

Abstract
Hypertrophic scar (HS) is a fibrotic disease in which excessive extracellular matrix forms due to the response of fibroblasts to tissue damage. Novel evidence suggests that microRNAs (miRNAs or miRs) may contribute to hypertrophic scarring; however, the role of miRNAs in HS formation remains unclear. In the present study, miR-26a was significantly downregulated in HS tissues and human HS fibroblasts (hHSFs) was detected by reverse transcription-quantitative analysis. TargetScan was used to predict that mothers against decapentaplegic homolog 2 (Smad2) is a potential target gene of miR-26a and a dual-luciferase reporter assay confirmed that Smad2 was a target gene of miR-26a. The expression of Smad2 was upregulated in HS tissues and hHSFs. Cell Counting Kit-8 and flow cytometry analyses demonstrated that the overexpression of miR-26a significantly suppressed the proliferation ability of hHSFs and the apoptotic rate of hHSFs was significantly upregulated in response to miR-26a mimic transfection. Furthermore, the expression of B-cell lymphoma-2 (Bcl-2)-associated X protein was increased and Bcl-2 expression was decreased following miR-26a mimic transfection. The expression of collagens I and III was significantly inhibited following treatment with miR-26a mimics in hHSF cells. Conversely, miR-26a inhibitors served an opposing role in hHSFs. Furthermore, Smad2 overexpression enhanced the expression of collagens I and c III; however, Smad2 silencing inhibited the expression of collagens I and c III. In conclusion, the results of the present study indicate that miR-26a inhibits HS formation by modulating proliferation and apoptosis ad well as inhibiting the expression of extracellular matrix-associated proteins by targeting Smad2.
Keywords: microRNA, hypertrophic scar, mothers against decapentaplegic homolog 2, extracellular matrix
Introduction
Skin wound healing is an intermittent process that occurs immediately following tissue damage and often results in scars (1). The ideal scar is thin, flat and almost invisible, known as a normal nutritional scar (2). Severe trauma and sometimes standard surgery with improper wound closure may result in the formation of atypical raised scars, known as hypertrophic scars (HSs) (3). HSs are characterized by excessive extracellular matrix deposition, including fibronectin, elastin, collagen, α-smooth muscle actin and matrix-directed proteases and protease inhibitors (4–6). At present, there are a number of treatment options available for HSs, including surgical resection, steroid injections and radiotherapy (7); however, there is gold standard treatment and the clinical behavior of HSs remains unclear. Some studies have reported that a variety of non-coding RNAs and growth factors are associated with the formation of HSs (8,9).
MicroRNAs (miRNAs or miRs) are evolutionary conserved non-coding RNAs of ~19–25 nucleotides that are widely expressed in animals and plants (10). miRNA can result in post-transcriptional mRNA degradation or the inhibition of translation by binding to the 3′ untranslated region (3′-UTR) of its target mRNA (11). miRNAs serve a key role in cell proliferation, cell death and organ development (12–14). It has previously been reported that inhibition of miR-26a may increase the apoptosis of primary cultured chronic lymphocytic leukemia cells by inhibiting phosphatase and tension protein homologues (15). In addition, miR-26a inhibits the transcription and replication of the hepatitis B virus by targeting zinc-binding proteins of host factor cysteine and histidine-rich domains (16). Previous studies have indicated that miR-26 may serve a key role in the growth and development of normal tissues, as well as in the pathogenesis of non-neoplastic diseases and tumorigenesis (17–19). HS formation is characterized by the excessive growth of dense fibrous tissue (20). Based on this, it was hypothesized in the present study that miR-26a may serve an important role in the formation of HS.
In the present study, the differential expression of miR-26a in HS tissues and paired normal skin (NS) tissues was investigated. Mothers against decapentaplegic homolog 2 (Smad2), one of the important members of the transforming growth factor β (TGF-β) pathway family, was identified as a target of miR-26a by luciferase reporter assay. Overexpression of miR-26a was demonstrated to inhibit proliferation and induce the apoptosis of HSF cells in vitro. In addition, miR-26a overexpression inhibited extracellular matrix deposition in human HS fibroblasts (hHSFs). miR-26a in hHSFs may be a novel potential therapeutic target for the treatment of HSs.
