

Abstract
Despite evident success in clarifying many important features of Alzheimer’s disease (AD) the efficient methods of its prevention and treatment are not yet available. The reasons are likely to be the fact that AD is a multifactorial and heterogeneous health disorder with multiple alternative pathways of disease development and progression. The availability of genetic data on individuals participated in longitudinal studies of aging health and longevity, as well as on participants of cross-sectional case-control studies allow for investigating genetic and non-genetic connections with AD and to link the results of these analyses with research findings obtained in clinical, experimental, and molecular biological studies of this health disorder. The objective of this paper is to perform GWAS of AD in several study populations and investigate possible roles of detected genetic factors in developing AD hallmarks and in other health disorders. The data collected in the Framingham Heart Study (FHS), Cardiovascular Health Study (CHS), Health and Retirement Study (HRS) and Late Onset Alzheimer’s Disease Family Study (LOADFS) were used in these analyses. The logistic regression and Cox’s regression were used as statistical models in GWAS. The results of analyses confirmed strong associations of genetic variants from well-known genes APOE, TOMM40, PVRL2 (NECTIN2), and APOC1 with AD. Possible roles of these genes in pathological mechanisms resulting in development of hallmarks of AD are described. Many genes whose connection with AD was detected in other studies showed nominally significant associations with this health disorder in our study. The evidence on genetic connections between AD and vulnerability to infection, as well as between AD and other health disorders, such as cancer and type 2 diabetes, were investigated. The progress in uncovering hidden heterogeneity in AD would be substantially facilitated if common mechanisms involved in development of AD, its hallmarks, and AD related chronic conditions were investigated in their mutual connection.
Keywords: Neurodegeneration, Neuroinflammation, Synapse loss, Neuronal death, Pleiotropy, Cancer, Diabetes, Amyloid cascade hypothesis, Response to infection, BBB
1. Introduction
Alzheimer’s disease (AD) is a progressive degeneration of the brain, inducing memory decline, learning impairment, language and behavioral disturbances, depressive symptoms, and personality changes, resulting in a marked decline in all mental activities and eventually in death. AD is most prevalent neurological health disorder in developed part of the world today. Recent estimates rank AD as the third cause of death for people older than 75 years. Despite substantial efforts to understand its causes and biological mechanisms the etiology of AD remains largely unknown. There are no medications that can notably influence rate of AD progression when it started. The multifactorial and heterogeneous nature of AD which is manifested in multiple and alternative pathways of its development and progression is likely to be responsible for this situation. This indicates that the improvement in current understanding of AD can be reached by uncovering alternative mechanisms of this health disorder.
The availability of genetic data collected for individuals participated in longitudinal studies of aging, health, and longevity opens a unique opportunity for studying genetic components of hidden heterogeneity using genome wide association studies of AD. Useful insights about the variety of biological mechanisms of AD can also be obtained from findings obtained in clinical, experimental and molecular biological studies of this health disorder. The non-genetic part of heterogeneity in AD can be evaluated from data on aging, health and longevity related traits collected in longitudinal and cross-sectional studies, Medicare Service Use files and other datasets. An important feature of aging-related health decline observed in longitudinal data is the dependence among chronic conditions detected in epidemiological studies. For example, AD negatively correlates with cancer. Such connections are likely to be caused by common genetic and non-genetic factors as well as by an increase in susceptibility to these disorders with increasing age.
Most of the information and ideas about potential mechanisms connecting the AD with its various biomarkers and risk factors comes from experimental studies of transgenic animal models and from studies of cell cultures, so it is often difficult to directly translate to humans. Still, such studies generated valuable hypotheses that can be further tested using human data, which may substantially improve current understanding of the mechanisms of AD. The availability of rich genetic, behavioral, environmental and other information collected in longitudinal human studies of aging, health and longevity over recent decades opens a unique opportunity for integrating the knowledge from animal and human studies for better understanding of AD. Particularly useful insights about biological pathways involved in AD can be obtained from integration of the results of genome-wide association studies (GWAS) of AD with research findings from clinical, experimental, epidemiological and population studies of AD and the aging-associated physiological and health decline.
In this paper we present the results of GWAS of AD using data from three longitudinal (CHS, FHS, HRS) and one case-control (LOADFS) human studies. Then we discuss how genes detected in our analyses are involved in mechanisms linking this health disorder with its hallmarks. We emphasize possible involvement of these genes in common biological processes related to health disorders other than AD, such as infectious diseases, cancer, and type 2 diabetes (T2D) - to better understand how exactly the detected genes may contribute to the development of AD, and whether the impact of these genes on AD is a part of their broader pleiotropic influence on organism’s vulnerability and resistance to stresses.
2. Data and methods
We used data from the Framingham Heart Study (FHS), Cardiovascular Health Study (CHS), Health and Retirement Study (HRS), and Late Onset Alzheimer Disease Family Study (LOADFS) (Lee et al., 2008), to identify genetic variants associated with AD in genome-wide association study (GWAS). Tables 1.1 and 1.2 provide brief description of these datasets.
Table 1.1.
The sample sizes (by gender) and the number of SNPs in four datasets before and after quality control (QC) procedure.
| By gender
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Dataset | Before QC
|
After QC
|
||||||
| #Samples | #Males | #Females | #SNPs | #Samples | #Males | #Females | #SNPs | |
| CHS | 5270 | 2252 | 3018 | 49,094 | 5018 | 2137 | 2881 | 33,328 |
| FHS | 3788 | 1651 | 2317 | 49,094 | 3651 | 1592 | 2059 | 35,259 |
| HRS | 9641 | 4120 | 5521 | 2,315,518 | 9631 | 4118 | 5513 | 1,329,158 |
| LOADFS | 4561 | 1666 | 2895 | 590,247 | 4561 | 1666 | 2895 | 551,330 |
Table 1.2.
The sample sizes in three datasets (by race) before and after quality control (QC) procedure.
| By race
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Dataset | Before QC
|
After QC
|
||||||
| #White | #Black | #Others | #White | #Black | #Others | |||
| CHS | 4434 | 807 | 29 | 4209 | 781 | 28 | ||
| HRS | 7968 | 1299 | 374 | 7960 | 1297 | 374 | ||
| LOADFS | 3894 | 251 | 271 | 145* | 3894 | 251 | 271 | 145* |
LOADFS has 145 individuals with missing race values.
More information about the FHS, CHS, HRS, and LOADFS data are given in (Dawber, 1980; Mahmood et al., 2014; D’Agostino et al., 1989; Tucker-Seeley et al., 2011; Lee et al., 2008), respectively. See also dbGaP (https://www.ncbi.nlm.nih.gov/gap).
In the logistic regression model, ‘cases’ corresponded to study participants with AD, and ‘controls’ corresponded to study subjects without AD. The Cox regression model was applied to the data where the information on the age at disease onset was available. To take family links in LOADFS into account in the logistic regression model, we used the GLIMMIX program in SAS. The year of birth, gender and race (when available) were used as observed covariates in the analyses.
To control for possible population stratification we calculated 20 principal components and used them as observed covariate (Price et al., 2006). In the HRS data, the genomic control was used to control for possible population stratification, and to avoid the inflation of association test statistics for both the logistic and Cox regressions (Price et al., 2006).
FHS and CHS CARe data used in this analyses were genotyped on the Illumina IBC chip including ~49 K SNPs in ~2000 candidate genes (for major complex diseases). HRS data were genotyped on the Illumina platform with ~2.5mln SNPs, and LOADFS data – on the Illumina platform with ~600 K SNPs. The quality control (QC) has been performed before running GWAS analyses. Individuals with > 5% missing SNPs we excluded. SNPs are kept only if the genotyping rate is higher than 95% and minor allele frequency (MAF) was higher than 1%. In addition, SNPs failed the Hardy-Weinberg test (p-value < 10−7) were also excluded.
3. Results
3.1. Genetics of AD: results from GWAS of FHS, CHS, HRS, and LOADFS data
Fig. 1 shows the QQ plot and Manhattan plot of the results of GWAS of AD using logistic regression applied to CHS, FHS, HRS, and LOADFS data.
Fig. 1.
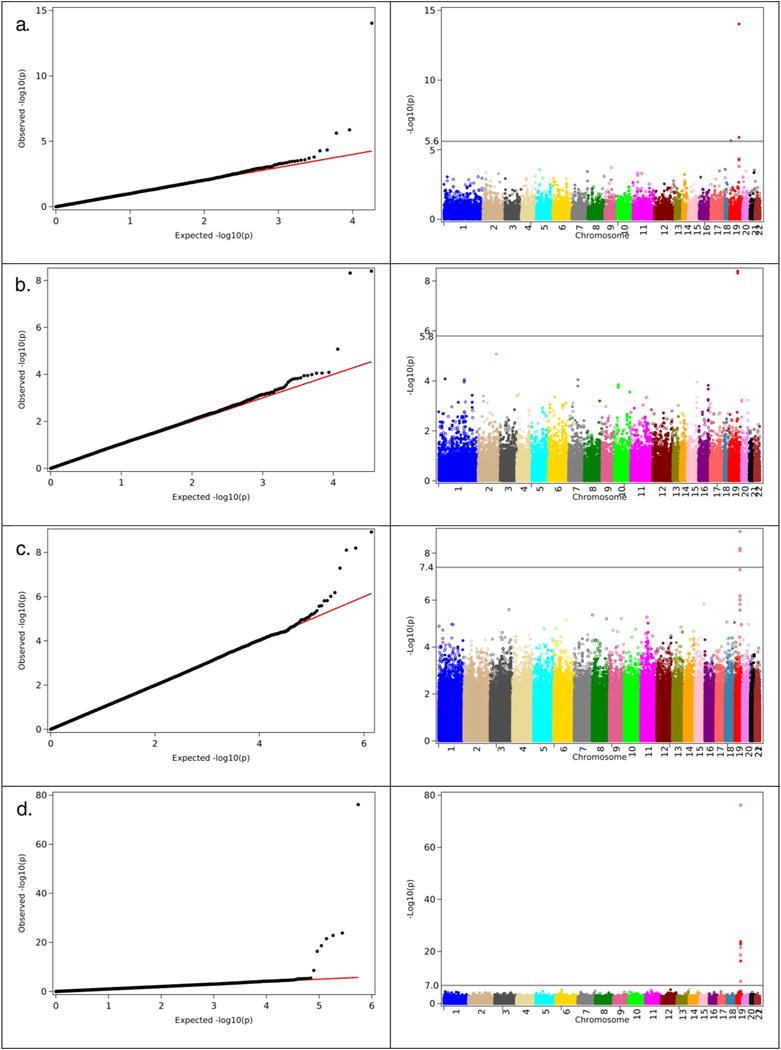
Left panel. The QQ-plots of the results of GWAS of Alzheimer’s disease obtained in the analyses using logistic regression (GLIMMIX in LOADFS) for male and females combined. a). CHS (case: 286; control: 4732); b). FHS (case: 308; control: 3343); c. HRS (case: 656; control: 8768); d). LOADFS (case: 2319; control: 2242). Right panel. Corresponding Manhattan plots for the same analysis as shown on the left panel.
One can see from this figure that in all four analyses the genome wide significant SNPs are located on chromosome 19. Fig. 2 shows the QQ plots and Manhattan plots of the results of GWAS of AD using Cox regression for CHS, FHS and HRS datasets. One can see from Figs. 1 and 2 that in all analyses, SNPs located on chromosome 19 show highly significant associations with AD. Note that CHS and FHS have overall smaller numbers of SNPs with highly significant associations. This is because the genotyping platforms in these datasets contain smaller number of SNPs than those for HRS and LOADFS data (see Data and methods). Tables 2.1 and 2.2 summarize results of these analyses for logistic and Cox regression statistical models, respectively. These tables show that the SNPs on chromosome 19 that demonstrated highly significant associations with AD in more than one dataset (these SNPs are shown in bold font in “SNP” column), and in both Cox and logistic regression analyses, are located in/near APOE, APOC1, TOMM40, and PVRL2 (NECTIN2) genes. Also shown in bold is rs769449, which is in strong LD (r2 = 0.82, D′ = 1) with rs429358 representing APOE e2/e3/e4 polymorphism.
