

Abstract
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterized by a destructive hyperplasia of synovium. Fibroblast-like synoviocytes (FLS) are a major component of synovial pannus and actively participate in the pathologic progression of RA. How rheumatoid FLS acquire and sustain such a uniquely aggressive phenotype remains poorly understood. We describe the current state of knowledge of the molecular alterations in rheumatoid FLS at the genomic, epigenomic, transcriptomic, proteomic, and metabolomic levels, which offers a comprehensive and complementary reconstruction of the potential pathways leading to rheumatoid pannus. Such data provide new pathologic insight and suggest means to more sensitively assess disease activity and response to therapy, as well as support new avenues for therapeutic development.
Introduction
The current paradigm of rheumatoid arthritis (RA) pathogenesis emphasizes the development of a systemic autoimmune response that leads to persistent synovial inflammation and progressive joint destruction. Autoimmunity may be triggered at mucosal surfaces and serologic evidence suggests that asymptomatic immune dysregulation can exist for many years before joint inflammation becomes clinically evident (1). The inflamed synovium ultimately becomes hyperplastic and develops into the pathologic hallmark of RA - a synovial pannus with invasive properties that comprises infiltrating leukocytes and germinal centers, proliferating fibroblast-like synoviocytes, and neo-vasculature. As the major component of hyperplastic pannus, fibroblast-like synoviocytes (FLS) actively participate in joint inflammation and in cartilage and bone destruction by both contact-dependent interactions and by the production of inflammatory mediators and matrix-degrading enzymes (2). The FLS in RA resist apoptosis, show increased adhesive and invasive properties, escape contact inhibition, and can seed, infiltrate, and damage joints in immunodeficient mice (3–6). Remarkably, this aggressive phenotype endures after multiple cell passages in vitro, suggesting that FLS are persistently, if not irreversibly, altered in the rheumatoid joint.
Pannus development has long been noted to be central to joint destruction but why it develops in RA but not in other autoimmune arthritides is unknown (7). The molecular basis of pannus formation and the pathways by which synovial fibroblasts achieve and maintain an aggressive phenotype, even after removal from the articular milieu, are not fundamentally understood but are so tumor-like in character as to have been compared to a locally invasive cancer (8,9). Indeed, there is evidence in rheumatoid FLS for both proto-oncogene activation and functional or mutational inactivation of tumor suppressor genes (2). The molecular signatures and biological networks that define the distinct pathology of rheumatoid FLS have not been systematically explored but a leading notion is that absent oncogenic transformation, such phenotypic maintenance results from self-perpetuating or re-entrant stimulatory pathways involving autocrine intermediation and sustained intracellular signaling.
Pannus supports immune cell recruitment, germinal center formation and localized autoantibody production, and may provide the necessary micro-environment to sustain autoimmunity and joint destruction. Closer knowledge of the pathways underlying the aggressive phenotype of FLS would facilitate experimental study and potential therapeutic targeting. RA remains incurable and whether current therapies adequately suppress synovial pathology is unclear. The connective tissue origin of FLS may render them intrinsically resistant to immunosuppression, but interfering in their proliferative and invasive character could add significantly to a more effective treatment regimen.
In this review, we describe the current state of knowledge of the molecular alterations in rheumatoid FLS at the genomic, epigenomic, transcriptomic, proteomic, and metabolomic levels. To date, molecular profiling in RA has been carried out mainly in immune cells. We suggest that a comprehensive and complementary investigation of “omics” data in rheumatoid FLS, in combination with datasets available in other cell types (10,11) – including from pre-malignant and malignant tissues, may enable the identification of the key molecular signatures for RA-specific joint destruction.
Genetic Control of Rheumatoid Synoviocytes
Genetic variation in germline DNA, particularly within loci that influence immune responsiveness, contributes to RA susceptibility and to the clinical expression of disease. HLA alleles that are essential for antigen presentation and T cell repertoire selection feature prominently in disease susceptibility in ethnically diverse populations that develop RA (12). Genetic variation in the HLA-DRB1 locus that defines a “shared epitope” - a 5 amino acid motif in the third allelic hypervariable region of the HLA-DRβ molecule, is the most highly associated with RA and may exist in almost 60% of subjects in some studies (13). Nevertheless, the genetic contribution of this and other germline alleles to RA susceptibility is tempered by observations in genetically related siblings; for instance, among monozygotic twins, the concordance rate of RA is only 15% (14). Such low concordance, as in other autoimmune diseases, has been interpreted as supporting the more important role of environmental or stochastic factors in ultimate disease development. The most well-described environmental factors for RA include smoking, stress, and infection (1). Smoking perturbs mucosal immunity and promotes protein modification (e.g. citrullination, carbamylation) that induces autoantigens that may directly trigger HLA shared epitope autoimmunity (15). Circulating and persistent anti-citrullinated protein autoantibodies (ACPAs) presage disease development by up to 10 years (16,17). Smoking also increases the clinical severity of RA in individuals with the HLA shared epitope (13,18). Repeated exposure to stress or to infection also may dysregulate inflammatory responses, lower immune tolerance, and trigger autoimmunity, although mechanisms that may be specific to RA resist precise definition.
Persistent but subclinical inflammatory activation of the synovium - mediated by ACPA or rheumatoid factor containing immune complexes together with localized inflammatory mediator expression, contributes to genotoxicity. Oncogenic mutation is well known to occur at sites of chronic inflammation; for instance, in inflammatory bowel disease, Barrett’s esophagitis, Helicobacter gastritis, chronic pancreatitis, hepatitis, and other conditions (19). Reactive oxygen and nitrogen species (ROS, RNS) are mutagenic and are persistently upregulated in rheumatoid FLS (Figure 1). There is evidence that somatic mutations occur in synoviocytes, which contribute to growth dysregulation and an invasive phenotype. Mutations in proto-oncogenes such as P53, RAF1, and in mitochondrial DNA have been reported in rheumatoid FLS; while these are insufficient for oncogenesis and malignant transformation, they deregulate DNA repair, perturb cell cycle control, and can activate mutS homolog (MSH) genes to increase microsatellite instability and further gene variation (20). Such changes increase synoviocyte survival in the face of inflammatory signals by bypassing cell-cycle arrest and promote an aggressive phenotype that contributes to pannus formation. Interestingly, synovial mutations in P53 show a pathologic spatial heterogeneity; they are detectable in intimal lining cells that are most closely apposed to areas of cartilage destruction (21). As in the case of the tumor microenvironment, it is conceivable that multiple and distinct populations of rheumatoid FLS emerge. Indeed, recent studies have identified distinct molecular profiles in FLSs from different anatomical sites (22–26). In concert with infiltrating immune cells, rheumatoid FLS sustain the signaling interactions necessary for autoimmunity and expression of the full phenotypic features of rheumatoid pannus.
Figure 1.
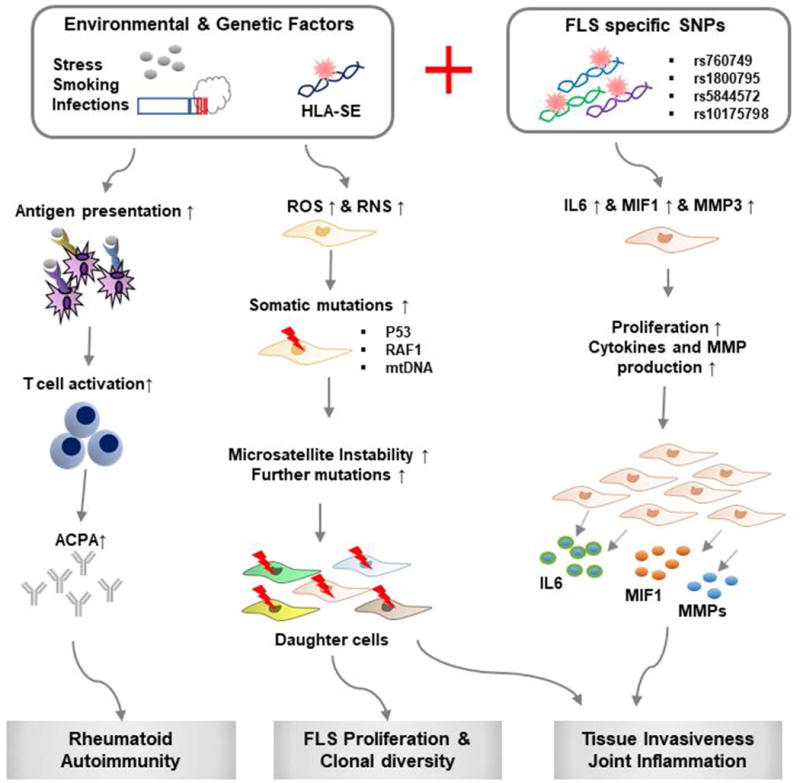
Schematic model of pathogenic progression of RA with the contribution of FLS-specific genetic factors. Abbreviations: FLS, fibroblast-like synoviocytes; HLA-SE, human leucocyte antigen shared epitope; ACPA, Anti-citrullinated peptide antibody; ROS, reactive oxygen species; RNS, reactive nitrogen species; SNP, single nucleotide polymorphism; mtDNA, mitochondrial DNA.