Materials and methods
Cell culture and tissue samples
In total, 20 paired HS and normal skin (NS) tissues were obtained during a scar excision and auto-skin grafting biopsy from 20 patients (age range, 17–58 years; 13 female, 7 male) in the Affiliated Hospital of Nantong University (Nantong, China) between February 2013 and December 2016. All patients were confirmed by clinical and pathological diagnosis. The inclusion criteria were as follows: the patients must not have systemic disease, must not have received hormonal drugs 3 months prior to surgery and must have a local scar without treatment in the form of radiation, injection or other topical drugs. All tissues were immediately stored in liquid nitrogen until use. The present study was approved by the Ethics Committee of the Affiliated Hospital of Nantong University (Nantong, China). Written informed consent was obtained from all patients.
The human embryonic skin fibroblasts CCC-ESF-1 (ESF) and human HS fibroblasts (hHSFs) were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA USA) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin-streptomycin solution (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Cells (5×104 cells per well) were seeded in a 6-well plate and incubated in a humidified incubator at 37°C for ~24 h in an atmosphere containing 5% CO2. To investigate the role of miR-26a in hHSFs, the 50 nM miR-26a mimic (5′-TTCAAGTAATCCAGGATAGGCT-3′), 100 nM miR-26a inhibitor (5′-AGCCTATCCTGGATTACTTGAA-3′), 50 nM negative control (NC; all Shanghai GenePharma, Co., Ltd., Shanghai, China), Smad2 plasmids, control plasmids, Smad2 siRNA, or control siRNA (Santa Cruz Biotechnology, Inc., Dallas, CA, USA) were transfected into hHSFs using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocols. Transfected cells were incubated at 37°C in an atmosphere containing 5% CO2. Following transfection for 48 h, the cells were subjected to following experiments.
RNA extraction and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from ESFs, hHSFs, HS tissues and NS tissues using TRIzol reagent (Thermo Fisher Scientific, Inc.), and cDNA was synthesized using the miScript Reverse Transcription kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's protocol. The RT conditions were as follows: 16°C for 30 min, 42°C for 30 min, 85°C for 5 min and hold at 4°C. qPCR was used to detect miR-26a and associated mRNA expression using a Bio-Rad machine (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and a SYBR Premix Ex TaqTM II (TliRNaseH Plus) kit (Takara Bio, Inc., Otsu, Japan). The Primer sequences used in qPCR are presented in Table I. The thermocycling conditions were: 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 15 sec and annealing/elongation at 60°C for 30 sec. The expression of miR-26a was analyzed using the 2−ΔΔCq method (21), normalizing to U6 expression.
Table I.
Primer sequences for polymerase chain reaction.
| Primer name | Sequence (5′-3′) |
|---|---|
| Smad2-Forward: | CGTCCATCTTGCCATTCACG |
| Smad2-Reverse: | CTCAAGCTCATCTAATCGTCCTG |
| Type I collagen-Forward: | CCCTGAGTGGAAGAGTGGAG |
| Type I collagen-Reverse: | GAGGCGTGAGGTCTTCTGTG |
| Type III collagen-Forward: | GGAGCTGGCTACTTCTCGC |
| Type III collagen-Reverse: | GGGAACATCCTCCTTCAACAG |
| miR-26a-Forward: | CAAGUAAUCCAGGAUAGG |
| miR-26a-Reverse: | GGCCAACCGCGAGAAGATGTTTTTTTTT |
| U6-Forward: | GGAGCGAGATCCCTCCAAAAT |
| U6-Reverse: | GGCTGTTGTCATACTTCTCATGG |
| GAPDH-Forward: | CTTTGGTATCGTGGAAGGACTC |
| GAPDH-Reverse: | GTAGAGGCAGGGATGATGTTCT |
miR, microRNA.
Cell proliferation assay
The Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) assay was used as a qualitative index of cell viability, which was based on the conversion of a water-soluble tetrazolium salt to a water-soluble formazan dye upon reduction by dehydrogenases in the presence of an electron carrier (22). To determine cell growth, ~5,000 hHSFs were seeded into the wells of 96-well plates in triplicate and transfected with miR-26a mimic, miR-26a inhibitor or NC. At 48 h post-cell transfection, 10 µl CCK-8 solution mixed with 90 µl RPMI-1640 was added to each well. After 4 h of incubation at 37°C, the absorbance was measured at 450 nm.