Fig. 2.
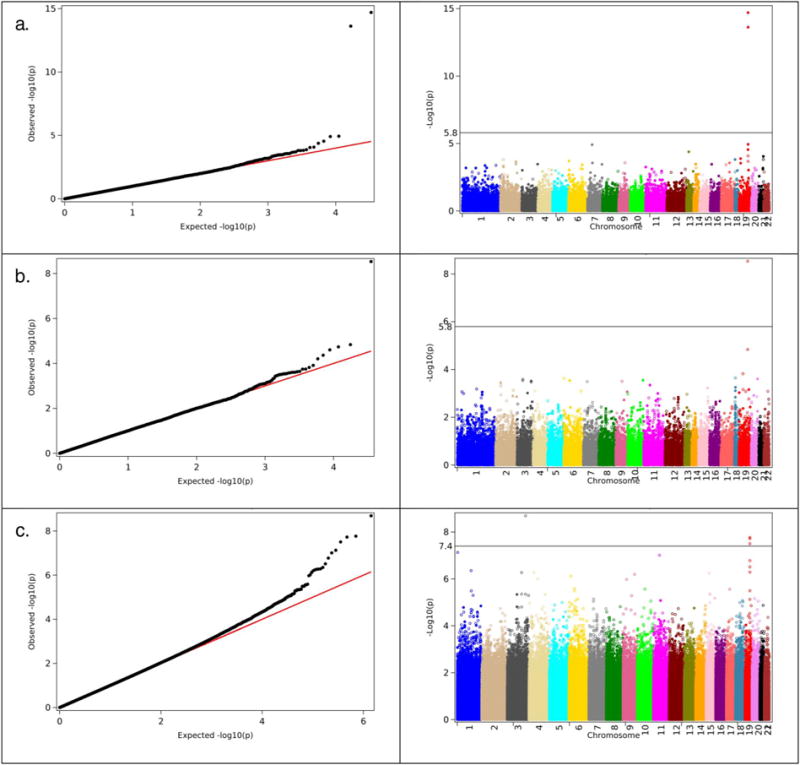
Left panel. The QQ-plots of the results of GWAS of Alzheimer’s disease obtained in the analyses using Cox regression. a). CHS; b). FHS; c). HRS. Right panel. Corresponding Manhattan plots for the same analysis on the left panel.
Table 2.1.
SNPs from the TOMM40, APOC1, APOE and PVRL2 (NECTIN2) genes that showed most significant associations in GWAS of Alzheimer’s disease in CHS, FHS, HRS and LOADFS, using logistic regression (GLIMMIX in LOADFS).
| Dataset | SNP | p-Value | OR | Chr | Minor | Major | MAF case | MAF contr | Gene | Location |
|---|---|---|---|---|---|---|---|---|---|---|
| CHS | rs2075650 | 9.32E–15 | 2.3 | 19 | G | A | 0.22 | 0.12 | TOMM40 | Intron (LD with rs429358, rs769449 in APOE) |
| rs405509 | 1.31E–06 | 1.5 | 19 | T | G | 0.53 | 0.43 | APOE | Upstream, promoter | |
| rs8106922 | 4.54E–05 | 0.7 | 19 | G | A | 0.3 | 0.39 | TOMM40 | Intron | |
| rs6859 | 5.34E–05 | 1.4 | 19 | A | G | 0.49 | 0.4 | PVRL2 | 3′-UTR | |
| rs769450 | 1.59E–04 | 0.7 | 19 | A | G | 0.33 | 0.41 | APOE | Intron | |
| FHS | rs2075650 | 4.02E–09 | 2.3 | 19 | G | A | 0.17 | 0.12 | TOMM40 | Intron (LD with rs429358, rs769449 in APOE) |
| rs12721046 | 4.87E–09 | 2.2 | 19 | A | G | 0.18 | 0.12 | APOC1 | Intron (LD with rs769449 and rs429358 in APOE) | |
| HRS | rs769449 | 3.00E–12 | 1.9 | 19 | A | G | 0.15 | 0.09 | APOE | Intron (strong LD with rs429358 in APOE; moderate LD with rs12721046 in APOC1, rs2075650 in TOMM40) |
| rs157582 | 3.32E–09 | 1.5 | 19 | T | C | 0.31 | 0.24 | TOMM40 | Intron | |
| rs283815 | 1.41E–08 | 1.5 | 19 | G | A | 0.31 | 0.25 | PVRL2 | 3′-UTR | |
| rs71352238 | 7.54E–08 | 1.6 | 19 | C | T | 0.17 | 0.12 | TOMM40 | Intron | |
| rs2075650 | 1.65E–07 | 1.5 | 19 | G | A | 0.18 | 0.13 | TOMM40 | Intron (LD with rs429358, rs769449 in APOE) | |
| LOADFS | rs2075650 | 2.86E–64 | 3.6 | 19 | G | A | 0.34 | 0.18 | TOMM40 | Intron (LD with rs429358 in APOE) |
| rs8106922 | 1.14E–23 | 0.5 | 19 | G | A | 0.26 | 0.37 | TOMM40 | Intron | |
| rs157580 | 1.50E–23 | 0.5 | 19 | G | A | 0.23 | 0.33 | TOMM40 | Intron (LD with rs405509, rs769449 near APOE) | |
| rs405509 | 1.50E–20 | 1.7 | 19 | T | G | 0.59 | 0.49 | APOE | Upstream, promoter | |
| rs6859 | 1.12E–18 | 1.7 | 19 | A | G | 0.52 | 0.42 | PVRL2 | 3′-UTR | |
| rs439401 | 5.17E–17 | 0.6 | 19 | T | C | 0.26 | 0.33 | APOE, APOC1 | Between genes |
The columns notations: dataset name, SNP name, p-value in logistic regression (GLIMMIX for LOADFS), Odds Ratio (OR) for logistic regression, chromosome (Chr), reference allele (Ref), alternative allele (Alt), minor allele frequencies (MAF) in the case and in control groups, gene’s name and location with LD information. (The ORs for LOADFS are from GLIMMIX regression.)
Table 2.2.
SNPs from the TOMM40, APOC1, APOE and PVRL2 (NECTIN2) genes that showed most significant associations in GWAS of AD in CHS, FHS and HRS using Cox regression.
| Dataset | SNP | p-Value | HR | Chr | Ref | Alt | MAF | Gene | Location |
|---|---|---|---|---|---|---|---|---|---|
| CHS | rs2075650 | 1.99E–15 | 2.3 | 19 | A | G | 0.12 | TOMM40 | Intron (LD with rs429358, rs769449 in APOE) |
| rs12721046 | 2.39E–14 | 2.2 | 19 | G | A | 0.08 | APOC1 | Intron (LD with rs769449 and rs429358 in APOE) | |
| rs405509 | 1.16E–05 | 1.4 | 19 | T | G | 0.47 | APOE | Upstream, promoter | |
| rs6859 | 2.87E–05 | 1.4 | 19 | A | G | 0.37 | PVRL2 | 3′-UTR | |
| rs8106922 | 8.34E–05 | 0.7 | 19 | A | G | 0.30 | TOMM40 | Intron | |
| FHS | rs12721046 | 2.93E–09 | 2.0 | 19 | G | A | 0.08 | APOC1 | Intron (LD with rs769449 and rs429358 in APOE) |
| rs2075650 | 1.46E–05 | 1.7 | 19 | A | G | 0.12 | TOMM40 | Intron (LD with rs429358, rs769449 in APOE) | |
| HRS | rs115881343 | 2.03E–09 | 0.5 | 19 | C | G,T | 0.03 | TOMM40 | Intron |
| rs76366838 | 1.71E–08 | 0.4 | 19 | G | A | 0.03 | TOMM40 | Intron | |
| rs769449 | 3.12E–08 | 1.7 | 19 | G | A | 0.06 | APOE | Intron (LD with rs12721046 in APOC1, rs2075650 in TOMM40, rs429358 in APOE) | |
| rs157582 | 1.68E–07 | 0.7 | 19 | C | T | 0.29 | TOMM40 | Intron | |
| rs283815 | 3.07E–07 | 0.7 | 19 | A | G | 0.30 | PVRL2 | Intron (LD with rs429358 in APOE) | |
| rs2075650 | 5.17E–07 | 1.5 | 19 | A | G | 0.12 | TOMM40 | Intron (LD with rs429358, rs769449 in APOE) | |
| rs71352238 | 3.19E–06 | 1.5 | 19 | T | C | 0.09 | TOMM40 | Intron |
The columns notations: dataset name, SNP name, p-value in Cox regression, Hazard Ratio (HR) for Cox regression, chromosome (Chr), reference allele (Ref), alternative allele (Alt), minor allele frequency (MAF), gene’s name and location with LD information.
4. Discussion
The results of GWAS of AD using HRS, CHS, FHS and LOADFS data confirmed strong association of genetic variation in APOE, APOC1, TOMM40, and PVRL2 (NECTIN2) genes with AD. Associations between SNPs in these genes and AD were also detected in earlier studies (Ortega-Rojas et al., 2016; Takei et al., 2009; Seripa et al., 2012; Bagnoli et al., 2013; Roses et al., 2016). Several top significant SNPs in our research showed associations with AD in more than one dataset (CHS, FHS, HRS or LOADFS), using either logistic or Cox model. At least three of these SNPs (rs2075650, rs405509, rs6859) have also been linked to AD in previous research (Harold et al., 2009; Wang et al., 2017; Logue et al., 2011), which supports their true association with AD in present study. These well confirmed associations justify a deeper look into functions of detected SNPs and genes to get insights into potential mechanisms of their connection with AD.
The histopathological manifestation of AD is typically characterized by the two hallmarks: extracellular senile plagues predominantly made up of beta amyloid (Aβ) (a peptide cleaved from the amyloid beta protein precursor (AβPP)), and intracellular neurofibrillary tangles (NFT) made of aggregates of the hyper-phosphorylated tau protein, which plays crucial role in the microtubules assembly and stabilization on neurons. The accumulation of Aβ inside neurons that may contribute to synaptic dysfunction and cognitive impairment, has also been observed in AD (Gouras et al., 2010; Takahashi et al., 2017). Other major features of AD include cerebral cortex atrophy with a loss of neurons and the connections between them, and glucose hypometabolism - reduction of the cerebral metabolic rate for glucose (CMRglc) (Mosconi et al., 2009; Ramos Bernardes da Silva Filho et al., 2017). During 25+ years of studies of AD with main focus on testing the amyloid cascade hypothesis, additional characteristics, biomarkers and risk factors of AD emerged, including mitochondrial dysfunction, oxidative stress, neuro-inflammation, synapse loss, air pollution, viral infection, impaired lipid transport and axonal repair, among others. All these factors may potentially play major roles in AD development or progression in different subsets of AD patients. However, consistent and well replicated in independent datasets, including in present study, findings of AD associated SNPs in/near APOE, APOC1, TOMM40 and PVRL2 (NECTIN2) genes located on chromosome 19 suggest that there may also exist a mechanism of AD, which is linked to biological functions of SNPs in these genes, potentially acting together.
Table 3 shows SNPs that have demonstrated genome-wide significant associations with AD in more than one dataset (CHS, FHS, HRS or LOADFS) using logistic and/or Cox model, that is, were replicated. We also included rs769449 in APOE gene. It was found to be significantly associated with AD in HRS data only (Tables 2.1, 2.2), however, this SNP is in LD with two top replicated SNPs from this study (rs2075650 in TOMM40 and rs12721046 in APOC1), and with rs429358, representing APOE e2/e3/e4 polymorphism.
Table 3.
