

Abstract
Through the study of structure–property relationships using a combination of experimental and computational analyses, a number of phenoxazine derivatives have been developed as visible light absorbing, organic photoredox catalysts (PCs) with excited state reduction potentials rivaling those of highly reducing transition metal PCs. Time-dependent density functional theory (TD-DFT) computational modeling of the photoexcitation of N-aryl and core modified phenoxazines guided the design of PCs with absorption profiles in the visible regime. In accordance with our previous work with N,N-diaryl dihydrophenazines, characterization of noncore modified N-aryl phenoxazines in the excited state demonstrated that the nature of the N-aryl substituent dictates the ability of the PC to access a charge transfer excited state. However, our current analysis of core modified phenoxazines revealed that these molecules can access a different type of CT excited state which we posit involves a core substituent as the electron acceptor. Modification of the core of phenoxazine derivatives with electron-donating and electron-withdrawing substituents was used to alter triplet energies, excited state reduction potentials, and oxidation potentials of the phenoxazine derivatives. The catalytic activity of these molecules was explored using organocatalyzed atom transfer radical polymerization (O-ATRP) for the synthesis of poly(methyl methacrylate) (PMMA) using white light irradiation. All of the derivatives were determined to be suitable PCs for O-ATRP as indicated by a linear growth of polymer molecular weight as a function of monomer conversion and the ability to synthesize PMMA with moderate to low dispersity (dispersity less than or equal to 1.5) and initiator efficiencies typically greater than 70% at high conversions. However, only PCs that exhibit strong absorption of visible light and strong triplet excited state reduction potentials maintain control over the polymerization during the entire course of the reaction. The structure–property relationships established here will enable the application of these organic PCs for O-ATRP and other photoredox-catalyzed small molecule and polymer syntheses.
Graphical abstract
INTRODUCTION
Increased interest in photoredox catalysis for small molecule and macromolecular synthesis during the past decade has led to the development of new catalytic transformations using mild conditions.1 Critical to these advancements has been the use of ruthenium complexes, which were first demonstrated as photoredox catalysts (PCs) several decades ago,2 and iridium complexes, both of which are capable of absorbing visible light to initiate electron or energy transfer reactions from their reactive photoexcited states. The impressive performance of these catalysts arises from their redox stability and photo-physical properties including visible light absorption and formation of excited states that can engage in both reductive and oxidative electron transfers.1,3 Despite the success of ruthenium and iridium-containing PCs, the use of these transition metal complexes in catalysis has some limitations. For example, the use of ruthenium and iridium-containing PCs in polymer synthesis can limit the application scope for the materials synthesized since these complexes can be challenging to remove from the polymer matrix leading to contamination of the final polymer product. Polymeric materials contaminated with transition metals may not be suitable for biomedical or electronic applications, which limits the broad use of these synthetic methods. As such, the development of organic PCs is desirable.4 While organic PCs that are strong excited state reductants have recently been reported,5 it is imperative to establish the structure–property relationships and molecular design principles that govern the photophysical and catalytic capabilities of these organic molecules to develop new PCs, expand their use in photoredox catalysis, and enable the development of new transformations.
Our work in the field of photoredox catalysis originated with our interest in organocatalyzed atom transfer radical polymerization (O-ATRP), a method to synthesize well-defined polymers using organic PCs activated by UV or visible light. The O-ATRP method originated with the application of UV light-absorbing N-phenyl phenothiazine,5b or visible light absorbing perylene5e as organic PCs for the polymerization of vinyl monomers. N-phenyl phenothiazine is proposed to access a highly reducing excited state and operate via an oxidative quenching pathway analogous to previously used iridium-catalyzed ATRP systems.6 Perylene was the first example of a visible light-absorbing PC for O-ATRP, but it is less efficient compared to these other PC families.5e We have also investigated N,N-diaryl dihydrophenazines5f and N-aryl phenoxazines5g as PCs for O-ATRP. N,N-Diaryl dihydrophenazines absorb visible light to access highly reducing excited states with computationally predicted triplet excited state reduction potentials (E0*T1,calc[2PC•+/3PC*]) that can exceed −2.0 V versus SCE. Furthermore, it has been shown that N,N-diaryl dihydrophenazines bearing electron poor or highly conjugated N-aryl substituents access charge transfer (CT) excited states, a property that is proposed to engender superior performance compared to related systems that do not access such states.7 The CT excited states of these molecules are analogous to the metal-to-ligand charge transfer (MLCT) excited states of transition metal complexes. Similar to how transition metal complexes transfer electron density from an electron-rich metal center to an electron-deficient ligand to access MLCT states, N,N-diaryl dihydrophenazines transfer electron density from the electron-rich tricyclic phenazine core to one or both N-aryl “ligand(s)” to access intramolecular CT excited states if the N-aryl substituents are sufficient electron acceptors (exhibit a low-lying π*). Following this work, we explored structurally similar N-aryl phenoxazines, which typically absorb UVA light to access excited states with similar reduction potentials to those of N,N-diaryl dihydrophenazines (E0*T1,calc ∼ −1.8 to −2.0 V vs SCE). Computational modeling of N-aryl phenoxazines predicted that derivatives bearing N-naphthyl substituents possess spatially separated singularly occupied molecular orbitals (SOMOs) in the triplet excited state, a feature that is characteristic of CT species.7 These N-naphthyl phenoxazine derivatives were found to be effective PCs for O-ATRP.
Despite the excellent performance of N-naphthyl phenoxazine PCs in O-ATRP, we aimed to design visible light-absorbing derivatives because UV light can initiate undesirable side reactions in ATRP.8 We reasoned that visible light-absorbing phenoxazines could be realized through extending the conjugation on the phenoxazine core via installation of biphenyl core substituents.5g For organic light-emitting diodes (OLEDs), core modified phenoxazines were reported for use as donor–acceptor molecules.9 Guided by this precedence for synthetic modification of the phenoxazine core, we synthesized visible light absorbing PC 1 via installation of biphenyl groups at the 3- and 7-positions of the phenoxazine core of 2 (Figure 1A). Indeed, PC 1 absorbs visible light and catalyzes the O-ATRP of methacrylate monomers to synthesize polymers of target MWs with improved control compared to the noncore modified parent compound (2). In addition, PC 1 exhibits robust catalytic performance for O-ATRP carried out in flow reactors10 and in varied reaction irradiation conditions.11 More recently, we demonstrated that PC 1 was also effective in catalyzing small molecule transformations such as atom transfer radical additions, substitution trifluoromethylations, and dual photoredox/nickel-catalyzed C–N and C–S cross-coupling reactions that were previously exclusive to transition metal complexes.12 Given that the phenoxazine core structure can be synthetically modified and that 1 exhibits excellent catalytic performance for numerous transformations, we envisioned a versatile scaffold for the development of superior organic PCs using core modified N-aryl phenoxazines.
Figure 1.
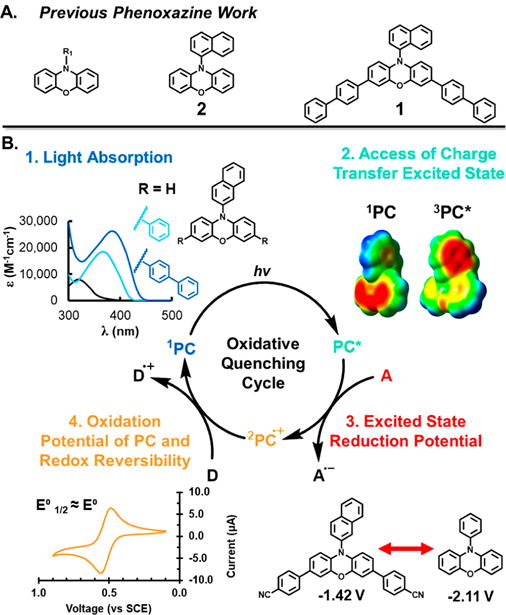
(A) Our previous work with phenoxazine PCs focused on noncore modified PCs and core modified PC 1. (B) Proposed general photoredox cycle that proceeds via an oxidative quenching pathway. 1PC: ground state PC; PC*: excited state PC, which can be either in the singlet (1PC*) or triplet (3PC*) excited state; 2PC•+: radical cation of the photoredox catalyst.
While our previous work with PC 1 demonstrated the ability to alter the absorption properties of N-aryl phenoxazines, broadening the reaction scope and increasing the selectivity of reactions catalyzed by these molecules may require further alteration of their redox properties. In photoredox catalysis employing ruthenium and iridium PCs, alteration of the redox properties of these catalysts has been achieved by changing the electronics of the ligand set.1,13 Analogous to transition metal PCs, we hypothesized that alteration of the electronics of the phenoxazine scaffold via modification of the N-aryl and core substituent(s) would enable the redox properties of the parent catalyst to be tuned. In this work, we investigate this hypothesis as well as characterize the absorption properties and ability of core modified phenoxazines to access CT excited states. We experimentally and computationally characterize the properties of four previously reported phenoxazines (PCs 1-3 and 6) and 15 new N-aryl core modified phenoxazines (PCs 4, 5, and 7-19) to identify structure–property relationships for tailoring organic PC properties (Figure 2). In addition, these new phenoxazine derivatives are employed as PCs for photoredox-catalyzed polymer synthesis via O-ATRP to demonstrate their catalytic activity and to understand how differences in PC properties manifest in differences in catalytic performance in O-ATRP. To the best of our knowledge, this is the first report that characterizes the structure–property relationships of phenoxazine derivatives in the context of photoredox catalysis, and we envision that the design principles established herein will encourage the discovery of photoredox-catalyzed transformations using these PCs.
Figure 2.
