

Abstract
Objective
To explore the role of lysosomal acid lipase (LAL) in macrophage cholesterol efflux and whole body reverse cholesterol transport (RCT).
Approach and Results
Immortalized peritoneal macrophages from lal−/− mice showed reduced expression of ABCA1 and ABCG1, reduced production of the regulatory oxysterol 27-hydroxycholesterol, and impaired suppression of cholesterol synthesis upon exposure to acetylated LDL (acLDL) when compared to lal+/+ macrophages. LAL-deficient mice also showed reduced hepatic ABCG5 and ABCG8 expression compared to lal+/+ mice. LAL-deficient macrophages loaded with [3H]-cholesteryl oleate-labeled acLDL showed impaired efflux of released [3H]-cholesterol to apolipoprotein A-I (apoA-I), with normalization of [3H]-cholesteryl ester levels and partial correction of ABCA1 expression and cholesterol efflux to apoA-I when treated with exogenous recombinant human LAL. LAL-deficient mice injected intraperitoneally with lal−/− macrophages cholesterol loaded and labeled in the same way exhibited only 1.55 ± 0.35 % total injected [3H]-cholesterol counts appearing in the feces over 48 h (n=30), compared to 5.38 ± 0.92 % in lal+/+ mice injected with labeled lal+/+ macrophages (n=27), p<0.001. To mimic the therapeutic condition of delivery of supplemental LAL in vivo, injection of labeled lal−/− macrophages into lal+/+ mice resulted in a significant increase in RCT (2.60 ± 0.46 % of 3H-cholesterol counts in feces at 48 hr (n=19), p<0.001 when compared to injection into lal−/− mice.
Conclusions
These results indicate a critical role for LAL in promoting both macrophage and whole body reverse cholesterol transport, and the ability of supplemental LAL to be taken up and correct RCT in vivo.
Keywords: lysosomal acid lipase, ATP-binding cassette transporter 1, macrophages, cholesterol, high density lipoprotein, reverse cholesterol transport
Subject Codes: 90, 112, 142, 143, 145
Introduction
Retention and uptake of low density (LDL) and other apolipoprotein B-containing lipoproteins by cells in the artery wall is the key driver of atherosclerosis development.1 Lipoprotein cholesterol ingested by artery wall cells including macrophages is primarily in the ester form, which must be hydrolyzed in and released from lysosomes to allow eventual removal of the excess cholesterol from cells. The sole lysosomal enzyme known to carry out this function is lysosomal acid lipase (LAL), encoded by the gene LIPA.2 The critical importance of LAL is indicated by the lethality of complete LAL deficiency (Wolman disease), and by the liver disease, lipoprotein abnormalities and premature atherosclerosis seen individuals with subtotal LAL deficiency (Cholesteryl Ester Storage Disease, CESD).3, 4 Modification of LAL by mannose-6-phosphate allows the uptake of exogenous LAL by cells and targeting to lysosomes,5 where it has been shown to correct the consequences of LAL deficiency at the cellular level.6, 7 Recombinant human LAL delivered intravenously to humans is lifesaving in Wolman Disease,8 and has been shown to rapidly correct the liver and lipoprotein abnormalities in individuals with CESD,9 indicating its efficient uptake in tissues from the circulation.
We previously reported that LAL deficiency in human skin fibroblasts leads to impaired upregulation of ABCA1 and ABCG1, two mediators of cholesterol efflux and high density lipoprotein (HDL) formation, following cholesterol loading, and correction of these defects following delivery of exogenous LAL to the cells.7 Although there is some suggestion of benefit of delivery of exogenous LAL in reducing atherosclerosis,10 the mechanism of this is not understood, including the role of LAL in promoting cholesterol efflux from macrophages and whole-body reverse cholesterol transport (RCT). Different from our results using LAL-deficient fibroblasts, a recent study reported macrophages generated from human pluripotent stem cells and lacking LIPA expression had a similar upregulation of ABCA1 in response to acetylated LDL loading as cells expressing LIPA.11 In the current studies we have utilized immortalized peritoneal macrophages from lal+/+ and lal−/− mice to study the effect of LAL deficiency and replacement on cholesterol metabolism in cultured macrophages, and the role of LAL in promoting whole body RCT from lipoprotein cholesteryl esters in macrophages to the plasma, liver and feces of lal+/+ and lal−/− mice. Our results indicate a major role for LAL in the regulation of macrophage and whole body RCT and the ability of supplemental LAL to correct these defects in LAL deficiency.
Materials and Methods
The authors declare that all supporting data are available within the article and its online supplement or available from the corresponding author upon request. Materials and additional Methods are available in the online-only Data Supplement.
Preparation of 3H-cholesteryl-oleate labeled acetylated LDL and isolation of apolipoprotein A-1 from plasma
LDL was isolated from pooled plasma from fasting, healthy donors by density gradient ultracentrifugation.12 Radiolabeling of LDL with 3H-cholesteryl oleate was performed as previously described,13 then acetylated according to the methods of Basu et al.14 Briefly, an equal volume of 3H-cholesteryl-oleate labeled LDL in saline solution (typically 4–5 ml) was mixed with an equal volume of a saturated sodium acetate solution and 1.5μl acetic anhydride per mg acLDL protein was added in 1μl aliquots over 1 hour, while stirring on ice and then dialyzed extensively in a saline-EDTA solution. Apolipoprotein A-1 (apoA-I) was purified from human plasma using Q-Sepharose Fast Flow chromatography as described.15
LAL-deficient mice
LAL null mice (lal−/−) were generated on a CF-1/129Sv genetic background as previously described.16
Cell Culture
Mouse wild-type (lal+/+) or LAL knockout mice (lal−/−) immortalized peritoneal macrophages were isolated by peritoneal lavage and engineered to have temperature-sensitive expression of SV40 large T-antigen under an IFN-γ inducible promoter, using methods described in Castoreno et al.17
Cholesterol efflux assay
Radiolabelled acLDL loading of macrophages and efflux to apoA-I were performed as described in the online only Data Supplement. Cellular lipids were separated by thin layer chromatography. Unesterified cholesterol (UC) and cholesteryl ester (CE) spots were located on plates using unlabeled carrier lipids stained with iodine gas, and radioactivity was quantified by liquid scintillation counting (LSC) 18 as previously described.7, 19 Medium UC and cell UC and CE were calculated as percent of total 3H-sterol (cell + medium).
Mevalonolactone incorporation assay
Using radiolabeled mevalonolactone, radioactivity in newly synthesized cholesterol was quantified by LSC as previously described 20 and normalized to cell proteins.
Measurement of 27-hydroxycholesterol production
The mass of 27-hydroxycholesterol (ng) was determined by HPLC tandem mass spectrometry by modification of a previously described method.21
Macrophage reverse cholesterol transport
Methods to measure macrophage to feces RCT were adapted from previous publications22–24 as described in the online only Data Supplement.
