

Abstract
Enteric Gram-negative bacteria, including Escherichia coli, biosynthesize and deploy the triscatecholate siderophore enterobactin (Ent) in the vertebrate host to acquire iron, an essential nutrient. We report that Ent–Cipro, a synthetic siderophore–antibiotic conjugate based on the native Ent platform that harbors an alkyl linker at one of the catechols with a ciprofloxacin cargo attached, affords targeted antibacterial activity against E. coli strains that express the pathogen-associated iroA gene cluster. Attachment of the siderophore to ciprofloxacin, a DNA gyrase inhibitor and broad-spectrum antibiotic that is used to treat infections caused by E. coli, generates an inactive prodrug and guides the antibiotic into the cytoplasm of bacteria that express the Ent uptake machinery (FepABCDG). Intracellular hydrolysis of the siderophore restores the activity of the antibiotic. Remarkably, Fes, the cytoplasmic Ent hydrolase expressed by all E. coli, does not contribute to Ent–Cipro activation. Instead, this processing step requires IroD, a cytoplasmic hydrolase that is only expressed by E. coli that harbor the iroA gene cluster and are predominantly pathogenic. In the uropathogenic E. coli UTI89 and CFT073, Ent–Cipro provides antibacterial activity comparable to unmodified ciprofloxacin. This work highlights the potential of leveraging and targeting pathogen-associated microbial enzymes in narrow-spectrum antibacterial approaches. Moreover, because E. coli include harmless gut commensals as well as resident microbes that can contribute to disease, Ent–Cipro may provide a valuable chemical tool for strain-selective modulation of the microbiota.
Graphical Abstract

Introduction
Bacterial infections, especially those caused by Gram-negative pathogens that have developed resistance against antibiotics in clinical use, pose a tremendous threat to global health and motivate investigations of new antibacterial strategies.1 Moreover, an increasing appreciation for the important role of the human microbiome in health and disease stimulates consideration of how this complex community can be preserved during antibiotic therapy, as well as the identification of strategies that can be used to modulate its composition to resolve pathological conditions such as inflammatory bowel disease.2,3 The design and utilization of narrow-spectrum antibiotics that target specific pathogens is one strategy to limit the spread of antibiotic resistance,4–7 and also provides chemical tools for fundamental studies that involve manipulating the microbiota. The activity spectrum of established broad-spectrum antibiotics, such as β-lactams or fluoroquinolones, can be narrowed by attaching a targeting moiety. For targeting Gram-negative bacteria such as pathogenic Escherichia coli, outer membrane receptors provide opportunities for selective recognition and intracellular delivery.8 These β-barrel proteins are involved in the uptake of various molecules that include essential nutrients.9
Transition metal ions are essential nutrients that bacterial pathogens must acquire from the host environment. To starve invading microbial pathogens, the human host reduces metal availability in a process termed ‘nutritional immunity.’10 Bacteria utilize a variety of metal acquisition systems to successfully colonize the host,11 and these machineries provide opportunities for new antibiotic strategies. One strategy to acquire Fe(III) involves the secretion of siderophores, which are low-molecular-weight iron chelators.12–14 Pathogenic strains are often characterized by the utilization of multiple siderophores, which contribute to virulence and enable them to outcompete other microbes colonizing the same niche.15,16
We have employed the native siderophore enterobactin (Ent, Figure 1A) for the targeted delivery of small-molecule cargo, including antibiotics, to Gram-negative bacteria that utilize this siderophore.17–19 Ent is a triscatecholate siderophore produced by various Gram-negative species including E. coli, Salmonella enterica, and Klebsiella pneumoniae. The extraordinary iron-binding affinity of the triscatecholate (Ka ~1049 M−1) enables bacteria to extract iron from host proteins.20,21 Ferric Ent is recognized by the outer membrane (OM) receptor FepA and transported into the periplasm with energy provided by a TonB-ExbB-ExbD complex (Figure 1B). FepB, a periplasmic binding protein, mediates translocation to the inner membrane (IM), and an ATP-binding cassette (ABC) transporter, FepCDG, transports the ferric siderophore across the IM. Cytoplasmic release of the bound iron requires hydrolysis of the trilactone by the esterase Fes, and is likely accompanied by reduction of the metal ion.22–24 Several Ent producers also biosynthesize and utilize salmochelins, C-glucosylated analogs of Ent (Figure 1A), to acquire iron and evade the Ent-scavenging host-defense protein lipocalin-2 (also termed “siderocalin”).25–27 The C-glycosyltransferase required for glucosylation of Ent is encoded by the pathogen-associated iroA gene cluster (iroBCDEN),28 along with additional transport proteins and hydrolase enzymes. In particular, the OM transporter IroN enables uptake of the ferric salmochelins,29,30 and the esterase IroD hydrolyzes ferric salmochelins for iron release (Figure 1B).22 An additional periplasmic hydrolase, IroE, is predicted to partially hydrolyze the salmochelin trilactone during export of the siderophores.22,26,31
Figure 1.