To date, investigation of somatic mutations in synovial fibroblasts has relied largely on candidate gene approaches derived from cancer data. Genome-wide association studies (GWAS) have uncovered a large number of single nucleotide polymorphisms (SNPs) associated with RA, however genetic markers that might be associated with synoviocyte phenotypes have not been comprehensively assessed (27). Only a few studies have identified genes whose variants may influence FLS function (28–32). The genomic locus of the SPAG16 gene (rs7607479) is associated with radiological progression in RA, and FLS encoding the SPAG16 minor allele secrete less matrix metalloproteinase-3 (28). Risk SNPs in synoviocyte DNA also have been reported that have not been annotated in leukocyte DNA (29). An SNP (rs10175798) proximal to the LBH gene may regulate synoviocyte proliferation (30), and an SNP (rs1800795) in the proximal promoter of IL6 influences IL-6 expression in synoviocytes but not in CD14+ monocytes (27). Recently, Yoo et al. reported distinct pathologic phenotypes among rheumatoid FLS encoding a high expression variant of the MIF gene (rs5844572) (32), which is a common risk allele for erosive RA (33,34). Rheumatoid FLS with high-expression MIF promoter variants exhibit an up-regulation of inflammatory and invasive phenotype, with enhanced recruitment of the MIF signaling receptor (CD44) into a functional signal transduction complex and alternative exon splicing to produce invasive CD44v3-v6 isoforms associated with oncogenic progression (28). Beyond activating invasion, MIF recapitulates other cardinal features of malignant transformation, including resistance to apoptosis, proliferative signaling, evasion of growth suppressors, and angiogenesis (35–37). An anti-MIF receptor (e.g. Milatuzumab) strategy could be rationally considered in RA, particularly in those individuals identified to carry high expression MIF alleles.
While further study is needed, synoviocyte-specific gene associations based on rheumatoid FLS DNA could allow for improved pathogenic insight and for identifying pathways to treat invasive pannus. One attraction of this approach would be pharmacologic targeting invasive or growth-regulating processes that could be non-overlapping in action or toxicity with concurrent immunosuppression.
Epigenetic Control of Rheumatoid Synoviocytes
Rheumatoid FLS exhibit unique pathologic characteristics that include: a) secretion of pro-inflammatory cytokines, prostanoids, and proangiogenic factors (7,38–40), b) enhanced proliferation and survival, and c) invasive character via the expression of MMPs and adhesion molecules that bind extracellular components such as integrins and CD44 (41–45). These features are cell intrinsic and their persistence in isolated cell culture has prompted a search for causative epigenetic modifications (Figure 2). Epigenetic modification through DNA methylation or histone modification (e.g. acetylation, methylation, and sumoylation) modulates DNA accessibility to transcriptional regulator complexes. Rheumatoid FLS DNA is broadly hypomethylated across the genome (46), histone acetylation is increased (47), and specific microRNAs (miRNAs) are upregulated (48).
Figure 2.
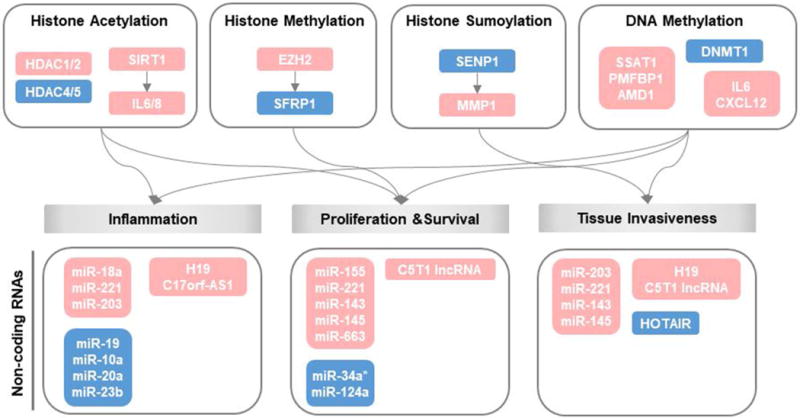
Epigenetic processes relevant to rheumatoid FLS. Genes in the red box indicate up-regulated processes and genes in the blue box are down-regulated processes. HDAC=histone deacetylase; SIRT1=Sirtuin1; IL=interleukin; TNF-α=tumor necrosis factor-α; EZH2=histone methyltransferase enhancer of zeste homologue 2; SFRP1=secreted frizzled-related protein 1; SENP1=SUMO-specific protease 1; MMP1=metalloproteinase 1; SSAT=spermidine/spermine N1-acetyltransferase; PMFBP1=polyamine-modulated factor 1-binding protein 1; AMD1=S-adenosyl methionine decarboxylase; DNMT1=DNA methyltransferase 1; CXCL12=chemokine (C-X-C motif) ligand 12.
Global genomic hypomethylation in particular is considered to have a role in RA pathogenesis, and studies have shown that the expression of DNA methyltransferase 1 (DNMT1) is reduced in rheumatoid FLS (46). There is increased expression of enzymes necessary for demethylation, including S-adenosyl methionine decarboxylase (AMD), spermidine/spermine N1-acetyltransferase (SSAT-1) and polyamine regulatory factor 1 binding protein 1 (PMFBP1), together with reduced expression of S-adenosyl methionine (SAM) and nuclear 5-methylcytosine (5-MeC) in rheumatoid FLS when compared to osteoarthritic FLS (49). Genome-wide profiling studies of epigenetic histone modifications remain limited however. Nakano et al. (50) described 1,859 differentially methylated loci in rheumatoid FLS, and with respect to specific target genes, a reduction in the methylation of promoters for the pro-inflammatory cytokines IL6 (51) and CXCL12 (52) have been reported (Figure 2). DNA hypomethylation could alter the expression of genes that are relevant to joint pathology, including those necessary for synovial invasiveness by enhancing migratory activity and extracellular matrix interactions (50).
TNF-α increases the expression of HDAC1 and nuclear HDAC activity in FLS, suggesting that the histone deacetylation observed in rheumatoid synovium results from inflammatory signaling (53). Niederer et al. (54) also demonstrated that the histone deacetylase, sirtuin 1 (SIRT1), is overexpressed in RA synovium and that expression levels are further increased by TNF-α stimulation. Sirtuin 1 in turn enhances production of the pro-inflammatory cytokines IL-6 and IL-8, and promotes synoviocyte survival (Figure 2). Other histone modifications such as methylation, ubiquitination, and sumoylation have been examined in rheumatoid synoviocytes. Trenkmann et al. demonstrated that the histone methyltransferase, enhancer of zeste homolog 2 (EZH2), is overexpressed (55) and identified secreted frizzled-related protein 1 (SFRP1), an inhibitor of Wnt signaling, as a downstream target of EZH2. These authors further showed that SFRP1 expression correlates with promoter occupation and with activating and silencing histone marks (55). Rheumatoid FLS have intrinsically high levels of the small ubiquitin-like modifier 1 (SUMO1) enzyme and consequently low levels of its unique protease SENP1 (56). Transient overexpression of SENP1 in FLS reduces H4 acetylation in the MMP1 promoter and MMP1 expression, which is consistent with the notion that histone sumoylation contributes to the local invasiveness of rheumatoid FLS (Figure 2).
Non-coding RNA (ncRNA), including microRNA (miRNA) and long noncoding RNA (lncRNA), is an epigenetic regulatory mechanism that imparts broad and long-term changes in gene expression. In Table 1, miRNAs and lncRNAs are categorized according to three major pathologic features of rheumatoid FLS: 1) inflammation, 2) proliferation and survival, and 3) tissue invasiveness, and the common functional roles for ncRNAs identified both in rheumatoid FLS and cancer cells are highlighted (57–60). miR-221, for instance, is overexpressed in both cell types; it promotes tissue invasiveness by upregulating MMPs in rheumatoid FLS (57,58,61) and it increases the migration of metastatic cancer cells (62,63). This similarity in expression and function supports the potential role of this miRNA in the tumor-like de-differentiation and invasive character of rheumatoid FLS. There are nevertheless limitations to such comparisons as the expression and role of ncRNAs are not always coincident between rheumatoid FLS and cancer cells. For example, miR-203 is upregulated in FLS and promotes MMP1 and IL-6 production, but it is down-regulated in several cancers (64–66) and it suppresses the proliferation and invasiveness of melanoma cells (67). Forced expression of homeobox antisense intergenic RNA (HOTAIR), a lncRNA, decreases MMP2 and MMP13 levels in rheumatoid FLS (68) but is elevated in expression in some types of cancer, where it reprograms chromatin to increase tumor invasiveness and metastasis (69). Additional discrepancies include miR-143 and miR-145, which are highly expressed in rheumatoid FLS (70). These two miRNAs increase NF-κB activity and IL-6 production by regulating IGFBP5 and promote FLS survival by suppressing SEMA3A (70), but they are down-regulated in multiple cancer types (71,72). The differential expression and functional role of ncRNAs thus raises the possibility that FLS possess distinct epigenetic regulatory mechanism(s) to sustain their proliferative and invasive properties. It is exceedingly rare for rheumatoid synovia to transform into cancer, suggesting that even with mutations in P53 or other proto-oncogenes, distinct cell- or tissue-specific regulatory mechanisms prevent oncogenic progression.
Table 1.