Statistical Analysis
Results were analyzed using GraphPad Prism version 5.0 for statistical significance between treatment groups. The normality of data for figures was determined by performing a D’Agostino & Pearson omnibus normality test (alpha=0.05) and by visual inspection of normality using Q-Q plots. Normally distributed data was analyzed using parametric tests, either a one way ANOVA with Bonferroni post-hoc comparisons or a Students T-test, as indicated in the figure legends. Data not normally distributed was analyzed using non-parametric tests, either a Kruskal-Wallis test with Dunn’s comparisons or a Mann Whitney test, as indicated in the figure legends. A p-value <0.05 was considered significant.
Results
ABCA1 expression and cholesterol efflux are impaired in macrophages lacking LAL and corrected by addition of exogenous LAL
Macrophages are a primary site of cholesterol deposition in atherosclerotic plaque, and the level of ABCA1 expression is the rate-limiting determinant of removal of excess cholesterol by apolipoprotein A-I (apoA-I) from these cells.25 To determine the role of LAL in regulating ABCA1 expression and other determinants of macrophage cholesterol efflux, we used immortalized peritoneal macrophages obtained from wild type (lal+/+) and LAL-deficient (lal−/−) mice. In contrast to the robust increase in ABCA1 expression in response to incubating lal+/+ macrophages with acLDL, lal−/− macrophages exhibited significantly reduced basal and acLDL-stimulated increase in ABCA1 mRNA and protein levels (Figure 1A and 1B). Incubation with recombinant human LAL (rhLAL) 1 hr prior to and during acLDL loading for 24 hours resulted in a significant increase in ABCA1 mRNA and protein levels in lal−/− macrophages (Figures 1A–1C). This is consistent with the role of LAL in hydrolyzing lipoprotein cholesteryl esters and the subsequent release of this cholesterol from lysosomes driving ABCA1 expression.7 In contrast, addition of supplemental rhLAL to lal+/+ macrophages stimulated no further increase in ABCA1 mRNA (Figure 1A) or protein (Figure 1C).
Figure 1. ABCA1 mRNA and protein are reduced in lal−/− mouse peritoneal macrophages and partially rescued by rhLAL.

Wild-type (lal+/+) and LAL knockout (lal−/−) mouse immortalized peritoneal macrophage cell lines were grown to confluence and supplemented without (-) or with (+) 50 μg/ml acetylated LDL (acLDL) for 24 hours. Some dishes of lal−/− cells were also treated with 5, 10 or 20 μg/ml of rhLAL 1 hour prior to and during acLDL loading. A. ABCA1 mRNA expression in the absence or presence of acLDL and 20 μg/ml rhLAL was measured by quantitative real time PCR, corrected for the housekeeping gene m-cyclophilin, and normalized relative to the lal+/+ control. B. ABCA1 protein levels were resolved by SDS-PAGE, detected by Western blotting using polyclonal antibodies against ABCA1 or protein disulfide isomerase (PDI) loading control, and normalized relative to the lal+/+ control. Error bars are shown as standard error of the mean (SEM) from (A) 6 or (C) 8 experiments using ± 20 μg/ml rhLAL (*p<0.05, Kruskal-Wallis test with Dunn’s comparisons, significantly different from lal+/+ with no acLDL, unless otherwise indicated).
Cholesterol efflux from macrophages is also mediated by ABCG1, apolipoprotein E (apoE), and scavenger receptor class B type 1 (SR-BI). Similar to ABCA1, lal−/− macrophages showed significantly lower ABCG1 mRNA than lal+/+ macrophages in the basal state and following acLDL loading (Figure 2). ApoE mRNA showed a non-significant trend to higher levels in lal−/− compared to lal+/+ macrophages in both the basal and acLDL-loaded conditions. No significant differences were seen in SR-BI mRNA in the basal condition or following acLDL loading of lal+/+ and lal−/− macrophages.
Figure 2. ABCG1 but not ApoE or SR-B1 expression is reduced in lal−/− macrophages.

Macrophages were grown in culture and incubated with or without 50 μg/ml acetylated LDL (acLDL) for 24 hours. Cell lysates were collected and mRNA levels were analyzed by quantitative real-time PCR as in Figure 1A (corrected for m-cyclophilin), using primers against ATP-binding cassette transporter G1 (ABCG1), apolipoprotein E (ApoE) or scavenger receptor B1 (SR-B1). Error bars are shown as SEM from 5 experiments (*p<0.05, Kruskal-Wallis test with Dunn’s comparisons, significantly different from lal+/+ with or without acLDL unless otherwise indicated for ABCG1 and ApoE, and one-way ANOVA with Bonferroni comparisons for SR-B1).
The role of LAL in promoting removal of excess cholesterol from macrophage foam cells was then determined following cholesterol loading with acLDL and incubation with apoA-I to determine ABCA1-dependent cholesterol efflux. Cells of both genotypes were loaded with 3H-cholesteryl oleate-labeled acLDL for 24 hours and 3H-UC counts in the medium and 3H-CE and 3H-UC counts in cells were quantified following a 24 hour incubation with or without 10 μg/ml apoA-I (Figure 3). Elevated levels of 3H-CE and low 3H-UC in lal−/− macrophages prior to addition of apoAI were recapitulated in lal+/+ macrophages treated with the lysosomal inhibitor chloroquine, confirming the dependence of this observation in lal−/− macrophages on deficiency of LAL activity (Supplemental Figure I). Lal+/+ macrophages exhibited a high level of efflux to the medium without addition of apoA-I (~15%), likely due to macrophage-secreted apoE (Supplemental Figure II) promoting cholesterol efflux, as previously reported,26, 27 and which is ABCA1-dependent.28 Addition of apoA-I to medium significantly further increased efflux in lal+/+ macrophages (Figure 3A). Efflux to medium alone was reduced to almost half of wild-type levels in lal−/− macrophages (Figure 3A). This is consistent with both the reduced cellular pool of 3H-UC available for efflux (Figure 3C) and reduced ABCA1 (Figure 1), both reducing the efflux potential of macrophage-secreted apoE (Supplemental Figure II). For the same reasons, addition of apoA-I did not significantly increase efflux from lal−/− macrophages (Figure 3A). Treatment of lal−/− cells with 10 μg/ml rhLAL significantly increased cholesterol efflux to apoA-I (Figure 3A), consistent with the upregulation of ABCA1 in these cells (Figure 1) and reduced availability of UC for efflux. As expected, lal−/− macrophages showed increased cellular 3H-CE and reduced 3H-UC when compared to lal+/+ macrophages. CE hydrolysis in lal−/− macrophages was corrected with a consequent increase in 3H-UC following addition of rhLAL (Figure 3B and C). This result is consistent with the ability of exogenous LAL to be taken up, delivered to lysosomes and mediate neutral lipid hydrolysis in both cultured cells6, 7 and in vivo.9
Figure 3. Rescue of impaired apoA-I-dependent cholesterol efflux in lal−/− macrophages by treatment with rhLAL.