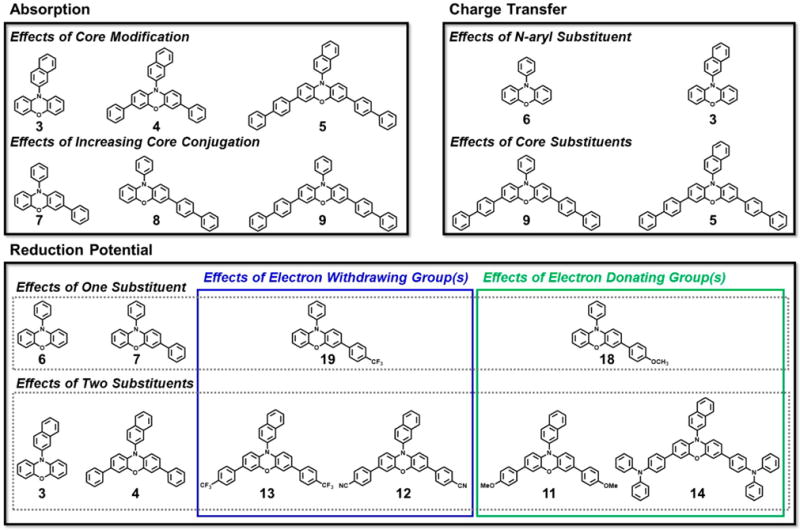
Schematic representation of the subsets of catalysts compared in this work to identify structure–property relationships. The absorption profiles of several phenoxazines were investigated to determine the effects that core modification has on photon absorption in these molecules in order to design visible light-absorbing derivatives (top left). The ability of noncore-modified and core-modified phenoxazines to access CT excited states was evaluated computationally and spectroscopically (top right). Alteration of the reduction potentials of N-aryl phenoxazines via mono- and dicore substitutions was explored; key comparisons within this group included investigating the effects of electron-withdrawing groups (highlighted in blue) and electron-donating groups (highlighted in green).
RESULTS AND DISCUSSIONS
Desirable PC features can be identified with guidance from the photophysical and redox processes involved in a photoredox-catalyzed oxidative quenching cycle (Figure 1B). PCs that absorb visible light are preferred over ultraviolet (UV) light absorbers as UV light can cause undesirable side reactions. Moreover, PCs that possess high molar extinction coefficients (ε) absorb light more efficiently which may increase the population of the reactive excited state catalyst. In general, upon light absorption, the PC is promoted to a singlet excited state (Sn, n ≥ 1), whereupon, in accordance with Kasha’s rule, it quickly relaxes via internal conversion (IC) to S1. From S1, triplet states (Tn, n ≥ 1) can be accessed via intersystem crossing (ISC). Similar to the singlet manifold, higher-energy triplets (Tn) relax via IC to the lowest-energy T1. Electron transfer processes pertinent to photoredox catalysis can occur from either the S1 or T1 state; however, the longer-lived T1 state is typically invoked to be responsible for photoredox catalysis when its initiation requires bimolecular collision events.
The ability to access an excited state with CT character in which electron density has been shifted from a donor moiety to an acceptor moiety in the molecule has been identified in both transition metal and organic PCs (Figure 1B). Polypyridyl ruthenium and iridium complexes are known to access metal to ligand charge transfer (MLCT) T1 excited states, where energy wasting charge recombination is slowed, leading to improved catalytic efficiency.2,14 Analogous to the MLCT states of transition metal complexes, several of the organic molecules that our group7 and others15 have investigated are capable of accessing CT excited states. Regardless of whether the PC exhibits CT character in the excited state, it can transfer an electron to a substrate acting as an electron acceptor (A) and enable further reaction. The ability of the photoexcited catalyst (PC*) to transfer an electron is partially dictated thermodynamically by the excited state reduction potential (E0*). Following the oxidative electron transfer reaction, the catalytic cycle is completed when the oxidized PC (radical cation [2PC•+]) is reduced back to the ground state (1PC) by an electron donor (D).
The four major steps of the catalytic cycle (Figure 1B): (1) light absorption, (2) access of a CT excited state, (3) excited state reduction potential, and (4) oxidation potential and redox reversibility were used as criteria to investigate how structural modification of organic excited state reductants impact PC properties. The fundamental questions guiding investigation of these catalyst properties were as follows: (I) How does core modification affect photoexcitation events in N-aryl phenoxazines and how does extending the conjugation of the phenoxazine core affect the absorption profiles of these molecules? (II) How does core modification affect the ability of N-aryl phenoxazines to access CT excited states? (III) How does alteration of the electronics of the core substituents affect the excited and ground state redox properties and redox reversibility of core modified phenoxazines? To answer these questions, we designed and investigated three series of phenoxazine derivatives (Figure 2). Comparison of PCs within each of these series was used to test our hypotheses corresponding to each of the aforementioned questions.
Light Absorption
In our initial design of PC 1, we hypothesized that installation of aryl core substituents would stabilize the π* orbitals involved in photon absorption, allowing for photoexcitation using lower-energy visible light (Figure 1A). This hypothesis was corroborated by observing that installation of biphenyl substituents onto the core of PC 2 red shifts the maximum wavelength of absorption (λmax,abs) from 323 to 388 nm (PC 1) with an absorption profile that tails into the visible regime. Very recently, a similar approach was applied to develop visible light absorbing phenothiazine-based PCs.16 Given that PCs that absorb visible light rather than UV light are more desirable for minimization of side reactions,1b,8 we were driven to better understand how these structural modifications fundamentally affect photon absorption. Herein, we disclose studies on how systematic core modification alters the energies of the π* orbitals of phenoxazine PCs, which in turn affects the wavelength of light absorbed and energies of the S1 and T1 states (Figure 2). Furthermore, the energies of these states dictate the reducing power of the PC in each respective state.
Time-dependent density functional theory (TD-DFT) calculations at the CAM-B3LYP/6-31+g(d,p) level of theory were employed to understand the orbitals involved in the λmax,abs of photoexcitation for phenoxazine derivatives with increasing aryl conjugation on the core, namely noncore-modified PC 3, phenyl core-modified PC 4, and biphenyl core-modified PC 5 (Figure 3). We note that 2-naphthalene is used as the N-aryl substituent for the PCs investigated in this work, rather than the 1-naphthalene substitution used previously, in order to expand our overall understanding of these types of systems, and the noncore-substituted derivative (3) was shown to synthesize polymers with low dispersity (Đ) via O-ATRP.5g
Figure 3.
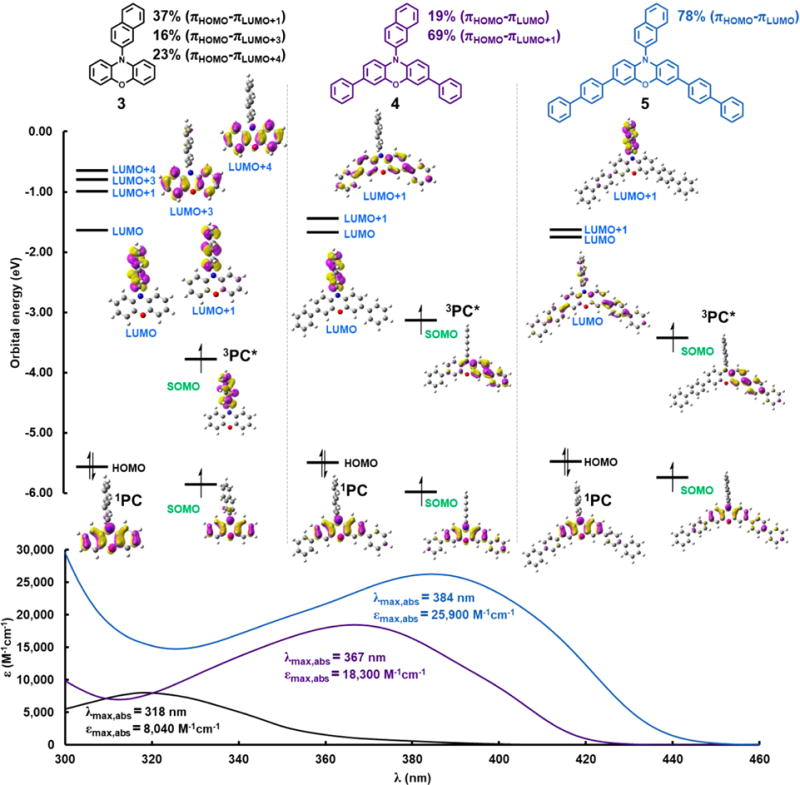
(Top) Time-dependent density functional theory calculations of orbitals involved in photoexcitation of PCs 3, 4, and 5 at their corresponding λmax, abs. Computationally predicted percentage contribution of corresponding orbitals involved in photoexcitation at the λmax, abs are also presented. The zero point on the orbital axis is defined as the electron in a vacuum. (Bottom) UV–vis spectra of each PC acquired in N,N-dimethylacetamide (DMAc) with the observed λmax, abs and εmax, abs shown.
As can be seen with PCs 3, 4, and 5, regardless of aryl core substituents, the highest occupied molecular orbital (HOMO) is localized on the phenoxazine core with lowest-energy excitation involving these πHOMO electrons (Figure 3). Further, these aryl core substituents do not appreciably perturb the energy of the πHOMO, as demonstrated by the similar calculated energies of −5.57 eV, −5.49 eV, and −5.46 eV for PCs 3, 4, and 5, respectively. However, aryl core substituents do significantly change the energies and nature of the low-lying πLUMO orbitals. For PC 3, which lacks core substituents, the energies of unoccupied π orbitals involved in photoexcitation are −0.99 eV (πLUMO+1), −0.79 eV (πLUMO+3), and −0.63 eV (πLUMO+4). PC 3 was experimentally determined to absorb primarily UVA light (λmax,abs = 318 nm, Figure 3), which is computationally predicted to be the result of excitation from πHOMO into the aforementioned high-lying orbitals, namely πLUMO+1, πLUMO+3, and πLUMO+4, with respective contributions of 37%, 16%, and 23%. In PC 4, core substitution with phenyl groups introduces a new lower-lying πLUMO+1 orbital (−1.43 eV) with character inclusive of the phenoxazine core’s π* with extended conjugation into the phenyl substituents. Excitation from πHOMO into this low-lying πLUMO+1 (69% contribution) is corroborated by the observed red shift of λmax,abs to 367 nm. A similar trend is observed for PC 5, but with a notably larger effect, where biphenyl substituents exert greater stabilization on the phenoxazine core’s π* to yield an even lower-lying πLUMO (−1.74 eV). Thus, the observed λmax,abs is red-shifted even further to 384 nm (contributed by 78% πHOMO–πLUMO transition). These analyses and observations highlight the importance of aryl core substituents for the design of visible light-absorbing PCs.