Differential expression of non-coding RNAs and their functional association in rheumatoid FLS versus cancers.
| Class | Gene Name | Expression in Rheumatoid FLS | Function in Rheumatoid FLS | Cancer Association | ||
|---|---|---|---|---|---|---|
| MicroRNAs | miR-10a | Down | Regulates NF-κB pathway and production of inflammatory cytokines | Mu et al, 2016 | Decreases cell growth in CML and promotes proliferation and survival in DLBCL | Agirre et al, 2008 Fan et al, 2016 |
| miR-19 | Down | Regulates TLR2, IL-6, IL-8 and MMP3 expression | Philippe et al, 2012 | Associated with unfavorable prognosis in patients with T-cell lymphoblastic lymphoma. | Xi et al, 2015 | |
| miR-20a | Down | Regulates IL-6, TNF and IL-1β expression | Philippe et al, 2013 | Plays a role in myeloid leukemic cell differentiation. | He et al, 2013 | |
| miR-23b | Down | Reduces arthritis in mouse model | Zhu et al, 2012 | Increases the cisplatin sensitivity in chondrosarcoma. | Huang et al, 2017 | |
| miR-34a* | Down | Regulates FLS survival | Niederer et al, 2012 | Plays a key role as a tumor suppressor in several cancers | Agostini et al, 2014 | |
| miR-124a | Down | Regulates FLS proliferation | Nakamachi et al, 2009 | Down-regulated in glioblastoma and is involved in migration and invasion. | Fowler et al, 2011 | |
| miR-18 | Up | Increases inflammatory cytokines, MMP-1 and NF-κB signaling | Trenkmann et al, 2013 | Associated with a poor prognosis, and involved in cancer cell proliferation and invasion | He, L. et al. 2005 O’Donnell et al, 2005 | |
| miR-155 | Up | Inhibits MMP-3 expression, proliferation and survival of FLS | Long et al, 2013 | Up-regulated in lymphomas and breast cancer | Iorio et al, 2005 Metzler et al, 2004 Eis et al, 2005 Kluiver et al, 2005 | |
| miR-203 | Up | Promotes MMP1 and IL-6 production | Stanczyk et al, 2011 | Frequently down-regulated in bladder, colon, lung, and skin cancers | Gottardo et al, 2007 Sonkoly et al, 2012 Deng et al, 2016 wang et al, 2014 | |
| miR-221 | Up | Promotes inflammatory cytokines, survival, migration and invasion of FLS | Pandis et al, 2012Yang et al, 2015 | Induces cell survival and promotes treatment resistance. Up-regulated in breast, colon, gastric, bone, and bladder cancers. | Zhao et al, 2013 M. Garofalo et al, 2012 | |
| miR-663 | Up | Regulates FLS proliferation and IL-6 production | Miao et al, 2015 | Down-regulated in gastric and pancreatic cancers. | Tili et al, 2010 Pan et al, 2010 | |
| miR-143/145 | Up | Drives FLS proliferations and promotes invasion | Hong et al, 2017 | Decreased abundance in breast, colon, prostate, cervical and lymphoid cancer cell lines. | Iorio et al, 2005 Michael et al, 2003 | |
| LncRNAs | HOTAIR | Down | Increased MMP-2 and MMP-13 expression | Song et al, 2015 | Correlates with diverse tumor metastasis by reprograming chromatin state. | Gupta et al, 2010 |
| C5T1lncRNA | Up | Regulates C5 mRNA expression | Messemaker et al, 2016 | NA | NA | |
| C17orf-AS1 | Up | Positively correlated with the level of CRP and the SDAI score. | Zhang et al, 2016 | NA | NA | |
LncRNA=long noncoding RNA; IL-6=interleukin-6; IL-8=interleukin-8; TNF=tumor necrosis factor; TLR2=toll-like receptor 2; MMP-1=matrix metalloproteinase 1; MMP-3=matrix metalloproteinase 3; MMP-13=matrix metalloproteinase 13; CRP=C-reactive protein; HOTAIR=homeobox antisense intergenic RNA; CML=chronic myeloid leukemia; DLBCL=Diffuse large B-cell lymphoma; NA=Not available.
Integrative Approaches for Defining Pathologic Signatures in Rheumatoid Synoviocytes
The development of high-throughput technologies such as microarray-based expression analyses and next generation sequencing have enabled the collection of an unprecedented quantity of information from clinical and biological samples. The integration of different “omics” data by statistical informatics also has the potential to provide for a more informative and holistic understanding of pathologic processes (24,27,73). Transcriptome, epigenome, and proteome data have been generated from FLS from independent studies, opening opportunities for defining signatures for rheumatoid pannus. Whitaker et al. recently performed an integrated analysis of SNPs identified by GWAS with transcriptome and methylome data derived from rheumatoid FLS (74). These authors identified a significant enrichment of “multi-evidence” genes (MEGs) related to RA pathogenicity, such as Cell Motility Protein-1 (ELMO1), which is overexpressed, promotes cell migration and invasion, and regulates Rac1 GTPase activity that is essential for cytoskeletal dynamics and motility.
For the purposes of this review, we performed a comparative analysis of available genomic, transcriptome, methylome, and proteome data (75–80) to identify genes and proteins altered in rheumatoid synoviocytes (Figure 3A). Notably, only a few genes were identified to coincide by simple pairwise comparison of these databases, with the exception of the differentially expressed and methylated genes (e.g. DEGs vs. DMGs). Proteome data would be expected to reflect transcriptome and methylome signatures, and it not evident why differentially regulated protein (DRPs) data from five independent studies do not overlap with DEGs and DMGs in this analysis. The queried databases may be compromised by individual cell- or data-specific profiles, and limited by disease stage (early versus long standing) (81), clinical activity, drug treatment, or selection bias due to the small number of tested samples. More precise and uniform clinical phenotyping, greater samples sizes, and an integrated biostatistical approach are desirable to resolve this individual variability.
Figure 3.
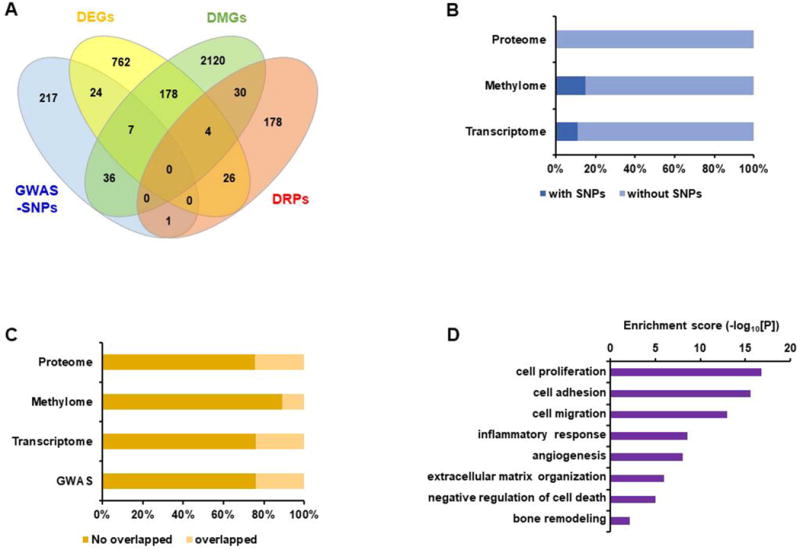
Comparative view of different omics profiles and pathologic signatures of rheumatoid FLS. Differentially expressed genes (DEGs) were first screened using transcriptome data produced for rheumatoid FLS and FLS of osteoarthritic patients (80). Second, the differentially methylated gene (DMGs) signature was obtained from Whitaker et al. (74). Finally, a list of 257 differentially regulated proteins (DRPs) with significant differences in expression or phosphorylation in rheumatoid FLS versus osteoarthritis FLS were found by combining DRPs from five independent studies (75–79). GWAS-SNPs, DEGs, DMGs, and DRPs were comapred to identify overlapping and unique lists of genes and proteins. A. The Venn diagram shows the number of common and distinct genes that are detected or differentially expressed in rheumatoid FLS by these comparisons. B. The stacked bar graph depicts the proportion of the overlapping genes harboring RA-associated SNPs in comparison with the proteome, methylome, or transcriptome of rheumatoid FLS. C. The stacked bar graph displays the proportion of the overlapping and non-overlapping genes between at least two different levels of omics data. D. Functional enrichment analysis of the overlapping genes between at least two different levels of omics.
To what degree might genetic variation at the DNA level impact the transcriptome, methylome, and proteome signatures of rheumatoid synoviocytes? We examined if SNPs identified from GWAS coincide with genes identified by other omics approaches (Figure 3B). As there is no GWAS catalog of synoviocyte DNA, our query was limited to SNP data from the peripheral leukocyte DNA (27). Less than 20% of SNPs coincided with genes or proteins discovered by the other omics profiles, indicating that the genetic variation represented by SNPs, or their tagged genes, do not contribute significantly to the pathologic signatures of rheumatoid FLS. A similar analysis performed after excluding the GWAS data also showed that fewer than 30% of genes coincided in a one-to-one comparison between two or more omics data (Figure 3C). These findings again suggest appreciable individual heterogeneity, bias, or inherent limitations in the gene/protein scoring methodologies.
To complete this multi-dimensional analytic approach, we selected 306 MEGs (Supplementary file) that were identified in two or more of the omics databases and conducted a functional enrichment analysis of the genes using DAVID software (ver. 6.8) (82). The synovial pathologies of inflammation, proliferation/survival, and invasiveness (cell adhesion, migration, and extracellular matrix organization) were enriched by this analysis (Figure 3D). To a certain extent, this finding supports an integrated MEG approach to analyzing such data, although the representational content and quality of the collected omics datasets remain a limiting issue (74). The goal nevertheless would be to apply multi-dimensional analysis of molecular profiling data to uncover disease-relevant signatures and facilitate the discovery of coherent pathways for pathologic insight and therapeutic targeting.
Metabolic Dysregulation and Metabolomics in Rheumatoid Synovium
It is reasonable to presume that the sustained activation of rheumatoid synoviocytes leads to an altered metabolic profile and a characteristic signature by metabolomic analysis. Metabolite changes have been described in the serum, urine, and synovial fluid of rheumatoid patients, and include both changes in amino acid and lipid metabolism and a dysregulation in glycolysis with an increase in lactate production (83–85) that has been attributed to the tumor-like phenomenon of aerobic glycolysis or the ‘Warburg effect’ (86). Aerobic glycolysis characterizes inflammatory signaling or rapid cell division and reflects systemic inflammation (87). Similar metabolic alterations have been reported in other autoimmune conditions (88).
