

Abstract
Objective
Deletion of microsomal (m) prostaglandin (PG) E synthase (S)-1, an anti-inflammatory target alternative to cyclooxygenase-2, attenuates injury-induced neointima formation in mice. This is attributable to the augmented levels of PGI2, a known restraint of the vascular response to injury, acting via I prostanoid receptor (IP). To examine the role of mPGES-1 derived PGE2 in vascular remodeling without the IP.
Approach and Results
Mice deficient in both IP and mPGES-1 (double knockout, DKO) and littermate controls (IP KO) were subjected to angioplasty-wire injury. Compared with the deletion of IP alone, coincident deletion of IP and mPGES-1 increased neointima formation, without affecting media area. Early pathological changes include impaired reendothelialization and increased leukocyte invasion in neointima. Endothelial cells (ECs), but not vascular smooth muscle cells, isolated from DKOs exhibited impaired cell proliferation. Activation of PGE2 receptor (EP)4 (and EP2, to a lesser extent), but not of EP1 or EP3, promoted EC proliferation. EP4 antagonism inhibited proliferation of mPGES-1-competent ECs, but not of mPGES-1-deficient ECs, which showed suppressed PGE2 production. EP4 activation inhibited leukocyte adhesion to ECs in vitro, promoted reendothelialization and limited neointima formation post injury in the mouse. Endothelium-restricted deletion of EP4 in mice suppressed reendothelialization, increased neointimal leukocytes, and exacerbated neointimal formation.
Conclusions
Removal of the IP receptors unmasks a protective role of mPGES-1 derived PGE2 in limiting injury-induced vascular hyperplasia. EP4, in the endothelial compartment, is essential to promote reendothelialization and restrain neointimal formation after injury. Activating EP4 bears therapeutic potential to prevent restenosis after percutaneous coronary intervention.
Keywords: prostaglandin E synthase-1, PGE2, EP4, endothelium, vascular remodeling
Subject Codes: Vascular Biology, Remodeling, Coronary Artery Disease, Endothelium, Stenosis
Graphical abstract
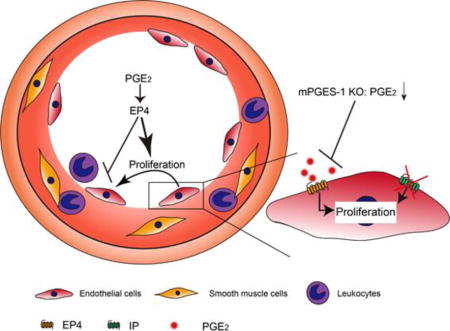
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) ameliorate pain, fever and inflammation by inhibiting cyclooxygenase (COX, two major isomers identified as COX-1 and COX-2), the rate-limiting enzyme in the synthetic cascade of prostanoids1. Clinical evidence shows that NSAIDs selective for COX-2 inhibition increase cardiovascular thrombotic events2-4. This is attributable to suppression of COX-2 derived prostaglandin (PG) I25. Microsomal (m) PGE synthase(S)-16 has emerged as a therapeutic target that is downstream of COX-27, 8. Deletion of mPGES-1, unlike COX-2 inhibition, does not predispose to thrombosis, due to augmented PGI2 (redirection of PGH2, the COX product, to PGI synthase), and retards atherogenesis independent of IP (the receptor for PGI2) deficiency9-11. Targeting mPGES-1 may avoid the cardiovascular risk associated with COX-2 inhibitors while preserving their analgesic and anti-inflammatory efficacy12. Accumulative evidence suggests that COX pathway components differentially modulate vascular responses to injury.
Celecoxib, a COX-2 selective NSAID, reduces in-stent late luminal loss in patients with coronary artery disease treated with aspirin plus clopidogrel after percutaneous coronary intervention (PCI)13. However, a follow-up trial confirmed an increased thrombotic risk with celecoxib despite the dual anti-platelet therapy14, which limits its clinical application. Tissue-specific deletion of mPGES-1, the underlying alternative target for COX-2, in endothelial cells (ECs) or vascular smooth muscle cells enhances neointima formation, whereas myeloid cell mPGES-1 deletion reduces vascular hyperplasia response to injury15. Our previous study shows that deletion of mPGES-1 attenuates injury-induced neointimal formation in mice, with sustained reduction in PGE2 and augmentation in PGI216. PGI2 signaling via the IP is known to restrain the neointimal formation17. In contrast to IP deletion, deletion of the thromboxane A2 receptor depresses this response17. The role of mPGES-1 derived PGE2 in the vascular remodeling that is independent of the IP signaling remains unknown.
PGE2 has four G-protein coupled receptors, EP1 through EP4, which mediates differential downstream signaling18. EP3 mediates vascular smooth muscle cell migration facilitating neointimal hyperplasia in mice19. EP4 protects against ischemia-reperfusion injury20, 21, hypertension22 or atherosclerosis23. EP4 promotes endothelial migration and angiogenesis24, 25. However, whether EP4 regulates injury-induced neointimal formation is unknown.
Here, we report that removal of the IP receptor uncovers a protective role of mPGES-1 derived PGE2 in endothelial repair and the vascular response to injury. Furthermore, deletion of endothelial EP4 exacerbates, whereas pharmacological activation of EP4 limits neointimal hyperplasia after wire injury.
Materials and Methods
Animal study
Mice deficient in mPGES-1 (gene: Ptges)26 and IP (gene: Ptgir)17 were obtained from Pfizer and the FitzGerald lab at the University of Pennsylvania, respectively. Both strains had been backcrossed to a C57BL/6 background for over 10 generations, and were used to derive IP/mPGES-1 double knockout (DKO) mice and littermate controls (IP KO) by intercrossing Ptgir−/− Ptges−/− with Ptgir −/− Ptges+/-. DKO mice develop normally without overt abnormalities. Global deletion of EP4 is perinatally lethal due to patent ductus arteriosus27. To circumvent this defect, endothelial-specific EP4 (gene: Ptger4) knockout mice were generated using a tamoxifen-CreERT2 strategy28. Briefly, C57BL/6 Ptger4flox/flox mice29 were intercrossed with Cdh5-promoter driven CreERT2 (Cdh5 (PAC)-CreERT2+) mice30, kindly provided by Ralf Adams. The resulting Ptger4f/f Cdh5-CreERT2+ and Ptger4f/f Cdh5-CreERT2− mice were then intercrossed to generate the animals used in this study— endothelial EP4 conditional-knockout (Ptger4f/f Cdh5-CreERT2+: abbreviated, cKO) and littermate controls (Ptger4f/f Cdh5-CreERT2−: abbreviated, Ctl). To induce endothelial EP4 deletion, tamoxifen (37.5 mg/ml dissolved in sunflower seed oil) was intraperitoneally injected into the experimental mice and littermate controls at a dose of 150 mg/kg/day for six days which were interrupted for three days after the third dose. Gene-modified mice used in this study were gender matched. The pooled data from both gender were used to show the gene specific effects in each study, and gender-specific sub-group analyses were provided in Supplemental Table I and II. Male C57BL/6 mice aged 6-8 weeks from the National Institutes for Food and Drug Control (Beijing, China) were used to determine the effect of AE1-329 (Gifted by ONO Pharmaceutical Co., Ltd., Osaka, Japan) or misoprostol (410004; purity, 98.9%; National Institutes for Food and Drug Control, Beijing, China) in vascular remodeling. All animal protocols were performed following the guidelines of the Institutional Animal Care and Use Committee, the Experimental Animal Center, Fuwai Hospital, National Center for Cardiovascular Diseases, China.
