

Abstract
By their ability to shatter quality of life for both patients and caregivers, neurodegenerative diseases are the most devastating of human disorders. Unfortunately, there are no effective or long-terms treatments capable of slowing down the relentless loss of neurons in any of these diseases. One impediment is the lack of detailed knowledge of the molecular mechanisms underlying the processes of neurodegeneration. While some neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease and Amyotrophic lateral sclerosis, are mostly sporadic in nature, driven by both environment and genetic susceptibility, many others, including Huntington’s disease, Spinocerebellar ataxias and Spinal-bulbar muscular atrophy, are genetically inherited disorders. Surprisingly, given their different roots and etiologies, both sporadic and genetic neurodegenerative disorders have been linked to disease mechanisms involving histone deacetylase (HDAC) proteins, which consists of 18 family members with diverse functions. While most studies have implicated certain HDAC subtypes in promoting neurodegeneration, a substantial body of literature suggests that other HDAC proteins can preserve neuronal viability. Of particular interest, however, is the recent realization that a single HDAC subtype can have both neuroprotective and neurotoxic effects. Diverse mechanisms, beyond transcriptional regulation have been linked to these effects, including deacetylation of non-histone proteins, protein-protein interactions, post-translational modifications of the HDAC proteins themselves and direct interactions with disease proteins. The roles of these HDACs in both sporadic and genetic neurodegenerative diseases will be discussed in the current review.
Keywords: Histone deacetylase, Neurodegenerative diseases, Neuroprotection, Huntington’s disease
Graphical Abstract
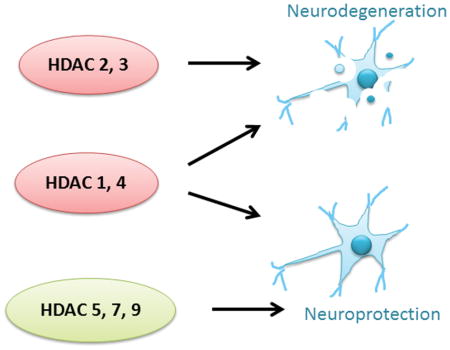
In our review we describe that some members of the histone deactylase (HDAC) family, such as HDACs 2 and 3, promote neurodegeneration whereas others, such as HDACs 5, 7 and 9 protect against it. HDACs 1 and 3 have both neuroprotective and neurotoxic roles. These actions are mediated through histone acetylation, non-histone acetylation, posttranslational modifications and protein-protein interactions. The roles of these HDACs in both sporadic and genetic neurodegenerative diseases are discussed in the current review.
INTRODUCTION
Studies over the past decade have implicated HDAC proteins in a wide range of neurodegenerative diseases, including polyglutamine disorders, Alzheimer’s disease, Parkinson’s disease, as well as other conditions associated with neuronal loss, such as ischemic stroke(D’Mello 2009; Sleiman et al. 2009; Thomas 2009). Accordingly, the use of inhibitors of these enzymes as a potential therapeutic option to treat these disorders has gained much recent attention. HDAC inhibitors have been shown to be effective in numerous different mouse models of neurodegenerative diseases (D’Mello 2009; Sleiman et al. 2009; Thomas 2009). However, HDAC enzymes comprise a large family of proteins and it has become clear that individual HDAC enzymes play vastly different biological roles in the brain, complicating the potentially diverse effects elicited by HDAC inhibitors. The design of subtype-selective HDAC inhibitors represents a major improvement to this line of therapy, however, it is essential to elucidate the distinct and varied roles of individual HDAC proteins, for their potential clinical utility to be fully realized(Millard et al. 2017; Thomas 2009). This is especially important, given that many HDAC family members have been shown to exhibit both neurotoxic and neuroprotective properties, as we will review herein. This review focuses on the neurotoxic and neuroprotective roles of classical (i.e. class I and II) HDACs in neurodegenerative disorders. The role of class II HDACs (aka sirtuins) in the pathology of neurodegeneration has been covered in other excellent reviews (Ajami et al. 2017; Donmez 2012; Jesko et al. 2017) and will not be discussed in this review.
THE HDAC FAMILY OF PROTEINS
HDAC proteins comprise an ancient enzyme family, conserved in evolution from yeast to plants and animals (Gregoretti et al. 2004; Yang and Seto 2008). In humans, there are 18 HDAC subtypes, which have been divided into four separate classes based on sequence and structural homology (Gregoretti et al. 2004; Grozinger et al. 1999). Class I HDAC proteins consist of HDACs 1, 2, 3 and 8, and show primarily nuclear localization (Wang et al. 2004). Class II HDACs can shuttle between the nucleus and cytoplasm and are further divided into two groups: class IIa, consisting of HDACs 4, 5, 7 and 9, and class IIb, consisting of HDACs 6 and 10 (Haberland et al. 2009; Xu et al. 2007; Yang and Seto 2008). Both class I and class II enzymes, typically referred to as “classical” HDACs, require zinc for catalytic activity. In contrast, the members of class III HDAC proteins, called the “sirtuins”, are structurally unrelated from classes I and II and require NAD+ for their enzymatic activity (Michan and Sinclair 2007; Sauve et al. 2006). HDAC11 is the sole member of class IV, and while sharing similar characteristics to HDACs in classes I and II, HDAC 11 is thought to differ in its physiological properties (Gao et al. 2002; Yang and Seto 2008). All of the classical HDACs, as well as HDAC11, are expressed in the brain, albeit at different levels depending on the region (Liu et al. 2008). Details on individual HDACs and their roles in neurodegenerative processes are discussed below.
HDAC MECHANISMS OF ACTION
The link between histone acetylation/deacetylation and transcriptional regulation was first realized more than 25 years ago by the identification of proteins with intrinsic histone acetylase and deacetylase activity (Kornberg and Lorch 1991). The initial founding members of the HDAC family were the human, HDAC1, and yeast Rpd3 proteins (Gregoretti et al. 2004; Yang and Seto 2008). Deletions of these genes resulted in increased acetylation levels in both core histones H3 and H4 (Rundlett et al. 1996). It is now well understood that regulation of histone acetylation by histone acetyltransferase (HAT) and HDAC enzymes, is a major driving force in the control of gene expression (Gregoretti et al. 2004; Yang and Seto 2008). Typically, increases in HAT activity promote acetylation of histone proteins leading to increased gene transcription by creating a more open conformation of chromatin, while HDAC activity involves removing the acetyl group from histones, which results in gene repression (Figure 1A). HDACs lack intrinsic DNA-binding activity and are recruited to target genes via their association with transcriptional activators and repressors, as well as their incorporation into large multiprotein complexes, often containing more than one HDAC subtype (Gregoretti et al. 2004; Haberland et al. 2009; Xu et al. 2007; Yang and Seto 2008).
Figure 1.

Different mechanisms of actions for HDAC proteins. A. Traditional effects of HDAC to alter histone acetylation and chromatin structure leading to changes in gene transcription. B. HDACs can also deacetylate non-histone proteins including transcription factors, such as Nuclear factor-kappa beta (NF-KB), Mitogen-activated protein kinase phosphatase 1 (MKP-1) and Neural-restrictive silencer factor (NRSF), among others. C. Examples of interactions of class II HDACs with regulatory proteins, including Heterochromatin protein 1 (HP1) and C-terminal-binding protein (CTBP). Class II HDACs, such as HDAC4 and HDAC5, can also interaction with class I HDACs, such as HDAC3, as part of transcriptional regulatory complexes. The nuclear receptor co-repressor 2/ silencing mediator for retinoid and thyroid hormone receptors (N-Cor/SMRT) complex is shown as an example.
Aside from their essential roles in gene transcription, HDAC proteins are now known to deacetylate a large and ever-growing number of non-histone proteins (Spange et al. 2009; Xu et al. 2007; Yang and Seto 2008) (Figure 1B). These include a variety of different transcription factors, structural proteins, ion channels, receptors and enzymes. Deacetylation of these proteins can have dramatic effects on their function, stability, sub-cellular localization, as well as their interactions with other proteins. For example, studies have shown that HDAC1, -2, and -3 deacetylate MAP kinase phosphatase (MKP1) and that this post-translational modification increases Mitogen-activated protein kinase (MAPK) and innate immune signaling (Jeong et al. 2014) (Figure 1B). Importantly, the acetylation status of transcription factors is known to alter DNA-binding properties and transcriptional activity. This means that HDAC proteins could exert their neurotoxic or neuroprotective actions through histone deacetylation and gene expression changes, or through a host of other mechanisms, some of which are described below and summarized in Figure 1. This also might explain why unbiased gene expression studies have revealed that treatment with HDAC inhibitors do not always lead to increased gene expression changes, but rather, include both increases and decreases in gene activity (Gardian et al. 2005; Thomas et al. 2008a).
Although it was initially believed that all classical HDACs possess catalytic activity, subsequent analyses revealed that class IIa HDACs have minimal deacetylase activity at best and do not associate with histone tails (Jones et al. 2008; Lahm et al. 2007; Parra 2015). This is because a catalytic tyrosine residue, which is conserved in all HDAC proteins and necessary for catalytic activity, is replaced by a histidine residue in class II HDACs. Therefore, any effects class II HDACs have on neurodegeneration and that are blocked by HDAC inhibitors are likely mediated through association with class I HDACs that would allow acquisition of deacetylase activity. For example, Fischle and colleagues demonstrated that the HDAC domains of HDAC4 and HDAC5 do not possess intrinsic enzymatic activity as isolated polypeptides but are associated with HDAC activity only by interacting with HDAC3, via the transcriptional corepressor N-CoR/SMRT (Fischle et al. 2002) (Figure 1C).
Class IIa HDACs could regulate neurodegeneration by deacetylase-independent mechanism possibly through interaction with non-HDAC proteins (Ma and D’Mello 2011a; Majdzadeh et al. 2008a; Rawat et al. 2016). Several of such proteins have been identified, most of which interact with the N-terminal half of the HDACs (Martin et al. 2007). For example, the regulatory domains of class IIa HDACs have been shown to interact with transcriptional repressors, such as heterochromatin protein 1 (HP1) and C-terminal-binding protein (CTBP) (Figure 1C). Through such interactions, these class IIa HDACs can function as adaptors to regulate multiple types of transcriptional regulators and regulate downstream gene transcription (Bertos et al. 2001; Sparrow et al. 1999; Wang et al. 2000; Zhou et al. 2000). Consistent with this concept are recent reports describing that some HDAC inhibitors protect against neurodegeneration independently of their deacetylase activity (Olson et al. 2015; Sleiman et al. 2014).