Siderophores and siderophore uptake machinery relevant to this work. (A) Structures of enterobactin (Ent) and salmochelin S4. (B) Cartoon depiction of the Ent and salmochelin transport and processing machinery in E. coli. IroD can also catalyze the hydrolysis of ferric Ent. The periplasmic esterase IroE is not shown. Reduction of Fe(III) to Fe(II) that occurs during release of the metal ion is not catalyzed by the hydrolases. DHBS, 2,3-dihydroxybenzoyl serine; Glc, glucose.
Some bacteria biosynthesize and deploy siderophores tethered to antibiotic moieties to target and kill competitors that express the requisite siderophore receptor.8,32 These “sideromycins” have inspired the design and chemical synthesis of many siderophore–drug conjugates to target bacterial siderophore uptake machinery for antibiotic delivery.33–37 Indeed, conjugation of antibiotics with periplasmic targets, such as β-lactams, to siderophores can afford significantly increased antibacterial activity against Gram-negative pathogens.18,38–46 However, to the best of our knowledge, attempts to employ this strategy for delivering antibiotics with cytoplasmic targets to Gram-negative strains have not yet succeeded. Conjugation of antibiotics with cytoplasmic targets, such as fluoroquinolones, to siderophores usually results in significantly attenuated activity of the drugs.47–60 Along these lines, we found that two Ent conjugates carrying the fluoroquinolone ciprofloxacin, Ent–PEG3–Cipro 1 and Ent–Cipro 2 (Chart 1), were inactive against E. coli K-12 as well as Pseudomonas aeruginosa PAO1, a Gram-negative opportunistic pathogen that cannot biosynthesize Ent but expresses a receptor for ferric Ent.17
Chart 1.

Chemical structures of the ciprofloxacin conjugates and hydrolytic products. For 3, only one possible regioisomer of the ester is shown.
Ciprofloxacin is widely used in the clinic to treat a number of Gram-negative bacterial infections, including urinary tract infections (UTIs).61 It inhibits the DNA gyrase, a topoisomerase that plays an important role during DNA replication and transcription.62 Despite our prior report and other failed attempts to identify siderophore–ciprofloxacin conjugates that exhibit growth inhibitory activity against Gram-negative bacteria, we continued to investigate whether ciprofloxacin can be a useful tool for studying siderophore-mediated antibiotic delivery to the bacterial cytoplasm. During these efforts, we revisited conjugates 1 and 2 and uncovered that alkyl-linked Ent–Cipro 2 exhibits potent antibacterial activity against select E. coli strains, including uropathogenic E. coli (UPEC).
Herein, we report this discovery and studies that decipher the uptake and fate of Ent–Cipro 2. This conjugate is delivered into the cytoplasm of E. coli where it acts as a prodrug that is activated by intracellular hydrolysis of the siderophore. This hydrolysis is catalyzed by the salmochelin esterase IroD that is only expressed in strains harboring the iroA gene cluster. These investigations reveal a new approach to convert broad-spectrum antibiotics into more selective therapeutics by exploiting siderophore processing machinery predominantly used by pathogenic strains.
Results
An Enterobactin–Ciprofloxacin Conjugate is Active Against Uropathogenic E. coli
The antibacterial activity of Ent–ciprofloxacin conjugates 1 and 2 (Chart 1) was evaluated against a panel of non-pathogenic and uropathogenic E. coli strains in a minimal M9 medium previously employed in studies of siderophore–β-lactam conjugates.19 Although the conjugates only differ in the linker between the antibiotic and siderophore, they exert strikingly different antibacterial activity under these conditions. Whereas the PEG3-linked conjugate 1 exhibits no growth inhibitory activity against any of the four tested strains (E. coli K-12, B, UTI89 and CFT073), the alkyl-linked Ent–Cipro 2 exerts high antibacterial activity against two UPEC strains, UTI89 and CFT073, with minimum inhibitory concentration (MIC) values similar to that of ciprofloxacin (0.1–1 μM) (Figures 2, S1, S2; Table S5). In agreement with our prior work,17 the growth of the non-pathogenic laboratory strains E. coli K-12 and B is not affected by Ent–Cipro 2. Given this potent and strain-specific antibacterial activity, Ent–Cipro 2 provides an excellent chemical tool to study the determinants for successful siderophore-mediated delivery of antibiotics into the cytoplasm of Gram-negative pathogens.
Figure 2.

Antibacterial activity of ferric Ent–Cipro 2 against non-pathogenic and pathogenic E. coli strains. (A) Laboratory strain E. coli K-12. (B) Laboratory strain E. coli B. (C) Uropathogenic E. coli UTI89. (D) Uropathogenic E. coli CFT073. All assays were performed in modified M9 medium (t = 20 h, T = 30 °C; mean ± SDM, n = 3). Ent–Cipro was pre-loaded with 0.9 equiv Fe(III); data for assays performed with apo Ent–Cipro are presented in Figure S2.