Femoral artery injury model
Femoral arteries were injured using a protocol as we described previously28. Briefly, a groin incision was made on one side of the anesthetized mouse. The femoral artery and its small branch between the rectus femoris and vastus medialis muscles were then carefully exposed and separated from the accompanying nerve and vein via blunt dissection. A 6-0 silk suture was then looped round the proximal femoral artery to stop the blood flow during the surgery. Another 6-0 silk suture was placed under the branch. A transverse arteriotomy was then made in the branch, and a flexible angioplasty wire (0.35 mm diameter; Cook Inc., IN, USA) was inserted into the femoral artery from the branch for a length no less than 5 mm toward the iliac artery. The wire was left in place for 3 min to dilate and denude the artery. Then, the wire was removed, the branch was ligated proximally with the 6-0 silk suture, and the blood flow in the femoral artery was restored by releasing the sutures for blood flow control. The skin incision was then closed with a 5-0 silk suture. Seven or twenty-eight days after injury, the arteries were harvested, embedded in paraffin and stained with haematoxylin and eosin (H&E) for determination of the severity of hyperplasia. In detail, cross-sections of the arteries were serially obtained for 10-13 levels at 150 μm intervals, and the sections with the most severe hyperplasia were used for comparisons. To evaluate reendothelialization, the sections from the arteries 7 days post-injury were immunostained with von Wllebrand Factor (vWF), an endothelial cell marker, and photographed with a CCD camera coupled to a microscope system (AXI0; Zeiss, Oberkochen, Germany). The segments along the vascular inner wall that were positive in vWF and the peripheral length of the vascular inner wall were measured using Image-Pro Plus 6.0 software (Media Cybernetics, MD, USA). The ratio of the total length of the vWF positive segments to the peripheral length was calculated to represent the extent of reendothelialization. To evaluate leukocytes infiltration/migration, representative sections from the arteries injured for 7 days were analyzed with H&E staining. Briefly, after capturing the images, intima leukocytes were objectively determined using a Hue (H) Saturation (S) Intensity (I)-based color selection strategy using Image-Pro Plus 6.0 software (Media Cybernetics, MD, USA).
Cell study
Endothelial cells
Mouse aorta endothelial cells (MAECs) were isolated as previously described28, 31. Briefly, aortas were harvested and cut into 1-2 mm2 sections. The aortic segments were attached to a petri dish by their luminal side, and were then cultured in a DMEM medium containing 20 % fetal bovine serum (FBS) and 100 ug/mL endothelial cell growth supplement (ECGS) for 5-7 days, to allow the outgrowth of endothelial cells. Then, the endothelial cells were passaged and cultured. MAECs in passage 2-6 were used in this study. Endothelial cell proliferation was compared in MAECs isolated from IP KO or DKO, in passage 2. Primary human microvascular endothelial cells (HMECs) were brought from ScienCell (6000; Carlsbad, CA, USA), and cultured in the same medium as that for MAEC culture.
Smooth muscle cells
Mouse aorta smooth muscle cells (MASMCs) were isolated from IP KOs and DKOs. Briefly, aortas were isolated, scratched for three times on the inner surfaces, and cut into 1-2 mm2 sections. The aortic segments were then attached to a petri dish by their luminal side, covered by a cover slid, and cultured in a DMEM medium containing 10 % FBS for 5-7 days. The MASMCs were then passaged and cultured. Proliferation of MASMCs in passage 2 was compared.
Cell proliferation
Cell growth was determined with a cell counting kit-8 (CCK-8; 40203ES60; Yeasen, Shanghai, China), following the manufacturer’s instructions, as previously described28. The assay allows multiple detection without obvious cell toxicity. Briefly, the cells were seeded onto a 96-well flat-bottomed plate. After the cells were attached, ECs were cultured in the medium containing 3% FBS for 6-8 hours. Afterwards, the culture medium was replaced with a 3% FBS medium-CCK-8 mixture (10:1 in volume). Thereafter, the cells were cultured in the mixture for no longer than 4 hours; absorbance at 450 nm was determined as the baseline. Then the cells were cultured in fresh 3% FBS medium with the indicated reagents for another 48 hours. The culture medium was then replaced by the medium-CCK-8 mixture, and the culture continued for the same time as above for determination of the absorbance at 450 nm. The change in absorbance between the two measurements was used to define the cell growth. To determine MASMC proliferation, cells were pre-starved in FBS-free serum for 24 hours, then cultured in a medium containing 1% FBS, with two sequencial incubations with CCK-8. The reagents and their concentrations used in the proliferation studies were as follows: AE1-329 (0.1-1 μmol/L; Gifted by ONO Pharmaceutical Co., Ltd., Osaka, Japan), Butaprost (1 μmol/L; 13740; Cayman Chemical, MI, USA), Sulprostone (1 μmol/L; 14765; Cayman), GW627368X (0.1-1 μmol/L; HY-16963; MedChemExpress, NJ, USA), L-798106 (1 μmol/L; 11129; Cayman), iloprost (1 μmol/L; 18215; Cayman), Cay10441 (10 μmol/L; 10005186; Cayman), PF-04418948 (1 μmol/L; S7211; Selleck), ONO-8130 (1 μmol/L; 19118; Cayman), SQ22536 (200 μmol/L; S8283; Selleck), H89 2HCl (10 μmol/L; S1582; Selleck), ESI-09 (10 μmol/L; 19130; Cayman), db-cAMP (3-100 μmol/L; D0260; Sigma, Darmstadt, Germany) and misoprostol (10 μmol/L; 410004; National Institutes for Food and Drug Control, Beijing, China).
Endothelium-leukocytes adhesion study
For endothelium-leukocyte adhesion assay, MAECs were seeded into the 96-well plate, pre-starved in the DMEM containing 3% FBS for 6-8 hours, and incubated with tested reagents for 2 hours. Leukocytes were gathered from mouse peritoneal. Briefly, 4% Brewer Modified Thioglycollate Medium (211716; BD Biosciences, NJ, USA) was injected into the mouse peritoneal (1 mL per mouse). Four to five hours later, the peritoneal leukocytes were washed out with 0.1% bovine serum albumin, centrifuged, and re-suspended in the 1640 medium containing 10% FBS. When the preparation of endothelial cells was completed, the culture medium with indicated drugs was replaced by the 1640 medium containing leukocytes (3×104/well). Endothelial cells and leukocytes were then co-cultured for 30 minutes. Later, the cells were rinsed once with 1640 medium containing Rhodamine 6G (200 μg/mL; 252433; Sigma, Darmstadt, Germany), followed by 3 washes with fresh 1640 medium. The fluorescent signals were finally detected by a microplate reader (Excitation wavelength: 560 nm, emitter wavelength: 560 nm; Infinite M200, Tecan, Hombrechtikon, Switzerland). The reagents and their concentrations used in this study were as follows: misoprostol (10 μmol/L; 410004; National Institutes for Food and Drug Control, Beijing, China), AE1-329 (1 μmol/L; Gifted by ONO Pharmaceutical Co., Ltd., Osaka, Japan), GW627368X (1 μmol/L; HY-16963; MedChemExpress, NJ, USA) and db-cAMP (30 μmol/L; D0260; Sigma, Darmstadt, Germany).
Immunofluorescence staining
Immunofluorescence staining was performed following the same protocol as we described previously28. Briefly, the paraffin sections (5 μm) were deparaffinized, rehydrated, and subjected to antigen retrieval using an EDTA antigen-retrieval water (PH 9.0; ZSGB-BIO, Beijing, China). Following 1-hour incubation with normal goat serum at room temperature, the samples were incubated with primary antibodies overnight at 4 °C, and subsequently stained with Alexa Fluor-488-coupled or Alexa Fluor-594-coupled secondary antibodies for 3 hours at room temperature. The sections were then mounted with a VectaShield medium containing DAPI to stain nuclei, and imaged using a Zeiss microscope system (AXI0; Zeiss) or a laser-scanning confocal microscope system (SP8; Leica). For determination of reendothelialization, images were analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Inc. Rockville, MD, USA). The primary antibodies and dilution fold used in the study were polyclonal anti-EP4 antibody (2.5 μg/mL; 101775; Cayman Chemical, MI, USA), rabbit polyclonal anti-vWF antibody (1 μg/mL; F3520; Sigma, Darmstadt, Germany), sheep polyclonal anti-vWF antibody (10 μg/mL; Ab11713; Abcam, Cambridge, UK), monoclonal anti-α-SMA antibody (5 μg/mL; A5228; Sigma, Darmstadt, Germany) and polyclonal anti-F4/80 antibody (2 μg/mL; Ab16911; Abcam, Cambridge, UK).