Biological roles of top SNPs (and genes) that showed significant associations with AD in more than one dataset (CHS, FHS, HRS or LOADFS) using logistic and/or Cox model.
| SNP | Closest gene (involved in, associated with) | LD with (0.4 ≤ r2; D′ > 0.7) | eQTL (SNP influences expression of) | SNP is in enhancer region in | SNP was previously associated with | References |
|---|---|---|---|---|---|---|
| rs2075650 | TOMM40 (protein precursors’ import into mitochondria, AD)a | rs429358 and rs769449 in APOE; SNPs in PVRL2 | TOMM40, PVRL2 | 12 tissues | Longevity, AD; LDLC | (Harold et al., 2009; Deelen et al., 2011; Holliday et al., 2013; Abe et al., 2015) |
| rs405509 | APOE (lipid transport, AD, stroke, cancer, longevity, hippocampus atrophy, response to viruses (HCV))b | SNPs in TOMM40; PVRL2 | APOE, PVRL2 | 10 tissues | Longevity, AD; cognitive impairment (interacts with APOE e4) | (Wang et al., 2017; Lu et al., 2014; Ryu et al., 2016; Ma et al., 2016) |
| rs8106922 | TOMM40 (protein precursors’ import into mitochondria, AD) | SNPs in APOE; PVRL2 | PVRL2 | blood | Longevity; TG | (Lin et al., 2016; Salakhov et al., 2014) |
| rs6859 | PVRL2 (adherens junctions, resistance to herpesviruses, cancer prognosis) | SNPs in PVRL2 | PVRL2, TOMM40 | 7 tissues | AD | (Logue et al., 2011) |
| rs12721046 | APOC1 (AD, cancer, HDL, VLDL, virus (HCV) infectivity) | rs769449 and rs429358 in APOE | LDL, HDL | (Kettunen et al., 2012; Musunuru et al., 2012) | ||
| rs769449 | APOE (lipid transport, AD, stroke, cancer, longevity, response to viruses (HCV)) | rs429358 in APOE, rs12721046 in APOC1, rs2075650 in TOMM40; SNPs in PVRL2 | 5 tissues | CSF tau levels, cognitive decline, LDL | (Cruchaga et al., 2013; Zhang and Pierce, 2014; Kettunen et al., 2012) |
Biological effects of SNPs were assessed using NCBI resources (PubMed, dbSNP, etc.), NHGRI-EBI Catalog of published GWAS (https://www.ebi.ac.uk/gwas), GRASP (https://grasp.nhlbi.nih.gov), and HaploReg v4.1 (Ward and Kellis, 2016) (http://archive.broadinstitute.org/mammals/haploreg).
LD with other genes column also shows LD with rs429358, one of two SNP defining APOE e2/e3/e4 polymorphism, and LD between individual SNPs shown in the table.
4.1. SNP rs2075650 is associated with AD in all datasets
The most consistent association with AD across our datasets, using Cox as well as logistic regression, was observed for the SNP rs2075650 (Tables 2.1 and 2.2). This SNP was previously linked to AD, longevity, cholesterol levels and macular degeneration (e.g., (Deelen et al., 2011; Shadyab et al., 2017; Holliday et al., 2013; Harold et al., 2009). It is located in intron of TOMM40 gene involved in protein precursors’ import into mitochondria. It is also in strong (r2 ≥ 0.92) LD with several SNPs in PVRL2 (NECTIN2) gene, and in moderate (r2 ~0.5) LD with SNPs rs769449 and rs429358 in APOE, representing APOE e2/e3/e4 polymorphism. Notably, the rs2075650 is also eQTL and may influence expression levels of TOMM40 and NECTIN2. Altogether, available information about this SNP (briefly summarized in Table 3) suggests that its phenotypic effects may not necessary be related to functions of TOMM40 itself, but may also be related to the functions of APOE and/or NECTIN2. This means that mechanism connecting rs2075650 and AD may potentially involve biological effects of any or all of these genes; for instance, it could be related to mitochondrial function via TOMM40 and APOE, to lipid transport via APOE, and to resistance to viral infection via APOE and NECTIN2 and their products (De Chiara et al., 2012; Bassendine et al., 2013; Mahley, 2016; Roses et al., 2016; Zeitlow et al., 2017). Comparing potential biological functions of rs2075650 with those of other top SNPs associated with AD in our study may help clarify mechanism connecting these SNPs with AD.
4.2. What do top AD-related SNPs have in common?
AD is a heterogeneous health disorder, which means that the pool of AD patients may contain cases of the different etiology. Some mechanisms, however, may be more prevalent than others and play role in majority of AD cases. If so, then one may expect a certain overlap between the biological effects of SNPs and genes associated with AD. In our study, five SNPs (rs2075650, rs6859, rs405509, rs12721046, and rs8106922) showed top genome-wide significant associations with AD in more than one dataset (CHS, FHS, HRS or LOADFS). Table 3 summarizes biological and health effects of these SNPs, as well as rs769449, based on current literature and results of HaploReg v4.1 (Ward and Kellis, 2016) tool for the analysis of SNPs regulatory effects.
One can see from this table that the selected SNPs and respective genes do share common functional features: Most are involved in lipid metabolism (especially in LDL cholesterol levels), response to viral infection, cancer and longevity. These overlaps in metabolic and health effects indicate that APOE, APOC1, TOMM40 and NECTIN2 might influence AD acting in concert (given that the top AD-associated SNPs in these genes are not in complete LD with each other). Studies of interaction between these genes support a possibility of AD mechanism that involves collective effects of genes on chromosome 19 that have been associated with the disease. For example, mutation in APOC1 in combination with APOE e4 serves as a potential risk factor for developing AD. Individuals carrying both APOE e4 and the APOC1 insertion allele had an approximately 66.49% increased risk of AD (Zhou et al., 2014).
Most SNPs shown in Table 3 (rs2075650, rs6859, rs405509, rs769449, rs8106922) are in LD with SNPs in PVRL2 (emphasized in bold font), and/or influence its expression as eQTLs, as well as located in enhancer regions of genome. This indicates that the SNPs associated with AD may contribute to AD through regulatory effects, by influencing transcription levels and resulting protein concentrations, without changes in protein structure. It is also important to note that while most SNPs from Table 3 are in LD with the SNPs in PVRL2 gene, the rs6859, which is actually located in PVRL2 gene, is not in LD with SNPs in the other genes. This indicates that PVRL2 (NECTIN2) may potentially be main gene functionally related to AD (of the top four AD-associated genes in this study: TOMM40, APOE, APOC1, PVRL2). PVRL2 involvement in AD could be, e.g., through its role in cell adhesion and brain’s susceptibility to viral infections, the latter was also suggested for APOE and APOC1 (Porcellini et al., 2010; De Chiara et al., 2012). This warrants further research into the role of ‘brain vulnerability to infection’ in AD development, and deserves a deeper look into functions of the detected genes.
PVRL2 (poliovirus receptor-related 2, formerly herpesvirus entry mediator B, HVEB), a.k.a. NECTIN2 (nectin, cell adhesion molecule 2), codes for a human plasma membrane glycoprotein involved in “adherens junction”. It also serves as an entry for certain mutant strains of herpes and pseudorabies viruses, and is involved in cell to cell spreading of these viruses. The fact that AD-associated SNPs in TOMM40 and APOE both influence expression of PVRL2 indicates that biological effects of PVRL2 may at least in part explain observed genetic associations with AD. PVRL2 is important for maintaining proper cell junctions and the extra-cellular matrix (ECM) structure. Proper cell junctions are in turn important to control BBB permeability and may protect brain from spreading the viral infection, which was suggested to play role in AD (Urosevic and Martins, 2008; Miklossy, 2011; Itzhaki et al., 2016; Itzhaki, 2016) PVRL2 and respective protein were also implemented in cancer (Oshima et al., 2013; Karabulut et al., 2016). The gene is also responsive to plasma cholesterol acting at endothelial sites of vascular inflammation and could potentially be a new therapeutic target for atherosclerosis prevention. The involvement of PVRL2 in lymphomagenesis has been previously suggested to play a role in the pathogenesis of acute myeloid leukemia (Graf et al., 2005) and in lymphomagenesis (Almire et al., 2007). Furthermore, gene expression profiling performed on hepatocellular carcinomas revealed an up-regulation of PVRL2, which was supposed to participate in the inhibit ion of apoptosis in hepatoma cells (Kurokawa et al., 2006; Logue et al., 2011). In GWAS of the late onset AD in African Americans, SNPs related to APOE, PVRL2, TOMM40 and APOC1 also showed genome-wide significant associations with AD (Logue et al., 2011). Importantly, the association of rs6859 of PVRL2 with AD remained statistically significant after adjusting for the effect of APOE, which supports its independent role in AD. Overall, available evidence suggests that PVRL2 is gene with highly pleiotropic effects on health-related phenotypes. It also has notable overlap in functions with other AD-related genes, which refers to its involvement in brain’s susceptibility to viral and bacterial infections.
4.3. Roles of APOE in Aβ clearance and neuro-inflammation
As mentioned, the SNP rs769449 in the APOE gene, found in this study, is in strong LD (r2 = 0.82; D′ = 1) with rs429358 of APOE e2/e3/e4 polymorphism. The rs769449 is also in moderate LDs (r2 ~0.5–0.6) with rs12721046 in APOC1 and rs2075650 in TOMM40, which are of the same strength as LDs between these SNPs and rs429358. This supports the possibility that rs769449 actually represents APOE e2/e3/e4 polymorphism in this study, and the genome-wide significant effect of rs769449 on AD that we observed in HRS data (Table 2.1) is probably related to that effect of APOE e4. The involvement of APOE gene in AD has been confirmed more than two decades ago. The product of this gene interacts with Aβ and that the presence of APOE e4 isoform of this gene in a person’s genome increases his/her risks of AD. In the brain apolipoprotein E (apoE) is synthesized predominantly by astrocytes (Koldamova et al., 2010). It is the primary transporter of cholesterol within the blood brain barrier (BBB). ApoE interacts with Aβ during its aggregation and deposition and together they influence risks of AD development. Many studies confirm the presence of such interaction, however, explanations of its nature differ considerably from one study to the next (LaDu et al., 1994; Hashimoto et al., 2012; Manelli et al., 2004; Cerf et al., 2011; Carter, 2005). Recently Verghese et al. (2013) provided evidence that the apoE proteins do not bind to soluble (i.e., monomeric) Aβ. Garai et al. (2014) found that apoE may interact with the Aβ oligomers and fibrils. The researchers emphasized considerable heterogeneity in the size and structures of the Aβ oligomers that are likely to influence such interaction. The presence of APOE e4 variant may also affect Aβ accumulation in brain. Studies of longitudinal cohorts showed that APOE e4 was significantly associated with lower CSF Aβ1–42, suggesting increased Aβ deposition in brain (Resnick et al., 2015). ApoE is involved in complex relationships with Aβ suitable to cross the BBB that may regulate Aβ clearance (Huynh et al., 2017). E.g., apoE4 animals showed reduced Aβ clearance across the BBB compared to apoE3 animals, linked to less efficient regulation by apoE4 isoform of lipoprotein receptor shedding (Bachmeier et al., 2014). Alternative pathways of Aβ clearance could be mediated by microglia (Morgan, 2009). Brains of AD patients carrying the APOE e4 allele were found to have increased density of Aβ deposits, limited capacity to clear Aβ and enhanced neuro-inflammation (Castellano et al., 2011). Despite an association between neurodegenerative disease and APOEe4 many APOEe4 non-carriers develop AD. Analyses of genetic associations of people without APOEe4 allele showed that rs2075650 in TOMM40 is still associated with AD (Bekris et al., 2011; Naj et al., 2014). After adjustment for age, sex, and APOEe4 status the association of rs157580 in TOMM40 with AD also remained statistically significant in Han Chinese cohort (Ma et al., 2013) suggesting APOEe4 independent mechanism of AD involving TOMM40.
