

Abstract
Ovarian cancer (OvCa) is characterized by widespread and rapid metastasis in the peritoneal cavity. Visceral adipocytes promote this process by providing fatty acids (FAs) for tumour growth. However, the exact mechanism of FA transfer from adipocytes to cancer cells remains unknown. This study shows that OvCa cells co-cultured with primary human omental adipocytes express high levels of the FA receptor, CD36, in the plasma membrane, thereby facilitating exogenous FA uptake. Depriving OvCa cells of adipocyte-derived FAs using CD36 inhibitors and short hairpin RNA knockdown prevented development of the adipocyte-induced malignant phenotype. Specifically, inhibition of CD36 attenuated adipocyte-induced cholesterol and lipid droplet accumulation and reduced intracellular reactive oxygen species (ROS) content. Metabolic analysis suggested that CD36 plays an essential role in the bioenergetic adaptation of OvCa cells in the adipocyte-rich microenvironment and governs their metabolic plasticity. Furthermore, the absence of CD36 affected cellular processes that play a causal role in peritoneal dissemination, including adhesion, invasion, migration and anchorage independent growth. Intraperitoneal injection of CD36-deficient cells or treatment with an anti-CD36 monoclonal antibody reduced tumour burden in mouse xenografts. Moreover, a matched cohort of primary and metastatic human ovarian tumours showed upregulation of CD36 in the metastatic tissues, a finding confirmed in three public gene expression datasets. These results suggest that omental adipocytes reprogram tumour metabolism through the upregulation of CD36 in ovarian cancer cells. Targeting the stromal-tumour metabolic interface via CD36 inhibition may prove to be an effective treatment strategy against OvCa metastasis.
Keywords: ovarian cancer, metastasis, omentum, adipocyte, CD36
Introduction
Unlike most solid tumours, ovarian cancer (OvCa) disseminates widely in the peritoneal cavity before giving rise to hematogenous or lymphatic metastasis. The rapid and clinically silent spread of OvCa has been attributed to the unique mechanism of transcoelomic metastasis, which ultimately involves attachment and growth of tumour cells on peritoneum-covered abdominal organs.28, 33, 53 Tumourous transformation of the omentum, a peritoneal apron-like organ, predominantly composed of adipocytes, is a hallmark of advanced stage OvCa.31 The omental metastasis is often the biggest tumour, larger than tumors in the ovary or the fallopian tube,50 which are currently considered to be the organs of origin for ovarian’ cancer.9, 17 Previously, our group showed that adipocytes promote OvCa growth through the provision of fatty acids (FA).37, 38, 47 However, the exact mechanism through which adipocyte-derived FAs are transported into OvCa cells and whether interference with FA trafficking can halt tumour progression, remains unknown.
FAs are translocated across the phospholipid bilayers of the plasma membrane through either passive diffusion or a saturable protein-mediated transport system. Several membrane-associated FA binding proteins and transporters reportedly facilitate the transport process, including CD36 (also known as FA translocase [FAT]), FATPs (Fatty Acid Transport Protein Family), and FABPpm (Plasma Membrane Fatty Acid Binding Protein). CD36, a transmembrane glycoprotein that belongs to the class B scavenger receptor family, is one of the most well-characterized members of this group.1 In addition to FA uptake, CD36 facilitates cholesterol uptake and transduces intracellular signaling that regulates the metabolic targeting of FAs.42 CD36 has also been implicated in other important cellular functions pertaining to tumour biology, including antigen presentation, inflammation, angiogenesis and cell adhesion (reviewed in42, 51).
Although tissue-specific expression of the FA transporters has been described in FA-utilizing organs, less is known about the expression profile of these receptors in cancerous tissues and the role of the tumour microenvironment in their distribution.52 Our objective in this study was to determine which FA receptors in the adipocyte-rich tumour microenvironment are involved in transporting FAs inside the OvCa cells and whether these receptors play a wider role in OvCa metastasis.
Results
Omental adipocytes induce expression of CD36
To identify the effect of human primary adipocytes (HPAs) on FA transporter and distribution, we studied a panel of OvCa cell lines14 with immunoblotting and immunofluorescence microscopy. We found that, except for CD36 (Figure 1a), the most prevalent FA transporters (FABPpm, FATP1, and FATP4)18, 55 were unaffected by HPA co-culturing (Figure 1b, c). The mutational status of p53, a regulator of lipid metabolism pathways39, and cellular immortalization had no effect on adipocyte-induced CD36 over-expression (Supplementary Figure S1a and S1b). HPA co-culture increased CD36 protein levels in cancer cells over time with maximum levels reached in 24 to 48 h (Figure 1a, Supplementary Figure S1c). Quantitative Real-Time Reverse Transcription-PCR (qRT-PCR) showed increased CD36 mRNA levels in all three OvCa cell lines in the presence of HPA (Supplementary Figure S1d). CD36 expression in OvCa cells was localized to the plasma membrane, as demonstrated by cellular fractionation (Figure 1d, Supplementary Figure S1e) and confocal microscopy (Figure 2a, Supplementary Video 1). It was apparent that the induction of CD36 was specific to adipocytes because other stromal cell types (fibroblast, mesothelial cells, macrophages) in the omentum did not induce CD36 expression (data not shown).
Figure 1. Adipocytes induce CD36 expression in ovarian cancer cells.
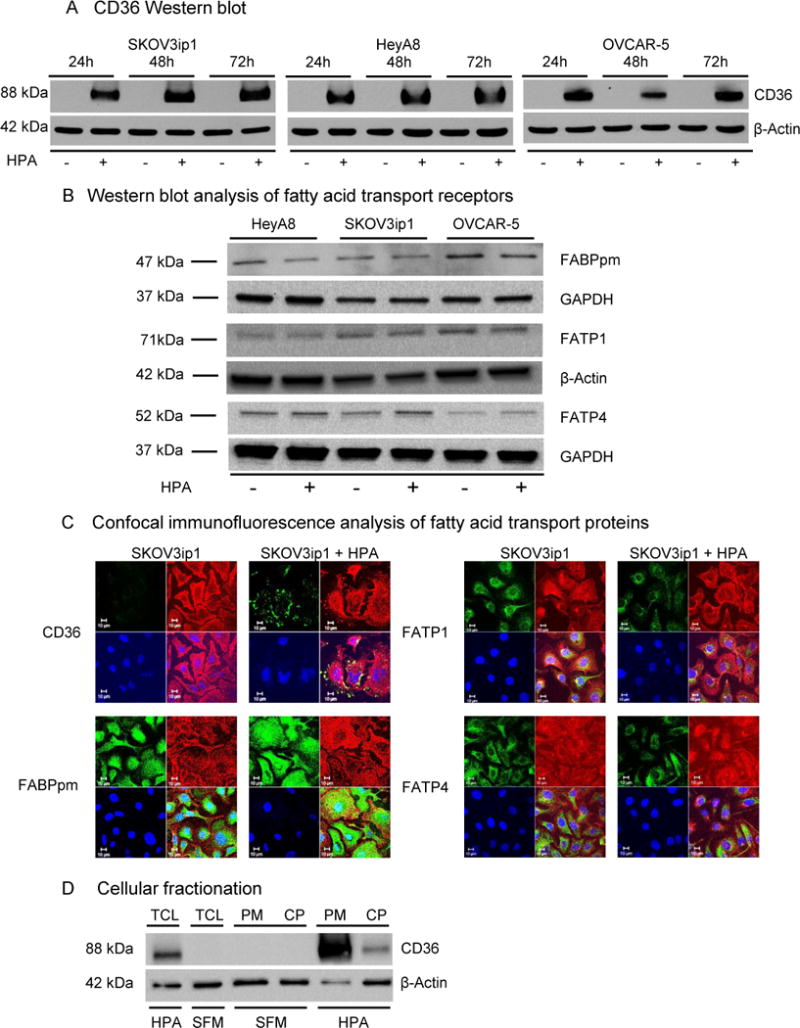
(a) Western blot analysis of CD36 protein expression in OvCa cell lines co-cultured with human primary adipocytes (HPA) at the indicated time points.
(b) Western blot analysis of FA transport proteins FABPpm, FATP1 and FATP4 in three different OvCa cell lines co-cultured with HPA for 24 h.
(c) Confocal immunofluorescent microscopy for the indicated fatty acid transporters in the absence (-) and presence (+) of HPA. Fatty acid transporters are detected using green fluorescence, plasma membrane using red fluorescence (Wheat Germ Agglutinin), cell nuclei are counterstained with Hoechst 33258 (blue).
(d) Subcellular localization of CD36 protein. OvCa cells were either co-cultured with HPA or were grown in serum free media (SFM). After 16 h, cells were either lysed to obtain total cellular lysate (TCL) or fractionated to cytoplasmic (CP) and plasma membrane (PM) fractions.
Figure 2. Inhibition of CD36 reduces adipocyte induced fatty acid uptake and lipid accumulation.
