2 h, the Cy5.5 fluorescence was measured by flow cytometry. For PMO–CPP conjugates, HeLa-654 cells were treated with 5 μM of the conjugate in media containing 10% FBS. After 22 h, the eGFP fluorescence was measured by flow cytometry. (B) The cargo dependency of a given CPP, normalized to the activity of the highest-performing CPP for each type of cargo. There is little relationship between the CPPs that led to the most cellular Cy5.5 fluorescence and the CPPs that led to the most eGFP fluorescence.
It is important to note that the PMO assay does not distinguish between cellular uptake and other downstream effects that influence exon skipping (e.g., nuclear delivery or mRNA splicing). The amount of eGFP fluorescence is not a measure of intracellular concentration, even though they are correlated. We focused on an activity measurement because we believe that, in the context of macromolecule delivery, the final functional output of a cargo represents the most relevant criterion for judging a CPP.
After benchmarking literature CPPs and noting the cargo dependency, we sought to develop a method to identify efficacious CPPs for PMO delivery. We hypothesized that we could leverage the consistency of our data set (identical concentrations, treatment times, serum-containing media, and assay) along with techniques from machine learning to build a predictive computational model for PMO delivery. While the algorithms we employed are standard in the computer science field, this is the first time they have been applied to the prediction task of peptides for antisense oligonucleotide delivery.
We chose to train a random forest classifier to select CPPs specific to PMO delivery (Figure 3A). Random forests are sets of decision trees, each fit to a randomly selected subset of features and training examples, and we selected this ensemble learning method due to its scalability and robustness to overfitting.35 We calculated 23 features for each peptide sequence. Two features were peptide molecular weight and sequence length (total number of residues). The theoretical net charge of the sequence was averaged across the five N-terminal residues, the five C-terminal residues, and the entire peptide sequence to give three features. The remaining 18 features were derived from six previously described amino acid physicochemical descriptors. These six descriptors were produced by factor analysis of 384 molecular properties calculated for 22 natural and 593 non-natural amino acids.36 For each peptide sequence, the six descriptors were also averaged across the five N-terminal residues, the five C-terminal residues, and the entire peptide sequence.
Figure 3.
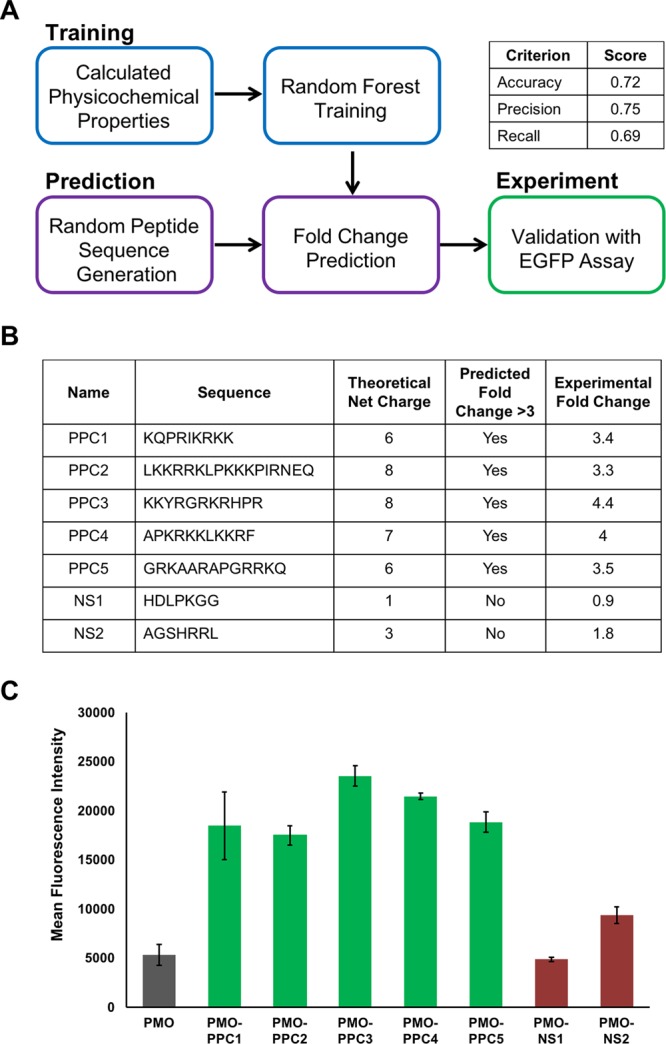
Random forest ensemble learning methods can be used to predict peptide sequences that facilitate PMO delivery. (A) Scheme of the workflow for the development of computationally derived peptide sequences for antisense delivery. One component of the workflow was random forest training using the properties of the CPPs in the library to build a model. Another component was randomly generating peptide sequences and using the model to predict the peptide activity. Lastly, select sequences were synthesized for experimental validation. Performance metrics for the model based on the test set are given in the table. (B) Table of five predicted PMO carriers (PPC) and two predicted negative sequences (NS). All five PMO–PPC conjugates exhibited above a 3-fold improvement in eGFP fluorescence, whereas both PMO–NS conjugates exhibited less than a 3-fold change with respect to PMO. (C) The mean fluorescence intensity of eGFP HeLa cells treated at a concentration of 5 μM with each of the PMO–PPCs and PMO–NSs in serum-containing media. Each individual experiment consisted of the average of three different wells with the same treatment conditions, and the experiment was repeated three times. The error bars represent the standard deviation across the experimental replicates.