In addition to red-shifting the λmax, abs, core modification also increases the εmax of PC 3 from 8040 M−1cm−1 to 18300 M−1 cm−1 (PC 4) and 25900 M−1cm−1 (PC 5), improving the efficiency of photoexcitation (Figure 3, bottom). This observation motivated us to determine trends in how structural modifications affect the efficiency of photon absorption in phenoxazine PCs. As a simple phenoxazine scaffold, phenyl-10-phenoxazine (PC 6) was decorated with a single phenyl (7) or biphenyl (8) core substituent. Systematically increasing the aryl conjugation on the core position increases the εmax value from 6580 M−1 cm−1 in PC 7, to 10300 M−1cm−1 in PC 8 (Figure 4A). A further increase of εmax was achieved with installation of a second biphenyl core substituent (PC 9) which more than doubles the εmax of PC 8 (εmax = 24000 M−1cm−1 for 9). These data are supported by higher values in the computationally predicted oscillator strength (f) for these PCs (Table S2).
Figure 4.

(A) UV–vis absorption spectra acquired in DMAc for N-phenyl phenoxazines. (B) UV–vis absorption spectra for N-2-naphthyl phenoxazines with electron-withdrawing, neutral, and donating core substituents. The light gray area indicates the UV regime of the electromagnetic spectrum.
Previously, we showed that N-2-naphthyl phenoxazine (3) exhibits superior catalytic performance in O-ATRP (as indicated by producing polymeric material with lower Đ, higher I*, and with a more linear growth of polymer molecular weight as a function of conversion) compared to N-phenyl phenoxazine (6), which we proposed is due to the ability of noncore-modified N-naphthyl phenoxazines to access CT excited states.5g The better performance of PC 3, motivated investigation of the effects of core modification on the absorption profiles of N-2-naphthyl phenoxazines (Figure 4B). Core modification with electron-donating 4-methoxy-phenyl groups (PC 11) leads to a 4 nm blue-shift relative to PC 4, while core modification with aryl electron-withdrawing substituents has the opposite effect, and the λmax,abs of PCs 12 (R= 4-cyanophenyl) and 13 (R= 4-trifluoromethylphenyl) are red-shifted 44 and 21 nm, respectively (Figure 4B and Figures S64–S66). In fact, the cyano-containing PC 12 exhibits the most red-shifted absorption spectrum of the phenoxazines investigated, being the only PC with a λmax,abs in the visible regime (λmax,abs = 411 nm, ε = 22,300 M−1cm−1). Exceptions to these trends are observed in the absorption profiles of PCs 14, 15, and 16 (Figures S67, S62, and S63, respectively, and Table S3). PC 14, which possesses electron-donating diphenyl amino core substituents, does not exhibit a blue shift in absorption compared to PC 4. Installation of more highly conjugated phenanthracenyl (15) and pyrenyl (16) core substituents does not lead to a red shift of the λmax,abs compared to PC 4. In these cases, in-depth evaluation of the destabilizing or stabilizing effects of aryl core-substituents on πcore (HOMO) and π*core (LUMOs) have to be considered to determine the net effect on λmax,abs. Ultimately, the design of core-modified phenoxazines enabled 12 visible light absorbing N-aryl phenoxazine PCs to be realized with red-shifted λmax,abss compared to noncore-modified derivatives, with one derivative exhibiting a λmax,abs = 411 nm (PC 12). In addition, these derivatives exhibit εmaxs typically greater than 10000 M−1cm−1, with one derivative exhibiting a εmax of 37700 M−1cm−1 (PC 14).
Access of Charge Transfer Excited State
Previously, we observed that N,N-diaryl dihydrophenazine PCs, which are structurally similar to N-aryl phenoxazines, are capable of accessing CT excited states.7 In the lowest excited state of these compounds, CT occurs from the electron-rich dihydrophenazine core (electron donor) to the N-aryl substituents (electron acceptor), given that the substituents possess a low-lying π* orbital (e.g., 2-naphthyl). We empirically observed that N,N-diaryl dihydrophenazine PCs that exhibit CT character perform better in O-ATRP, which we posit is due, in part, to minimization of unproductive back electron transfer.7 In our previous work with N-aryl phenoxazines, we observed a similar trend. Specifically, PCs with N-naphthyl substituents (PCs 1, 2, and 3) are computationally predicted to possess spatially separated SOMOs in the triplet excited state (for the SOMOs of PC 3 see Figure 3), and these PCs (1, 2, and 3) perform better in O-ATRP compared to those that do not exhibit this feature (PC 6).5g However, experimental characterization of the nature of CT in phenoxazine PCs was only recently investigated using PC 1.12 In the current work, CT character in core and noncore modified N-phenyl and N-2-naphthyl phenoxazines is explored.
To investigate the nature of the excited states of N-aryl phenoxazines, the properties of 3PC*, 1PC*, and 1PC for 3 and 6 were explored. Electrostatic potential (ESP)-mapped electron density diagrams were used to predict the distribution of electron density for PCs 3 and 6 in their ground and triplet excited states (Figure 5A). In PC 6, similar electron density distributions (depicted in red) are observed in both 1PC and 3PC*, which indicates a lack of CT character. We reason that the N-phenyl substituent in PC 6 does not possess a π* that is low enough in energy to accept an electron from the phenoxazine core in the excited state, thus preventing CT. On the contrary, PC 3, which possesses a lower-lying 2-naphthyl π* orbital, exhibits a shift in electron density for 1PC versus the 3PC*, suggesting that the T1 state has CT character with transfer of electron density to the 2-naphthyl substituent and depletion on the phenoxazine core.
Figure 5.
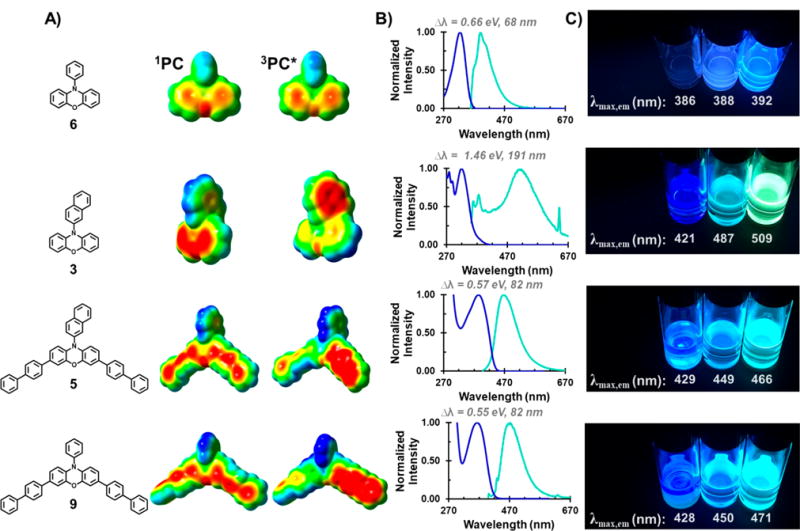
(A) Electrostatic potential (ESP)-mapped electron density of phenoxazine PCs 3, 5, 6, and 9 are shown for the singlet ground state (left) and triplet excited state (right). (B) Overlays of the absorption profiles acquired in DMAc (dark blue) and emission profiles acquired in DMAc (turquoise) with the experimentally determined Stokes Shifts (Δλ) are shown for each PC. (C) Photographs of the PCs dissolved in solvents with increasing polarity, as indicated by their increasing Reichardt parameter (ET(30) in kcal/mol). From left to right: 1-hexene (ET(30) = 32.4 kcal/mol), THF (ET(30) = 37.4 kcal/mol), and DMAc (ET(30) = 42.9 kcal/mol). The maximum wavelength of emission (λmax,em) of PCs 3, 5, 6, and 9 acquired in each of these solvents is shown below the corresponding scintillation vial in the photograph. For the full emission spectra of PCs 3, 5, 6, and 9 in these solvents, please see Figures S89–S92.
For these same molecules, fluorescence studies enable exploration of CT character (Figure 5B). The observed emission in PCs 3 and 6 is likely from the S1 state since the emission is not quenched by the presence of oxygen, a known triplet quencher. On one hand, PC 6 exhibits a small Stokes shift, Δλ (difference between its λmax,abs [324 nm] and λmax,em [392 nm]), in DMAc [Δλ = 68 nm (0.66 eV)] with a sharp and featured emission peak. In contrast, PC 3 exhibits a larger Stokes shift in DMAc [Δλ = 191 nm (1.46 eV)] and a broad and featureless emission profile, a hallmark of a CT emission.7,15 These data suggest that the S1 state of PC 3, possessing significant CT character, is qualitatively different from that of PC 6.
Further support for the assignment of CT character in the excited state is the observance of fluorescence solvatochromism with changing solvent polarity.17 The intramolecular transfer of electron density in CT molecules typically creates a larger molecular dipole in the excited state (compared to the ground state), which is more stabilized in polar solvents, leading to lower energy emission. To qualitatively and quantitatively determine the presence or absence of solvatochromic behavior in PCs 6 and 3, the emission of these PCs was studied in three solvents (1-hexene, tetrahydrofuran [THF], and DMAc) of varying polarity, as indicated by their different Reichardt polarity parameters, ET(30), expressed in kcal per mole [ET(30) = 32.4, 37.4, 42.9, respectively, Figure 5C].17 While PC 3 clearly exhibits solvatochromic behavior, as indicated by a change in the color of its emission in different solvents, PC 6 qualitatively appears to emit the same color. Moreover, the wavelength of emission for PC 6 was measured in these three solvents and found to shift by no more than 6 nm or 0.05 eV (Figure S89). In contrast, the λmax,em for PC 3 red shifts 66 nm (0.40 eV) going from 1-hexene to THF and another 22 nm (0.11 eV) going from THF to DMAc (Figure S90). The emission profiles, Stokes shifts, and solvatochromic behavior of PCs 6 and 3 are analogous to that of N,N-diphenyl and N,N-dinaphthyl dihydrophenazines, respectively, suggesting that noncore modified N-aryl phenoxazines must also be designed with an N-aryl substituent possessing a low-lying π* to yield PCs with accessible CT states.