Growth Conditions and Iron Loading Influence Antibacterial Activity of Ent–Cipro 2
The siderophore biosynthesis and transport machinery is controlled by the ferric uptake regulator (Fur) and expressed under low-Fe conditions.63 Thus, studies of siderophore–antibiotic conjugates are often performed employing a growth medium, such as Mueller Hinton Broth (MHB), supplemented with the iron chelator 2,2′-dipyridyl (DP). Guided by our prior studies of the antibacterial activity of Ent-β-lactam conjugates against E. coli,18,19 we performed the antibacterial activity assays in MHB (50%) with and without 200 μM DP, but observed no growth inhibitory activity for Ent–Cipro 2 against E. coli under these conditions (Figure S12). Antibacterial activity was only observed in the modified M9 medium (Figures 2, S2), a low-iron growth medium (0.6 μM iron content; Table S4) that is not supplemented with an iron chelator such as DP. Thus, all subsequent studies with conjugate 2 were performed employing this medium. Next, we supplemented the M9 medium with iron (10 or 50 μM). As expected, this supplement attenuates the antibacterial activity of conjugate 2 because the expression of siderophore uptake machinery is repressed when sufficient iron is available (Figure S3).63 In contrast, the addition of iron has negligible effect on the antibacterial activity of ciprofloxacin. These results indicate that both iron levels and medium composition influence the antibacterial activity of Ent–Cipro 2, and that these variables should be examined when assaying the growth inhibitory activity of siderophore–antibiotic conjugates.
In addition, siderophore–antibiotic conjugates can be prepared and employed as either apo or Fe(III)-bound molecules. To determine whether iron pre-loading affects the antibacterial activity of Ent–Cipro 2, we treated E. coli with the apo conjugate or the ferric complex, and observed that the Fe(III)-bound conjugate exhibits higher antibacterial activity than the apo conjugate against the UPEC strains (Figure S2). Because the OM siderophore receptors recognize and transport the ferric siderophores, iron pre-loading of the conjugate may facilitate rapid binding at the receptor and thus uptake. In the case of the non-pathogenic strains K-12 and B, incubation with apo conjugate 2 causes growth inhibition at high concentrations (i.e., 10 μM), indicating that the siderophore causes an iron-withholding effect when present in excess to the iron content of the growth medium. Moreover, this starvation effect suggests that the uptake of Ent–Cipro 2 into these strains or the release of iron from the siderophore after uptake is impeded.
Ent–Cipro Enters E. coli Through the Ent Uptake Machinery
A major difference among the strains employed in this study is that the UPEC strains UTI89 and CFT073 harbor the iroA gene cluster, whereas the laboratory strains K-12 and B do not.64–66 Thus, both UTI89 and CFT073 produce and transport Ent as well as salmochelins. The observation that Ent–Cipro 2 is only active against the UPEC strains may suggest that it requires the salmochelin transporter IroN and that uptake into the non-pathogenic strains that only express FepA is impaired. To probe the cellular uptake pathway of Ent–Cipro 2, we employed the UPEC strain CFT073 and generated six transporter mutants (Table S2). The fepA, iroN, and ihA mutants and the fepA iroN double mutant were employed to probe recognition and transport across the OM. The fepC mutant (ATPase) and the fepD fepG double mutant (IM translocase) were used to investigate transport across the IM. Antibacterial activity assays with the OM transporter mutants reveal that deletion of iroN does not affect the activity of the conjugate (Figures 3, S5). Moreover, single deletion of fepA or ihA, an additional catecholate receptor expressed in E. coli CFT073,66,67 does not attenuate the activity of 2. Only double deletion of fepA and iroN abolishes the activity of Ent–Cipro 2, indicating that the conjugate can enter E. coli CFT073 through both FepA and IroN. This observation is in agreement with prior studies of Ent–β-lactam conjugates, which revealed that both FepA and IroN provide transport into the periplasm.19 In contrast, any deletion of components of the IM transporter FepCDG abolishes the antibacterial activity of Ent–Cipro 2 (Figures 3, S6). This observation indicates that the conjugate is transported into the cytoplasm by FepCDG, and that cytoplasmic delivery is required for E. coli growth inhibition. Overall, these studies demonstrate that Ent–Cipro 2 crosses two membranes and enters the E. coli cytoplasm through the Ent uptake machinery FepABCDG. IroN provides a second conduit for the conjugate to cross the OM of CFT073, but is not essential.
Figure 3.