NEUROTOXIC AND/OR NEUROPROTECTIVE PROPERTIES OF HDACS
The discovery of distinct neurotoxic or neuroprotective roles of individual HDACs has deepened our understanding of this family of proteins. However, the recent realization that a single HDAC can have both neuroprotective and neurodegenerative effects appears especially confusing. The basis of this is still unclear, but likely depends on context-specific and tissue/cell-specific effects, whereby different mechanisms are at play. For example, emerging evidence implicates several different mechanisms in the actions of HDACs, including protein-protein interactions, cellular localization (i.e. nucleus vs. cytoplasm), cell-type specificity, alternative splicing or post-translational modifications. Below, we describe the individual HDAC subtypes followed by the roles, in some cases opposing, for each HDAC in the regulation of neuronal survival in general, as well as in specific neurodegenerative diseases. We focus primarily on those HDAC subtypes showing strong expression in the CNS, as these are the ones that have been more consistently implicated in CNS diseases.
HDAC1
HDAC1 is perhaps the most widely studied members of the HDAC family. HDAC1 exhibits a high degree of homology to HDAC2 and has a high degree of functional overlap with HDAC2 for many biological processes (Gregoretti et al. 2004; Tsai and Seto 2002). However, it has become evident from knockout studies that HDAC1 and HDAC2 also have distinct and non-redundant biological functions (Brunmeir et al. 2009). HDAC1 is mostly localized to the nucleus; however, studies have also demonstrated that HDAC1 is expression in the cytoplasm (Jia et al. 2012). HDAC1 shows widespread expression throughout the brain, but is most abundantly expressed in the cerebellum, followed by amygdala and hippocampus (Broide et al. 2007a; Thomas 2009). HDAC1 is expressed primarily in neurons but it is also expressed in glial cells, including astrocytes (Broide et al. 2007a; Kalinin et al. 2013) and oligodendrocytes(Ye et al. 2009). Several lines of evidence have indicated that HDAC1 can have both neurotoxic and neuroprotective roles, which appear to depend, in part, by its cellular localization, non-histone acetylation and/or its interacting partners (Table 1).
Table 1.
Summary of the neuroprotective and/or neurotoxic properties of HDAC proteins. Mechanisms of action were categorized broadly into the following groups: Protein-protein interactions, Post-translational modifications, Non-histone acetylation, Histone-acetylation, Deacetylase-independent. Note: for some studies summarized in the text, specific mechanisms for the individual HDAC proteins were not known, hence are not shown here. N/A, not applicable.
Neurotoxic effects of HDAC1
Previous studies in cultured neurons have shown that neurotoxicity by HDAC1 depends on its export from the nucleus into the cytoplasm, resulting in disruption of axonal transport and mitochondrial dysfunction (Kim et al. 2010). The toxic effect of cytosolic HDAC1 on axonal transport was shown to be due to its ability to bind to motor proteins (i.e. kinesin heavy chains members 2A and 5) and α-tubulin, disrupting their ability to form complexes with cargo proteins (Kim et al. 2010)). Interestingly, cytosolic HDAC1 has been detected in the brains of patients with multiple sclerosis, an autoimmune disease linked to demyelination of axons, and in animal models of demyelination (Kim et al. 2010).
Other studies demonstrated that nuclear export of HDAC1 was dependent on post-translational modifications of HDAC1, whereby phosphorylation at Ser421 and Ser423 residues promoted nuclear export (Zhu et al. 2017). In that same study, the authors show that decreasing HDAC1 levels by genetic ablation was neuroprotective against acute neurotoxicity in hippocampal slices. The protective effect of Hdac1 ablation was also detected in CA3 neurons in mice, which were more resistant to the excitotoxic damage induced by intraventricular injection of kainic acid (Zhu et al. 2017).
HDAC1/2-mediated acetylation of p53 has also been implicated in its neurotoxic effects (Jacob et al. 2011; Lebrun-Julien and Suter 2015). Studies conducted on retinal ganglion cells and Schwaan cells have shown neuroprotective effects when both the Hdac1 and Hdac2 genes were ablated (Jacob et al. 2011; Lebrun-Julien and Suter 2015). Specific Hdac1/2 ablation inhibited this apoptotic pathway by impairing the crucial acetylation status of p53 and reducing PUMA expression, thereby contributing to the ensuing enhanced neuroprotection. In another study, a newly identified but relatively uncharacterized HDAC1/HDAC2-selective inhibitor, K560, had protective effects in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neuronal death in both in vitro and in vivo Parkinson’s disease models (Choong et al. 2016). Treatment with K560 caused a sustained increase in expression of XIAP (X-linked inhibitor of apoptosis) an antiapoptotic protein, leading to beneficial effects (Choong et al. 2016).
HDAC1 has also been linked to the pathology of Huntington’s disease, an autosomal dominant, progressive neurodegenerative disease. Treatment of Huntington’s disease mice with HDAC1/HDAC3-selective inhibitors was shown to elicit beneficial effects on disease phenotypes (Jia et al. 2012; Thomas et al. 2008b), although it is not entirely clear whether this was due to inhibition of HDAC1 or HDAC3, or possibly both. HDAC1 alone has been linked to Huntington’s disease pathogenesis by a mechanism involving promoting autophagy and clearance of the mutant form of the huntingtin protein (Jeong et al. 2009; Ravikumar et al. 2004; Yamamoto et al. 2006). This was found to occur at a specifically identified lysine residue, Lys444 whereby acetylation of Lys444 facilitated trafficking of mutant huntingtin into autophagosomes, thereby promoting its clearance (Jeong et al. 2009). HDAC1 was implicated in this effect from knockdown studies of Hdac1 in cultured striatal and cortical neurons and in a transgenic C. elegans model of Huntington’s disease in which also showed increased Lys444 acetylation, improved clearance of mutant huntingtin protein and reduced neurodegeneration (Jeong et al. 2009). HDAC1 can also interact with HDAC3 to promote toxicity as observed in neurons primed to die both in vitro and in HD mice (Bardai et al. 2012a).
Recent studies have implicated increased levels of HDAC1/2 in synaptic dysfunction and behavioral deficits in Angelman syndrome, a neurodevelopmental disorder characterized by severe intellectual and developmental deficits caused by the loss of function of maternally inherited ubiquitin protein ligase E3A (UBE3A). Both HDAC1 and HDAC2 protein levels are robustly increased in Angelman syndrome Angelman syndrome mice along with decreased acetylation of histone H3/H4 (Jamal et al. 2017). Significant improvement in social, cognitive and motor impairment was described in Angelman syndrome mice after pharmacological inhibition with sodium valproate, an inhibitor selective for Class I HDACs (Jamal et al. 2017).
Neuroprotective effects of HDAC1
Neuroprotective roles for HDAC1 have also been reported (Morrison et al. 2006a),(Dobbin et al. 2013). While its association with HDAC3 results in a neurotoxic complex, HDAC1 interacts with histone deacetylase-related protein (HDRP) to confer neuroprotective effects in cultured neurons (Morrison et al. 2006a). This effect was related to a mechanism involving histone deacetylation of the c-Jun gene promoter and its repression of downstream genes (Morrison et al. 2006a). Another binding partner of HDAC1 is sirtuin 1 (SIRT1), a class III HDAC, which has well established neuroprotective effects (Donmez and Outeiro 2013; Outeiro et al. 2008). Interaction between HDAC1 and SIRT1 is thought to protect neurons by preserving genomic stability (Dobbin et al. 2013). Additionally, HDAC1 interacts with FUS, an RNA/DNA-binding protein mutation of which cause familial amyotrophic lateral sclerosis and frontotemporal lobar degeneration, to regulate DNA damage response and repair in neurons (Qiu et al. 2014; Wang et al. 2013). This is thought to be an important mechanism in Amyotrophic Lateral Sclerosis, whereby disease-causing mutations of FUS result in reduced association of FUS with HDAC1 (Qiu et al. 2014; Wang et al. 2013). Consistent with a neuroprotective role, downregulation of HDAC1 leads to DNA damage in both cultured neurons and in vivo models of neurodegeneration (Kim et al. 2008). It is possible the reduced association with FUS facilitates the translocation of HDAC1 to the cytoplasm of degenerating neurons (Kim et al. 2008).
HDAC2
HDAC2, although intimately associated with HDAC1 as described above, also shows distinct roles with regards to neurotoxicity. Interestingly, HDAC2 is expressed at higher levels in the brain compared to HDAC1. It is most abundantly expressed in the hippocampus and cerebellum, followed by cortex and amygdala (Broide et al. 2007b; Thomas 2009). It is primarily localized in the nucleus, where it is thought to exert its effects in a more traditional manner, via regulating acetylation of histone proteins and ensuing changes in gene expression (Table 1). Past literature supports mostly a neurotoxic role for HDAC2 in the brain, as described below.
Neurotoxic effects of HDAC2
HDAC2 has been implicated in several different neurodegenerative diseases, as well as cognitive impairment, which is common to many of these disorders. Niemann-Pick type C disease is a fatal neurodegenerative disorder characterized by the accumulation of free cholesterol in lysosomes. Studies in the past five years have indicated that inhibition of HDACs is a potential treatment for this disease (Contreras et al. 2016), with HDAC2 being primarily implicated. HDAC2 levels and activity were shown to be increased in Niemann-Pick type C neuronal models and in Npc1(−/−) mice (Contreras et al. 2016). Neuronal gene repression in these disease models is mediated by the c-Abl/HDAC2 signaling pathway, whereby c-Abl tyrosine kinase activity increases HDAC2-induced neuronal gene repression of key synaptic genes (Contreras et al. 2016).