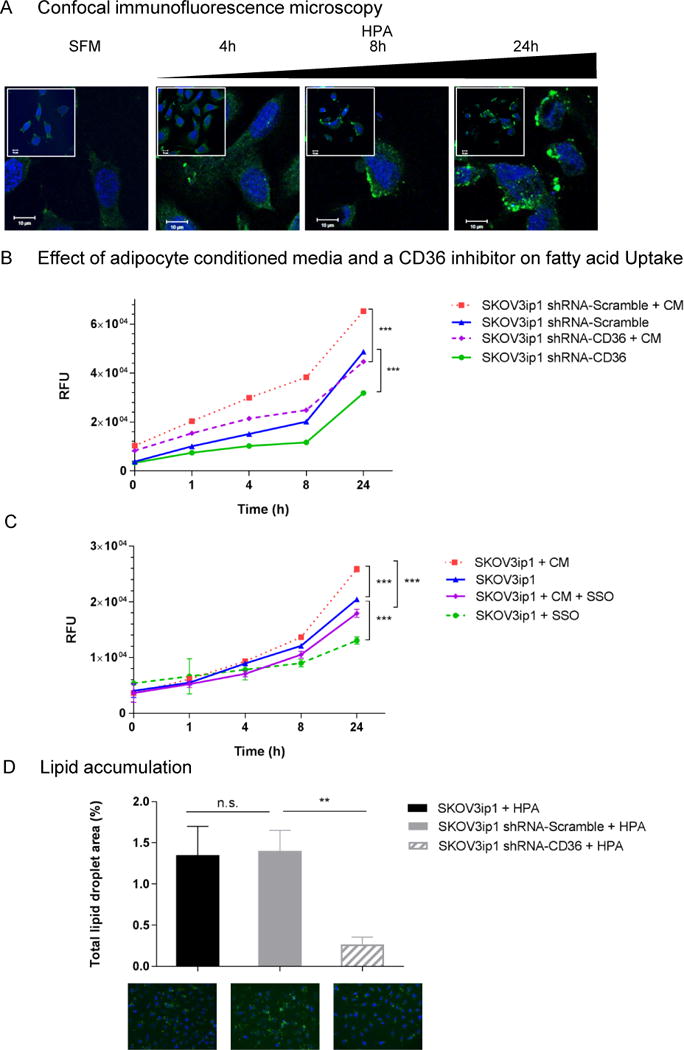
(a) Confocal immunofluorescence microscopy to detect CD36. SKOV3ip1 cells were cultured alone or with HPA for the indicated times and fixed on glass coverslips followed by incubation with a CD36 primary antibody and an Alexa-488 conjugated secondary antibody (green). Nuclei were counterstained with Hoechst 33258 (blue). Scale bars, 10 μm.
(b and c) Effects of adipocyte conditioned media (CM) and the CD36 specific inhibitor Sulfo-N-Succinimidyl Oleate (SSO) on parental (SKOV3ip1), scramble control (SKOV3ip1 shRNA-Scramble) and CD36 shRNA transduced cells (SKOV3ip1 shRNA-CD36) over time. Cancer cells (2.5×105) were serum-starved and when indicated incubated with adipocyte-CM and/or the irreversible CD36 inhibitor, SSO. FA transport was measured with a fluorescently labeled fatty acid (BODIPY-FA) and a nontoxic cell-impermeable quenching agent (Q-Red.1) at the indicated time points. Exclusion of the quenching reagent from the cells results in an increase in fluorescent signal intensity originating from intracellular BODIPY-FA. Changes in intracellular FA levels are quantified with a fluorescent plate reader as described in the materials and methods. ***p<0.001.
(d) Intracellular lipid accumulation in parental (SKOV3ip1), scramble control (SKOV3ip1 shRNA-Scramble) and CD36 shRNA transduced cells (SKOV3ip1 shRNA-CD36) cultured with HPA. After 16 hrs. of co-culturing, neutral lipid droplets (LD) were stained with BODIPY 493/503 and total LD area was calculated with Image J. Bars represent the mean ± s.e.m. from 3 independent experiments (n.s., not significant, **p<0.01) Representative immunofluorescent images of cellular LD accumulation (LD in green, BODIPY 493/503 and nuclei in blue, Hoechst 33258) are shown.
CD36 expression is associated with increased fatty acid uptake and lipid accumulation
The induction of CD36 at the plasma membrane upon HPA co-culture (Figure 2a) coincided with increased cellular FA uptake as measured using a fluorescently-labeled FA analog (Figure 2b). Adipocyte-conditioned media (CM) stimulated a significant increase in FA uptake in SKOV3ip1 cells, which was suppressed by the CD36 inhibitor,20 sulfo-N-succinimidyl oleate (SSO) (Figure 2b).
To investigate the impact of CD36 inhibition on metabolic and tumourigenic properties of OvCa cells, we generated stable populations of SKOV3ip1 cells expressing lentivirus-derived short hairpin RNA (shRNA) targeting CD36. The selected CD36 shRNA silenced both constitutive and adipocyte-induced CD36 expression (Supplementary Figure S2a, b). The proliferation and viability rates of the altered SKOV3ip1 cells were comparable to the parental control (Supplementary Figure S2c, d). Abrogation of CD36 expression in SKOV3ip1 cells using a shRNA led to lower basal and adipocyte CM-induced FA uptake (Figure 2c). Reduced CD36 expression and consequent reduced FA uptake resulted in lower lipid droplet (LD) accumulation, as shown using immunofluorescent staining for neutral lipids (Figure 2d). Collectively, these results point to a role for CD36 in adipocyte-induced FA uptake and lipid accumulation in OvCa cells.
Omental adipocyte-induced gene expression signature is associated with CD36 functions
To understand the molecular mechanisms leading to CD36 activation in OvCa cells and to identify pathways behind the observed metabolic changes with adipocyte stimulation, we performed an unbiased global gene expression analysis. HPA co-culturing altered 502 differentially expressed genes (DEG) in SKOV3ip1 cells; of these, 278 and 224 genes were up- and down- regulated, respectively. The top up- and down-regulated genes are shown in Table 1 and the distributions of the fold changes and p-values for each DEG are shown in Supplementary Figure S3 as a volcano plot. Consistent with our previous results identifying IL-8 as the most abundantly secreted cytokine responsible for omental metastasis, we observed a striking (44-fold) upregulation of the interleukin (IL)-8 gene37 upon HPA co-culturing. Further establishing a link between adipocyte biology and OvCa metastasis, IPA analysis predicted over-representation of several cellular functions integral to tumour survival and metastasis (Table 2 and Supplementary Table S1).37, 38
Table 1.
Top ten up- and down- regulated genes in ovarian cancer cells co-cultured with adipocytes
| Number | Symbol | Entrez Gene Name | P-value | False Discovery Rate (q-value) |
|---|---|---|---|---|
| UPREGULATED | ||||
| 1 | IL8 | interleukin 8 | 3.08E-04 | 1.60E-02 |
| 2 | IFITM1 | interferon induced transmembrane protein 1 | 5.10E-06 | 5.68E-03 |
| 3 | ISG15 | ISG15 ubiquitin-like modifier | 3.95E-04 | 1.60E-02 |
| 4 | IFI27 | interferon, alpha-inducible protein 27 | 1.79E-04 | 1.60E-02 |
| 5 | MX1 | myxovirus (influenza virus) resistance 1, interferon-inducible protein p78 (mouse) | 7.29E-04 | 1.60E-02 |
| 6 | IFI6 | interferon, alpha-inducible protein 6 | 6.96E-04 | 1.60E-02 |
| 7 | OAS3 | 2′-5′-oligoadenylate synthetase 3, 100kDa | 2.24E-04 | 1.60E-02 |
| 8 | IFIT1 | interferon-induced protein with tetratricopeptide repeats 1 | 6.46E-04 | 1.60E-02 |
| 9 | SOD2 | superoxide dismutase 2, mitochondrial | 4.52E-04 | 1.60E-02 |
| 10 | IRF9 | interferon regulatory factor 9 | 6.38E-06 | 5.68E-03 |
| DOWNREGULATED | ||||
| 1 | GPR56 | G protein-coupled receptor 56 | 9.28E-04 | 1.66E-02 |
| 2 | ALDH3A1 | aldehyde dehydrogenase 3 family, member A1 | 3.88E-04 | 1.60E-02 |
| 3 | EGR1 | early growth response 1 | 1.15E-02 | 3.05E-02 |
| 4 | TM7SF2 | transmembrane 7 superfamily member 2 | 7.30E-05 | 1.42E-02 |
| 5 | MMP7 | matrix metallopeptidase 7 (matrilysin, uterine) | 5.17E-04 | 1.60E-02 |
| 6 | ACSS2 | acyl-CoA synthetase short-chain family member 2 | 9.90E-04 | 1.70E-02 |
| 7 | CLIC3 | chloride intracellular channel 3 | 3.33E-03 | 2.18E-02 |
| 8 | HES2 | hairy and enhancer of split 2 (Drosophila) | 2.85E-03 | 2.10E-02 |
| 9 | UCP2 | uncoupling protein 2 (mitochondrial, proton carrier) | 1.00E-03 | 1.71E-02 |
| 10 | CGNL1 | cingulin-like 1 | 9.18E-04 | 1.66E-02 |
Table 2.