The CPP sequences were classified as either positive or negative examples based on whether or not they exhibited above a 3-fold change in eGFP fluorescence with respect to the unconjugated PMO. Forty-four sequences were used as the training set for the random forest model. The other 20 sequences were held out to serve as a test set to evaluate the degree to which the model properly fit the data and could successfully predict the exon skipping activity of a sequence. The performance metrics of the model are shown in Figure 3A.
After testing our model computationally, we sought to validate it experimentally. Random peptide sequences were generated by selecting a peptide length and amino acid composition with probability proportional to the distribution observed in the training data set from the CPP library. Of the random peptides, we selected five positive sequences predicted to lead to above a 3-fold increase in eGFP fluorescence and two negative sequences (NSs). We selected more positive sequences as our goal was to develop novel peptide sequences for PMO delivery, which we have termed predicted PMO carriers (PPCs). These PPCs were synthesized by solid-phase peptide synthesis, conjugated to PMO IVS2-654, and purified by RP-HPLC.
We tested the PPCs in the eGFP assay, and all five PPCs had a greater than 3-fold change in cellular fluorescence with respect to unconjugated PMO (Figure 3B,C). In fact, PMO–PPC3 increased fluorescence 4.4-fold, which is a larger increase than 80% of the literature CPPs. The negative sequences demonstrated less than a 3-fold change, indicating that our model could accurately discriminate between positive and negative sequences.
To demonstrate that our model is sensitive to the effects of cargo on CPP effectiveness, we next assessed whether or not our novel PPCs were CPPs with regard to small-molecule fluorophore delivery. We prepared variants of our PPCs labeled with Cy5.5, rather than the PMO, and measured the uptake by flow cytometry (Figure S1). Here, the trends were completely different, as cells treated with Cy5.5-NS2 exhibited fluorescence similar to cells treated with our positive control Cy5.5–YTA-4 and twice the fluorescence of cells treated with Cy5.5–PPC1. These data parallel our observations of the literature CPPs and suggest that our computational model is truly specific for PMO-based cargo, providing further evidence that CPPs must be chosen in the context of the cargo of interest.
Next, we sought to characterize the mechanisms by which our PMO–PPC conjugates are internalized into cells, focusing on PPC3 and PPC5. All mechanistic studies were conducted at concentrations of 5 μM to remain consistent with the conditions with which we evaluated our PMO–PPC library. The experiments were performed in a pulse-chase format, in which the cells were preincubated in a particular treatment condition for 30 min followed by addition of PMO–PPC. After 3 h, the media was exchanged for fresh media that did not contain the conjugate, and the cells were allowed to incubate for an additional 22 h at 37 °C.