4.4. APOC1 encodes a member of the apolipoprotein C1 family
This gene is expressed primarily in the liver and also in the brain. The encoded protein plays a central role in high density lipoprotein (HDL) and very low density lipoprotein (VLDL) metabolism. This protein has also been shown to inhibit cholesteryl ester transfer protein in plasma. Alternative splicing and the use of alternative promoters results in multiple transcript variants. The GWAS of T2D and AD (Gao et al., 2016) identified six SNPs (rs111789331, rs12721046, rs12721051, rs4420638, rs56131196, and rs66626994) related to the APOC1 gene that showed pleiotropic associations with these health disorders. Among them rs12721046, located in the intron area of APOC1 showed genome-wide significant association with AD in our analyses (FHS data in Table 2.1, and CHS and FHS data in Table 2.2). This SNP is in high LD with rs769449, also found in our study, as well as with rs429358 (Table 2.2), which suggests that it’s effect might be through APOEe4.
4.5. Connection with human longevity
The genetic variants from the same four genes (and often the same SNPs) were detected in GWAS of human longevity (Ang et al., 2008; Deelen et al., 2011; Nebel et al., 2011; Lu et al., 2014; Garatachea et al., 2014; Garatachea et al., 2015; Deelen et al., 2014; Lin et al., 2016; Nebel et al., 2011; Shadyab et al., 2017). The connection of this set of genes with human longevity suggests that they might play some roles in risks of other diseases or in biological processes that influence such risks. Indeed, a number of studies linked these genes with multiple phenotypes including lipid traits (Teslovich et al., 2010; Chang and Chang, 2017), rate of information processing (Lyall et al., 2014; Smith et al., 2010), cardiovascular risk (Smith et al., 2010), inflammation (Rebeck, 2017), cancer (Slattery et al., 2005; Watson et al., 2003), type 2 diabetes (El-Lebedy et al., 2016). These facts indicate that detected genes are likely to have pleiotropic associations with other health disorders. Such pleiotropy is manifested as dependence among chronic conditions at the population level. The results of epidemiologic studies confirm this conjecture. The dependence between AD and other diseases such as cancer and T2D have been detected and described in a number of studies. It is likely that not only genes detected with high level of statistical significance but also genes whose variants showed less significant associations with AD are involved in development of these health disorders. The existence of connections between AD and other health disorders suggests that better understanding AD can be reached in studies of systemic mechanisms of aging related health decline which involve several pathological conditions.
4.6. TOMM40 and mitochondrial dysfunction
Mutations in TOMM40 have a high potential to result in mitochondria dysfunction – a novel hallmark of AD (Grimm et al., 2016). TOMM40 encodes the Tom40 a channel-forming subunit of the translocase of the outer mitochondrial membrane (TOM complex), which plays a role in cytoplasmic peptide and protein transport into the mitochondria. Tom40 pores play the central role in the TOM complex that is made of three Tom40 subunits, which are connected to one another by Tom22 subunits. Each Tom40 subunit forms a pore across the membrane through which precursor proteins can pass from the cytosol into the space between the outer and inner mitochondrial membranes. Tom5, Tom6 and Tom7 subunits are located around the periphery of the pores. About 1500 nuclear-encoded mitochondrial proteins are imported from the cytosol to mitochondria through the TOM complex. The results of experimental studies suggest that altered expression of Tom40 may be linked to a spectrum of neurological diseases (Gottschalk et al., 2014). In particular, APP may thwart the TOM40 pore, inhibiting import of proteins needed for normal mitochondrial functioning. Recent study provided arguments that connection between TOMM40 and AD might be a side effect of the evolutionary development of cognitive function in humans (Larsen et al., 2017).
Strong connection between genetic variants from TOMM40 gene and AD has been detected in a number of genetic association studies of this health disorder. The meta-analysis of polymorphisms in TOMM40 gene (Bao et al., 2016) showed that minor allele of rs157580 (TOMM40 intron area) was significantly associated with a reduced risk of AD whereas minor allele of rs2075650 was significantly associated with an increased risk of AD (TOMM40 intron area). The case-control study of AD in Canadian population confirmed the association of rs2075650 with this disorder (Omoumi et al., 2014). The depression is a risk factor for AD. The connection between rs2075650 and depression has been studied in (McFarquhar et al., 2014). It was found that rs2075650 G allele was a significant risk factor for lifetime depression and, in depressed subjects it was a significant predictor of low extraversion. However, the analyses performed by (He et al., 2016) did not show significant association between rs2075650 and AD risk in Chinese population. The genetic analyses of AD using Italian data showed that TOMM40 gene does not have an APOE independent effect on the risk of developing AD.
To explain the role of mitochondria dysregulation in development of AD the mitochondrial cascade hypothesis has been proposed (Swerdlow and Khan, 2009; Swerdlow, 2011; Swerdlow, 2016). This hypothesis states that, in the sporadic AD, mitochondrial dysfunction is the primary event that causes Aβ deposition, synaptic degeneration, NFTs formation, and neuronal death. Indeed, there is a bulk of studies showing that mitochondrial dysfunction is a common event in AD. Although this theory looks plausible, some problems remains. The dysfunction of mitochondria due to relevant TOMM40 SNPs should lead to inadequate ATP (and/or ROS) production, both of which could negatively affect the neuron and glia metabolism in the bearers of relevant SNPs, possibly accelerating death of respective cell types. If the mitochondrial dysfunction decreases ATP output, then it should also negatively affect global rate and bulk of protein phosphorylation, including tau phosphorylation necessary for NFT formation.
4.7. Other mechanism linking AD and genes found in this study: Involvement of genes regulating cellular stress responses
A number of experimental and molecular biological studies provide evidence about strong involvement of genes regulating cellular stress response in the development of AD and other health disorders. Although most of such genes were not detected with the high level of statistical significance in GWAS of AD, many of them showed nominally significant associations (p < 0.05). This fact may indicate variability of stressors and other genetic and non-genetic factors affecting cellular stress response outcomes. Cell may have different fate, depending on duration of stress and interactions between pathways promoting specific responses.
4.8. Evidence of connection between AD and T2D
The connection of AD and other dementias with T2D has been widely discussed in the literature (Sun and Alkon, 2006; Bornstein et al., 2014; Alam et al., 2016; Abbatecola et al., 2011; Alam et al., 2016; Barbagallo and Dominguez, 2014; Bosco et al., 2011; Correia et al., 2012; Dai and Kamal, 2014; De Felice et al., 2014). The AD and T2D have comparable pathological features related to the abnormal behavior: of the β-amyloid in the brain in case of AD and of the islet amyloid derived from islet amyloid polypeptide in the pancreas in T2D. Although the biological mechanism that links the progression of T2D and AD is not completely understood the growing evidence supports the concept that AD is a metabolic health disorder that is caused by progressive inability of the brain’s to properly respond to insulin and insulin-like growth factor (IGF) stimulation to utilize glucose. The connection between T2D and AD is also manifested in several similarities between the developments of two pathologies (Steen et al., 2005). These include uncontrolled glucose metabolism, glucose toxicity, a direct effect of insulin on amyloid metabolism, oxidative stress, abnormal protein processing, stimulation of inflammatory pathways and inflammation, hypercholesterolemia, dyslipidemia, impaired central nervous response to the adipose tissue-derived hormone leptin, increased oxidative stress and production of advanced glycation end products (Dar et al., 2014).
In addition to similarities between T2D and AD, described above, studies often provide controversial evidence about effects of different factors on disease development. For example, some studies claim that insulin deficiency may cause AD, suggesting that inhibiting the insulin-degrading enzyme (IDE) or the use of other treatments that increase insulin level in the brain could slow down the disease. Other studies provide evidence that hyperinsulinemia causes the disease which implies that treatments increasing insulin level would exacerbate the disease. Further studies are needed to integrate findings linking T2D and AD and provide better understanding of biological mechanisms linking these health disorders.
4.9. AD and cancer
Many recent studies of connection between cancer and AD revealed an inverse association between these disorders (Yashin et al., 2009; Ukraintseva et al., 2010; Akushevich et al., 2013). The quantitative meta-analysis of cohort studies suggested that individuals diagnosed with AD had a decreased risk for incident cancer by 42%, and patients with a history of cancer had a 37% decreased risk of AD. This study demonstrated an inverse association between cancer and AD (Ma et al., 2014). Small inverse associations between cancer and AD were detected in (Schmidt et al., 2017). The authors concluded that these associations might be caused by ascertainment bias due to decreased awareness of non-melanoma skin cancers (NMSC) in persons with undiagnosed early cognitive impairment or by confounding from a more neuroprotective lifestyle among persons with NMSC. The obesity-related mechanism may be involved in trade-off between cancer and AD (Nixon, 2017). The leptin and adiponectin produced by adipose tissues may be responsible for this property. Leptin has cancer-stimulating and AD-inhibiting actions while, in contrast, adiponectin has cancer-inhibiting and AD-stimulating properties. These opposing actions, mediated through p53, Wnt, and other signaling pathways, may account for the inverse cancer/AD relationship.
Recently it was found that the use of androgen-deprivation therapy (ADT) – the preferred first-line treatment for advanced prostate cancer is associated with an increased risk of developing AD (Jhan et al., 2017). Cancer survivors have reduced chances of developing AD and a lower burden of neurofibrillary tangle deposition. This conclusion resulted from studying the risks of developing AD among participants with and without a history of cancer at autopsy (Yarchoan et al., 2017). At autopsy, participants with a history of cancer had significantly fewer paired helical filament (PHF) tau tangles (p < 0.001) than participants without a history of cancer, but similar levels of Aβ. Comprehensive review of cellular pathways with roles in cancers, cell survival, growth, proliferation, development, aging, and also contributing to AD disease showed the possibility of inverse relationship between AD and cancer (Shafi, 2016). Many factors that are upregulated in any cancer to sustain growth and survival are downregulated in AD contributing to neuro-degeneration. However, cancer did not provide protection from AD in retrospective cohort study using data from the Utah Population Database (Hanson et al., 2017). The authors concluded that taking mortality selection in heterogeneous population into account may explain biased associations.
Note that SNPs from most of genes involved in pleiotropic associations described above did not reach genome wide level of statistical significance in GWAS of AD in our study. Recently the association with AD has been tested in case-control studies for 695 candidate genes (Sun et al., 2014), however, their roles in risks of other health disorders remain unclear.
To test the presence of genetic variants having pleiotropic effects on diseases discussed above we performed GWAS of AD, cancer and T2D using HRS data and identified genetic variants that have pleiotropic effects on these health disorders. In these analyses for each pair of diseases three sets of genetic variants emerged. They include SNPs whose minor alleles have positive, negative and opposite effects on corresponding health disorders. The results of these analyses are shown in Fig. 3.
Fig. 3.
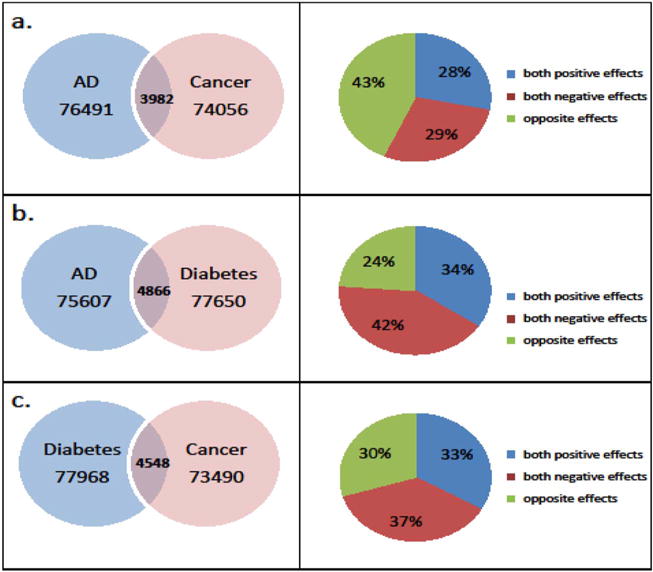
Left panel. The Venn diagrams of intersection of SNPs obtained from GWAS analyses on HRS white individuals using logistic regression: a). between Alzheimer’s/dementia (AD) and cancers (non-skin); b). between Alzheimer’s/dementia (AD) and diabetes; c). between cancers (non-skin) and diabetes. Right panel. Distribution of the overlapped SNPs by the effects corresponding to the left panel.
