

Abstract
Cancer vaccine aims to invoke antitumor adaptive immune responses to detect and eliminate tumors. However, the current dendritic cells (DCs)-based cancer vaccines have several limitations that are mostly derived from the ex vivo culture of patient DCs. To circumvent the limitations, direct activation and maturation of host DCs using antigen-carrying materials, without the need for isolation of DCs from patients, are required. In this study, we demonstrate the synthesis of extra-large pore mesoporous silica nanoparticles (XL-MSNs) and their use as a prophylactic cancer vaccine through the delivery of cancer antigen and danger signal to host DCs in the draining lymph nodes. Extra-large pores of approximately 25 nm and additional surface modification of XL-MSNs resulted in significantly higher loading of antigen protein and toll-like receptor 9 (TLR9) agonist compared with conventional small-pore MSNs. In vitro study showed the enhanced activation and antigen presentation of DCs and increased secretion of proinflammatory cytokines. In vivo study demonstrated efficient targeting of XL-MSNs co-delivering antigen and TLR9 agonist to draining lymph nodes, induction of antigen-specific cytotoxic T lymphocytes (CTLs), and suppression of tumor growth after vaccination. Furthermore, significant prevention of tumor growth after tumor rechallenge of the vaccinated tumor-free mice resulted, which was supported by a high level of memory T cells. These findings suggest that mesoporous silica nanoparticles with extra-large pores can be used as an attractive platform for cancer vaccines.
Short abstract
Cancer vaccine based on extra-large pore mesoporous silica nanoparticles was developed. The strong antigen-specific immune responses obtained after vaccination significantly prevented tumor growth even after tumor rechallenge.
Introduction
Cancer vaccine, a type of immunotherapy, aims to invoke antitumor adaptive immune responses to detect and eliminate tumors.1−4 The basic strategy of cancer vaccine treatment involves dendritic cells (DCs), the most potent professional antigen-presenting cells. DCs can uptake extracellular antigens and become mature forms that present epitopes as a complex with major histocompatibility complex (MHC) type I on the cell surface. The antigen-processed DCs migrate to lymph nodes to prime downstream immune effectors such as antigen-specific cytotoxic T lymphocytes (CTLs) through signaling between epitope–MHC I complex and T-cell receptors and costimulatory signaling between CD80/86 on DCs and CD28 on naïve T cells.5−7 Successful development of anticancer vaccines is centered on how to most effectively modulate DCs to promote the desired adaptive immune responses.
The conventional DC-based cancer vaccine is based on ex vivo culture and maturation of DCs derived from monocytes isolated from patient blood. The immature DCs that are differentiated from monocytes in the presence of a cytokine mixture are pulsed with tumor antigen together with an activating signal. Finally, the antigen-presenting mature DCs are injected back into the patient’s body, where they migrate to lymph nodes and invoke antigen-specific adaptive immunity that includes CTLs.8 However, the current DC-based cancer vaccines have several limitations that are mostly derived from the ex vivo culture of patient DCs, such as the requirement for expensive and laborious processes, a large burden to patients, and low therapeutic efficacy.9−12 To circumvent these limitations, a method for direct activation and maturation of host DCs using antigen-carrying materials, without the need for isolation of DCs from patients, has been recently researched.13−20
Mesoporous silica nanoparticles (MSNs) are a highly promising material platform for cancer vaccine because of their outstanding characteristics as delivery carriers, including tunable pore structure, easy modification of surface chemistry, and intrinsic biocompatibility.21 MSNs acted as an adjuvant and immunization of MSNs loaded with cancer antigen markedly inhibited the development of challenged and rechallenged cancer.22,23 Hollow MSNs loaded with both model tumor antigen and LPS or poly I:C adjuvant were demonstrated to prevent tumor growth.24,25 From the carrier point of view, however, the previously reported small pore size of around 3 nm for conventional MSNs has a limitation for loading the relatively large target molecules required in the formulation of cancer vaccine, such as antigenic proteins, cytokines, and whole tumor lysate. Although there have been recent reports on the preparation of large-pore MSNs using cosolvent,26 block-co-polymer27 and swelling agent,28 preparation of discrete large-pore MSNs with a high colloidal stability remains a challenge. We recently reported the synthesis of extra-large pore mesoporous silica nanoparticles (XL-MSNs) with 20–30 nm mesopores and their application to high loading of IL-4 cytokine delivery for macrophage modulation.29 The large pores of XL-MSNs are beneficial for loading of protein antigen with high molecular weight, and their surface modification for controlling the loading and release of diverse guest molecules is facile, which allows us to apply XL-MSNs for cancer immunotherapy by co-delivery of high amounts of biomolecules for modulating immune cells. Although the previous studies based on MSNs on co-delivery of antigen or/and adjuvant showed meaningful results that led tumor suppression and immune response,22−25 the higher loading capacity of XL-MSNs could allow less or minimal introduction of mesoporous silica materials into the body to activate the immune system, which could be a potential benefit in the clinical trials.
In this study, we demonstrate the use of XL-MSNs as a cancer vaccine through the delivery of protein antigen and danger signal to host DCs (Scheme 1). Extra-large pores of approximately 25 nm and additional surface modification of XL-MSNs resulted in significantly higher loading of antigenic protein and toll-like receptor 9 (TLR9) agonist compared with conventional small-pore MSNs. In vitro culture of bone-marrow-derived DCs (BMDCs) in the presence of XL-MSNs loaded with antigen and TLR9 agonist led to enhanced DC activation and antigen presentation and increased secretion of proinflammatory cytokines. An in vivo study demonstrated efficient targeting to draining lymph nodes, induction of antigen-specific CTLs, enhanced suppression of tumor growth after vaccination, and prevention of tumor growth after rechallenge of cancer cells into vaccinated mice due to a significant generation memory T cells.