Ingenuity Pathway Analysis predicted top five diseases/functions categories increased or decreased in ovarian cancer cells co-cultured with adipocytes
| Number | Diseases or Functions Annotation | P-value | Z-score |
|---|---|---|---|
| UPREGULATED | |||
| 1 | Cell survival | 5.05E-09 | 3.540 |
| 2 | Cell viability | 1.49E-07 | 3.467 |
| 3 | Proliferation of cells | 2.52E-19 | 3.260 |
| 4 | Cytokinesis of tumor cell lines | 1.69E-12 | 2.889 |
| 5 | Metastasis of tumor | 3.08E-05 | 2.859 |
| DOWNREGULATED | |||
| 1 | Infection of mammalia | 8.83E-05 | −3.901 |
| 2 | Organismal death | 2.57E-09 | −3.820 |
| 3 | Replication of VSV | 6.92E-05 | −2.750 |
| 4 | Synthesis of lipid | 3.84E-05 | −2.403 |
| 5 | Morphology of lymphatic system component | 1.79E-04 | −2.378 |
We found that genes regulating global lipid metabolism and cholesterol synthesis were strongly inhibited (Figure 3a, 3b, Supplementary Table S2). Two important hubs in this network include the down-regulation of Acetyl-CoA carboxylase (ACACA), the rate-limiting enzyme in FA synthesis,49 and the up-regulation of nuclear factor-kappa B (NFκB), a crucial player in cancer metastasis and oxidative stress (Figure 3b).32 Transcription factor analysis revealed that key regulators of lipogenic genes, including the Sterol Regulatory Element Binding Transcription Factors (SREBPF1 and SREBPF2), were strongly inhibited by HPA stimulation (Supplementary Table S2), whereas transcription factors (e.g., STAT1, HIF-1α, FOXO1, and PPARγ coactivator-related 1) known to be involved in the regulation of CD36 activation and plasma membrane recruitment, were activated.4, 8, 33 In summary, we observed an overlap between the adipocyte-induced transcriptional gene signature characterized by the induction of pro-inflammatory cytokine secretion, oxidative stress, enrichment of pathways affecting lipid and cholesterol synthesis, and the previously reported signaling pathways downstream of CD36.7, 21, 29
Figure 3. Microarray analysis of ovarian cancer cells with adipocyte co-culturing.
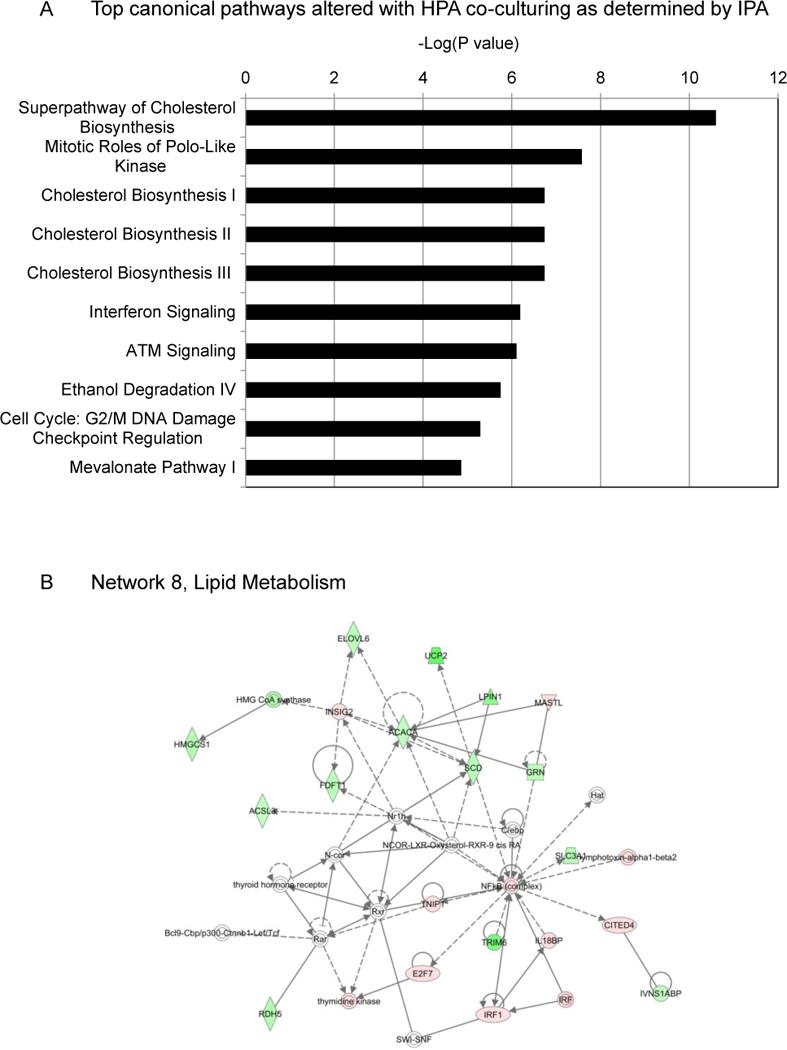
(a) Canonical pathways altered by human primary adipocyte (HPA) co-culturing. Pathways are ranked based on their significance (p-value) calculated using the right-tailed Fisher’s exact test. The p-value for each pathway is indicated by the bar and is expressed as -1 times the log of the p-value.
(b) Ingenuity Network Analysis. Network 8, changes in lipid metabolism in response to HPA stimulation. Gene products are represented as nodes and biological relationships between two nodes as a line. Red and green indicate up- and down- regulation, respectively. The color intensity correlates with the degree of change. Genes in white did not exhibit significant changes in expression.
Cellular energy homeostasis, intra-cellular cholesterol and ROS content are regulated by CD36
The response to increased energy needs in muscle tissue is coordinated by the interaction of AMP-activated protein kinase (AMPK) and CD36.48 We had previously shown that AMPK is activated in OvCa cells with HPA co-culture.37 In this study, we observed a very robust increase in p-AMPK when CD36 shRNA cells were co-cultured with HPA (Figure 4a). This suggests that while CD36 expression suppresses AMPK activation in OvCa cells in a similar manner to the process observed in muscle cells, exogenous FAs can acutely activate AMPK in both a CD36 dependent and independent fashion.48
Figure 4. CD36 depletion affects cellular metabolism.
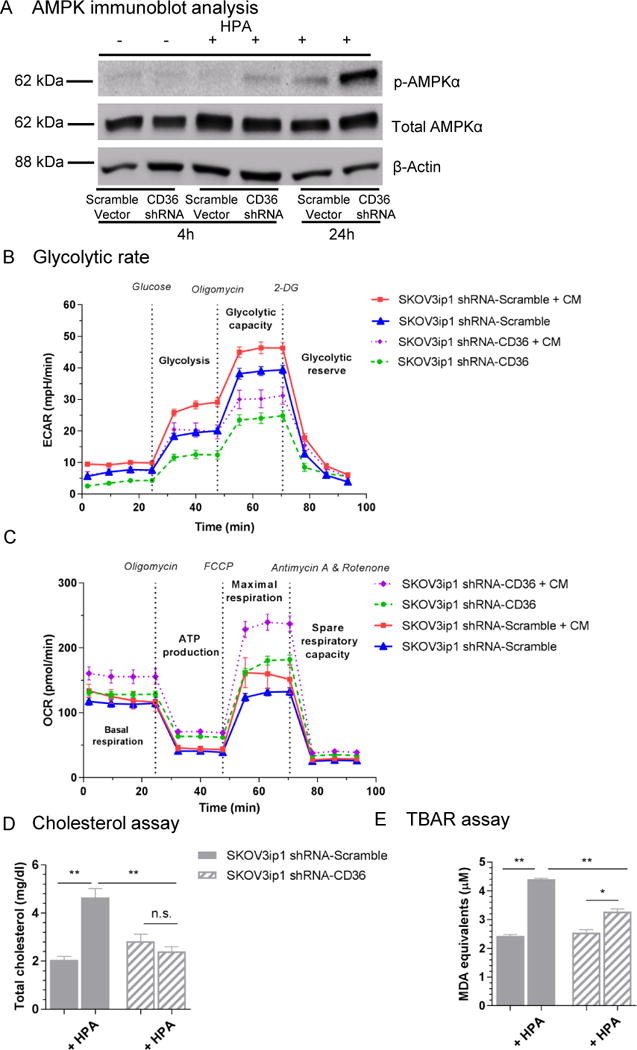
(a) Western blot analysis of total AMP-activated protein kinase (total AMPKα) and phospho-AMPK (p-AMPKα) levels in scramble control and CD36 shRNA transduced cells (CD36 shRNA) in the absence (-) and presence (+) of human primary adipocytes (HPA) at the indicated time points. The p-AMPK antibody recognizes T172 in the catalytic subunit of AMPK.
(b) Glycolytic function. Scramble control (SKOV3ip1 shRNA-Scramble) and CD36 shRNA transduced cells (SKOV3ip1 shRNA-CD36) were incubated with or without adipocyte conditioned media (CM). Extracellular acidification (ECAR) rate was measured with the Seahorse XF24 Analyzer. Data are means ± s.e.m. of seven replicates.
(c) Basal oxygen consumption rate. Scramble control (SKOV3ip1 shRNA-Scramble) and CD36 shRNA transduced cells (SKOV3ip1 shRNA-CD36) were incubated with or without adipocyte conditioned media (CM). OCR, oxidative phosphorylation and ATP production were measured with the Seahorse XF24 Analyzer. Data are means ± s.e.m. of four replicates.
(d) Cholesterol assay. Intracellular cholesterol accumulation was measured by the cholesterol oxidation reaction method using fluorescence in the absence and presence (+ HPA) of human primary adipocytes. All data shown are means, ± s.e.m. (n=3) **p<0.01, n.s., not significant. Data are representative of three independent experiments.
(e) TBAR assay. Oxidized lipid production was quantified with fluorometric measurement of malondialdehyde (MDA) formation in the absence and presence (+ HPA) of human primary adipocytes. All data shown are means, ± s.e.m. (n=3) **p<0.01, *p<0.05. Data are representative of three independent experiments.