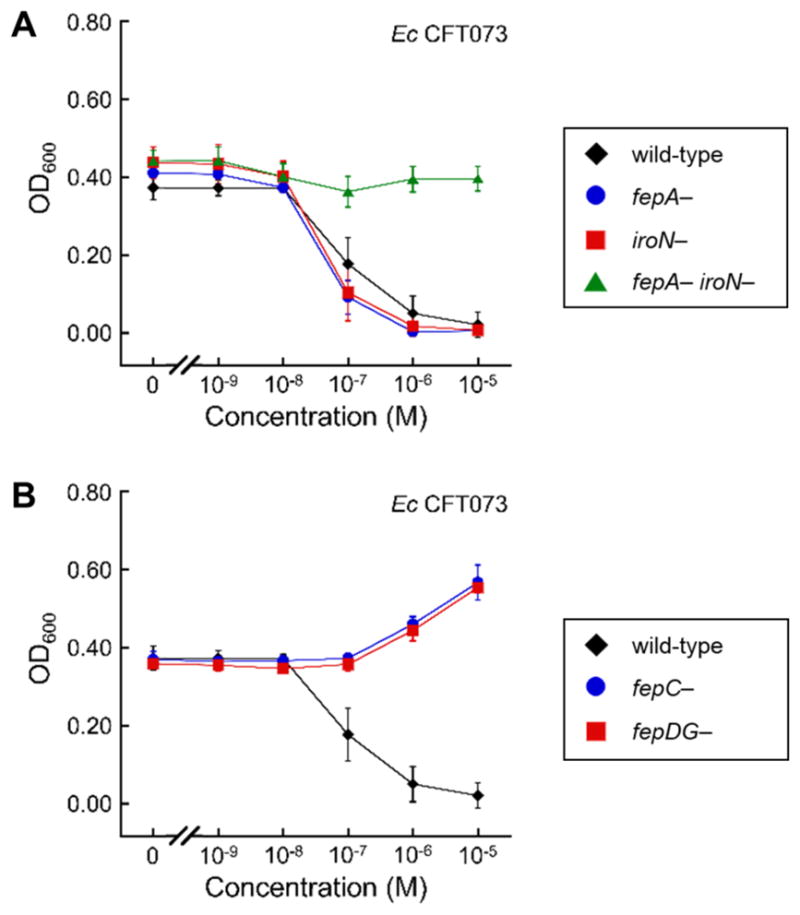
Antibacterial activity of ferric Ent–Cipro 2 against siderophore transporter mutants of E. coli CFT073. (A) Wild-type and OM transporter knock-out mutants. (B) Wild-type and IM transporter knock-out mutants. All assays were performed in modified M9 medium (t = 20 h, T = 30 °C; mean ± SDM, n = 3). Ent–Cipro was pre-loaded with 0.9 equiv Fe(III); data for assays performed with apo Ent–Cipro are presented in Figures S5, S6.
The low MIC values obtained for Ent–Cipro 2 against UPEC suggest that the conjugate is not significantly outcompeted by endogenous siderophores for binding at the siderophore receptors, at least under the assay conditions. Nevertheless, the conjugate exhibits slightly increased antibacterial activity against an entC mutant of E. coli CFT073, which is unable to biosynthesize Ent (Figure S8). When the UPEC strains are co-treated with Ent–Cipro 2 and Ent (1:1 molar ratio), growth promotion is observed at high concentrations (Figure S4), suggesting that the conjugate can be outcompeted by the native siderophore for binding at the receptor when high concentrations of the endogenous siderophore are present.
Ent–Cipro Requires Intracellular Hydrolysis to Exert Antibacterial Activity
The uptake studies with the OM receptor mutants indicate that Ent–Cipro 2 likely enters the cytoplasm of all E. coli strains through FepABCDG. Thus, we questioned if the ferric siderophore processing machinery could account for the different susceptibilities of the strains. All E. coli express the cytoplasmic ferric Ent hydrolase Fes, and strains that harbor the iroA gene cluster also express the salmochelin hydrolase IroD in this locale. We therefore prepared two hydrolase-deficient strains of CFT073 (a fes mutant and an iroD mutant), and examined the susceptibility of these mutants to Ent–Cipro 2. Whereas deletion of the Ent hydrolase Fes does not significantly affect its activity, deletion of the salmochelin hydrolase IroD abolishes the antibacterial activity of Ent–Cipro 2 (Figures 4, S7). Moreover, complementation of E. coli K-12 with iroD makes this strain susceptible to the conjugate (Figures 4, S9). Taken together, these results demonstrate that IroD plays a crucial role for the antibacterial activity of Ent–Cipro 2, and indicate that the conjugate acts as a prodrug that is activated in the cytoplasm by IroD-catalyzed hydrolysis of the siderophore. Consistent with these observations, incubation of Ent–Cipro 2 with purified IroD shows that the enzyme catalyzes the hydrolysis of the attached siderophore (Figures 5, S15, S18). Hydrolysis of the siderophore trilactone of 2 proceeds similarly to that of Ent and results in release of 2,3-dihydroxybenzoyl serine (DHBS), as well as formation of (DHBS)2–Cipro 3 and ultimately the monocatecholate DHBS–Cipro 4 (Chart 1, Scheme S1). Overall, it appears that Ent–Cipro 2 must be hydrolyzed in the E. coli cytoplasm to exert antibacterial activity, that Fes cannot perform this function sufficiently, and that the requirement for IroD confers selectivity to Ent–Cipro 2 such that it only exhibits antibacterial activity against E. coli strains that harbor the iroA gene cluster. E. coli include harmless gut commensal organisms, pathogens and pathobionts (i.e., organisms that are generally harmless but can cause disease in some circumstances),68 but because the iroA gene cluster is associated with pathogenicity,26 Ent–Cipro 2 targets E. coli strains that are problematic for human health.