Western blot analysis
For Western blot analysis, the cells were lysed in a RIPA buffer that contained protease inhibitors (4693116001, Roche, Basel, Switzerland). After centrifugation (15800 g, 10 min), cell lysates were mixed with a loading buffer, fractionated with 10% SDS-PAGE, and transferred onto polyvinylidene fluoride membranes. The membranes were then probed with the primary poly-clonal antibodies against EP4 (2.5 μg/mL; 101775; Cayman, Michigan, USA) overnight at 4 °C, followed by incubation with goat anti-rabbit secondary antibody for 1 hour at room temperature. Finally, the membranes were incubated with ECL luminous liquid (P1020; PPLYGEN, Beijing, China), and signals from immunoreactive bands were visualized using a FluorChem System (ProteinSimple, CA, USA).
Prostanoid determination
PGE2 and PGI2 levels were determined via measuring their major urinary metabolites, tetranor-PGEM and 2,3-dinor-6-keto-PGF1α by liquid (L) chromatography (C)–tandem mass (M) spectrometry (S), as previously described32.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 software (GraphPad Software Inc., San Diego, California, USA). When only two groups were involved, Student’s two-tailed unpaired t-test was used for comparison. Comparisons of multiple groups were made using a one-way ANOVA analysis. Tukey’s or Bonferroni’s post hoc tests were performed for the data with equal variances, while Dunnett’s post hoc tests were used for data with unequal variances. And Bartlett’s tests were used for analyzing the variances of data. Results are expressed as mean ± SEM. Differences were considered statistically significant at p<0.05.
Results
1. mPGES-1 derived PGE2 protects against wire injury-induced neointimal formation in IP-deficient mice
Deletion of mPGES-1 in mice enhances PGI2 production, which leads to the attenuated vascular remodeling16. To elucidate a role of mPGES-1 derived PGE2 in vascular remodeling, we crossed mPGES-1 KOs with IP KOs to generate double KO (DKO) mice and littermate IP KO mice as control. The mice were subjected to wire-injury (endothelial denudation) of the femoral artery for 28 days. On this IP deficient background, mPGES-1 deletion increased neointima area by ~84%, and also increased the ratio of intimal to medial area, with media thickness unaltered (Figure 1A-D). This reveals a protective role of mPGES-1 derived PGE2 in the vascular response to injury. Urinary metabolite of PGE2 was reduced in DKO mice, while PGI2 metabolite was enhanced (Figure 1E & F).
Figure 1. IP deletion reveals a protective role of mPGES-1 derived PGE2 against wire-injury-induced neointimal formation.
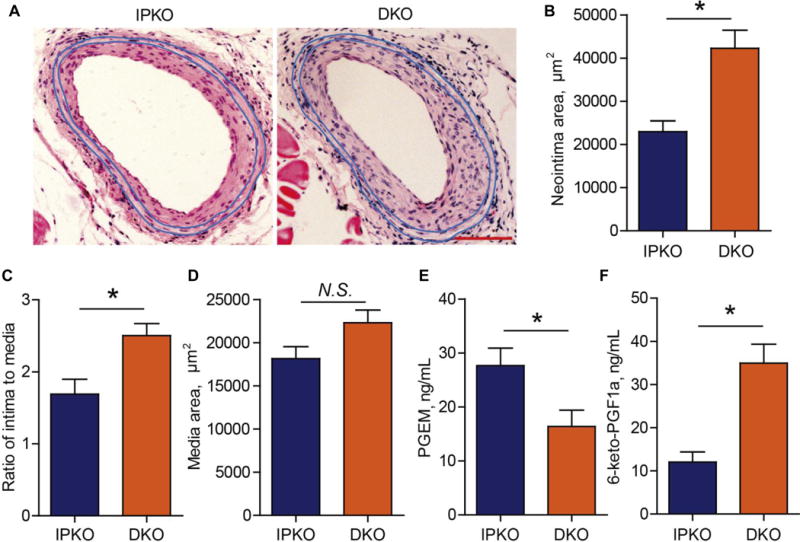
DKO (Ptgir −/− Ptges −/−) and littermate IPKO (Ptgir −/−) mice were subjected to wire-injury at femoral arteries, and the vessels were harvested at 28 days after injury and quantified for neointima formation. Representative images of hematoxylin and eosin (H&E) staining are shown (A; Bar= 100 μm). Neointimal area (B), ratio of intima to media (C) and media thickness (D) were determined (n= 14 IPKO, 16 DKO). PGE2 (E) and PGI2 (F) metabolites in urine were determined by HPLC-MS/MS as detailed in the Method (n= 8). *P<0.05; Student’s unpaired t-test.
To explore the underlying mechanism, vessels were harvested at 7 days after surgery for histology examination. Again, the neointimal area and the ratio of intima to media, but not the medial area, were significantly enhanced in DKO (Figure 2A & B). Intima leukocytes, mainly macrophages (positive for F4/80 staining), were increased in the DKOs (Figure 2C, D and Supplemental Figure I). Expression of α-SMA was mainly detected in smooth muscle cells (SMCs) in the media, but rarely detected in the neointima area regardless genotypes (Figure 2E). When von Wllebrand Factor (vWF, an endothelial marker) was stained, a striking reduction in the number of endothelial cells (ECs) was observed in the DKOs (Figure 2D & F), reflecting suppressed reendothelialization after the wire denudation injury.
Figure 2. Deletion of mPGES-1 suppresses reendothelialization after endothelial denudation injury in IP deficient mice.
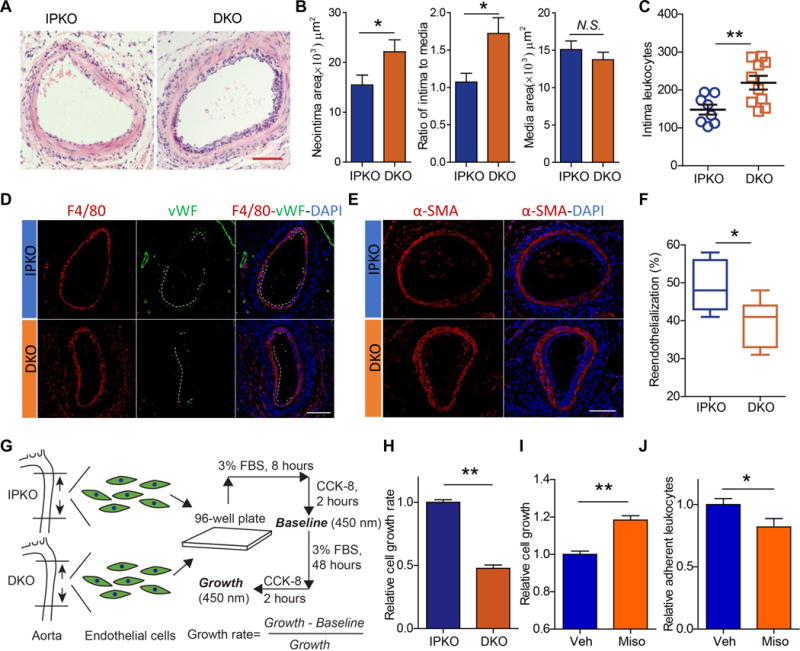
Injured femoral arteries of DKO and IPKO were harvested at 7 days after surgery. Neointima formation and leukocytes infiltration were evaluated on H&E stained sections, with representative H&E images shown (A; Bar= 100 μm). Neointimal area, ratio of intima to media and media thickness were quantified (B; n= 14 IPKO, 16 DKO). Number of neointima leukocytes was quantified by analyzing the H&E stained tissue sections with IPP software (C; n= 8 IPKO, 10 DKO). Injured vessels were immunostained for F4/80 (a marker of macrophage, red) and vWF (an EC marker, green) (D; Bar= 100 μm) and for α-SMA (a SMCs marker) (E; Bar= 100 μm). DAPI stains nuclei in blue. The number of vWF-positive cells was quantified to determine reendothelialization (F; n=7). ECs were isolated from the descending aortae of IPKO and DKO mice, and their cell proliferation was analyzed in vitro (G) and compared (H). Calculation of the EC proliferation rate was based on the difference in cell number before vs after treatment with 3% FBS for 48 hours. Misoprostol, a PGE analogue at 10 μmol/L, promoted endothelial proliferation (I, n=9 from two independent experiments) and inhibited endothelium-leukocytes adhesion (J, n=9 from three independent experiments). *P<0.05, **P<0.01; Student’s unpaired t-test.