A number of studies have reported altered metabolism in rheumatoid FLS with implications for inflammatory joint damage (89-92). Garcia-Carbonell et al. showed that platelet-derived growth factor (PDGF) increases glucose metabolism and glucose transporter 1 expression in rheumatoid synoviocytes (90), and they demonstrated that inhibiting glycolysis mitigated inflammatory cytokine secretion, proliferation, and migration by FLS and decreased joint damage in vivo (90). A parallel study showed that hypoxic stimuli drive synoviocytes to upregulate glycolysis, promote invasion, and stimulate angiogenic pathways, and that inhibition of glycolysis decreases inflammatory cytokine secretion, the migration and invasion of rheumatoid FLS, and endothelial tube formation (91). Inflammation and ROS-induced mitochondrial DNA damage can mutate the mitochondrial genome, leading to electron transport and respiratory chain dysfunction (93). Mitochondrial dysfunction in turn promotes ROS production and energy depletion, which if unresolved may activate autophagy and cell death pathways (94). Metabolic intermediates generated through the mitochondrial tricarbocylic acid cycle differ between RA patients and healthy individuals (89). Among the 306 MEGs defined in the two-way database comparisons, genes or gene products were selected using DAVID gene-KEGG pathway information to identify those associated with energy metabolism (Table 2, Supplementary Figure S1). Increased expression of nicotinamide phosphoribosyltransferase (NAMPT) was noted, which maintains NAD levels under stress conditions and is increased in expression rheumatoid FLS and in the joints and serum of mice with collagen-induced arthritis (95).
Table 2.
Metabolic genes and protein products altered in rheumatoid FLS and their association with cancer.*
| Related Metabolic Pathways | Genes | Protein | Cancer Association |
|---|---|---|---|
| Pentose phosphate pathway | GPX7 | Glutathione peroxidase7 | Downregulation in gastric cancer cell lines (127) whereas over-expression in hepatocellular carcinoma tissue (128) |
| MGST2 | Microsomal glutathione S-transferase 2 | Common variation in psoriasis vulgaris in Chinese population (129) | |
| NAMPT | Nicotinamide phosphoribosyl transferase | Increased intracellular expression has been reported in primary colorectal cancer, involved in angiogenesis by activation of extracellular signal-regulated kinase 1/2 and inducing VEGF and MMP2/9 (130-132) | |
| NNMT | Nicotinamide methyltransferase | Regulates hepatic nutrient metabolism (133), associated with obesity, diabetes (134), non-small cell lung cancer (135), colorectal cancer (136) and oral carcinoma cells (137) | |
| Glycolysis | HK2 | Hexokinase 2 | Mutated in cancer cells, harnesses OXPHOS ATP production to drive glycolysis (138) |
| IDH1 | Isocitratedehydrogenase1 | Oncogenic mutations in fibrosarcoma cells (139), chondrosarcoma (140), acute myelogenous leukemia, low-grade glioma, and secondary glioblastoma (141), promote cancer cell growth and resistance to targeted therapy. | |
| ACO1 | Aconitase 1 | Associated with the survival of patients with pancreatic cancer (142) | |
| TPI1 | Triosephosphate isomerase 1 | Decreased expression in hepatocellular carcinoma, suppresses growth, migration and invasion of hepatocellular cancer cells (143) | |
| PGD | Phosphogluconate dehydrogenase | Upregulated in cancer cells, lipogenesis and tumor growth, tumor cell migration (144-146) | |
| Glycogen pathway | UGP2 | UDP-glucose pyrophosphorylase 2 | Progression and poor prognostic biomarker of gallbladder cancer (147) |
| PYGB | glycogen phosphorylase B | Common variants are associated with type 2 diabetes (148) | |
| Triacylglycerol synthesis and degradation | FADS1 | Fatty acid desaturase 1 | Associated with colorectal cancer risk in East Asians (149) |
| SCD | Stearoyl-CoA desaturase | Suppresses tumor cell proliferation and induce apoptosis in neoplastic lesions when inhibited (150) | |
| Amino acid metabolism | ANPEP | Alanyl aminopeptidase | Known as CD13, inhibition of CD4+CD25+ T cells ameliorates acute colitis in mice (151) |
ATP= Adenosine triphosphate; MMP=matrix metalloproteinase; OXHOS= Oxidative phosphorylation; VEGF=vascular endothelial growth factor.
For reference detail, see Supplementary references.
Abnormalities in choline metabolism are a feature of oncogenesis and tumor progression (96). Choline kinase, which catalyzes phosphatidyl choline biosynthesis, is expressed in RA synovium, and is induced by stimulation with TNF or PDGF (97). Inhibition of choline kinase represses the aggressive behavior of rheumatoid FLS and attenuates disease in mice with serum transfer arthritis (97).
The current data suggest a dysregulation in synoviocyte metabolism in RA with a shift away from oxidative phosphorylation and toward aerobic glycolysis that is reminiscent of transformed cells. Presumably, this phenotype ensues from chronic inflammatory stimulation by cytokines, ROS, and hypoxia, and dysregulated metabolism correlates with mitochondrial dysfunction and DNA damage. Such metabolic abnormalities likely support the invasive phenotype of the rheumatoid synovium and may offer diagnostic biomarker and therapeutic opportunities for targeting rheumatoid FLS without affecting the overall immune repertoire.
Diagnostic Applications
The hyperplasia and de-differentiation of FLS within the rheumatoid joint is accompanied by a neo-angiogenic response to produce the fibro-vascular changes characteristic of pannus. Unique morphologic features of grossly inflamed synovial tissues, such as the edematous villous projections in the joint cavity, can be visualized by ultrasound (US) and magnetic resonance image (MRI). Grey scale and Doppler US measure synovial proliferation and increased vascularity, respectively (Figure 4A, 4B). Synovial hypertrophy can be defined by US as hyper-echoic intraarticular tissue that is non-displaceable and poorly compressible, and with an enhanced Doppler signal (98). Notably, US color Doppler imaging shows positive correlation with quantitative immunohistochemical staining for von Willebrand factor, CD3, CD68, and Ki67 in synovial biopsies (99). MRI employing gadolinium contrast-enhancement permits for the visualization of synovitis as a thickened synovial compartment with above-normal post-gadolinium enhancement (Figure 4C) (100).
Figure 4.
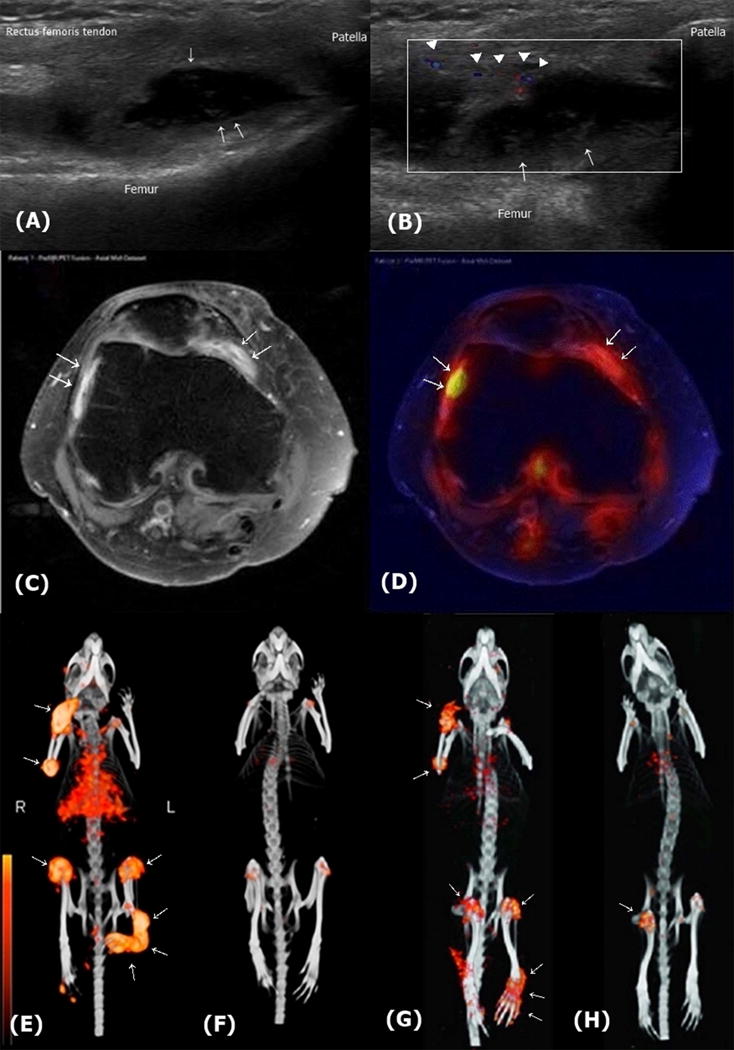
Identification of early synovitis in RA patients or in mice collagen-induced arthritis (CIA) by US, MRI, and SPECT. (A, B). US findings of synovial proliferation in an RA patient. A. Longitudinal grey-scale. US image of the suprapatellar recess in the knee joint shows marked synovial thickening with papillary projection (arrows), accompanied with synovial fluid. B. Power Doppler US image shows increased vascularization (arrowheads). (C and D). MRI and PET findings of early synovitis in a RA patient. C. Axial contrast-enhanced MRI of a patient with RA. Synovial hyperplasia is visualized by signal enhancement on T1-weighted image (arrows). D. Co-localization of metabolic activity and infiammation in vivo using axial hybrid PET/MRI fusion. Increased synovial infiammation is co-localized with altered metabolic activity (arrows). Adapted with permission from ref. 91 (Reproduced from Dysregulated bioenergetics: a key regulator of joint inflammation, Biniecka M, Canavan M, McGarry T, Gao W, McCormick J, Cregan S, et al., 75, 2192-200, 2016 with permission from BMJ Publishing Group Ltd) (E though H). 3D SPECT/CT scans using anti-fibroblast activation protein (FAP) antibody for detection of hyperplastic FLSs. Mice were injected with 15 MBq of 111In-labeled FAP antibody (111In-28H1). After 24 hrs, radiotracer uptake (arrows) is clearly visible in the infiamed joints of the mouse with CIA (E), but not in the control mouse (F). 3D SPECT/CT scans using 99mTc-labeled FAP antibody also revealed that increased radiotracer uptake in the joints of an arthritic mouse (G) were reduced by treatment with prednisolone (H). E and F, from adapted with permission from ref. 103 (This research was originally published in JNM. Laverman P, van der Geest T, Terry SY, Gerrits D, Walgreen B, Helsen MM, et al. Immuno-PET and Immuno-SPECT of Rheumatoid Arthritis with Radiolabeled Anti-Fibroblast Activation Protein Antibody Correlates with Severity of Arthritis. J Nucl Med 2015;56:778-83. © by the Society of Nuclear Medicine and Molecular Imaging, Inc.). G and H are adapted with permission from ref. 104 (This research was originally published in JNM. van der Geest T, Laverman P, Gerrits D, Walgreen B, Helsen MM, Klein C, et al. Liposomal Treatment of Experimental Arthritis Can Be Monitored Noninvasively with a Radiolabeled Anti-Fibroblast Activation Protein Antibody. J Nucl Med 2017;58:151-5 © by the Society of Nuclear Medicine and Molecular Imaging, Inc.).
