

Abstract
Objective
To determine whether white matter changes influence progression of cognitive decline in individuals with clinically diagnosed Alzheimer disease (AD) and differing biomarker profiles.
Methods
Two hundred thirty-six individuals from the Alzheimer's Disease Neuroimaging Initiative database with clinical diagnoses of cognitively normal older adult (older controls [OCs]), mild cognitive impairment, and AD were studied. Support vector machine experiments were first performed to determine the utility of various biomarkers for classifying individuals by clinical diagnosis. General linear models were implemented to assess the relationships between CSF measures of β-amyloid 1–42, phosphorylated tau181p, and MRI-based white matter signal abnormality (WMSA) volumes and cognitive decline. Analyses were performed across all patients as well as within subgroups of individuals that were defined by clinical cutoff points for both CSF measures.
Results
CSF biomarkers alone classified individuals with AD vs OCs with 82% accuracy, and the addition of WMSA did not enhance this. Both CSF biomarkers as well as WMSA volume significantly contributed to predicting cognitive decline in executive and memory domains when assessed across all 236 individuals. In individuals with pathologic levels of both CSF biomarkers, WMSA only significantly contributed to models of future executive function decline. In individuals with subpathologic CSF biomarker levels (levels similar to those in OC individuals), WMSA significantly contributed to prediction of memory decline and were the sole significant predictor of executive function decline.
Conclusions
WMSA hold additional predictive power regarding cognitive progression in older individuals and are most effective as biomarkers in individuals who are cognitively impaired but do not fit the expected CSF biomarker profile of AD.
Dementia refers to a varied collection of cognitive impairment syndromes whose underlying causes are often difficult to elucidate. Of these, the most common is Alzheimer disease (AD), which is pathologically diagnosed based on abnormal amyloid and tau pathology but is also highly comorbid with additional pathologic processes.1,2 To date, clinical trials that target amyloid and tau have been largely unsuccessful, suggesting that other factors that affect cognition either independently or in concert with amyloid and tau are at play and warrant deeper consideration.3
CSF levels of β-amyloid 1–42 (Aβ42) and tau phosphorylated at threonine 181 (p-tau181p) are two of the most clinically utilized biomarkers of AD4–6 and are related to the presence of Aβ plaques and tau neurofibrillary tangles in the cerebral cortex.7 Autopsy studies, however, have demonstrated the presence of AD pathologies in the cerebral cortices of individuals who had fully intact cognitive functioning, as well as a lack thereof in individuals who passed away with a clinical diagnosis of AD.8,9
There is a strong body of evidence suggestive of a cerebrovascular component to AD.9–11 White matter signal abnormalities (WMSA) as seen on MRI are used as a surrogate marker of cerebrovascular damage in vivo12,13 and are associated with cognitive decline and dementia.14,15 Previous work ranges from suggesting a synergistic role of WMSA and CSF biomarkers on cognition to no relationship between the two.9,16–28 Of note, it is unclear whether WMSA have a prominent role in future cognitive decline and how they are related to CSF biomarkers in this context.
The purpose of this study was to assess the relationships among WMSA, Aβ42, and p-tau181p and their combined effect on cognitive decline. We assessed the ability of these markers to classify clinical diagnoses as well as to predict longitudinal changes in cognition. Finally, we assessed the relationships of Aβ42, p-tau181p, WMSA, and cognitive decline in a specific subgroup of these individuals who demonstrated a “healthy” CSF profile.
Methods
Participants
Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu) and are described here as directed by ADNI. Two hundred thirty-six individuals were used in the analyses; 61 of these had a diagnosis of AD, 56 were cognitively healthy age-matched older controls (OCs), and 119 had a diagnosis of amnestic mild cognitive impairment (MCI). Diagnoses were based on clinical assessment that included standard neuropsychological test batteries and interviews, and were made independently of CSF biomarker values. Within the 36-month duration of the ADNI-1 study, 64 of the patients with MCI converted to a diagnosis of AD, creating a further stratification of individuals with MCI who converted to having AD within 36 months (MCI-C; n = 64) and those who did not convert (MCI-NC; n = 55). All individuals underwent genetic screening for the presence of 0, 1, or 2 APOE ε4 alleles. Demographic data, such as age, sex, years of education, history of hypertension, history of endocrine-metabolic disorder, composite memory scores (ADNI-MEM), and composite executive function scores (ADNI-EF), were additionally acquired from the ADNI database (table 1).