To pursue a potential impact on endothelial repair, ECs were isolated from IP KOs and DKOs, and their capacity for proliferation was evaluated in vitro (Figure 2G). The EC proliferation was significantly impaired in the DKOs (Figure 2H), revealing a proliferative effect of PGE2 signaling in ECs. Further, treatment of ECs with misoprostol, a PGE analogue, promoted EC proliferation in vitro (Figure 2I), and also decreased leukocyte adhesion to endothelial monolayer in vitro (Figure 2J).
Primary aortic smooth muscle cells were also isolated from IP KOs and DKOs. No difference in cell proliferation was detected between the two groups (Supplemental Figure II).
2. EP4 activation promotes endothelial cell proliferation and reduces endothelium-leukocyte adhesion
Receptors that might mediate the effect of PGE2 on EC proliferation were then studied in vitro. ECs from DKO mice were treated with agonists33, 34 for the EP1/3 (sulprostone, EC50=0.42 nmol/L for EP3, also a weak agonist for EP1), EP2 (butaprost, EC50=32 nmol/L) or EP4 (AE1-329, EC50=3.1 nmol/L) receptors, all at a concentration of 1 μmol/L. AE1-329, but not sulprostone or butaprostpromoted proliferation in the DKO ECs (Figure 3A). Similar effects were observed with wild type ECs (Figure 3B). We further tested each drug at mutiple concentrations for cell proliferating activity (Supplemental Figure III). AE1-329 dose-dependently promoted EC proliferation, while butaprost showed a pro-proliferative effect at a higher concentration, i.e., 10 μmol/L. AE1-329 enhanced EC proliferation in either absence or presence of IL-1β, a stimulus to PGE2 production (Figure 3C & Supplemental Figure IVA). Conversely, GW627368X (a selective EP4 antagonist) markedly inhibited EC proliferation under IL-1β stimulation (Figure 3D). When mPGES-1 KO ECs were used, where PGE2 production was depressed (Supplemental Figure IVB), the effect of EP4 agonism, but not that of EP4 antagonism, remained significant (Figure 3E). Thus, endogenous PGE2 derived from EC mPGES-1 plays an active role in promoting proliferation of the IL-1β stimulated ECs, which can be blocked by antagonism of EP4, but not of other PGE2 receptors (Figure 3F). This further confirms the proliferative effect of EP4 activation in ECs (Figure 3A & B).
Figure 3. PGE2 promoted in vitro endothelial cell proliferation via EP4/cAMP/PKA signaling.
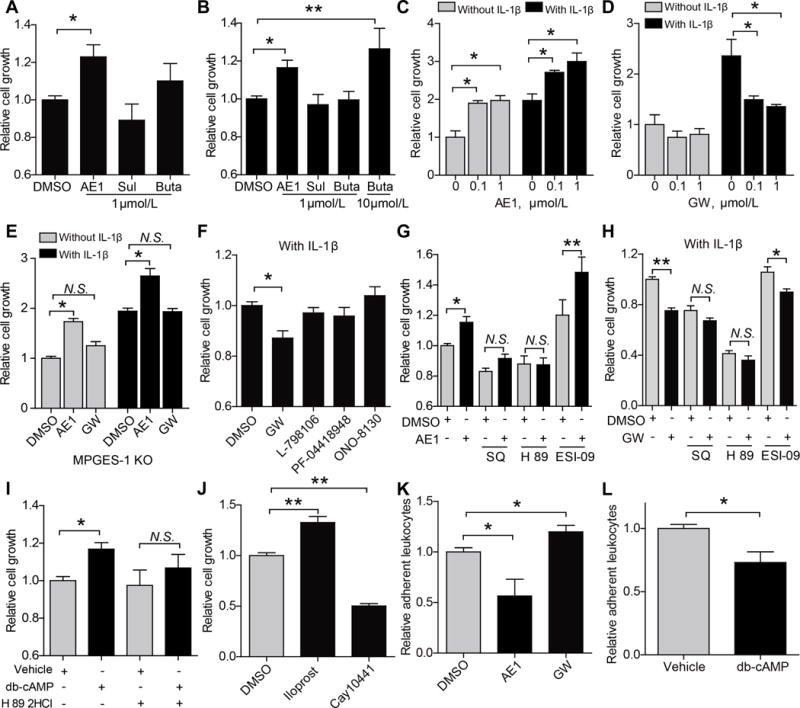
Mouse aortic endothelial cells (MAECs) were used for the study of cell proliferation in vitro. Cells from DKO (A) and wildtype (WT) mice (B) were stimulated with agonists for PGE2 receptors [AE1-329 (AE1), an EP4 agonist; Sulprostone (Sul), an EP1/3 agonist; Butaprost (Buta), an EP2 agonist], and relative proliferation is shown. WT ECs were treated with AE1 of different concentrations, with or without IL-1β (10 ng/mL), and proliferation was determined (C). GW627368X (GW, an EP4 antagonist) suppressed proliferation of IL-1β-stimulated MAECs (D). Effects of AE1 or GW on the proliferation of Ptges −/− ECs, stimulated with IL-1β (10 ng/mL) or not, were shown (E). Under IL-1β (10 ng/mL) stimulation, GW (an EP4 antagonist) but not L-798106 (an EP3 antagonist), PF-04418948 (an EP2 antagonist) or ONO-8130 (an EP1 antagonist), all at a concentration of 1 μmol/L, inhibited endothelial cell proliferation (F). The pro-proliferative effect of AE1 (G) and the anti-proliferative effect of GW (H) was prevented by SQ (SQ22536, an adenylate cyclase inhibitor; 200 μmol/L) or H 89 (H 89 2HCl, a PKA inhibitor; 10 μmol/L), but not by ESI-09 (an EPAC inhibitor; 10 μmol/L). Db-cAMP (I; a cell permeable cAMP analogue, 30 μmol/L) promoted endothelial proliferation, and this was also blunted by H89 2HCl (a PKA inhibitor). Endothelial proliferation was stimulated by iloprost (an IP agonist, 1 μmol/L) and inhibited by Cay10441 (an IP antagonist, 10 μmol/L) (J). The adhesion of leukocytes to ECs was inhibited by the EP4 agonist, AE1-329, and was promoted by GW, an EP4 antagonist (K). Db-cAMP treatment reduced leukocyte adhesion to ECs (L). All results are from at least three independent data sets. *P<0.05. **P<0.01; One-way ANOVA was used for data comparisons with Bonferroni’s (A, G-K), Dunnett’s (B) or Turkey’s (C-F) post-test. Student’s unpaired t-test was used in L.