US and MRI are powerful diagnostic tools for the early diagnosis of synovial disease and predict radiographic progression and joint erosion in early undifferentiated arthritis (101,102). These techniques do not assess functional abnormalities however, which may have added diagnostic or prognostic utility. Positron emission tomography (PET) scanning is sensitive to glucose-dependent metabolic processes (Figure 4D) (91). The combination of immuno-PET or immuno-single photon (SPECT) imaging using radiolabeled anti-fibroblast activation protein (FAP) antibody visualizes arthritic joints with high resolution and accurately measures the severity of inflammation and therapeutic responses in murine arthritis (Figure 4E-H) (103,104).
Prospects for Therapeutic Strategies Targeting Rheumatoid Synovium and FLS
Newly approved therapies that target the immune system offer effective disease control, albeit with risk of infection and malignancy. While clinical remission is now more readily achieved, an appreciable percentage of patients experience incomplete responses or refractory disease. There also is no cure for RA despite continuing clinical trial efforts with new and combination immunotherapies. Pharmacologic targeting of the rheumatoid pannus may be considered to be an attractive strategy for several reasons. The chronic inflammatory micro-environment driven by FLS may perpetuate autoimmunity by ongoing autoantigen presentation, co-stimulatory cytokine production, and the maintenance of autoantibody-producing germinal centers. Recurrent or refractory disease may depend on immunopathologic interactions that are spatially coordinated within pannus stroma, and such effects may be analogous to the tumor microenvironment, although in the case of RA the interactions are immunostimulatory rather than immunosuppressive. Synovial-based interventions could offer an approach toward more effective and long-term disease control by disrupting synoviocyte-specific immunopathology. Moreover, interference with proliferating and invasive synoviocytes may prevent the destruction of articular cartilage and subchondral bone by mechanisms that do not contribute to systemic immunosuppression and adverse events.
One approach that has been pursued - by analogy to anti-tumor pharmacology, is to suppress synovial angiogenesis. Notably, one effect of anti-TNF therapy is to reduce serum concentrations of the angiogenic mediator vascular endothelial growth factor (VEGF) (105) and to decrease synovial blood flow and vascularity (106). Therapeutic inhibition of the VEGF pathway has been studied in pre-clinical models of inflammatory arthritis under the hypothesis that vasculogenesis is essential for the expansion and maintenance of pannus tissue. The anti-VEGF antibody Avastin reduces arthritis and synovial injury to a similar degree as TNF inhibition when studied in a collagen-induced arthritis rat model (107). Interference with VEGF receptor action by neutralizing antibody or by inhibiting downstream kinase signaling also ameliorates disease in pre-clinical models (108,109), and similar treatment effects were reported with the angiogenesis inhibitor endostatin (110).
Given the wealth of evidence implicating FLS in perpetuating synovial inflammation and destroying articular cartilage and subchondral bone, could FLS be selectively targeted? Ideally, unique or cell specific markers for these cells could be employed for an immune targeting or cell depletion strategy. We explored the omics databases to identify genes that show specificity for expression in rheumatoid FLS (Table 3, Supplementary Figure S2). Examination of the Genotype-Tissue Expression (GTEx) database using criteria that: 1) the gene is increased in expression in rheumatoid FLS and has potential pathologic significance, and 2) is rarely expressed in other cell types yielded several candidates. FAP (fibroblast activation protein) and GREM1 (gremlin 1) were classified as highly specific for rheumatoid FLS, CDH11 (cadherin-11) and THY1 (thymocyte differentiation antigen) 1 as moderately specific, and intracellular adhesion molecule-1 (ICAM1) and CD55 as having low cell specificity. GREM1 is an antagonist of bone morphogenetic protein and regulates the survival, proliferation, migration, and invasion of rheumatoid FLS (111). FAP encodes a type II membrane serine protease that is highly expressed in rheumatoid synovia and contributes to the myofibroblastic phenotype of synioviocytes (26,112). Notably, CDH11, which mediates homophilic cell adhesion, has been well studied in synovium for its ability to regulate synovial morphogenesis and fibroblast pro-inflammatory function. Cadherin-11 gene deficient mice have hypoplastic synovia and show reduced inflammation and cartilage erosion when challenged in an inflammatory arthritis model. Cadherin-11 is minimally expressed in immune cells (113) and anti-cadherin-11 antibody reduces arthritis severity in mice (114). A humanized monoclonal antibody targeting cadherin-11 has been evaluated in a phase I tolerability study (115) (Table 4).
Table 3.
Selected gene or gene product expressed by FLS.
| Marker | Fibroblast Specificity* | Pathologic Role in RAs | Ref |
|---|---|---|---|
| Surface proteins | |||
| FAP | Highly specific | 95-kDa cell surface-bound type II transmembrane serine protease. Expressed by fibroblastic cells in the area of active tissue remodeling. Associated with myofibroblast phenotype of FLS. Co-localized with MMP-1, MMP-13, and CD44 splice variants. |
Bauer et al. 2006 |
| CDH11 | Moderate specific | Overexpressed in the synovium of RA pannus, mainly at the synovial lining cells. Also expressed by osteoblasts. Regulates cell-to-cell contact between FLS Promotes invasive capacity of FLS Cadherin-11 deficient mice are resistant to inflammatory arthritis. Anti-cadherin-11 monoclonal antibody ameliorates arthritis in the K/BxN model. |
Kiener et al. 2006 and 2009; Valencia et al, 2004; Noss et al. 2011; Lee et al.2007 |
| THY-1 (CD90) | Moderate Specific | 35-kDa cell-surface glycoprotein. Expressed on brain cells, fibroblasts, activated endothelial cells. Soluble Thy-1 is elevated at the local inflammatory sites (i.e. synovial fluid). |
Saalbach et al. 1999 |
| VCAM-1 (CD106) | Less specific | Expressed on FLS within the intimal layer of normal, osteoarthritis, and RA synovium. Regulates cell-to-cell contact between FLS and macrophages Mediates endothelial progenitor cells adhesion to FLS Neutralizing VCAM-1 antibody reduces adhesion endothelial progenitor cell to FLS. |
Wilkinson et al. 1993; Siverman et al. 2007 |
| CD44 | Less specific | Mediates cell–cell adhesion and cell–matrix interactions. Implicated in cellular homing, tumor invasiveness, metastasis, and angiogenesis. Intracytoplasmic phosphorylated by MIF-CD74 signal transduction. |
Yoo et al. 2016 |
| ICAM-1 (CD54) | Non-specific | Expressed on endothelial cells and leukocytes. Mediates cell-to-cell contact between FLS and macrophages. |
Krenn et al. 1997 |
| DAF (CD55) | Non-specific | 70-kDa GPI-anchored membrane protein and protect cells from complement-mediated damage. CD55 is also a cellular ligand for CD97. CD55-deficient mice have reduced arthritis. |
Hock et al. 2010; Hamann et al. 1999 |
| Intracellular proteins | |||
| GREM1 | Highly specific | 20.7 kDa protein belonging to the subfamily of bone morphogenetic protein antagonists. Expressed in rheumatoid FLS, synovial tissue, and synovial fluid Essential to survival, migration, and invasion of rheumatoid FLS |
Han et al. 2016 |
| UDPGD | Less specific | Expressed preferentially by intimal lining fibroblasts Reflects hyaluronan synthesis ability. |
CDH11=Cadherin-11; DAF=decay-accelerating factor; FAP=Fibroblast activation protein; FLS=fibroblast like synoviocytes; GPI=glycosylphosphatidylinositol; GREM1=Gremlin1; ICAM-1=intracellular adhesion molecule-1; kDa=kiloDalton; MIF=migration inhibitory factor; MMP=matrix metalloproteinase; THY1= thymocyte differentiation antigen 1; UDPGD=Uridine diphospho-glucose 6-dehydrogenase; VCAM-1=vascular cell adhesion molecule-1;
Specificity was determined based on the number of cells and tissues that are significantly expressed in various tissues and cell types in Genotype-Tissue Expression (GTEx) database. See Supplementary Figure S2 for details.
Table 4.