After gaining an understanding of the nature of CT in 3 and 6, we were motivated to determine if core modification affects the ability of N-aryl phenoxazines to access CT excited states (Figure 5). To do this, we performed a similar analysis of PCs 5 and 9, which are core modified derivatives of PCs 3 and 6, respectively. We initially hypothesized that the excited states of these core-modified derivatives would mimic that of the parent molecules such that PC 5 accesses an excited state with CT character and exhibits similar emission to PC 3 while PC 9 exhibits no CT character. However, in comparing PC 5 in the ground state and T1 state, the ESP diagrams show that electron density has moved from the phenoxazine core in the ground state to one of the biphenyl core substituents in the triplet excited state. This observation is qualitatively different from PC 3, in which CT is predicted to be toward the N-naphthyl substituent in the triplet excited state. PC 5 exhibits bathochromic emission behavior similar to PC 3 but to a lesser extent. For PC 5, its λmax,em red shifts from 1-hexene [ET(30) = 32.4 kcal/mol] to THF [ET(30) = 37.4 kcal/mol] by 20 nm and exhibits another 17 nm red shift in DMAc [ET(30) = 42.9 kcal/mol, Figure S91]. In addition, PC 5 exhibits a smaller Δλ (82 nm, 0.57 eV in DMAc) than PC 3, suggesting a smaller energy difference between the initially excited Sn state and the relaxed S1 state. In concert, these findings refute our original hypothesis by indicating that PC 5 can access a CT excited state that is qualitatively distinct from PC 3. This conclusion is corroborated by the emission data of PC 9. To our surprise, the λmax,em, Δλ, and solvatochromic behavior of PC 9 is similar to that of PC 5, indicating that it can access a CT excited state despite bearing an N-phenyl group rather than an N-naphthyl group (Figure 5 and Figure S92). These observations support the notion that photon emission in PCs 5 and 9 is CT in nature but is tied to the phenoxazine core and its biphenyl substituents.
Excited State Reduction Potential
An inherent challenge in the design of visible light absorbing PCs lies in lowering the energy of photoexcitation without sacrificing excited state reducing power. Ideally, one would like to lower the energy of the Sn state for visible excitation while maintaining similar energies for the S1 or T1 state required for excited state redox chemistry with similar reactivity. This principle was applied to design visible light absorbing phenoxazine PCs that are still strongly reducing in their excited states. For example, noncore-modified PC 3 absorbs in the UV regime but core-modified PC 5 absorbs in the visible regime, indicating that photoexcitation is to higher energy Sn states for PC 3 compared to PC 5. Since the T1 state is typically assigned as the reactive species in photoredox-catalyzed transformations requiring bimolecular collisions, the triplet excited state reduction potentials of these PCs (E0*T1,calc) were computationally calculated (Figure 6). The E0*T1, calc was determined to be −1.90 V versus SCE5g for UV-light absorbing PC 3 and somewhat lower (−1.70 V vs SCE) for visible light absorbing PC 5 (Figure 6). Even though the E0*T1, calc is lower for PC 5, this value is still on par with some of the most reducing transition metal PCs such as fac-Ir(ppy)3, which exhibits a triplet excited state reduction potential of −1.73 V versus SCE.1b This example demonstrates that the strong reducing power of N-aryl phenoxazines in the excited state can still be maintained after core modification is used to lower the energy of light required for photoexcitation.
Figure 6.
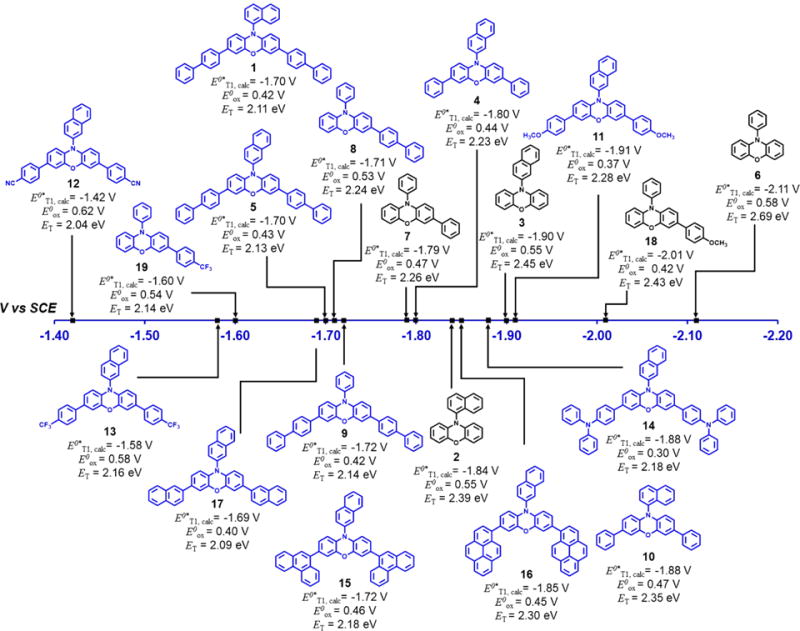
Structures, calculated triplet excited state reduction potentials (E0*T1,calc[PC•+/3PC*]), calculated oxidation potential of 2PC•+ (E0ox=E0[2PC•+/1PC]), and calculated triplet energies (ET) of phenoxazines investigated in this study. Catalysts colored in gray are UVA light absorbing and catalysts colored in blue are visible light absorbing.
In addition to designing visible light absorbing PCs that are strong excited state reductants, we sought to design PCs with a range of excited state reduction potentials. The triplet excited state reduction potential (E0*T1) is defined as E0*−[2PC•+/3PC*] = E0[2PC•+/1PC] − ET, where ET is the triplet energy (the difference in energy between T1 excited state and S0 ground state) and E0ox = E0[2PC•+/1PC] is the potential to oxidize 1PC to 2PC•+. Thus, for catalysts with similar triplet energies, those that are easier to oxidize will be more strongly reducing in the excited state. We envisioned that the excited state reduction potentials (E0*s) of N-aryl core modified phenoxazines could be tuned through alteration of the electronics of the core substituents which would in turn alter the values of E0ox, the singlet energy (ES), or ET and lead to alteration of the singlet and triplet E0*s.
To investigate the range of E0*s that can be achieved with the phenoxazine PC scaffold, core-modified PCs bearing electron-donating and electron-withdrawing groups (4, 5, and 7–19) were synthesized and their triplet excited state reduction potentials (E0*T1, calc) were calculated using computationally determined values for ET and E0ox (Figure 6). It is important to note that aryl core substituents were chosen for the design of these derivatives in pursuit of making them visible light absorbers (vide supra). Core modification of PCs 6 and 3 with aryl substituents (4, 5, 7–9, and 11–19) lowers the computationally calculated ETs compared to the parent compounds but with ETs still exceeding 2.0 eV in all cases (Figure 6). Core modification of PCs 6 and 3 with electron-withdrawing trifluoromethyl (13, 19) or cyano (12) substituents, or highly conjugated (15, 16) substituents, yields PCs with lower ETs that are also more difficult to oxidize. Consequently, these PCs exhibit more positive values of E0*T1, calc rendering them less reactive PCs in the excited state. We reason that derivatives bearing electron-withdrawing groups exhibit these redox properties because electron-withdrawing core substituents shift electron density away from the phenoxazine core making them more challenging to oxidize to the 2PC•+. Conversely, installation of electron-donating methoxy (11, 18) or diphenyl amino (14) core substituents leads to PCs with higher ETs and are easier to oxidize (exhibit lower E0ox values), making them stronger excited state reductants (as indicated by their more negative E0*s).
In addition to computationally calculating the triplet excited state reduction potentials, we experimentally determined the singlet excited state reduction potentials (E0*S1, exp[2PC•+/1PC*]) using a combination of fluorescence spectroscopy and electrochemistry (Table 1). This analysis is particularly useful for PCs capable of accessing CT excited states in which the donor and acceptor likely exhibit minimal electronic coupling as the S1 and T1 states in these systems are nearly degenerate, such that the singlet E0*S1 approximates the triplet E0*T1.7,18 The singlet energies determined from fluorescence emission (which correspond to emission from the S1 state and are denoted as ES1,exp in Table 1) were found to be smaller than the computationally calculated T1 energies in two cases (for PC 10 and PC 16). We note that the differences in these cases are 0.02 and 0.10 eV, which is within the expected error of ±0.2 eV for DFT calculations. We also note that while S1 and T1 energies are close to degenerate for species that exhibit extensive charge transfer, the S1 and T1 energy splitting gets increasing larger as the extent of CT decreases.
Table 1.