Table 1.
Demographic information of individuals in the 3 diagnostic groups

Standard protocol approvals, registrations, and patient consents
Each participating ADNI site (1) received approval from an ethical standards committee on human experimentation before study initiation, and (2) obtained written informed consent for research from all individuals participating in the study.
Neuropsychological MRI acquisition
All data were acquired on a 1.5-tesla scanner at rigorously validated sites, which all followed a previously described standardized protocol.29 The protocol included a high-resolution, T1-weighted, sagittal volumetric magnetization-prepared rapid-acquisition gradient echo sequence and axial proton density–weighted/T2-weighted fast spin echo sequence. The ADNI MRI core optimized the acquisition parameters of these sequences for each make and model of scanner included in the study. All scanner sites were required to pass a strict scanner validation test before authorization to scan ADNI participants. In addition, each scan of ADNI participants included a scan of the phantom, which was required to pass strict validation tests.
MRI preprocessing
Cortical reconstruction and volumetric segmentation was performed using FreeSurfer's standard recon-all stream (surfer.nmr.mgh.harvard.edu, version 5.1). The technical details of these procedures are described in prior publications.30–34 Automatic segmentation and volumetric measurement of WMSA was done using a FreeSurfer-based validated tool described in our previous work that uses T1-weighted, T2-weighted, and proton density–weighted images as inputs.35
CSF biomarker data
In the ADNI study, participants receive a lumbar puncture at their baseline visit to obtain CSF for assays of Aβ42, total tau, and p-tau181p. Sample collection and analysis procedures are described in detail in reference 36. In the present study, CSF biomarker quadrants were created across all 238 individuals based on the median Aβ42 and p-tau181p values in the participants with MCI (figure 1). For Aβ42, this median value was 144 pg/mL, and for p-tau181p, this median value was 35 pg/mL. Individuals who were classified in Q1 as demonstrated in figure 1 had pathologic levels of both CSF biomarkers, while individuals in Q3 had normal levels of both CSF biomarkers, and based on these values alone, would be expected to present as healthy older individuals. While there is no definitive consensus for clinical cutoff points for CSF biomarkers in determining AD diagnosis, these 2 values are reflective of what is typically used in the clinical setting.37–39 In these data, it is important to note that ADNI diagnosis of individuals is done independently of the CSF biomarker profile.
Figure 1. CSF biomarker data for all 236 study individuals.
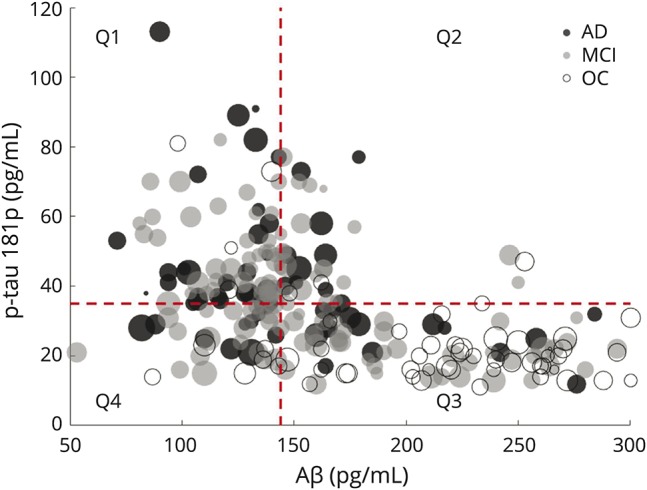
The radius of each point reflects the total white matter signal abnormality burden for that individual. Horizontal and vertical dashed lines are MCI-derived median cutoff points for each of the 2 CSF biomarkers, which reflect clinical standard values, dividing the entire study population into 4 quadrants, with the upper left-hand quadrant deemed quadrant 1 (Q1) and the lower right-hand quadrant deemed quadrant 3 (Q3). Individuals in Q1 demonstrate the most pathologic burdens of both CSF biomarkers, and individuals in Q3 fall into the normal range of both CSF biomarkers and would be expected to be cognitively healthy based on these values. Aβ = β-amyloid; AD = Alzheimer disease; MCI = mild cognitive impairment; OC = older control; p-tau181p = tau phosphorylated at threonine 181.
