

Abstract
Here we present the development of self-regulated “breathing” polymersome nanoreactors that show temporally programmable biocatalysis induced by a chemical fuel. pH-sensitive polymersomes loaded with horseradish peroxidase (HRP) and urease were developed. Addition of an acidic urea solution (“fuel”) endowed the polymersomes with a transient size increase and permeability enhancement, driving a temporal “ON” state of the HRP enzymatic catalysis; subsequent depletion of fuel led to shrinking of the polymersomes, resulting in the catalytic “OFF” state. Moreover, the nonequilibrium nanoreactors could be reinitiated several cycles as long as fuel was supplied. This feedback-induced temporal control of catalytic activity in polymersome nanoreactors provides a platform for functional nonequilibrium systems as well as for artificial organelles with precisely controlled adaptivity.
Inspired by biology, chemists have created a plethora of synthetic adaptive or life-like materials with temporal control over structure and function by employing chemical and enzymatic reaction networks. These materials are endowed with unique features because of their out-of-equilibrium state, compared to conventional equilibrium systems that demonstrate thermodynamic stability.1 Although nonequilibrium systems have been well studied, these are mostly related to structural changes, whereas systems with biomimetic self-adaptive function are less common. In this regard, fuel-driven functional operations under temporal control are thought to play a key role in bridging man-made materials and biology in terms of natural organization and function. For example, DNA-based systems have paved the way for the development of autonomous molecular walkers and nanomotors mediated by fuel and catalytic units.2
Polymeric vesicles, or polymersomes, are versatile compartments that have been developed for a range of applications, including nanoreactors.3 To regulate the transport of substrate across the polymer membrane barrier, stimuli-responsive polymersomes that can sense changes in the environment have been designed and constructed.4
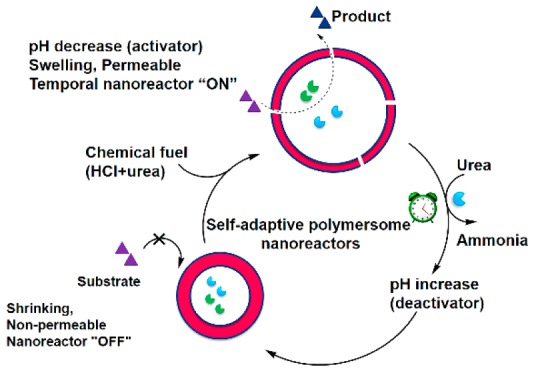
The bell-shaped pH-activity curve of urease coupled with its ability to produce base has provided a platform to attain temporal control over pH feedback systems.5 Recently, we have shown the construction of a self-regulated and time-programmed “breathing” microgel that uses chemical fuels to keep the system in an out-of-equilibrium state.6 Here we report an artificial organelle with precisely controlled adaptive dynamic behavior and enzymatic activity. The envisioned feedback-induced temporal control of the polymersome nanoreactors is depicted in Figure 1a. Urease, which controls the pH change, and horseradish peroxidase (HRP), which acts as model enzyme, are encapsulated in the polymersomes. First, polymersome nanoreactors are dispersed in a high pH buffer. This causes the polymersomes to shrink due to deprotonation of the pH-sensitive polymers. In this situation, the nanoreactors are nonpermeable and substrates cannot pass the polymer membranes, giving rise to a nanoreactor “OFF” state (left). Addition of chemical fuel (HCl and urea) results in a fast pH decrease, thereby increasing the size of the polymersomes. The swelling leads to increased polymer membrane permeability, turning the nanoreactor in the “ON” state as the substrate is able to penetrate into the polymersomes (above). Over time, a gradual increase in pH occurs through the conversion of urea into ammonia. Thus, polymersomes return to their initial shrunken state and the enzyme catalysis automatically changes to “OFF” again. A continuous addition of chemical fuels to the system allows the “ON” and “OFF” process to be reversible.
Figure 1.