Redox Properties of Core Modified Phenoxazines
| PC | λmax,em (nm)a | ES1, exp (eV)b | ET1, calc (eV)c | E1/2 (2PC•+/1PC) (V vs SCE)d | E0ox (2PC•+/1PC) (V vs SCE)c | E0*S1, exp (2PC•+/1PC*) (V vs SCE)e | E0*T1, calc (2PC•+/3PC*) (V vs SCE)c |
|---|---|---|---|---|---|---|---|
| N-phenyl PCs | |||||||
| 7 | 427 | 2.90 | 2.26 | 0.65 | 0.47 | −2.25 | −1.79 |
| 8 | 472 | 2.63 | 2.24 | 0.64 | 0.53 | −1.99 | −1.71 |
| 9 | 471 | 2.63 | 2.14 | 0.62 | 0.42 | −2.01 | −1.72 |
| 18 | 412 | 3.01 | 2.43 | 0.60 | 0.42 | −2.41 | −2.01 |
| 19 | 476 | 2.60 | 2.14 | 0.69 | 0.54 | −1.91 | −1.60 |
| N-(1-naphthyl) PCs | |||||||
| 1f | 506 | 2.45 | 2.11 | 0.65 | 0.42 | −1.80 | −1.70 |
| 10 | 532 | 2.33 | 2.35 | 0.64 | 0.47 | −1.69 | −1.88 |
| N-(2-naphthyl) PCs | |||||||
| 4 | 514 | 2.41 | 2.23 | 0.64 | 0.44 | −1.77 | −1.80 |
| 5 | 466 | 2.66 | 2.13 | 0.63 | 0.43 | −2.03 | −1.70 |
| 11 | 532 | 2.33 | 2.28 | 0.52 | 0.37 | −1.81 | −1.91 |
| 12 | 508 | 2.44 | 2.04 | 0.69 | 0.62 | −1.75 | −1.42 |
| 13 | 467 | 2.65 | 2.16 | 0.72 | 0.58 | −1.93 | −1.58 |
| 14g | 522 | 2.37 | 2.18 | 0.54 | 0.30 | −1.83 | −1.88 |
| 15g | 489 | 2.53 | 2.18 | 0.67 | 0.46 | −1.86 | −1.72 |
| 16g | 564 | 2.20 | 2.30 | 0.66 | 0.45 | −1.54 | −1.85 |
| 17 | 466 | 2.66 | 2.09 | 0.63 | 0.40 | −2.03 | −1.69 |
Emission wavelength measured using fluorescence spectroscopy in DMAc. See the Supporting Information for more details.
Singlet energies were calculated using the maximum wavelength of emission.
DFT calculations performed at the uM06/6-311+G(d,p)//uM06/6-31+G(d,p) level of theory with CPCM-described solvation in aqueous solvent.
All measurements were performed in a 3-compartment electrochemical cell with an Ag/AgNO3 reference electrode in MeCN (0.01 M) and 0.1 M NBu4PF6 electrolyte solution. DMAc was used to solvate the PCs and in the working electrode compartment, while platinum was used as both the working and counter electrodes. E (V vs SCE) = E (V vs Ag/AgNO3 [0.01 M]) + 0.298 V.
Singlet excited state reduction potentials were calculated using the singlet energies (estimated from the maximum wavelength of emission) and the E1/2.
Values for the properties of PC 1 were taken from ref 12.
Due to the extensive molecular structure, frequency calculations of these compounds were computed at the uM06/6-31G(d,p)/CPCM-H2O level of theory.
The experimentally determined singlet excited state reduction potential (E0*S1, exp) values were found to follow the same trends as the computationally calculated triplet reduction potentials (E0*T1, calc). Namely, PCs bearing electron-donating core substituents were more strongly reducing in the singlet excited state and typically more easily oxidized in the ground state while the opposite trends were observed for PCs bearing electron-withdrawing core substituents. We note that the E1/2 (2PC•+/1PC) values determined computationally (E0ox) are systematically ∼0.2 eV less positive than the values determined experimentally, which demonstrates the utility of these computational calculations as a predictive tool for this application. From the computational and experimental data, we demonstrate that core-modified derivatives exhibit a wide range of excited state reduction potentials with experimentally determined E0*S1,exp values spanning from −1.54 V to −2.25 V versus SCE and computationally predicted E0*T1,calc values spanning from −1.42 V to −2.01 V versus SCE. Notably, E0*S1,exp values of PCs 11 and 14 were experimentally determined to be −1.81 V and −1.83 V versus SCE, respectively, with E0*T1,calc values of −1.91 V and −1.88 V vs SCE, respectively, making them some of the strongest visible light absorbing organic excited state reductants reported to date. For example, these PCs are on par with highly reducing organic PCs such as UV-absorbing phenyl phenothiazine (E0*S1,exp = −2.10 V vs SCE)4c and visible light-absorbing N,N-diaryl dihydrophenazines (E0*T1,calc ∼ −2.2 V vs SCE).5f
Oxidation Potential of PC and Redox Reversibility
After the photoexcited catalyst reduces a substrate in an oxidative quenching catalytic cycle, the PC is oxidized to a radical cation, 2PC•+. To complete the catalytic cycle, the 2PC•+ must be reduced to regenerate 1PC. Therefore, the efficiency of the catalytic cycle is reliant to a large degree on the ability of the 2PC•+ to engage in rapid and reversible electron transfer reactions. Previously, it was shown that PC 1 exhibits a reversible cyclic voltammogram in DMAc for the 2PC•+/1PC redox couple and that electron transfer reactions involving this PC and the electrode are diffusion-limited.12 To evaluate electron transfer reactions involving the phenoxazine derivatives explored in this work, CV was performed to analyze their oxidation potentials (Table 1) and redox reversibility (Figure 7 and Figures S43–57). The oxidation potential of each PC was approximated from its half wave potential, where E0ox[2PC•+/1PC] ≈ E1/2[2PC•+/1PC].19 In particular, we sought to determine whether installation of electron-donating, electron-withdrawing, or highly conjugated core substituents on phenoxazine PCs affects the reversibility of electron transfer processes. Additionally, the cyclic voltammograms of the PCs were acquired at different scan rates to gain insight into the kinetics of electron transfer processes involving these PCs.
Figure 7.
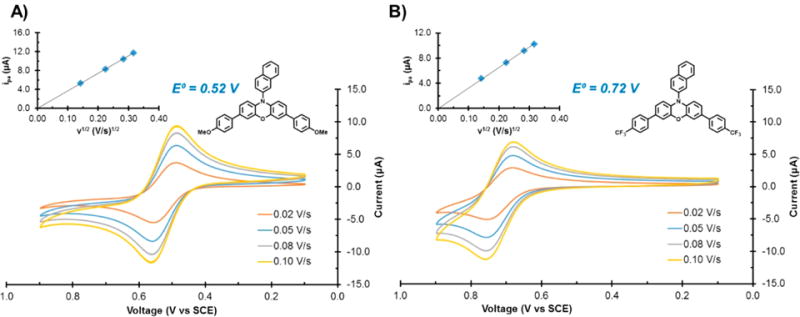
Cyclic voltammograms (CVs) of (A) PCs 11 and (B) 13 acquired at different scan rates 0.02 V/s, 0.05 V/s, 0.08 V/s, and 0.10 V/s.
PCs with electron-donating core substituents transfer electron density to the oxidized phenoxazine core and thus stabilize formation of the 2PC•+, such that they are more easily oxidized (E1/2= ∼ 0.50 to 0.60 V vs SCE for PCs 11 and 14) compared to those bearing electronically neutral core substituents or no core substituents (E1/2 = ∼0.60 to 0.69 V vs SCE for PCs 4, 5, 7–10, and 15–18). These PCs are observed to exhibit reversible cyclic voltammograms when acquired in DMAc (Figures S43–S48 and Figures S51–S56). A representative example is the cyclic voltammogram of PC 11, in which the difference between the anodic and cathodic peak potentials (ΔEp) is 75 mV (compared to the theoretical value of 59 mV for a reversible system) and the ratio of peak anodic current (ipa) to peak cathodic current (ipc) is 1.07 (compared to a theoretical value of 1 for a reversible system) when acquired with a scan rate of 0.05 V/s (Figure 7A, blue trace). The reversibility of this redox couple is maintained at different scan rates (0.02 V/s to 0.10 V/s) across which the ΔEp (65–78 mV) and ipa/ipc values are also consistent (1.06–1.07).
Although 2PC•+’s with electron-withdrawing core substituents (PCs 12, 13, and 19) are more destabilized (E1/2 > 0.69), reversible CVs are still observed for these species (Figures S49, S50, and S57). For example, the most oxidizing 2PC•+ (the radical cation of PC 13) exhibits a reversible CV with a ΔEp of 78 mV in DMAc (Figure 7B). The ipa/ipc could not be accurately determined for this catalyst in DMAc due to the proximity of its oxidation potential to that of the solvent window. Therefore, CV with PC 13 was also conducted in THF for which the ipa/ipc is 1.10 at the same scan rate of 0.05 V/s (Figure S57). To evaluate whether electron transfer reactions between the PC and the electrode are faster than the rate of diffusion of the PC, the cyclic voltammograms were acquired at different scan rates ranging from 0.02 V/s to 0.10 V/s. A linear relationship between the ipa and the square root of the scan rate (ν1/2) is observed for both PCs 11 and 13 (Figure 7, insets). This observation indicates that the electron transfer process involving these PCs and the electrode is fast and diffusion-limited. Overall, these studies demonstrate that regardless of the nature of the substituents installed onto the phenoxazine core, all of the derivatives explored herein engage in reversible and diffusion-limited electron transfer reactions with an electrode.
Application of Core Modified Phenoxazines as PCs in O-ATRP
After establishing structure–property relationships for the phenoxazines presented in this work, we aimed to better understand how differences in the phenoxazines’ properties manifest in differences in catalytic performance in O-ATRP. We first compared the performance of noncore modified phenoxazines 3 and 6 (Figure 6) and then extended this comparison to core-modified derivatives of these molecules. Next, we compared the performance of all the N-2-naphthyl core modified phenoxazines (4, 5, and 11–17, Figure 6) in O-ATRP. Through this study, we provide insight for the development of improved O-ATRP catalysts (vide infra).