Classification
Support vector machine (SVM) classification experiments were conducted to determine whether CSF biomarkers and WMSA volumes contained information sufficient to predict diagnoses. In these experiments, we implemented a k-fold cross-validation scheme in which we used 90% of patients to train an SVM to classify between 2 diagnostic groups, and then tested this SVM on the remaining 10% of patients. For each SVM paradigm, this process was repeated 1,000 times with a random training subset of 90% of patients and tested on the remaining 10%. The average accuracy of all 1,000 iterations is the classification accuracy for a given paradigm.
Cognitive measures
To assess cognition in participants in the current study, we utilized the ADNI-MEM and ADNI-EF composite scores of memory and executive functioning that are available in the ADNI dataset (table 1). These scores were created and rigorously validated in previous works.40,41 Briefly, the ADNI-MEM score is a composite score of results from the Rey Auditory Verbal Learning Test, Alzheimer’s Disease Assessment Scale–Cognitive, Mini-Mental State Examination, and Logical Memory neuropsychological examinations. This composite memory score has been shown to detect changes in memory functioning over time in individuals with MCI and AD, and is also a strong predictor of conversion from MCI to AD.40 The ADNI-EF score is a composite score of results from Trails A and B, Category Fluency, Clock Drawing, Wechsler Adult Intelligence Scale–Revised Digit Symbol Substitution, and backward Digit Span examinations. Similarly to the ADNI-MEM composite score, ADNI-EF has been shown to be a strong predictor of AD conversion in an MCI cohort, is associated with MRI-derived measures of structures involved in frontal systems, and is associated with baseline CSF measures of Aβ42, total tau, and p-tau181p.41
Statistical analyses
To assess the relationships among CSF biomarkers, WMSA, and continuous measures of cognition, we used a general linear model (GLM) with cognitive scores as the dependent variables in each separate model. Significance values are reported for each independent variable in each different model to reflect that variable's contribution to the model. For each model, residual sum of squares (RSS) is reported as a measure of model performance.
Data availability
All data used in this article are available in the ADNI public data repository. Anonymized patient identification numbers from the ADNI cohort used in this article are available by request from any qualified investigator.
Results
Biomarker relationships
The Pearson product moment correlation coefficient was computed between each pair of measures and demonstrated that p-tau181p and Aβ42 are highly correlated across all individuals (r = −0.4757, p < 0.0001), and this relationship remained significant at p < 0.01 when assessing each diagnostic subgroup individually. There was no observed significant correlation between Aβ42 and total WMSA across all 236 individuals (r = −0.0559, p = 0.39) and none existed in any of the diagnostic subgroups. There was a significant correlation between p-tau181p and total WMSA (r = −0.1206, p = 0.06) across all 236 individuals at baseline. This relationship was not statistically significant in the AD or OC subgroups alone but was in the MCI cohort (r = −0.3307, p < 0.001). Figure 1 demonstrates the relationships among Aβ42, p-tau181p, and WMSA across all 236 individuals in all 3 diagnostic groups at baseline.
Diagnostic classification
SVM experiments were conducted to classify individuals with AD and OCs and then again to classify individuals with MCI-C and MCI-NC using different combinations of CSF biomarkers and WMSA. Adding total WMSA volume to classification paradigms consisting of CSF biomarkers only incrementally increased accuracies when classifying between individuals with AD and OCs, and showed no improvement when classifying between individuals with MCI-C and MCI-NC. CSF biomarkers alone achieved a maximum classification accuracy of 82% when classifying between individuals with AD and OCs, which increased to 83% when additionally using WMSA. CSF biomarkers alone were only able to classify individuals with MCI-C vs MCI-NC at an accuracy of 59%, which reduced to 57% when additionally adding WMSA. When using WMSA alone as a classifying feature, 60% accuracy was achieved for AD vs OCs and 56% accuracy was achieved for MCI-C vs MCI-NC.