First, we compared eGFP fluorescence after 3 h of treatment at 4 °C vs 37 °C. We found that for PMO alone and both PMO–PPC conjugates, eGFP fluorescence decreased when treatment occurred at 4 °C, suggesting that energy-dependent mechanisms play a role in the uptake of our conjugates (Figure 4A). One plausible explanation for the residual fluorescence in the 4 °C condition is that the cells incubate for an additional 22 h at 37 °C after treatment, so any conjugate that binds to the surface of the cells during treatment at 4 °C may be subsequently internalized and trigger eGFP expression.
Figure 4.

The PMO–PPC conjugates engage endocytic mechanisms in their uptake into cells. (A) Effect of temperature on PMO–PPC activity. eGFP HeLa cells were incubated at 4 °C for 30 min before incubation with either PMO or PMO–PPC conjugates at 4 °C at a concentration of 5 μM. After 3 h, the treatment media was replaced with fresh, untreated media, and the cells were allowed to grow for an additional 22 h at 37 °C. CO refers to cell-only. (B) Effect of cytochalasin D on PMO–PPC activity. eGFP HeLa cells were treated with cytochalasin D for 30 min in serum-containing media before the addition of either PMO or PMO–PPC conjugates at a concentration of 5 μM. After 3 h, the treatment media was replaced with fresh, untreated media, and the cells were allowed to grow for an additional 22 h. The cells were analyzed for eGFP fluorescence by flow cytometry, and the results are shown in terms of the mean fluorescence intensity. These experiments were conducted on a plate alongside several other inhibitors, and the full results are in the Supporting Information.
We also treated the HeLa eGFP cells with a panel of endocytosis disruptors and assessed the effects on internalization (Figure 4B and Figure S2). While eGFP fluorescence was relatively unchanged after preincubation with many of the inhibitors, preincubation with cytochalasin D led to a notable decrease in eGFP fluorescence. Cytochalasin D binds to the barbed, fast growing ends of actin microfilaments, which prevents assembly and disassembly of actin monomers.37 This affects not only the cytoskeleton of the cell, but also the ability of the membrane to ruffle and reorganize to facilitate macropinocytosis. While it is possible that the decrease in eGFP fluorescence is due to effects downstream in the exon skipping pathway, these results suggest that macropinocytosis plays a significant role in the internalization of our conjugates.
Concluding Remarks
Serendipity is not necessary to discover new cell-penetrating peptide sequences for macromolecule delivery. Although many CPPs have been discovered using peptide fragments from natural proteins, computational models challenge the notion that CPP sequences must be found in nature. Here, we generated a random forest classifier that could accurately predict whether or not conjugation of a given peptide sequence would increase PMO activity 3-fold. Our model enabled the discovery of five completely novel sequences that increased PMO activity, and accurately discriminated between active and inactive sequences. A BLAST search revealed that these new sequences are not found in nature.
One key component of our computational model is the standardized assay conditions used in the data set for training. Multiple experimental variables influence cellular uptake, including treatment concentration and the presence of serum. To enable the accurate classification of multiple CPPs, the peptides must be tested under similar conditions. Additionally, as noted previously in the CPP field, the computational models are only as valuable as the data used to train a given model. By testing a PMO–CPP library under standardized conditions, we obtained a high-quality training set to improve the accuracy of the predictions from the model.
The second key component of our model is the type of cargo utilized to assess cell penetration. Functional macromolecules have diverse chemical structures, mechanisms of action, and sites of activity inside of cells. Although many CPPs are investigated using only an attached fluorophore as a metric of their efficacy, our experiments indicated that fluorophore studies have little predictive value with regard to the optimal CPP for macromolecular delivery. Therefore, to develop an optimal computational model, the training set should involve CPPs tested in the appropriate context. For our experiments, we investigated a library of PMO–CPP conjugates to focus on the context of improving PMO delivery and promoting exon skipping.