Apoptosis assay
In order to study the effect of miR-26a on cell apoptosis, hHSFs were transfected with miR-26a mimic, miR-26a inhibitor or NC for 48 h. Cells were subsequently stained with Annexin V-fluorescein isothiocyanate/propidium iodide (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer's protocol for 15 min at room temperature in the dark. Cell apoptosis was then detected by flow cytometry. Data were collected using a flow cytometer and analyzed using WinMDI version 2.5 software (Purdue University Cytometry Laboratories; http://www.cyto.purdue.edu/flowcyt/software/Catalog.htm).
Dual-luciferase reporter assay
To confirm direct target binding, the wild type (WT-Smad2) and mutant (MUT-Smad2) 3′-UTR of Smad2 (identified by TargetScan; http://www.targetscan.org) was cloned into a pmiR-RB-ReportTM dual luciferase reporter gene plasmid vector (Guangzhou RiboBio Co., Ltd., Guangzhou, China). hHSFs were co-transfected with 100 ng WT-Smad2 or 100 ng MUT-Smad2 and 50 nM miR-26a or its control (50 nM; miR-C) vector using Lipofectamine® 2000 according to the manufacturer's protocols. After 48 h, luciferase activity was analyzed using the Dual-Luciferase Assay system (Promega Corporation, Madison, WI, USA) according to the manufacturer's protocol and normalized to Renilla luciferase activity.
Western blot analysis
hHSFs were transfected with miR-26a mimic, miR-26a inhibitor or NC for 48 h, cells were collected and total proteins were extracted in 40 mM Tris-HCl (pH 7.4) containing 150 mM NaCl and 1% (v/v) Triton X-100, supplemented with protease inhibitors (Auragene Bioscience Co., Changsha, China). The protein concentration was determined using a bicinchoninic acid protein assay (Pierce; Thermo Fisher Scientific, Inc.). Equal amounts of protein (30 µg/lane) were resolved by 10% SDS-PAGE and then transferred to a polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA, USA). Following blocking with 5% skimmed milk in tris-buffered saline with Tween-20 at room temperature for 1.5 h, membranes were probed with antibodies against Smad2 (1:1,000; cat. no. 8685; Cell Signaling Technology, Inc., Danvers, MA, USA), collagen (Col) I (1:1,000; cat. no. ab34710; Abcam, Cambridge, UK), Col III (1:1,000; cat. no. ab7778; Abcam), Bcl-2 (B-cell lymphoma-2; 1:1,000; cat. no. 4223; Cell Signaling Technology, Inc.), Bax (Bcl-2-associated X protein; 1:1,000; cat. no. 5023; Cell Signaling Technology, Inc.) and β-actin (1:5,000; cat. no. 4970; Cell Signaling Technology, Inc.). Following three washes with Tris-buffered saline/Tween-20, membranes were incubated with anti-rabbit Immunoglobulin G horseradish peroxidase-coupled secondary antibody (cat no. 7074; 1:1,000; Cell Signaling Technology). Immunoreactive bands were visualized using the enhanced chemiluminescence detection system (Applygen Technologies, Inc., Beijing, China). Data were analyzed using WinMDI version 2.5 software (Purdue University Cytometry Laboratories).
Statistical analysis
SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA) was used to analyze the data. Values are expressed as the mean ± standard deviation of experiments performed in triplicate. Data were analyzed by one-way analysis of variance followed by a post hoc Tukey's test or a Student's t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
miR-26a is downregulated in HS tissues and hHSFs
A total of 20 HS tissues and 20 paired normal skin (NS) were used in the present study. Compared with NS, the expression of miR-26a was significantly decreased in HS tissues (Fig. 1A). In addition, the expression of miR-26a in human embryonic skin fibroblasts CCC-ESF-1 and hHSFs was assessed; the expression of miR-26a was significantly lower in hHSFs compared with the CCC-ESF-1 cells (Fig. 1B).
Figure 1.

Expression of miR-26a in HS. Relative miR-26a expression in (A) HS and paired NS tissues, (B) CCC-ESF-1 cells and hHSFs, and (C) Con, NC, mimic and inhibitor transfection groups. Data are presented as the mean ± standard deviation. &&P<0.01 vs. NS; ##P<0.01 vs. CCC-ESF-1; **P<0.01 vs. Con. miR, microRNA; HS, hypertrophic scar; NS, normal skin; hHSFs, human HS fibroblasts; NC, negative control; Con, control, normal hHSFs; inhibitor, cells transfected with miR-26a inhibitor; mimic, cells transfected with miR-26a mimic.