Spinal muscular atrophy is an autosomal-recessive and fatal motor neuron disease caused by mutation of the survival motor neuron (SMN1) gene (Lorson et al. 2010; Nurputra et al. 2013). As a result of a duplication of the SMN locus, humans express a second SMN gene, SMN2, which also encodes the SMN protein. Since Spinal muscular atrophy patients lack SMN1-derived protein, increasing expression of the SMN2 gene has been achieved by pharmacological inhibition of HDACs, with evidence pointing to HDAC2 as being the target of these HDAC inhibitors (Kernochan et al. 2005; Mohseni et al. 2016). Importantly, the administration of HDAC inhibitors has been found to ameliorate disease phenotype in Spinal muscular atrophy mouse models (Avila et al. 2007; Riessland et al. 2010).
Genetic knockdown studies showed that heterozygous Hdac2+/− mice displayed less retinal degeneration than wild-type mice in a mouse model of ischemic retinal injury (Fan et al. 2013). In this study, HDAC2 protein was primarily localized to the nucleus, where it likely acts to alter gene expression (Fan et al. 2013). HDAC2 activity accounted for approximately 35% of the total HDAC activities in the retina, suggesting that treatment with class I HDAC inhibitors could reduce ischemic retinal injury due to inhibition, in part, of this subtype (Fan et al. 2013).
Several studies have demonstrated detrimental effects associated with HDAC2 in the context of cognitive impairment. In one study, overexpression of HDAC2 in the hippocampus was associated with hypo-acetylation of specific lysine residues, Lys12 and Lys5, on histone H4 (Guan et al. 2009). This effect was accompanied by decreased synapse number and synaptic plasticity, resulting in impaired memory formation. The mechanism implicated in this effect was the binding of HDAC2 to the promoters of synaptic-plasticity-related genes, thereby negatively regulating their transcription (Guan et al. 2009). Another study showed that HDAC2 associates with, and reduces the histone acetylation of, genes important for learning and memory (Graff et al. 2012). In one study, knocking down Hdac2 by shRNA restored the structural synaptic plasticity and memory impairments in the CK-p25 mouse model of Alzheimer’s disease (Graff et al. 2012). Alzheimer’s disease is the most prevalent neurodegenerative disease associated with cognitive impairment. These findings underline the important roles of HDAC2-regulated chromatin modification in regulating synaptic plasticity and memory formation contributing to cognitive impairment, which is relevant for a wide range of neurodegenerative disorders.
HDAC3
HDAC3 is the third HDAC identified in mammals by sequence homology with HDAC1 and HDAC2 (Gregoretti et al. 2004; Yang and Seto 2008). Unlike the other Class I HDACs, which are normally nuclear proteins, HDAC3 localizes to both the nucleus and cytoplasm (Takami and Nakayama 2000). Germ-line deletion of Hdac3 is lethal, indicating a requirement for proper embryonic development (Bhaskara et al. 2008; Montgomery et al. 2008). HDAC3 is found in many tissues throughout the body, especially the brain (Mahlknecht et al. 1999). It is the most highly expressed class I HDAC in the brain with greatest expression in the hippocampus, cortex, and cerebellum, but also shows moderate levels of expression in other important brain regions, including the striatum, amygdala and hypothalamus (Broide et al. 2007a; Thomas 2009). HDAC3 is predominantly expressed in neurons, but studies have also shown expression in glial cells, including astrocytes and oligodendrocytes (Broide et al. 2007a; Debacker et al. 2012; Shen et al. 2005). A large body of evidence from cell culture and in vivo models indicates that HDAC3 promotes neurodegeneration, as described below. Further, it has been implicated as a major target for the neuroprotective effects resulting from pharmacological HDAC inhibitors. The toxic effects of HDAC3 are associated with several different mechanisms, including histone acetylation, nuclear translocation, post-translational modifications and direct interactions with disease proteins (Table 1).
Neurotoxic effects of HDAC3
The first reports of neurotoxic effects of HDAC3 were from the D’Mello lab, who showed that overexpression of HDAC3 induced cell death in cortical and cerebellar granule neurons (Bardai and D’Mello 2011). Toxicity was also seen in the HT22 neuroblastoma cell line, but not in primary kidney fibroblasts or HEK293 cells, indicating neuronal specificity for the toxic effects of HDAC3 (Bardai and D’Mello 2011). Confirming its requirement for neuronal death, shRNA-mediated suppression of HDAC3 expression protected against oxidative stress and potassium deprivation-induced cell death (Bardai and D’Mello 2011). In this study, HDAC3-induced neurotoxicity requires phosphorylation by Glycogen synthase kinase-3 beta (GSK3β), a kinase inhibited by Protein kinase B/Akt, which is widely implicated in the promotion of neurodegeneration (Bardai and D’Mello 2011; Bardai et al. 2013a). Interestingly, HDAC1 participation was also required for neurotoxicity by HDAC3, as knockdown of HDAC1 was shown to reduce the neurotoxic effects of HDAC3 (Bardai et al. 2012b). Both HDAC1 and HDAC3 are believed to be transcriptional repressors. One target of HDAC3-mediated repression could be the cell cycle inhibitory protein, Cdkn1a (p21Cip1/Waf1)(Knutson et al. 2008; Trivedi et al. 2008; Wilson et al. 2006), which is known to have has strong neuroprotective activity (Harms et al. 2007; Langley et al. 2008; Louis Sam Titus et al. 2017; Mallick and D’Mello 2014). Consistent with the above studies, HDAC3 does inhibit p21Cip1/Waf1 in non-neuronal systems (Knutson et al. 2008; Trivedi et al. 2008; Wilson et al. 2006).
HDAC3 is also important in the context of several different neurodegenerative diseases. In Huntington’s disease, knockdown of HDAC3 in mutant huntingtin-overexpressing cortical neurons protects them against cell death (Bardai et al. 2013b). In R6/2 transgenic mice, HDAC3 has been shown to interact with HDAC1 in the striatum and the cortex (Bardai et al. 2012c), two brain regions that suffer neuronal loss in Huntington’s disease. This interaction was found to coincide with the onset of motor deficits (Bardai et al. 2013b). Consistent with these results are those showing that administration of chemical inhibitors selective for HDAC1/HDAC3, or HDAC3 alone, reduced neuropathology and improved behavioral performance in R6/2 and N171-82Q transgenic mouse models of Huntington’s disease (Jia et al. 2012; Jia et al. 2015; Jia et al. 2016; Suelves et al. 2017; Thomas et al. 2008c; Thomas 2014). Alterations in histone acetylation and ensuing gene expression were implicated in these effects (Jia et al. 2012; Jia et al. 2015; Jia et al. 2016; Thomas et al. 2008c; Thomas 2014). Protective effects of an HDAC3-selective inhibitor, RGFP966, in yet another Huntington’s disease mouse model, HdhQ111 knock-in mice, was also demonstrated by a different laboratory (Suelves et al. 2017). Genetic reduction of HDAC3 using Hdac3 (+/−) heterozygous mice has been reported, and was not found to ameliorate disease phenotypes when crossed with R6/2 transgenic mice (Moumne et al. 2012). However, the overall protein levels of HDAC3 were only reduced 20%, hence it is likely that the reduction of HDAC3 in the Hdac3+/− mice was not sufficient to affect the HD phenotype (Moumne et al. 2012).
HDAC3 has been shown to interact with wild-type huntingtin protein, but not with the mutant form (Bardai et al. 2013b). Although not interacting directly with HDAC3, mutant huntingtin promotes the disassociation of HDAC3 from the normal version of the protein permitting it to interact with HDAC1, leading to neurodegeneration (Bardai et al. 2013b). The significance of the interaction between HDAC3 and normal huntingtin remains to be investigated but is likely to be necessary for its contributions to proper brain development and functioning of the nervous system. Indeed, genetic ablation of either the HTT gene or HDAC3 has severe effects on brain development and function (Norwood et al. 2014; Saudou and Humbert 2016).
In other neurodegenerative diseases, pharmacological inhibition of HDAC3, also with RGFP966, protected CA1 hippocampal neurons against oligomeric beta-amyloid-induced impairment of synaptic plasticity (Krishna et al. 2016), having implications to Alzheimer’s disease. HDAC3 has also been linked to Parkinson’s disease, a progressive neurodegenerative disease characterized by the loss of dopaminergic neurons in the substantia nigra (Olanow and Tatton 1999). Leucine-rich repeat kinase-2 (LRRK2), mutations of which are the most common genetic cause of both familial and sporadic Parkinson’s disease (Mata et al. 2006), binds to HDAC3 and phosphorylates it at Ser424, which resulted in a stimulation of HDAC3 activity (Han et al. 2017). Additionally, LRRK2-mediated toxicity following 6-hydroxydopamine treatment increased HDAC3 phosphorylation and its nuclear localization (Han et al. 2017).
Death of injury-induced retinal ganglion neurons in mice was also associated with the translocation of HDAC3 to the nucleus (Schmitt et al. 2014). Consistent with a role for nuclear HDAC3 has been implicated in neuronal loss in rats subjected to ischemic stroke and in cultured cortical neurons subjected to oxygen-glucose deprivation (OGD) (Yang et al. 2016). Administration of the HDAC3-selective inhibitor, RGFP966, depleted HDAC3 from the nucleus and protected against neuronal loss both in vivo and in cultured neurons (Yang et al. 2016). A separate study described a substantial increase in HDAC3 expression soon after the induction of ischemic stroke in mice (Chen et al. 2012). In this study, it was found that knockdown of HDAC3 using shRNA methods protected cortical neurons from OGD-induced death (Chen et al. 2012). Thus, reduction of HDAC3 activity either pharmacologically or using shRNA-mediated knockdown was shown to be protective against ischemic stroke 102,103.
Recent studies provide evidence that HDAC3 might contribute to the pathogenesis of other inherited polyglutamine diseases, such as the spinocerebellar ataxias (SCAs) (Underwood and Rubinsztein 2008). Like Huntington’s disease, six of the SCAs (1, 2, 3, 7, 6 and 17) are caused by an expanded CAG repeat mutation in the coding regions of the relevant disease genes (Underwood and Rubinsztein 2008). HDAC3 has been shown to interact with disease proteins for three of these, SCA1, 3 and 7, which are caused by mutations in the ATXN1, ATXN3 and ATXN7 genes, respectively (Underwood and Rubinsztein 2008). These autosomal-dominantly inherited disorders are characterized by aggregation of the mutant polyQ-expanded protein, nuclear inclusions and repeat-dependent neurotoxicity (Paulson et al. 1997; Underwood and Rubinsztein 2008).