Combining these two components leads to a computational model that enables exploration of the design space for cell-penetrating peptides. Although many, potentially infinite, peptide sequences may facilitate PMO delivery, our model can be employed to select sequences based on certain desirable parameters. For example, peptide sequences with a large number of arginine residues can lead to toxicity in vivo and so, in our computational model, sequence space can be restricted to sequences containing fewer than three arginine residues. Similar approaches can be utilized for identifying and avoiding peptide sequences that are immunogenic (e.g., by referencing a database of immunogenic sequences), or peptide sequences that will be synthetically challenging (e.g., peptides with multiple β-branched residues). We envision that using our computational model, vast numbers of putative sequences can be tested in silico, reducing experimental burden and drastically increasing the chemical space that can be investigated. Then, only the optimized sequences that meet the desired criteria can be evaluated experimentally.
Moving forward, we seek to understand the generalizability of our current computational model. If our model extends to clinically relevant PMO sequences, it could serve as a valuable resource to optimize therapeutic PMO–peptide conjugates. Additionally, we will investigate the utility of our computational model for improving the delivery of different classes of antisense oligonucleotides. Understanding the strengths and limitations of computational prediction will be critical for applying machine learning to the challenge of intracellular delivery. We envision that careful experimental design coupled with the appropriate machine learning algorithm will significantly increase the portfolio of CPPs for macromolecule delivery.
Experimental Section
Peptide Synthesis
Peptides were synthesized using either manual flow peptide synthesis or various iterations of an automated flow peptide synthesizer.38,39 For detailed methods on peptide synthesis and purification, please see the Supporting Information.
PMO Azide Synthesis
PMO IVS2-654 was provided by Sarepta Therapeutics. The sequence is shown in Figure 2A. To conjugate the azide to the 3′ end, PMO IVS2-654 was dissolved in DMSO (53 mM). To the solution was added 4 equiv of 5-azidopentanoic acid activated with HBTU and 4 equiv of DIEA dissolved in DMF. The reaction proceeded for 25 min before being quenched with water and ammonium hydroxide. The ammonium hydroxide was used to hydrolyze any ester formed during the course of the reaction. After 1 h, the solution was diluted and purified by reversed-phase HPLC using a linear gradient from 2% to 60% B over 58 min. Mobile phase A: water. Mobile phase B: acetonitrile. For LC-MS characterization, please see Supporting Information.
PMO Peptide Conjugation
PMO peptide conjugates were synthesized by copper-catalyzed azide alkyne cycloaddition using a copper bromide catalyst in DMF. Under nitrogen gas, a mixture of peptide alkyne (1.1 μmol), PMO azide (0.95 μmol), and copper bromide (0.05 mmol) was dissolved in 1 mL of DMF, vortexed, and allowed to react for 1 h. The reaction was quenched with the addition of 10 mL of 50 mM Tris (pH 8). Our optimized purification procedure utilized reversed-phase HPLC with a linear gradient from 5 to 45% B over 20 min. Mobile phase A: 100 mM ammonium acetate pH 7.2 in water. Mobile phase B: acetonitrile. For additional purification procedures and LC-MS characterization of all PMO-peptide conjugates, please see the Supporting Information.
Fluorophore Conjugation
Cy5.5 azide was conjugated to peptide alkyne by copper-catalyzed azide alkyne cycloaddition using copper sulfate and ascorbic acid. Briefly, 0.5 μmol of peptide alkyne was dissolved in 200 μL of 50:50 t-butanol/water in a 1.7 mL microcentrifuge tube. The following solutions were added in order: 10 μL of 50 mM Cy5.5 azide in DMSO, 100 μL of 500 mM Tris pH 8 in water, 50 μL of 100 mM copper(II) sulfate in water, 10 μL of 10 mM Tris(benzyltriazolylmethyl)amine (TBTA) in DMSO, 530 μL of 50:50 t-butanol/water, and 100 μL of 1 M ascorbic acid in water. After 1 h, the reactions were purified by reverse-phase HPLC using a linear gradient from 5 to 45% B over 80 min. Mobile phase A: water with 0.1% TFA. Mobile phase B: acetonitrile with 0.1% TFA. For LC-MS characterization of Cy5.5–PPC conjugates, please see the Supporting Information.