Table 1S (in Supplementary materials) summarizes the results of these analyses for ten most significant SNPs detected in GWAS for each health disorder and for each intersection with both positive, both negative and the opposite genetic associations with different pairs of selected diseases (AD and cancer, AD and T2D, cancer and T2D). These results show that two SNPs related to detected genes on chromosome 19 have pleiotropic associations with selected health disorders. Specifically, rs34095326 (in the intron of TOMM40) has negative associations with both AD and cancer; rs405697 has negative association with AD and positive association with T2D. SNP rs34095326 SNP is in strong LD (D′ = 1) with rs15782 in Table 2.1, and rs405697 is in strong LD with rs405509 (D′ = 0.98), rs769450 (D′ = 0.99), and rs157580 (D′ = 0.94) (see Table 2.1). Two more SNPs from chromosome 19 showed pleotropic associations with selected diseases: rs8109796 is positively associated with AD and negatively associated with cancer; rs10411527 is negatively associated with both AD and T2D. However, these SNPs are not related to any specific gene. Note that other SNPs shown in Table 1S showed only nominal levels of statistical significance in GWAS for specific health disorders. Therefore, many of detected connections are likely to be false-positive. More analyses that involve the results of clinical, experimental, and molecular biological studies are needed to separate the true-positive pleiotropic associations with selected diseases from the false-positive ones.
5. Conclusions
The results of GWAS of AD in our study confirm findings from earlier genetic studies of this health disorder. The literature summarizing results of clinical, experimental, and molecular biological studies provides evidence that these genes are highly pleiotropic and participate in signaling and metabolic pathways linked with the origin and progression of AD, as well as several other major health disorders such as cancer, T2D and viral infections. This fact suggests that studying common components of the biological mechanisms may be mutually beneficial for better understanding the etiology of each health disorder.
One hypothesis that converge effects of APOE, APOC1 and PVRL2 (NECTIN2) on AD could be that proper maintenance of cell junctions and ECM structure relevant to functions of PVRL2 may help prevent the spread of viral infection, and such infection could be a common risk factor for AD and all-cause mortality. Proper cell junctions are important to control brain blood barrier (BBB) permeability and protect the brain from infection, especially from viruses, which may play a role in AD (Itzhaki, 2016). Performance of lipid transport particles, such as LDL and HDL, depends on APOE and APOC1, which may be also involved in host resistance to infection since they can modulate virus life cycle and secretion (Chiba-Falek et al., 2012). Thus, host resistance to infection could potentially contribute to a common mechanism for the involvement of most of the identified genes (APOE, APOC1, PVRL2) in AD.
Some studies also provide evidence about involvement of genes responsible for regulation of cellular stress response in development of AD and other health disorders (Bell et al., 2016; Viana et al., 2012; Shah et al., 2017). Although most of such genes were not detected with high level of statistical significance in GWAS of AD many of them showed nominally significant associations (p < 0.05) with this health disorder. These results confirm that AD is likely to be a highly heterogeneous phenotypic trait.
Aging process is likely to be the major factor responsible for the similarity of pathological pathways involved in the development of AD and other diseases of the elderly by increasing organism’s vulnerability to different kinds of disturbances (Fulop, 2016). Some of these stresses being transformed to cellular levels induce cellular stress response. The pathological development might be the consequence of the cellular stress response, in which stress is persistent, or damage is not possible to repair or compensate to functional level. The cellular stress response genes are involved in development of cancer and other aging related chronic pathologies (Cunard, 2015; Oakes and Papa, 2015; Alasiri et al., 2017). This means that better understanding AD requires integration of the research results obtained in genetic association studies with findings obtained in clinical, experimental, and molecular biological studies of aging, as well in studies of other health disorders.
Supplementary Material
Acknowledgments
The analyses described in this paper were supported by the National Institutes of Health/National Institute of Aging (NIH/NIA) grants R01AG046860, P01AG043352, P30AG034424. The FHS project is conducted and supported by the National Heart, Lung and Blood Institute (NHLBI) in collaboration with Boston University (N01 HC25195). The FHS data used for the analyses were obtained through the database of Genotypes and Phenotypes (dbGaP). The authors acknowledge the investigators that contributed the phenotype and genotype data for this study. This manuscript was not prepared in collaboration with investigators of the FHS and does not necessarily reflect the opinions or views of the FHS, Boston University or the NHLBI. The CHS project was supported by contract numbers N01-HC-85079 through N01-HC-85086, N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133, grant number U01 HL080295 from the National Heart, Lung, and Blood Institute, with additional contribution from the National Institute of Neurological Disorders and Stroke. Additional support was provided through R01 AG-15928, R01 AG-20098, and AG-027058 from the National Institute on Aging, R01 HL-075366 from the National Heart, Lung and Blood Institute, and the University of Pittsburgh Claude D. Pepper Older Americans Independence Center P30-AG-024827. A full list of principal CHS investigators and institutions can be found at http://www.chs-nhlbi.org/pi.htm. The Health and Retirement Study data is sponsored by the National Institute on Aging (grant number U01AG009740) and is conducted by the University of Michigan. HRS Restricted Data Access #2014-008. The Health and Retirement Study genetic data is sponsored by the National Institute on Aging (grant numbers U01AG009740, RC2AG036495, and RC4AG039029) and was conducted by the University of Michigan. LOADFS: Funding support for the “Genetic Consortium for Late Onset Alzheimer’s Disease” was provided through the Division of Neuroscience, NIA. The Genetic Consortium for Late Onset Alzheimer’s Disease includes a genome-wide association study funded as part of the Division of Neuroscience, NIA. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by Genetic Consortium for Late Onset Alzheimer’s Disease.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.exger.2017.10.020.
References
- Abbatecola AM, Olivieri F, Corsonello A, Antonicelli R, Corica F, Lattanzio F. Genome-wide association studies: is there a genotype for cognitive decline in older persons with type 2 diabetes? Curr Pharm Des. 2011;17(4):347–356. doi: 10.2174/138161211795164239. [DOI] [PubMed] [Google Scholar]
- Abe S, Tokoro F, Matsuoka R, Arai M, Noda T, Watanabe S, Horibe H, Fujimaki T, Oguri M, Kato K, Minatoguchi S, Yamada Y. Association of genetic variants with dyslipidemia. Mol Med Rep. 2015;12(4):5429–5436. doi: 10.3892/mmr.2015.4081. [DOI] [PubMed] [Google Scholar]
- Akushevich I, Kravchenko J, Ukraintseva S, Arbeev K, Kulminski A, Yashin AI. Morbidity risks among older adults with pre-existing age-related diseases. Exp Gerontol. 2013;48(12):1395–1401. doi: 10.1016/j.exger.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam F, Islam MA, Sasongko TH, Gan SH. Type 2 diabetes mellitus and Alzheimer’s disease: bridging the pathophysiology and management. Curr Pharm Des. 2016;22(28):4430–4442. doi: 10.2174/1381612822666160527160236. [DOI] [PubMed] [Google Scholar]
- Alasiri G, Fan LY, Zona S, Goldsbrough IG, Ke HL, Auner HW, Lam EW. ER stress and cancer: The FOXO forkhead transcription factor link. Mol Cell Endocrinol. 2017 doi: 10.1016/j.mce.2017.05.027. (pii: S0303-7207(17)30296-4) [Epub ahead of print] Review. [DOI] [PubMed] [Google Scholar]
- Almire C, Bertrand P, Ruminy P, Maingonnat C, Wlodarska I, Martin-Subero JI, Siebert R, Tilly H, Bastard C. PVRL2 is translocated to the TRA@ locus in t (14;19)(q11;q13)-positive peripheral T-cell lymphomas. Genes Chromosomes Cancer. 2007;46(11):1011–1018. doi: 10.1002/gcc.20490. [DOI] [PubMed] [Google Scholar]
- Ang LS, Cruz RP, Hendel A, Granville DJ. Apolipoprotein E, an important player in longevity and age-related diseases. Exp Gerontol. 2008;43(7):615–622. doi: 10.1016/j.exger.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Bachmeier C, Shackleton B, Ojo J, Paris D, Mullan M, Crawford F. Apolipoprotein E isoform-specific effects on lipoprotein receptor processing. NeuroMolecular Med. 2014;16(4):686–696. doi: 10.1007/s12017-014-8318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnoli S, Piaceri I, Tedde A, Bessi V, Bracco L, Sorbi S, Nacmias B. TOMM40 polymorphisms in Italian Alzheimer’s disease and frontotemporal dementia patients. Neurol Sci. 2013;34(6):995–998. doi: 10.1007/s10072-013-1425-6. [DOI] [PubMed] [Google Scholar]
- Bao J, Wang XJ, Mao ZF. Associations between genetic variants in 19p13 and 19q13 regions and susceptibility to Alzheimer disease: a meta-analysis. Med Sci Monit. 2016;22:234–243. doi: 10.12659/MSM.895622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbagallo M, Dominguez LJ. Type 2 diabetes mellitus and Alzheimer’s disease. World J Diabetes. 2014;5(6):889–893. doi: 10.4239/wjd.v5.i6.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassendine MF, Sheridan DA, Bridge SH, Felmlee DJ, Neely RD. Lipids and HCV. Semin Immunopathol. 2013;35(1):87–100. doi: 10.1007/s00281-012-0356-2. [DOI] [PubMed] [Google Scholar]
- Bekris LM, Galloway NM, Millard S, Lockhart D, Li G, Galasko DR, Farlow MR, Clark CM, Quinn JF, Kaye JA, Schellenberg GD, Leverenz JB, Seubert P, Tsuang DW, Peskind ER, Yu CE. Amyloid precursor protein (APP) processing genes and cerebrospinal fluid APP cleavage product levels in Alzheimer’s disease. Neurobiol Aging. 2011;32(3):556.e513–523. doi: 10.1016/j.neurobiolaging.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell MC, Meier SE, Ingram AL, Abisambra JF. PERK-opathies: an endoplasmic reticulum stress mechanism underlying neurodegeneration. Curr Alzheimer Res. 2016;13(2):150–163. doi: 10.2174/1567205013666151218145431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein NM, Brainin M, Guekht A, Skoog I, Korczyn AD. Diabetes and the brain: issues and unmet needs. Neurol Sci. 2014;35(7):995–1001. doi: 10.1007/s10072-014-1797-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J Cell Mol Med. 2011;15(9):1807–1821. doi: 10.1111/j.1582-4934.2011.01318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter DB. The interaction of amyloid-beta with ApoE. Subcell Biochem. 2005;38:255–272. doi: 10.1007/0-387-23226-5_13. [DOI] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3(89):89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerf E, Gustot A, Goormaghtigh E, Ruysschaert JM, Raussens V. High ability of apolipoprotein E4 to stabilize amyloid-beta peptide oligomers, the pathological entities responsible for Alzheimer’s disease. FASEB J. 2011;25(5):1585–1595. doi: 10.1096/fj.10-175976. [DOI] [PubMed] [Google Scholar]