To elucidate EP4 downstream signaling, SQ22536 [an adenylyl cyclase (AC) inhibitor], H 89 2HCl (a PKA inhibitor) and ESI-09 (an EPAC inhibitor) were used. Treatment with SQ22536 or H 89 2HCl, but not with ESI-09, abrogated the pro-proliferation effect of AE1-329 (Figure 3G) and the anti-proliferation effect of GW627368X (Figure 3H), indicating that cAMP-PKA axis underlies the enhanced EC proliferation driven by EP4 activation. Consistent with this finding, Db-cAMP (a cell permeable cAMP analogue) and forskolin (a potent AC activator) both promoted EC proliferation, and such proliferative effects were blunted by PKA inhibition with H89 2HCl or PKI (Figure 3I, and Supplemental Figure V & VI). PGI2 is known to elevate cAMP signaling via IP receptor1. Indeed, iloprost (1 μmol/L), an IP agonist, promoted endothelial proliferation, whereas Cay10441 (10 μmol/L), an IP antagonist inhibited this response (Figure 3J), indicating a proliferative effect of PGI2 on ECs.
The effect of EP4 on endothelium-leukocyte adhesion was also studied in vitro. Incubation of a monolayer of ECs with AE1-329, the EP4 selective agonist, significantly suppressed endothelium-leukocyte adhesion, while inhibition of endothelial EP4 by GW627368X enhanced the leukocyte adhesion to ECs (Figure 3K). This is consistent with a cAMP mediated effect, since Db-cAMP (a cell permeable cAMP analogue, 30 μmol/L) treatment similarly reduced the leukocyte adhesion (Figure 3L).
3. Endothelium-restricted deletion of EP4 impairs reendothelization and exacerbates neointimal formation
The role of endothelial EP4 in vascular remodeling was then examined in mice deficient in this receptor only in ECs (Figure 4A) as confirmed by significant suppression of EP4 protein expression in ECs (Figure 4B & C). The mice were subjected to the wire-injury and studied 28 days later. The neointimal area and the ratio of intimal to medial area were both enhanced in the mice lacking endothelial EP4, while the medial area was unchanged (Figure 4D-G). Seven days after vascular injury, reendothelialization was significantly suppressed in mice lacking EP4 only in ECs (Figure 5A & B). This coincided with an increase in the number of intimal leukocytes and enhanced neointimal formation in cKOs (Figure 5C-E).
Figure 4. Induced deletion of endothelial EP4 promotes neointimal formation.
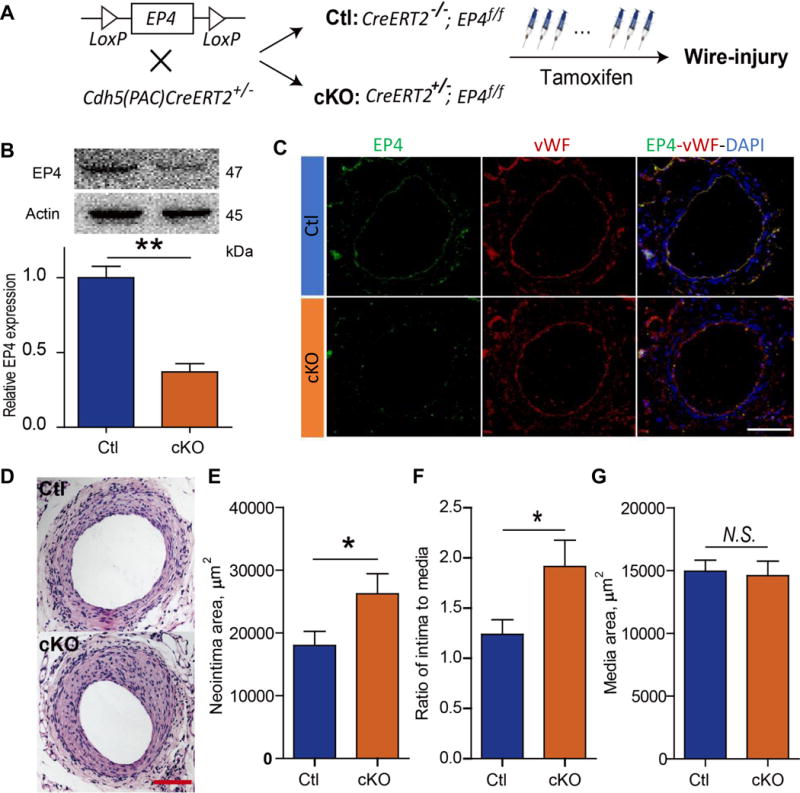
Postnatal deletion of endothelial EP4 gene was induced in mice by tamoxifen treatment (A). Expression of EP4 in the primary ECs isolated from cKO and Ctl mice was detected with Western blot (B), and its expression in femoral arteries was determined by immunofluorescent staining (C; Bar= 100 μm). Representative images are shown (EP4, green; vWF, red). The injured vessels, harvested at 28 days after surgery, were stained with H&E, and representative images are shown (D; Bar= 100 μm). Neointimal area (E), ratio of intima to media (F) and medial thickness (G) were quantified. n= 14 Ctl, 10 cKO. *P<0.05. **P<0.01; Student’s unpaired t-test.
Figure 5. Induced deletion of endothelial EP4 impaired reendothelialization.
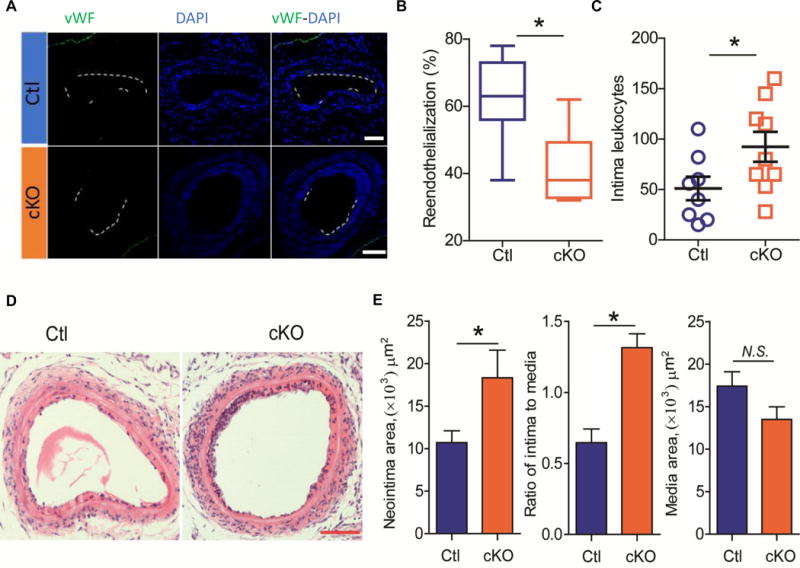
Injured femoral arteries of Ctl and cKO mice were harvested at 7 days after wire-injury. Endothelial cells were immunostained for vWF (green) and quantified for vascular coverage (reendothelialization) (A & B, respectively; n= 7 Ctl, 6 cKO). Neointimal formation and leukocyte infiltration were evaluated with H&E staining. The number of neointimal leukocytes was quantified (C; n= 8 Ctl, 9 cKO). Representative H&E images are shown (D; Bar= 100 μm). Neointimal area, ratio of intima to media and media thickness were statistically quantified (E; n= 8). DAPI stains nuclei in blue. *P<0.05; Student’s unpaired t-test.
4. EP4 activation protects against wire-injury induced neointimal formation
Systemic administration of AE1-329, the EP4 agonist, also ameliorated neointimal formation without affecting media thickness (Figure 6A & B). Misoprostol, which promotes endothelial proliferation in vitro (Figure 2I), attenuated neointimal formation (Figure 6A & C) and promoted reendothelialization at 7 days after vascular injury (Figure 6D & E) in vivo. Misoprostol also decreased leukocyte infiltration (Supplemental Figure VII).
Figure 6. Pharmacological activation of EP4 promoted endothelial repair and protected against neointimal formation.