While these PCs are likely capable of catalyzing a range of transformations, we originally designed them for application in O-ATRP. A suitable PC for O-ATRP must be both strongly reducing in the excited state and sufficiently stable and oxidizing as 2PC•+ in order to synthesize well-defined polymers (Figure 8).20 Specifically, O-ATRP initiators and the alkyl bromide polymer chain-end group possess reduction potentials from ∼ −0.6 V to −0.8 V vs SCE, requiring a strong excited state reductant for the reaction to occur without addition of sacrificial electron donors.21 Additionally, the 2PC•+ must be sufficiently stable and more oxidizing than the propagating radical of the polymer chain end, for which E0 ∼ −0.8 V vs SCE, to deactivate the propagating chain.5f Since these catalysts likely operate via an outer-sphere electron transfer (O-SET) pathway (more specifically that of dissociative electron transfer),5g Marcus theory predicts that the redox properties of these PCs (and those of the electron donor or acceptor) will directly relate to the rates of activation and deactivation in O-ATRP (Figure 8). Importantly, the synthesis of well-defined polymers via any controlled radical polymerization requires a low concentration of radical species in solution, and in ATRP this is maintained by a fast rate of deactivation relative to the rates of activation and propagation.22 If an O-ATRP PC possesses the required redox properties for efficient reversible deactivation then the polymerization is expected to proceed with linear reaction kinetics that are first-order with respect to monomer consumption and a linear growth of polymer molecular weight (MW) as a function of monomer conversion. As such, polymers of target MWs (quantified by initiator efficiency, I* = Mn(theo)/Mn(actual), where Mn is the number-average MW) and low dispersity (Đ = Mw/Mn, where Mw is the weight-average MW) can be synthesized.22 Advances in controlled radical polymerization methodologies such as ATRP over the last few decades have enabled the synthesis of polymers with near quantitative I*’s with moderate (Đ < 1.3) to low (Đ < 1.1) dispersity.23 Guided by these standards, the success of these phenoxazines to efficiently catalyze O-ATRP was evaluated. In particular, it was determined whether O-ATRP employing these PCs demonstrates linear growth of polymer MW as a function of conversion and produces polymeric material with Đ < 1.3 and high I* (for an ideal O-ATRP I* = 100%).
Figure 8.
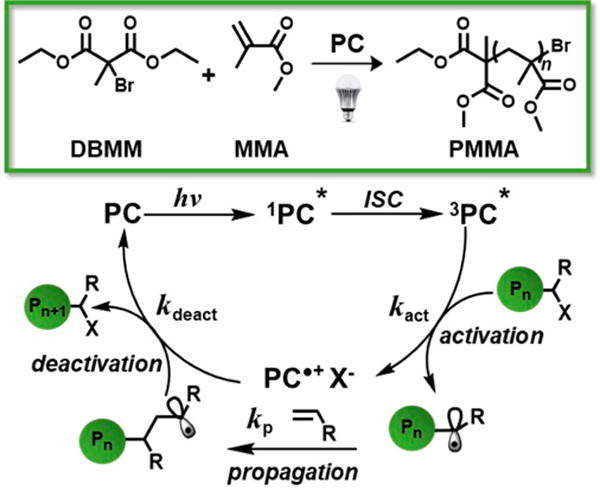
Reaction scheme for the polymerization of MMA (top) and proposed mechanism of O-ATRP using alkyl halide initiators (bottom) are shown. In the proposed mechanism of O-ATRP, initiation occurs once the excited state organic PC reduces the alkyl halide bond of the initiator or halide-capped polymer chain-end (Figure 8). This reaction generates a carbon-centered radical on the initiator or polymer chain end that can propagate via reaction with monomers. Meanwhile, the same reduction event also yields a bromide anion, which we propose associates with the oxidized 2PC•+ to form the 2PC•+Br− ion pair.7a We hypothesize that this ion pair is responsible for deactivating the polymer chain by reinstalling the halide end group and regenerating the ground state 1PC, which can reenter the catalytic cycle.
Previously, we observed that the O-ATRP of methyl methacrylate (MMA) catalyzed by N-phenyl phenoxazine (6) using UV-light yields polymers of less predictable MW (I* = 137%) and relatively high Đ (Đ = 1.48) compared to N-2-naphthyl phenoxazine 3 (I* = 77%, Đ = 1.11). The comparatively better performance of PC 3 in O-ATRP is further exemplified by the linear growth of polymer MW with respect to monomer conversion with close agreement to the theoretical growth of MW for a well-controlled polymerization (Figure 9A).15 In contrast, O-ATRP catalyzed by 6 exhibits less linear growth of polymer MW throughout the polymerization with more appreciable deviation from theoretical MW values (Figure 9B). Notably, the O-ATRP catalyzed by 6 suffers from poor control at low monomer conversions (at 24% conversion I* = 41%). In addition, the Đ of the PMMA synthesized using PC 6 is higher and more erratic throughout the polymerization. The superior performance of PC 3 compared to PC 6 in O-ATRP demonstrates the importance of the N-aryl substituent in designing noncore-modified N-aryl phenoxazine PCs. Due to the similar absorption and redox properties of PCs 3 and 6, we posit that the significant difference in their catalytic performance is a result of differences in the nature of these molecules in the excited state. Namely, the N-naphthyl-substituted PC (3) exhibits a polar, CT excited state while the N-phenyl substituted PC (6) exhibits a less polar, locally excited state (vide supra). Future photophysical investigation will be aimed at understanding the connection between the ability of these molecules to access CT excited states and their catalytic performance in O-ATRP.
Figure 9.

Plots of growth of the experimentally measured number-average molecular weight (Mn) as a function of monomer conversion (blue squares) with theoretical values (blue dotted line). The dispersity of each corresponding sample is shown as a function of monomer conversion (orange ◆). Data for the aliquots of each polymerization taken after 8 h are shown above each plot. All polymerizations (A–D) were run with [1000]:[10]:[1] of [MMA]: [DBMM]:[PC] in an equal volume of DMAc as that of MMA. Polymerizations catalyzed by (A) PC 3 and (B) PC 6 were performed in a UV-nail apparatus, while polymerizations catalyzed by PC 5 (C) and PC 9 (D) were run in the presence of white light LEDs. The polymerization data acquired for PC 3 were acquired from ref 5g. Additional experimental details are provided in the Supporting Information.
After furthering our understanding of the performance of noncore-modified phenoxazines 3 and 6 in O-ATRP, we compared biphenyl core-modified derivatives of these molecules (5 and 9). Characterization of the absorption properties, nature of the excited state, and redox properties of these molecules revealed that the nature of the core substituents, rather than the N-aryl substituent, more greatly dictates the properties of these PCs. In particular, 5 and 9 exhibit similar absorption profiles (λmax = 389 and 389 nm, respectively, and ε = 25900 and 24000 M−1cm−1, respectively), and in contrast to their parent molecules, both absorb in the visible regime. Investigation of the nature of 5 and 9 in the excited state revealed that they both access similar CT excited states which likely involve one of their biphenyl core substituents. This property again stands in contrast to the noncore modified molecules for which 3 (the parent molecule of 5) exhibits CT involving the N-naphthyl substituent and 6 (the parent compound of 9) does not exhibit CT character in the excited state. Furthermore, these core-modified derivatives exhibit redox properties that are very similar to each other but distinct from that of 3 and 6 (Table 1).
Given the similar overall properties of PCs 5 and 9, we hypothesized that they would exhibit similar performance in O-ATRP, as quantified by the parameters described above. To test this hypothesis, the O-ATRP of MMA was performed employing 5 and 9 as catalysts in the presence of white light LEDs (Figure 9, panels C and D, for emission of white light LEDs see Figure S41). O-ATRP with both PCs demonstrates linear growth of polymer MW with respect to monomer conversion with close agreement to theoretical MW values throughout the entire polymerization. As a result, 5 and 9 are able to synthesize PMMA with nearly quantitative I* at high monomer conversions, a marked improvement over the performance of PCs 3 and 6. In addition, O-ATRP catalyzed by these core-modified derivatives exhibits a lowering of Đ throughout the polymerization, with Đ values rivaling that of PC 3 at high conversions. In summary, the performance of 5 and 9 remains similar to each other through the entire polymerization and is distinct from the performance of their parent molecules. Together, these studies highlight the importance of determining which structural motifs lead to improved catalytic properties and performance within each subset of noncore-modified and core-modified N-aryl phenoxazines.
To further investigate our overarching hypothesis that catalyst structure dictates catalyst properties (specifically their absorption and redox properties) which in turn affects catalyst performance, the N-2-naphthyl core-modified phenoxazines (4, 5, and 11–17) were used as catalysts for O-ATRP. Specifically, these phenoxazines were employed for the O-ATRP of MMA using the alkyl bromide initiator diethyl 2-bromo-2-methylmalonate (DBMM) and white light LEDs (see scheme in Figure 8). These O-ATRP conditions were previously found to be the optimal batch reactor conditions for N-aryl phenoxazine PCs.5g Analysis of the polymerization results at reasonably high conversions revealed that all of the PCs investigated were able to synthesize PMMA with typically moderate to low Đ and I* ≥ 77% (Table 2). These data demonstrate that the phenoxazines explored are all competent PCs for O-ATRP. However, closer analysis of the evolution of these parameters throughout the entire polymerization reveals more stark discrepancies in the catalytic performance of these molecules.
Table 2.
O-ATRP Results from Employing Visible Light Absorbing Core Modified Phenoxazines for the Polymerization of Methyl Methacrylate (MMA)a
| PC | conv (%) | Mn(theo) (kDa)b | Mn (kDa)b | Mw (kDa)b | Đc | I*d (%) |
|---|---|---|---|---|---|---|
| 3e | 80 | 8.26 | 10.7 | 11.9 | 1.11 | 77 |
| 4 | 69 | 7.16 | 8.38 | 12.1 | 1.45 | 85 |
| 5 | 81 | 8.36 | 8.02 | 9.24 | 1.15 | 105 |
| 11 | 80 | 8.26 | 8.07 | 10.9 | 1.36 | 102 |
| 12 | 85 | 8.76 | 8.11 | 10.6 | 1.30 | 108 |
| 13 | 87 | 8.96 | 9.08 | 11.7 | 1.29 | 99 |
| 14 | 76 | 7.86 | 8.66 | 10.3 | 1.19 | 91 |
| 15 | 75 | 7.76 | 9.11 | 13.7 | 1.50 | 85 |
| 16 | 67 | 6.96 | 7.25 | 8.75 | 1.21 | 96 |
| 17 | 82 | 8.46 | 8.98 | 10.4 | 1.17 | 94 |
All polymerizations were conducted using the initiator diethyl 2-bromo-2-methylmalonate (DBMM) in a ratio of 1000:10:1 of MMA:DBMM:PC with DMAc as a solvent.