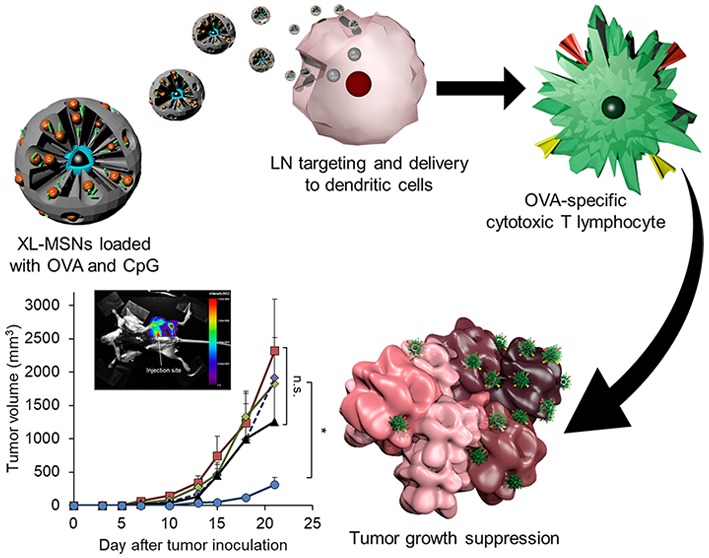
Scheme 1. Schematic Illustration of the Overall Vaccination Process Using Extra-Large Pore Mesoporous Silica Nanoparticles (XL-MSNs) Loaded with Antigen and TLR9 Agonist To Invoke Antigen-Specific Cytotoxic T Lymphocytes (CTLs) To Suppress Tumor Growth.

Results and Discussion
XL-MSNs were synthesized using a method based on our previous reports on the synthesis of uniform, discrete MSNs with 3 nm small pores30,31 with modification of the synthetic conditions to enlarge the mesopores.29 To prepare large mesopores while maintaining high colloidal stability in MSNs, we employed a large amount of ethyl acetate, an organic additive, as a pore expansion agent in the presence of cethyltrimethylammonium bromide (CTAB)-stabilized iron oxide nanoparticles and seed material in a silica sol–gel reaction. XL-MSNs showed extra-large, deep, furrow-like mesopores in transmission electron microscopy (TEM, Figure 1a) and scanning electron microscopy (SEM, Figure 1b). In contrast, conventional small-pore MSNs (S-MSNs) showed 3 nm small mesopores on the TEM image (Figure S1a). The pore size and pore volume of XL-MSNs were determined by N2 sorption (Figure 1c). Pore size distribution showed that XL-MSNs had bimodal pores that peaked at 3.2 nm and around 25 nm in the range between 10 and 30 nm, whereas S-MSNs had a single 3.2 nm peak (Figure S1b). The Brunauer–Emmett–Teller (BET) surface area and pore volume of XL-MSNs and S-MSNs were 686 and 768 m2 g–1, and 1.05 and 1.16 cm3 g–1, respectively (Figure S2). The size of the XL-MSNs was controllable from 100 to 200 nm by changing the amount of silica precursors (Figure S3). When the amount of the organic solvent, ethyl acetate, was increased, the morphology of the pore structure changed to become wider and thinner (Figure S4). We assume that the extra-large mesopores are constructed by what organic additive, the ethyl acetate, performs expanded emulsion in templating CTAB micelles.
Figure 1.

(a) TEM and (b) SEM images of extra-large pore mesoporous silica nanoparticles (XL-MSNs). (c) Nitrogen sorption isotherms and the corresponding pore size distribution obtained from adsorption branch of XL-MSNs. (d) Surface zeta potential of XL-MSNs-OH and XL-MSNs-NH2. The loading of (e) ovalbumin (OVA) and (f) CpG oligonucleotide in small-pore mesoporous silica nanparticles (S-MSNs) and XL-MSNs with initial hydroxyl surface group or with modified amine groups (n = 4).
To investigate the loading efficiency in XL-MSNs, we tested the adsorption of two biomacromolecules, ovalbumin (OVA) and CpG oligodeoxynucleotide, both important components in the formulation of cancer vaccine. The OVA protein was used as a model antigen. The unmethylated CpG oligodeoxynucleotide is a sequence of viral DNA that acts as an agonist to TLR9 in dendritic cells to enhance the expression of CD86, a costimulatory molecule required for the priming of CTLs together with the antigenic peptide–MHC I complex.32 As OVA is slightly negatively charged,23 and CpG is negatively charged33 in physiological conditions, to enhance the loading of both molecules, we modified the surface of XL-MSNs with amino groups via silane chemistry34,35 and tested the adsorption of OVA and CpG in XL-MSNs and S-MSNs with and without surface modification. After the amine modification, the surface zeta potential of amine-modified XL-MSNs (XL-MSNs-NH2) was shifted from −10.5 mV to +12.8 mV (Figure 1d), but the particle size and pore morphology were maintained (Figure S5a), representing successful surface modification with amino groups. Fourier transform infrared (FTIR) spectroscopy of the XL-MSNs-NH2 showed the existence of an N–H stretching peak (Figure S5b) and X-ray photoelectron spectroscopy (XPS) also determined the amine group and 2.285 atomic % of 1s nitrogen in the XL-MSNs-NH2 (Figure S5c). Loading of OVA and CpG in XL-MSNs and S-MSNs with and without surface modification shows that XL-MSNs-NH2 had the highest loading capacity compared with other conditions for both OVA (Figure 1e) and CpG (Figure 1f). This is probably due to the synergistic effect of enlarged pores and the positively charged surface in XL-MSNs-NH2. The cumulative releases of OVA and CpG from XL-MSN-NH2 were obtained over time (Figure S6), representing a sustained release of OVA and CpG with 56 and 27% release in 50 h, respectively. The slower release of CpG compared to OVA is presumably due to a higher negative charge of CpG and their stronger electrostatic interaction with positively charged XL-MSN-NH2.