Baseline cognitive performance
To assess the relationships among CSF biomarkers, WMSA, and cognitive functioning, 5 separate GLM experiments were conducted across all patients in all 3 diagnostic groups, using baseline ADNI-MEM as a dependent variable. These 5 experiments were then repeated using baseline ADNI-EF as a dependent variable. As expected based on the studies in which ADNI-MEM and ADNI-EF were created and validated, both CSF biomarkers demonstrated significant contributions to the models. Of note, however, the addition of WMSA into both models with baseline ADNI-MEM and ADNI-EF as outcome measures yielded a significant WMSA model coefficient (p < 0.001) for both cognitive domains.
Continuous measures of cognitive functioning: Longitudinal analyses
We next assessed the relationships among CSF biomarkers, WMSA, and cognitive changes over a 1-year duration using GLM experiments. The results of these experiments are reported in table 2. Only p-tau181p demonstrated a significant contribution to predicting a 1-year change in ADNI-MEM. The additions of Aβ42 and WMSA did not significantly decrease the RSS, and neither variable rendered a significant model coefficient. When assessing 1-year changes in ADNI-EF, however, all 3 independent variables (Aβ42, p-tau181p, and WMSA) significantly contributed to the model.
Table 2.
Results of general linear model experiments conducted using 1-year changes in memory scores and 1-year changes in executive function scores as dependent variables across all study patients, regardless of diagnosis

Quadrant structural differences
When assessing structural differences between individuals with impairment (MCI + AD) in Q1 (those with pathologic levels of CSF biomarkers) vs those in Q3 (normal levels of CSF biomarkers), hippocampal volume showed no significant difference (p = 0.50, figure 2, A and B). Individuals with impairment in these 2 quadrants did, however, differ in total WMSA volume (p < 0.01, figure 2C), with those in Q3 having higher overall WMSA burdens. WMSA burden between individuals with impairment and OCs in Q3 was also assessed, and figure 2 shows significantly higher WMSA in the Q3 individuals with impairment than in Q3 OC individuals (p < 0.01).
Figure 2. Hippocampal volumes, example coronal slices, and WMSA volumes.

(A) Hippocampal volumes between Q1 and Q3 in individuals with impairment (MCI + AD). (B) Example coronal slices with hippocampal view for individuals with AD in both Q1 and Q3. (C) WMSA volumes between Q1 and Q3 in individuals with impairment (MCI + AD). (D) WMSA volumes between OCs and individuals with impairment in Q3. **Significantly different at p < 0.01. AD = Alzheimer disease; ICV = intracranial volume; MCI = mild cognitive impairment; OC = older control; Q = quadrant; WMSA = white matter signal abnormality.
Quadrant genetic differences
The 2 quadrants of interest showed strikingly different genetic profiles. Of the individuals with impairment in Q1, 29% were homozygous APOE ε4 carriers (APOE ε4 +/+), but only 2% of individuals with impairment in Q3 were homozygous carriers. This imbalance was also seen in heterozygous APOE ε4 carriers whereby 49% of Q1 individuals with impairment fit this profile and only 29% of individuals with impairment in Q3 did.
Biomarkers and cognitive outcomes
When assessing 1-year changes in ADNI-MEM in Q1, there were surprisingly no significant relationships with either CSF biomarker or with WMSA. However, when assessing 1-year change in ADNI-EF in Q1, only WMSA demonstrated a significant coefficient, and adding both CSF biomarkers into the model did not significantly decrease the RSS (table 3). In Q3, when each of the CSF biomarkers and WMSA were tested individually in modeling 1-year changes in ADNI-MEM, WMSA alone showed the lowest RSS and most significant model coefficient. Aβ42 did not show a significant relationship with 1-year change in ADNI-MEM in any model configuration. For 1-year changes in ADNI-EF in Q3, only WMSA demonstrated a significant relationship with cognitive outcome (table 3).