Computational Design
Random forest classifier hyperparameters were optimized through grid search with classification accuracy estimated with 3-fold cross validation. The selected number of features per tree, number of trees, and maximum tree depth were 11, 50, and 20, respectively. The change in accuracy from varying select hyperparameters or removing each feature is shown in the Supporting Information. Performance metrics from classifier evaluation on a held-out test set of 20 sequences are given in Figure 2A.
The performance metrics are defined below, where TP refers to true positive, TN refers to true negative, FP refers to false positive, and FN refers to false negative.
Flow Cytometry
For testing the library of PMO–CPP conjugates, flow cytometry analysis of GFP fluorescence was conducted as previously described.27 For testing the PMO–PPC conjugates, HeLa 654 cells were maintained in MEM supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin-streptomycin at 37 °C and 5% CO2. Stocks of each PMO–PPC conjugate were prepared in phosphate-buffered saline (PBS). The concentration of the stocks was determined by measuring the absorbance at 260 nm and using an extinction coefficient of 168 700 L mol–1 cm–1. Cells were incubated with each respective conjugate at a concentration of 5 μM in MEM supplemented with 10% FBS and 1% penicillin-streptomycin for 22 h at 37 °C and 5% CO2. Next, the treatment media was aspirated. The cells were incubated with trypsin-EDTA 0.25% for 15 min at 37 °C and 5% CO2, washed 1× with PBS, and resuspended in PBS with 2% FBS and 2 μg/mL propidium iodide.
Flow cytometry analysis was carried out on a BD LSRII flow cytometer. Gates were applied to the data to ensure that cells that were highly positive for propidium iodide or exhibited forward/side scatter readings that were sufficiently different from the main cell population were excluded. Each histogram contained at least 10 000 gated events. Representative histograms are shown in the Supporting Information.
Inhibitor Experiments
To inhibit a variety of endocytic mechanisms, a pulse-chase experiment was performed. Briefly, HeLa 654 cells were plated at a density of 5000 cells per well in a 96-well plate in MEM supplemented with 10% FBS and 1% penicillin-streptomycin. The next day, the cells were treated with each inhibitor at the indicated concentration. After 30 min, PMO–peptide conjugate was added to each well at a concentration of 5 μM. After incubation at 37 °C and 5% CO2 for 3 h, the treatment media was replaced with fresh media (containing neither inhibitor nor PMO–peptide), and the cells were allowed to grow for another 22 h at 37 °C and 5% CO2. For the 4 °C experiments, the day after plating, the cells were preincubated for 30 min at 4 °C, followed by the addition of PMO–peptide conjugate to each well at a concentration of 5 μM. After incubation at 4 °C for 3 h, the treatment media was replaced with fresh media, and the cells were allowed to grow for another 22 h at 37 °C and 5% CO2. Sample preparation and flow cytometry were then performed as described above. Each histogram contains at least 2000 gated events, with the exception of treatment with 20 μM cytochalasin D and 200 nM wortmannin.
Acknowledgments
This work was supported by Sarepta Therapeutics, Cambridge, MA. J.M.W. and R.L.H. are supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1122374. C.M.F. is supported by the David H. Koch Graduate Fellowship Fund and by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health under Award Number F30HD093358. The authors acknowledge the Swanson Biotechnology Center Flow Cytometry Facility at the Koch Institute for advice and the use of their flow cytometers. We thank Mark Simon, Dan Dunkelmann, and Alex Mijalis for assistance with peptide synthesis. We also thank Ethan Evans, Dr. Alan Beggs, and Dr. Guriq Basi for helpful discussions and comments during manuscript preparation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00098.
Methods for peptide synthesis, flow cytometry analysis of fluorophore-labeled PPCs, flow cytometry analysis of PPCs in the presence of endocytosis inhibitors, information on random forest classifier development and the importance of individual features, representative flow cytometry histograms, and LC-MS analysis of peptide conjugates (PDF)
Author Contributions
# J.M.W. and C.M.F. contributed equally to this work.
The authors declare the following competing financial interest(s): GJH and MY are employees of Sarepta Therapeutics.