New candidates for therapeutic agents targeting rheumatoid FLS*
| Efficacy in animal model | Development stage | Potential adverse effects | |
|---|---|---|---|
| Targeting specific surface molecule | |||
| Cadherin-11 antagonistic antibodies | Cadherin-11-Fc fusion protein and anti- cadherin-11 monoclonal antibody ameliorated K/BxN serum transfer arthritis activity (115). | RG6125: (SDP051 formerly, Roche): completed phase I trial in RA (115) | Not reported |
| Cell cycle inhibitors | |||
| Cyclin-dependent kinase (CDK) inhibitor | Pan-CDK inhibitor (118) and selective CDK 4/6 inhibitor (117) suppressed arthritis dose dependently in murine CIA model. Immune response to type II collagen was not impaired. Synergistic anti-arthritic effect with cytokine blockade (Etanercept, IL-6 blocker) |
Palbociclib: (selective CDK 4/6 inhibitor, Pfizer) approved by the U.S. FDA for the treatment of metastatic breast cancer. | Neutropenia Gastrointestinal adverse Events (152) |
| Epigenetic regulators | |||
| SSAT-1 inhibitor | Diminazene aceturate restored DNA methylation and reduced the adhesion ability, MMP1 production, and invasiveness of rheumatoid FLS in SCID mice model (124). | Diminazene aceturate: the drug of choice for treatment animal Trypanosomiasis (153). | Not reported |
| Histone deacetylase inhibitor | Givinostat (125), SAHA, MS-275 (154), Trichostatin A (155) ameliorated synovial inflammation and joint destruction in murine arthritis model. MS-275 and SAHA decreased serum IL-6 and IL-1beta level (156). MS-275 suppressed cytokine-induced metalloproteinase expression in cartilage cells (157). |
Givinostat: showed therapeutic benefit with excellent safety profile on phase II, open-label trial in JIA (126) SAHA (Vorinostat): approved by U.S. FDA for the treatment of cutaneous T cell lymphoma. MS-275 (Entinostat): being studied phase II trial on Hodgkin’s lymphoma, breast cancer, and lung cancer. |
Self-limited respiratory or gastrointestinal disturbances, QTc prolongation (126) Fatigue, diarrhea, nausea, anorexia, anemia and thrombocytopenia (158) Nausea, anorexia, thrombocytopenia, anemia, neutropenia, hypophosphatemia (159, 160) |
CDK=cyclin-dependent kinase; CIA=collagen induced arthritis; IL=interleukin; FLS=fibroblast-like synoviocytes; MMP=matrix metalloproteinase; JIA=juvenile idiopathic arthritis; QTc=corrected QT interval; RA=rheumatoid arthritis; SAHA=suberoylanilide hydroxamic acid; SCID=severe combined immunodeficiency; SSAT-1=Spermidine/spermine N1-acetyltransferase 1
For reference detail, see Supplementary references.
As synoviocyte proliferation features prominently in synovial pathology, interference in cell cycle regulation may be a promising therapeutic intervention. Small molecule inhibitors of cyclin-dependent kinases (CDKs), which regulate cell cycle progression at the G1/S phase transition, have been developed as anti-cancer therapies (116) (Table 4). Both Alvocidib, a pan-CDK inhibitor, and Palbociclib, a selective CDK 4/6 inhibitor, ameliorate arthritis in a murine model (117,118). Combination therapy with cytokine blockade and Palbociclib also shows a synergistic anti-arthritic effect (117) (Table 4). Rheumatoid synovium has been targeted by radionuclide synovectomy (119). In one approach, a colloidal solution of a β-emitting radioactive agent is injected intra-articularly so that the particles are phagocytized by the synovial lining cells, which concentrates the β-energy within pannus (120). Yttrium90 citrate has been the most widely studied agent (120,121) but phosphate32, rhenium186, and erbium169 citrate also have been tested (122). Although the injected radionuclide affects immune cells within the synovium, the localized activity of the radionuclide may curtail stromal hyperplasia without influencing systemic immunity. In a related approach employing non-radionuclides, cadmium (Cd) was tested for its cytotoxic action on synoviocytes. Cd-injection into rat joints improved arthritis, reduced inflammatory cell recruitment, and protected from bone/cartilage destruction. The authors suggest that the limited systemic dispersion of Cd and auspicious safety profile support exploration of this approach (123).
Pharmacologic strategies directed at DNA methylation and histone modification are under increasing study in different diseases, and hold interest given the likely epigenetic basis for the sustained aggressive phenotype of rheumatoid synoviocytes (124). Targeting synoviocyte-specific proteins or the cell cycle could inhibit proliferative pathways in early or established RA (117), whereas targeting epigenetic modification could additionally affect asymptomatic autoimmunity. Neidhart et al. have proposed that inhibition of the polyamine recycling pathway with diminazene aceturate, an inhibitor of the SSAT-1 that increases cellular 5-MeC and DNMT-1 levels, may restore DNA methylation in rheumatoid synovium. This agent alone or in combination with SAM reduces adhesive character and MMP1 expression, as well as the invasiveness of synoviocytes implanted into immunodeficient mice (124,125). Pharmacologic HDAC inhibitors are under investigation as anti-neoplastic agents and have been evaluated in preclinical and clinical studies in RA (Table 4). Inhibition of HDAC activity by various HDAC inhibitors, such as givinostat, suberoylanilide hydroxamic acid, MS-275, Trichostatin A, and FK228, ameliorates joint inflammation and prevents cartilage and bone destruction in experimental models of inflammatory arthritis (125). Notably, a clinical trial of givinostat in patients with systemic-onset juvenile idiopathic arthritis resistant to standard treatment has been reported recently and showed therapeutic benefit with an excellent safety profile (Table 4) (126). If acquired mutations feature prominently in the perpetuation of synovial aggressiveness, approaches to selectively kill or incapacitate rheumatoid FLS may prove difficult. All such approaches nevertheless will benefit from a closer molecular definition of RA disease “subphenotypes”, and a precision-based therapeutic approach similar to what is being achieved in anti-cancer pharmacology ultimately may be attainable.
Conclusions
While definitive human genetic or experimental data remain to be acquired, germline or acquired mutations in synoviocytes likely contribute to RA susceptibility, progression from pre-RA to frank disease, and clinical severity. Genetic factors, working over time and in concert with environmental factors, contribute to the epigenetic alterations that appear responsible for the sustained and aggressive phenotype of rheumatoid FLS. Defining pathologic relationships among RA risk alleles, profiling databases, and pathologic synoviocyte phenotypes may enable a better understanding of the molecular basis for the aggressive transformation of rheumatoid FLS. Limitations in the current omics databases with respect to annotation, sample sources, and data quality presently restrict any comprehensive or complementary reconstruction of pathways that may provide pathologic insight and avenues for therapeutic development. Nevertheless, the thoughtful application and integration of genomic, epigenomic, proteomic and metabolomic technologies should ultimately yield these insights. The immunopathologic contribution of rheumatoid synoviocytes to eventual joint destruction appears highly significant and the stromal origin of these cells may render them intrinsically resistant to current immunotherapies. Similar limitations exist in our therapeutic approaches to inflammatory fibrosing disorders such as systemic sclerosis, where activated connective tissue cells have a central pathologic role. If RA synovium delivers cues for perpetuating autoimmunity, then the therapeutic targeting of synoviocytes may offer a powerful approach for long-term remission (if not cure) and the possibility of intervention in the most disabling and morbid features of the disease.
Search strategy and selection criteria
We searched PubMed using terms “fibroblast” and “synoviocytes”, “fibroblast-like synoviocytes”. Studies relevant to rheumatoid arthritis were identified using the term “rheumatoid arthritis”. Our search included 2,779 articles published since 1980 through June 2017.
Supplementary Material
Acknowledgments
Gratitude is expressed for support from the US National Institutes of Health (AR049610, AR050498) and the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2014R1A6A3A04054066, 2014R1A2A1A11049812, 2015R1A3A2032927).