Ataxin-1, the disease protein for SCA1, has been found to interact selectively with HDAC3 in transfected cells (Venkatraman et al. 2014). Interestingly and in contrast to mutant Htt, the HDAC3-ataxin 1 interaction was not polyglutamine-dependent (Venkatraman et al. 2014), which could suggest that inhibition of HDAC3 in this context may interfere with normal ataxin-1 regulatory properties. Indeed, ablating the HDAC3 gene selectively in Purkinje cells of SCA1 (154Q/2Q), knock-in mice resulted not in beneficial effects, but in early onset ataxia, and progressive degeneration (Venkatraman et al. 2014).
HDAC3 has also been shown to interact with ataxin-3, which is mutated in SCA3, also known as Machado-Joseph disease (Kawaguchi et al. 1994; Riess et al. 2008). While interacting with both normal and polyQ-expanded ataxin-3 in cultured cells and in the human pons, normal ataxin-3-containing protein complexes showed increased HDAC activity, whereas polyQ-expanded ataxin-3-containing complexes had reduced HDAC activity in specified chromatin regions (Evert et al. 2006). Although the broadly-acting HDAC inhibitor, sodium butyrate, was found to reverse transcriptional downregulation and ameliorate ataxic symptoms in a transgenic mouse model of SCA3 (Chou et al. 2011), the consequences of selective HDAC3 inhibition, however, are less clear; it is possible that disruption of the HDAC3 repressor complex by polyQ-expanded ataxin-3 may liberate HDAC3 to elicit toxic effects in neurons, as observed in Huntington’s disease models (Bardai et al. 2013a). Finally, HDAC3 was found to interact and stabilize both normal and polyQ-expanded ataxin-7(Chou et al. 2011). HDAC3 expression and interaction with ataxin-7 was elevated in neurons and glia of the cerebellum in SCA7 transgenic mice in a polyQ-dependent manner (Duncan et al. 2013).
HDAC4
HDAC4 is expressed widely in the brain with highest expression in the cerebellum, hippocampus and olfactory bulb (Broide et al. 2007b; Thomas 2009). It is a protein essential for development, as evident in Hdac4 knock-out mice, which die in early postnatal life (Vega et al. 2004). HDAC4 is normally expressed in the cytoplasm, but is also known to exhibit signal-dependent shuttling between the cytoplasm and nucleus, which is regulated in part by calcium/calmodulin-dependent kinase-mediated phosphorylation (McKinsey et al. 2000). Confusingly, the cellular localization of HDAC4 differentially contributes to its protective versus toxic effects; with some studies showing, that nuclear HDAC4 is protective in one disease model, and toxic in another. Interestingly, recent studies demonstrated that, in fact, HDAC4 has no deacetylase activity in the brain (Mielcarek et al. 2013b). This finding suggests that HDAC4 exerts its effects via interactions with other HDAC proteins or other regulatory proteins. HDAC4 was first shown to be a neuroprotective protein; however, further studies have demonstrated neurotoxic properties as well, as discussed below.
Neuroprotective effects of HDAC4
Neuroprotective effects of HDAC4 were reported over a decade ago (Majdzadeh et al. 2008b). In early studies, overexpression of HDAC4 protected both cultured cortical neurons and cerebellar granule neurons against low potassium-induced apoptosis (Majdzadeh et al. 2008b). This HDAC4-mediated neuroprotection did not require its HDAC catalytic domain and was not inhibited by chemical inhibitors of HDACs. Also in that study, the authors showed that the cerebellum of HDAC4 knockout mice displayed extensive degeneration of Purkinje neurons (Majdzadeh et al. 2008b). Neuroprotective roles for HDAC4 have also been demonstrated in other studies that described that HDAC4 promoted the survival of retinal neurons during development and protected photoreceptors in a mouse model of retinal degeneration (Chen and Cepko 2009). The protective action of HDAC4 was suggested by these authors to be dependent on the activity of hypoxia-inducible factor 1 alpha (Chen and Cepko 2009). A more recent study described that HDAC4 protects neurons from death induced by a blockade of neuronal activity or by reduction of NMDA activity (Chen et al. 2014). Nuclear translocation of HDAC4 is observed after ischemic stroke and this has been suggested to have a protective effect in promoting neuronal recovery and remodeling (Kassis et al. 2015; Kassis et al. 2016).
In other studies, HDAC4 was found to be necessary for the ability of DNJB6 proteins, a subclass of heat shock proteins, to reduce polyglutamine aggregation and toxicity (Hageman et al. 2010). This finding clearly has relevance for the entire class of polyglutamine disorders. Again, HDAC4 did not regulate the acetylation status of the DNAJB6 proteins suggesting that this cooperative neuroprotection was HDAC-independent or via an interaction with another HDAC subtype (Hageman et al. 2010).
Neurotoxic effects of HDAC4
In contrast to the findings mentioned above, several other studies have reported that HDAC4 participates in promoting neurodegeneration. Although the nuclear translocation of HDAC4 was shown to be protective in ischemic stroke (Kassis et al. 2015; Kassis et al. 2016), nuclear accumulation of HDAC4 was found to be associated with neuronal cell death in a mouse model of Ataxia Telangectasia (Herrup et al. 2013; Li et al. 2012). This effect-required de-phosphorylation of HDAC4 by the protein phosphatase 2A, whose activity is enhanced in this disease (Li et al. 2012). In another study, genetic reduction of HDAC4 resulted in elevated expression of brain-derived neurotrophic factor (BDNF), which led to reduced disease symptoms and increase life span in the R6/2 transgenic mouse model of Huntington’s disease (Mielcarek et al. 2013a). Again, in contrast to that finding, cytoplasmic HDAC4 was shown to be particularly neurotoxic, whereby HDAC4 was found to associate with huntingtin protein in the cytoplasm, and co-localized with cytoplasmic inclusions (Mielcarek et al. 2013a). Huntington’s disease-related global transcriptional alteration and nuclear aggregation of huntingtin were not affected by HDAC4 reduction (Mielcarek et al. 2013a). In another study, a reduction in the level of HDAC4 in the cortex of R6/2 transgenic mice was observed after administration of SAHA, a broadly-acting HDAC inhibitor that has protective effects in disease mice (Mielcarek et al. 2011).
Translocation of HDAC4 to the nucleus was also associated with detrimental effects in cultured cerebellar granule neurons in response to low-potassium or excitotoxic glutamate conditions (Bolger and Yao 2005). Also in that study, treatment with the neuronal survival factor BDNF suppresses HDAC4 nuclear translocation, whereas a proapoptotic Calcium/calmodulin-dependent protein kinase inhibitor stimulated HDAC4 nuclear accumulation (Bolger and Yao 2005).
HDAC5
HDAC5 is abundantly expressed throughout the brain and peripheral tissues (Broide et al. 2007a). HDAC5 is known to undergo nuclear-cytoplasmic shuttling and to be a critical transcriptional regulator (McKinsey et al. 2000). Although compelling evidence for the involvement HDAC5 in regulating neurodegeneration is lacking, there are studies implicating HDAC5 in neuroprotective effects. For example, HDAC5 has been shown to play a pivotal role in stimulating axonal regrowth after injury (Cho and Cavalli 2012; Cho et al. 2013; Whalley 2014). This required the export of HDAC5 from the nucleus and interaction with filamin A in the axons (Cho et al. 2013; Cho et al. 2015). Other studies showed that injury to sensory axons resulted in HDAC5 phosphorylation by protein kinase C, which promoted axon regeneration (Cho and Cavalli 2012; Cho et al. 2013).
HDAC5 was also investigated in the role of memory function and Alzheimer’s disease pathogenesis in a mouse model (Agis-Balboa et al. 2013). Loss of HDAC5 was found to impair memory function, but did not affect pathogenesis, in a mouse model for amyloid pathology (Agis-Balboa et al. 2013). These findings suggest a novel role for HDAC5 in memory consolidation and suggests that selective HDAC inhibitors for the treatment of Alzheimer’s disease should avoid targeting HDAC5.
HDAC6
HDAC6 has two features that distinguishes it from all the other HDACs: it is exclusively cytoplasmic and it has two catalytic domains (Gregoretti et al. 2004). Initially described to be a specific alpha-tubulin deacetylase, HDAC6 was later found to deacetylate other substrates, including tau, heat shock protein 90, the actin binding protein cortactin, the tumor suppressor macrophage stimulating 1 and the beta-catenin transcription factor (Cook et al. 2014; Kekatpure et al. 2009; Li et al. 2016; Li et al. 2008; Zhang et al. 2007). Both the neuroprotective and neurotoxic effects of HDAC6 have been associated with altered acetylation of target proteins, not a result of histone acetylation and ensuing effects on chromatin (Table 1).
Neuroprotective effects of HDAC6
Earlier studies examining the role of HDAC6 in the regulation of degeneration concluded that it played a protective role, acting by enhancing the clearance of potentially harmful misfolded proteins and protein aggregates(Guthrie and Kraemer 2011; Pandey et al. 2007). This action of HDAC6 was accomplished through a variety of different mechanisms. Through a C-terminal zinc finger domain (ZnF-UBP) HDAC6 binds mono- and polyubiquinated misfolded proteins and, through association with dynein motor proteins, transports these potentially toxic proteins via microtubules to form aggresomes sequestering them (Kawaguchi et al. 2003; Pandey et al. 2007). One such potentially toxic protein that HDAC6 has been found to transport to aggresomes is misfolded and polyubiquitinated DJ-1 (mutations of DJ-1 cause a genetic form of Parkinson’s disease) (Olzmann et al. 2007). The eventual clearance of autophagosomes through lysosomal fusion also was shown to involve HDAC6 (Lee et al. 2010a). In addition to ubiquitinated proteins, HDAC6 can promote the clearance of ubiquitinated mitochondria through mitophagy (Lee et al. 2010b). Other studies have shown that HDAC6 can promote clearance of misfolded proteins by stimulating the synthesis of heat shock proteins through the activation of heat shock transcription factor 1 (HSF1) (Boyault et al. 2007). Through activation of HSF1 and the expression of heat-shock proteins, HDAC6 was found to prevent alpha-synuclein aggregates in cultured cells (Du et al. 2014). Inhibition of HDAC6 enhanced alpha-synuclein aggregation and toxicity (Du et al. 2014). Not surprisingly, HDAC6 inhibition in mice was reported to upregulate α-synuclein oligomers levels and exacerbate nigrostriatal dopamine neurodegeneration (Du et al. 2014). In addition to stimulating chaperone activity through HSF1 activation, HDAC6 can stimulate the chaperone activity of HSP90 by directly deacetylating it (Bali et al. 2005; Kovacs et al. 2005).