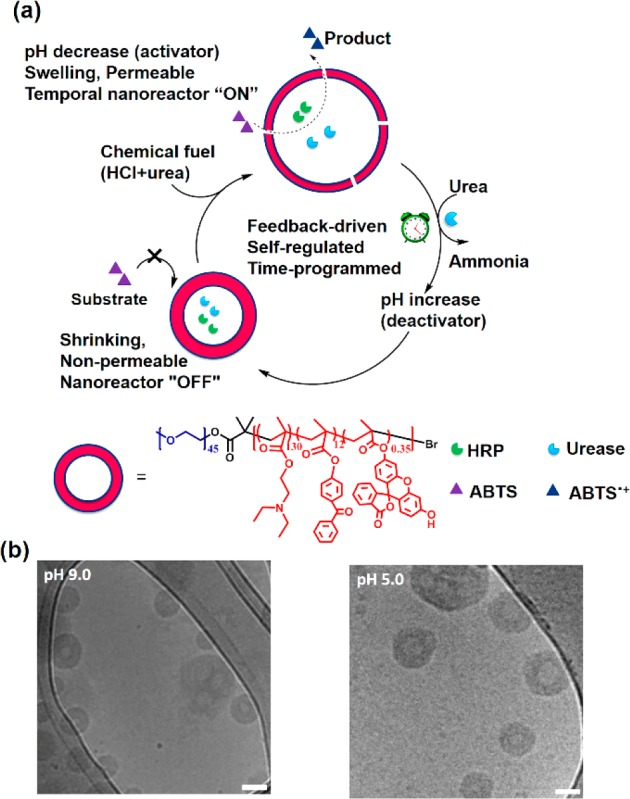
(a) Schematic overview of feedback-induced temporal control of polymersome nanoreactors. (b) Cryo-TEM images of polymersomes at pH 9.0 (left) and pH 5.0 (right). All scale bars are 100 nm.
To realize this aim, amphiphilic copolymers consisting of poly(ethylene glycol) (PEG) and pH-responsive poly[2-(diethylamino) ethyl methacrylate (PDEAEMA) were synthesized. Poly[2-hydroxy-4-(methacryloyloxy) benzophenone] (PBMA) was used as a photo-cross-linkable block, and a fluorescein group was introduced as a fluorescence read-out platform (Supporting Information, Scheme S1, Figure S1 and Figure S2). Block copolymer mPEG45-b-P(DEAEMA130-co-BMA12-co-FMA0.35) was self-assembled into pH-responsive polymersomes by the nanoprecipitation method and subsequently cross-linked.7 During the formation of polymersomes, HRP and urease were coencapsulated. Urease was labeled with Rhodamine-B (RhB) before encapsulation, which did not affect the high activity of the enzyme (Figure S3). From HRP’s characteristic Soret band with a maximum at 403 nm4g and RhB’s absorbance at 550 nm, the urease and HRP loading content was determined to be 25% and 14%, respectively, after disassembly of the polymersomes (Supporting Information, Figure S4). Asymmetric flow field-flow fractionation (AF4, settings in Table S1), was used to determine particle morphology. The ratio between the radius of gyration (Rg) and the hydrodynamic radius (Rh) was close to 1, indicative of vesicle formation (Figure S5).8 Transmission electron microscopy (TEM) and cryogenic TEM images of the polymersomes confirmed a vesicular structure at both basic and acidic conditions (Figure 1 b, and Figure S6). Dynamic light scattering (DLS) measurements revealed an Rh increase from 102 to 183 nm when the pH was decreased from 9 to 5 (Figure S7). The average Rh was plotted against solution pH and reversibility upon pH variation was observed during multiple cycles of swelling and shrinking (Figure S8, and Figure S9).