The effects of miR-26a gain and loss-of-function on HS formation were investigated in the present study. hHSFs were transfected with miR-26a mimic, miR-26a inhibitor or NC. The transfection efficiency in each cell line was examined by RT-qPCR. miR-26a expression was significantly increased in cells transfected with miR-26a mimic and significantly decreased in those transfected with miR-26a inhibitor (Fig. 1C).
miR-26a directly targets Smad2
Targetscan was used to predict the targets of miR-26a and Smad2 was identified as a potential miR-26a target gene (Fig. 2A). The putative binding sites in the Smad2 3′-UTR evolved in a considerable number of species. To verify the binding site, a dual luciferase reporter assay was performed and the results indicated that cotransfection withmiR-26a significantly decreased the luciferase activity in hHSFs transfected with WT-Smad2 compared with in the miR-C group, whereas cotransfection with MUT-Smad2 inhibited the miR-26a-mediated downregulation in luciferase activity (Fig. 2B).
Figure 2.

Smad2 is a target gene of miR-26a. (A) TargetScan was used to predict the interaction between miR-26a and 3′untranslated region of Smad2. (B) Luciferase activity was measured by dual-luciferase assay. Relative Smad2 mRNA expression in (C) HS tissues and (D) CCC-ESF-1 cells and hHSFs were determined by reverse transcription-quantitative polymerase chain reaction. (E) Effects of miR-26a on Smad2 (E) protein and (F) mRNA expression in hHSFs Data are presented as the mean + standard deviation. **P<0.01 vs. miR-C, &&P<0.01 vs. NS, ##P<0.01 vs. CCC-ESF-1, $$ P<0.01 vs. Co. Smad2, mothers against decapentaplegic homolog 2; miR, microRNA; HS, hypertrophic scar; hHSFs, human HS fibroblasts; Smad2-MUT, Smad2 3′UTR with a mutation in the miR-26a binding site; Smad2-WT, wild type Smad2 3′UTR; Con, control group, cells without any treatment; inhibitor, cells transfected with miR-26a inhibitor; mimic, cells transfected with miR-26a mimic; NC, negative control group, cells transfected with the negative control vector.
The expression of Smad2 in HS tissues and hHSFs was assessed using RT-qPCR. The results demonstrated that Smad2 expression was significantly upregulated compared in HS compared with NS tissues (Fig. 2C) and in hHSFs compared with CCC-ESF-1 cells (Fig. 2D). Furthermore, following transfection with the miR-26a mimics, the mRNA of Smad2 expression in hHSFs was detected. The results revealed that Smad2 levels were markedly decreased following miR-26a overexpression (Fig. 2E). Furthermore, Smad2 protein expression was significantly downregulated in cells transfected with miR-26a-mimic, whereas it was significantly upregulated in cells transfected with the miR-26a inhibitor (Fig. 2F).
miR-26a suppresses hHSF cell proliferation and induces cell apoptosis in vitro
At 48 h following transfection, the cell proliferation ability was measured using a CCK-8 assay. The results revealed that transfection with miR-26a mimics significantly suppressed hHSF cell proliferation. Conversely, cell proliferation was promoted in the miR-26a inhibitor group (Fig. 3).
Figure 3.
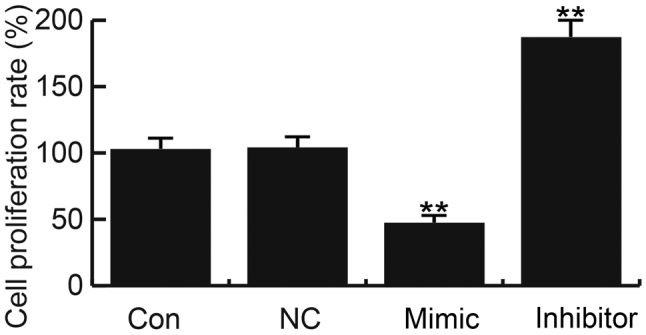
Effects of miR-26a on hHSF cell proliferation. At 48 h post-transfection, a Cell Counting Kit-8 assay was conducted to determine the effects of miR-26a on hHSF cell proliferation. Data are presented as the mean ± standard deviation. **P<0.01 vs. Con. miR, microRNA; hHSFs, human hypertrophic scar fibroblasts; Con, control group, cells without any treatment; inhibitor, cells transfected with miR-26a inhibitor; mimic, cells transfected with miR-26a mimic; NC, negative control group, cells transfected with the negative control vector.