Neurotoxic roles for HDAC6
Several other studies suggest that HDAC6 can promote neurodegenerative disease pathology (Bali et al. 2005; Du et al. 2014; Kovacs et al. 2005). Treatment of cultured cortical neurons with HDAC6 selective inhibitors was found to protect them against oxidative stress-induced death and to promote neurite extension (Rivieccio et al. 2009). HDAC6-mediated deacetylation of tubulin was found to negatively affect the recruitment of kinesin and dynein motor complexes leading to the impairment of axonal transport (Dompierre et al. 2007). Conversely, loss of HDAC6 activity was found to enhance microtubule stability, axonal transport and microtubule-mediated mitochondrial transport, effects that all promote neuronal survival (Chen et al. 2010; Dompierre et al. 2007; Kim et al. 2012). In addition to tubulin, HDAC6 can deacetylate the tau protein. This action results in reduction of tau clearance which promotes aggregation and toxicity (Cook et al. 2014). Reduction of HDAC6 by genetic ablation or pharmacological inhibition was found to prevent cognitive impairment in Alzheimer’s disease mice (Govindarajan et al. 2013). Because amyloid plaque burden was not affected, the beneficial actions were thought to be due to effects on tau.
Detrimental effects of HDAC6 have also been implicated in other neurodegenerative diseases. Pharmacological inhibition of HDAC6 improved mitochondrial trafficking, axonal transport and BDNF release in mutant huntingtin overexpressing neurons (Dompierre et al. 2007). Although not delaying disease onset, HDAC6 deletion extended the survival of SOD1(G93A) mice and maintained motor axon integrity indicating a role for HDAC6 in Amyolateral Sclerosis pathogenesis (Taes et al. 2013).
While most previous studies have utilized tubacin to inhibit HDAC6, other inhibitors have been recently developed with better pharmacological properties (Wang et al. 2017). These HDAC6-selective inhibitors have been shown to ameliorate disease phenotypes in mouse models of Alzheimer’s disease (Zhang et al. 2014), Charcot-Marie-Tooth disease (Benoy et al. 2017) and in cell culture models of Huntington’s disease (Guedes-Dias et al. 2015) and tauopathy (Cook et al. 2014), confirming earlier findings. Based on their well demonstrated beneficial effects, HDAC6 inhibitors may represent a promising therapeutic approach against different neurodegenerative diseases. A complicating issue though that warrants further investigation, however, is that genetic knockdown of HDAC6 has been described to have different effects in Huntington’s disease mouse models. In one study using the R6/2 mouse model, HDAC6 knockdown did not modify disease progression (Bobrowska et al. 2011). However, another study using the related R6/1 described no effect on motor deficits but an exacerbation of some disease phenotypes was observed with HDAC6 deletion (Ragot et al. 2015). Surprisingly, given the worsening of symptoms, tubulin acetylation as well as BDNF expression was found to be increased in this study (Ragot et al. 2015).
HDACs 7, 8, 9,10 and 11
In contrast to class I and IIa HDACs, HDACs 7, 8, 9 and 10 are expressed at relatively low levels in the brain (Broide et al. 2007b; Thomas 2009). There is limited information on the role of these family members in relation to neurodegenerative conditions. For example, there have been no studies to date linking HDAC8 or HDAC10 to neurodegenerative diseases. However, a few studies have demonstrated neuroprotective effects of HDACs 7, 9 and 11, which are mentioned below.
In cultured neurons, overexpression of HDAC7 can protect against death by inhibiting the expression of c-Jun (Ma and D’Mello 2011b). Interestingly, HDAC inhibitors failed to reduce neuroprotection and a catalytically-dead form of HDAC7 was fully protective indicating that neuroprotection by HDAC7 is independent of deacetylase activity (Ma and D’Mello 2011b). In other studies, researchers described that R6/2 Huntington’s disease mice hemizygous for Hdac7 deletion do not show any amelioration of disease phenotype (Benn et al. 2009). While this suggests that HDAC7 is not involved in Huntington’s disease pathogenesis, a compensatory effect of other HDACs resulting from HDAC7 reduction cannot be ruled out. Also possible is that a more complete reduction of HDAC7 is necessary for a beneficial effect in this aggressive mouse model of disease.
While the effects of HDAC9 on neurodegeneration are unclear, HDRP, a truncated form of HDAC9 resulting from alternative splicing, protects cultured neurons from death (Morrison et al. 2006b). As with HDAC7, protection was found to be due to the inhibition of c-Jun gene transcription through deacetylation of the c-Jun promoter (Morrison et al. 2006b). Although completely lacking a catalytic domain, the inhibitory effect on the c-Jun promoter is mediated through the recruitment of HDAC1. Consistently, neuroprotection by HDRP is reduced by treatment with HDAC inhibitors (Morrison et al. 2006b). It has been suggested that whether HDAC1 protects or promotes neuronal death is dependent on whether it interacts with HDRP or HDAC3 (Bardai et al. 2012a). Besides c-Jun, HDRP can protect neurons through interaction with the amino enhancer of split, a member of the Groucho/GRG family of proteins (Zhang et al. 2008).
Based on proteins that interact with it, HDAC11 has been suggested to be involved in the regulation of survival of motor neuron complex-dependent splicing (Joshi et al. 2013). In T-cells, downregulation of HDAC11 results in mis-splicing of ataxin-10(Joshi et al. 2013), the disease protein for SCA10, a neurodegenerative disorder characterized by cerebellar dysfunctions and seizures, but not a polyglutamine disease like the other SCA diseases.
Conclusions
Our review covers the roles of classical HDACs (classes I, II and IV) in the regulation of neuronal death and survival, and their known associations with neurodegenerative pathologies. In some cases, HDACs have been described to both promote and protect against neurodegeneration. This could be due to several reasons, including differences in contexts or models used, as well as different mechanisms of actions. These depend on the cellular localization and post-translational modifications of the individual HDAC, the acetylation of histone and non-histone proteins or their diverse binding partners. Also becoming clear is that at least some HDACs can be expressed as different isoforms. For example, HDAC5, HDAC7 and HDAC9 can be each produced as two separate isoforms (Di Giorgio and Brancolini 2016; Yang and Seto 2008), introducing another variability that has not yet been adequately explored. While there is substantial evidence supporting the use of HDAC inhibitors as a potential therapeutic option for many of the discussed neurodegenerative diseases, one must strongly consider the specificity of these agents for the different HDAC family members.
Acknowledgments
This work was supported by NIH grant NS040408 to Santosh D’Mello.
Abbrebiations
- CTBP
C-terminal-binding protein
- GSK3β
Glycogen synthase kinase-3 beta
- SOD1
superoxide dismutase-1
- HDAC
histone deactylase
- HP1
heterochromatin protein 1
- HSF1
heat shock factor-1
- LRRK2
Leucine-rich repeat kinase-2
- MKP-1
Mitogen-activated protein kinase phosphatase
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NRSF
Neural-restrictive silencer factor
- N-Cor/SMRT
The nuclear receptor co-repressor 2/ silencing mediator for retinoid and thyroid hormone receptors complex
- NF-κB
Nuclear factor-kappa B, PolyQ, polyglutamine
- SCA
spinocerebellar ataxia, UBE3A, ubiquitin protein ligase E3A (UBE3A)
- XIAP
X-linked inhibitor of apoptosis
- ZnF-UBP
N-Cor/SMRT, The nuclear receptor co-repressor 2/ silencing mediator for retinoid and thyroid hormone receptors complex C-terminal zinc finger domain
Footnotes
conflict of interest disclosure. The authors have no conflict of interest.