Next, we determined the bioenergetic profile of CD36 wild-type and deficient OvCa cancer cells in response to HPA co-culture. Extracellular acidification rate (ECAR) increased in response to adipocyte CM in both parental and knockdown cells. However, glycolytic rate and maximum glycolytic capacity were impaired in the CD36 shRNA cells (Figure 4b). Inhibition of CD36 led to higher levels of both basal and maximal mitochondrial respiration, as indicated by the increased oxygen consumption rate (OCR) (Figure 4c). These data suggest that in tumour cells, CD36 expression, in addition to enhancing FA uptake, enhances anaerobic glucose metabolism while suppressing glucose oxidation.
Following up on the major pathways identified by the microarray analysis, we measured the effect of HPA stimulation on intracellular cholesterol content. At baseline, we found no statistically significant difference between total cellular cholesterol levels in parental and CD36 shRNA cells. Upon adipocyte stimulation, intracellular cholesterol content increased significantly, but was abrogated in the CD36 shRNA cells (p<0.01) (Figure 4d). These data, together with evidence of the down-regulation of genes of lipid and cholesterol synthesis, are consistent with the concept that CD36 expression in tumour cells shifts metabolism from production of endogenous lipids to reliance on exogenous FA and cholesterol. Previously reported data in other model systems have shown that CD36 can regulate inflammatory mediators and reactive oxygen species (ROS).11, 13 We therefore investigated whether adipocyte-induced lipid influx results in CD36-mediated lipotoxicity.5 For this purpose, we utilized the thiobarbituric acid-reactive (TBAR) assay, which measures malondialdehyde (MDA), a product of lipid peroxidation and an indicator of ROS. HPA co-culture increased the generation of TBARs in wild-type CD36 cells by 81%, while CD36shRNA cells showed a more modest 29% increase in response to HPA (p<0.01) (Figure 4e).
CD36 regulates in vitro tumourigenicity and in vivo metastasis
Before it was thought of as a FA transporter, CD36 was identified as a glycoprotein receptor (glycoprotein IIIB/IV) for thrombospondin-1 (TSP-1),6 and collagen.22 As such, it was found to be involved in cellular migration and adhesion in a variety of cell types. In our model system, inhibition of CD36 expression significantly reduced both baseline and adipocyte-stimulated invasion and migration (Figure 5a), as well as adhesion of OvCa cells to two (type I collagen: 51% and laminin: 39%, p<0.01) of the most common extracellular matrix components of the peritoneum (Figure 5b).58 Furthermore, silencing CD36 expression resulted in significantly fewer colonies (142 vs. 27, p<0.01) in a soft agar assay (Figure 5c).
Figure 5. Silencing of CD36 impairs invasion, migration, adhesion and clonogenic capacity of SKOV3ip1 cells in vitro.
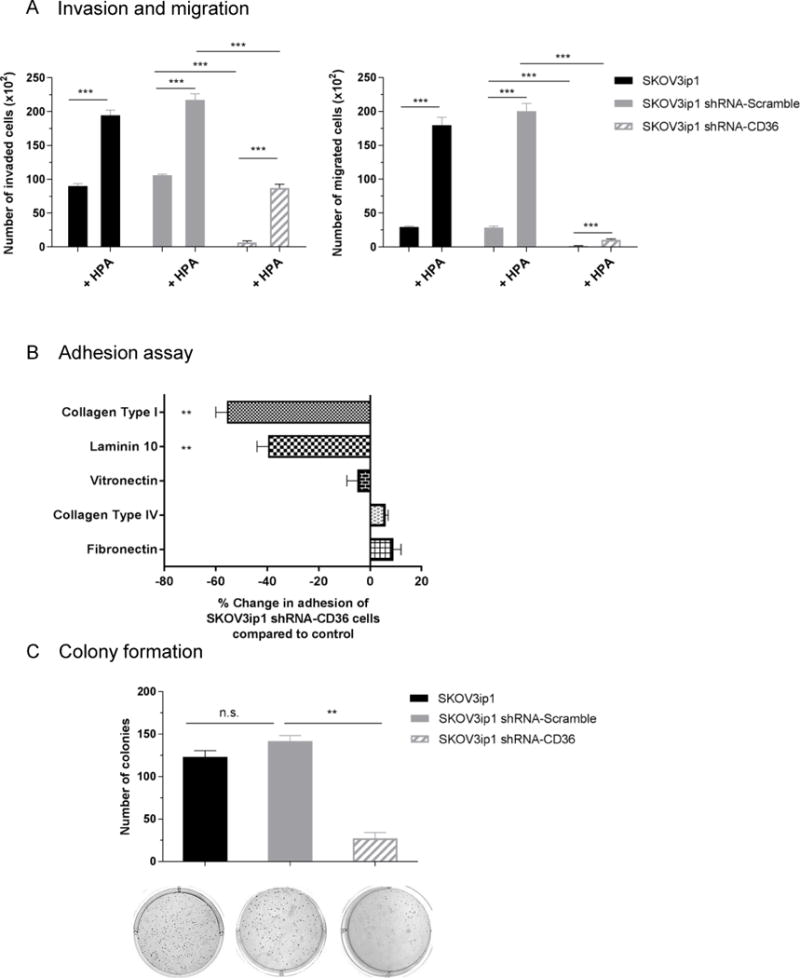
(a) Invasion (left) and migration (right) of parental (SKOV3ip1), scramble vector (SKOV3IP1 shRNA-Scramble) and CD36 shRNA-1 transduced (SKOV3ip1 shRNA-CD36) cells towards, serum free media (SFM) or primary human adipocytes (HPA). Invaded and migrated cells were counted with Image-J. Data are representative of three independent experiments performed in triplicates. ***p<0.001.
(b) Adhesion of SKOV3ip1 shRNA-CD36 cells to the indicated extracellular matrix proteins. Fluorescently labeled SKOV3ip1 cells were plated onto 96-well plates coated with the indicated ECM components. Plates were washed and the number of adherent cells were quantified by measuring fluorescence intensity and a standard curve. Percent change in adhesion of CD36 shRNA transduced cells compared to scrambled vector and normalized to poly-d-lysine coated wells. Bars represent the means ± s.e.m. from two independent experiments (n=5). **p<0.01.
(c) Colony formation. SKOv3ip1 cells (4,000) were plated onto soft agar; after 33 days, colonies were stained with crystal violet and counted. The data are representative of three independent experiments and columns represent the mean from three different wells. **p<0.01, n.s., not significant. Representative images of stained colonies are shown.
To understand the in vivo tumourigenic potential of CD36-deficient cancer cells we injected shRNA-mediated CD36 knockdown cells intraperitoneally into female athymic BALB/c nude mice. Animals injected with CD36 shRNA cells developed significantly smaller (tumour weight: 0.12 vs. 1.19 g, p<0.01) and fewer tumours (number of metastatic nodules: 18 vs. 188, p<0.001) compared to scrambled shRNA controls (Figure 6a). In order to further assess the possible therapeutic utility of CD36-inhibition, another set of mice was treated with daily intraperitoneal injections of a functionally neutralizing anti-CD36 antibody or its isotype control.40 We again observed a significant reduction in tumour burden (tumour weight: 1.12 vs. 0.63g, p<0.01 and number of metastatic nodules: 164 vs. 104, p<0.01, Figure 6b). We also tested the anti-tumour activity of the specific CD36 inhibitor, SSO, and observed a modest tumour reduction. However, in contrast to prior reports,10, 35 we observed significant in vivo toxicity (data not shown).
Figure 6. CD36 inhibition reduces in vivo tumour growth.
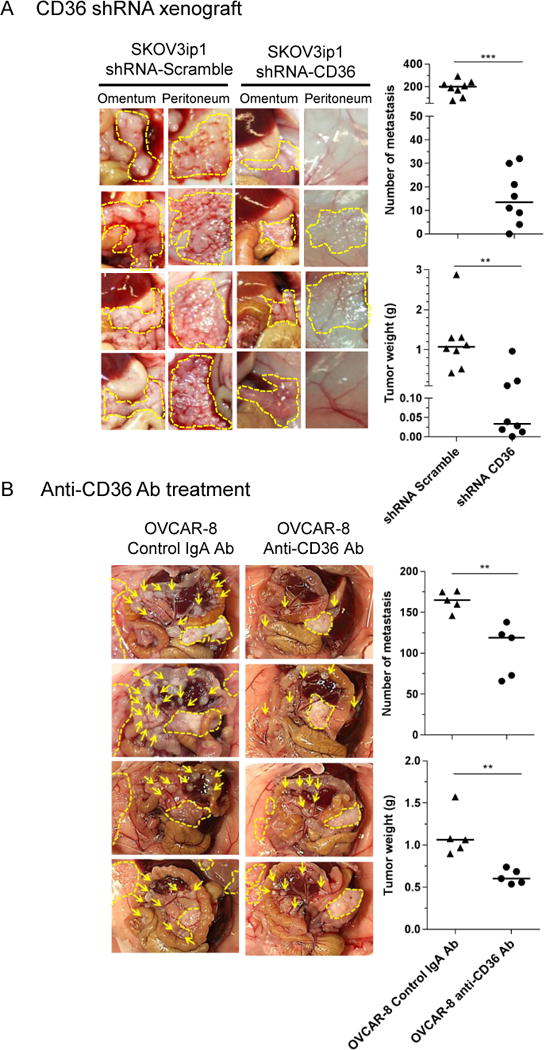
Xenograft models of metastasis. Female athymic mice were injected i.p with (a) 1×106 scrambled vector or CD36 shRNA transduced SKOV3ip1cells and were sacrificed after 32 days or (b) 5×106 OVCAR-8 cells. One week following tumour inoculation, animals were treated for 4 weeks with a daily injection of anti-CD36 monoclonal antibody or its isotype control. At the conclusion of the experiments, tumours were counted, excised, and weighed. The number of tumour metastases (top) and tumour weight (bottom) were determined. Each point represents an individual animal and the horizontal bar is the mean (***p<0.001, **p<0.01). Representative images of omental and peritoneal tumour nodules are shown from four different animals. Arrows show mesenteric tumour nodules, dashed lines highlight omental and peritoneal metastases.