Figure 4.

Antibacterial activity of ferric Ent–Cipro 2 against siderophore hydrolase mutants of E. coli CFT073 and E. coli K-12. (A) Wild-type and hydrolase knock-out mutants of E. coli CFT073. (B) E. coli K-12(DE3), wild-type and complemented with iroD. All assays were performed in modified M9 medium (t = 20 h, T = 30 °C; mean ± SDM, n = 3). Ent–Cipro was pre-loaded with 0.9 equiv Fe(III); data for assays performed with apo Ent–Cipro are presented in Figures S7, S9.
Figure 5.

Hydrolysis of the ferric siderophores by IroD. (A) Hydrolysis of ferric Ent to linear Ent (lin.), (DHBS)2, and DHBS. (B) Hydrolysis of ferric Ent–Cipro 2 to (DHBS)2–Cipro 3, DHBS–Cipro 4, and DHBS. (C) Hydrolysis of ferric Ent–PEG3–Cipro 1 to (DHBS)2–PEG3–Cipro (dim.), DHBS–PEG3–Cipro (mon.), and DHBS. Analytical HPLC traces (316 nm absorption) from enzymatic activity assays performed with 100 μM ferric siderophore (pre-loaded with 1 equiv Fe(III)) and 0.15 μM IroD in 75 mM Tris-HCl, pH 8.0. Data from additional time points, assays with apo siderophores, and control assays are presented in Figures S13–S18; molecular structures of the compounds are presented in Chart 1 and Scheme S1.
Siderophore Conjugation Attenuates DNA Gyrase Inhibitory Activity of Ciprofloxacin
When ciprofloxacin inhibits DNA gyrase, the fluoroquinolone moiety intercalates into the DNA that is bound by the gyrase, and the carboxyl group of the antibiotic participates in a metal-ion–water bridge that stabilizes the drug–enzyme–DNA complex.69–72 Whereas modification and neutralization of the carboxyl moiety result in inactivation, some structural modifications at the piperazinyl moiety are tolerated without significant loss of antibacterial activity.62 However, attachment of large residues at the piperazine impairs binding at the DNA–gyrase complex. The inhibition of E. coli DNA gyrase by Ent–Cipro 2 was tested by monitoring the negative supercoiling of plasmid DNA in the presence of the enzyme. Consistent with prior reports of siderophore–ciprofloxacin conjugates,47,56,58 the DNA gyrase inhibitory activity of Ent–Cipro 2 (IC50: 70 μM) is ≈280-fold lower than that of unmodified ciprofloxacin (IC50: 0.25 μM) (Table S8, Figure S21). The final hydrolytic product DHBS–Cipro 4 (IC50: 20 μM) exhibits somewhat enhanced antibacterial activity relative to Ent–Cipro 2, but ≈80-fold lower activity than unmodified ciprofloxacin. These results indicate that modification of the piperazinyl moiety of ciprofloxacin with an alkyl linker appended to Ent or DHBS attenuates its ability to inhibit DNA gyrase. Moreover, it is difficult to reconcile the IC50 values obtained from this assay and the MIC values obtained for Ent–Cipro 2 against UPEC. Together, these data may indicate that hydrolysis product 4 is not the active species that inhibits DNA gyrase, that the active species inhibits a target other than DNA gyrase, or that multiple targets including DNA gyrase exist. Alternatively, siderophore-mediated delivery could result in an increased accumulation of DHBS–Cipro 4 in the cytoplasm compared to ciprofloxacin, which may compensate for its lower DNA gyrase inhibitory activity.
The Intact Siderophore is Required for Antibacterial Activity
Given that IroD and thus hydrolysis of conjugate 2 is essential for its antibacterial activity, we questioned whether its hydrolytic products would exert inhibitory activity against all strains independent of IroD expression. We performed large-scale degradation assays of Ent–Cipro 2 to obtain sufficient quantities of (DHBS)2–Cipro 3 and DHBS–Cipro 4 for bacterial susceptibility testing against the UPEC strains as well as E. coli K-12 and B. Neither (DHBS)2–Cipro 3 nor DHBS–Cipro 4 exhibits growth inhibitory activity against the four strains evaluated (Figures 6, S10, S11). Moreover, pre-loading of (DHBS)2–Cipro 3 with Fe(III) (0.9 equiv) to enable formation of an [Fe(3)(H2O)2]2− complex, or addition of Fe(III) (0.9 equiv) and DHBS (1.0 equiv) to form [Fe(3)(DHBS)]5−, which structurally resembles ferric Ent–Cipro 2, does not result in growth inhibition. Lastly, growth inhibition does not occur for the CFT073 entC mutant (Figure S8), indicating that displacement by the endogenous siderophore at the Ent receptors does not significantly affect the activity of the DHBS conjugates. In total, these observations suggest that the truncated siderophore conjugate may not be well recognized or transported by the siderophore uptake machinery, and demonstrate that the intact Ent scaffold is required for the antibacterial activity of the ciprofloxacin conjugate.