hHSF cell apoptosis was detected at 48 h post-transfection with miR-26a mimics, miR-26a inhibitor or NC. Flow cytometry analysis demonstrated that the number of cells in early (Q3 in Fig. 4) and late apoptosis (Q2 in Fig. 4) was increased in hHSFs transfected with miR-26a mimic compared with the control groups (Fig. 4). Conversely, apoptosis was decreased in miR-26a inhibitor transfected hHSFs compared with control cells (Fig. 4A and B). Additionally, pro-apoptotic (Bax) and anti-apoptotic proteins (Bcl-2) were detected using western blotting. Transfection with miR-26a mimics promoted Bax expression and inhibited Bcl-2 expression compared with the control cells (Fig. 3C). Transfection with miR-26a had the opposite effect (Fig. 4C). Collectively, these data indicate that miR-26a may inhibit cell proliferation and promote cell apoptosis in hHSFs in vitro.
Figure 4.

Effects of miR-26a on hHSF cell apoptosis. (A and B) At 48 h post-transfection, flow cytometry was used to assess the effect of miR-26a on hHSF cell apoptosis. (C) Effects of miR-26a on the protein expression levels of Bcl-2 and Bax were analyzed by western blotting. Data are presented as the mean ± standard deviation. *P<0.05 and **P<0.01 vs. Con. miR, microRNA; hHSFs, human hypertrophic scar fibroblasts; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; Con, control group, cells without any treatment; FITC, fluorescein isothiocyanate; inhibitor, cells transfected with miR-26a inhibitor; mimic, cells transfected with miR-26a mimic; NC, negative control group, cells transfected with the negative control vector; PI, propidium iodide.
miR-26a suppresses extracellular matrix deposition in hHSFs
To further investigate the molecular mechanism of miR-26a, western blotting and RT-qPCR were performed to detect Col I and Col III in hHSFs transfected with miR-26a mimics or miR-26a inhibitor. miR-26a mimics decreased the protein and mRNA expression of Col I and Col III; the reverse was observed when cells were transfected with the miR-26a inhibitor (Fig. 5). To determine whether modulation of Smad2 affects HS formation, hHSFs cells were transfected with Smad2 plasmids or Smad2 siRNA. The results demonstrated that Smad2 overexpression may increase the expression of Col I and Col III; however, Smad2 silencing significantly downregulated Col I and Col III expression (Fig. 6). These results suggest that miR-26a may suppress HS formation via the targeting Smad2 and regulating Col I and Col III expression.
Figure 5.

Effects of miR-26a on Col I and Col III expression in hHSFs. At 48 h post-transfection, Col I and Col III expression was assessed using (A) western blotting and (B and C) reverse transcription-quantitative polymerase chain reaction. Data are presented as the mean ± standard deviation. **P<0.01 vs. Con. miR, microRNA; Col, collagen; hHSFs, human hypertrophic scar fibroblasts; con, control group, cells without any treatment; inhibitor, cells transfected with miR-26a inhibitor; mimic, cells transfected with miR-26a mimic; NC, negative control group, cells transfected with the negative control vector.
Figure 6.

Effects of Smad2 on Col I and Col III expression in hHSFs. hHSFs cells were transfected with Smad2-plasmid or Smad2-siRNA. Smad2, Col I and Col III expression were determined by (A) western blotting and (B-D) reverse transcription-quantitative polymerase chain reaction at 48 h post-transfection. Data are presented as the mean ± standard deviation. *P<0.05 and **P<0.01 vs. Con-p; #P<0.05 and ##P<0.01 vs. Con-s. Smad2, mothers against decapentaplegic homolog 2; Col, collagen; hHSFs, human hypertrophic scar fibroblasts; Smad2-siRNA, cells transfected with Smad2-small interfering RNA; Con-p, cells transfected with control plasmid; con-s, cells transfected with control siRNA; plasmid, cells transfected with Smad2-plasmid.
Discussion
miR-26a may serve a key role in the growth and development of normal tissues, as well as the pathogenesis of non-neoplastic diseases and tumorigenesis (17–19,23,24). HS formation is characterized by excessive growth of dense fibrous tissue, which is caused by severe heat or traumatic injury to the skin (20). Abnormal expression of miRNAs may contribute to the management of skin fibrotic diseases (25). A recent study investigated the role of miR-26a in scar formation following glaucoma filtration surgery and the findings suggested that miR-26a served an essential role in filtering tract scar formation (26); however, these results were limited to the role of miR-26a in postoperative scar formation. The present study was conducted to investigate the role of miR-26a in HS.