- Chang TY, Chang C. ApoE and lipid homeostasis in Alzheimer’s disease: introduction to the thematic review series. J Lipid Res. 2017;58(5):823. doi: 10.1194/jlr.R075697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba-Falek O, Linnertz C, Guyton J, Gardner SD, Roses AD, McCarthy JJ, Patel K. Pleiotropy and allelic heterogeneity in the TOMM40-APOE genomic region related to clinical and metabolic features of hepatitis C infection. Hum Genet. 2012;131(12):1911–1920. doi: 10.1007/s00439-012-1220-0. [DOI] [PubMed] [Google Scholar]
- Correia SC, Santos RX, Carvalho C, Cardoso S, Candeias E, Santos MS, Oliveira CR, Moreira PI. Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012;1441:64–78. doi: 10.1016/j.brainres.2011.12.063. [DOI] [PubMed] [Google Scholar]
- Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, Benitez BA, Jeng AT, Skorupa T, Carrell D, Bertelsen S, Bailey M, McKean D, Shulman JM, De Jager PL, Chibnik L, Bennett DA, Arnold SE, Harold D, Sims R, Gerrish A, Williams J, Van Deerlin VM, Lee VM, Shaw LM, Trojanowski JQ, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD, Peskind ER, Galasko D, Fagan AM, Holtzman DM, Morris JC, Goate AM. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron. 2013;78(2):256–268. doi: 10.1016/j.neuron.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunard R. Endoplasmic reticulum stress in the diabetic kidney, the good, the bad and the ugly. J Clin Med. 2015;4(4):715–740. doi: 10.3390/jcm4040715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Agostino RB, Kannel WB, Belanger AJ, Sytkowski PA. Trends in CHD and risk factors at age 55–64 in the Framingham Study. Int J Epidemiol. 1989;18(3 Suppl 1):S67–72. [PubMed] [Google Scholar]
- Dai Y, Kamal MA. Fighting Alzheimer’s disease and type 2 diabetes: pathological links and treatment strategies. CNS Neurol Disord Drug Targets. 2014;13(2):271–282. doi: 10.2174/18715273113126660134. [DOI] [PubMed] [Google Scholar]
- Dar TA, Sheikh IA, Ganie SA, Ali R, Singh LR, Gan SH, Kamal MA, Zargar MA. Molecular linkages between diabetes and Alzheimer’s disease: current scenario and future prospects. CNS Neurol Disord Drug Targets. 2014;13(2):290–298. doi: 10.2174/18715273113126660135. [DOI] [PubMed] [Google Scholar]
- Dawber TR. The Framingham Study: The Epidemiology of Atherosclerotic Disease. Harvard University Press; Cambridge, MA: 1980. [Google Scholar]
- De Chiara G, Marcocci ME, Sgarbanti R, Civitelli L, Ripoli C, Piacentini R, Garaci E, Grassi C, Palamara AT. Infectious agents and neurodegeneration. Mol Neurobiol. 2012;46(3):614–638. doi: 10.1007/s12035-012-8320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Lourenco MV, Ferreira ST. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement. 2014;10(1 Suppl):S26–32. doi: 10.1016/j.jalz.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Deelen J, Beekman M, Uh HW, Helmer Q, Kuningas M, Christiansen L, Kremer D, van der Breggen R, Suchiman HE, Lakenberg N, van den Akker EB, Passtoors WM, Tiemeier H, van Heemst D, de Craen AJ, Rivadeneira F, de Geus EJ, Perola M, van der Ouderaa FJ, Gunn DA, Boomsma DI, Uitterlinden AG, Christensen K, van Duijn CM, Heijmans BT, Houwing-Duistermaat JJ, Westendorp RG, Slagboom PE. Genome-wide association study identifies a single major locus contributing to survival into old age; the APOE locus revisited. Aging Cell. 2011;10(4):686–698. doi: 10.1111/j.1474-9726.2011.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deelen J, Beekman M, Uh HW, Broer L, Ayers KL, Tan Q, Kamatani Y, Bennet AM, Tamm R, Trompet S, Guethbjartsson DF, Flachsbart F, Rose G, Viktorin A, Fischer K, Nygaard M, Cordell HJ, Crocco P, van den Akker EB, Bohringer S, Helmer Q, Nelson CP, Saunders GI, Alver M, Andersen-Ranberg K, Breen ME, van der Breggen R, Caliebe A, Capri M, Cevenini E, Collerton JC, Dato S, Davies K, Ford I, Gampe J, Garagnani P, de Geus EJ, Harrow J, van Heemst D, Heijmans BT, Heinsen FA, Hottenga JJ, Hofman A, Jeune B, Jonsson PV, Lathrop M, Lechner D, Martin-Ruiz C, McNerlan SE, Mihailov E, Montesanto A, Mooijaart SP, Murphy A, Nohr EA, Paternoster L, Postmus I, Rivadeneira F, Ross OA, Salvioli S, Sattar N, Schreiber S, Stefansson H, Stott DJ, Tiemeier H, Uitterlinden AG, Westendorp RG, Willemsen G, Samani NJ, Galan P, Sorensen TI, Boomsma DI, Jukema JW, Rea IM, Passarino G, de Craen AJ, Christensen K, Nebel A, Stefansson K, Metspalu A, Magnusson P, Blanche H, Christiansen L, Kirkwood TB, van Duijn CM, Franceschi C, Houwing-Duistermaat JJ, Slagboom PE. Genome-wide association meta-analysis of human longevity identifies a novel locus conferring survival beyond 90 years of age. Hum Mol Genet. 2014;23(16):4420–4432. doi: 10.1093/hmg/ddu139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Lebedy D, Raslan HM, Mohammed AM. Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc Diabetol. 2016;15:12. doi: 10.1186/s12933-016-0329-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop T. Biological research into aging: from cells to clinic. Biogerontology. 2016;17(1):1–6. doi: 10.1007/s10522-016-9633-0. [DOI] [PubMed] [Google Scholar]
- Gao L, Cui Z, Shen L, Ji HF. Shared genetic etiology between type 2 diabetes and Alzheimer’s disease identified by bioinformatics analysis. J Alzheimers Dis. 2016;50(1):13–17. doi: 10.3233/JAD-150580. [DOI] [PubMed] [Google Scholar]
- Garai K, Verghese PB, Baban B, Holtzman DM, Frieden C. The binding of apolipoprotein E to oligomers and fibrils of amyloid-beta alters the kinetics of amyloid aggregation. Biochemistry. 2014;53(40):6323–6331. doi: 10.1021/bi5008172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garatachea N, Emanuele E, Calero M, Fuku N, Arai Y, Abe Y, Murakami H, Miyachi M, Yvert T, Verde Z, Zea MA, Venturini L, Santiago C, Santos-Lozano A, Rodriguez-Romo G, Ricevuti G, Hirose N, Rabano A, Lucia A. ApoE gene and exceptional longevity: insights from three independent cohorts. Exp Gerontol. 2014;53:16–23. doi: 10.1016/j.exger.2014.02.004. [DOI] [PubMed] [Google Scholar]
- Garatachea N, Marin PJ, Santos-Lozano A, Sanchis-Gomar F, Emanuele E, Lucia A. The ApoE gene is related with exceptional longevity: a systematic review and meta-analysis. Rejuvenation Res. 2015;18(1):3–13. doi: 10.1089/rej.2014.1605. [DOI] [PubMed] [Google Scholar]
- Gottschalk WK, Lutz MW, He YT, Saunders AM, Burns DK, Roses AD, Chiba-Falek O. The broad impact of TOM40 on neurodegenerative diseases in aging. J Park Dis Alzheimers Dis. 2014;1(1) doi: 10.13188/2376-922X.1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 2010;119(5):523–541. doi: 10.1007/s00401-010-0679-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf M, Reif S, Hecht K, Kroell T, Nuessler V, Schmetzer H. Expression of poliovirus receptor-related proteins PRR1 and PRR2 in acute myeloid leukemia: first report of surface marker analysis, contribution to diagnosis, prognosis and implications for future therapeutical strategies. Eur J Haematol. 2005;75(6):477–484. doi: 10.1111/j.1600-0609.2005.00539.x. [DOI] [PubMed] [Google Scholar]
- Grimm A, Friedland K, Eckert A. Mitochondrial dysfunction: the missing link between aging and sporadic Alzheimer’s disease. Biogerontology. 2016;17(2):281–296. doi: 10.1007/s10522-015-9618-4. [DOI] [PubMed] [Google Scholar]
- Hanson HA, Horn KP, Rasmussen KM, Hoffman JM, Smith KR. Is cancer protective for subsequent Alzheimer’s Disease Risk? Evidence from the Utah Population Database. J Gerontol B Psychol Sci Soc Sci. 2017;72(6):1032–1043. doi: 10.1093/geronb/gbw040. http://dx.doi.org/10.1093/geronb/gbw040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, Heun R, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Serrano-Pozo A, Hori Y, Adams KW, Takeda S, Banerji AO, Mitani A, Joyner D, Thyssen DH, Bacskai BJ, Frosch MP, Spires-Jones TL, Finn MB, Holtzman DM, Hyman BT. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J Neurosci. 2012;32(43):15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Li C, Yang Y, Li Y, Wang Y, Yang H, Jin T, Chen S. Meta-analysis of the rs2075650 polymorphism and risk of Alzheimer disease. Aging Clin Exp Res. 2016;28(5):805–811. doi: 10.1007/s40520-015-0489-y. [DOI] [PubMed] [Google Scholar]
- Holliday EG, Smith AV, Cornes BK, Buitendijk GH, Jensen RA, Sim X, Aspelund T, Aung T, Baird PN, Boerwinkle E, Cheng CY, van Duijn CM, Eiriksdottir G, Gudnason V, Harris T, Hewitt AW, Inouye M, Jonasson F, Klein BE, Launer L, Li X, Liew G, Lumley T, McElduff P, McKnight B, Mitchell P, Psaty BM, Rochtchina E, Rotter JI, Scott RJ, Tay W, Taylor K, Teo YY, Uitterlinden AG, Viswanathan A, Xie S, Vingerling JR, Klaver CC, Tai ES, Siscovick D, Klein R, Cotch MF, Wong TY, Attia J, Wang JJ. Insights into the genetic architecture of early stage age-related macular degeneration: a genome-wide association study meta-analysis. PLoS ONE. 2013;8(1):e53830. doi: 10.1371/journal.pone.0053830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh TV, Davis AA, Ulrich JD, Holtzman DM. Apolipoprotein E and Alzheimer’s disease: the influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J Lipid Res. 2017;58(5) doi: 10.1194/jlr.R075481. http://dx.doi.org/10.1194/jlr.R075481. Epub 2017 Feb 27. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki RF. Herpes and Alzheimer’s disease: subversion in the central nervous system and how it might be halted. J Alzheimers Dis. 2016;54(4):1273–1281. doi: 10.3233/JAD-160607. [DOI] [PubMed] [Google Scholar]
- Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, Bullido MJ, Carter C, Clerici M, Cosby SL, Del Tredici K, Field H, Fulop T, Grassi C, Griffin WS, Haas J, Hudson AP, Kamer AR, Kell DB, Licastro F, Letenneur L, Lovheim H, Mancuso R, Miklossy J, Otth C, Palamara AT, Perry G, Preston C, Pretorius E, Strandberg T, Tabet N, Taylor-Robinson SD, Whittum-Hudson JA. Microbes and Alzheimer’s disease. J Alzheimers Dis. 2016;51(4):979–984. doi: 10.3233/JAD-160152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhan JH, Yang YH, Chang YH, Guu SJ, Tsai CC. Hormone therapy for prostate cancer increases the risk of Alzheimer’s disease: a nationwide 4-year longitudinal cohort study. Aging Male. 2017:1–6. doi: 10.1080/13685538.2016.1271782. [DOI] [PubMed] [Google Scholar]