See the Supporting Information for more experimental details.
All calculated by Mw/Mn.
Calculated by Mn(theoretical)/Mn(GPC).
Analysis of the high conversion data points shown in Table 2 demonstrates that most of the PCs synthesize polymeric material with moderate to low Đ (Đ < 1.3) except for PCs 4 (Đ = 1.45), 11 (Đ = 1.36), and 15 (Đ = 1.50). To determine whether the latter three cases of high Đ represented outliers of these polymerization data sets, we analyzed the Đ of the PMMA synthesized by these three PCs at different conversions throughout the polymerization (Figures S101, S108, and S104, respectively). Indeed, for these PCs polymer Đ is greater than 1.4 at all conversions, indicating poor control over the polymerization. For the other PCs, half of them (5, 14, and 17) synthesize PMMA with Đ that never exceeds 1.4 throughout the polymerization (Figures S102, S109, and S103, respectively) while the other half (12, 13, and 16) exhibit relatively high Đ (Đ > 1.4) at low conversions (conversion <40%, Figures S107, S106, and S105, respectively).
We posit that efficient photon absorption is key for successful catalytic performance in O-ATRP since this characteristic partially dictates the population of the catalyst in the excited state present early in the reaction. Sufficient population of PC* at early reactions times is likely necessary for fast and uniform initiation and rapid buildup of the deactivating species (2PC•+), which is necessary to control the polymerization.11 As such, we attribute the inability of PCs 4, 11, and 15 to synthesize polymers with moderate to low Đ to their poor absorption of light emitted by the white light LEDs used for the reactions. In particular, 4, 11, and 15 exhibit the most blue-shifted absorption profiles (λmaxs = 367, 363, and 355, respectively) of the PCs investigated. While we recognize that the performance of a PC is dictated by a combination of its photophysical and electrochemical properties, these PCs clearly exhibit poorer control of polymer Đ despite their different redox properties (Table 1).
Further analysis of the catalytic performance of phenoxazines with stronger visible light absorption (5, 12–14, and 16–17) reveals another trend. At high monomer conversions, these PCs synthesize PMMA with moderate to low Đ (Đ ranges from 1.30 to 1.15) and nearly quantitative I* (I* ranges from 94% to 108%), indicating good control over the polymerization. However, comparison of the growth of polymer molecular weight as a function of conversion for these O-ATRP reactions allows us to further sort them into two distinct categories based on their performance and properties. Specifically, PCs 5, 14, and 17 exhibit very linear growth of polymer molecular weight as a function of conversion, with trendlines that agree well with the trendline for the theoretical growth of polymer molecular weight (Figures S102, S109, and S103, respectively). On the other hand, PMMA synthesized by PCs 12, 13, and 16 approaches ideal molecular weights only at high conversions. This phenomenon can clearly be seen by the convergence of the trendline for experimentally determined growth of polymer molecular weight as a function of conversion with that of the theoretical trendline for the polymerizations using these PCs (Figures S107, S106, and S105, respectively). Again, we suggest that these distinct differences in catalytic performance are a manifestation of the properties of the PCs. PCs 5, 14, and 17 exhibit stronger excited state reduction potentials (E0*T1, calc = −1.70, −1.88, and −1.69 V, respectively) than PCs 12 and 13 (E0*T1, calc = −1.42 V and −1.58 V, respectively) which bear electron-withdrawing core substituents. Thus, we posit that the redox properties of PCs 5, 14, and 17 are in a more ideal range for the polymerization of MMA. Given that the PCs with strong excited state reduction potentials and strong visible light absorption performed the best overall of the PCs investigated, our data suggests that PCs that exhibit these properties execute O-ATRP more efficiently for methacrylate monomers.
The case of PC 16 is interesting since this PC absorbs strongly in the visible regime compared to derivatives 4, 11, and 15 and is predicted to exhibit a relatively strong triplet excited state reduction potential (E0*T1, calc = −1.85 V) but suffers from poor control early in the polymerization, similar to PCs 12 and 13. While the performance of this PC appears to be an anomaly, the singlet excited state reduction potential for this PC was experimentally determined to be −1.54 V, which is evidence that the PC must be less reducing in the triplet excited state (i.e., have a more positive potential than −1.54 V). As such, the triplet excited state reduction potential of PC 16 is likely to be on par with those of PCs 12 and 13. However, the effects of the properties of this PC on its performance in O-ATRP warrant further future investigation since the performance of these PCs arises from a multitude of factors.
CONCLUSION
Investigation of the properties of a new family of core-modified phenoxazines revealed several design principles that we foresee will enable the use of these organic PCs in a range of photoredox-catalyzed transformations. For the phenoxazine PC properties explored in this work, the core substituents played a greater role than the N-aryl substituent in dictating the catalysts’ properties. This observation was further supported by the similar performance in O-ATRP of catalysts with the same core substituents but different N-aryl substituents (Figure 9, PC 5 vs PC 9). To design N-aryl phenoxazine catalysts that absorb in the visible regime, this study suggests that two aryl core substituents should be installed (Figure 4A, PC 8 vs PC 9), and to maximize visible light absorption, the substituents of choice are biphenyl groups or aryl electron-withdrawing groups (Figure 4B).
For the design of phenoxazine catalysts that can access CT excited states, more highly conjugated groups such as N-naphthyl substituents are required for noncore modified phenoxazines, as no CT character is observed for N-phenyl phenoxazine. However, the presence of core substituents can be used to alter the nature of these molecules in the excited state. In particular, we found that even N-phenyl phenoxazines can access CT excited states if suitable core substituents are installed (Figure 5, e.g. PC 6 vs PC 9). These differences in the excited states of core-modified phenoxazines compared to their parent N-aryl phenoxazines warrants future photophysical investigation, as the nature of these molecules in the excited state may greatly affect their catalytic performance.
To design phenoxazine catalysts with different reduction potentials that retain redox reversibility, aryl electron-donating or electron-withdrawing groups can be installed (Figure 6). In particular, installation of 4-methoxy phenyl core substituents (PC 11) yielded the most highly reducing catalyst, and installation of 4-cyano phenyl substituents (PC 12) yielded the least strongly reducing catalyst of the dicore-modified PCs investigated. Notably, we established that through functionalization at the phenoxazine core, a wide range of excited state reduction potentials can be achieved (e.g., E0*T1,calc values spanning from −1.42 V to −2.01 V vs SCE.). Thus, with this degree of tunability, we envision that core-modified phenoxazine PCs are promising in existing applications5g,10–12 and enable new synthetic methodologies with improved selectivity to be developed.
Lastly, the application of these molecules as PCs for the O-ATRP of methyl methacrylate revealed their competency as catalysts for this transformation, with most derivatives synthesizing polymeric material with dispersity typically less than 1.3 and initiator efficiencies ≥95% at high conversions. However, despite their good performance at high conversions, only three of the PCs explored (5, 14, and 17) maintain good control throughout the entire polymerization. We posit that the superior performance of these three PCs compared to the other derivatives investigated is due to their strong absorption of visible light and strong excited state reduction potentials. Ongoing work is aimed at improving our understanding of the capabilities and limitations of these PCs for O-ATRP.
Supplementary Material
Acknowledgments
This work was supported by Colorado State University and the University of Colorado Boulder. Research reported in this publication was supported by the National Institute of General Medical Sciences (Award R35GM119702) of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Acknowledgement is made to the donors of The American Chemical Society Petroleum Research Fund for partial support of this research (56501-DNI7) and the Advanced Research Projects Agency-Energy (DE-AR0000683). B.G.M. and S.M.S. acknowledge support from NSF GRFPs. C.-H.L. acknowledges NIH’s F32 postdoctoral fellowship support (F32GM122392). We gratefully acknowledge the use of XSEDE supercomputing resources (NSF ACI-1053575).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b12074.
Materials and methods, experimental procedures, NMR compound characterization, control experiments, cyclic voltammetry data, spectroscopic data, computational modeling data, computational modeling details, and supplementary polymerization data (PDF)
ORCID
Chern-Hooi Lim: 0000-0003-1823-6305
Niels H. Damrauer: 0000-0001-8337-9375
Garret M. Miyake: 0000-0003-2451-7090
Notes
The authors declare no competing financial interest.