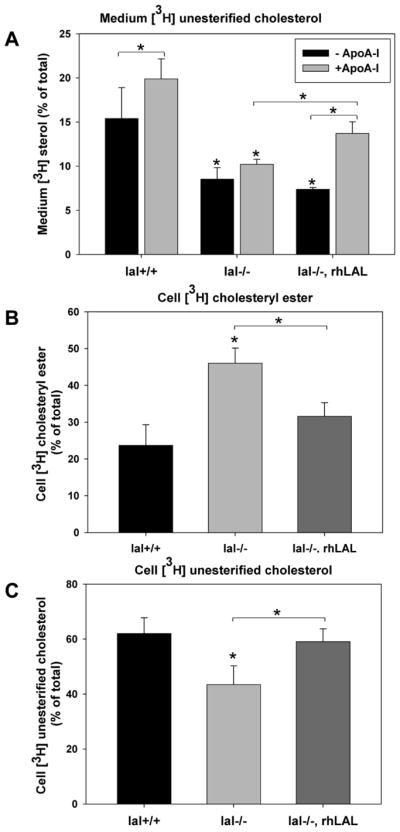
Immortalized mouse peritoneal macrophages were grown to confluence and loaded with 50 μg/ml 3H-CE-labeled acetylated LDL for 24 hours and then washed and incubated in the presence (+ ApoA-I) or absence (− ApoA-I) of 10μg/ml purified apolipoprotein A–I for 24 hours. Where indicated 20 μg/ml rhLAL was added 1 hour prior to and during acLDL loading. A. Medium was removed and 3H-cholesterol was quantified by LSC. B, C. From cells that were not incubated with apoA-I, cellular lipids were extracted, cellular 3H-cholesteryl esters and 3H-unesterified cholesterol were separated by thin layer chromatography and counted by LSC. Uptake of 3H-CE-acLDL was 2 times higher in lal−/− than lal+/+ macrophages (73462±8662 versus 36447±4823 cpm/mg cell protein, respectively; avg±SEM, 3 experiments with n=3 replicates each). Measurements are expressed as the percent of total 3H counts (medium + cell). Error bars are shown as SEM from 7 experiments (*p<0.05, One-way ANOVA with Bonferroni comparisons, significantly different from lal+/+, unless otherwise indicated).
Dysregulation of post-lysosomal cholesterol metabolism in lal−/− macrophages and correction by exogenous LAL
Reduced release of lipoprotein-derived cholesterol from lysosomes in LAL-deficient human fibroblasts has previously been shown to impair downstream regulatory events including suppression of cholesterol synthesis29 and upregulation of ABCA1 expression.7 To further investigate the importance of LAL in regulating cholesterol homeostasis in macrophages we measured production of 27-hydroxycholesterol, a key endogenous LXR agonist oxysterol upregulating ABCA1 expression in the artery wall,30 and new cholesterol synthesis from the cholesterol precursor mevalonate, before and after addition of acLDL. Formation of 27-hydroxycholesterol was similar in both lal+/+ and lal−/− macrophages in the basal state (0.37±0.06 and 0.54±0.08 ng/mg cell protein respectively, mean±SEM of 7 experiments, p=0.11). 27-hydroxycholesterol synthesis rose significantly in lal+/+ but was unaffected in lal−/− macrophages following incubation with acLDL (Figure 4A). Addition of rhLAL resulted in a similar fold increase in 27-hydroxycholesterol synthesis in lal−/− as seen in lal+/+ macrophages treated with acLDL. Incubation with acLDL for 24 hr resulted in a modest but significant reduction of new cholesterol synthesis from mevalonate in lal+/+ macrophages (Figure 4B). Lal−/− macrophages showed significantly higher new cholesterol synthesis in the basal condition and after loading with acLDL compared to lal+/+ macrophages. Addition of rhLAL to lal−/− macrophages reduced their synthesis of new cholesterol down to a level similar to lal+/+ cells (Figure 4B). These two results confirm the dysregulation of cholesterol homeostasis in lal−/− macrophages, and provide a likely reason for impaired upregulation of ABCA1 following addition of acLDL, failure to increase formation of the key LXR agonist 27-hydroxycholesterol in the presence of LAL deficiency. Supplementation of exogenous rhLAL corrects these pathways as reflected in the increased ABCA1 expression, correction of cholesterol efflux to apoA-I by lal−/− macrophages, and suppression of de novo cholesterol synthesis.
Figure 4. Impaired regulation of oxysterol production and cholesterol synthesis in lal−/− macrophages and correction with rhLAL.

A. Immortalized mouse peritoneal macrophages were grown to confluence and treated ± 50 μg/mL acLDL for 24 hrs. In addition, lal−/− macrophages were also pretreated with 10 μg/mL rhLAL 1 hour prior to and during acLDL loading. Cell monolayers were then washed and equilibrated for an additional 24 hrs in medium alone. Following lipid extraction, the mass of 27-hydroxycholesterol was determined by HPLC tandem mass spectrometry and normalized to cell proteins. Results are indicated as fold change relative to non–lipid loaded controls. B. Immortalized mouse peritoneal macrophages were grown to confluence and loaded with 0.5 μCi/mL 3H-Mevalonolactone ± 50 μg/mL acLDL. In addition, LAL−/− macrophages were pretreated with 10 μg/mL rhLAL 1 hour prior to and during acLDL loading. After 24 hours cell monolayers were washed and lipids extracted by thin layer chromatography. New cholesterol synthesized was determined by LSC and normalized to cell proteins. Results are indicated as fold change relative to the lal+/+ control. Error bars are shown as SEM from (A) 6 experiments (n=3 replicates each) or (B) 3 experiments (n=3 replicates each) [*p<0.05, Kruskal-Wallis test with Dunn’s comparisons (A) or one-way ANOVA with Bonferroni comparisons (B), significantly different from the lal+/+ control, unless otherwise indicated].