We next tested intracellular delivery of OVA as a model antigen protein using XL-MSNs. Different concentrations of XL-MSNs labeled with rhodamine B isothiocyanate (RITC) were incubated with bone-marrow-derived dendritic cells (BMDCs) for 12 h. Flow cytometry analysis revealed that the cellular uptake of XL-MSNs increased with higher incubation concentration (Figure 2a). When RITC-labeled XL-MSNs loaded with fluorescein isothiocyanate (FITC)-labeled OVA and Cy5-labaled CpG were incubated with BMDCs, confocal microscope imaging clearly showed that the intracellular fluorescence of XL-MSNs, OVA, and CpG were observed and they were overlapped, representing that XL-MSNs delivered OVA and CpG into the intracellular region successfully (Figure 2b). As the amine-modified MSNs could contribute endosomal escape due to the proton sponge effect,36−38 cross-presentation of exogenous antigen into MHC class I could be enhanced. The concentration of XL-MSNs tested for cellular uptake was found to have low cytotoxicity (Figure 2c). Taken together, these results indicate that the XL-MSNs could be used as nanocarriers for intracellular delivery of a large amount of antigen and adjuvant without significant toxicity.
Figure 2.

(a) Flow cytometry analysis of bone-marrow-derived dendritic cells (BMDCs) cultured with different amounts of RITC-labeled XL-MSNs. (b) Fluorescent image of a BMDC incubated with RITC-labeled XL-MSNs loaded with FITC-labeled OVA protein and Cy5-labeled CpG. The scale bars in all panels indicate 10 μm. (c) Cytotoxicity of XL-MSNs at various concentrations.
To enhance expression of costimulatory molecules on DCs, we adsorbed CpG adjuvant on amino-modified XL-MSNs together with OVA antigen and performed flow cytometry and enzyme-linked immunosorbent assay (ELISA) for analysis of cell surface markers and cytokine secretion, respectively. The flow cytometry data showed that co-delivery of OVA and CpG resulted in higher CD11c+CD86+ cell populations of BMDCs compared with other conditions, where CD11c is a representative DC surface marker and CD86 is a costimulatory molecule that shows increased expression upon DC activation39 (Figure 3a). This result shows that incubation of BMDCs with XL-MSNs loaded with antigen and adjuvant led to a shift of DC phenotype from the immature to the mature state with upregulation of costimulatory molecules. Furthermore, XL-MSNs also led to mature DC, like adjuvant, which probably induced a synergetic immune stimulus effect on DCs along with CpG. Next, we investigated the expression of antigenic peptide (SIINFEKL)-major histocompatibility complex class I (MHC I) complexes on the DCs (Figure 3b). Compared with the OVA-only condition, XL-MSNs loaded with OVA generated higher levels of antigen-presenting DCs presumably due to enhanced intracellular uptake of OVA by XL-MSNs. When CpG was additionally incorporated into XL-MSNs, the highest level of antigen-presenting DCs was observed, demonstrating that XL-MSNs coloaded with antigen and TLR9 agonist resulted in the highest generation of antigen-presenting DCs in vitro.
Figure 3.

(a) Activated CD11c+CD86+ BMDCs (n = 4) and (b) BMDCs presenting antigenic SIINFEKL peptide on the MHC-molecule (n = 4) analyzed by flow cytometry. (c) Secreted TNF-α and IL-12 from BMDCs measured by ELISA (n = 4). Error bars, mean ± s.d. *P < 0.05.
Secretion of proinflammatory cytokines is another indicator of maturation of DCs and is critical for induction of CTLs.40 Mature DCs show higher secretion levels of interleukin 12 (IL-12) and tumor necrosis factor alpha (TNF-α) than immature DCs.41 Because IL-12 preferentially induces type 1 T helper (Th1) cells and TNF-α play an important role in the antitumor immune response, efficient secretion of IL-12 and TNF-α from mature DCs is helpful to overcome cancer. The amounts of TNF-α and IL-12 secreted by BMDCs incubated with different samples were measured by ELISA. Although the soluble OVA induced slightly increased levels of the cytokines, both XL-MSNs loaded with OVA and XL-MSNs coloaded with OVA and CpG showed significantly higher cytokine levels (Figure 3c). Taken together, these results suggest that XL-MSNs loaded with antigen and TLR 9 agonist have the potential to achieve enhanced DC maturation and antigen presentation.
The migration of antigen-carrying nanoparticles to lymph nodes (LNs) is essential for efficient induction of DC-T cell interactions and the subsequent antigen-specific CTL response to tumor in vivo.42,43 To investigate LN targeting of subcutaneously injected XL-MSNs, RITC-labeled XL-MSNs were injected into the left flank (Figure 4a) and footpad (Figure S7) of a C57BL/6 mouse, and the mouse was analyzed under fluorescent imaging. Strong fluorescent signals were observed in the draining LNs close to both injection sites, indicating that the injected XL-MSNs were transported to LNs from the injection site, probably via lymphatic flow due to their small size and high colloidal stability. Previous reports showed that nanoparticles smaller than 100 nm have a greater chance of being transported to LNs via lymphatic flow, whereas larger particles with submicron size mostly remain at the injection site.42,44 Successful delivery of antigens to DCs by targeting of XL-MSNs in LNs is advantageous for antigen presentation to naïve T cells in the LNs.45
Figure 4.

(a) Fluorescent images of mouse injected with RITC-labeled XL-MSNs subcutaneously on abdomen region, showing targeting of XL-MSNs to the draining lymph node (white dotted circle). (b) OVA-specific and (c) intracellular cytokine secreting CTLs in the spleens of vaccinated mice measured in flow cytometry (n = 6). Error bars, mean ± s.d. *P < 0.05. (d) Proliferation of CFSE-labeled OVA-specific CD8+ T cells in the lymph node (red line: XL-MSN + OVA + CpG, black line: control).