Our preliminary study demonstrated that miR-26a expression was significantly reduced in HS tissues compared with paired NS tissues and a similar result was observed in hHSFs. Therefore, hHSFs were selected for further analysis.
Smad is a conserved protein in eukaryotes and contains three subtypes (27). Smad2 is a tumor suppressor that is able to regulate chromosome 18q21-22, which is a receptor-activated Smad (28). TGF-β1 is the most important fibroblast-forming cytokine and serves roles in cell differentiation, proliferation and apoptosis. The TGF-β1 signal is transduced mainly by downstream molecules Smad2 and Smad3 (28). Following trauma, TGF-β1 is firstly released from platelets to the site of injury, then dose-dependent chemotaxis of fibroblasts, monocytes, neutrophils and other cells occur (29). Smad2/3-TGF-β1 signals are translocated from the cytoplasm to the nucleus and to regulate matrix protein production (including fibronectin and collagen), target gene transcription, and extracellular matrix (ECM) synthesis and degradation (30,31). The results of the present study suggest that Smad2 is a target gene of miR-26a and the expression of Smad2 was upregulated both in HS tissues and HSFs. Additionally, Smad2 was significantly reduced in hHSF cells transfected with miR-26a mimics; however, opposing results were observed within hHSF cells transfected with the miR-26a inhibitor.
The role of miR-26a in hHSFs was also investigated in the present study. miR-26a mimic transfection significantly suppressed cell proliferation and induced cell apoptosis in HSFs. As expected, transfection with the miR-26a inhibitor had the opposite effect. These findings may provide a novel insight into how miR-26a may regulate hHSF cell proliferation and apoptosis by inhibiting Smad2 expression.
Increased collagen synthesis and alterations are considered to be the main features of HS formation (32,33). It has previously been reported that the expression of Col I and Col III in hHSFs and HS tissues is significantly higher compared with NS tissues (34). Overexpression of fibroblasts, in particular Col I and Col III, is responsible for keloid and HS formation (35). In the present study, miR-26a mimics and inhibitors reduced and enhanced Col I and Col III expression, respectively. Moreover, the current study demonstrated that Smad2 overexpression and silencing enhanced and inhibited the expression of Col I and Col III, respectively. The data indicated that miR-26a regulates Col I and Col III expression by targeting Smad2, suggesting that miR-26a acts as an antifibrotic factor in HSFs. Based on these findings, miR-26a and Smad2 may serve an important role in tissue repair, which may be associated with inflammatory processes. However, further investigation is required to verify these results.
In conclusion, the results of the present study demonstrate that miR-26a reduces the expression of ECM-associated proteins by targeting Smad2, which may affect hypertrophic scarring by inhibiting cell proliferation and inducing HSF apoptosis. Thus, the present study provided evidence that miR-26a may serve as a novel potential therapeutic target for the treatment of hypertrophic scarring.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The analyzed data sets generated during the present study are available from the corresponding author on reasonable request.
Authors' contributions
JQ and YW designed the study. JQ, YL, KH, YZ and XZ were responsible for the data collection and analysis. All authors collaborated to interpret results and develop the manuscript. All authors have read and approved this manuscript.