Expression of genes affecting hepatic and fecal cholesterol secretion and efflux in lal-deficient mice
To further investigate the role of LAL in regulating genes involved in RCT, mRNA levels for ABCA1, ABCG5, ABCG8 and SRBI were determined in livers isolated from lal+/+ and lal−/− mice. Hepatic ABCA1 and SR-BI mRNA levels were not significantly different between genotypes (Figure 5A), consistent with lack of LXR responsiveness of these genes in mouse liver31. Hepatocyte LXR-dependent genes ATP-binding cassette transporter G5 and G8 (ABCG5/G8)32 showed significantly lower mRNA levels in lal−/− compared to lal+/+ mouse liver (Figure 5A), as previously reported33; these results were confirmed at the protein level (Figure 5B). No sex differences were seen in expression of these genes in both lal+/+ or lal−/− mice, consistent with previous studies showing lack of major differences between male and female lal−/− mice.33, 34 Despite normal hepatic mRNA levels, levels of ABCA1 protein were lower in the livers of lal−/− mice (Figure 5B); ABCA1 protein was also lower in the spleen, kidney, as well as skin fibroblasts of lal−/− compared to lal+/+ mice (Supplemental Figure III). Intestinal ABCA1 protein levels were not found to be reduced in lal−/− compared to lal+/+ mice (data not shown); similar results have been found previously, depending on the segment of intestine assessed (S. Turley, personal communication). While lal−/− mouse liver Cyp7A1 mRNA was found to be reduced, we found no reduction in Cyp7A1 protein levels in lal−/− when compared to livers of lal+/+ mice (data not shown). This finding is consistent with equivalent rates of bile acid secretion in lal+/+ and lal−/− mice reported previously.33
Figure 5. Differential RCT gene expression in lal+/+ and lal−/− mouse liver.

A. Livers were dissected from mice, homogenized, and RNA was extracted. Quantitative real-time PCR was performed using primers against ATP-binding cassette transporters A1, G5, G8, and SR-B1 as described in methods. mRNA levels were corrected for the housekeeping gene m-cyclophilin. Error bars are shown as SEM (*p<0.05, Student T-test, significantly different compared to lal+/+, n = 3 males and 3 females for each group.) B. Livers from mice were homogenized in maltoside buffer with protease inhibitors and proteins resolved in SDS-PAGE gels followed by immunoblotting for the indicated proteins. Results are representative of 3 individual mice, n=2 experiments.
Dependence of macrophage to feces RCT on LAL
Lastly we used an established model24 to determine the role of LAL in promoting transport of cholesterol from macrophages to feces in lal+/+ or lal−/− mice. Lal+/+ or lal−/− macrophages were grown in monoculture and loaded with radiolabeled 3H-CE-acLDL for 24 hours, suspended in saline and injected intraperitoneally into mice of the same genotype. A similar level of total 3H-CE radiolabel was injected into each mouse. Lal−/− mice injected with lal−/− macrophages showed a significant reduction in 3H-cholesterol in plasma 24 and 48 hours after injection when compared to lal+/+ mice injected with lal+/+ macrophages, (0.61 ± 0.06% of total 3H counts injected at 48 hours, n= 30 lal−/− mice compared to 3.06 ± 0.32% of injected label at 48 hours, n= 27 lal+/+ mice) (Figure 6A). Despite this, no significant difference in liver 3H-sterol was observed between the lal+/+ into +/+ vs. lal−/− into −/− mice, when expressed as either percentage of total cpm injected (Figure 6B) or cpm/mg liver protein (not shown). This likely reflects accumulation of 3H-cholesterol that does reach the liver in lal−/− mice due to decreased expression of ABCG5 and ABCG8 (Figure 5) and therefore reduced excretion from the liver into bile in these mice. Consistent with this, over 48 hours, lal−/− mice injected with lal−/− macrophages showed reduced appearance of 3H-cholesterol in feces (1.55 ± 0.35% of total CPM injected) compared to the lal+/+ macrophage into lal+/+ mouse group (5.38 ± 0.92% of injected label) (Figure 6C). In order to mimic the therapeutic condition of uptake of supplemental LAL by LAL-deficient macrophages, 3H-CE-labeled lal−/− macrophages were injected into lal+/+ mice. This resulted in significantly increased appearance of 3H-sterol in plasma (1.36 ± 0.08%) and feces (2.60 ± 0.46%) (n=19 mice) at 48 hours when compared to lal−/− macrophages injected into lal−/− mice (Figures 6A, C). 3H-sterol levels in the liver were significantly lower in lal+/+ mice injected with lal−/− macrophages (5.29 ± 0.60%, n=19) compared to lal−/− mice injected with lal−/− macrophages (16.54 ± 3.40%, n= 30) (Figure 6B). These results indicate that LAL from the lal+/+ mice was taken up by the injected lal−/− macrophages, with partial correction of 3H-CE hydrolysis and efflux of 3H-cholesterol from these macrophages, consistent with our in vitro rhLAL treatment experiments (Figure 3A and B). The reduced hepatic 3H-sterol in this condition compared to lal+/+ macrophages injected into lal+/+ mice is likely a reflection of the intermediate level of 3H-cholesterol efflux from the lal−/− macrophages injected into lal+/+ mice but efficient delivery of this cholesterol to the feces in the lal+/+ liver expressing normal levels of ABCG5 and ABCG8, also resulting in significantly higher fecal 3H-sterols than in lal−/− mice injected with lal−/− macrophages (Figure 6C).
Figure 6. Impaired macrophage reverse cholesterol transport in LAL knockout mice.

Macrophages (lal+/+ or lal−/−) were loaded with 50 μg/ml 3H-CE-acLDL for 24 hours and injected into lal+/+ or lal−/− mice as indicated. A. At 24 and 48 hrs post-injection blood was drawn by cardiac puncture into tubes containing anticoagulant and centrifuged at low speed to collect plasma. B. Liver samples were collected at 48 hours C. Feces were collected and combined during the 48 hour time course. Liver (B) and feces (C) were homogenized in water or a 1:1 water/ethanol mixture, respectively. 3H-Sterols in aliquots of plasma or homogenate were quantified by LSC. The lal+/+ into +/+ group consisted of 27 mice (n=12 males, n= 15 females), the lal−/− into −/− group consisted of 30 mice (n=16 males, n=14 females), and lal−/− into +/+ group consisted of 19 mice (n=8 males, n=11 females). Data are expressed as percent of the total CPM injected per mouse. Error bars are shown as SEM (*p<0.05, Kruskal-Wallis test with Dunn’s comparisons, significantly different compared to lal+/+ into lal+/+, unless otherwise indicated.)
Discussion
Lysosomal acid lipase is the sole lysosomal enzyme mediating hydrolysis of cholesteryl esters derived from endocytosed lipoproteins and also stored cholesteryl esters delivered to lysosomes by lipophagy. In the current studies we provide evidence for a critical role of LAL in driving expression of ABCA1 in macrophages and multiple tissues, cholesterol efflux from cells, and excretion of cholesterol from the liver into the feces. In addition, we show that reduced cholesterol flux from lysosomes due to LAL deficiency results in impaired oxysterol generation and failure to suppress de novo cholesterol synthesis in macrophages following cholesterol loading with acLDL, and correction of these defects following treatment with supplemental LAL. These results extend our previous findings of the impaired ABCA1 expression and cholesterol efflux machinery in fibroblasts of patients with LAL deficiency7, and indicate a major role for LAL as a regulator of macrophage cholesterol homeostasis and whole body reverse cholesterol transport.