Figure 6.

Antibacterial activity of the hydrolytic products of Ent–Cipro 2, (DHBS)2–Cipro 3 and DHBS–Cipro 4, against uropathogenic E. coli strains. (A) E. coli UTI89. (B) E. coli CFT073. All assays were performed in modified M9 medium (t = 20 h, T = 30 °C; mean ± SDM, n = 3). Ent–Cipro and (DHBS)2–Cipro were pre-loaded with 0.9 equiv Fe(III); data for assays performed with additional strains and apo conjugate are presented in Figures S10, S11.
Discussion
Herein, we show that conjugation of the broad-spectrum antibiotic ciprofloxacin to Ent narrows the activity spectrum of the antibiotic and affords selective growth inhibition of E. coli strains that harbor the iroA gene cluster, and in particular UPEC. We elucidate that Ent–Cipro 2 is delivered into the cytoplasm of E. coli strains through the Ent transport machinery FepABCDG (Figure 7). Moreover, the attached siderophore abolishes the antibacterial activity of ciprofloxacin and the conjugate needs to be hydrolyzed after uptake to activate the prodrug. This hydrolysis step requires the salmochelin esterase IroD that is only expressed in strains harboring the iroA gene cluster. Many UPEC isolates carry this gene cluster within pathogenicity islands, and its genes are expressed in vivo during urinary tract infection (UTI).73,74 Thus, it is possible that Ent–Cipro 2 may be effective in vivo in an UTI model while minimally perturbing the commensal microbiota. This conjugate, and more broadly IroD-catalyzed prodrug activation, may also have utility for targeting other infectious Gram-negative strains that harbor the iroA gene cluster and cause diseases that range from food-borne illness to pneumonia and sepsis, including other pathogenic E. coli, S. enterica and K. pneumoniae.19,26 Moreover, some E. coli are associated with inflammatory disease and dysbiosis in the gut and “precision editing” of the gut microbiome is a topic of current interest.75 We posit that Ent–Cipro 2 and other siderophore–antibiotic conjugates may provide useful tools for manipulating the gut microbiome.
Figure 7.

Proposed model for the antibacterial action of Ent–Cipro 2.
The current work can be compared and contrasted to our reported studies of siderophore-mediated β-lactam delivery to the E. coli periplasm.18,19 β-Lactam antibiotics such as ampicillin and amoxicillin inhibit cell wall biosynthesis by inhibiting penicillin-binding proteins in the periplasm. In this prior work, we established that Ent–β-lactam conjugates are transported into E. coli via the OM transporters FepA and IroN and exert antibacterial activity in the periplasm. Moreover, we demonstrated that substitution of the Ent for salmochelin S4 (Figure 1) provided targeted delivery of β-lactams into E. coli that express IroN. This strategy of antibiotic targeting relies on conjugate recognition and transport by OM siderophore receptors expressed by pathogenic E. coli.19 The current work also targets E. coli that harbor the iroA cluster, but the selectivity of Ent–Cipro 2 is based on the encoded cytoplasmic siderophore processing machinery.
To the best of our knowledge, Ent–Cipro 2 is the first reported synthetic siderophore–antibiotic conjugate carrying a cytoplasmic antibiotic that exhibits antibacterial activity against Gram-negative pathogens comparable to that of the unmodified antibiotic. The requirement of intracellular processing and release of the antibiotic is reminiscent of the naturally occurring albomycins, where intracellular release of the tRNA synthetase inhibitor from the ferrichrome-like siderophore is essential for the antibacterial activity.76 Intracellular release of antibiotics from synthetic conjugates has been attempted by introducing cleavable linkers; however, these prior investigations uncovered limitations. Ester linkages were employed to enable release of the attached antibiotic by intracellular esterase- or acid-catalyzed hydrolysis, but the hydrolytic lability of these ester bonds resulted in premature cleavage of the conjugates before uptake into the cells.48,49,51,53,59,77–79 Another approach utilizing a trimethyl-lock linker based on a reduction-triggered cleavage mechanism did not yield a conjugate with high antibacterial activity.60 Ent–Cipro 2 benefits from utilizing a native siderophore in that the cleavage of the conjugate is associated with the endogenous siderophore processing machinery, which may be advantageous to linkers that rely on separate cleavage mechanisms. On the other hand, a linker that enables release of the unmodified antibiotic may provide a conjugate with higher activity, as long as negligible premature cleavage before entry into the cytoplasm occurs and recognition and transport of the conjugate are not impeded by the linker.