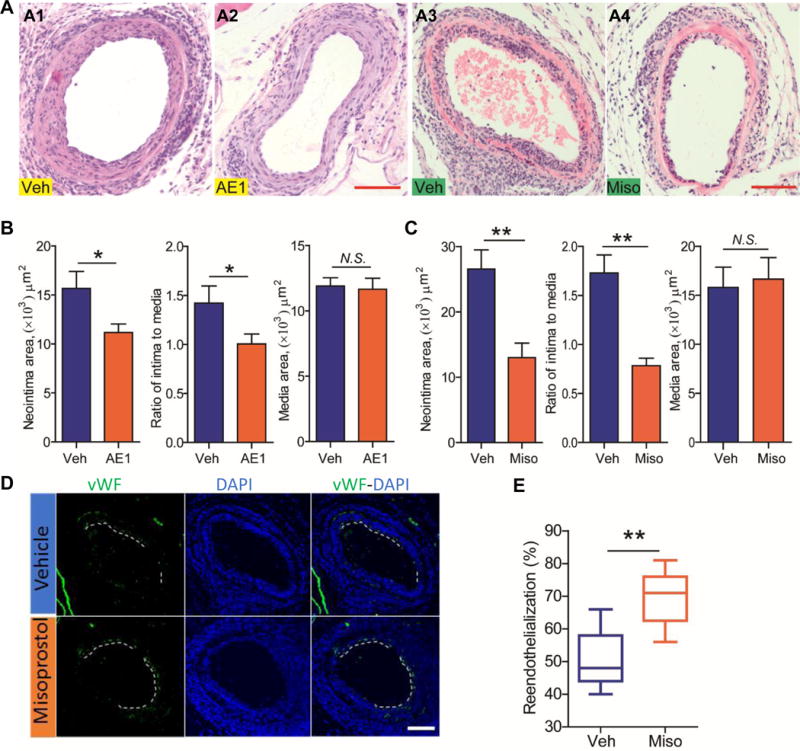
Following vascular injury, C57BL/6 mice were i.p. injected with vehicle (Veh), or with AE1-329 (AE1, an EP4 selective agonist) at a dose of 0.3 mg/kg/day for 28 days (n= 12). Injured vessels were stained with H&E, with representative images shown (A; A1, A2). Neointimal area, ratio of intima to media, media thickness was quantified (B). Another batch of mice, after vascular injury, were treated via i.p. injection with vehicle (Veh), or with misoprostol (Miso, a PGE analogue) at a dose of 100 μg/Kg, three times a day, for a total of 7 days (n= 7 Veh, 5 Miso). Injured vessels were stained with H&E, with representative images shown (A; A3, A4). Neointimal area, ratio of intima to media, media thickness was quantified (C). Reendothelialization was examined by immunostaining of vWF and quantified (D & E) as detailed previously. *P<0.05, **P<0.01; Student’s unpaired t-test.
5. EP4 critically involves in human endothelial cell proliferation
Activation of EP4 by AE1-329, but not of other PGE2 receptors, promoted proliferation of human primary endothelial cells (Figure 7A). Conversely, among antagonists of the four EPs, only EP4 blockade by GW627368X inhibited the EC proliferation (Figure 7B).
Figure 7. EP4 signaling participated in the proliferation of human endothelial cells.
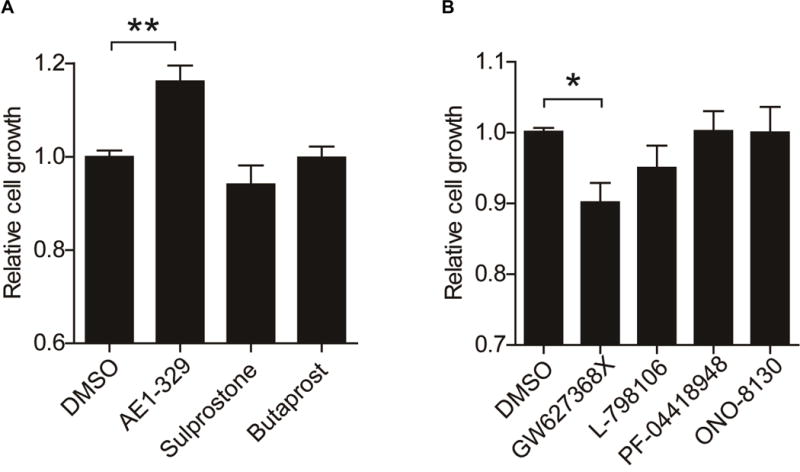
Human microvascular endothelial cells were treated with 1 μmol/L of agonists (A) or antagonist (B) of the receptors of PGE2, and relative proliferation was determined. Agonists: AE1-329 (EP4), sulprostone (EP1/3) and butaprost (EP2). Antagonist: GW627368X (EP4), L-798106 (EP3), PF-04418948 (EP2) and ONO-8130 (EP1). Both results were from three independent data sets. *P<0.05, **P<0.01; One-way ANOVA with Dunnett’s post-test was used for data comparisons.
Discussion
Here we report a novel mechanism that regulates vascular responses to injury in mice: PGE2 acts via endothelial EP4 to promote reendothelialization and restrain neointimal formation after wire induced vascular injury. A similar mechanism promotes proliferation of human endothelial cells in vitro. These observations raise the possibility that EP4 agonists may have value as adjunctive therapy in patients undergoing percutaneous revascularization.
We have previously reported that augmented biosynthesis of PGI2 explains the reduced neointimal response to injury in mPGES-1 KO, raising the possibility that inhibitors of this enzyme will have a more favorable cardiovascular profile than NSAIDs targeting COX-2 that depress PGI2 biosynthesis16. Here, removal of the PGI2 receptor reveals a second therapeutic opportunity as mPGES-1 derived PGE2 can limit neointimal formation after vascular injury (Figures 1&2). Indeed, we show in mice that pharmacological activation of EP4 confers protection against the injury-induced intima hyperplasia (Figure 6). Mechanistically, these beneficial effects are at least mediated through a support of EC repair by mPGES-1 derived PGE2 and EP4 (Figures 2&6). We also observed an increased leukocytes infiltration (Figure 2C). This is consistent with a suppressing effect of EP4 activation on leukocyte adhesion (Figure 2J and 3K). Previous studies22, 35-39 indicate that the PGE2/EP4 axis inhibits mobilization of inflammatory cells and their response to inflammation. These studies highlights a possible contribution of the PGE2/EP4 axis on bone marrow microenvironment and on inflammatory cells per se in mediating the increased leukocyte infiltration to the injured vessels observed in the present study. Here, we discovered that EP4 in the endothelial compartment also mediates endothelial-leukocytes adhesion (Figure 5). There was no discernible difference in the neointimal SMCs (Figure 2E) or in the proliferation of VSMCs isolated from the mPGES-1 deficient mice (Supplemental Figure II). In cultured ECs, activation of EP4 (and perhaps EP2, but not EP1 and EP3), via cAMP-PKA pathway, promotes endothelial proliferation in vitro (Figure 3), consistent with the observed suppression of neointimal formation after injury by such ligands in vivo. In line with this mechanistic hypothesis, deletion of the endothelial EP4 impaired reendothelialization and exacerbated neointimal formation after injury (Figure 4&5). Furthermore, EP4 activation promoted reendothelialization and inhibited neointima formation in mice (Figure 6). Therefore, EC EP4 mediated endothelial repair underscores a key mechanism that restrains the vascular hyperplasia after denudation injury. Promotion of endothelialization by activating endothelial EP4 represents a novel strategy to limit restenosis after PCI40. The mechanism of mPGES-1 derived PGE2 in vascular remodeling is schematically illustrated in Supplemental Figure VIII.
Development of mPGES-1 inhibitors has now reached clinical stage. Interestingly, an augmented production of PGI2 was observed with suppression of mPGES-1 derived PGE27. This is consistent with our initial observation with mPGES-1 KO mice11. Potential elevation of PG products due to substrate rediversion may depend on specific tissue/cell or disease pathology10. Their significance in mediating the cardiovascular profile of mPGES-1 inhibition relative to COX-2 inhibition, warrants future study. Our study here suggests that augmented PGI2 may balance a vascular risk of suppressing PGE2-EP4 axis when mPGES-1 inhibition is pursued under pathologic insult.