Using immortalized peritoneal macrophages, we found reduced basal and lipoprotein-stimulated ABCA1 and ABCG1 expression and reduced apoAI-dependent cholesterol efflux in lal−/− compared to lal+/+ macrophages, with partial correction of ABCA1 expression and cholesterol efflux from these cells by addition of rhLAL (Figures 1–3). rhLAL supplementation resulted in normalization of elevated radiolabeled cholesteryl esters and an increase in free cholesterol in lal−/− macrophages. This correlated directly with correction of production of 27-hydroxycholesterol, produced in mitochondria by sterol-27-hydroxylase and a necessary driver of ABCA1 expression in macrophages via activation of the nuclear receptor liver X receptor on the promoter region of ABCA1,35 in lal−/− macrophages. These results provide evidence that LAL is critical in regulating both the expression of ABCA1 by generating lysosomally-derived cholesterol for 27-hydroxycholesterol production, and by providing a substrate for ABCA1-dependent cholesterol efflux from macrophages. We also found an inability of acLDL loading to suppress new cholesterol synthesis in lal−/− macrophages, and correction of this defect following addition of rhLAL. These results are consistent with a similar inability of LDL loading to suppress new cholesterol synthesis in LAL-deficient human fibroblasts,29 and further demonstrate the critical role of lysosomally-derived cholesterol in regulating downstream cholesterol metabolism in cells.
Further evidence of impaired LXR-dependent gene expression in LAL deficiency is the low level of ABCG5 and ABGC8 mRNA and protein, previously shown to be LXR-responsive,32 seen in lal−/− compared to lal+/+ mouse liver (Figure 5) and as previously described at the mRNA level.33 We did not find reduced ABCA1 mRNA in lal−/− mouse liver, consistent with a previous report33 and the known lack of LXR dependence of hepatic ABCA1 expression in mice.31 Reduced ABCA1 protein was seen in the liver of lal−/− mice, however, possibly due to the high level of Kupffer cells, a type of macrophage, in the liver of these mice,34 or possibly more rapid degradation of ABCA1 protein in the plasma membrane of liver cells in the highly fatty lal−/− mouse liver. The lack of significant reduction of plasma HDL-C in lal−/− mice,34 however, suggests that hepatic ABCA1, a major determinant of circulating HDL levels,36 is still functional in these mice.
In order to determine the role of LAL in promoting whole body RCT from macrophages to feces, we injected lal+/+ and lal−/− macrophages loaded with acLDL radiolabeled in the cholesterol moiety of CE intraperitoneally into mice of the same genotype. In this method the injected macrophages have been shown to remain in the peritoneal cavity over the duration of the experiment,24, 37 meaning cholesterol efflux from these cells is dependent on HDL and apoAI that diffuse into the peritoneal cavity from the circulation, and on LAL produced by the macrophages or other cells in this cavity or diffusing in from the circulation. All mice were injected with equivalent levels of labeled 3H-CE-acLDL in macrophages; based on a two-fold higher uptake of acLDL by lal−/− compared to lal+/+ macrophages (legend Figure 3) and an approximate 2-fold higher CE mass in lal−/− macrophages loaded in this way,38 normalization of injected 3H-CE into the mice provided an equivalent pool of macrophage CE mass available for hydrolysis by LAL, efflux and movement through the RCT pathway in all mice. Injection of lal−/− macrophages into lal−/− mice resulted in only 1/5th the level of 3H-cholesterol in plasma at 48 hrs compared to injection of labeled lal+/+ macrophages into lal+/+ mice (Figure 6A). Despite this, there was a similar level of 3H-cholesterol in the livers of lal−/− and lal+/+ mice at 48 hrs. Therefore, although efflux of cholesterol from the injected lal−/− macrophages is markedly reduced, 3H-cholesterol that is delivered to the livers of lal−/− mice is likely held up there due to reduced expression of ABCG5 and ABCG8 (Figure 5), necessary to transport hepatic cholesterol into bile,39 with subsequent reduced appearance of 3H-cholesterol in the feces of lal−/− mice (Figure 6C). Similar levels of SR-BI expression by livers of lal+/+ and lal−/− mice (Figure 5A) suggests accumulation of 3H-cholesterol in lal−/− mouse livers is not due to increased SR-BI-dependent hepatic uptake of 3H-cholesterol in these mice. The equivalent levels of bile acid excretion in lal−/− and lal+/+ mice previously reported by the Turley lab33 further suggests that accumulation of 3H-cholesterol in the liver of lal−/− mice is due to reduced expression of ABCG5 and ABCG8 and not reduced bile acid synthesis or secretion in these mice. Injection of lal−/− macrophages into the peritoneal cavity of lal+/+ mice, performed in order to mimic the therapeutic condition of providing supplemental LAL in human LAL deficiency, resulted in significantly higher appearance of 3H-cholesterol in plasma at 48 hrs compared to lal−/− mice injected with lal−/− macrophages. This effect can be assumed to be due to uptake by the lal−/− macrophages of LAL produced by intraperitoneal cells or diffusing into the peritoneal cavity of the lal+/+ mice. The significantly lower accumulation of 3H-cholesterol in the liver and increased appearance of cholesterol in the feces in this condition is a reflection of normal hepatic ABCG5 and ABCG8 expression in the lal++ mice, allowing efficient hepatic RCT of 3H-cholesterol once it is released by the lal−/− macrophages and delivered to the plasma and liver.
These findings have significant implications for the ability of supplemental LAL to correct RCT in humans with LAL deficiency. In addition to the marked reduction of hepatic fat content and liver transaminase levels seen within weeks of initiation of intravenous rhLAL treatment, this treatment significantly raises low HDL-C levels in these individuals.9 This finding is consistent with hepatic ABCA1, a major predictor of plasma HDL-C levels36 and an LXR-responsive gene in humans,40, 41 being corrected by rescue of hepatic LAL activity with this treatment. The ability of circulating LAL to diffuse into the artery wall would also be expected to correct macrophage LAL activity at least partially, and along with the increased plasma HDL-C level result in reduced arterial macrophage cholesterol accumulation and plaque burden. Correction of hepatic ABCG5 and ABCG8 expression with this treatment would also enhance RCT in LAL-deficient individuals.