To investigate whether XL-MSNs can enhance CTL immune responses, we immunized C57BL/6 mice with soluble OVA, a mixture of soluble OVA and CpG, XL-MSNs loaded with OVA, or XL-MSNs coloaded with OVA and CpG. One week after immunization, the strength of the CD8+ T-cell response was probed by analyzing the frequency of tetramer+ CD8+ T cells (Figure 4b) and intracellular IFN-γ+ in CD8+ T cells (Figure 4c) in the splenocytes. Immunization with XL-MSNs coloaded with OVA and CpG induced proliferation of antigen-specific CD8+ T cells and IFN-γ secreting CD8+ T cells more efficiently than immunization with the mixture of soluble OVA and CpG. To evaluate antigen-specific CD8+ T-cell expansion in vivo, we adoptively transferred CFSE-labeled OT-I T-cells into control mice and mice immunized with the XL-MSNs coloaded with OVA and CpG. T cells in OT-I transgenic mice have T-cell receptors (TCRs) which recognize OVA peptide (SIINFEKL)-MHC I complex expressed on APCs to proliferate.46 LNs were isolated from recipient mice after 7 days adoptive transfer and CFSE-labeled OT-I T cells was analyzed by flow cytometry (Figure 4d). The significant dilution of CFSE signal intensity found in LNs of immunized mice indicated the enhanced T-cell proliferation, revealing that the immunization with the XL-MSNs-loaded with OVA and CpG successfully induced DCs expressing SIINFEKL-MHC I complex.
To demonstrate the potential of XL-MSNs as cancer vaccine, we investigated their tumor protective activity. Mice (C57BL/6, female, 6 weeks old) were subcutaneously vaccinated twice, with a 1-week interval between injections, with soluble OVA, a mixture of soluble OVA and CpG, XL-MSNs loaded with OVA, or XL-MSNs coloaded with OVA and CpG. One week after the second vaccination, OVA-expressing melanoma (B16-OVA, 1 × 106 cells/mouse) cells were inoculated subcutaneously into the right flank of the mice, and tumor development was monitored (Figure 5a). Soluble OVA alone or mixture of soluble OVA and CpG, which mimic conventional soluble vaccines, led to a slight suppression of tumor compared with the control, indicating that the antigen presentation by soluble vaccination was not sufficient to boost up the antigen-specific adaptive immune responses. Although vaccination with XL-MSNs + OVA resulted in smaller tumor size on average at latest time point compared to control, soluble OVA, OVA + CpG, there was no significant difference from other groups. In contrast, XL-MSNs coloaded with OVA and CpG led to significant inhibition of tumor growth compared with all other groups (Figure 5b). XL-MSNs coloaded with OVA and CpG resulted in the highest survival compared with other groups at the end of the study (Figure S8). These results represent that XL-MSNs could successfully deliver both antigenic information and TLR agonist into DCs and the resulting antigen-presenting DCs activated the antigen-specific adaptive immune response systemically. Previous studies on cancer immunotherapy using small pore MSNs have attempted multiple vaccination at least three times to demonstrate a strong prophylactic effect.23−25 In this study, however, the prophylactic vaccinations were given only two times with longer interval between vaccinations than previous studies, which resulted in comparable antitumor effect. Higher loading of both antigen and CpG in large mesopores of XL-MSNs based on their electrostatic interactions on the amine-surface led to successful delivery of immunological information without a significant loss prior to uptake into DCs, which probably affected to the induction of comparable tumor suppression even with less vaccination.
Figure 5.

(a) Schedule of prophylactic vaccination. Mice were vaccinated with soluble OVA, a mixture of OVA and CpG, XL-MSNs loaded with OVA, or amine-modified XL-MSNs loaded with OVA and CpG. (b) Tumor growth until day 21 after tumor inoculation (n = 8). Error bars, mean ± s.e.m. *P < 0.05. (c) Memory T cell population of CD4 and CD8 T cells in the spleens of vaccinated mice measured in flow cytometry (n = 5). Error bars, mean ± s.d. *P < 0.05. (d) Tumor-free mice (n = 4) after vaccination with XL-MSNs coloaded with OVA and CpG and the subsequent inoculation of B16-OVA tumor cells were rechallenged with the 1 × 106 of B16-OVA cells 15 days after the first inoculation. Error bars, mean ± s.d. *P < 0.05.
To further investigate if immune memory effect was induced after vaccination with XL-MSNs coloaded with OVA and CpG, the population of memory T cells was accessed by analyzing central memory T cells (TCM) and effector memory T cells (TEM) subset at 1 week after immunization (Figure 5c). TCM (CD44hiCD62L+) stays in secondary lymphoid organs has little or no function of effector, but continuously mediate proliferation and differentiation of effector cell, whereas TEM (CD44hiCD62L–) migrate to immune response site and show rapid effector function.47,48 The results showed that significantly higher CD4+ and CD8+ TEM cells and higher CD4+ TCM were generated in vaccination group than control mice. Based on this result, we designed a tumor rechallenge experiment in which naïve control mice or tumor-free mice after first prophylactic vaccination experiment were inoculated with additional B16-OVA cells (1 × 106 cells/mouse) and the tumor growth was monitored over time (Figure 5d). The significant resistance to the tumor growth upon tumor rechallenge was observed in vaccinated tumor-free mice group compared to fast tumor growth in the control group, suggesting that the memory T cells generated by immunization led to the suppression of tumor growth upon second challenge of tumor cells. Taken together, these results demonstrated that the XL-MSNs system facilitated co-delivery of OVA and CpG to DCs in LNs after immunization and subsequently induced a strong antigen-specific adaptive immune response to prevent tumor development.
Conclusions
In summary, we synthesized mesoporous silica nanoparticles with extra-large pores and tunable pore structure and particle size, which resulted in a high loading capacity of large biomolecules. Amine-modified XL-MSNs showed significantly higher loading of ovalbumin, a model protein antigen, and CpG oligonucleotide, a TLR9 agonist. The XL-MSNs successfully delivered the antigen protein and TLR9 agonist into the cytosol and led to enhanced maturation and antigen presentation of DCs. Subcutaneously injected XL-MSNs were transported from the injection site to LNs in the animal study. Finally, vaccination with XL-MSNs loaded with antigen and TLR9 agonist substantially stimulated adaptive immune responses including antigen-specific cytotoxic T cells and subsequently suppressed tumor growth in a prophylactic tumor model. Our findings, combined with the known biodegradability and tunable physicochemical properties of mesoporous silica nanoparticles, suggest that mesoporous silica nanoparticles can be used as an attractive platform for cancer immunotherapy in the future.