The histologies of the xenograft tumours were similar on hematoxylin and eosin (H&E) staining (Supplementary Figure S4a). The differential growth rate was not caused by reduced proliferation and/or increased apoptosis as staining of excised tumour nodules with the proliferation marker (Ki-67) and TUNNEL assay were not significantly different (Supplementary Figure S4a). The absence of CD36 was accompanied by a reduction in microvessel density (MVD) observed in both SKOV3ip1 and OVCAR8 xenograft mouse models (Supplementary Figure S4a and S4b). However, reduced MVD was not associated with increased intra-tumoural hypoxia as indicated by low indexes of the endogenous surrogate marker CA IX (Supplementary Figure S4c). Extracellular matrix deposition, assayed by automated image analysis after fibronectin immunostaining (Supplementary Figure S4d) and Masson’s Trichrome staining (Supplementary Figure S4e) were found to be comparable between control and CD36 antibody treated tumours.
CD36 is up-regulated in advanced stage ovarian cancer patients
Because CD36 modulated in vitro and in vivo tumour growth and metastasis, we examined its presence in paired, primary tumour (PT) and omental metastasis tissue samples from ten patients with high-grade serous OvCa (Supplementary Table S3). To obtain a homogenous cancer cell population, tissues were laser micro-dissected (Supplementary Figure S5a) followed by the assessment of CD36 mRNA expression. In seven of the ten samples evaluated, CD36 expression increased in the metastatic samples when compared to their PT counterparts (p<0.05, Wilcoxon signed rank test, Figure 7a). Using the Oncomine expression analysis tool, we validated these findings in three publically-available datasets3, 45, 56 that provided matched quantitative gene expression data from human PT and either (OM) or peritoneal metastases samples (Figure 7b). Consistent with the in vitro, in vivo, and ex vivo experiments, a significant up-regulation of CD36 was observed in all three datasets (***p < 0.001). Survival analysis revealed that high CD36 expression is associated with reduced progression-free and overall survival (Supplementary Figure S5b).
Figure 7. Relative CD36 mRNA levels are increased in ovarian cancer metastasis.
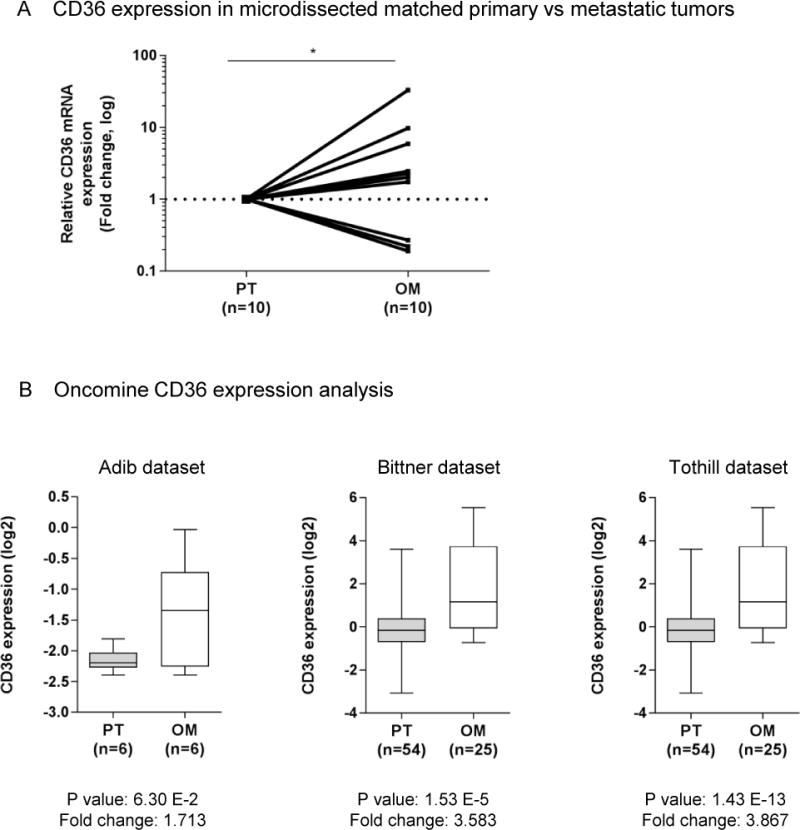
(a) Scatter diagram showing quantitative Real-time PCR comparison of relative CD36mRNA levels from laser micro dissected high grade serous human ovarian tumours and their corresponding omental metastases (n=10). The log ratio of paired metastatic and primary OvCa samples is plotted, >1 is higher in metastasis (n=7) and <1 is lower in metastasis (n=3). p<0.05, obtained by paired Wilcoxon Signed Ranks test.
(b) Oncomine analysis of CD36 expression in primary and metastatic high grade serous ovarian carcinomas. Oncomine datasets containing primary tumours as well as omental metastasis (top row) or peritoneal metastasis (bottom row) were queried for CD36 expression. Box legend: + inside box represents mean value, bar inside box represents median value, upper bar represents maximum of distribution, p-values and fold changes are indicated below the x-axis of each graph. All p-values represent a student’s t-test. Data are publicly available on Oncomine, and citations are included in the Supplementary Data.
Discussion
A recently described hallmark of cancer metabolism is the ability of cancer cells to acquire nutrients from the tumour microenvironment.41 Our previous work showed that OvCa cells obtain FA from surrounding adipocytes,37, 38 while Kamphorst et al. found that, through oncogenic signaling, hypoxic tumour cells could increase their FA import by scavenging FAs from serum phospholipids.23 Furthermore, several murine models combining in vitro, ex vivo, and in vivo studies suggested that, in obesity, enhanced vascularity and lipid transport increase the metastatic success of OvCa.30 However, none of these studies, which describe the cellular effects of external lipid acquisition on cancer cells, have elucidated the exact mechanism(s) for lipid transport.
Here we report that lipids are transported into OvCa cells through CD36, an integral membrane FA receptor. Omental adipocytes induce CD36 expression on cancer cells and subsequently FA uptake. The mechanism described is very similar to the physiologic process involved in peripheral FA acquisition in muscle and adipose tissues, which strongly relies on CD36. Consequently, CD36 knockout mice have defective FA uptake in myocardial, skeletal muscle and adipose tissue, and impaired triglyceride synthesis.12 Despite its role in enhancing FA uptake, CD36 is not absolutely required for the process.42 FAs from adipocytes can diffuse across the plasma membrane, although passive uptake is inefficient in meeting the FA requirement of most tissues, most likely because it is not coupled with metabolic targeting.24
Our findings suggest that omental adipocytes alter tumour metabolism to accommodate exogenous supplies of FA and cholesterol and that induction of CD36 on tumour cells is important for this metabolic remodeling. CD36 expression reduces glucose oxidation by tumour cells and shifts metabolism to reliance on the uptake and storage of exogenous lipids while endogenous lipid synthesis is inhibited (Figure 3A and Supplementary Table S2). CD36-mediated FA uptake is a likely factor in the inhibition of oxidative glucose metabolism,44 while modulation of PGC-1 signaling by CD36 might play a role in inhibition of cholesterol biosynthesis.46 CD36 interaction with the lipogenic pathway was noted in mice with stearoyl-CoA desaturase-1 (SCD1) deficiency and is consistent with cross regulation between FA uptake and FA synthesis.16
Co-culture with adipocytes not only induced CD36 expression, but also led to a substantial increase in the number and size of LDs and intracellular cholesterol content. A number of recent studies in various solid tumours have recognized LDs, the storage sites of excessive lipids and cholesterol, as functional organelles that influence tumour growth, aggressiveness and chemotherapy response.2, 15, 19 Conversely, we show that loss of CD36 expression confers protection from adipocyte-induced LD accumulation and reduces oxidative stress.
Our finding of overexpression of CD36 mRNA in omental metastasis samples is in accordance with a recent report showing that the CD36 protein is more frequent and prominent in visceral OvCa metastases, relative to the primary tumour and normal tissue.57 Potentially supporting a broad role for CD36 in epithelial cancers, Pascual et al. found that CD36 expression is correlated strongly with poor survival in patients with squamous cell lung carcinoma, bladder, or luminal A breast cancers.40 In this study, high expression of CD36 characterized the head and neck cancer stem cells that drive metastasis. The metastatic potential of the subpopulation of CD36+ metastasis-initiating cells was dependent on the ability of CD36 to internalize FAs, as well as on external FAs or a high-fat diet. Inhibiting CD36 expression blocked both lymphatic and hematogenous metastasis, which together with our observations specific to the transcoelomic metastatic process, implies that CD36 has a universal role in tumour progression and metastasis.40
Due to the multi-functional nature of the CD36 receptor and its diverse collection of ligands, we cannot exclude the possibility that mechanisms other than FA transport may be involved in its pro-tumourigenic role.36 For example, in liver cancer, Nath et al. showed that inhibition of CD36 suppressed the epithelial-mesenchymal transition and tumourigenicity via Wnt/β -catenin and TGF-β signaling pathways.34 Consistent with the recognized function of CD36 as a collagen receptor, we found that CD36 inhibition led to reduced adhesion on collagen I matrices.54 This aspect of CD36 function may be of particular importance during intra-peritoneal dissemination, since type I collagen is abundantly expressed in the omental basement membrane where OvCa cells preferentially attach.25 Reduced adhesion of the CD36shRNA cells to laminin constitutes a novel observation that warrants further investigation.