We also found reduced ABCA1 protein levels in the spleen and kidney of lal−/− mice (Supplemental Figure III). While these findings may not directly impact the results of our RCT studies, reduced expression of ABCA1 in the spleen has been shown to enhance chemokine production by splenic macrophages,42 which could contribute to the atherogenic phenotype in LAL deficiency. Reduced renal ABCA1 expression could potentially exacerbate the increased catabolism of apoA1 seen in ABCA1 deficiency.43
In addition to reduced ABCA1 expression, reduced expression of ABCG1 would also reduce cholesterol efflux by macrophages, and would be expected to be corrected with LAL supplementation. ApoE and SR-BI expression were not reduced in lal−/− macrophages, consistent with previous reports of the lack of LXR-responsiveness of these genes in human and mouse macrophages.44–46 We observed a significant increase in ApoE mRNA expression in lal−/− cells treated with acLDL loading compared to untreated lal+/+ cells, possibly as an LXR-independent compensation to the cholesterol loading.47 Cholesterol efflux from lal−/− macrophages was nevertheless impaired (Figure 3A), consistent with the significantly reduced UC pool available for efflux (Figure 3C), and that apoE and SR-B1 are not able to compensate for the reduced activities of ABCA1 and ABCG1 caused by LAL deficiency.
Limitations of this study include the absence of a test of RCT from lal+/+ macrophages injected into lal−/− mice. While this condition would allow examination of the specific roles of macrophage and hepatic LAL in promoting RCT, it would not represent a relevant therapeutic condition where LAL is deficient in all cells of the body. The amount of LAL secreted by lal+/+ macrophages in the peritoneal cavity would not be expected to have a significant systemic effect, including in the liver, compared to LAL replacement delivered intravenously in humans.9 We also noted a significant level of residual hydrolysis of acLDL in lal−/− macrophages. This is apparently non-lysosomal, as incubation of lal−/− macrophages with chloroquine did not further reduce CE hydrolysis in the cells (Supplemental Figure I). Despite this finding, this level of hydrolysis did not appreciably affect ABCA1 regulation, further supporting the conclusion that release of lysosomally-derived UC is a key regulator of regulatory oxysterol production and ABCA1 expression, but not cholesterol derived from non-lysosomal CE hydrolysis.
A very recent publication has also found that knockdown of LIPA in macrophages derived from human pluripotent stem cells abolished apoAI-mediated efflux of cholesterol derived from 3H-CE-acLDL, but did not find a relationship between LIPA and expression of ABCA1 in these cells.11 The reasons for this difference from those in the current studies are not clear. Our results using human fibroblasts,7 immortalized mouse peritoneal macrophages and multiple tissues in lal−/− mice are consistent in showing a clear relationship between LAL activity and ABCA1 expression, and are also consistent with our previous findings of the critical role of lysosomally-derived cholesterol in regulating ABCA1 in Niemann Pick Disease Type C.20, 48
In conclusion, these results implicate LAL as a key regulator of macrophage cholesterol metabolism including expression of ABCA1 and ABCG1, and an essential driver of whole body RCT. Our results clearly demonstrate a beneficial role of LAL supplementation in a deficient state for transport of lipoprotein-derived cholesterol in peripheral lipid-loaded macrophages back to the liver for excretion. This suggests that treatment with rhLAL would increase reverse cholesterol transport and potentially reduce the elevated risk of premature atherosclerosis in LAL-deficient individuals.
Supplementary Material
Highlights.
Macrophage LXR-dependent genes ABCA1 and ABCG1 and hepatic ABCG5 and ABCG8 show reduced expression in lysosomal acid lipase (LAL)-deficient mice
LAL deficiency leads to impairment of post-lysosomal regulation of cholesterol metabolism in macrophages
Whole body reverse cholesterol transport is impaired in LAL-deficient mice, and partially corrected by supplementation with exogenous LAL
Acknowledgments
The authors would like to thank Julia Kong and Tatjana Bozin for helpful assistance, and Josephine Francis for creation of the graphical abstract.
Sources of Funding: Canadian Institutes for Health Research operating grant MOP 79532 and project grant PJT 148883 to GAF, National Institutes of Health operating grant HL087001 to HD and Heart and Stroke Foundation of Canada Doctoral Research Award to KLB.
Disclosures: GAF was a consultant to Alexion Pharmaceuticals, the maker of sebelipase alfa, a recombinant form of human LAL, in 2017. GAG was a consultant to Alexion Pharmaceuticals from 2015–2017. GAG and HD are inventors of patents related to the therapeutic uses of human LAL.
Abbreviations
- ABCA1
ATP-binding cassette transporter A1
- ABCG1
ATP-binding cassette transporter G1
- ABCG5
ATP-binding cassette transporter G5
- ABCG8
ATP-binding cassette transporter G8
- AcLDL
acetylated low density lipoprotein
- CE
cholesteryl ester
- CESD
cholesteryl ester storage disease
- HDL
high density lipoprotein
- LAL
lysosomal acid lipase
- LALD
lysosomal acid lipase deficiency
- LSC
liquid scintillation counting
- LXR
liver X receptor
- RCT
reverse cholesterol transport
- rhLAL
recombinant human lysosomal acid lipase protein
- SEM
standard error of the mean
- SR-B1
scavenger receptor class B type1
- UC
unesterified cholesterol
References
- 1.Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation. 2007;116:1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 2.Dubland JA, Francis GA. Lyosomal acid lipase: At the crossroads of normal and atherogenic cholesterol metabolism. Front Cell Dev Biol. 2015;3:1–11. doi: 10.3389/fcell.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernstein DL, Hulkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease. Journal of hepatology. 2013;58:1230–1243. doi: 10.1016/j.jhep.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 4.Grabowski GA, Du H, Charnas L. Lysosomal acid lipase deficiency: The wolman disease/cholesteryl ester storage disease spectrum. The Online Metabolic and Molecular Bases of Inherited Disease. 2016 [Google Scholar]
- 5.Du H, Levine M, Ganesa C, Witte DP, Cole ES, Grabowski GA. The role of mannosylated enzyme and the mannose receptor in enzyme replacement therapy. American journal of human genetics. 2005;77:1061–1074. doi: 10.1086/498652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown MS, Sobhani MK, Brunschede GY, Goldstein JL. Restoration of a regulatory response to low density lipoprotein in acid lipase-deficient human fibroblasts. The Journal of biological chemistry. 1976;251:3277–3286. [PubMed] [Google Scholar]