Materials and Methods
Materials and Antibodies
Rhodamine B isothiocyanate (RITC), (3-aminopropyl)trimethoxysilane (APTMS), cethyltrimethylammonium bromide (CTAB), ammonium hydroxide, tetraethylorthosilicate (TEOS), and ovalbumin (OVA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). HCl, methanol, and ethyl acetate were purchased from SamChun Chemical (Seoul, Korea). Bovine serum albumin (BSA) was purchased from Millipore (Billerica, MA, USA), CpG oligodeoxynucleotide was purchased from Bioneer (Daejeon, Korea). Recombinant murine GM-CSF was purchased from Peprotech (Rocky Hill, NJ, USA). Antibodies against the following proteins were used: CD11c-APC, MHC class-II-FITC, CD86-Vioblue, MHC class-I(H-2Kb)/SIINFEKL-PE-Vio770, CD3e-FITC, CD4-PE-Vio770, CD8-APC, IFN-γ-FITC, CD44-Vioblue, and CD62L-PE were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany) and H-2Kb/SIINFEKL tetramer-PE was purchased from MBL life science (Woburn, MA). IL-12 and TNF-α ELISA kits were purchased from BD science.
Synthesis of XL-MSNs and RITC-labeled XL-MSNs
The synthetic procedure was based on the previously reported methods to prepare MSNs.29,30 Iron oxide nanoparticles prepared by heat-up process based on iron-oleate complex49 were dispersed in chloroform in a concentration of 6.0 mg Fe/mL that was measured by an inductively coupled plasma mass spectrometer (ICP-MS, Varian 820-MS, Varian). 0.5 mL of iron oxide nanoparticles in chloroform were mixed into 10 mL of 0.055 M CTAB aqueous solution under vigorous stirring for 30 min. The mixture was heated to 60 °C and kept at this temperature with stirring for 15 min. After being cooled down to room temperature, the resulting solution was added to a mixed solvent of 95 mL of DI water, 5 mL of methanol, 3 mL of ammonium hydroxide, and 20 mL of ethyl acetate with stirring, and then 300 μL of TEOS was added to the reaction solution. The reaction solution was stirred for 12 h. The as-synthesized XL-MSNs were washed three times with excess ethanol and stored in 40 mL ethanol. To extract CTAB, the pore template micelle, the as-synthesized XL-MSNs solution was adjusted to pH 1.6 and stirred for 3 h at 60 °C. After being washed three times with ethanol, the XL-MSNs were dispersed in 20 mL ethanol. The resulting XL-MSNs can be also supplied from Porous Nanoparticle Bank (PNB 210005). To prepare RITC-labeled XL-MSNs, 50 μL of RITC-APTMS stock solution, that was prepared by reacting 0.02 mmol RITC with 0.2 mmol APTMS (molar ratio = 1:10) in 0.75 mL ethanol (anhydrous, 99.9%) under dark conditions for 1 day, was added along with TEOS in the reaction procedure described above. To modify the surface functional groups of the XL-MSNs to amine groups, 20 mL of stock solution of XL-MSNs in ethanol were diluted 2-fold, and 23 μL of APTMS was added with stirring. After reaction for 12 h, the resulting solution was washed three times with excess ethanol and stored in 10 mL of ethanol.
Loading of OVA and CpG in XL-MSNs
The intact or amine-modified XL-MSNs in ethanol were centrifuged, washed three times with PBS, and dispersed in PBS. To load OVA, the samples (400 μL, 6.25 mg mL–1 in PBS) were mixed with OVA (400 μL, 7.5 mg mL–1 in PBS), and the mixture was incubated on the rotator at room temperature for 2 h. The mixture was centrifuged at 11000 rpm for 5 min and washed three times with PBS. During the washing, whole supernatant was separately collected from each sample. The unloaded OVA in the collected supernatants was measured using a UV–vis spectrometer (Varioskan LUX, Thermo Scientific) at absorbance 280 nm. To load CpG, the samples (250 μL, 4 mg mL–1 in PBS) were mixed with CpG (250 μL, 20 mg μL–1 in PBS), and the resulting solution was rotated at room temperature for 1 h. After adsorption was complete, the mixture was centrifuged at 11 000 rpm for 5 min, washed three times, and the supernatants were collected for measurement by UV–vis spectrometer. The amount of CpG in the supernatant was measured at a wavelength of 260 nm. The loading amounts of OVA and CpG were calculated by subtraction of unloaded amounts from the initial amounts of molecules mixed with MSN samples.
In Vitro Cell Cytotoxicity Test
HCT-116 cells (4 × 104 cells well–1) were initially seeded in 96-well plates in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin and cultured in 5% CO2 at 37 °C for 12 h. Different amounts of XL-MSNs were added to the cells and incubated for 24 and 72 h. The cells were incubated with MTT (500 μg mL–1) at 37 °C for 3 h, and the culture supernatant was removed. MTT formazan was solubilized with DMSO, and absorbance at 560 nm was measured.
Isolation and Culture of Bone-Marrow-Derived Dendritic Cells
Bone-marrow-derived dendritic cells (BMDCs) were prepared from the hind limbs of C57BL/6 mice according to the previous report with a slight modification.50 The BMDC culture media was RPMI media supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, 0.05 mM β-mercaptoethanol, and 20 ng mL–1 granulocyte-macrophage colony-stimulating factor (GM-CSF). The number of cells was adjusted to 2 × 106 cells mL–1 in culture media, and 10 mL of the adjusted cell suspension was added to a 100 mm Petri dish and cultured in a CO2 incubator (37 °C, 5% CO2). After 3 days, another 10 mL of the fresh culture media was added to each of the prepared Petri dishes. After a further 3 days, 7–8 mL of cells suspended in media was collected, centrifuged at 450 g for 5 min at 4 °C, re suspended in 10 mL of fresh culture media, and returned to the original Petri dish.