- Karabulut M, Gunaldi M, Alis H, Afsar CU, Karabulut S, Serilmez M, Akarsu C, Seyit H, Aykan NF. Serum nectin-2 levels are diagnostic and prognostic in patients with colorectal carcinoma. Clin Transl Oncol. 2016;18(2):160–171. doi: 10.1007/s12094-015-1348-1. [DOI] [PubMed] [Google Scholar]
- Kettunen J, Tukiainen T, Sarin AP, Ortega-Alonso A, Tikkanen E, Lyytikainen LP, Kangas AJ, Soininen P, Wurtz P, Silander K, Dick DM, Rose RJ, Savolainen MJ, Viikari J, Kahonen M, Lehtimaki T, Pietilainen KH, Inouye M, McCarthy MI, Jula A, Eriksson J, Raitakari OT, Salomaa V, Kaprio J, Jarvelin MR, Peltonen L, Perola M, Freimer NB, Ala-Korpela M, Palotie A, Ripatti S. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat Genet. 2012;44(3):269–276. doi: 10.1038/ng.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova R, Fitz NF, Lefterov I. The role of ATP-binding cassette transporter A1 in Alzheimer’s disease and neurodegeneration. Biochim Biophys Acta. 2010;1801(8):824–830. doi: 10.1016/j.bbalip.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa Y, Honma K, Takemasa I, Nakamori S, Kita-Matsuo H, Motoori M, Nagano H, Dono K, Ochiya T, Monden M, Kato K. Central genetic alterations common to all HCV-positive, HBV-positive and non-B, non-C hepatocellular carcinoma: a new approach to identify novel tumor markers. Int J Oncol. 2006;28(2):383–391. [PubMed] [Google Scholar]
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269(38):23403–23406. [PubMed] [Google Scholar]
- Larsen PA, Lutz MW, Hunnicutt KE, Mihovilovic M, Saunders AM, Yoder AD, Roses AD. The Alu neurodegeneration hypothesis: A primate-specific mechanism for neuronal transcription noise, mitochondrial dysfunction, and manifestation of neurodegenerative disease. Alzheimers Dement. 2017;13(7):828–838. doi: 10.1016/j.jalz.2017.01.017. http://dx.doi.org/10.1016/j.jalz.2017.01.017. Epub 2017 Feb 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Cheng R, Graff-Radford N, Foroud T, Mayeux R. Analyses of the National Institute on Aging Late-Onset Alzheimer’s Disease Family Study: implication of additional loci. Arch Neurol. 2008;65(11):1518–1526. doi: 10.1001/archneur.65.11.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Zhang Y, Yan D, Liao X, Gong G, Hu J, Fu Y, Cai W. Association of common variants in TOMM40/APOE/APOC1 region with human longevity in a Chinese population. J Hum Genet. 2016;61(4):323–328. doi: 10.1038/jhg.2015.150. [DOI] [PubMed] [Google Scholar]
- Logue MW, Schu M, Vardarajan BN, Buros J, Green RC, Go RC, Griffith P, Obisesan TO, Shatz R, Borenstein A, Cupples LA, Lunetta KL, Fallin MD, Baldwin CT, Farrer LA. A comprehensive genetic association study of Alzheimer disease in African Americans. Arch Neurol. 2011;68(12):1569–1579. doi: 10.1001/archneurol.2011.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Guan H, Gong B, Liu X, Zhu R, Wang Y, Qian J, Zhou T, Lan X, Wang P, Lin Y, Ma S, Lin H, Zhu X, Chen R, Zhu X, Shi Y, Yang Z. Genetic variants in PVRL2-TOMM40-APOE region are associated with human longevity in a Han Chinese population. PLoS ONE. 2014;9(6):e99580. doi: 10.1371/journal.pone.0099580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyall DM, Harris SE, Bastin ME, Munoz Maniega S, Murray C, Lutz MW, Saunders AM, Roses AD, Valdes Hernandez Mdel C, Royle NA, Starr JM, Porteous DJ, Wardlaw JM, Deary IJ. Are APOE varepsilon genotype and TOMM40 poly-T repeat length associations with cognitive ageing mediated by brain white matter tract integrity? Transl Psychiatry. 2014;e449:4. doi: 10.1038/tp.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma LL, Yu JT, Wang HF, Meng XF, Tan CC, Wang C, Tan L. Association between cancer and Alzheimer’s disease: systematic review and meta-analysis. J Alzheimers Dis. 2014;42(2):565–573. doi: 10.3233/JAD-140168. [DOI] [PubMed] [Google Scholar]
- Ma XY, Yu JT, Wang W, Wang HF, Liu QY, Zhang W, Tan L. Association of TOMM40polymorphisms with late-onset Alzheimer’s disease in a Northern Han Chinesepopulation. Neuromolecular Med. 2013;15(2):279–287. doi: 10.1007/s12017-012-8217-7. http://dx.doi.org/10.1007/s12017-012-8217-7. Epub 2013 Jan 4. [DOI] [PubMed] [Google Scholar]
- Ma C, Zhang Y, Li X, Zhang J, Chen K, Liang Y, Chen Y, Liu Z, Zhang Z. Is there a significant interaction effect between apolipoprotein E rs405509 T/T and epsilon4 genotypes on cognitive impairment and gray matter volume? Eur J Neurol. 2016;23(9):1415–1425. doi: 10.1111/ene.13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med (Berl) 2016;94(7):739–746. doi: 10.1007/s00109-016-1427-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood SS, Levy D, Vasan RS, Wang TJ. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet. 2014;383(9921):999–1008. doi: 10.1016/S0140-6736(13)61752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manelli AM, Stine WB, Van Eldik LJ, LaDu MJ. ApoE and Abeta1-42 interactions: effects of isoform and conformation on structure and function. J Mol Neurosci. 2004;23(3):235–246. doi: 10.1385/JMN:23:3:235. [DOI] [PubMed] [Google Scholar]
- McFarquhar M, Elliott R, McKie S, Thomas E, Downey D, Mekli K, Toth ZG, Anderson IM, Deakin JF, Juhasz G. TOMM40 rs2075650 may represent a new candidate gene for vulnerability to major depressive disorder. Neuropsychopharmacology. 2014;39(7):1743–1753. doi: 10.1038/npp.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy J. Emerging roles of pathogens in Alzheimer disease. Expert Rev Mol Med. 2011;13:e30. doi: 10.1017/S1462399411002006. [DOI] [PubMed] [Google Scholar]
- Morgan D. The role of microglia in antibody-mediated clearance of amyloid-beta from the brain. CNS Neurol Disord Drug Targets. 2009;8(1):7–15. doi: 10.2174/187152709787601821. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Mistur R, Switalski R, Brys M, Glodzik L, Rich K, Pirraglia E, Tsui W, De Santi S, de Leon MJ. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology. 2009;72(6):513–520. doi: 10.1212/01.wnl.0000333247.51383.43. http://dx.doi.org/10.1212/01.wnl.0000333247.51383.43. Epub 2008 Nov 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Romaine SP, Lettre G, Wilson JG, Volcik KA, Tsai MY, Taylor HA, Jr, Schreiner PJ, Rotter JI, Rich SS, Redline S, Psaty BM, Papanicolaou GJ, Ordovas JM, Liu K, Krauss RM, Glazer NL, Gabriel SB, Fornage M, Cupples LA, Buxbaum SG, Boerwinkle E, Ballantyne CM, Kathiresan S, Rader DJ. Multi-ethnic analysis of lipid-associated loci: the NHLBI CARe project. PLoS ONE. 2012;7(5):e36473. doi: 10.1371/journal.pone.0036473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Jun G, Reitz C, Kunkle BW, Perry W, Park YS, Beecham GW, Rajbhandary RA, Hamilton-Nelson KL, Wang LS, Kauwe JS, Huentelman MJ, Myers AJ, Bird TD, Boeve BF, Baldwin CT, Jarvik GP, Crane PK, Rogaeva E, Barmada MM, Demirci FY, Cruchaga C, Kramer PL, Ertekin-Taner N, Hardy J, Graff-Radford NR, Green RC, Larson EB, St George-Hyslop PH, Buxbaum JD, Evans DA, Schneider JA, Lunetta KL, Kamboh MI, Saykin AJ, Reiman EM, De Jager PL, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Martin ER, Haines JL, Mayeux RP, Farrer LA, Schellenberg GD, Pericak-Vance MA, Albert MS, Albin RL, Apostolova LG, Arnold SE, Barber R, Barnes LL, Beach TG, Becker JT, Beekly D, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Cantwell LB, Cao C, Carlson CS, Carney RM, Carrasquillo MM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cribbs DH, Crocco EA, DeCarli C, DeKosky ST, Dick M, Dickson DW, Duara R, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Kramer JH, LaFerla FM, Lah JJ, Leverenz JB, Levey AI, Li G, Lieberman AP, Lin CF, Lopez OL, Lyketsos CG, Mack WJ, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Murrell JR, Olichney JM, Pankratz VS, Parisi JE, Paulson HL, Peskind E, Petersen RC, Pierce A, Poon WW, Potter H, Quinn JF, Raj A, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosen HJ, Rosenberg RN, Sano M, Schneider LS, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Tanzi RE, Thornton-Wells TA, Trojanowski JQ, Troncoso JC, Valladares O, Van Deerlin VM, Van Eldik LJ, Vardarajan BN, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Wishnek S, Woltjer RL, Wright CB, Younkin SG, Yu CE, Yu L. Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease: a genome-wide association study. JAMA Neurol. 2014;71(11):1394–1404. doi: 10.1001/jamaneurol.2014.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebel A, Kleindorp R, Caliebe A, Nothnagel M, Blanche H, Junge O, Wittig M, Ellinghaus D, Flachsbart F, Wichmann HE, Meitinger T, Nikolaus S, Franke A, Krawczak M, Lathrop M, Schreiber S. A genome-wide association study confirms APOE as the major gene influencing survival in long-lived individuals. Mech Ageing Dev. 2011;132(6–7):324–330. doi: 10.1016/j.mad.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Nixon DW. The inverse relationship between cancer and Alzheimer’s Disease: A possible mechanism. Curr Alzheimer Res. 2017;14(8):883–893. doi: 10.2174/1567205014666170216152905. http://dx.doi.org/10.2174/1567205014666170216152905. [DOI] [PubMed] [Google Scholar]
- Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoumi A, Fok A, Greenwood T, Sadovnick AD, Feldman HH, Hsiung GY. Evaluation of late-onset Alzheimer disease genetic susceptibility risks in a Canadian population. Neurobiol Aging. 2014;35(4):936.e935–912. doi: 10.1016/j.neurobiolaging.2013.09.025. [DOI] [PubMed] [Google Scholar]
- Ortega-Rojas J, Morales L, Guerrero E, Arboleda-Bustos CE, Mejia A, Forero D, Lopez L, Pardo R, Arboleda G, Yunis J, Arboleda H. Association analysis of polymorphisms in TOMM40, CR1, PVRL2, SORL1, PICALM, and 14q32.13 regions in Colombian Alzheimer disease patients. Alzheimer Dis Assoc Disord. 2016;30(4):305–309. doi: 10.1097/WAD.0000000000000142. [DOI] [PubMed] [Google Scholar]
- Oshima T, Sato S, Kato J, Ito Y, Watanabe T, Tsuji I, Hori A, Kurokawa T, Kokubo T. Nectin-2 is a potential target for antibody therapy of breast and ovarian cancers. Mol Cancer. 2013;12:60. doi: 10.1186/1476-4598-12-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcellini E, Carbone I, Ianni M, Licastro F. Alzheimer’s disease gene signature says: beware of brain viral infections. Immun Ageing. 2010;7:16. doi: 10.1186/1742-4933-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Ramos Bernardes da Silva Filho S, Oliveira Barbosa JH, Rondinoni C, Dos Santos AC, Garrido Salmon CE, da Costa Lima NK, Ferriolli E, Moriguti JC. Neuro-degeneration profile of Alzheimer’s patients: A brain morphometry study. Neuroimage Clin. 2017;15:15–24. doi: 10.1016/j.nicl.2017.04.001. http://dx.doi.org/10.1016/j.nicl.2017.04.001. eCollection 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeck GW. The role of APOE on lipid homeostasis and inflammation in normal brains. J Lipid Res. 2017;58(8):1493–1499. doi: 10.1194/jlr.R075408. http://dx.doi.org/10.1194/jlr.R075408. Epub 2017 Mar 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick SM, Bilgel M, Moghekar A, An Y, Cai Q, Wang MC, Thambisetty M, Prince JL, Zhou Y, Soldan A, Wong DF, O’Brien RJ, Ferrucci L, Albert MS. Changes in Abeta biomarkers and associations with APOE genotype in 2 longitudinal cohorts. Neurobiol Aging. 2015;36(8):2333–2339. doi: 10.1016/j.neurobiolaging.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roses A, Sundseth S, Saunders A, Gottschalk W, Burns D, Lutz M. Understanding the genetics of APOE and TOMM40 and role of mitochondrial structure and function in clinical pharmacology of Alzheimer’s disease. Alzheimers Dement. 2016;12(6):687–694. doi: 10.1016/j.jalz.2016.03.015. [DOI] [PubMed] [Google Scholar]