Table 3.
Results of general linear model experiments conducted within the Q1 and Q3 CSF biomarker quadrants using 1-year changes in ADNI-MEM and ADNI-EF as outcome measures

Discussion
The results presented in this work provide new insights into the role that WMSA have in the clinical presentation of AD. Along with these new insights, our results confirm much of what is already known regarding the clinical-pathologic mismatch seen in AD diagnosis. The results of our classification experiments reflect real-world clinical misdiagnosis rates of AD that have been reported to be as high as 25% in the literature.8,42 Based on previous studies of WMSA and cognitive decline, we hypothesized that WMSA would hold predictive power for changes in continuous cognition measurements.25,43 Across all patients, regardless of diagnosis, WMSA showed a strong relationship with a 1-year change in EF but they were not a robust predictor of memory decline. Particularly in the individuals whose cognitive profiles do not match their CSF pathologic profiles (e.g., individuals with impairment in Q3), however, we demonstrate that WMSA provide added information that is predictive of future cognitive decline in both memory and executive functioning domains. Our findings combined with others' suggest that other factors must be at play for the manifestation of AD-like symptoms, and that as the burden of classic AD pathologies increases, the impact of WMSA on cognitive function decreases. This also suggests, however, that WMSA are most important when classic AD pathologies are low and that WMSA may be critically linked to AD conversion, a concept supported by the previous works of others.9,28,44 To this end, our findings suggest that therapeutic studies focused on the peri-conversion period of the disease process may be most vulnerable to WMSA influence.
Reflective of the accuracy levels that were revealed by our classification experiments, 25% of individuals with an AD diagnosis fell into the biomarker quadrant whose p-tau181p and Aβ42 levels were in a normal clinical range (Q3). Similarly, 34% of individuals with MCI fell into this quadrant. These individuals presented an opportunity for deeper study and possible discovery of novel biomarkers to explain their cognitive symptoms. In these individuals, WMSA were the strongest predictor of a 1-year change in memory when assessed against the CSF biomarkers, and were the only predictor of a 1-year change in executive function. Because all of the individuals with MCI-C in this study converted to AD within 3 years of the time that CSF measurements were taken, our findings suggest that this 3-year time window is not a sensitive period for Aβ42 and p-tau181p in the CSF, and this is supported by the work of others.5 These findings are particularly important in the context of choosing participants for clinical trials in which therapeutics are aimed at classic AD pathologies, as it is more likely that WMSA or a related process is the primary cause of the cognitive clinical presentation in individuals similar to those in Q3 of our study.
Two interesting findings that add to the complexity of the current picture are the lack of difference in hippocampal volumes of individuals with impairment in Q1 and Q3, and the increase in APOE ε4 prevalence in Q1 individuals with impairment over Q3. While hippocampal atrophy is typically considered to be a hallmark biomarker of AD, it is also associated with other unrelated neurodegenerative conditions such as hippocampal sclerosis, and it is possible that the individuals with impairment in Q3 reflect this process. It is also possible, however, that this AD-like hippocampal atrophy is due to the same upstream process in all individuals with impairment, but that the cognitive symptom manifestation necessary for a clinical AD diagnosis requires a second hit.45 Under this hypothesis, the second hit could be the development of tau and amyloid pathologies or it could be a cerebrovascular process such as those that lead to WMSA. The low prevalence of APOE ε4 in the Q3 individuals with impairment further suggests that these individuals are protected from the development of classic AD pathologies. Such complexities between and misalignments of pathologic and cognitive presentations may be explained by the notion of cognitive reserve18,46,47 and warrants future study into whether there are common risk factors among individuals in each given quadrant that may explain their susceptibility to a unique manifestation of pathologic findings. For example, there may be particular aspects of cognitive reserve (e.g., level of education) that protect an individual from the development of classic AD pathologies, and if there are other aspects of cognitive reserve that protect an individual from the development of WMSA, then the combination of these protective mechanisms may lower an individual's risk of cognitive decline and development of AD altogether.