In Vitro Intracellular Delivery and Cellular Uptake
BMDCs (1 × 106 cells) were precultured in 6-well plates for 12 h and incubated with different amounts of RITC-labeled XL-MSNs (0–500 μg) or RITC-labeled XL-MSNs loaded with FITC-OVA and Cy5-CpG for 12h. After incubation, the nuclei were stained by DAPI, and the colored fluorescence in cells was detected under fluorescence microscopy (Dragonfly 302, Andor) and flow cytometry (MACSQuant VYB, Miltenyi Biotec).
In Vitro BMDC Activation, Antigen Expression and Cytokine Secretion Measurement
To determine BMDC activation and antigen expression, (1) 10 μg of OVA, (2) 10 mg of XL-MSNs loaded with 10 μg of OVA, and (3) 10 mg of XL-MSNs loaded with 10 μg of OVA and 0.1 μg of CpG were incubated with 1 × 106 BMDCs in 6-well plates for 12 h and analyzed by flow cytometry. The supernatant of cell culture media was collected, and TNF-α and IL-12 concentrations in the supernatant were measured by ELISA.
Adoptive Transfer Study
2 × 107 splenocytes were isolated from donor OT-I mice, labeled with CFSE and adoptively transferred intravenously into the recipient mice. On day 1, recipient mice were vaccinated subcutaneously 100 mg of XL-MSNs loaded with 100 μg of OVA and 1 μg of CpG. 7 days after vaccination, recipient mice were sacrificed, and LN was isolated and analyzed for T-cell expansion by flow cytometry.
In Vivo Cytotoxic T Lymphocyte (CTL) Response
(1) 100 μg of OVA, (2) 100 μg of OVA and 0.1 μg of CpG, (3) 100 mg of XL-MSNs loaded with 100 μg of OVA, and (4) 100 mg of XL-MSNs loaded with 100 μg of OVA and 1 μg of CpG were administered to mice by subcutaneous injection. Spleens were isolated 1 week after injection and homogenized. The isolated CTL were stained by anti-CD8, IFN-γ antibodies, and H-2Kb SIINFEKL-tetramer and analyzed by flow cytometry.
In Vivo Immune Memory Effect Study
100 mg of XL-MSNs loaded with 100 μg of OVA and 1 μg of CpG were administered to mice by subcutaneous injection. Spleens were isolated 1 week after injection and homogenized. The isolated T lymphocytes were stained by anti-CD4, CD8, CD44, and CD62L antibodies and analyzed by flow cytometry.
In Vivo Vaccination and Tumor Rechallenge
The first prophylactic vaccination was administered to 6 weeks old female C57BL/6 mice (DBL, Korea) by subcutaneous injection. After 1 week, the second vaccination was performed 1 week prior to tumor implantation. On day 14, B16-OVA tumor cells (1 × 106) were inoculated into the right flanks of C57BL/6 mice. The tumor volumes were measured with a caliper every 3 days until the 21th day after tumor implantation. The tumor volume was calculated using the following formula: tumor volume (mm3) = length × (width)2/2. For tumor rechallenge, tumor-free mice at day 15 were rechallenged with 1 × 106 B16-OVA tumor cells in the opposite flank and the tumor volumes were measured until the 2 weeks after rechallenge. All animal experiments were conducted by following the guidelines of National Institutes of Health (NIH), with the approval of SKKU School of Pharmacy Institutional Animal Care and Use Committee.
Acknowledgments
This work was supported by grants funded by the National Research Foundation (NRF) under the Ministry of Science, ICT & Future Planning, Republic of Korea (2014M3A9B8023471, 2010-0027955), and a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI17C0076).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00035.
TEM images, and other supporting figures and tables (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Finn O. J. Cancer vaccines: between the idea and the reality. Nat. Rev. Immunol. 2003, 3, 630–641. 10.1038/nri1150. [DOI] [PubMed] [Google Scholar]
- Kim J.; Mooney D. J. In vivo modulation of dendritic cells by engineered materials: towards new cancer vaccines. Nano Today 2011, 6, 466–477. 10.1016/j.nantod.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler G.; Schuler-Thurner B.; Steinman R. M. The use of dendritic cells in cancer immunotherapy. Curr. Opin. Immunol. 2003, 15, 138–147. 10.1016/S0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- Steinman R. M.; Banchereau J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- Pardoll D. M. Cancer vaccines. Nat. Med. 1998, 4, 525–531. 10.1038/nm0598supp-525. [DOI] [PubMed] [Google Scholar]
- Heath W. R.; Carbone F. R. Cross–presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 2001, 19, 47–64. 10.1146/annurev.immunol.19.1.47. [DOI] [PubMed] [Google Scholar]