References
- Agis-Balboa RC, Pavelka Z, Kerimoglu C, Fischer A. Loss of HDAC5 impairs memory function: implications for Alzheimer’s disease. J Alzheimers Dis. 2013;33:35–44. doi: 10.3233/JAD-2012-121009. [DOI] [PubMed] [Google Scholar]
- Ajami M, Pazoki-Toroudi H, Amani H, Nabavi SF, Braidy N, Vacca RA, Atanasov AG, Mocan A, Nabavi SM. Therapeutic role of sirtuins in neurodegenerative disease and their modulation by polyphenols. Neurosci Biobehav Rev. 2017;73:39–47. doi: 10.1016/j.neubiorev.2016.11.022. [DOI] [PubMed] [Google Scholar]
- Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- Bardai FH, D’Mello SR. Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3beta. J Neurosci. 2011;31:1746–1751. doi: 10.1523/JNEUROSCI.5704-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Price V, Zaayman M, Wang L, D’Mello SR. Histone Deacetylase-1 (HDAC1) Is a Molecular Switch between Neuronal Survival and Death. J Biol Chem. 2012a;287:35444–35453. doi: 10.1074/jbc.M112.394544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Price V, Zaayman M, Wang L, D’Mello SR. Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J Biol Chem. 2012b;287:35444–35453. doi: 10.1074/jbc.M112.394544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Price V, Zaayman M, Wang L, D’Mello SR. Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J Biol Chem. 2012c;287:35444–35453. doi: 10.1074/jbc.M112.394544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Verma P, Smith C, Rawat V, Wang L, D’Mello SR. Disassociation of histone deacetylase-3 from normal huntingtin underlies mutant huntingtin neurotoxicity. J Neurosci. 2013a;33:11833–11838. doi: 10.1523/JNEUROSCI.5831-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Verma P, Smith C, Rawat V, Wang L, D’Mello SR. Disassociation of histone deacetylase-3 from normal huntingtin underlies mutant huntingtin neurotoxicity. J Neurosci. 2013b;33:11833–11838. doi: 10.1523/JNEUROSCI.5831-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn CL, Butler R, Mariner L, Nixon J, Moffitt H, Mielcarek M, Woodman B, Bates GP. Genetic knock-down of HDAC7 does not ameliorate disease pathogenesis in the R6/2 mouse model of Huntington’s disease. PLoS One. 2009;4:e5747. doi: 10.1371/journal.pone.0005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoy V, Vanden Berghe P, Jarpe M, Van Damme P, Robberecht W, Van Den Bosch L. Development of Improved HDAC6 Inhibitors as Pharmacological Therapy for Axonal Charcot-Marie-Tooth Disease. Neurotherapeutics. 2017;14:417–428. doi: 10.1007/s13311-016-0501-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertos NR, Wang AH, Yang XJ. Class II histone deacetylases: structure, function, and regulation. Biochem Cell Biol. 2001;79:243–252. [PubMed] [Google Scholar]
- Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, Hiebert SW. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008;30:61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrowska A, Paganetti P, Matthias P, Bates GP. Hdac6 knock-out increases tubulin acetylation but does not modify disease progression in the R6/2 mouse model of Huntington’s disease. PLoS One. 2011;6:e20696. doi: 10.1371/journal.pone.0020696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger TA, Yao TP. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J Neurosci. 2005;25:9544–9553. doi: 10.1523/JNEUROSCI.1826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, Garrido C, Yao TP, Vourc’h C, Matthias P, Khochbin S. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–2181. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ. Distribution of histone deacetylases 1–11 in the rat brain. J Mol Neurosci. 2007a;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ. Distribution of histone deacetylases 1–11 in the rat brain. J Mol Neurosci. 2007b;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- Brunmeir R, Lagger S, Seiser C. Histone deacetylase HDAC1/HDAC2-controlled embryonic development and cell differentiation. Int J Dev Biol. 2009;53:275–289. doi: 10.1387/ijdb.082649rb. [DOI] [PubMed] [Google Scholar]
- Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323:256–259. doi: 10.1126/science.1166226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Owens GC, Makarenkova H, Edelman DB. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One. 2010;5:e10848. doi: 10.1371/journal.pone.0010848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang Y, Modrusan Z, Sheng M, Kaminker JS. Regulation of neuronal gene expression and survival by basal NMDA receptor activity: a role for histone deacetylase 4. J Neurosci. 2014;34:15327–15339. doi: 10.1523/JNEUROSCI.0569-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YT, Zang XF, Pan J, Zhu XL, Chen F, Chen ZB, Xu Y. Expression patterns of histone deacetylases in experimental stroke and potential targets for neuroprotection. Clin Exp Pharmacol Physiol. 2012;39:751–758. doi: 10.1111/j.1440-1681.2012.05729.x. [DOI] [PubMed] [Google Scholar]
- Cho Y, Cavalli V. HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 2012;31:3063–3078. doi: 10.1038/emboj.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Park D, Cavalli V. Filamin A is required in injured axons for HDAC5 activity and axon regeneration. J Biol Chem. 2015;290:22759–22770. doi: 10.1074/jbc.M115.638445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Sloutsky R, Naegle KM, Cavalli V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell. 2013;155:894–908. doi: 10.1016/j.cell.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choong CJ, Sasaki T, Hayakawa H, Yasuda T, Baba K, Hirata Y, Uesato S, Mochizuki H. A novel histone deacetylase 1 and 2 isoform-specific inhibitor alleviates experimental Parkinson’s disease. Neurobiol Aging. 2016;37:103–116. doi: 10.1016/j.neurobiolaging.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Chou AH, Chen SY, Yeh TH, Weng YH, Wang HL. HDAC inhibitor sodium butyrate reverses transcriptional downregulation and ameliorates ataxic symptoms in a transgenic mouse model of SCA3. Neurobiol Dis. 2011;41:481–488. doi: 10.1016/j.nbd.2010.10.019. [DOI] [PubMed] [Google Scholar]
- Contreras PS, Gonzalez-Zuniga M, Gonzalez-Hodar L, Yanez MJ, Dulcey A, Marugan J, Seto E, Alvarez AR, Zanlungo S. Neuronal gene repression in Niemann-Pick type C models is mediated by the c-Abl/HDAC2 signaling pathway. Biochim Biophys Acta. 2016;1859:269–279. doi: 10.1016/j.bbagrm.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Stankowski JN, Carlomagno Y, Stetler C, Petrucelli L. Acetylation: a new key to unlock tau’s role in neurodegeneration. Alzheimers Res Ther. 2014;6:29. doi: 10.1186/alzrt259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debacker K, Frizzell A, Gleeson O, Kirkham-McCarthy L, Mertz T, Lahue RS. Histone deacetylase complexes promote trinucleotide repeat expansions. PLoS Biol. 2012;10:e1001257. doi: 10.1371/journal.pbio.1001257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio E, Brancolini C. Regulation of class IIa HDAC activities: it is not only matter of subcellular localization. Epigenomics. 2016;8:251–269. doi: 10.2217/epi.15.106. [DOI] [PubMed] [Google Scholar]
- D’Mello SR. Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 2009;22:513–524. doi: 10.1358/dnp.2009.9.1428871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbin MM, Madabhushi R, Pan L, Chen Y, Kim D, Gao J, Ahanonu B, Pao PC, Qiu Y, Zhao Y, Tsai LH. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci. 2013;16:1008–1015. doi: 10.1038/nn.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez G. The neurobiology of sirtuins and their role in neurodegeneration. Trends Pharmacol Sci. 2012;33:494–501. doi: 10.1016/j.tips.2012.05.007. [DOI] [PubMed] [Google Scholar]
- Donmez G, Outeiro TF. SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol Med. 2013;5:344–352. doi: 10.1002/emmm.201302451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Wang F, Zou J, Le W, Dong Q, Wang Z, Shen F, Yu L, Li Y. Histone deacetylase 6 regulates cytotoxic alpha-synuclein accumulation through induction of the heat shock response. Neurobiol Aging. 2014;35:2316–2328. doi: 10.1016/j.neurobiolaging.2014.04.029. [DOI] [PubMed] [Google Scholar]
- Duncan CE, An MC, Papanikolaou T, Rugani C, Vitelli C, Ellerby LM. Histone deacetylase-3 interacts with ataxin-7 and is altered in a spinocerebellar ataxia type 7 mouse model. Mol Neurodegener. 2013;8 doi: 10.1186/1750-1326-8-42. 42-1326-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evert BO, Araujo J, Vieira-Saecker AM, de Vos RA, Harendza S, Klockgether T, Wullner U. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J Neurosci. 2006;26:11474–11486. doi: 10.1523/JNEUROSCI.2053-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Alsarraf O, Dahrouj M, Platt KA, Chou CJ, Rice DS, Crosson CE. Inhibition of HDAC2 protects the retina from ischemic injury. Invest Ophthalmol Vis Sci. 2013;54:4072–4080. doi: 10.1167/iovs.12-11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem. 2002;277:25748–25755. doi: 10.1074/jbc.M111871200. [DOI] [PubMed] [Google Scholar]
- Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem. 2005;280:556–563. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- Govindarajan N, Rao P, Burkhardt S, Sananbenesi F, Schluter OM, Bradke F, Lu J, Fischer A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol Med. 2013;5:52–63. doi: 10.1002/emmm.201201923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, Su SC, Samiei A, Joseph N, Haggarty SJ, Delalle I, Tsai LH. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483:222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci U S A. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedes-Dias P, de Proenca J, Soares TR, Leitao-Rocha A, Pinho BR, Duchen MR, Oliveira JM. HDAC6 inhibition induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. Biochim Biophys Acta. 2015;1852:2484–2493. doi: 10.1016/j.bbadis.2015.08.012. [DOI] [PubMed] [Google Scholar]
- Guthrie CR, Kraemer BC. Proteasome inhibition drives HDAC6-dependent recruitment of tau to aggresomes. J Mol Neurosci. 2011;45:32–41. doi: 10.1007/s12031-011-9502-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman J, Rujano MA, van Waarde MA, Kakkar V, Dirks RP, Govorukhina N, Oosterveld-Hut HM, Lubsen NH, Kampinga HH. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol Cell. 2010;37:355–369. doi: 10.1016/j.molcel.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Han KA, Shin WH, Jung S, Seol W, Seo H, Ko C, Chung KC. Leucine-rich repeat kinase 2 exacerbates neuronal cytotoxicity through phosphorylation of histone deacetylase 3 and histone deacetylation. Hum Mol Genet. 2017;26:1–18. doi: 10.1093/hmg/ddw363. [DOI] [PubMed] [Google Scholar]
- Harms C, Albrecht K, Harms U, Seidel K, Hauck L, Baldinger T, Hubner D, Kronenberg G, An J, Ruscher K, Meisel A, Dirnagl U, von Harsdorf R, Endres M, Hortnagl H. Phosphatidylinositol 3-Akt-kinase-dependent phosphorylation of p21(Waf1/Cip1) as a novel mechanism of neuroprotection by glucocorticoids. J Neurosci. 2007;27:4562–4571. doi: 10.1523/JNEUROSCI.5110-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, Li J, Chen J. The role of ATM and DNA damage in neurons: upstream and downstream connections. DNA Repair (Amst) 2013;12:600–604. doi: 10.1016/j.dnarep.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob C, Christen CN, Pereira JA, Somandin C, Baggiolini A, Lotscher P, Ozcelik M, Tricaud N, Meijer D, Yamaguchi T, Matthias P, Suter U. HDAC1 and HDAC2 control the transcriptional program of myelination and the survival of Schwann cells. Nat Neurosci. 2011;14:429–436. doi: 10.1038/nn.2762. [DOI] [PubMed] [Google Scholar]