- 7.Bowden KL, Bilbey NJ, Bilawchuk LM, Boadu E, Sidhu R, Ory DS, Du H, Chan T, Francis GA. Lysosomal acid lipase deficiency impairs regulation of abca1 gene and formation of high density lipoproteins in cholesteryl ester storage disease. The Journal of biological chemistry. 2011;286:30624–30635. doi: 10.1074/jbc.M111.274381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones SA, Rojas-Caro S, Quinn AG, Friedman M, Marulkar S, Ezgu F, Zaki O, Gargus JJ, Hughes J, Plantaz D, Vara R, Eckert S, Arnoux JB, Brassier A, Le Quan Sang KH, Valayannopoulos V. Survival in infants treated with sebelipase alfa for lysosomal acid lipase deficiency: An open-label, multicenter, dose-escalation study. Orphanet journal of rare diseases. 2017;12:25. doi: 10.1186/s13023-017-0587-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burton BK, Balwani M, Feillet F, Baric I, Burrow TA, Camarena Grande C, Coker M, Consuelo-Sanchez A, Deegan P, Di Rocco M, Enns GM, Erbe R, Ezgu F, Ficicioglu C, Furuya KN, Kane J, Laukaitis C, Mengel E, Neilan EG, Nightingale S, Peters H, Scarpa M, Schwab KO, Smolka V, Valayannopoulos V, Wood M, Goodman Z, Yang Y, Eckert S, Rojas-Caro S, Quinn AG. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. The New England journal of medicine. 2015;373:1010–1020. doi: 10.1056/NEJMoa1501365. [DOI] [PubMed] [Google Scholar]
- 10.Du H, Schiavi S, Wan N, Levine M, Witte DP, Grabowski GA. Reduction of atherosclerotic plaques by lysosomal acid lipase supplementation. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:147–154. doi: 10.1161/01.ATV.0000107030.22053.1e. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Shi J, Hachet MA, Xue C, Bauer RC, Jiang H, Li W, Tohyama J, Millar J, Billheimer J, Phillips MC, Razani B, Rader DJ, Reilly MP. Crispr/cas9-mediated gene editing in human ipsc-derived macrophage reveals lysosomal acid lipase function in human macrophages-brief report. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:2156–2160. doi: 10.1161/ATVBAHA.117.310023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung BH, Wilkinson T, Geer JC, Segrest JP. Preparative and quantitative isolation of plasma lipoproteins: Rapid, single discontinuous density gradient ultracentrifugation in a vertical rotor. Journal of lipid research. 1980;21:284–291. [PubMed] [Google Scholar]
- 13.Sattler W, Stocker R. Greater selective uptake by hep g2 cells of high-density lipoprotein cholesteryl ester hydroperoxides than of unoxidized cholesteryl esters. Biochem J. 1993;294(Pt 3):771–778. doi: 10.1042/bj2940771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basu SK, Goldstein JL, Anderson GW, Brown MS. Degradation of cationized low density lipoprotein and regulation of cholesterol metabolism in homozygous familial hypercholesterolemia fibroblasts. Proc Natl Acad Sci U S A. 1976;73:3178–3182. doi: 10.1073/pnas.73.9.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boadu E, Bilbey NJ, Francis GA. Cellular cholesterol substrate pools for adenosine-triphosphate cassette transporter a1-dependent high-density lipoprotein formation. Curr Opin Lipidol. 2008;19:270–276. doi: 10.1097/MOL.0b013e3282feea99. [DOI] [PubMed] [Google Scholar]
- 16.Du H, Duanmu M, Witte D, Grabowski GA. Targeted disruption of the mouse lysosomal acid lipase gene: Long-term survival with massive cholesteryl ester and triglyceride storage. Human molecular genetics. 1998;7:1347–1354. doi: 10.1093/hmg/7.9.1347. [DOI] [PubMed] [Google Scholar]
- 17.Castoreno AB, Wang Y, Stockinger W, Jarzylo LA, Du H, Pagnon JC, Shieh EC, Nohturfft A. Transcriptional regulation of phagocytosis-induced membrane biogenesis by sterol regulatory element binding proteins. Proc Natl Acad Sci U S A. 2005;102:13129–13134. doi: 10.1073/pnas.0506716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Trotha KT, Heun R, Schmitz S, Lutjohann D, Maier W, Kolsch H. Influence of lysosomal acid lipase polymorphisms on chromosome 10 on the risk of alzheimer’s disease and cholesterol metabolism. Neuroscience letters. 2006;402:262–266. doi: 10.1016/j.neulet.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Francis GA, Oram JF, Heinecke JW, Bierman EL. Oxidative tyrosylation of hdl enhances the depletion of cellular cholesteryl esters by a mechanism independent of passive sterol desorption. Biochemistry. 1996;35:15188–15197. doi: 10.1021/bi9618169. [DOI] [PubMed] [Google Scholar]
- 20.Choi HY, Karten B, Chan T, Vance JE, Greer WL, Heidenreich RA, Garver WS, Francis GA. Impaired abca1-dependent lipid efflux and hypoalphalipoproteinemia in human niemann-pick type c disease. The Journal of biological chemistry. 2003;278:32569–32577. doi: 10.1074/jbc.M304553200. [DOI] [PubMed] [Google Scholar]
- 21.Burkard I, Rentsch KM, von Eckardstein A. Determination of 24s- and 27-hydroxycholesterol in plasma by high-performance liquid chromatography-mass spectrometry. Journal of lipid research. 2004;45:776–781. doi: 10.1194/jlr.D300036-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Naik SU, Wang X, Da Silva JS, Jaye M, Macphee CH, Reilly MP, Billheimer JT, Rothblat GH, Rader DJ. Pharmacological activation of liver x receptors promotes reverse cholesterol transport in vivo. Circulation. 2006;113:90–97. doi: 10.1161/CIRCULATIONAHA.105.560177. [DOI] [PubMed] [Google Scholar]
- 23.Wang MD, Franklin V, Sundaram M, Kiss RS, Ho K, Gallant M, Marcel YL. Differential regulation of atp binding cassette protein a1 expression and apoa-i lipidation by niemann-pick type c1 in murine hepatocytes and macrophages. The Journal of biological chemistry. 2007;282:22525–22533. doi: 10.1074/jbc.M700326200. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein a-i promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 25.Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, Maeda N, Parks JS. Increased cellular free cholesterol in macrophage-specific abca1 knock-out mice enhances pro-inflammatory response of macrophages. The Journal of biological chemistry. 2008;283:22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang WY, Gaynor PM, Kruth HS. Apolipoprotein e produced by human monocyte-derived macrophages mediates cholesterol efflux that occurs in the absence of added cholesterol acceptors. The Journal of biological chemistry. 1996;271:28641–28646. doi: 10.1074/jbc.271.45.28641. [DOI] [PubMed] [Google Scholar]