The feedback-driven pH regulation of the polymersomes was first investigated. The polymersome solution was brought to pH 9.0 (N-cyclohexyl-2-aminoethanesulfonic acid (CHES) buffer, 10 mM), under which conditions urease is inactive. Upon addition of a concentrated solution of urea and HCl, the pH of the polymersome solution was instantaneously brought down to 5.0, at which point urease was activated. This was reflected by a gradual increase in pH due to the conversion of urea into ammonia. The final pH value leveled off at approximately the original level, as the urease activity was self-diminished at high pH. By changing [urea] from 2 to 10 mM (urease level constantly maintained at 30 U/mL), the pH recovery time was decreased (Figure 2a). More interestingly, the out-of-equilibrium situation was reinitiated by the repeated addition of chemical fuel over five cycles (Figure S10). The dynamic pH regulation behavior was also validated using fluorescence spectroscopy, as fluorescein incorporated in the polymersome membranes could change its fluorescence intensity concurrently with the change in pH (Figure 2b, Figures S11 and S12).
Figure 2.
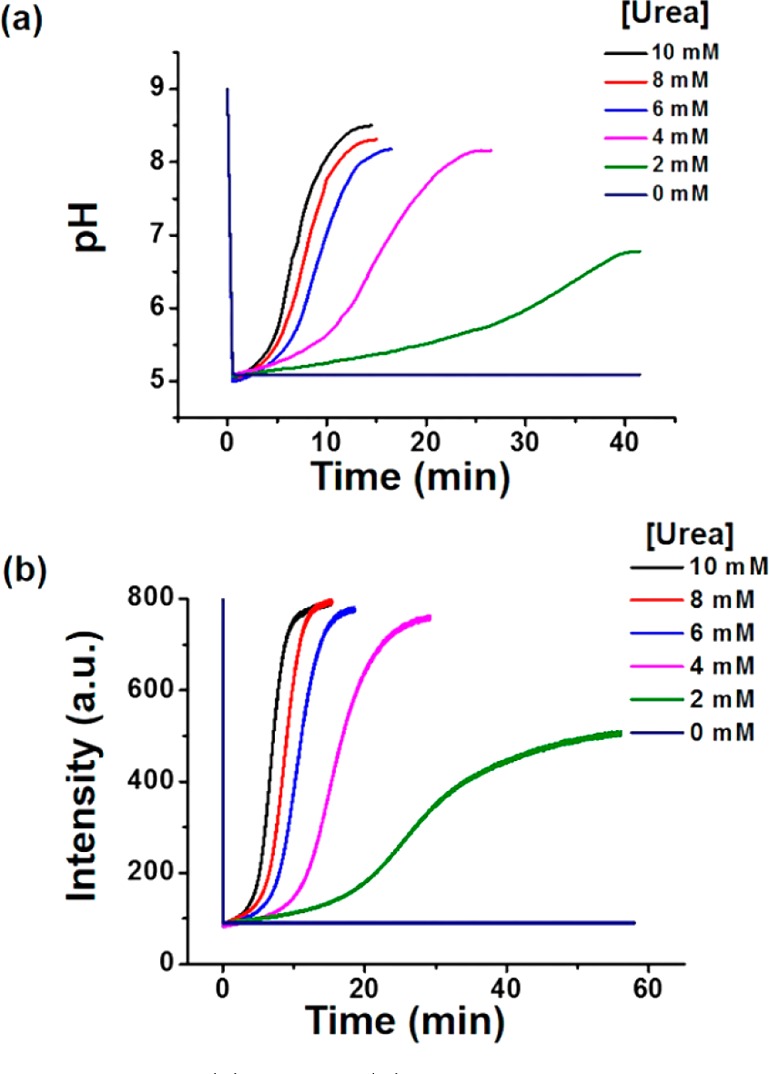
Plots of the (a) pH and (b) fluorescence intensity switch of the polymersome nanoreactor solution against time in a nonequilibrium cycle for different initial urea concentrations. The concentration of urease in the polymersomes is 30 U/mL.