In conclusion, we have previously provided evidence that inhibition of mPGES-1 may offer an approach adjunctive to PCI to limit restenosis due to its capacity to augment PGI2. Here, we provide evidence that PGE2 formation might be targeted to achieve the same objective, here by activating the EP4 receptor in endothelial cells. Given that restenosis remains a restraint on the clinical effectiveness of PCI40, 41, the provision of pre-clinical data consistent with two distinct strategies by which this might be mitigated is timely.
Study limitations
The mechanistic exploration of this study was mainly focused on endothelial proliferation/repair by mPGES-1 derived PGE2 and EP receptors. Whether impaired reendothelialization process might allow enhanced exposure to certain thrombotic components that may also contribute to the vascular responses, warrants future study. This study was conducted with mice of C57BL/6 background. Confirming this study with other mouse strains or other species more relevant to human pathophysiology would help increase the translational potential of the mechanism discovered here.
Supplementary Material
Highlights.
Deletion of mPGES-1, on a background of IP depletion, exacerbates wire-injury induced neointima formation, with early impairment of reendothelialization.
Activation of EP4 or EP2 promotes endothelial cell proliferation, and endothelium-restricted deletion of EP4 impairs reendothelialization and increases neointima formation.
EP4 agonism promotes reendothelialization and attenuates neointima formation, raising a possibility that EP4 agonism may prevent restenosis after percutaneous coronary intervention.
Acknowledgments
We thank Yuuki Nagao from ONO Pharmaceutical Co., Ltd. for supplying the research materials and for reviewing the manuscript. We thank Xu Zhang from Tianjin Medical University for measuring prostaglandins.
Funding Sources: This work was supported by the National Natural Science Foundation of China (81570269 and 81370222), Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2017-12M-1-008, 2016-I2M-1-003/005), a research award from the National 1000-Talent Plan of China and research funds from Fuwai Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences, with all to MW, by the fund from National Heart, Lung, and Blood Institute (HL117798) to G.A.F., and by the National Natural Science Foundation of China (81703517) to HH.
Abbreviations
- AE1
AE1-329
- Buta
Butaprost
- CCK-8
Cell counting kit-8
- cKO
Endothelial EP4 conditional-knockout mice
- COX
Cyclooxygenase
- Ctl
Littermate controls for cKO
- DKO
Double knockout
- ECGS
Endothelial cell growth supplement
- ECs
Endothelial cells
- EP1, 2, 3, 4
Prostaglandin E receptor 1, 2, 3, 4
- FBS
Fetal bovine serum
- GW
GW627368X
- H&E
Haematoxylin and Eosin
- H 89
H 89 2HCl
- HMECs
Human microvascular endothelial cells
- IL-1β
Interleukin 1β
- i.p.
Intraperitoneal
- IP
I prostanoid receptor
- IP KO
I prostanoid receptor knockout
- MAECs
Mouse aorta endothelial cells
- Miso
Misoprostol
- mPGES-1
Microsomal prostaglandin E synthase-1
- NSAIDs
Nonsteroidal anti-inflammatory drugs
- PCI
Percutaneous coronary intervention
- PG
Prostaglandin
- PGI2
Prostacyclin
- SMA
Smooth muscle actin
- SQ
SQ22536
- Sul
Sulprostone
- Veh
Vehicle
- VSMC
Vascular smooth muscle cell
- vWF
von Willebrand Factor
Footnotes
Conflict of Interest Disclosures
Patent application (CN201710293360.9) is pending.
References
- 1.Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50(Suppl):S423–428. doi: 10.1194/jlr.R800094-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coxib, traditional NTC. Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, Bombardier C, Cannon C, Farkouh ME, FitzGerald GA, Goss P, Halls H, Hawk E, Hawkey C, Hennekens C, Hochberg M, Holland LE, Kearney PM, Laine L, Lanas A, Lance P, Laupacis A, Oates J, Patrono C, Schnitzer TJ, Solomon S, Tugwell P, Wilson K, Wittes J, Baigent C. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: Meta-analyses of individual participant data from randomised trials. Lancet. 2013;382:769–779. doi: 10.1016/S0140-6736(13)60900-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, Anderson WF, Zauber A, Hawk E, Bertagnolli M. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071–1080. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 4.Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A, Konstam MA, Baron JA. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–1102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 5.Grosser T, Yu Y, FitzGerald GA. Emotion recollected in tranquility: Lessons learned from the cox-2 saga. Ann Rev Med. 2010;61:17–33. doi: 10.1146/annurev-med-011209-153129. [DOI] [PubMed] [Google Scholar]
- 6.Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin e synthase: A microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin Y, Smith CL, Hu L, Campanale KM, Stoltz R, Huffman LG, Jr, McNearney TA, Yang XY, Ackermann BL, Dean R, Regev A, Landschulz W. Pharmacodynamic comparison of ly3023703, a novel microsomal prostaglandin e synthase 1 inhibitor, with celecoxib. Clin Pharmacol Ther. 2016;99:274–284. doi: 10.1002/cpt.260. [DOI] [PubMed] [Google Scholar]
- 8.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin e synthase-1: A novel therapeutic target. Pharmacol Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 9.Tang SY, Monslow J, G RG, Todd L, Pawelzik SC, Chen L, Lawson J, Pure E, FitzGerald GA. Cardiovascular consequences of prostanoid i receptor deletion in microsomal prostaglandin e synthase-1-deficient hyperlipidemic mice. Circulation. 2016;134:328–338. doi: 10.1161/CIRCULATIONAHA.116.022308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M, Zukas AM, Hui Y, Ricciotti E, Pure E, FitzGerald GA. Deletion of microsomal prostaglandin e synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci U S A. 2006;103:14507–14512. doi: 10.1073/pnas.0606586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin e synthase-1, and cardiovascular function. The Journal of clinical investigation. 2006;116:1391–1399. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang M, FitzGerald GA. Cardiovascular biology of microsomal prostaglandin e synthase-1. Trends Cardiovasc Med. 2010;20:189–195. doi: 10.1016/j.tcm.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koo BK, Kim YS, Park KW, Yang HM, Kwon DA, Chung JW, Hahn JY, Lee HY, Park JS, Kang HJ, Cho YS, Youn TJ, Chung WY, Chae IH, Choi DJ, Oh BH, Park YB, Kim HS. Effect of celecoxib on restenosis after coronary angioplasty with a taxus stent (corea-taxus trial): An open-label randomised controlled study. Lancet. 2007;370:567–574. doi: 10.1016/S0140-6736(07)61295-1. [DOI] [PubMed] [Google Scholar]
- 14.Kang HJ, Oh IY, Chung JW, Yang HM, Suh JW, Park KW, Kwon TK, Lee HY, Cho YS, Youn TJ, Koo BK, Kang WY, Kim W, Rha SW, Bae JH, Chae IH, Choi DJ, Kim HS. Effects of celecoxib on restenosis after coronary intervention and evolution of atherosclerosis (mini-corea) trial: Celecoxib, a double-edged sword for patients with angina. Eur Heart J. 2012;33:2653–2661. doi: 10.1093/eurheartj/ehs001. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, Yang G, Xu X, Grant G, Lawson JA, Bohlooly YM, FitzGerald GA. Cell selective cardiovascular biology of microsomal prostaglandin e synthase-1. Circulation. 2013;127:233–243. doi: 10.1161/CIRCULATIONAHA.112.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang M, Ihida-Stansbury K, Kothapalli D, Tamby MC, Yu Z, Chen L, Grant G, Cheng Y, Lawson JA, Assoian RK, Jones PL, Fitzgerald GA. Microsomal prostaglandin e2 synthase-1 modulates the response to vascular injury. Circulation. 2011;123:631–639. doi: 10.1161/CIRCULATIONAHA.110.973685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, Lawson JA, FitzGerald GA. Role of prostacyclin in the cardiovascular response to thromboxane a2. Science. 2002;296:539–541. doi: 10.1126/science.1068711. [DOI] [PubMed] [Google Scholar]