Supplementary Material
References
- Fu A.; Tang R.; Hardie J.; Farkas M. E.; Rotello V. M. Promises and Pitfalls of Intracellular Delivery of Proteins. Bioconjugate Chem. 2014, 25 (9), 1602–1608. 10.1021/bc500320j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart M. P.; Sharei A.; Ding X.; Sahay G.; Langer R.; Jensen K. F. In Vitro and Ex Vivo Strategies for Intracellular Delivery. Nature 2016, 538 (7624), 183–192. 10.1038/nature19764. [DOI] [PubMed] [Google Scholar]
- Buckley D. L.; Crews C. M. Small-Molecule Control of Intracellular Protein Levels through Modulation of the Ubiquitin Proteasome System. Angew. Chem., Int. Ed. 2014, 53 (9), 2312–2330. 10.1002/anie.201307761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano R. L. The Delivery of Therapeutic Oligonucleotides. Nucleic Acids Res. 2016, 44 (14), 6518–6548. 10.1093/nar/gkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D. G.; Yin H.; Kauffman K. J. Delivery Technologies for Genome Editing. Nat. Rev. Drug Discovery 2017, 16 (6), 387. 10.1038/nrd.2016.280. [DOI] [PubMed] [Google Scholar]
- Copolovici D. M.; Langel K.; Eriste E.; Langel Ü. Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 2014, 8 (3), 1972–1994. 10.1021/nn4057269. [DOI] [PubMed] [Google Scholar]
- Cell-Penetrating Peptides; Langel Ü., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, 2011; Vol. 683. [DOI] [PubMed] [Google Scholar]
- Agrawal P.; Bhalla S.; Usmani S. S.; Singh S.; Chaudhary K.; Raghava G. P. S.; Gautam A. CPPsite 2.0: A Repository of Experimentally Validated Cell-Penetrating Peptides. Nucleic Acids Res. 2016, 44 (D1), D1098–D1103. 10.1093/nar/gkv1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milletti F. Cell-Penetrating Peptides: Classes, Origin, and Current Landscape. Drug Discovery Today 2012, 17 (15–16), 850–860. 10.1016/j.drudis.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Soomets U.; Lindgren M.; Gallet X.; Hällbrink M.; Elmquist A.; Balaspiri L.; Zorko M.; Pooga M.; Brasseur R.; Langel Ü. Deletion Analogues of Transportan. Biochim. Biophys. Acta, Biomembr. 2000, 1467 (1), 165–176. 10.1016/S0005-2736(00)00216-9. [DOI] [PubMed] [Google Scholar]
- Morris M. C.; Vidal P.; Chaloin L.; Heitz F.; Divita G. A New Peptide Vector for Efficient Delivery of Oligonucleotides into Mammalian Cells. Nucleic Acids Res. 1997, 25 (14), 2730–2736. 10.1093/nar/25.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels D. S.; Schepartz A. Intrinsically Cell-Permeable Miniature Proteins Based on a Minimal Cationic PPII Motif. J. Am. Chem. Soc. 2007, 129 (47), 14578–14579. 10.1021/ja0772445. [DOI] [PubMed] [Google Scholar]
- Tünnemann G.; Ter-Avetisyan G.; Martin R. M.; Stöckl M.; Herrmann A.; Cardoso M. C. Live-Cell Analysis of Cell Penetration Ability and Toxicity of Oligo-Arginines. J. Pept. Sci. 2008, 14 (4), 469–476. 10.1002/psc.968. [DOI] [PubMed] [Google Scholar]
- Sanders W. S.; Johnston C. I.; Bridges S. M.; Burgess S. C.; Willeford K. O. Prediction of Cell Penetrating Peptides by Support Vector Machines. PLoS Comput. Biol. 2011, 7 (7), e1002101. 10.1371/journal.pcbi.1002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam A.; Chaudhary K.; Kumar R.; Sharma A.; Kapoor P.; Tyagi A.; Raghava G. P. S. In Silico Approaches for Designing Highly Effective Cell Penetrating Peptides. J. Transl. Med. 2013, 11, 74. 10.1186/1479-5876-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holton T. A.; Pollastri G.; Shields D. C.; Mooney C. CPPpred: Prediction of Cell Penetrating Peptides. Bioinformatics 2013, 29 (23), 3094–3096. 10.1093/bioinformatics/btt518. [DOI] [PubMed] [Google Scholar]