Next, polymersome size evolution upon addition of the acidic urea solution was investigated by DLS. As expected, the polymersomes first quickly increased to a maximum size of Rh = 182 nm, followed by an autonomous size recovery, until they finally reached the starting situation (Rh = 101 nm) (Figure S13). The spontaneous expansion and contraction of these polymersomes endows them with a “breathing” feature. In contrast to other reported “breathing” vesicles with changeable sizes,9 our system follows a self-regulated mechanism. In addition, this polymersome size evolution process was perfectly modulated by varying concentrations of the substrate (Figure 3). Thus, an increase in substrate gives rise to a decrease in the swelling lifetimes and an acceleration of the rate of polymersome “breathing”. Figure 3c shows average induction periods within five cycles. The expansion-contraction process was reinitiated with controllable lifetimes by urea supply over five cycles, and the pH and size modulations followed almost a similar trend during each cycle (Figure 2d).
Figure 3.
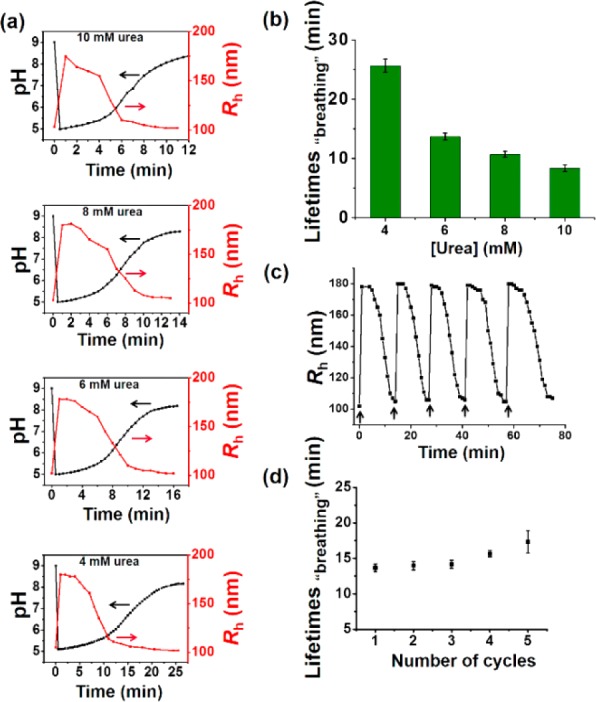
(a) Influence of the urea concentration on the size changes of the polymersome nanoreactor solution. Urea concentrations from top to bottom: 10, 8, 6, and 4 mM. (b) Average transient periods of polymersome “breathing” as a function of the urea concentration. (c) Reversible Rh changes of the polymersomes in time following repeated additions of chemical fuel (6 mM urea). (d) Average transient periods of “breathing” within five cycles (6 mM urea in all cases). Concentrations of urease in the polymersomes are 30 U/mL.
As the self-regulated “breathing” behavior of the polymersomes is correlated to a temporal change in membrane permeability, these compartments are attractive as adaptive nanoreactors with automatically modulated catalysis. First, the effect of pH on the activity of free and encapsulated HRP was investigated, using an ABTS assay. As shown in Figure S14, HRP displays pH-dependent activity, with a slight increase from pH 5.0 to 6.0, and a gradual decrease from pH 6.0 to 9.0. When HRP was encapsulated in the responsive polymersomes, nearly the same activity profile was observed in the pH range from 5.0 to 6.0. However, the HRP activity significantly dropped below 5% when the pH was increased above 6.5, while the corresponding free HRP activity remained above 40% (Figure S15). This enzyme activity difference between free HRP and encapsulated HRP can be ascribed to the pH-triggered polymersome permeability change. At high pH value, the PDEAEMA block is deprotonated, therefore the polymersome membranes are fully hydrophobic, and substrates cannot diffuse into the polymersome lumen. When the pH is below the pKa (around 7.0) of PDEAEMA, the polymersome membranes change to a swollen and permeable state, which allows for the catalytic reaction to occur.