Taken together, we interpret our results to suggest the following. Individuals with impairment in Q1 embody the classic AD pathologic profile with high levels of CSF p-tau181p, low CSF Aβ42, and prevalent APOE ε4. These pathologies are the main drivers of memory loss leading to a clinical AD diagnosis in Q1. In these individuals, tau and amyloid have a saturating effect on memory loss and WMSA do not further contribute to impairment. In the Q3 individuals with impairment who lack classic CSF biomarkers, however, WMSA are the dominating pathology that drives memory loss, and their effect can be appreciated in the absence of p-tau181p and Aβ42 saturation. This is further supported by the finding that Q3 individuals with impairment have a greater WMSA burden than those in Q1, which would presumably have a greater effect on memory. Across all individuals and within quadrants, WMSA demonstrate a longitudinal relationship with executive function. This has been reported in other studies as well,48,49 and perhaps reflects a robust and general association between WMSA and cognitive function that exists even in the presence of other strong neurodegenerative pathologies.
The results presented in this work hold clinical utility in understanding patients who present with AD-like cognitive symptoms but do not have remarkable CSF biomarker profiles. The more immediate value of this work, however, may relate to the researchers who study AD and conduct clinical trials for therapeutic interventions in that it demonstrates an important role of white matter disease in AD. The results of this work suggest that the combined relationship between WMSA and CSF biomarkers of AD should be assessed when designing novel therapeutic interventions. In addition, this work may aid in the task of screening individuals for enrollment in specific clinical trials, which is a current challenge as the relationships between complex biomarker profiles and future cognitive decline are not well understood. Future work should address the more immediate clinical utility of such results by aiming to create disease-related thresholds of white matter disease in the context of other pathologies and risk factors for clinical assessment.
Glossary
- Aβ42
β-amyloid 1–42
- AD
Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- ADNI-EF
Alzheimer's Disease Neuroimaging Initiative–executive function
- ADNI-MEM
Alzheimer's Disease Neuroimaging Initiative–memory
- GLM
general linear model
- MCI
mild cognitive impairment
- MCI-C
mild cognitive impairment–conversion
- MCI-NC
mild cognitive impairment–no conversion
- OC
older control
- p-tau181p
tau phosphorylated at threonine 181
- Q
quadrant
- RSS
residual sum of squares
- SVM
support vector machine
- WMSA
white matter signal abnormality
Author contributions
Emily Lindemer: study concept and design, analysis and interpretation. Douglas Greve: analysis and interpretation. Bruce Fischl: critical revision of the manuscript for important intellectual content. David Salat: study supervision. Teresa Gomez-Isla: critical revision of the manuscript for important intellectual content.
Study funding
Study funded by NIH (T32EB001680, T90DA022759/R90DA023427, 2R01NR010827-06A1).