In summary, CD36 inhibition effectively reduced microenvironment derived FA uptake in OvCa cells, diminishing adipocyte-mediated invasion and migration, and in vitro and in vivo tumour growth. The present work, supported by in vivo experiments, upholds the idea that CD36 inhibition effectively de-couples the interplay between OvCa cells and their supporting omental microenvironment by preventing the metabolic remodeling that allows OvCa cells to thrive in a milieu rich in FA and cholesterol.
Materials and Methods
Cell lines
OVCAR-5, OVCAR-8, and CAOV-3 were purchased from American Type Culture Collection. SKOV3ip1 and HEYA8 were received from Dr. Gordon B. Mills (MD Anderson Cancer Center). The KURAMOCHI cell line was obtained from the Japanese Collection of Research Bioresources Cell Bank and the TYKNU cell line was obtained from Osaka University Graduate School of Medicine. The cell lines were validated by short tandem repeat DNA fingerprinting using the AmpFℓSTR Identifier kit (Applied Biosystems, Carlsbad, CA) and compared with known American Type Culture Collection fingerprints, the Cell Line Integrated Molecular Authentication database (CLIMA), and the University of Texas, MD Anderson Cancer Center fingerprint database.
Immunoblotting
Immunoblotting was performed as previously described.37 The following dilutions of antibodies were used: goat anti-CD36 (AF1955, 1:1,000, R&D Systems, Minneapolis, MN), mouse anti-FABPpm and rabbit anti-FATP1 (NBP1-47469, NBP1-89264 and 1:2,000, Novus Biologicals, Littleton, CO), mouse anti-FATP4 (H00010999-M01, 1:1,000, Abnova, Walnut, CA), mouse anti-actin (A5441, 1:5,000, Sigma-Aldrich, St. Louis, MO) and rabbit anti-GAPDH and anti p-AMPK (14C10, 1:2,000 and 2535 1:1,000, Cell Signaling Technology, Danvers, MA). Extraction of cytoplasmic and plasma membrane fractions was performed with a detergent-based selective extraction protocol (Mem-PER Plus Membrane Protein Extraction Kit, Pierce Biotechnology, Rockford, IL).
Quantitative Real-Time RT-PCR
Quantitative RT-PCR was performed using the 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA) with probes for CD36 (Hs00169627_m1, 77bp) and GAPDH (Hs00266705_gl, 74bp) (Applied Biosystems). Total RNA was isolated and transcribed into cDNA using a high capacity cDNA kit (Applied Biosystems). Relative levels of mRNA gene expression were calculated using the 2-ΔΔCt method.43
Immunofluorescence
Cells were plated on glass coverslips, fixed with 4% paraformaldehyde (PFA) and incubated overnight at 4°C with primary antibodies against CD36 (rabbit, HPA002018, 1:200, Sigma-Aldrich), FABPpm (mouse, 1:200, Novus Biologicals) FATP1 (mouse, 1:200, Novus Biologicals), FATP-4 (mouse, 1:200, Abnova). For plasma membrane labeling, Alexa Fluor 594 wheat germ agglutinin conjugate (W11262, Molecular Probes, Eugene, OR) was used. Samples were stained with Alexa Fluor 488 (A11059, A11001 and A11008, 1:2,000, Molecular Probes) conjugated secondary antibodies. After nuclei counterstain with Hoechst, coverslips were imaged with an inverted laser scanning confocal microscope (Zeiss LSM 510 equipped with a 63× oil immersion objective, Thornwood, NY). Z-stack confocal images were taken at 30-s intervals using AIM (version 3.2, Zeiss), and images were compiled using Zeiss LSM Image Browser (versions, 4.2, Zeiss). For epifluorescent microscopy, images were taken with a Zeiss Axiovert 100 microscope.
Invasion, migration and adhesion
Invasion and migration were measured using transwell chambers as previously described.25 Briefly, 4×105 cells were added to the upper chamber and allowed to migrate for 12 h or invade into collagen-coated (15μg) membranes for 24 h at 37°C. The number of migrated and invaded cells was counted in five (200×) random fields of view. Cellular adhesion was assayed in 96-well plates pre-coated with extracellular matrix proteins (5mg/ml). Collagen I (rat tail), fibronectin, and vitronectin were purchased from BD Biosciences (San Jose, CA), laminin was obtained from Millipore (Billerica, MA). Fluorescently labeled (10 mM 5- Chloromethylfluorescein Diacetate (CMFDA, Molecular Probes) cancer cells (5×104 cells/well) were allowed to adhere for 1 h. The number of adherent cells was determined with using a standard curve and was expressed in reference to Poly-D-Lysine coated wells.
Soft agar assay
Anchorage-independent colony formation was assessed using a two-layer soft agar assay. Cells (2×103) were re-suspended in culture media, mixed in with the top agar layer (0.4%) and plated over a solidified 1.5 ml nutrient feeder agar (0.8 %) underlay in 6-well plates. Plates were incubated for 21 days, after which colonies were stained with 0.005% crystal violet and counted using the GeneSys software colony-counting algorithm at medium sensitivity.
Lipid droplet accumulation
Intracellular LDs were measured using the cell permeable fluorescent lipophilic dye, BODIPY 493/503 (Molecular Probes). After overnight incubation, cells (2×105) were fixed in 4% PFA for 15min and incubated for 10 min with 1 mg/ml BODIPY 493/503. Following nuclei counterstain with Hoechst, digital images were obtained and the ImageJ software (http://imagej.nih.gov/ij/) was used to quantify LD areas, which were normalized to the numbers of nuclei per field. Average intensities were calculated by analyzing five randomly chosen fields.
Thiobarbituric acid reactive substance assay
Oxidized lipid production was assessed by measuring the formation of fluorescent MDA-TBA product (TBARS assay, Cayman Chemical, Ann Arbor, Michigan). Cells (1×107) were harvested and sonicated over ice for 15 seconds. After addition of SDS and the TBA substrate, homogenates were boiled for 1 h and the reaction was stopped. After centrifugation for 10 min at 1600 × g (4°C), the supernatant was transferred to a black bottom 96-well plate and fluorescence was read using a Tecan Safire2 microplate reader (excitation 530 nm/emission 550 nm) and compared to a MDA reference standard.
Total and esterified cholesterol assay
Intracellular total cholesterol content was determined with a coupled enzyme assay (EnzyChromTM AF Cholesterol Assay, Bioassay Systems, Hayward, CA) that results in a fluorometric product, proportional to the cholesterol present. 5×105 cells were plated in 6-well plates and cellular lipid content was extracted with methanol: chloroform (1:2) using a glass homogenizer. Insoluble materials were removed with centrifugation (13,000 × g for 10 min) and the organic phase was vacuum dried. Lipids were dissolved in 50 μL of the assay buffer and the cholesterol concentration was compared to a reference standard.
Fatty acid uptake
Cancer cells (2.5×105) were serum starved for 24 h and then incubated with and without adipocyte-CM for 4 h in 96-well plates. When indicated, 50 μM of the specific irreversible CD36 inhibitor, SSO (Santa Cruz Biotechnology, Dallas, TX) was added to the wells. After extracellular FAs were washed away, BODIPY-dodecanoic acid fluorescent FA analog (BODIPY 558/568 C12) and a nontoxic cell-impermeable quenching agent, Q-Red.1 were added to the wells at the indicated time points according to the manufacturer’s instructions (QBT Fatty Acid Uptake Assay Kit, Molecular Molecular Devices, Sunnyvale CA). Measurements of FA uptake were calculated from average fluorescence intensity from triplicate wells at excitation 480 nm and emission 520 nm using a Tecan Safire2 microplate reader.
Bioenergetics assay
The Seahorse XF96 Extracellular Flux Analyzer (Seahorse Biosciences, Billerica, MA) was used to obtain measurements of OCR and ECAR. OvCa cells (2.5×105 cells/well) were plated onto the XF96 cell plate 24 h before the assay. On the day of the assay, media were changed to assay media (serum, glucose and bicarbonate free, containing 2 mM Glutamine) or adipocyte CM, and incubated for 1 hour in a non-CO2 incubator at 37°C. 25 μl of Glucose (10 mM final concentration), oligomycin (3μM final concentration) and 2-DG (0.1 M final concentration) were added to the appropriate injection ports. For measurement of the mitochondrial capacity, drugs were injected to the final concentration as 2 μg/ml of oligomycin, 2.5 μM of carbonyl cyanide-p trifluoromethoxyphenylhydrazone (FCCP) and 2 μM of antimycin A.