The impact of structural modifications on the transmembrane transport of the conjugate as well as the exact mechanism of action of Ent–Cipro 2 warrant further investigations. Notably, the hydrolytic product DHBS–Cipro 4 exhibits an IC50 value for inhibition of the DNA gyrase (20 μM) significantly higher than the MIC value of Ent–Cipro 2 for the growth inhibition of the UPEC strains (0.1–1 μM). At this point, it is not apparent whether DHBS–Cipro 4 is the actual active species or if the molecule is further metabolized during or after hydrolysis of the siderophore. Future investigations in a cellular context should inform whether cytoplasmic accumulation occurs to a greater extent for DHBS-Cipro 4 than for ciprofloxacin, which could account for the similar MIC values of these compounds despite the differing IC50 values for DNA gyrase inhibition. Moreover, further studies designed to identify the cellular target(s) of Ent–Cipro 4 should inform whether DNA gyrase is the actual target inhibited by the conjugate.
Our studies emphasize that delivery of siderophore–antibiotic conjugates into the cytoplasm of Gram-negative bacteria strongly depends on the molecular structure of the conjugate. Although PEG linkers are commonly employed because they provide flexibility and spatial separation between different functional moieties, our prior and current observations suggest that this linker may impede translocation across the IM. Given that the structurally-related Ent–PEG3–β-lactam conjugates exhibited high antibacterial activity,18,19 it is possible that the PEG3-linked conjugate 1 is transported across the OM by FepA/IroN but may get trapped in the periplasm. If conjugate 1 was transported into the cytoplasm, we would expect an antibacterial activity similar to that of Ent–Cipro 2 based on the observation that hydrolysis by IroD proceeds comparably (Figures 5, S14, S17) and that the hydrolytic product exhibits similar inhibitory activity against DNA gyrase (Table S8, Figure S21). However, the length or hydrophilicity of the PEG3 linker could affect further intracellular processing of the hydrolytic products or inhibition of an as-yet unidentified target.
Despite the fact that intracellular hydrolysis of Ent–Cipro 2 is essential for antibacterial activity, conjugation of ciprofloxacin to the hydrolytic fragments of Ent abolishes the activity of the antibiotic, indicating that a truncated siderophore likely impedes delivery into the cytoplasm. Currently, the extent to which (DHBS)2–Cipro 3 and DHBS–Cipro 4 are taken up into the cells is unknown. In addition to the Ent transport machinery, uptake into the periplasm could be mediated by the DHBS receptors (Fiu, Cir), as previously shown for β-lactam–catechol conjugates.39 However, transport across both the OM and IM may be hampered or fast efflux through the siderophore export machinery or multi-drug efflux pumps may occur, and could prevent cytoplasmic accumulation of the conjugates to an amount sufficient for growth inhibition. Overall, we reason that intact native siderophores are preferable for antibiotic delivery to ensure strong recognition by the siderophore transporters and to minimize outcompetition by endogenous siderophores, which was observed previously for a β-lactam conjugate with a truncated siderophore.80
The development of antibiotic resistance in microbial pathogens is a serious concern for existing therapies as well as any new approach to treat microbial infections. We believe that future studies that follow resistance to Ent–Cipro 2 in E. coli CFT073 and UTI89 will be informative. For siderophore–antibiotic conjugates, resistance can occur via OM transporter mutations or downregulation of siderophore uptake systems.81,82 Ent–Cipro 2 can enter E. coli through FepA and IroN, and both transporters are important virulence factors that become upregulated during urinary tract infection.73,74,83 It is possible that this feature makes resistance from OM transporter mutations less likely because both transporters would need to acquire loss-of-function mutations. For pathogenic strains that rely on Ent or the salmochelins for iron acquisition in the vertebrate host, we reason that the loss or mutation of OM receptors that would prevent siderophore-based targeting is unlikely. These organisms require iron for replication, and loss or mutation of OM transporters such that the ferric siderophores are no longer transported would attenuate growth in vivo.
In closing, this contribution uncovers that the siderophore uptake and processing machinery can be leveraged to convert a broad-spectrum antibiotic into a narrow-spectrum antibiotic. More broadly, this work highlights the potential of targeting pathogen-associated microbial enzymes in narrow-spectrum antibacterial approaches. We look forward to examining whether IroD-catalyzed hydrolysis can activate Ent-based conjugates harboring other cytoplasmic warheads. We also hope that this study inspires new strategies that harness virulence-associated mechanisms for combating infectious disease, limiting the spread of antibiotic resistance and preserving the vitally important microbiota.