Disclosure
E. Lindemer and D. Greve report no disclosures relevant to the manuscript. B. Fischl has a financial interest in CorticoMetrics, a company whose medical pursuits focus on brain imaging and measurement technologies. Dr. Fischl’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. D. Salat and T. Gomez-Isla report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Clifford RJJ, Knopman DS, Jagust WJ, et al. Update on hypothetical model of Alzheimer's disease biomarkers. Lancet Neurol 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol 2010;120:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartzokis G. Alzheimer's disease as homeostatic responses to age-related myelin breakdown. Neurobiol Aging 2011;32:1341–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 5.Jack CR Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arai H, Terajima M, Miura M, et al. Tau in cerebrospinal fluid: a potential diagnostic marker in Alzheimer's disease. Ann Neurol 1995;38:649–652. [DOI] [PubMed] [Google Scholar]
- 7.Clark CM, Xie S, Chittams J, et al. Cerebrospinal fluid tau and beta-amyloid. Arch Neurol 2003;60:1696–1702. [DOI] [PubMed] [Google Scholar]
- 8.Kawas CH, Kim RC, Sonnen JA, Bullain SS, Trieu T. Multiple pathologies are common and related to dementia in the oldest-old. Neurology 2015;85:535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toledo JB, Arnold SE, Raible K, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's Coordinating Centre. Brain 2013;136:2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol 2010;120:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drachman DA. The amyloid hypothesis, time to move on: amyloid is the downstream result, not cause, of Alzheimer's disease. Alzheimers Dement 2014;10:372–380. [DOI] [PubMed] [Google Scholar]
- 12.Benedictus MR, Binnewijzend MAA, Kuijer JPA, et al. Brain volume and white matter hyperintensities as determinants of cerebral blood flow in Alzheimer's disease. Neurobiol Aging 2014;35:2665–2670. [DOI] [PubMed] [Google Scholar]
- 13.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013;12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frisoni GB, Galluzzi S, Pantoni L, Filippi M. The effect of white matter lesions on cognition in the elderly: small but detectable. Nat Clin Pract Neurol 2007;3:620–627. [DOI] [PubMed] [Google Scholar]
- 15.Wolf H, Ecke GM, Bettin S, Dietrich J, Gertz HJ. Do white matter changes contribute to the subsequent development of dementia in patients with mild cognitive impairment? A longitudinal study. Int J Geriatr Psychiatry 2000;15:803–812. [DOI] [PubMed] [Google Scholar]
- 16.Provenzano F, Muraskin J, Tosto G, et al. White matter hyperintensities and cerebral amyloidosis: necessary and sufficient for clinical expression of Alzheimer disease? JAMA Neurol 2013;70:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grimmer T, Faust M, Auer F, et al. White matter hyperintensities predict amyloid increase in Alzheimer's disease. Neurobiol Aging 2012;33:2766–2773. [DOI] [PubMed] [Google Scholar]
- 18.Hertze J, Palmqvist S, Minthon L, Hansson O. Tau pathology and parietal white matter lesions have independent but synergistic effects on early development of Alzheimer's disease. Dement Geriatr 2013;3:113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pascoal TA, Dadar M, Manitsirikul S, et al. Association between apolipoprotein A-I levels and white matter hyperintensities depends on CSF tau levels in a high-risk cohort of aging cognitively normal persons: the Prevent-Alzheimer's Disease Study. Alzheimers Dement 2015;11:P103. [Google Scholar]
- 20.Bendlin BB, Carlsson CM, Johnson SC, et al. CSF t-tau/Aβ42 predicts white matter microstructure in healthy adults at risk for Alzheimer's disease. PLoS One 2012;7:e37720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maruyama M, Matsui T, Tanji H, et al. Cerebrospinal fluid tau protein and periventricular white matter lesions in patients with mild cognitive impairment: implications for 2 major pathways. Arch Neurol 2004;61:716–720. [DOI] [PubMed] [Google Scholar]
- 22.Rutten-Jacobs LCA, de Leeuw FE, Geurts-van Bon L, et al. White matter lesions are not related to β-amyloid deposition in an autopsy-based study. Curr Gerontol Geriatr Res 2011;2011:826862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YF, Wang H, Chu Y, Huang YC, Su MY. Regional quantification of white matter hyperintensity in normal aging, mild cognitive impairment, and Alzheimer's disease. Dement Geriatr Cogn Disord 2006;22:177–184. [DOI] [PubMed] [Google Scholar]