Ethics approval and consent to participate
The present study was approved by the Ethics Committee of the Affiliated Hospital of Nantong University (Nantong, China). Written informed consent was obtained from all patients.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Tyack ZF, Pegg S, Ziviani J. Postburn dyspigmentation: Its assessment, management, and relationship to scarring-a review of the literature. J Burn Care Rehabil. 1997;18:435–440. doi: 10.1097/00004630-199709000-00012. [DOI] [PubMed] [Google Scholar]
- 2.Das S, Baker AB. Biomaterials and nanotherapeutics for enhancing skin wound healing. Front Bioeng Biotechnol. 2016;4:82. doi: 10.3389/fbioe.2016.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gauglitz GG, Korting HC, Pavicic T, Ruzicka T, Jeschke MG. Hypertrophic scarring and keloids: Pathomechanisms and current and emerging treatment strategies. Mol Med. 2011;17:113–125. doi: 10.2119/molmed.2009.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sidgwick GP, Iqbal SA, Bayat A. Altered expression of hyaluronan synthase and hyaluronidase mRNA may affect hyaluronic acid distribution in keloid disease compared with normal skin. Exp Dermatol. 2013;22:377–379. doi: 10.1111/exd.12147. [DOI] [PubMed] [Google Scholar]
- 5.Meyer LJ, Russell SB, Russell JD, Trupin JS, Egbert BM, Shuster S, Stern R. Reduced hyaluronan in keloid tissue and cultured keloid fibroblasts. J Invest Dermatol. 2000;114:953–959. doi: 10.1046/j.1523-1747.2000.00950.x. [DOI] [PubMed] [Google Scholar]
- 6.Syed F, Ahmadi E, Iqbal SA, Singh S, McGrouther DA, Bayat A. Fibroblasts from the growing margin of keloid scars produce higher levels of collagen I and III compared with intralesional and extralesional sites: Clinical implications for lesional site-directed therapy. Br J Dermatol. 2011;164:83–96. doi: 10.1111/j.1365-2133.2010.10048.x. [DOI] [PubMed] [Google Scholar]
- 7.Zuccaro J, Ziolkowski N, Fish J. A systematic review of the effectiveness of laser therapy for hypertrophic burn scars. Clin Plast Surg. 2017;44:767–779. doi: 10.1016/j.cps.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Kashiyama K, Mitsutake N, Matsuse M, Ogi T, Saenko VA, Ujifuku K, Utani A, Hirano A, Yamashita S. miR-196a downregulation increases the expression of type I and III collagens in keloid fibroblasts. J Invest Dermatol. 2012;132:1597–1604. doi: 10.1038/jid.2012.22. [DOI] [PubMed] [Google Scholar]
- 9.Li P, He QY, Luo CQ. Overexpression of miR-200b inhibits the cell proliferation and promotes apoptosis of human hypertrophic scar fibroblasts in vitro. J Dermatol. 2014;41:903–911. doi: 10.1111/1346-8138.12600. [DOI] [PubMed] [Google Scholar]
- 10.Chen Z, Li J, Tian L, Zhou C, Gao Y, Zhou F, Shi S, Feng X, Sun N, Yao R, et al. MiRNA expression profile reveals a prognostic signature for esophageal squamous cell carcinoma. Cancer Lett. 2014;350:34–42. doi: 10.1016/j.canlet.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 11.Lee HJ. Exceptional stories of microRNAs. Exp Biol Med (Maywood) 2013;238:339–343. doi: 10.1258/ebm.2012.012251. [DOI] [PubMed] [Google Scholar]
- 12.Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 13.McCarthy JJ. The MyomiR network in skeletal muscle plasticity. Exerc Sport Sci Rev. 2011;39:150–154. doi: 10.1097/JES.0b013e31821c01e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heinrich EM, Dimmeler S. MicroRNAs and stem cells: Control of pluripotency, reprogramming, and lineage commitment. Circ Res. 2012;110:1014–1022. doi: 10.1161/CIRCRESAHA.111.243394. [DOI] [PubMed] [Google Scholar]
- 15.Zou ZJ, Fan L, Wang L, Xu J, Zhang R, Tian T, Li JY, Xu W. miR-26a and miR-214 down-regulate expression of the PTEN gene in chronic lymphocytic leukemia, but not PTEN mutation or promoter methylation. Oncotarget. 2015;6:1276–1285. doi: 10.18632/oncotarget.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao F, Xu G, Zhou Y, Wang L, Xie J, Ren S, Liu S, Zhu Y. MicroRNA-26b inhibits hepatitis B virus transcription and replication by targeting the host factor CHORDC1 protein. J Biol Chem. 2014;289:35029–35041. doi: 10.1074/jbc.M114.589978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao S, Ye X, Xiao L, Lian X, Feng Y, Li F, Li L. MiR-26a inhibits prostate cancer progression by repression of Wnt5a. Tumour Biol. 2014;35:9725–9733. doi: 10.1007/s13277-014-2206-4. [DOI] [PubMed] [Google Scholar]