Next, we sought to develop dissipative polymersome nanoreactors with out-of-equilibrium catalytic behavior. For this purpose, polymersomes were loaded with both HRP and urease. Initially, the samples were incubated in pH 9.0 buffer, and no absorbance at 416 nm was observed, suggesting the catalytic “OFF” state due to the nonpermeable polymersome membrane. When acidic urea was applied to the system, the HRP enzymatic reaction first became activated and subsequently turned dormant (Figure 4a). This self-controlled catalysis behavior of the nanoreactors was caused by the temporally programmed “breathing” feature and permeability modulation of the polymersome membranes.
Figure 4.
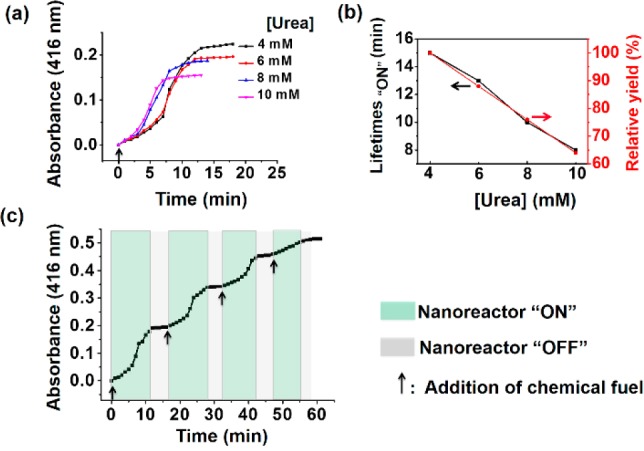
(a) UV absorbance at 416 nm of the oxidation of ABTS by nanoreactors upon the addition of different concentrations of urea. (b) Lifetimes (black line) of the nanoreactor “ON” state and relative yield (red line) as a function of urea concentration. (c) Reversible nanoreactor “ON–OFF” modulation in time following repeated additions of 6.0 mM urea. Experimental conditions: Urease, 30 U/mL; HRP, 10 U/mL; ABTS, 8 mM; H2O2, 5 mM.
As the concentration of urea determines the lifetime of the membrane shrinking and swelling process, we reasoned that we could tune the yield of the ABTS oxidation process. Samples with different urea concentration (4, 6, 8, and 10 mM) underwent a gradual increase in the absorbance at 416 nm after addition of chemical fuel, which leveled off to a constant value. The higher the concentration of urea, the faster the catalysis reached its end point. Most interestingly, an approximately linear relationship between the concentration of added urea and the nanoreactor “ON” lifetimes as well as the final yield was observed (Figure 4b).
As this fuel-driven temporal nanoreactor is in principle a switchable process, the self-adaptive cycle was refueled by the consecutive introduction of urea. In a control experiment where the free HRP-based enzymatic reaction was performed, the absorbance over time (60 min) at 416 nm was linear, which confirms that the amount of substrate is not limiting in the study (Figure S16). Identical to polymersome swelling and shrinking behavior, three consecutive nanoreactor “OFF–ON–OFF” cycles could be achieved, supporting the above mechanism of feedback-driven membrane permeability modulation (Figure 4c). We need to note, however, that the efficiency of the self-regulated catalytic reaction decreased after three cycles, whereas the reversible polymersome expansion and contraction could be performed for at least six cycles. The reversible pH changes of the enzyme-filled polymersomes in the presence of ABTS clearly shows that the product of the HRP reaction does not have an effect on the lifetime of the polymersome nanoreactors (Figure S17). So, one possible reason is that the activity of HRP is affected by continuous pH regulation and accumulation of waste (ammonia and CO2).