- Cresswell P.; Ackerman A. L.; Giodini A.; Peaper D. R.; Wearsch P. A. Mechanisms of MHC class–I restricted antigen processing and cross-presentation. Immunol. Rev. 2005, 207, 145–157. 10.1111/j.0105-2896.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- Jahnisch H.; Fussel S.; Kiessling A.; Wehner R.; Zastrow S.; Bachmann M.; Rieber E. P.; Wirth M. P.; Schmitz M. Dendritic cell-based immunotherapy for prostate cancer. Clin. Dev. Immunol. 2010, 2010, 517493. 10.1155/2010/517493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R. H.; Kroll A. V.; Zhang L. Nanoparticle–Based Manipulation of Antigen–Presenting Cells for Cancer Immunotherapy. Small 2015, 11, 5483–5496. 10.1002/smll.201501284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa E.; Nair S. K.; Lyerly H. K. Immunotherapy of cancer with dendritic-cell-based vaccines. Cancer Immunol. Immunother. 1998, 46, 82–87. 10.1007/s002620050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacken P. J.; de Vries I. J. M.; Torensma R.; Figdor C. G. Dendritic–cell immunotherapy: from ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- Banchereau J.; Palucka A. K. Dendritic cells as therapeutic vaccines against cancer. Nat. Rev. Immunol. 2005, 5, 296–306. 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- Moon J. J.; Suh H.; Bershteyn A.; Stephan M. T.; Liu H.; Huang B.; Sohail M.; Luo S.; Um S. H.; Khant H.; Goodwin J. T.; Ramos J.; Chiu W.; Irvine D. J. Interbilayer–crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat. Mater. 2011, 10, 243–251. 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Li Y.; Jiao J.; Hu H. Alpha-alumina nanoparticles induce efficient autophagy–dependent cross-presentation and potent antitumour response. Nat. Nanotechnol. 2011, 6, 645–650. 10.1038/nnano.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuai R.; Ochyl L. J.; Bahjat K. S.; Schwendeman A.; Moon J. J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017, 16, 489–496. 10.1038/nmat4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamachari Y.; Salem A. K. Innovative strategies for co–delivering antigens and CpG oligonucleotides. Adv. Drug Delivery Rev. 2009, 61, 205–217. 10.1016/j.addr.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine D. J.; Hanson M. C.; Rakhra K.; Tokatlian T. Synthetic nanoparticles for vaccines and immunotherapy. Chem. Rev. 2015, 115, 11109–11146. 10.1021/acs.chemrev.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Xu Y.; Tian Y.; Chen C.; Wang C.; Jiang X. Functional nanomaterials can optimize the efficacy of vaccines. Small 2014, 10, 4505–4520. 10.1002/smll.201401707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Li W. A.; Choi Y.; Lewin S. A.; Verbeke C. S.; Dranoff G.; Mooney D. J. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat. Biotechnol. 2015, 33, 64–72. 10.1038/nbt.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Li X.; Ito A.; Sogo Y.; Ohno T. Particle-size-dependent toxicity and immunogenic activity of mesoporous silica–based adjuvants for tumor immunotherapy. Acta Biomater. 2013, 9 (7), 7480–7489. 10.1016/j.actbio.2013.03.031. [DOI] [PubMed] [Google Scholar]
- Tang F.; Li L.; Chen D. Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery. Adv. Mater. 2012, 24, 1504–1534. 10.1002/adma.201104763. [DOI] [PubMed] [Google Scholar]
- Mahony D.; Cavallaro A. S.; Stahr F.; Mahony T. J.; Qiao S. Z.; Mitter N. Mesoporous Silica Nanoparticles Act as a Self–Adjuvant for Ovalbumin Model Antigen in Mice. Small 2013, 9, 3138–3146. 10.1002/smll.201300012. [DOI] [PubMed] [Google Scholar]
- Wang X.; Li X.; Yoshiyuki K.; Watanabe Y.; Sogo Y.; Ohno T.; Tsuji N. M.; Ito A. Comprehensive Mechanism Analysis of Mesoporous–Silica–Nanoparticle-Induced Cancer Immunotherapy. Adv. Healthcare Mater. 2016, 5, 1169–1176. 10.1002/adhm.201501013. [DOI] [PubMed] [Google Scholar]
- Wang X.; Li X.; Ito A.; Watanabe Y.; Sogo Y.; Tsuji N. M.; Ohno T. Stimulation of in vivo antitumor immunity with hollow mesoporous silica nanospheres. Angew. Chem., Int. Ed. 2016, 55, 1899–1903. 10.1002/anie.201506179. [DOI] [PubMed] [Google Scholar]
- Wang X.; Li X.; Ito A.; Yoshiyuki K.; Sogo Y.; Watanabe Y.; Yamazaki A.; Ohno T.; Tsuji N. M. Hollow Structure Improved Anti–Cancer Immunity of Mesoporous Silica Nanospheres In Vivo. Small 2016, 12 (26), 3510–3515. 10.1002/smll.201600677. [DOI] [PubMed] [Google Scholar]
- Niu D.; Ma Z.; Li Y.; Shi J. Synthesis of core–shell structured dual-mesoporous silica spheres with tunable pore size and controllable shell thickness. J. Am. Chem. Soc. 2010, 132, 15144–15147. 10.1021/ja1070653. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Xu P.; Chen H.; Li Y.; Bu W.; Shu Z.; Li Y.; Zhang J.; Zhang L.; Pan L.; Cui X.; Hua Z.; Wang J.; Zhang L.; Shi J. Colloidal HPMO Nanoparticles: Silica–Etching Chemistry Tailoring, Topological Transformation, and Nano–Biomedical Applications. Adv. Mater. 2013, 25, 3100–3105. 10.1002/adma.201204685. [DOI] [PubMed] [Google Scholar]
- Kim M.; Na H.; Kim Y.; Ryoo S.; Cho H. S.; Lee K. E.; Jeon H.; Ryoo R.; Min D. Facile synthesis of monodispersed mesoporous silica nanoparticles with ultralarge pores and their application in gene delivery. ACS Nano 2011, 5, 3568–3576. 10.1021/nn103130q. [DOI] [PubMed] [Google Scholar]
- Kwon D.; Cha B. G.; Cho Y.; Min J.; Park E.; Kang S.; Kim J. Extra-Large Pore Mesoporous Silica Nanoparticles for Directing in Vivo M2 Macrophage Polarization by Delivering IL–4. Nano Lett. 2017, 17, 2747–2756. 10.1021/acs.nanolett.6b04130. [DOI] [PubMed] [Google Scholar]
- Kim J.; Lee J. E.; Lee J.; Yu J. H.; Kim B. C.; An K.; Hwang Y.; Shin C.; Park J.; Kim J.; Hyeon T. Magnetic fluorescent delivery vehicle using uniform mesoporous silica spheres embedded with monodisperse magnetic and semiconductor nanocrystals. J. Am. Chem. Soc. 2006, 128, 688–689. 10.1021/ja0565875. [DOI] [PubMed] [Google Scholar]