- Jamal I, Kumar V, Vatsa N, Shekhar S, Singh BK, Sharma A, Jana NR. Rescue of altered HDAC activity recovers behavioural abnormalities in a mouse model of Angelman syndrome. Neurobiol Dis. 2017;105:99–108. doi: 10.1016/j.nbd.2017.05.010. [DOI] [PubMed] [Google Scholar]
- Jeong H, Then F, Melia TJ, Jr, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong Y, Du R, Zhu X, Yin S, Wang J, Cui H, Cao W, Lowenstein CJ. Histone deacetylase isoforms regulate innate immune responses by deacetylating mitogen-activated protein kinase phosphatase-1. J Leukoc Biol. 2014;95:651–659. doi: 10.1189/jlb.1013565. [DOI] [PubMed] [Google Scholar]
- Jesko H, Wencel P, Strosznajder RP, Strosznajder JB. Sirtuins and Their Roles in Brain Aging and Neurodegenerative Disorders. Neurochem Res. 2017;42:876–890. doi: 10.1007/s11064-016-2110-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Morris CD, Williams RM, Loring JF, Thomas EA. HDAC inhibition imparts beneficial transgenerational effects in Huntington’s disease mice via altered DNA and histone methylation. Proc Natl Acad Sci U S A. 2015;112:E56–64. doi: 10.1073/pnas.1415195112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Pallos J, Jacques V, Lau A, Tang B, Cooper A, Syed A, Purcell J, Chen Y, Sharma S, Sangrey GR, Darnell SB, Plasterer H, Sadri-Vakili G, Gottesfeld JM, Thompson LM, Rusche JR, Marsh JL, Thomas EA. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol Dis. 2012;46:351–361. doi: 10.1016/j.nbd.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Wang Y, Morris CD, Jacques V, Gottesfeld JM, Rusche JR, Thomas EA. The Effects of Pharmacological Inhibition of Histone Deacetylase 3 (HDAC3) in Huntington’s Disease Mice. PLoS One. 2016;11:e0152498. doi: 10.1371/journal.pone.0152498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Altamura S, De Francesco R, Gallinari P, Lahm A, Neddermann P, Rowley M, Serafini S, Steinkuhler C. Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg Med Chem Lett. 2008;18:1814–1819. doi: 10.1016/j.bmcl.2008.02.025. [DOI] [PubMed] [Google Scholar]
- Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, Cristea IM. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol. 2013;9:672. doi: 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinin S, Polak PE, Lin SX, Braun D, Guizzetti M, Zhang X, Rubinstein I, Feinstein DL. Dimethyl fumarate regulates histone deacetylase expression in astrocytes. J Neuroimmunol. 2013;263:13–19. doi: 10.1016/j.jneuroim.2013.07.007. [DOI] [PubMed] [Google Scholar]
- Kassis H, Shehadah A, Chopp M, Roberts C, Zhang ZG. Stroke Induces Nuclear Shuttling of Histone Deacetylase 4. Stroke. 2015;46:1909–1915. doi: 10.1161/STROKEAHA.115.009046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassis H, Shehadah A, Li C, Zhang Y, Cui Y, Roberts C, Sadry N, Liu X, Chopp M, Zhang ZG. Class IIa histone deacetylases affect neuronal remodeling and functional outcome after stroke. Neurochem Int. 2016;96:24–31. doi: 10.1016/j.neuint.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet. 1994;8:221–228. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- Kekatpure VD, Dannenberg AJ, Subbaramaiah K. HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J Biol Chem. 2009;284:7436–7445. doi: 10.1074/jbc.M808999200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kernochan LE, Russo ML, Woodling NS, Huynh TN, Avila AM, Fischbeck KH, Sumner CJ. The role of histone acetylation in SMN gene expression. Hum Mol Genet. 2005;14:1171–1182. doi: 10.1093/hmg/ddi130. [DOI] [PubMed] [Google Scholar]
- Kim C, Choi H, Jung ES, Lee W, Oh S, Jeon NL, Mook-Jung I. HDAC6 inhibitor blocks amyloid beta-induced impairment of mitochondrial transport in hippocampal neurons. PLoS One. 2012;7:e42983. doi: 10.1371/journal.pone.0042983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, Guan JS, Lee BH, Moy LY, Giusti P, Broodie N, Mazitschek R, Delalle I, Haggarty SJ, Neve RL, Lu Y, Tsai LH. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–817. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Shen S, Dietz K, He Y, Howell O, Reynolds R, Casaccia P. HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat Neurosci. 2010;13:180–189. doi: 10.1038/nn.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Chyla BJ, Amann JM, Bhaskara S, Huppert SS, Hiebert SW. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008;27:1017–1028. doi: 10.1038/emboj.2008.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. Irresistible force meets immovable object: transcription and the nucleosome. Cell. 1991;67:833–836. doi: 10.1016/0092-8674(91)90354-2. [DOI] [PubMed] [Google Scholar]
- Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Krishna K, Behnisch T, Sajikumar S. Inhibition of Histone Deacetylase 3 Restores Amyloid-beta Oligomer-Induced Plasticity Deficit in Hippocampal CA1 Pyramidal Neurons. J Alzheimers Dis. 2016;51:783–791. doi: 10.3233/JAD-150838. [DOI] [PubMed] [Google Scholar]
- Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, Sambucini S, Bottomley MJ, Lo Surdo P, Carfi A, Koch U, De Francesco R, Steinkuhler C, Gallinari P. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007;104:17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, D’Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, Ratan RR. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci. 2008;28:163–176. doi: 10.1523/JNEUROSCI.3200-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun-Julien F, Suter U. Combined HDAC1 and HDAC2 Depletion Promotes Retinal Ganglion Cell Survival After Injury Through Reduction of p53 Target Gene Expression. ASN Neuro. 2015;7 doi: 10.1177/1759091415593066. Print 2015 May-Jun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao TP. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010a;29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol. 2010b;189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen J, Ricupero CL, Hart RP, Schwartz MS, Kusnecov A, Herrup K. Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat Med. 2012;18:783–790. doi: 10.1038/nm.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Fang R, Liu B, Shi H, Wang Y, Zhang W, Zhang X, Ye L. Deacetylation of tumor-suppressor MST1 in Hippo pathway induces its degradation through HBXIP-elevated HDAC6 in promotion of breast cancer growth. Oncogene. 2016;35:4048–4057. doi: 10.1038/onc.2015.476. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang X, Polakiewicz RD, Yao TP, Comb MJ. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J Biol Chem. 2008;283:12686–12690. doi: 10.1074/jbc.C700185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Hu Q, Kaufman A, D’Ercole AJ, Ye P. Developmental expression of histone deacetylase 11 in the murine brain. J Neurosci Res. 2008;86:537–543. doi: 10.1002/jnr.21521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Rindt H, Shababi M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet. 2010;19:R111–8. doi: 10.1093/hmg/ddq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis Sam Titus ASC, Yusuff T, Cassar M, Thomas E, Kretzschmar D, D’Mello SR. Reduced Expression of Foxp1 as a Contributing Factor in Huntington’s Disease. J Neurosci. 2017;37:6575–6587. doi: 10.1523/JNEUROSCI.3612-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, D’Mello SR. Neuroprotection by histone deacetylase-7 (HDAC7) occurs by inhibition of c-jun expression through a deacetylase-independent mechanism. J Biol Chem. 2011a;286:4819–4828. doi: 10.1074/jbc.M110.146860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, D’Mello SR. Neuroprotection by histone deacetylase-7 (HDAC7) occurs by inhibition of c-jun expression through a deacetylase-independent mechanism. J Biol Chem. 2011b;286:4819–4828. doi: 10.1074/jbc.M110.146860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlknecht U, Emiliani S, Najfeld V, Young S, Verdin E. Genomic organization and chromosomal localization of the human histone deacetylase 3 gene. Genomics. 1999;56:197–202. doi: 10.1006/geno.1998.5645. [DOI] [PubMed] [Google Scholar]
- Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D’Mello SR. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008a;68:1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D’Mello SR. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008b;68:1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick S, D’Mello SR. JAZ (Znf346), a SIRT1-interacting protein, protects neurons by stimulating p21 (WAF/CIP1) protein expression. J Biol Chem. 2014;289:35409–35420. doi: 10.1074/jbc.M114.597575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Kettmann R, Dequiedt F. Class IIa histone deacetylases: regulating the regulators. Oncogene. 2007;26:5450–5467. doi: 10.1038/sj.onc.1210613. [DOI] [PubMed] [Google Scholar]
- Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M, Benn CL, Franklin SA, Smith DL, Woodman B, Marks PA, Bates GP. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS One. 2011;6:e27746. doi: 10.1371/journal.pone.0027746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M, Landles C, Weiss A, Bradaia A, Seredenina T, Inuabasi L, Osborne GF, Wadel K, Touller C, Butler R, Robertson J, Franklin SA, Smith DL, Park L, Marks PA, Wanker EE, Olson EN, Luthi-Carter R, van der Putten H, Beaumont V, Bates GP. HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 2013a;11:e1001717. doi: 10.1371/journal.pbio.1001717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M, Seredenina T, Stokes MP, Osborne GF, Landles C, Inuabasi L, Franklin SA, Silva JC, Luthi-Carter R, Beaumont V, Bates GP. HDAC4 does not act as a protein deacetylase in the postnatal murine brain in vivo. PLoS One. 2013b;8:e80849. doi: 10.1371/journal.pone.0080849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard CJ, Watson PJ, Fairall L, Schwabe JW. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol Sci. 2017;38:363–377. doi: 10.1016/j.tips.2016.12.006. [DOI] [PubMed] [Google Scholar]
- Mohseni J, Al-Najjar BO, Wahab HA, Zabidi-Hussin ZA, Sasongko TH. Transcript, methylation and molecular docking analyses of the effects of HDAC inhibitors, SAHA and Dacinostat, on SMN2 expression in fibroblasts of SMA patients. J Hum Genet. 2016;61:823–830. doi: 10.1038/jhg.2016.61. [DOI] [PubMed] [Google Scholar]
- Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, Richardson JA, Bassel-Duby R, Olson EN. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest. 2008;118:3588–3597. doi: 10.1172/JCI35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, Olson EN, D’Mello SR. Neuroprotection by histone deacetylase-related protein. Mol Cell Biol. 2006a;26:3550–3564. doi: 10.1128/MCB.26.9.3550-3564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, Olson EN, D’Mello SR. Neuroprotection by histone deacetylase-related protein. Mol Cell Biol. 2006b;26:3550–3564. doi: 10.1128/MCB.26.9.3550-3564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moumne L, Campbell K, Howland D, Ouyang Y, Bates GP. Genetic knock-down of HDAC3 does not modify disease-related phenotypes in a mouse model of Huntington’s disease. PLoS One. 2012;7:e31080. doi: 10.1371/journal.pone.0031080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norwood J, Franklin JM, Sharma D, D’Mello SR. Histone deacetylase 3 is necessary for proper brain development. J Biol Chem. 2014;289:34569–34582. doi: 10.1074/jbc.M114.576397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurputra DK, Lai PS, Harahap NI, Morikawa S, Yamamoto T, Nishimura N, Kubo Y, Takeuchi A, Saito T, Takeshima Y, Tohyama Y, Tay SK, Low PS, Saito K, Nishio H. Spinal muscular atrophy: from gene discovery to clinical trials. Ann Hum Genet. 2013;77:435–463. doi: 10.1111/ahg.12031. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson’s disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- Olson DE, Sleiman SF, Bourassa MW, Wagner FF, Gale JP, Zhang YL, Ratan RR, Holson EB. Hydroxamate-based histone deacetylase inhibitors can protect neurons from oxidative stress via a histone deacetylase-independent catalase-like mechanism. Chem Biol. 2015;22:439–445. doi: 10.1016/j.chembiol.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD, Chin LS. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J Cell Biol. 2007;178:1025–1038. doi: 10.1083/jcb.200611128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Marques O, Kazantsev A. Therapeutic role of sirtuins in neurodegenerative disease. Biochim Biophys Acta. 2008;1782:363–369. doi: 10.1016/j.bbadis.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Batlevi Y, Baehrecke EH, Taylor JP. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy. 2007;3:643–645. doi: 10.4161/auto.5050. [DOI] [PubMed] [Google Scholar]
- Parra M. Class IIa HDACs - new insights into their functions in physiology and pathology. FEBS J. 2015;282:1736–1744. doi: 10.1111/febs.13061. [DOI] [PubMed] [Google Scholar]
- Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, Tang AA, Oldham MC, Wang H, Shorter J, Filiano AJ, Roberson ED, Tourtellotte WG, Chen B, Tsai LH, Huang EJ. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest. 2014;124:981–999. doi: 10.1172/JCI72723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragot A, Pietropaolo S, Vincent J, Delage P, Zhang H, Allinquant B, Leinekugel X, Fischer A, Cho YH. Genetic deletion of the Histone Deacetylase 6 exacerbates selected behavioral deficits in the R6/1 mouse model for Huntington’s disease. Brain Behav. 2015;5:e00361. doi: 10.1002/brb3.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rawat V, Goux W, Piechaczyk M, D’Mello SR. c-Fos Protects Neurons Through a Noncanonical Mechanism Involving HDAC3 Interaction: Identification of a 21-Amino Acid Fragment with Neuroprotective Activity. Mol Neurobiol. 2016;53:1165–1180. doi: 10.1007/s12035-014-9058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riess O, Rub U, Pastore A, Bauer P, Schols L. SCA3: neurological features, pathogenesis and animal models. Cerebellum. 2008;7:125–137. doi: 10.1007/s12311-008-0013-4. [DOI] [PubMed] [Google Scholar]
- Riessland M, Ackermann B, Forster A, Jakubik M, Hauke J, Garbes L, Fritzsche I, Mende Y, Blumcke I, Hahnen E, Wirth B. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet. 2010;19:1492–1506. doi: 10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- Rivieccio MA, Brochier C, Willis DE, Walker BA, D’Annibale MA, McLaughlin K, Siddiq A, Kozikowski AP, Jaffrey SR, Twiss JL, Ratan RR, Langley B. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc Natl Acad Sci U S A. 2009;106:19599–19604. doi: 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundlett SE, Carmen AA, Kobayashi R, Bavykin S, Turner BM, Grunstein M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc Natl Acad Sci U S A. 1996;93:14503–14508. doi: 10.1073/pnas.93.25.14503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Humbert S. The Biology of Huntingtin. Neuron. 2016;89:910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- Schmitt HM, Pelzel HR, Schlamp CL, Nickells RW. Histone deacetylase 3 (HDAC3) plays an important role in retinal ganglion cell death after acute optic nerve injury. Mol Neurodegener. 2014;9 doi: 10.1186/1750-1326-9-39. 39-1326-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J Cell Biol. 2005;169:577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleiman SF, Basso M, Mahishi L, Kozikowski AP, Donohoe ME, Langley B, Ratan RR. Putting the ‘HAT’ back on survival signalling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin Investig Drugs. 2009;18:573–584. doi: 10.1517/13543780902810345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleiman SF, Olson DE, Bourassa MW, Karuppagounder SS, Zhang YL, Gale J, Wagner FF, Basso M, Coppola G, Pinto JT, Holson EB, Ratan RR. Hydroxamic acid-based histone deacetylase (HDAC) inhibitors can mediate neuroprotection independent of HDAC inhibition. J Neurosci. 2014;34:14328–14337. doi: 10.1523/JNEUROSCI.1010-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41:185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- Sparrow DB, Miska EA, Langley E, Reynaud-Deonauth S, Kotecha S, Towers N, Spohr G, Kouzarides T, Mohun TJ. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 1999;18:5085–5098. doi: 10.1093/emboj/18.18.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suelves N, Kirkham-McCarthy L, Lahue RS, Gines S. A selective inhibitor of histone deacetylase 3 prevents cognitive deficits and suppresses striatal CAG repeat expansions in Huntington’s disease mice. Sci Rep. 2017;7:6082-017-05125-2. doi: 10.1038/s41598-017-05125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taes I, Timmers M, Hersmus N, Bento-Abreu A, Van Den Bosch L, Van Damme P, Auwerx J, Robberecht W. Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum Mol Genet. 2013;22:1783–1790. doi: 10.1093/hmg/ddt028. [DOI] [PubMed] [Google Scholar]
- Takami Y, Nakayama T. N-terminal region, C-terminal region, nuclear export signal, and deacetylation activity of histone deacetylase-3 are essential for the viability of the DT40 chicken B cell line. J Biol Chem. 2000;275:16191–16201. doi: 10.1074/jbc.M908066199. [DOI] [PubMed] [Google Scholar]
- Thomas EA. Involvement of HDAC1 and HDAC3 in the Pathology of Polyglutamine Disorders: Therapeutic Implications for Selective HDAC1/HDAC3 Inhibitors. Pharmaceuticals (Basel) 2014;7:634–661. doi: 10.3390/ph7060634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EA. Focal nature of neurological disorders necessitates isotype-selective histone deacetylase (HDAC) inhibitors. Mol Neurobiol. 2009;40:33–45. doi: 10.1007/s12035-009-8067-y. [DOI] [PubMed] [Google Scholar]
- Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, Gao F, Fitzgerald KM, Borok JF, Herman D, Geschwind DH, Gottesfeld JM. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci U S A. 2008a;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, Gao F, Fitzgerald KM, Borok JF, Herman D, Geschwind DH, Gottesfeld JM. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci U S A. 2008b;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, Gao F, Fitzgerald KM, Borok JF, Herman D, Geschwind DH, Gottesfeld JM. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci U S A. 2008c;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi CM, Lu MM, Wang Q, Epstein JA. Transgenic overexpression of Hdac3 in the heart produces increased postnatal cardiac myocyte proliferation but does not induce hypertrophy. J Biol Chem. 2008;283:26484–26489. doi: 10.1074/jbc.M803686200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SC, Seto E. Regulation of histone deacetylase 2 by protein kinase CK2. J Biol Chem. 2002;277:31826–31833. doi: 10.1074/jbc.M204149200. [DOI] [PubMed] [Google Scholar]
- Underwood BR, Rubinsztein DC. Spinocerebellar ataxias caused by polyglutamine expansions: a review of therapeutic strategies. Cerebellum. 2008;7:215–221. doi: 10.1007/s12311-008-0026-z. [DOI] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson JA, Karsenty G, Olson EN. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Venkatraman A, Hu YS, Didonna A, Cvetanovic M, Krbanjevic A, Bilesimo P, Opal P. The histone deacetylase HDAC3 is essential for Purkinje cell function, potentially complicating the use of HDAC inhibitors in SCA1. Hum Mol Genet. 2014;23:3733–3745. doi: 10.1093/hmg/ddu081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AH, Kruhlak MJ, Wu J, Bertos NR, Vezmar M, Posner BI, Bazett-Jones DP, Yang XJ. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol Cell Biol. 2000;20:6904–6912. doi: 10.1128/mcb.20.18.6904-6912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Yan-Neale Y, Zeremski M, Cohen D. Transcription regulation by histone deacetylases. Novartis Found Symp. 2004;259:238–45. discussion 245–8, 285–8. [PubMed] [Google Scholar]
- Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IR, Huang EJ, Tsai LH. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci. 2013;16:1383–1391. doi: 10.1038/nn.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XX, Wan RZ, Liu ZP. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur J Med Chem. 2017 doi: 10.1016/j.ejmech.2017.10.040. [DOI] [PubMed] [Google Scholar]
- Whalley K. Neural repair: export duties for HDAC5. Nat Rev Neurosci. 2014;15:4–5. doi: 10.1038/nrn3649. [DOI] [PubMed] [Google Scholar]
- Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol. 2006;172:719–731. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Wu Q, Zhang L, Feng L. Inhibition of Histone Deacetylase 3 (HDAC3) Mediates Ischemic Preconditioning and Protects Cortical Neurons against Ischemia in Rats. Front Mol Neurosci. 2016;9:131. doi: 10.3389/fnmol.2016.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, Hsieh J, Bassel-Duby R, Olson EN, Lu QR. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci. 2009;12:829–838. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Liu C, Wu J, Tao JJ, Sui XL, Yao ZG, Xu YF, Huang L, Zhu H, Sheng SL, Qin C. Tubastatin A/ACY-1215 improves cognition in Alzheimer’s disease transgenic mice. J Alzheimers Dis. 2014;41:1193–1205. doi: 10.3233/JAD-140066. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen HM, Jaramillo E, Wang L, D’Mello SR. Histone deacetylase-related protein inhibits AES-mediated neuronal cell death by direct interaction. J Neurosci Res. 2008;86:2423–2431. doi: 10.1002/jnr.21680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, Yao TP, Lane WS, Seto E. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Richon VM, Rifkind RA, Marks PA. Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc Natl Acad Sci U S A. 2000;97:1056–1061. doi: 10.1073/pnas.97.3.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Vidaurre OG, Adula KP, Kezunovic N, Wentling M, Huntley GW, Casaccia P. Subcellular Distribution of HDAC1 in Neurotoxic Conditions Is Dependent on Serine Phosphorylation. J Neurosci. 2017;37:7547–7559. doi: 10.1523/JNEUROSCI.3000-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]