- Dobchev D. A.; Mager I.; Tulp I.; Karelson G.; Tamm T.; Tamm K.; Janes J.; Langel U.; Karelson M. Prediction of Cell-Penetrating Peptides Using Artificial Neural Networks. Curr. Comput.-Aided Drug Des. 2010, 6 (2), 79–89. 10.2174/157340910791202478. [DOI] [PubMed] [Google Scholar]
- Hansen M.; Kilk K.; Langel Ü. Predicting Cell-Penetrating Peptides. Adv. Drug Delivery Rev. 2008, 60 (4), 572–579. 10.1016/j.addr.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Derossi D.; Calvet S.; Trembleau A.; Brunissen A.; Chassaing G.; Prochiantz A. Cell Internalization of the Third Helix of the Antennapedia Homeodomain Is Receptor-Independent. J. Biol. Chem. 1996, 271 (30), 18188–18193. 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- Fischer P. m.; Zhelev N. z.; Wang S.; Melville J. e.; Fåhraeus R.; Lane D. p. Structure–activity Relationship of Truncated and Substituted Analogues of the Intracellular Delivery Vector Penetratin. J. Pept. Res. 2000, 55 (2), 163–172. 10.1034/j.1399-3011.2000.00163.x. [DOI] [PubMed] [Google Scholar]
- Gomez J. A.; Gama V.; Yoshida T.; Sun W.; Hayes P.; Leskov K.; Boothman D.; Matsuyama S. Bax-Inhibiting Peptides Derived from Ku70 and Cell-Penetrating Pentapeptides. Biochem. Soc. Trans. 2007, 35 (4), 797–801. 10.1042/BST0350797. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Melikov K.; Vives E.; Ramos C.; Verbeure B.; Gait M. J.; Chernomordik L. V.; Lebleu B. Cell-Penetrating Peptides A Reevaluation of the Mechanism of Cellular Uptake. J. Biol. Chem. 2003, 278 (1), 585–590. 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- LaRochelle J. R.; Cobb G. B.; Steinauer A.; Rhoades E.; Schepartz A. Fluorescence Correlation Spectroscopy Reveals Highly Efficient Cytosolic Delivery of Certain Penta-Arg Proteins and Stapled Peptides. J. Am. Chem. Soc. 2015, 137 (7), 2536–2541. 10.1021/ja510391n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia J. S.; Stan R. V.; Dowdy S. F. Transducible TAT-HA Fusogenic Peptide Enhances Escape of TAT-Fusion Proteins after Lipid Raft Macropinocytosis. Nat. Med. 2004, 10 (3), 310–315. 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- Schmidt S.; Adjobo-Hermans M. J. W.; Wallbrecher R.; Verdurmen W. P. R.; Bovée-Geurts P. H. M.; van Oostrum J.; Milletti F.; Enderle T.; Brock R. Detecting Cytosolic Peptide Delivery with the GFP Complementation Assay in the Low Micromolar Range. Angew. Chem., Int. Ed. 2015, 54 (50), 15105–15108. 10.1002/anie.201505913. [DOI] [PubMed] [Google Scholar]
- Kang S.-H.; Cho M.-J.; Kole R. Up-Regulation of Luciferase Gene Expression with Antisense Oligonucleotides: Implications and Applications in Functional Assay Development†. Biochemistry 1998, 37 (18), 6235–6239. 10.1021/bi980300h. [DOI] [PubMed] [Google Scholar]