- 18.Sugimoto Y, Narumiya S. Prostaglandin e receptors. The Journal of biological chemistry. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, Zou F, Tang J, Zhang Q, Gong Y, Wang Q, Shen Y, Xiong L, Breyer RM, Lazarus M, Funk CD, Yu Y. Cyclooxygenase-2-derived prostaglandin e(2) promotes injury-induced vascular neointimal hyperplasia through the e-prostanoid 3 receptor. Circ Res. 2013;113:104–114. doi: 10.1161/CIRCRESAHA.113.301033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao CY, Yuhki K, Hara A, Fujino T, Kuriyama S, Yamada T, Takayama K, Takahata O, Karibe H, Taniguchi T, Narumiya S, Ushikubi F. Prostaglandin e2 protects the heart from ischemia-reperfusion injury via its receptor subtype ep4. Circulation. 2004;109:2462–2468. doi: 10.1161/01.CIR.0000128046.54681.97. [DOI] [PubMed] [Google Scholar]
- 21.Hishikari K, Suzuki J, Ogawa M, Isobe K, Takahashi T, Onishi M, Takayama K, Isobe M. Pharmacological activation of the prostaglandin e2 receptor ep4 improves cardiac function after myocardial ischaemia/reperfusion injury. Cardiovascular research. 2009;81:123–132. doi: 10.1093/cvr/cvn254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang MZ, Yao B, Wang Y, Yang S, Wang S, Fan X, Harris RC. Inhibition of cyclooxygenase-2 in hematopoietic cells results in salt-sensitive hypertension. The Journal of clinical investigation. 2015;125:4281–4294. doi: 10.1172/JCI81550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Babaev VR, Chew JD, Ding L, Davis S, Breyer MD, Breyer RM, Oates JA, Fazio S, Linton MF. Macrophage ep4 deficiency increases apoptosis and suppresses early atherosclerosis. Cell metabolism. 2008;8:492–501. doi: 10.1016/j.cmet.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao R, Redha R, Macias-Perez I, Su Y, Hao C, Zent R, Breyer MD, Pozzi A. Prostaglandin e2-ep4 receptor promotes endothelial cell migration via erk activation and angiogenesis in vivo. The Journal of biological chemistry. 2007;282:16959–16968. doi: 10.1074/jbc.M701214200. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Daaka Y. Pge2 promotes angiogenesis through ep4 and pka cgamma pathway. Blood. 2011;118:5355–5364. doi: 10.1182/blood-2011-04-350587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson PJ, Carty TJ, Perez JR, Audoly LP. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin e synthase. Proc Natl Acad Sci U S A. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen M, Camenisch T, Snouwaert JN, Hicks E, Coffman TM, Anderson PA, Malouf NN, Koller BH. The prostaglandin receptor ep4 triggers remodelling of the cardiovascular system at birth. Nature. 1997;390:78–81. doi: 10.1038/36342. [DOI] [PubMed] [Google Scholar]
- 28.Hao H, Hu S, Chen H, Bu D, Zhu L, Xu C, Chu F, Huo X, Tang Y, Sun X, Ding BS, Liu DP, Hu S, Wang M. Loss of endothelial cxcr7 impairs vascular homeostasis and cardiac remodeling after myocardial infarction: Implications for cardiovascular drug discovery. Circulation. 2017;135:1253–1264. doi: 10.1161/CIRCULATIONAHA.116.023027. [DOI] [PubMed] [Google Scholar]
- 29.Schneider A, Guan Y, Zhang Y, Magnuson MA, Pettepher C, Loftin CD, Langenbach R, Breyer RM, Breyer MD. Generation of a conditional allele of the mouse prostaglandin ep4 receptor. Genesis. 2004;40:7–14. doi: 10.1002/gene.20048. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, Adams RH. Ephrin-b2 controls vegf-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Z, Subramanian P, Sevilmis G, Globke B, Soehnlein O, Karshovska E, Megens R, Heyll K, Chun J, Saulnier-Blache JS, Reinholz M, van Zandvoort M, Weber C, Schober A. Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing cxcl1 from the endothelium. Cell metabolism. 2011;13:592–600. doi: 10.1016/j.cmet.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 32.Song WL, Lawson JA, Wang M, Zou H, FitzGerald GA. Noninvasive assessment of the role of cyclooxygenases in cardiovascular health: A detailed hplc/ms/ms method. Methods in enzymology. 2007;433:51–72. doi: 10.1016/S0076-6879(07)33003-6. [DOI] [PubMed] [Google Scholar]
- 33.Markovic T, Jakopin Z, Dolenc MS, Mlinaric-Rascan I. Structural features of subtype-selective ep receptor modulators. Drug Discov Today. 2017;22:57–71. doi: 10.1016/j.drudis.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 34.Woodward DF, Jones RL, Narumiya S. International union of basic and clinical pharmacology. Lxxxiii: Classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev. 2011;63:471–538. doi: 10.1124/pr.110.003517. [DOI] [PubMed] [Google Scholar]
- 35.Hoggatt J, Mohammad KS, Singh P, Hoggatt AF, Chitteti BR, Speth JM, Hu P, Poteat BA, Stilger KN, Ferraro F, Silberstein L, Wong FK, Farag SS, Czader M, Milne GL, Breyer RM, Serezani CH, Scadden DT, Guise TA, Srour EF, Pelus LM. Differential stem- and progenitor-cell trafficking by prostaglandin e2. Nature. 2013;495:365–369. doi: 10.1038/nature11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawano Y, Fukui C, Shinohara M, Wakahashi K, Ishii S, Suzuki T, Sato M, Asada N, Kawano H, Minagawa K, Sada A, Furuyashiki T, Uematsu S, Akira S, Uede T, Narumiya S, Matsui T, Katayama Y. G-csf-induced sympathetic tone provokes fever and primes antimobilizing functions of neutrophils via pge2. Blood. 2017;129:587–597. doi: 10.1182/blood-2016-07-725754. [DOI] [PubMed] [Google Scholar]
- 37.McGonigle TA, Dwyer AR, Greenland EL, Scott NM, Keane KN, Newsholme P, Goodridge HS, Zon LI, Pixley FJ, Hart PH. Pge2 pulsing of murine bone marrow cells reduces migration of daughter monocytes/macrophages in vitro and in vivo. Experimental hematology. 2017;56:64–68. doi: 10.1016/j.exphem.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang EH, Shvartz E, Shimizu K, Rocha VZ, Zheng C, Fukuda D, Shi GP, Sukhova G, Libby P. Deletion of ep4 on bone marrow-derived cells enhances inflammation and angiotensin ii-induced abdominal aortic aneurysm formation. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:261–269. doi: 10.1161/ATVBAHA.110.216580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang EH, Shimizu K, Christen T, Rocha VZ, Shvartz E, Tesmenitsky Y, Sukhova G, Shi GP, Libby P. Lack of ep4 receptors on bone marrow-derived cells enhances inflammation in atherosclerotic lesions. Cardiovascular research. 2011;89:234–243. doi: 10.1093/cvr/cvq262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finn AV, Vorpahl M, Ladich E, Virmani R. Future directions in stenting. Expert Rev Cardiovasc Ther. 2010;8:1–6. doi: 10.1586/erc.09.144. [DOI] [PubMed] [Google Scholar]
- 41.Otsuka F, Finn AV, Yazdani SK, Nakano M, Kolodgie FD, Virmani R. The importance of the endothelium in atherothrombosis and coronary stenting. Nat Rev Cardiol. 2012;9:439–453. doi: 10.1038/nrcardio.2012.64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.