- 18.Shen W, Song M, Liu J, Qiu G, Li T, Hu Y, Liu H. MiR-26a promotes ovarian cancer proliferation and tumorigenesis. PLoS One. 2014;9:e86871. doi: 10.1371/journal.pone.0086871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian H, Yang C, Yang Y. MicroRNA-26a inhibits the growth and invasiveness of malignant melanoma and directly targets on MITF gene. Cell Death Discov. 2017;3:17028. doi: 10.1038/cddiscovery.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolfram D, Tzankov A, Pülzl P, Piza-Katzer H. Hypertrophic scars and keloids-a review of their pathophysiology, risk factors, and therapeutic management. Dermatol Surg. 2009;35:171–181. doi: 10.1111/j.1524-4725.2008.34406.x. [DOI] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Han SB, Shin YJ, Hyon JY, Wee WR. Cytotoxicity of voriconazole on cultured human corneal endothelial cells. Antimicrob Agents Chemother. 2011;55:4519–4523. doi: 10.1128/AAC.00569-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J, Liu QG. The role of miR-26 in tumors and normal tissues (Review) Oncol Lett. 2011;2:1019–1023. doi: 10.3892/ol.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B, Liu XX, He JR, Zhou CX, Guo M, He M, Li MF, Chen GQ, Zhao Q. Pathologically decreased miR-26a antagonizes apoptosis and facilitates carcinogenesis by targeting MTDH and EZH2 in breast cancer. Carcinogenesis. 2011;32:2–9. doi: 10.1093/carcin/bgq209. [DOI] [PubMed] [Google Scholar]
- 25.Babalola O, Mamalis A, Lev-Tov H, Jagdeo J. The role of microRNAs in skin fibrosis. Arch Dermatol Res. 2013;305:763–776. doi: 10.1007/s00403-013-1410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang WH, Deng AJ, He SG. A key role of microRNA-26a in the scar formation after glaucoma filtration surgery. Artif Cells Nanomed Biotechnol. 2017;7:1–7. doi: 10.1080/21691401.2017.1415212. [DOI] [PubMed] [Google Scholar]
- 27.Hao R, Chen L, Wu JW, Wang ZX. Structure of drosophila Mad MH2 domain. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:986–990. doi: 10.1107/S1744309108033034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2011;114:4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- 29.Froese AR, Shimbori C, Bellaye PS, Inman M, Obex S, Fatima S, Jenkins G, Gauldie J, Ask K, Kolb M. Stretch-induced activation of transforming growth factor-β1 in pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194:84–96. doi: 10.1164/rccm.201508-1638OC. [DOI] [PubMed] [Google Scholar]
- 30.Cao S, Xiao L, Rao JN, Zou T, Liu L, Zhang D, Turner DJ, Gorospe M, Wang JY. Inhibition of Smurf2 translation by miR-322/503 modulates TGF-β/Smad2 signaling and intestinal epithelial homeostasis. Mol Biol Cell. 2014;25:1234–1243. doi: 10.1091/mbc.E13-09-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin K, Yin W, Wang Y, Zhou L, Liu Y, Yang G, Wang J, Lu J. MiR-206 suppresses epithelial mesenchymal transition by targeting TGF-β signaling in estrogen receptor positive breast cancer cells. Oncotarget. 2016;7:24537–24548. doi: 10.18632/oncotarget.8233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuo J, Chen Z, Zhong X, Lan W, Kuang Y, Huang D. FBP1 is highly expressed in human hypertrophic scars and increases fibroblast proliferation, apoptosis, and collagen expression. Connect Tissue Res. 2017;31:1–9. doi: 10.1080/03008207.2017.1311327. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Chen L, Cao C, Yan H, Zhou B, Gao Y, Li Q, Li J. The long non-coding RNA LncRNA8975-1 is upregulated in hypertrophic scar fibroblasts and controls collagen expression. Cell Physiol Biochem. 2016;40:326–334. doi: 10.1159/000452548. [DOI] [PubMed] [Google Scholar]
- 34.Zhou R, Zhang Q, Zhang Y, Fu S, Wang C. Aberrant miR-21 and miR-200b expression and its pro-fibrotic potential in hypertrophic scars. Exp Cell Res. 2015;339:360–366. doi: 10.1016/j.yexcr.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Fan C, Dong Y, Xie Y, Su Y, Zhang X, Leavesley D, Upton Z. Shikonin reduces TGF-β1-induced collagen production and contraction in hypertrophic scar-derived human skin fibroblasts. Int J Mol Med. 2015;36:985–991. doi: 10.3892/ijmm.2015.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The analyzed data sets generated during the present study are available from the corresponding author on reasonable request.