References (Refer to supplementary materials for reference after 85)
- 1.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 2.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9:24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lefevre S, Knedla A, Tennie C, Kampmann A, Wunrau C, Dinser R, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15:1414–20. doi: 10.1038/nm.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bucala R, Ritchlin C, Winchester R, Cerami A. Constitutive production of inflammatory and mitogenic cytokines by rheumatoid synovial fibroblasts. J Exp Med. 1991;173:569–74. doi: 10.1084/jem.173.3.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baier A, Meineckel I, Gay S, Pap T. Apoptosis in rheumatoid arthritis. Curr Opin Rheumatol. 2003;15:274–9. doi: 10.1097/00002281-200305000-00015. [DOI] [PubMed] [Google Scholar]
- 6.Lafyatis R, Remmers EF, Roberts AB, Yocum DE, Sporn MB, Wilder RL. Anchorage-independent growth of synoviocytes from arthritic and normal joints. Stimulation by exogenous platelet-derived growth factor and inhibition by transforming growth factor-beta and retinoids. J Clin Invest. 1989;83:1267–76. doi: 10.1172/JCI114011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orr C, Vieira-Sousa E, Boyle DL, Buch MH, Buckley CD, Canete JD, et al. Synovial tissue research: a state-of-the-art review. Nat Rev Rheumatol. 2017;13:463–75. doi: 10.1038/nrrheum.2017.115. [DOI] [PubMed] [Google Scholar]
- 8.Fassbender HG, Simmling-Annefeld M. The potential aggressiveness of synovial tissue in rheumatoid arthritis. J Pathol. 1983;139:399–406. doi: 10.1002/path.1711390314. [DOI] [PubMed] [Google Scholar]
- 9.Zvaifler NJ. Relevance of the stroma and epithelial-mesenchymal transition (EMT) for the rheumatic diseases. Arthritis Res Ther. 2006;8:210. doi: 10.1186/ar1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat Med. 2017;23:938–44. doi: 10.1038/nm.4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo Y, Walsh AM, Fearon U, Smith MD, Wechalekar MD, Yin X, et al. CD40L-Dependent Pathway Is Active at Various Stages of Rheumatoid Arthritis Disease Progression. J Immunol. 2017;198:4490–501. doi: 10.4049/jimmunol.1601988. [DOI] [PubMed] [Google Scholar]
- 12.Holoshitz J. The rheumatoid arthritis HLA-DRB1 shared epitope. Curr Opin Rheumatol. 2010;22:293–8. doi: 10.1097/BOR.0b013e328336ba63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gregersen PK, Silver J, Winchester RJ. The Shared Epitope Hypothesis - an Approach to Understanding the Molecular-Genetics of Susceptibility to Rheumatoid-Arthritis. Arthritis Rheum. 1987;30:1205–13. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 14.Silman AJ, Macgregor A, Holligan S, Ollier WER, Thomson W, Carthy D. Concordance Rates for Rheumatoid-Arthritis in Twins - Results of a Nationwide Study. Arthritis Rheum. 1992;35:S47–S. doi: 10.1093/rheumatology/32.10.903. [DOI] [PubMed] [Google Scholar]
- 15.Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, et al. A new model for an etiology of rheumatoid arthritis. Arthritis Rheum. 2006;54:38–46. doi: 10.1002/art.21575. [DOI] [PubMed] [Google Scholar]
- 16.Mankia K, Emery P. Preclinical Rheumatoid Arthritis: Progress Toward Prevention. Arthritis Rheumatol. 2016;68:779–88. doi: 10.1002/art.39603. [DOI] [PubMed] [Google Scholar]
- 17.Orr C, Najm A, Biniecka M, McGarry T, Ng CT, Young F, et al. Synovial Immunophenotype and Anti-Citrullinated Protein Antibodies in RA patients: Relationship to treatment response and radiological prognosis. Arthritis Rheumatol. 2017 doi: 10.1002/art.40218. [DOI] [PubMed] [Google Scholar]
- 18.Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L, Rheumatoid EI. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004;50:3085–92. doi: 10.1002/art.20553. [DOI] [PubMed] [Google Scholar]
- 19.Tak PP, Zvaifler NJ, Green DR, Firestein GS. Rheumatoid arthritis and p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today. 2000;21:78–82. doi: 10.1016/s0167-5699(99)01552-2. [DOI] [PubMed] [Google Scholar]
- 20.Lee SH, Chang DK, Goel A, Boland CR, Bugbee W, Boyle DL, et al. Microsatellite instability and suppressed DNA repair enzyme expression in rheumatoid arthritis. J Immunol. 2003;170:2214–20. doi: 10.4049/jimmunol.170.4.2214. [DOI] [PubMed] [Google Scholar]
- 21.Yamanishi Y, Boyle DL, Rosengren S, Green DR, Zvaifler NJ, Firestein GS. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc Natl Acad Sci U S A. 2002;99:10025–30. doi: 10.1073/pnas.152333199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imamura F, Aono H, Hasunuma T, Sumida T, Tateishi H, Maruo S, et al. Monoclonal expansion of synoviocytes in rheumatoid arthritis. Arthritis Rheum. 1998;41:1979–86. doi: 10.1002/1529-0131(199811)41:11<1979::AID-ART13>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 23.Frank-Bertoncelj M, Trenkmann M, Klein K, Karouzakis E, Rehrauer H, Bratus A, et al. Epigenetically-driven anatomical diversity of synovial fibroblasts guides joint-specific fibroblast functions. Nat Commun. 2017;8:14852. doi: 10.1038/ncomms14852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ai R, Hammaker D, Boyle DL, Morgan R, Walsh AM, Fan S, et al. Joint-specific DNA methylation and transcriptome signatures in rheumatoid arthritis identify distinct pathogenic processes. Nat Commun. 2016;7:11849. doi: 10.1038/ncomms11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ospelt C, Frank-Bertoncelj M. Why location matters - site-specific factors in rheumatic diseases. Nat Rev Rheumatol. 2017;13:433–42. doi: 10.1038/nrrheum.2017.96. [DOI] [PubMed] [Google Scholar]
- 26.Choi IY, Karpus ON, Turner JD, Hardie D, Marshall JL, de Hair MJH, et al. Stromal cell markers are differentially expressed in the synovial tissue of patients with early arthritis. PLoS One. 2017;12:e0182751. doi: 10.1371/journal.pone.0182751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walsh AM, Whitaker JW, Huang CC, Cherkas Y, Lamberth SL, Brodmerkel C, et al. Integrative genomic deconvolution of rheumatoid arthritis GWAS loci into gene and cell type associations. Genome Biol. 2016;17 doi: 10.1186/s13059-016-0948-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knevel R, Klein K, Somers K, Ospelt C, Houwing-Duistermaat JJ, van Nies JA, et al. Identification of a genetic variant for joint damage progression in autoantibody-positive rheumatoid arthritis. Ann Rheum Dis. 2014;73:2038–46. doi: 10.1136/annrheumdis-2013-204050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitaker JW, Boyle DL, Bartok B, Ball ST, Gay S, Wang W, et al. Integrative omics analysis of rheumatoid arthritis identifies non-obvious therapeutic targets. PLoS One. 2015;10:e0124254. doi: 10.1371/journal.pone.0124254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ekwall AK, Whitaker JW, Hammaker D, Bugbee WD, Wang W, Firestein GS. The Rheumatoid Arthritis Risk Gene LBH Regulates Growth in Fibroblast-like Synoviocytes. Arthritis Rheumatol. 2015;67:1193–202. doi: 10.1002/art.39060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noss EH, Watts GF, Zocco D, Keller TL, Whitman M, Blobel CP, et al. Evidence for cadherin-11 cleavage in the synovium and partial characterization of its mechanism. Arthritis Res Ther. 2015;17:126. doi: 10.1186/s13075-015-0647-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoo SA, Leng L, Kim BJ, Du X, Tilstam PV, Kim KH, et al. MIF allele-dependent regulation of the MIF coreceptor CD44 and role in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2016;113:E7917–e26. doi: 10.1073/pnas.1612717113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baugh JA, Chitnis S, Donnelly SC, Monteiro J, Lin X, Plant BJ, et al. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 2002;3:170–6. doi: 10.1038/sj.gene.6363867. [DOI] [PubMed] [Google Scholar]
- 34.Radstake TR, Sweep FC, Welsing P, Franke B, Vermeulen SH, Geurts-Moespot A, et al. Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis Rheum. 2005;52:3020–9. doi: 10.1002/art.21285. [DOI] [PubMed] [Google Scholar]
- 35.Pakozdi A, Amin MA, Haas CS, Martinez RJ, Haines GK, 3rd, Santos LL, et al. Macrophage migration inhibitory factor: a mediator of matrix metalloproteinase-2 production in rheumatoid arthritis. Arthritis Res Ther. 2006;8:R132. doi: 10.1186/ar2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lacey D, Sampey A, Mitchell R, Bucala R, Santos L, Leech M, et al. Control of fibroblast-like synoviocyte proliferation by macrophage migration inhibitory factor. Arthritis Rheum. 2003;48:103–9. doi: 10.1002/art.10733. [DOI] [PubMed] [Google Scholar]
- 37.Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, et al. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci U S A. 2003;100:9354–9. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nozawa-Inoue K, Harada F, Magara J, Ohazama A, Maeda T. Contribution of synovial lining cells to synovial vascularization of the rat temporomandibular joint. J Anat. 2016;228:520–9. doi: 10.1111/joa.12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kennedy A, Ng CT, Biniecka M, Saber T, Taylor C, O’Sullivan J, et al. Angiogenesis and blood vessel stability in inflammatory arthritis. Arthritis Rheum. 2010;62:711–21. doi: 10.1002/art.27287. [DOI] [PubMed] [Google Scholar]
- 40.Izquierdo E, Canete JD, Celis R, Santiago B, Usategui A, Sanmarti R, et al. Immature blood vessels in rheumatoid synovium are selectively depleted in response to anti-TNF therapy. PLoS One. 2009;4:e8131. doi: 10.1371/journal.pone.0008131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Filer A, Antczak P, Parsonage GN, Legault HM, O’Toole M, Pearson MJ, et al. Stromal transcriptional profiles reveal hierarchies of anatomical site, serum response and disease and identify disease specific pathways. PLoS One. 2015;10:e0120917. doi: 10.1371/journal.pone.0120917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGarry T, Veale DJ, Gao W, Orr C, Fearon U, Connolly M. Toll-like receptor 2 (TLR2) induces migration and invasive mechanisms in rheumatoid arthritis. Arthritis Res Ther. 2015;17:153. doi: 10.1186/s13075-015-0664-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sipila K, Haag S, Denessiouk K, Kapyla J, Peters EC, Denesyuk A, et al. Citrullination of collagen II affects integrin-mediated cell adhesion in a receptor-specific manner. Faseb j. 2014;28:3758–68. doi: 10.1096/fj.13-247767. [DOI] [PubMed] [Google Scholar]