In summary, we have presented a self-adaptive polymersome nanoreactor that drives a catalytic reaction out-of-equilibrium by temporal control of the permeability of the polymersome membranes. This is achieved by implementing the urease-based pH feedback system in HRP-filled polymersomes, which induces “breathing” behavior in the nanoreactor. Compared to the previously reported classical nanoreactors that operate in a thermodynamically controlled one-way transition, the nonequilibrium nanoreactor functions with an “ON–OFF” switch in a biocatalytically controlled fashion. Hence the catalytically active period of HRP in the nanoreactors is governed by the amount of substrate present to steer the urease cycle, and the reversible nature is demonstrated for several cycles by refueling the system. The principle of feedback-induced temporal control of nanoreactors is an important addition to the area of nonequilibrium systems. We anticipate that the basic design rules in this work will promote engineering of artificial organelles with adaptive features.
Acknowledgments
The authors acknowledge the Dutch Science Foundation (VICI grant), the ERC Advanced grant Artisym 694120, the Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035) and the European Union’s Horizon 2020 research and innovation programme Marie Sklodowska-Curie Innovative Training Networks Nanomed, (No. 676137) for funding. We also thank Imke Pijpers and Alex Mason for help with Cryo-TEM measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b02387.
Synthesis, methods, and supporting data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Heinen L.; Walther A. Soft Matter 2015, 11, 7857. 10.1039/C5SM01660F. [DOI] [PubMed] [Google Scholar]; b Merindol R.; Walther A. Chem. Soc. Rev. 2017, 46, 5588. 10.1039/C6CS00738D. [DOI] [PubMed] [Google Scholar]; c della Sala F.; Neri S.; Maiti S.; Chen J. L.; Prins L. J. Curr. Opin. Biotechnol. 2017, 46, 27. 10.1016/j.copbio.2016.10.014. [DOI] [PubMed] [Google Scholar]; d van Roekel H. W.; Rosier B. J.; Meijer L. H.; Hilbers P. A.; Markvoort A. J.; Huck W. T.; de Greef T. F. Chem. Soc. Rev. 2015, 44, 7465. 10.1039/C5CS00361J. [DOI] [PubMed] [Google Scholar]; e Boekhoven J.; Hendriksen W. E.; Koper G. J.; Eelkema R.; van Esch J. H. Science 2015, 349, 1075. 10.1126/science.aac6103. [DOI] [PubMed] [Google Scholar]; f Van Ravensteijn B. G.; Hendriksen W. E.; Eelkema R.; Van Esch J. H.; Kegel W. K. J. Am. Chem. Soc. 2017, 139, 9763. 10.1021/jacs.7b03263. [DOI] [PubMed] [Google Scholar]; g Debnath S.; Roy S.; Ulijn R. V. J. Am. Chem. Soc. 2013, 135, 16789. 10.1021/ja4086353. [DOI] [PubMed] [Google Scholar]; h Tena-Solsona M.; Rieß B.; Grötsch R. K.; Löhrer F. C.; Wanzke C.; Käsdorf B.; Bausch A. R.; Müller-Buschbaum P.; Lieleg O.; Boekhoven J. Nat. Commun. 2017, 8, 15895. 10.1038/ncomms15895. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Dhiman S.; Jain A.; Kumar M.; George S. J. J. Am. Chem. Soc. 2017, 139, 16568. 