- Kim J.; Kim H. S.; Lee N.; Kim T.; Kim H.; Yu T.; Song I. C.; Moon W. K.; Hyeon T. Multifunctional uniform nanoparticles composed of a magnetite nanocrystal core and a mesoporous silica shell for magnetic resonance and fluorescence imaging and for drug delivery. Angew. Chem., Int. Ed. 2008, 47, 8438–8441. 10.1002/anie.200802469. [DOI] [PubMed] [Google Scholar]
- Klinman D. M. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol. 2004, 4, 249–259. 10.1038/nri1329. [DOI] [PubMed] [Google Scholar]
- Kumar M. R.; Bakowsky U.; Lehr C. Preparation and characterization of cationic PLGA nanospheres as DNA carriers. Biomaterials 2004, 25, 1771–1777. 10.1016/j.biomaterials.2003.08.069. [DOI] [PubMed] [Google Scholar]
- Hartono S. B.; Gu W.; Kleitz F.; Liu J.; He L.; Middelberg A. P.; Yu C.; Lu G. Q.; Qiao S. Z. Poly-L-lysine functionalized large pore cubic mesostructured silica nanoparticles as biocompatible carriers for gene delivery. ACS Nano 2012, 6, 2104–5117. 10.1021/nn2039643. [DOI] [PubMed] [Google Scholar]
- Kim T.; Kim M.; Eltohamy M.; Yun Y.; Jang J.; Kim H. Efficacy of mesoporous silica nanoparticles in delivering BMP–2 plasmid DNA for in vitro osteogenic stimulation of mesenchymal stem cells. J. Biomed. Mater. Res., Part A 2013, 101, 1651–1660. 10.1002/jbm.a.34466. [DOI] [PubMed] [Google Scholar]
- Slowing I.; Trewyn B. G.; Lin V. S.-Y. Effect of surface functionalization of MCM–41–type mesoporous silica nanoparticles on the endocytosis by human cancer cells. J. Am. Chem. Soc. 2006, 128, 14792–14793. 10.1021/ja0645943. [DOI] [PubMed] [Google Scholar]
- Huang D. M.; Hung Y.; Ko B. S.; Hsu S. C.; Chen W. H.; Chien C. L.; Tsai C. P.; Kuo C. T.; Kang J. C.; Yang C. S.; Mou C. Y.; Chen Y. C. Highly efficient cellular labeling of mesoporous nanoparticles in human mesenchymal stem cells: implication for stem cell tracking. FASEB J. 2005, 19, 2014–2016. 10.1096/fj.05-4288fje. [DOI] [PubMed] [Google Scholar]
- Huang X.; Zhuang J.; Teng X.; Li L.; Chen D.; Yan X.; Tang F. The promotion of human malignant melanoma growth by mesoporous silica nanoparticles through decreased reactive oxygen species. Biomaterials 2010, 31, 6142–6153. 10.1016/j.biomaterials.2010.04.055. [DOI] [PubMed] [Google Scholar]
- Fong L.; Engleman E. G. Dendritic cells in cancer immunotherapy. Annu. Rev. Immunol. 2000, 18, 245–273. 10.1146/annurev.immunol.18.1.245. [DOI] [PubMed] [Google Scholar]
- Märten A.; Ziske C.; Schöttker B.; Renoth S.; Weineck S.; Buttgereit P.; Schakowski F.; von Rücker A.; Sauerbruch T.; Schmidt-Wolf I. G. Interactions between dendritic cells and cytokine–induced killer cells lead to an activation of both populations. J. Immunother. 2001, 24, 502–510. 10.1097/00002371-200111000-00007. [DOI] [PubMed] [Google Scholar]
- Onishi H.; Kuroki H.; Matsumoto K.; Baba E.; Sasaki N.; Kuga H.; Tanaka M.; Katano M.; Morisaki T. Monocyte-derived dendritic cells that capture dead tumor cells secrete IL–12 and TNF−α through IL–12/TNF−α/NF−κB autocrine loop. Cancer Immunol. Immunother. 2004, 53, 1093–1100. 10.1007/s00262-004-0568-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy S. T.; van der Vlies A. J.; Simeoni E.; Angeli V.; Randolph G. J.; O’Neil C. P.; Lee L. K.; Swartz M. A.; Hubbell J. A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- Hotaling N. A.; Tang L.; Irvine D. J.; Babensee J. E. Biomaterial strategies for immunomodulation. Annu. Rev. Biomed. Eng. 2015, 17, 317–349. 10.1146/annurev-bioeng-071813-104814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy S. T.; Rehor A.; Schmoekel H. G.; Hubbell J. A.; Swartz M. A. In vivo targeting of dendritic cells in lymph nodes with poly (propylene sulfide) nanoparticles. J. Controlled Release 2006, 112, 26–34. 10.1016/j.jconrel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Figdor C. G.; de Vries I. J. M.; Lesterhuis W. J.; Melief C. J. Dendritic cell immunotherapy: mapping the way. Nat. Med. 2004, 10, 475–480. 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- Jung S.; Unutmaz D.; Wong P.; Sano G.; De los Santos K.; Sparwasser T.; Wu S.; Vuthoori S.; Ko K.; Zavala F.; Pamer E. G.; Littman D. R.; Lang R. A. In vivo depletion of CD11c dendritic cells abrogates priming of CD8 T cells by exogenous cell-associated antigens. Immunity 2002, 17, 211–220. 10.1016/S1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzierska K.; Valkenburg S. A.; Doherty P. C.; Davenport M. P.; Venturi V. Use it or lose it: establishment and persistence of T cell memory. Front. Immunol. 2012, 3, 357. 10.3389/fimmu.2012.00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F.; Geginat J.; Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- Park J.; An K.; Hwang Y.; Park J.; Noh H.; Kim J.; Park J.; Hwang N.; Hyeon T. Ultra–large–scale syntheses of monodisperse nanocrystals. Nat. Mater. 2004, 3, 891–895. 10.1038/nmat1251. [DOI] [PubMed] [Google Scholar]
- Lutz M. B.; Kukutsch N.; Ogilvie A. L.; Rößner S.; Koch F.; Romani N.; Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 1999, 223, 77–92. 10.1016/S0022-1759(98)00204-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.