- 24.Lindemer ER, Greve DN, Fischl B, Augustinack JC, Salat DH; Alzheimer's Disease Neuroimaging Initiative. Differential regional distribution of juxtacortical white matter signal abnormalities in aging and Alzheimer's disease. J Alzheimers Dis 2017;57:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brickman AM, Provenzano FA, Muraskin J, et al. Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Arch Neurol 2012;69:1621–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brickman AM, Zahodne LB, Guzman VA, et al. Reconsidering harbingers of dementia: progression of parietal lobe white matter hyperintensities predicts Alzheimer's disease incidence. Neurobiol Aging 2015;36:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McAleese KE, Walker L, Graham S, et al. Parietal white matter lesions in Alzheimer's disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol 2017;134:459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snowdon DA, Greiner LH, Mortimer JA, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease study. J Am Med Assoc 1997;277:813–817. [PubMed] [Google Scholar]
- 29.Jack CR Jr, Bernstein MA, Fox NC, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 2008;27:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–355. [DOI] [PubMed] [Google Scholar]
- 31.Destrieux C, Fischl B, Dale A, Halgren E. Automatic parcellation of human cortical gyri and sulci using standard anatomical nomenclature. Neuroimage 2010;53:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA 2000;97:11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischl B, Liu A, Dale AM. Automated manifold surgery: constructing geometrically accurate and topologically correct models of the human cerebral cortex. IEEE Trans Med Imaging 2001;20:70–80. [DOI] [PubMed] [Google Scholar]
- 34.Fischl B, Salat DH, Van Der Kouwe A, et al. Sequence-independent segmentation of magnetic resonance images. Neuroimage 2004;23:S69–S84. [DOI] [PubMed] [Google Scholar]
- 35.Lindemer ER, Salat DH, Smith EE, Nguyen K, Fischl B, Greve DN. White matter signal abnormality quality differentiates mild cognitive impairment that converts to Alzheimer's disease from nonconverters. Neurobiol Aging 2015;36:2447–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andreasen N, Vanmechelen E, Van de Voorde A, et al. Cerebrospinal fluid tau protein as a biochemical marker for Alzheimer's disease: a community based follow up study. J Neurol Neurosurg Psychiatry 1998;64:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andreasen N, Minthon L, Davidsson P, et al. Evaluation of CSF-tau and CSF-Aβ42 as diagnostic markers for Alzheimer disease in clinical practice. Arch Neurol 2001;58:373–379. [DOI] [PubMed] [Google Scholar]
- 39.Schoonenboom NSM, Pijnenburg YAL, Mulder C, et al. Amyloid beta(1–42) and phosphorylated tau in CSF as markers for early-onset Alzheimer disease. Neurology 2004;62:1580–1584. [DOI] [PubMed] [Google Scholar]
- 40.Crane PK, Carle A, Gibbons LE, et al. Development and assessment of a composite score for memory in the Alzheimer's Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav 2012;6:502–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibbons LE, Carle AC, Mackin RS, et al. A composite score for executive functioning, validated in Alzheimer's Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav 2012;6:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White LR, Edland SD, Hemmy LS, et al. Neuropathologic comorbidity and cognitive impairment in the Nun and Honolulu-Asia Aging Studies. Neurology 2016;86:1000–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tosto G, Zimmerman ME, Carmichael OT, Brickman AM. Predicting aggressive decline in mild cognitive impairment: the importance of white matter hyperintensities. JAMA Neurol 2014;71:872–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chui HC, Zarow C, Mack WJ, et al. Cognitive impact of subcortical vascular and Alzheimer's disease pathology. Ann Neurol 2006;60:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu X, Raina AK, Perry G, Smith MA. Alzheimer's disease: the two-hit hypothesis. Lancet Neurol 2004;3:219–226. [DOI] [PubMed] [Google Scholar]
- 46.Stern Y. Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurol 2012;11:1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brickman AM, Siedlecki KL, Muraskin J, et al. White matter hyperintensities and cognition: testing the reserve hypothesis. Neurobiol Aging 2011;32:1588–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Groot JC, de Leeuw FE, Oudkerk M, Hofman A, Jolles J, Breteler MM. Cerebral white matter lesions and subjective cognitive dysfunction: the Rotterdam Scan Study. Neurology 2001;56:1539–1545. [DOI] [PubMed] [Google Scholar]
- 49.Smith EE, Salat DH, Jeng J, et al. Correlations between MRI white matter lesion location and executive function and episodic memory. Neurology 2011;76:1492–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used in this article are available in the ADNI public data repository. Anonymized patient identification numbers from the ADNI cohort used in this article are available by request from any qualified investigator.