Xenograft model
All animal experiments were approved by the Institutional Animal Care and Use Committee and were in accordance with the University of Chicago’s policies on the care, welfare and treatment of laboratory animals. SKOV3ip1 cells (1×106) and OVCAR-8 cells (5×106) were injected intraperitoneally into 7-week-old female athymic nude mice (Harlan Teklad, Indianapolis, IN). Mice were randomly assigned into experimental or control groups, blinding was not possible. Group size was determined based on the results of pilot studies; all animals were included in the analysis. In the CD36 shRNA experiment (n=8 mice per group), thirty-two days after injection the mice were sacrificed and the tumour colonies were counted, collected, and weighed. In the anti-CD36 antibody experiment (n=5 mice per group), one week following tumour inoculation, intraperitoneal daily treatment with 10ug anti-CD36 monoclonal antibody (mouse, JC63.1, Cayman Chemical, Ann Arbor, MI) or the IgA isotype control (mouse, ab37322, Abcam) was initiated and continued for 4 weeks. All antibodies were azide-free with no added preservatives. Excised tumour tissues were stained with hematoxylin and eosin (H&E), Masson’s trichrome or incubated with antibodies against Ki-67 (RM-9106-S0, Thermo Scientific, Waltham, MA), CD31 (ab28364, Abcam), Anti-Carbonic Anhydrase IX (CA IX, ab 15086, Abcam) and Fibronectin (F3648, Sigma-Aldrich). Protein detection was completed using the Vectastain ABC Kit (Vector Laboratories, Burlingame, CA). For determination of apoptosis, the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (Millipore) was used. Images were taken using the AxioVision imaging system in five random fields per sample at 200× magnification and analyzed using the ImageJ software. For determination of Ki67 and TUNEL staining, the number of positive cells was recorded as a percentage of the total number of cells within a field of view. For MVD, the average number of positively stained (CD31+) blood vessels per field of view was counted. Tumour hypoxia was quantified by adding the CA IX staining intensity scores (1= weak or undetectable, 2=moderate, 3=strong staining) and the percentage scores of stained tumour cells (1= <5 %, 2= 5–50%, 3 >50 % positive cells). Only a membranous staining pattern was considered positive. Quantification of fibronectin protein expression and collagen content was performed using Aperio ImageScope and Spectrum software as described previously.27
Human tissue samples
After obtaining an informed consent human omentum and tumour samples were collected from patients undergoing surgery at the University of Chicago. All specimens were obtained in accordance with a protocol approved by the Institutional Review Board (IRB). Cryosections from ten fresh frozen high-grade serous primary ovarian tumours and their corresponding omental metastasis were mounted on nuclease free polyethylene naphthalate (PEN)-membrane covered slides (Zeiss MicroImaging). Slides were stained and evaluated by a gynecologic oncology pathologist. Tumour cells were microdissected into microcentrifuge caps (AdhesiveCaps, Zeiss MicroImaging) with a pulsed low heat ultraviolet-A (337nm nitrogen) laser microscope system (PALM MicroBeam, P.A.L.M. Microlaser Technologies GmbH, Bernried, Germany). Total RNA was harvested with the RNeasy® Micro Kit (Qiagen, Valencia, CA) and concentration and purity was tested with a spectrophotometer.
Proliferation
The proliferation of OvCa cells was measured using the CyQuant cell proliferation assay kit as described previously.25
Short hairpin RNA CD36 targeting
Knockdown of CD36 was achieved through the use of pLKO.1-puro shRNA lentiviral transduction particles obtained from the MissionTM shRNA library (Sigma-Aldrich). Five different shRNA constructs (TRCN0000056998-TRCN0000057002) were tested together with a non-targeting (scrambled) shRNA (SHC002) control. In brief, cells were plated in 96-well plates (1×104/well) and were incubated overnight with the appropriate lentiviral particles (MOI=5) in the presence of hexadimethrine bromide (final concentration: 10 μg/ml). Cells were then selected with 0.5 μg/ml puromycin for two weeks and resistant colonies were picked, expanded and assayed for knockdown efficiency.
Oligonucletodie microarray analysis
Total RNA was isolated using the RNeasy Mini kit and RNase-free DNase Set (Qiagen).26 RNA quality and integrity were determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA). Biotinylated cRNA was prepared using the Illumina TotalPrep RNA Amplification Kit (Ambion, Inc., Austin, TX), starting with 200 ng total RNA. Biotin-labeled cRNA (750 ng) from three biological replicates was hybridized to an Illumina HumanHT-12_V4 Expression BeadChip microarray (Illumina, San Diego, CA) at the University of Chicago Functional Genomics Core Facility. Arrays were scanned on an Illumina’s BeadStation 500GX Genetic Analysis System. Data were extracted using GenomeStudio Software (v2011.1, Illumina) and the raw probe intensities were background subtracted, log2 transformed and Quantile normalized. ANOVA and multiple testing corrections were used to detect DEGs. Fold change was calculated by the average log2 expression differences and were considered significant at p<0.05, fold change ≥2, with a false discovery rate of 5% or less. Volcano plots were generated in R with the use of Student’s t test and generic plot function. The array data have been deposited in Gene Expression Omnibus (GEO; accession number GSE104237)
Canonical pathway and biological network analysis
The differentially expressed gene data were uploaded into IPA (Ingenuity® System, http://www.ingenuity.com, version 8.6, Redwood City, CA) to identify top canonical pathways and molecular networks associated with candidate genes. Fisher’s Exact test was used to calculate p-values.
Differential gene expression analysis
CD36 expression was analyzed with the Oncomine (version 3.0) web-based data-mining platform (www.oncomine.org) in three publicly-available high grade serous epithelial ovarian carcinoma datasets3, 45, 56. Datasets with information on primary and omental/peritoneal metastasis samples were interrogated for evidence of differential expression of CD36.
Statistical Analysis
Data were expressed as means ± SEM. Statistical analyses were performed using two-tailed, unpaired t-test or one-way ANOVA (GraphPad Prism, version 7.01, La Jolla, CA). Matched primary and metastases samples were compared with the paired Wilcoxon signed-ranks test. Significant differences were denoted as follows: p < 0.05 (*), < 0.01 (**), and < 0.001 (***). Results from pilot studies were utilized to determine the adequate sample size for all experiments. No statistical method was used to determine sample size.
Supplementary Material
Acknowledgments
This work was supported by a grant from Bears Care, the charitable beneficiary of the Chicago Bears Football Club (SD Yamada and E Lengyel) by National Cancer Institute grant DK033301 (N Abumrad) and by CA 169604 (E Lengyel) and the Foundation for Women`s Cancer, Amgen Ovarian Cancer Research Grant (A Ladanyi). We thank Chunling Zhang and the Center for Research Informatics at the University of Chicago for their bioinformatics support on the microarray data.
Abbreviations
- OvCa
Ovarian Cancer
- FA
Fatty Acid
- ROS
Reactive Oxygen Species
- CD36/FAT
Fatty Acid Translocase
- FATP
Fatty Acid Transport Protein
- FABPpm
Plasma Membrane Fatty Acid Binding Protein
- HPA
Human Primary Omental Adipoytes
- qRT-PCR
Quantitative Real-Time Reverse Transcription-PCR (qRT-PCR)
- CM
Conditioned Media
- SSO
Sulfo-N-succinimidyl Oleate
- shRNA
short hairpin RNA
- LD
Lipid Droplet
- DEG
Differentially Expressed Genes
- IL
Interleukin
- IPA
Ingenuity Pathway Analysis
- ACACA
Acetyl-CoA Carboxylase
- NF-kappa B
Nuclear Factor-Kappa B
- SBERBF
Sterol Regulatory Element Binding Transcription Factor
- AMPK
AMP-activated protein kinase
- ECAR
Extracellular Acidification Rate
- OCR
Oxygen Consumption Rate
- TBAR
Thiobarbituric acid-reactive substance
- MDA
Malondialdehyde
- TSP-1
Thrombospondin-1
- MVD
Microvessel Density
- PT
Primary Tumour
- SCD-1
Stearoyl-CoA Desaturase-1
- PFA
Paraformaldehyde
Footnotes
Conflict of interests
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website.