10.1021/jacs.7b07469. [DOI] [PubMed] [Google Scholar]; j Jalani K.; Dhiman S.; Jain A.; George S. J. Chem. Sci. 2017, 8, 6030. 10.1039/C7SC01730H. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Hao X.; Sang W.; Hu J.; Yan Q. ACS Macro Lett. 2017, 6, 1151. 10.1021/acsmacrolett.7b00649. [DOI] [PubMed] [Google Scholar]; l Maiti S.; Fortunati I.; Ferrante C.; Scrimin P.; Prins L. J. Nat. Chem. 2016, 8, 725. 10.1038/nchem.2511. [DOI] [PubMed] [Google Scholar]
- a Yin P.; Yan H.; Daniell X. G.; Turberfield A. J.; Reif J. H. Angew. Chem., Int. Ed. 2004, 43, 4906. 10.1002/anie.200460522. [DOI] [PubMed] [Google Scholar]; b Lund K.; Manzo A. J.; Dabby N.; Michelotti N.; Johnson-Buck A.; Nangreave J.; Taylor S.; Pei R.; Stojanovic M. N.; Walter N. G.; et al. Nature 2010, 465, 206. 10.1038/nature09012. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cha T.-G.; Pan J.; Chen H.; Salgado J.; Li X.; Mao C.; Choi J. H. Nat. Nanotechnol. 2014, 9, 39. 10.1038/nnano.2013.257. [DOI] [PubMed] [Google Scholar]; d Wickham S. F.; Endo M.; Katsuda Y.; Hidaka K.; Bath J.; Sugiyama H.; Turberfield A. J. Nat. Nanotechnol. 2011, 6, 166. 10.1038/nnano.2010.284. [DOI] [PubMed] [Google Scholar]; e Yehl K.; Mugler A.; Vivek S.; Liu Y.; Zhang Y.; Fan M.; Weeks E. R.; Salaita K. Nat. Nanotechnol. 2016, 11, 184. 10.1038/nnano.2015.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Che H.; van Hest J. C. J. Mater. Chem. B 2016, 4, 4632. 10.1039/C6TB01163B. [DOI] [PubMed] [Google Scholar]; b Palivan C. G.; Goers R.; Najer A.; Zhang X.; Car A.; Meier W. Chem. Soc. Rev. 2016, 45, 377. 10.1039/C5CS00569H. [DOI] [PubMed] [Google Scholar]; c Tanner P.; Baumann P.; Enea R.; Onaca O.; Palivan C.; Meier W. Acc. Chem. Res. 2011, 44, 1039. 10.1021/ar200036k. [DOI] [PubMed] [Google Scholar]; d Schoonen L.; van Hest J. Adv. Mater. 2016, 28, 1109. 10.1002/adma.201502389. [DOI] [PubMed] [Google Scholar]; e Buddingh’ B. C.; van Hest J. C. Acc. Chem. Res. 2017, 50, 769. 10.1021/acs.accounts.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Marguet M.; Bonduelle C.; Lecommandoux S. Chem. Soc. Rev. 2013, 42, 512. 10.1039/C2CS35312A. [DOI] [PubMed] [Google Scholar]
- a Hu J.; Zhang G.; Liu S. Chem. Soc. Rev. 2012, 41, 5933. 10.1039/c2cs35103j. [DOI] [PubMed] [Google Scholar]; b Feng A.; Yuan J. Macromol. Rapid Commun. 2014, 35, 767. 10.1002/marc.201300866. [DOI] [PubMed] [Google Scholar]; c Kim K. T.; Meeuwissen S. A.; Nolte R. J.; van Hest J. C. Nanoscale 2010, 2, 844. 10.1039/b9nr00409b. [DOI] [PubMed] [Google Scholar]; d Peters R. J.; Louzao I.; van Hest J. C. Chem. Sci. 2012, 3, 335. 10.1039/C2SC00803C. [DOI] [Google Scholar]; e Hu X.; Zhang Y.; Xie Z.; Jing X.; Bellotti A.; Gu Z. Biomacromolecules 2017, 18, 649. 10.1021/acs.biomac.6b01704. [DOI] [PubMed] [Google Scholar]; f Le Meins J.-F.; Sandre O.; Lecommandoux S. Eur. Phys. J. E: Soft Matter Biol. Phys. 2011, 34, 14. 10.1140/epje/i2011-11014-y. [DOI] [PubMed] [Google Scholar]; g Spulber M.; Najer A.; Winkelbach K.; Glaied O.; Waser M.; Pieles U.; Meier W.; Bruns N. J. Am. Chem. Soc. 2013, 135, 9204. 