- Sazani P.; Kang S.-H.; Maier M. A.; Wei C.; Dillman J.; Summerton J.; Manoharan M.; Kole R. Nuclear Antisense Effects of Neutral, Anionic and Cationic Oligonucleotide Analogs. Nucleic Acids Res. 2001, 29 (19), 3965–3974. 10.1093/nar/29.19.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerton J.; Weller D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev. 1997, 7 (3), 187–195. 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- Moulton H. M.; Nelson M. H.; Hatlevig S. A.; Reddy M. T.; Iversen P. L. Cellular Uptake of Antisense Morpholino Oligomers Conjugated to Arginine-Rich Peptides. Bioconjugate Chem. 2004, 15 (2), 290–299. 10.1021/bc034221g. [DOI] [PubMed] [Google Scholar]
- Wu R. P.; Youngblood D. S.; Hassinger J. N.; Lovejoy C. E.; Nelson M. H.; Iversen P. L.; Moulton H. M. Cell-Penetrating Peptides as Transporters for Morpholino Oligomers: Effects of Amino Acid Composition on Intracellular Delivery and Cytotoxicity. Nucleic Acids Res. 2007, 35 (15), 5182–5191. 10.1093/nar/gkm478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abes R.; Moulton H. M.; Clair P.; Yang S.-T.; Abes S.; Melikov K.; Prevot P.; Youngblood D. S.; Iversen P. L.; Chernomordik L. V.; et al. Delivery of Steric Block Morpholino Oligomers by (R-X-R)4 Peptides: Structure–activity Studies. Nucleic Acids Res. 2008, 36 (20), 6343–6354. 10.1093/nar/gkn541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Saleh A. F.; Betts C.; Camelliti P.; Seow Y.; Ashraf S.; Arzumanov A.; Hammond S.; Merritt T.; Gait M. J.; et al. Pip5 Transduction Peptides Direct High Efficiency Oligonucleotide-Mediated Dystrophin Exon Skipping in Heart and Phenotypic Correction in Mdx Mice. Mol. Ther. 2011, 19 (7), 1295–1303. 10.1038/mt.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuss P. J.; Arzumanov A. A.; Williams D. L.; Gait M. J. Parallel Synthesis and Splicing Redirection Activity of Cell-Penetrating Peptide Conjugate Libraries of a PNA Cargo. Org. Biomol. Chem. 2013, 11 (43), 7621–7630. 10.1039/c3ob41659c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H.; Moroz E.; Castagner B.; Leroux J.-C. Activatable Cell Penetrating Peptide–Peptide Nucleic Acid Conjugate via Reduction of Azobenzene PEG Chains. J. Am. Chem. Soc. 2014, 136 (37), 12868–12871. 10.1021/ja507547w. [DOI] [PubMed] [Google Scholar]
- Breiman L. Random Forests. Mach. Learn. 2001, 45 (1), 5–32. 10.1023/A:1010933404324. [DOI] [Google Scholar]
- Liang G.; Liu Y.; Shi B.; Zhao J.; Zheng J. An Index for Characterization of Natural and Non-Natural Amino Acids for Peptidomimetics. PLoS One 2013, 8 (7), e67844. 10.1371/journal.pone.0067844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier M. F.; Criquet P.; Pantaloni D.; Korn E. D. Interaction of Cytochalasin D with Actin Filaments in the Presence of ADP and ATP. J. Biol. Chem. 1986, 261 (5), 2041–2050. [PubMed] [Google Scholar]
- Simon M. D.; Heider P. L.; Adamo A.; Vinogradov A. A.; Mong S. K.; Li X.; Berger T.; Policarpo R. L.; Zhang C.; Zou Y.; et al. Rapid Flow-Based Peptide Synthesis. ChemBioChem 2014, 15 (5), 713–720. 10.1002/cbic.201300796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijalis A. J.; Thomas D. A. III; Simon M. D.; Adamo A.; Beaumont R.; Jensen K. F.; Pentelute B. L. A Fully Automated Flow-Based Approach for Accelerated Peptide Synthesis. Nat. Chem. Biol. 2017, 13 (5), 464–466. 10.1038/nchembio.2318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.