References
- 1.Abumrad NA, Sfeir Z, Connelly MA, Coburn C. Lipid transporters: membrane transport systems for cholesterol and fatty acids. Curr Opin Clin Nutr Metab Care. 2000;3:255–262. doi: 10.1097/00075197-200007000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N, Robbs BK, Oliveira SS, et al. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008;68:1732–1740. doi: 10.1158/0008-5472.CAN-07-1999. [DOI] [PubMed] [Google Scholar]
- 3.Adib TR, Henderson S, Perrett C, Hewitt D, Bourmpoulia D, Ledermann J, et al. Predicting biomarkers for ovarian cancer using gene-expression microarrays. Brit J Cancer. 2004;90:686–692. doi: 10.1038/sj.bjc.6601603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agrawal S, Febbraio M, Podrez E, Cathcart MK, Stark GR, Chisolm GM. Signal transducer and activator of transcription 1 is required for optimal foam cell formation and atherosclerotic lesion development. Circulation. 2007;115:2939–2947. doi: 10.1161/CIRCULATIONAHA.107.696922. [DOI] [PubMed] [Google Scholar]
- 5.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asch AS, Barnwell J, Silverstein RL, Nachman RL. Isolation of the thrombospondin membrane receptor. J Clin Invest. 1987;79:1054–1061. doi: 10.1172/JCI112918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baranova IN, Bocharov AV, Vishnyakova TG, Kurlander R, Chen Z, Fu D, et al. CD36 is a novel serum amyloid A (SAA) receptor mediating SAA binding and SAA-induced signaling in human and rodent cells. Journal of Biological Chemistry. 2010;285:8492–8506. doi: 10.1074/jbc.M109.007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bastie CC, Nahle Z, McLoughlin T, Esser K, Zhang W, Unterman T, et al. FoxO1 stimulates fatty acid uptake and oxidation in muscle cells through CD36-dependent and -independent mechanisms. Journal of Biological Chemistry. 2005;280:14222–14229. doi: 10.1074/jbc.M413625200. [DOI] [PubMed] [Google Scholar]
- 9.Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC, Jr, Beral V, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nature reviews Cancer. 2015;15:668–679. doi: 10.1038/nrc4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng JJ, Li JR, Huang MH, Ma LL, Wu ZY, Jiang CC, et al. CD36 is a co-receptor for hepatitis C virus E1 protein attachment. Sci Rep. 2016;6:21808. doi: 10.1038/srep21808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G, et al. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci. 2005;25:2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coburn CT, Knapp FF, Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. Journal of Biological Chemistry. 2000;275:32523–32529. doi: 10.1074/jbc.M003826200. [DOI] [PubMed] [Google Scholar]
- 13.Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, et al. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coscia F, Watters KM, Curtis M, Eckert MA, Chiang CY, Tyanova S, et al. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nature Communications. 2016;7:12645. doi: 10.1038/ncomms12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Gonzalo-Calvo D, Lopez-Vilaro L, Nasarre L, Perez-Olabarria M, Vazquez T, Escuin D, et al. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: a molecular and clinicopathological study. BMC Cancer. 2015;15:460. doi: 10.1186/s12885-015-1469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobrzyn P, Sampath H, Dobrzyn A, Miyazaki M, Ntambi JM. Loss of stearoyl-CoA desaturase 1 inhibits fatty acid oxidation and increases glucose utilization in the heart. Am J Physiol Endocrinol Metab. 2008;294:E357–364. doi: 10.1152/ajpendo.00471.2007. [DOI] [PubMed] [Google Scholar]
- 17.Eckert MA, Pan S, Hernandez KM, Loth RM, Andrade J, Volchenboum SL, et al. Genomics of ovarian cancer progression reveals diverse metastatic trajectories including intraepithelial metastasis to the fallopian tube. Cancer Discovery. 2016 doi: 10.1158/2159-8290.CD-16-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehehalt R, Füllekrun J, Pohl J, Ring A, Herrmann T, Stremmel W. Translocation of long chain fatty acids across the plasma membrane-lipid rafts and fatty acid transport proteins. Molecular and Cellular Biochemistry. 2006;284:135–140. doi: 10.1007/s11010-005-9034-1. [DOI] [PubMed] [Google Scholar]
- 19.Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand V, Olivares O, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2015;112:2473–2478. doi: 10.1073/pnas.1421601112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harmon CM, Abumrad NA. Binding of sulfosuccinimidyl fatty acids to adipocyte membrane proteins: isolation and amino-terminal sequence of an 88-kD protein implicated in transport of long-chain fatty acids. The Journal of membrane biology. 1993;133:43–49. doi: 10.1007/BF00231876. [DOI] [PubMed] [Google Scholar]
- 21.Janabi M, Yamashita S, Hirano K, Sakai N, Hiraoka H, Matsumoto K, et al. Oxidized LDL-induced NF-kappa B activation and subsequent expression of proinflammatory genes are defective in monocyte-derived macrophages from CD36-deficient patients. Arterioscler Thromb Vasc Biol. 2000;20:1953–1960. doi: 10.1161/01.atv.20.8.1953. [DOI] [PubMed] [Google Scholar]
- 22.Janabi M, Yamashita S, Hirano K, Matsumoto K, Sakai N, Hiraoka H, et al. Reduced adhesion of monocyte-derived macrophages from CD36-deficient patients to type I collagen. Biochem Biophys Res Commun. 2001;283:26–30. doi: 10.1006/bbrc.2001.4718. [DOI] [PubMed] [Google Scholar]
- 23.Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A. 2013;110:8882–8887. doi: 10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kazantzis M, Stahl A. Fatty acid transport proteins, implications in physiology and disease. Biochim Biophys Acta. 2012;1821:852–857. doi: 10.1016/j.bbalip.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kenny HA, Krausz T, Yamada SD, Lengyel E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells. International Journal of Cancer. 2007;121:1463–1472. doi: 10.1002/ijc.22874. [DOI] [PubMed] [Google Scholar]
- 26.Kenny HA, Leonhardt P, Ladanyi A, Yamada SD, Montag AG, Im HK, et al. Targeting the urokinase plasminogen activator receptor inhibits ovarian cancer metastasis. Clinical Cancer Research. 2011;17:459–471. doi: 10.1158/1078-0432.CCR-10-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kenny HA, Chiang CY, White EA, Schryver EM, Habis M, Romero IL, et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J Clin Invest. 2014;124:4614–4628. doi: 10.1172/JCI74778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lengyel E. Ovarian cancer development and metastasis. American Journal of Pathology. 2010;177:1053–1064. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li W, Febbraio M, Reddy SP, Yu DY, Yamamoto M, Silverstein RL. CD36 participates in a signaling pathway that regulates ROS formation in murine VSMCs. J Clin Invest. 2010;120:3996–4006. doi: 10.1172/JCI42823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, Metzinger MN, Lewellen KA, Cripps SN, Carey KD, Harper EI, et al. Obesity Contributes to Ovarian Cancer Metastatic Success through Increased Lipogenesis, Enhanced Vascularity, and Decreased Infiltration of M1 Macrophages. Cancer Res. 2015;75:5046–5057. doi: 10.1158/0008-5472.CAN-15-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66:271–289. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 32.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mwaikambo BR, Yang C, Chemtob S, Hardy P. Hypoxia up-regulates CD36 expression and function via hypoxia-inducible factor-1- and phosphatidylinositol 3-kinase-dependent mechanisms. Journal of Biological Chemistry. 2009;284:26695–26707. doi: 10.1074/jbc.M109.033480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep. 2015;5:14752. doi: 10.1038/srep14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naville D, Duchampt A, Vigier M, Oursel D, Lessire R, Poirier H, et al. Link between intestinal CD36 ligand binding and satiety induced by a high protein diet in mice. PLoS One. 2012;7:e30686. doi: 10.1371/journal.pone.0030686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nergiz-Unal R, Rademakers T, Cosemans JM, Heemskerk JW. CD36 as a multiple-ligand signaling receptor in atherothrombosis. Cardiovascular & hematological agents in medicinal chemistry. 2011;9:42–55. doi: 10.2174/187152511794182855. [DOI] [PubMed] [Google Scholar]
- 37.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt M, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature Medicine. 2011;17:1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipocyte tissue and adipocytes support tumorigenesis and metastasis. Biochimica et Biophysica Acta. 2013;1831:1533–1541. doi: 10.1016/j.bbalip.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parrales A, Iwakuma T. p53 as a Regulator of Lipid Metabolism in Cancer. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17122074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2016 doi: 10.1038/nature20791. [DOI] [PubMed] [Google Scholar]
- 41.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell metabolism. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annual review of nutrition. 2014;34:281–303. doi: 10.1146/annurev-nutr-071812-161220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pfaffl M. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 45.Rhodes D, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. Oncomine: A cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodrigue-Way A, Caron V, Bilodeau S, Keil S, Hassan M, Levy E, et al. Scavenger receptor CD36 mediates inhibition of cholesterol synthesis via activation of the PPARgamma/PGC-1alpha pathway and Insig1/2 expression in hepatocytes. FASEB J. 2014;28:1910–1923. doi: 10.1096/fj.13-240168. [DOI] [PubMed] [Google Scholar]
- 47.Romero IL, Mukherjee A, Kenny HA, Litchfield L, Lengyel E. Molecular Pathways: Trafficking of Metabolic Resources in the Tumor Microenvironment. Clinical Cancer Research. 2015;21:680–686. doi: 10.1158/1078-0432.CCR-14-2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Samovski D, Sun J, Pietka T, Gross RW, Eckel RH, Su X, et al. Regulation of AMPK activation by CD36 links fatty acid uptake to beta-oxidation. Diabetes. 2015;64:353–359. doi: 10.2337/db14-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santos CR, Schulze A. Lipid metabolism in cancer. FEBS Journal. 2012;279:2610–2623. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 50.Sehouli J, Senyuva F, Fotopoulou C, Neumann U, Denkert C, Lichtenegger W, et al. Intra-abdominal tumor dissemination pattern and surgical outcome in 214 patients with primary ovarian cancer. Journal of Surgical Oncology. 2009;99:424–427. doi: 10.1002/jso.21288. [DOI] [PubMed] [Google Scholar]
- 51.Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Science Signaling. 2009;2:1–9. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su X, Abumrad NA. Cellular fatty acid uptake: A pathway under construction. Trends in endocrinology and metabolism: TEM. 2009;20:72–77. doi: 10.1016/j.tem.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan D, Agarwal R, Kaye SB. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet. 2006;7:925–934. doi: 10.1016/S1470-2045(06)70939-1. [DOI] [PubMed] [Google Scholar]
- 54.Tandon NN, Lipsky RH, Burgess WH, Jamieson GA. Isolation and characterization of platelet glycoprotein IV (CD36) Journal of Biological Chemistry. 1989;264:7570–7575. [PubMed] [Google Scholar]
- 55.Thompson BR, Loho S, Bernlohr DA. Fatty acid flux in adipocytes: The in’s and out’s of fat cell lipid trafficking. Molecular and Cellular Endocrinology. 2010;318:24–33. doi: 10.1016/j.mce.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clinical Cancer Research. 2008;14:5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 57.Wang S, Blois A, El Rayes T, Liu JF, Hirsch MS, Gravdal K, et al. Development of a prosaposin-derived therapeutic cyclic peptide that targets ovarian cancer via the tumor microenvironment. Science translational medicine. 2016;8:329ra334. doi: 10.1126/scitranslmed.aad5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Witz CA, Montoya-Rodriguez IA, Cho S, Centonze VE, Bonewald L, Schenken RS. Composition of the extracellular matrix of the peritoneum. Journal of the Society for Gynecologic Investigation. 2001;8:299–304. doi: 10.1016/s1071-5576(01)00122-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.