- 27.Smith JD, Miyata M, Ginsberg M, Grigaux C, Shmookler E, Plump AS. Cyclic amp induces apolipoprotein e binding activity and promotes cholesterol efflux from a macrophage cell line to apolipoprotein acceptors. The Journal of biological chemistry. 1996;271:30647–30655. doi: 10.1074/jbc.271.48.30647. [DOI] [PubMed] [Google Scholar]
- 28.Huang ZH, Fitzgerald ML, Mazzone T. Distinct cellular loci for the abca1-dependent and abca1-independent lipid efflux mediated by endogenous apolipoprotein e expression. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:157–162. doi: 10.1161/01.ATV.0000193627.12516.1d. [DOI] [PubMed] [Google Scholar]
- 29.Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. The Journal of biological chemistry. 1975;250:8487–8495. [PubMed] [Google Scholar]
- 30.Bjorkhem I, Andersson O, Diczfalusy U, Sevastik B, Xiu RJ, Duan C, Lund E. Atherosclerosis and sterol 27-hydroxylase: Evidence for a role of this enzyme in elimination of cholesterol from human macrophages. Proc Natl Acad Sci U S A. 1994;91:8592–8596. doi: 10.1073/pnas.91.18.8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang W, Ma Y, Jia L, Ioannou YA, Davies JP, Yu L. Niemann-pick c1-like 1 is required for an lxr agonist to raise plasma hdl cholesterol in mice. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:448–454. doi: 10.1161/ATVBAHA.107.160465. [DOI] [PubMed] [Google Scholar]
- 32.Repa JJ, Berge KE, Pomajzl C, Richardson JA, Hobbs H, Mangelsdorf DJ. Regulation of atp-binding cassette sterol transporters abcg5 and abcg8 by the liver x receptors alpha and beta. The Journal of biological chemistry. 2002;277:18793–18800. doi: 10.1074/jbc.M109927200. [DOI] [PubMed] [Google Scholar]
- 33.Aqul A, Lopez AM, Posey KS, Taylor AM, Repa JJ, Burns DK, Turley SD. Hepatic entrapment of esterified cholesterol drives continual expansion of whole body sterol pool in lysosomal acid lipase-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2014;307:G836–847. doi: 10.1152/ajpgi.00243.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du H, Heur M, Duanmu M, Grabowski GA, Hui DY, Witte DP, Mishra J. Lysosomal acid lipase-deficient mice: Depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. Journal of lipid research. 2001;42:489–500. [PubMed] [Google Scholar]
- 35.Fu X, Menke JG, Chen Y, Zhou G, MacNaul KL, Wright SD, Sparrow CP, Lund EG. 27-hydroxycholesterol is an endogenous ligand for liver x receptor in cholesterol-loaded cells. The Journal of biological chemistry. 2001;276:38378–38387. doi: 10.1074/jbc.M105805200. [DOI] [PubMed] [Google Scholar]
- 36.Timmins JM, Lee JY, Boudyguina E, Kluckman KD, Brunham LR, Mulya A, Gebre AK, Coutinho JM, Colvin PL, Smith TL, Hayden MR, Maeda N, Parks JS. Targeted inactivation of hepatic abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoa-i. The Journal of clinical investigation. 2005;115:1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang MD, Franklin V, Marcel YL. In vivo reverse cholesterol transport from macrophages lacking abca1 expression is impaired. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1837–1842. doi: 10.1161/ATVBAHA.107.146068. [DOI] [PubMed] [Google Scholar]
- 38.Schlager S, Vujic N, Korbelius M, Duta-Mare M, Dorow J, Leopold C, Rainer S, Wegscheider M, Reicher H, Ceglarek U, Sattler W, Radovic B, Kratky D. Lysosomal lipid hydrolysis provides substrates for lipid mediator synthesis in murine macrophages. Oncotarget. 2017;8:40037–40051. doi: 10.18632/oncotarget.16673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu L, Hammer RE, Li-Hawkins J, Von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH. Disruption of abcg5 and abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci U S A. 2002;99:16237–16242. doi: 10.1073/pnas.252582399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Menke JG, Macnaul KL, Hayes NS, Baffic J, Chao YS, Elbrecht A, Kelly LJ, Lam MH, Schmidt A, Sahoo S, Wang J, Wright SD, Xin P, Zhou G, Moller DE, Sparrow CP. A novel liver x receptor agonist establishes species differences in the regulation of cholesterol 7alpha-hydroxylase (cyp7a) Endocrinology. 2002;143:2548–2558. doi: 10.1210/endo.143.7.8907. [DOI] [PubMed] [Google Scholar]
- 41.Zelcer N, Hong C, Boyadjian R, Tontonoz P. Lxr regulates cholesterol uptake through idol-dependent ubiquitination of the ldl receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Westerterp M, Murphy AJ, Wang M, Pagler TA, Vengrenyuk Y, Kappus MS, Gorman DJ, Nagareddy PR, Zhu X, Abramowicz S, Parks JS, Welch C, Fisher EA, Wang N, Yvan-Charvet L, Tall AR. Deficiency of atp-binding cassette transporters a1 and g1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circulation research. 2013;112:1456–1465. doi: 10.1161/CIRCRESAHA.113.301086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mulya A, Lee JY, Gebre AK, Boudyguina EY, Chung SK, Smith TL, Colvin PL, Jiang XC, Parks JS. Initial interaction of apoa-i with atp binding cassette transporter a1 (abca1) impacts in vivo metabolic fate of nascent hdl. Journal of lipid research. 2008 doi: 10.1194/jlr.M800241-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cignarella A, Engel T, von Eckardstein A, Kratz M, Lorkowski S, Lueken A, Assmann G, Cullen P. Pharmacological regulation of cholesterol efflux in human monocyte-derived macrophages in the absence of exogenous cholesterol acceptors. Atherosclerosis. 2005;179:229–236. doi: 10.1016/j.atherosclerosis.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 45.Beyea MM, Heslop CL, Sawyez CG, Edwards JY, Markle JG, Hegele RA, Huff MW. Selective up-regulation of lxr-regulated genes abca1, abcg1, and apoe in macrophages through increased endogenous synthesis of 24(s),25-epoxycholesterol. The Journal of biological chemistry. 2007;282:5207–5216. doi: 10.1074/jbc.M611063200. [DOI] [PubMed] [Google Scholar]
- 46.Park Y, Pham TX, Lee J. Lipopolysaccharide represses the expression of atp-binding cassette transporter g1 and scavenger receptor class b, type i in murine macrophages. Inflamm Res. 2012;61:465–472. doi: 10.1007/s00011-011-0433-3. [DOI] [PubMed] [Google Scholar]
- 47.Basu SK, Ho YK, Brown MS, Bilheimer DW, Anderson RG, Goldstein JL. Biochemical and genetic studies of the apoprotein e secreted by mouse macrophages and human monocytes. The Journal of biological chemistry. 1982;257:9788–9795. [PubMed] [Google Scholar]
- 48.Boadu E, Choi HY, Lee DW, Waddington EI, Chan T, Asztalos B, Vance JE, Chan A, Castro G, Francis GA. Correction of apolipoprotein a-i-mediated lipid efflux and high density lipoprotein particle formation in human niemann-pick type c disease fibroblasts. The Journal of biological chemistry. 2006;281:37081–37090. doi: 10.1074/jbc.M606890200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.