References
- 1.(a) Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617–1622. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]; (b) Prier CK, Rankic DA, Macmillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schultz DM, Yoon TP. Science. 2014;343:985. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yoon TP, Stephenson CRJ. Adv Synth Catal. 2014;356:2739. [Google Scholar]; (e) Corrigan N, Shanmugam S, Xu J, Boyer C. Chem Soc Rev. 2016;45:6165–6212. doi: 10.1039/c6cs00185h. [DOI] [PubMed] [Google Scholar]; (f) Chen M, Zhong M, Johnson JA. Chem Rev. 2016;116:10167–10211. doi: 10.1021/acs.chemrev.5b00671. [DOI] [PubMed] [Google Scholar]; (g) Shaw MH, Twilton J, MacMillan DWC. J Org Chem. 2016;81:6898–6926. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Michaudel Q, Kottisch V, Fors BP. Angew Chem, Int Ed. 2017;56:9670–9679. doi: 10.1002/anie.201701425. [DOI] [PubMed] [Google Scholar]
- 2.(a) Hedstrand DM, Kruizinga WM, Kellogg RM. Tetrahedron Lett. 1978;19:1255–1258. [Google Scholar]; (b) van Bergen TJ, Hedstrand DM, Kruizinga WH, Kellogg RM. J Org Chem. 1979;44:4953–4962. [Google Scholar]; (c) Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]
- 3.(a) Sauvage JC, Collin JP, Chambron JC, Guillerez S, Coudret C, Balzani V, Barigelletti F, De Cola L, Flamigni L. Chem Rev. 1994;94:993–1019. [Google Scholar]; (b) Damrauer NH, Cerullo G, Yeh A, Boussie TR, Shank CV, McCusker JK. Science. 1997;275:54–57. doi: 10.1126/science.275.5296.54. [DOI] [PubMed] [Google Scholar]
- 4.(a) Higgins RF, Fatur SM, Shepard SG, Stevenson SM, Boston DJ, Ferreira EM, Damrauer NH, Rappé AK, Shores MP. J Am Chem Soc. 2016;138:5451–5464. doi: 10.1021/jacs.6b02723. [DOI] [PubMed] [Google Scholar]; (b) Kainz QM, Matier CD, Bartoszewicz A, Zultanski SL, Peters JC, Fu GC. Science. 2016;351:681–684. doi: 10.1126/science.aad8313. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Romero NA, Nicewicz DA. Chem Rev. 2016;116:10075–10166. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; (d) Joshi-Pangu A, Levesque F, Roth HG, Oliver SF, Campeau LC, Nicewicz D, DiRocco DA. J Org Chem. 2016;81:7244–7249. doi: 10.1021/acs.joc.6b01240. [DOI] [PubMed] [Google Scholar]
- 5.(a) Poelma SO, Burnett GL, Discekici EH, Mattson KM, Treat NJ, Luo Y, Hudson ZM, Shankel SL, Clark PG, Kramer JW, Hawker CJ, Read de Alaniz J. J Org Chem. 2016;81:7155–7160. doi: 10.1021/acs.joc.6b01034. [DOI] [PubMed] [Google Scholar]; (b) Treat NJ, Sprafke H, Kramer JW, Clark PG, Barton BE, Read de Alaniz J, Fors BP, Hawker CJ. J Am Chem Soc. 2014;136:16096–16101. doi: 10.1021/ja510389m. [DOI] [PubMed] [Google Scholar]; (c) Discekici EH, Luo Y, Hawker J, Hudson ZM, Treat NJ, Poelma SO, Mattson KM, de Alaniz JR. Chem Commun. 2015;51:11705–11708. doi: 10.1039/c5cc04677g. [DOI] [PubMed] [Google Scholar]; (d) Pan X, Fang C, Fantin M, Malhotra N, So WY, Peteanu LA, Isse AA, Gennaro A, Liu P, Matyjaszewski K. J Am Chem Soc. 2016;138:2411–2425. doi: 10.1021/jacs.5b13455. [DOI] [PubMed] [Google Scholar]; (e) Miyake GM, Theriot JC. Macromolecules. 2014;47:8255–8261. [Google Scholar]; (f) Theriot JC, Lim CH, Yang H, Ryan MD, Musgrave CB, Miyake GM. Science. 2016;352:1082–1086. doi: 10.1126/science.aaf3935. [DOI] [PubMed] [Google Scholar]; (g) Pearson RM, Lim CH, McCarthy BG, Musgrave CB, Miyake GM. J Am Chem Soc. 2016;138:11399–11407. doi: 10.1021/jacs.6b08068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Fors BP, Hawker CJ. Angew Chem, Int Ed. 2012;51:8850–8853. doi: 10.1002/anie.201203639. [DOI] [PubMed] [Google Scholar]; (b) Treat NJ, Fors BP, Kramer JW, Christianson M, Chiu C-Y, Read de Alaniz J, Hawker CJ. ACS Macro Lett. 2014;3:580–584. doi: 10.1021/mz500242a. [DOI] [PubMed] [Google Scholar]
- 7.(a) Lim CH, Ryan MD, McCarthy BG, Theriot JC, Sartor SM, Damrauer NH, Musgrave CB, Miyake GM. J Am Chem Soc. 2017;139:348–355. doi: 10.1021/jacs.6b11022. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ryan MD, Theriot JC, Lim CH, Yang H, Lockwood AG, Garrison NG, Lincoln SR, Musgrave CB, Miyake GM. J Polym Sci, Part A: Polym Chem. 2017;55:3017–3027. doi: 10.1002/pola.28574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Frick E, Anastasaki A, Haddleton DM, Barner-Kowollik C. J Am Chem Soc. 2015;137:6889–6896. doi: 10.1021/jacs.5b03048. [DOI] [PubMed] [Google Scholar]; (b) Ribelli GG, Konkolewicz D, Bernhard S, Matyjaszewski K. J Am Chem Soc. 2014;136:13303–13312. doi: 10.1021/ja506379s. [DOI] [PubMed] [Google Scholar]
- 9.(a) Zhu Y, Babel A, Jenekhe SA. Macromolecules. 2005;38:7983–7991. [Google Scholar]; (b) Zhu Y, Kulkarni AP, Wu P, Jenekhe SA. Chem Mater. 2008;20:4200–4211. [Google Scholar]
- 10.Ramsey BL, Pearson RM, Beck LR, Miyake GM. Macromolecules. 2017;50:2668–2674. doi: 10.1021/acs.macromol.6b02791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Buss BL, Beck LR, Miyake GM. Polym Chem. 2018 doi: 10.1039/C7PY01833A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ryan MD, Pearson RM, French TA, Miyake GM. Macromolecules. 2017;50:4616–4622. doi: 10.1021/acs.macromol.7b00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du Y, Pearson RM, Lim CH, Sartor SM, Ryan MD, Yang H, Damrauer NH, Miyake GM. Chem Eur J. 2017;23:10962–10968. doi: 10.1002/chem.201702926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Bock CR, Conner JA, Gutierrez AR, Meyer TJ, Whitten DG, Sullivan BP, Nagle JK. J Am Chem Soc. 1979;101:4815–4824. [Google Scholar]; (b) Top Curr Chem. 2007:281. [Google Scholar]; (c) Dixon IM, Collin J, Sauvage J, Flamigni L, Encinas S, Barigelletti F. Chem Soc Rev. 2000;29:385–391. [Google Scholar]
- 14.Arias-rotondo DM, McCusker JK. Chem Soc Rev. 2016;45:5803–5820. doi: 10.1039/c6cs00526h. [DOI] [PubMed] [Google Scholar]
- 15.(a) Benniston AC, Harriman A, Li P, Rostron JP, van Ramesdonk HJ, Groeneveld MM, Zhang H, Verhoeven JW. J Am Chem Soc. 2005;127:16054–16064. doi: 10.1021/ja052967e. [DOI] [PubMed] [Google Scholar]; (b) Tanaka H, Shizu K, Nakanotani H, Adachi C. Chem Mater. 2013;25:3766–3771. [Google Scholar]; (c) Fukuzumi S, Kotani H, Ohkubo K, Ogo S, Tkachenko NV, Lemmetyinen H. J Am Chem Soc. 2004;126:1600–1601. doi: 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]
- 16.(a) Zhao Y, Gong H, Jiang K, Yan S, Lin J, Chen M. Macromolecules. 2018;51:938–946. [Google Scholar]; (b) Gong H, Zhao Y, Shen X, Lin J, Chen M. Angew Chem, Int Ed. 2018;57:333–337. doi: 10.1002/anie.201711053. [DOI] [PubMed] [Google Scholar]
- 17.Reichardt C. Chem Rev. 1994;94:2319–2358. [Google Scholar]
- 18.(a) Lee J, Shizu K, Tanaka H, Nakanotani H, Yasuda T, Kaji H, Adachi C. J Mater Chem C. 2015;3:2175–2181. [Google Scholar]; (b) Uoyama H, Goushi K, Shizu K, Nomura H, Adachi C. Nature. 2012;492:234–238. doi: 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]; (c) Tao Y, Yuan K, Chen T, Xu P, Li H, Chen R, Zheng C, Zhang L, Huang W. Adv Mater. 2014;26:7931–7958. doi: 10.1002/adma.201402532. [DOI] [PubMed] [Google Scholar]
- 19.Roth HG, Romero NA, Nicewicz DA. Synlett. 2016;27:714–723. [Google Scholar]
- 20.(a) Theriot JC, McCarthy BG, Lim CH, Miyake GM. Macromol Rapid Commun. 2017;38:1700040. doi: 10.1002/marc.201700040. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pan X, Fang C, Fantin M, Malhotra N, So WY, Peteanu LA, Isse AA, Gennaro A, Liu P, Matyjaszewski K. J Am Chem Soc. 2016;138:2411–2425. doi: 10.1021/jacs.5b13455. [DOI] [PubMed] [Google Scholar]
- 21.Isse AA, Lin CY, Coote ML, Gennaro A. J Phys Chem B. 2011;115:678–684. doi: 10.1021/jp109613t. [DOI] [PubMed] [Google Scholar]
- 22.(a) Matyjaszewski K, Xia J. Chem Rev. 2001;101:2921–2990. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]; (b) Coessens V, Pintauer T, Matyjaszewski K. Prog Polym Sci. 2001;26:337–377. [Google Scholar]; (c) Ouchi M, Terashima T, Sawamoto M. Chem Rev. 2009;109:4963–5050. doi: 10.1021/cr900234b. [DOI] [PubMed] [Google Scholar]
- 23.(a) Matyjaszewski K. Macromolecules. 2012;45:4015–4039. [Google Scholar]; (b) Nicolas J, Guillaneuf Y, Lefay C, Bertin D, Gigmes D, Charleux B. Prog Polym Sci. 2013;38:63–235. [Google Scholar]; (c) Moad G, Rizzardo E, Thang S, Chong YK, Postma A. Polymer. 2005;46:8458–8468. [Google Scholar]; (d) Kamigaito M, Ando T, Sawamoto M. Chem Rev. 2001;101:3689–3746. doi: 10.1021/cr9901182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.