- Ryu S, Atzmon G, Barzilai N, Raghavachari N, Suh Y. Genetic landscape of APOE in human longevity revealed by high-throughput sequencing. Mech Ageing Dev. 2016;155:7–9. doi: 10.1016/j.mad.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salakhov RR, Goncharova IA, Makeeva OA, Golubenko MV, Kulish EV, Kashtalap VV, Barbarash OL, Puzyrev VP. TOMM40 gene polymorphism association with lipid profile. Genetika. 2014;50(2):222–229. [PubMed] [Google Scholar]
- Schmidt SA, Ording AG, Horvath-Puho E, Sorensen HT, Henderson VW. Non-melanoma skin cancer and risk of Alzheimer’s disease and all-cause dementia. PLoS ONE. 2017;12(2):e0171527. doi: 10.1371/journal.pone.0171527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seripa D, Bizzarro A, Pilotto A, Palmieri O, Panza F, D’Onofrio G, Gravina C, Archetti S, Daniele A, Borroni B, Padovani A, Masullo C. TOMM40, APOE, and APOC1 in primary progressive aphasia and frontotemporal dementia. J Alzheimers Dis. 2012;31(4):731–740. doi: 10.3233/JAD-2012-120403. [DOI] [PubMed] [Google Scholar]
- Shadyab AH, Kooperberg C, Reiner AP, Jain S, Manson JE, Hohensee C, Macera CA, Shaffer RA, Gallo LC, LaCroix AZ. Replication of genome-wide association study findings of longevity in White, African American, and Hispanic Women: The Women’s Health Initiative. J Gerontol A Biol Sci Med Sci. 2017;72(10):1401–1406. doi: 10.1093/gerona/glw198. http://dx.doi.org/10.1093/gerona/glw198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafi O. Inverse relationship between Alzheimer’s disease and cancer, and other factors contributing to Alzheimer’s disease: a systematic review. BMC Neurol. 2016;16(1):236. doi: 10.1186/s12883-016-0765-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SZA, Zhao D, Hussain T, Yang L. The role of unfolded protein response and mitogen-activated protein kinase signaling in neurodegenerative diseases with special focus on prion diseases. Front Aging Neurosci. 2017;9:120. doi: 10.3389/fnagi.2017.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery ML, Sweeney C, Murtaugh M, Ma KN, Potter JD, Levin TR, Samowitz W, Wolff R. Associations between apoE genotype and colon and rectal cancer. Carcinogenesis. 2005;26(8):1422–1429. doi: 10.1093/carcin/bgi088. [DOI] [PubMed] [Google Scholar]
- Smith EN, Chen W, Kahonen M, Kettunen J, Lehtimaki T, Peltonen L, Raitakari OT, Salem RM, Schork NJ, Shaw M, Srinivasan SR, Topol EJ, Viikari JS, Berenson GS, Murray SS. Longitudinal genome-wide association of cardiovascular disease risk factors in the Bogalusa heart study. PLoS Genet. 2010;6(9):e1001094. doi: 10.1371/journal.pgen.1001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J Alzheimers Dis. 2005;7(1):63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- Sun MK, Alkon DL. Links between Alzheimer’s disease and diabetes. Drugs Today (Barc) 2006;42(7):481–489. doi: 10.1358/dot.2006.42.7.973588. [DOI] [PubMed] [Google Scholar]
- Sun L, Hu C, Zheng C, Huang Z, Lv Z, Huang J, Liang S, Shi X, Zhu X, Yuan H, Yang Z. Gene-gene interaction between CETP and APOE polymorphisms confers higher risk for hypertriglyceridemia in oldest-old Chinese women. Exp Gerontol. 2014;55:129–133. doi: 10.1016/j.exger.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH. Brain aging, Alzheimer’s disease, and mitochondria. Biochim Biophys Acta. 2011;1812(12):1630–1639. doi: 10.1016/j.bbadis.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH. Bioenergetics and metabolism: a bench to bedside perspective. J Neurochem. 2016;139(Suppl. 2):126–135. doi: 10.1111/jnc.13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: an update. Exp Neurol. 2009;218(2):308–315. doi: 10.1016/j.expneurol.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Nagao T, Gouras GK. Plaque formation and the intraneuronal accumulation of beta-amyloid in Alzheimer’s disease. Pathol Int. 2017;67(4):185–193. doi: 10.1111/pin.12520. [DOI] [PubMed] [Google Scholar]
- Takei N, Miyashita A, Tsukie T, Arai H, Asada T, Imagawa M, Shoji M, Higuchi S, Urakami K, Kimura H, Kakita A, Takahashi H, Tsuji S, Kanazawa I, Ihara Y, Odani S, Kuwano R. Genetic association study on in and around the APOE in late-onset Alzheimer disease in Japanese. Genomics. 2009;93(5):441–448. doi: 10.1016/j.ygeno.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, Chambers J, Orho-Melander M, Melander O, Johnson T, Li X, Guo X, Li M, Cho YS, Go MJ, Kim YJ, Lee JY, Park T, Kim K, Sim X, Ong RTH, Croteau-Chonka DC, Lange LA, Smith JD, Song K, Zhao JH, Yuan X, Luan JA, Lamina C, Ziegler A, Zhang W, Zee RYL, Wright AF, Witteman JCM, Wilson JF, Willemsen G, Wichmann HE, Whitfield JB, Waterworth DM, Wareham NJ, Waeber G, Vollenweider P, Voight BF, Vitart V, Uitterlinden AG, Uda M, Tuomilehto J, Thompson JR, Tanaka T, Surakka I, Stringham HM, Spector TD, Soranzo N, Smit JH, Sinisalo J, Silander K, Sijbrands EJG, Scuteri A, Scott J, Schlessinger D, Sanna S, Salomaa V, Saharinen J, Sabatti C, Ruokonen A, Rudan I, Rose LM, Roberts R, Rieder M, Psaty BM, Pramstaller PP, Pichler I, Perola M, Penninx BWJH, Pedersen NL, Pattaro C, Parker AN, Pare G, Oostra BA, O’Donnell CJ, Nieminen MS, Nickerson DA, Montgomery GW, Meitinger T, McPherson R, McCarthy MI, McArdle W, Masson D, Martin NG, Marroni F, Mangino M, Magnusson PKE, Lucas G, Luben R, Loos RJF, Lokki ML, Lettre G, Langenberg C, Launer LJ, Lakatta EG, Laaksonen R, Kyvik KO, Kronenberg F, Koenig IR, Khaw KT, Kaprio J, Kaplan LM, Johansson A, Jarvelin MR, Janssens ACJW, Ingelsson E, Igi W, Hovingh GK, Hottenga JJ, Hofman A, Hicks AA, Hengstenberg C, Heid IM, Hayward C, Havulinna AS, Hastie ND, Harris TB, Haritunians T, Hall AS, Gyllensten U, Guiducci C, Groop LC, Gonzalez E, Gieger C, Freimer NB, Ferrucci L, Erdmann J, Elliott P, Ejebe KG, Doering A, Dominiczak AF, Demissie S, Deloukas P, de Geus EJC, de Faire U, Crawford G, Collins FS, Chen Y-dI, Caulfield MJ, Campbell H, Burtt NP, Bonnycastle LL, Boomsma DI, Boekholdt SM, Bergman RN, Barroso I, Bandinelli S, Ballantyne CM, Assimes TL, Quertermous T, Altshuler D, Seielstad M, Wong TY, Tai ES, Feranil AB, Kuzawa CW, Adair LS, Taylor HA, Jr, Borecki IB, Gabriel SB, Wilson JG, Holm H, Thorsteinsdottir U, Gudnason V, Krauss RM, Mohlke KL, Ordovas JM, Munroe PB, Kooner JS, Tall AR, Hegele RA, Kastelein JJP, Schadt EE, Rotter JI, Boerwinkle E, Strachan DP, Mooser V, Stefansson K, Reilly MP, Samani NJ, Schunkert H, Cupples LA, Sandhu MS, Ridker PM, Rader DJ, van Duijn CM, Peltonen L, Abecasis GR, Boehnke M, Kathiresan S. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker-Seeley RD, Li Y, Sorensen G, Subramanian SV. Lifecourse socioeconomic circumstances and multimorbidity among older adults. BMC Public Health. 2011;11:313. doi: 10.1186/1471-2458-11-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukraintseva SV, Arbeev KG, Akushevich I, Kulminski A, Arbeeva L, Culminskaya I, Akushevich L, Yashin AI. Trade-offs between cancer and other diseases: do they exist and influence longevity? Rejuvenation Res. 2010;13(4):387–396. doi: 10.1089/rej.2009.0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urosevic N, Martins RN. Infection and Alzheimer’s disease: the APOE epsilon4 connection and lipid metabolism. J Alzheimers Dis. 2008;13(4):421–435. doi: 10.3233/jad-2008-13407. [DOI] [PubMed] [Google Scholar]
- Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, Holtzman DM. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci U S A. 2013;110(19):E1807–1816. doi: 10.1073/pnas.1220484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana RJ, Nunes AF, Rodrigues CM. Endoplasmic reticulum enrollment in Alzheimer’s disease. Mol Neurobiol. 2012;46(2):522–534. doi: 10.1007/s12035-012-8301-x. [DOI] [PubMed] [Google Scholar]
- Wang S, Guan L, Luo D, Liu J, Lin H, Li X, Liu X. Gene-gene interaction between PPARG and APOE gene on late-onset Alzheimer’s disease: a case-control study in Chinese Han population. J Nutr Health Aging. 2017;21(4):397–403. doi: 10.1007/s12603-016-0794-y. [DOI] [PubMed] [Google Scholar]
- Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44(D1):D877–881. doi: 10.1093/nar/gkv1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MA, Gay L, Stebbings WS, Speakman CT, Bingham SA, Loktionov A. Apolipoprotein E gene polymorphism and colorectal cancer: gender-specific modulation of risk and prognosis. Clin Sci (Lond) 2003;104(5):537–545. doi: 10.1042/CS20020329. [DOI] [PubMed] [Google Scholar]
- Yarchoan M, James BD, Shah RC, Arvanitakis Z, Wilson RS, Schneider J, Bennett DA, Arnold SE. Association of cancer history with Alzheimer’s disease dementia and neuropathology. J Alzheimers Dis. 2017;56(2):699–706. doi: 10.3233/JAD-160977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashin AI, Ukraintseva SV, Akushevich IV, Arbeev KG, Kulminski A, Akushevich L. Trade-off between cancer and aging: what role do other diseases play? Evidence from experimental and human population studies. Mech Ageing Dev. 2009;130(1–2):98–104. doi: 10.1016/j.mad.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlow K, Charlambous L, Ng I, Gagrani S, Mihovilovic M, Luo S, Rock DL, Saunders A, Roses AD, Gottschalk WK. The biological foundation of the genetic association of TOMM40 with late-onset Alzheimer’s disease. Biochim Biophys Acta. 2017;1863(11):2973–2986. doi: 10.1016/j.bbadis.2017.07.031. http://dx.doi.org/10.1016/j.bbadis.2017.07.031. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Pierce BL. Genetic susceptibility to accelerated cognitive decline in the US Health and Retirement Study. Neurobiol Aging. 2014;35(6):1512.e1511–1518. doi: 10.1016/j.neurobiolaging.2013.12.021. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Zhao F, Lv ZP, Zheng CG, Zheng WD, Sun L, Wang NN, Pang S, de Andrade FM, Fu M, He XH, Hui J, Jiang W, Yang CY, Shi XH, Zhu XQ, Pang GF, Yang YG, Xie HQ, Zhang WD, Hu CY, Yang Z. Association between APOC1 polymorphism and Alzheimer’s disease: a case-control study and meta-analysis. PLoS ONE. 2014;9(1):e87017. doi: 10.1371/journal.pone.0087017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.