10.1021/ja404175x. [DOI] [PubMed] [Google Scholar]; h Edlinger C.; Einfalt T.; Spulber M.; Car A.; Meier W.; Palivan C. G. Nano Lett. 2017, 17, 5790. 10.1021/acs.nanolett.7b02886. [DOI] [PubMed] [Google Scholar]; i Einfalt T.; Goers R.; Dinu I. A.; Najer A.; Spulber M.; Onaca-Fischer O.; Palivan C. G. Nano Lett. 2015, 15, 7596. 10.1021/acs.nanolett.5b03386. [DOI] [PubMed] [Google Scholar]; j Kim K. T.; Cornelissen J. J.; Nolte R. J.; van Hest J. Adv. Mater. 2009, 21, 2787. 10.1002/adma.200900300. [DOI] [Google Scholar]; k Yan Q.; Wang J.; Yin Y.; Yuan J. Angew. Chem., Int. Ed. 2013, 52, 5070. 10.1002/anie.201300397. [DOI] [PubMed] [Google Scholar]
- a Hu G.; Pojman J. A.; Scott S. K.; Wrobel M. M.; Taylor A. F. J. Phys. Chem. B 2010, 114, 14059. 10.1021/jp106532d. [DOI] [PubMed] [Google Scholar]; b Russell A. J.; Erbeldinger M.; DeFrank J. J.; Kaar J.; Drevon G. Biotechnol. Bioeng. 2002, 77, 352. 10.1002/bit.10152. [DOI] [PubMed] [Google Scholar]; c Heuser T.; Weyandt E.; Walther A. Angew. Chem., Int. Ed. 2015, 54, 13258. 10.1002/anie.201505013. [DOI] [PubMed] [Google Scholar]; d Heinen L.; Walther A. Chem. Sci. 2017, 8, 4100. 10.1039/C7SC00646B. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Heuser T.; Merindol R.; Loescher S.; Klaus A.; Walther A. Adv. Mater. 2017, 29, 1606842. 10.1002/adma.201606842. [DOI] [PubMed] [Google Scholar]; f Okamoto Y.; Ward T. R. Angew. Chem., Int. Ed. 2017, 56, 10156. 10.1002/anie.201702181. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Jee E.; Bánsági T.; Taylor A. F.; Pojman J. A. Angew. Chem., Int. Ed. 2016, 55, 2127. 10.1002/anie.201510604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che H.; Buddingh B. C.; van Hest J. C. M. Angew. Chem., Int. Ed. 2017, 56, 12581. 10.1002/anie.201706716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gaitzsch J.; Appelhans D.; Wang L.; Battaglia G.; Voit B. Angew. Chem., Int. Ed. 2012, 51, 4448. 10.1002/anie.201108814. [DOI] [PubMed] [Google Scholar]; b Iyisan B.; Kluge J. r.; Formanek P.; Voit B.; Appelhans D. Chem. Mater. 2016, 28, 1513. 10.1021/acs.chemmater.5b05016. [DOI] [Google Scholar]; c Gaitzsch J.; Appelhans D.; Gräfe D.; Schwille P.; Voit B. Chem. Commun. 2011, 47, 3466. 10.1039/c0cc05355d. [DOI] [PubMed] [Google Scholar]
- a Brewer A. K.; Striegel A. M. Analyst 2011, 136, 515. 10.1039/C0AN00738B. [DOI] [PubMed] [Google Scholar]; b Abdelmohsen L. K. E. A.; Williams D. S.; Pille J.; Ozel S. G.; Rikken R. S. M.; Wilson D. A.; van Hest J. C. M. J. Am. Chem. Soc. 2016, 138, 9353. 10.1021/jacs.6b03984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yu S.; Azzam T.; Rouiller I.; Eisenberg A. J. Am. Chem. Soc. 2009, 131, 10557. 10.1021/ja902869q. [DOI] [PubMed] [Google Scholar]; b Yan Q.; Zhou R.; Fu C.; Zhang H.; Yin Y.; Yuan J. Angew. Chem. 2011, 123, 5025. 10.1002/ange.201100708. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.