- 44.Croft AP, Naylor AJ, Marshall JL, Hardie DL, Zimmermann B, Turner J, et al. Rheumatoid synovial fibroblasts differentiate into distinct subsets in the presence of cytokines and cartilage. Arthritis Res Ther. 2016;18:270. doi: 10.1186/s13075-016-1156-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Filer A, Ward LSC, Kemble S, Davies CS, Munir H, Rogers R, et al. Identification of a transitional fibroblast function in very early rheumatoid arthritis. Ann Rheum Dis. 2017 doi: 10.1136/annrheumdis-2017-211286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009;60:3613–22. doi: 10.1002/art.25018. [DOI] [PubMed] [Google Scholar]
- 47.Huber LC, Brock M, Hemmatazad H, Giger OT, Moritz F, Trenkmann M, et al. Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum. 2007;56:1087–93. doi: 10.1002/art.22512. [DOI] [PubMed] [Google Scholar]
- 48.Stanczyk J, Pedrioli DAL, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of microRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58:1001–9. doi: 10.1002/art.23386. [DOI] [PubMed] [Google Scholar]
- 49.Karouzakis E, Gay RE, Gay S, Neidhart M. Increased recycling of polyamines is associated with global DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2012;64:1809–17. doi: 10.1002/art.34340. [DOI] [PubMed] [Google Scholar]
- 50.Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis. 2013;72:110–7. doi: 10.1136/annrheumdis-2012-201526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nile CJ, Read RC, Akil M, Duff GW, Wilson AG. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 2008;58:2686–93. doi: 10.1002/art.23758. [DOI] [PubMed] [Google Scholar]
- 52.Karouzakis E, Rengel Y, Jungel A, Kolling C, Gay RE, Michel BA, et al. DNA methylation regulates the expression of CXCL12 in rheumatoid arthritis synovial fibroblasts. Genes Immun. 2011;12:643–52. doi: 10.1038/gene.2011.45. [DOI] [PubMed] [Google Scholar]
- 53.Kawabata T, Nishida K, Takasugi K, Ogawa H, Sada K, Kadota Y, et al. Increased activity and expression of histone deacetylase 1 in relation to tumor necrosis factor-alpha in synovial tissue of rheumatoid arthritis. Arthritis Res Ther. 2010;12:R133. doi: 10.1186/ar3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niederer F, Ospelt C, Brentano F, Hottiger MO, Gay RE, Gay S, et al. SIRT1 overexpression in the rheumatoid arthritis synovium contributes to proinflammatory cytokine production and apoptosis resistance. Ann Rheum Dis. 2011;70:1866–73. doi: 10.1136/ard.2010.148957. [DOI] [PubMed] [Google Scholar]
- 55.Trenkmann M, Brock M, Gay RE, Kolling C, Speich R, Michel BA, et al. Expression and function of EZH2 in synovial fibroblasts: epigenetic repression of the Wnt inhibitor SFRP1 in rheumatoid arthritis. Ann Rheum Dis. 2011;70:1482–8. doi: 10.1136/ard.2010.143040. [DOI] [PubMed] [Google Scholar]
- 56.Maciejewska-Rodrigues H, Karouzakis E, Strietholt S, Hemmatazad H, Neidhart M, Ospelt C, et al. Epigenetics and rheumatoid arthritis: the role of SENP1 in the regulation of MMP-1 expression. J Autoimmun. 2010;35:15–22. doi: 10.1016/j.jaut.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 57.Yang S, Yang Y. Downregulation of microRNA221 decreases migration and invasion in fibroblastlike synoviocytes in rheumatoid arthritis. Mol Med Rep. 2015;12:2395–401. doi: 10.3892/mmr.2015.3642. [DOI] [PubMed] [Google Scholar]
- 58.Stuhlmuller B, Kunisch E, Franz J, Martinez-Gamboa L, Hernandez MM, Pruss A, et al. Detection of oncofetal h19 RNA in rheumatoid arthritis synovial tissue. Am J Pathol. 2003;163:901–11. doi: 10.1016/S0002-9440(10)63450-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mu N, Gu J, Huang T, Zhang C, Shu Z, Li M, et al. A novel NF-kappaB/YY1/microRNA-10a regulatory circuit in fibroblast-like synoviocytes regulates inflammation in rheumatoid arthritis. Sci Rep. 2016;6:20059. doi: 10.1038/srep20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Agirre X, Jimenez-Velasco A, San Jose-Eneriz E, Garate L, Bandres E, Cordeu L, et al. Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol Cancer Res. 2008;6:1830–40. doi: 10.1158/1541-7786.MCR-08-0167. [DOI] [PubMed] [Google Scholar]
- 61.Pandis I, Ospelt C, Karagianni N, Denis MC, Reczko M, Camps C, et al. Identification of microRNA-221/222 and microRNA-323-3p association with rheumatoid arthritis via predictions using the human tumour necrosis factor transgenic mouse model. Ann Rheum Dis. 2012;71:1716–23. doi: 10.1136/annrheumdis-2011-200803. [DOI] [PubMed] [Google Scholar]
- 62.Zhao GY, Cai CK, Yang TT, Qiu XC, Liao B, Li W, et al. MicroRNA-221 Induces Cell Survival and Cisplatin Resistance through PI3K/Akt Pathway in Human Osteosarcoma. Plos One. 2013;8 doi: 10.1371/journal.pone.0053906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garofalo M, Quintavalle C, Romano G, Croce CM, Condorelli G. miR221/222 in Cancer: Their Role in Tumor Progression and Response to Therapy. Curr Mol Med. 2012;12:27–33. doi: 10.2174/156652412798376170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gottardo F, Liu CG, Ferracin M, Calin GA, Fassan M, Bassi P, et al. Micro-RNA profiling in kidney and bladder cancers. Urol Oncol. 2007;25:387–92. doi: 10.1016/j.urolonc.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 65.Deng B, Wang B, Fang J, Zhu X, Cao Z, Lin Q, et al. MiRNA-203 suppresses cell proliferation, migration and invasion in colorectal cancer via targeting of EIF5A2. Sci Rep. 2016;6:28301. doi: 10.1038/srep28301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang N, Liang H, Zhou Y, Wang C, Zhang S, Pan Y, et al. miR-203 suppresses the proliferation and migration and promotes the apoptosis of lung cancer cells by targeting SRC. PLoS One. 2014;9:e105570. doi: 10.1371/journal.pone.0105570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang X, Sun Y, Han S, Zhu W, Zhang H, Lian S. MiR-203 inhibits melanoma invasive and proliferative abilities by targeting the polycomb group gene BMI1. Biochem Biophys Res Commun. 2015;456:361–6. doi: 10.1016/j.bbrc.2014.11.087. [DOI] [PubMed] [Google Scholar]
- 68.Huh YH, Lee G, Song WH, Koh JT, Ryu JH. Crosstalk between FLS and chondrocytes is regulated by HIF-2alpha-mediated cytokines in arthritis. Exp Mol Med. 2015;47:e197. doi: 10.1038/emm.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong BK, You S, Yoo SA, Park D, Hwang D, Cho CS, et al. MicroRNA-143 and -145 modulate the phenotype of synovial fibroblasts in rheumatoid arthritis. Exp Mol Med. 2017;49:e363. doi: 10.1038/emm.2017.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–70. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 72.Michael MZ, O’Connor SM, Pellekaan NGV, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–91. [PubMed] [Google Scholar]
- 73.Ntougkos E, Chouvardas P, Roumelioti F, Ospelt C, Frank-Bertoncelj M, Filer A, et al. Genomic Responses of Mouse Synovial Fibroblasts During Tumor Necrosis Factor-Driven Arthritogenesis Greatly Mimic Those in Human Rheumatoid Arthritis. Arthritis Rheumatol. 2017;69:1588–600. doi: 10.1002/art.40128. [DOI] [PubMed] [Google Scholar]
- 74.Whitaker JW, Shoemaker R, Boyle DL, Hillman J, Anderson D, Wang W, et al. An imprinted rheumatoid arthritis methylome signature reflects pathogenic phenotype. Genome Med. 2013;5:40. doi: 10.1186/gm444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dasuri K, Antonovici M, Chen K, Wong K, Standing K, Ens W, et al. The synovial proteome: analysis of fibroblast-like synoviocytes. Arthritis Res Ther. 2004;6:R161–8. doi: 10.1186/ar1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bo GP, Zhou LN, He WF, Luo GX, Jia XF, Gan CJ, et al. Analyses of differential proteome of human synovial fibroblasts obtained from arthritis. Clin Rheumatol. 2009;28:191–9. doi: 10.1007/s10067-008-1013-y. [DOI] [PubMed] [Google Scholar]
- 77.Zhang H, Fan LY, Zong M, Sun LS, Lu L. Proteins related to the functions of fibroblast-like synoviocytes identified by proteomic analysis. Clin Exp Rheumatol. 2012;30:213–21. [PubMed] [Google Scholar]
- 78.Katano M, Kurokawa MS, Matsuo K, Masuko K, Suematsu N, Okamoto K, et al. Phosphoproteome analysis of synoviocytes from patients with rheumatoid arthritis. Int J Rheum Dis. 2017 doi: 10.1111/1756-185X.12997. [DOI] [PubMed] [Google Scholar]
- 79.Wang JG, Xu WD, Zhai WT, Li Y, Hu JW, Hu B, et al. Disorders in angiogenesis and redox pathways are main factors contributing to the progression of rheumatoid arthritis: a comparative proteomics study. Arthritis Rheum. 2012;64:993–1004. doi: 10.1002/art.33425. [DOI] [PubMed] [Google Scholar]
- 80.You S, Yoo SA, Choi S, Kim JY, Park SJ, Ji JD, et al. Identification of key regulators for the migration and invasion of rheumatoid synoviocytes through a systems approach. Proc Natl Acad Sci U S A. 2014;111:550–5. doi: 10.1073/pnas.1311239111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ai R, Whitaker JW, Boyle DL, Tak PP, Gerlag DM, Wang W, et al. DNA Methylome Signature in Synoviocytes From Patients With Early Rheumatoid Arthritis Compared to Synoviocytes From Patients With Longstanding Rheumatoid Arthritis. Arthritis Rheumatol. 2015;67:1978–80. doi: 10.1002/art.39123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 83.Young SP, Kapoor SR, Viant MR, Byrne JJ, Filer A, Buckley CD, et al. The impact of inflammation on metabolomic profiles in patients with arthritis. Arthritis Rheum. 2013;65:2015–23. doi: 10.1002/art.38021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gobelet C, Gerster JC. Synovial fluid lactate levels in septic and non-septic arthritides. Ann Rheum Dis. 1984;43:742–5. doi: 10.1136/ard.43.5.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mobasheri A, Rayman MP, Gualillo O, Sellam J, van der Kraan P, Fearon U. The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2017;13:302–11. doi: 10.1038/nrrheum.2017.50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.