Supplementary Material
Acknowledgments
We thank the NIH (Grants 1R21AI126465 and 1R01AI114625) for financial support; Prof. L. Cegelski for providing E. coli UTI89; Prof. K. T. Hughes for providing plasmid pJK611; Dr. Dean Gia Nguyen for technical assistance. W. N. acknowledges the German National Academy of Sciences Leopoldina for a postdoctoral fellowship (LPDS 2015-08). M. R. holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Footnotes
Notes: E.M.N. holds a patent related to this work.
Complete experimental methods, Tables S1–S8, Figures S1–S30. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. World Health Organization (WHO); Geneva: 2017. [Google Scholar]
- 2.Blaser MJ. Nat Rev Immunol. 2017;17:461–463. doi: 10.1038/nri.2017.77. [DOI] [PubMed] [Google Scholar]
- 3.Ni J, Wu GD, Albenberg L, Tomov VT. Nat Rev Gastroenterol Hepatol. 2017;14:573–584. doi: 10.1038/nrgastro.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clatworthy AE, Pierson E, Hung DT. Nat Chem Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 5.Barczak AK, Hung DT. Curr Opin Microbiol. 2009;12:490–496. doi: 10.1016/j.mib.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis K. Nat Rev Drug Discov. 2013;12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 7.Brown ED, Wright GD. Nature. 2016;529:336–343. doi: 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- 8.Wencewicz TA, Miller MJ. Topics in Medicinal Chemistry. Springer Berlin Heidelberg; Berlin, Heidelberg: 2017. Sideromycins as Pathogen-Targeted Antibiotics. [Google Scholar]
- 9.Schauer K, Rodionov DA, de Reuse H. Trends Biochem Sci. 2008;33:330–338. doi: 10.1016/j.tibs.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 10.Hood MI, Skaar EP. Nat Rev Microbiol. 2012;10:525–537. doi: 10.1038/nrmicro2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer LD, Skaar EP. Annu Rev Genet. 2016;50:67–91. doi: 10.1146/annurev-genet-120215-035146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miethke M, Marahiel MA. Microbiol Mol Biol Rev. 2007;71:413–451. doi: 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hider RC, Kong X. Nat Prod Rep. 2010;27:637–657. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 14.Chu BC, Garcia-Herrero A, Johanson TH, Krewulak KD, Lau CK, Peacock RS, Slavinskaya Z, Vogel HJ. Biometals. 2010;23:601–611. doi: 10.1007/s10534-010-9361-x. [DOI] [PubMed] [Google Scholar]
- 15.Holden VI, Bachman MA. Metallomics. 2015;7:986–995. doi: 10.1039/c4mt00333k. [DOI] [PubMed] [Google Scholar]
- 16.Wilson BR, Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Trends Mol Med. 2016;22:1077–1090. doi: 10.1016/j.molmed.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng T, Bullock JL, Nolan EM. J Am Chem Soc. 2012;134:18388–18400. doi: 10.1021/ja3077268. [DOI] [PubMed] [Google Scholar]
- 18.Zheng T, Nolan EM. J Am Chem Soc. 2014;136:9677–9691. doi: 10.1021/ja503911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chairatana P, Zheng T, Nolan EM. Chem Sci. 2015;6:4458–4471. doi: 10.1039/c5sc00962f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loomis LD, Raymond KN. Inorg Chem. 1991;30:906–911. [Google Scholar]
- 21.Raymond KN, Dertz EA, Kim SS. Proc Natl Acad Sci U S A. 2003;100:3584–3588. doi: 10.1073/pnas.0630018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin H, Fischbach MA, Liu DR, Walsh CT. J Am Chem Soc. 2005;127:11075–11084. doi: 10.1021/ja0522027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abergel RJ, Zawadzka AM, Hoette TM, Raymond KN. J Am Chem Soc. 2009;131:12682–12692. doi: 10.1021/ja903051q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miethke M, Hou J, Marahiel MA. Biochemistry. 2011;50:10951–10964. doi: 10.1021/bi201517h. [DOI] [PubMed] [Google Scholar]
- 25.Fischbach MA, Lin H, Zhou L, Yu Y, Abergel RJ, Liu DR, Raymond KN, Wanner BL, Strong RK, Walsh CT, Aderem A, Smith KD. Proc Natl Acad Sci U S A. 2006;103:16502–16507. doi: 10.1073/pnas.0604636103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischbach MA, Lin H, Liu DR, Walsh CT. Nat Chem Biol. 2006;2:132–138. doi: 10.1038/nchembio771. [DOI] [PubMed] [Google Scholar]
- 27.Raffatellu M, George MD, Akiyama Y, Hornsby MJ, Nuccio SP, Paixao TA, Butler BP, Chu H, Santos RL, Berger T, Mak TW, Tsolis RM, Bevins CL, Solnick JV, Dandekar S, Bäumler AJ. Cell Host Microbe. 2009;5:476–486. doi: 10.1016/j.chom.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischbach MA, Lin H, Liu DR, Walsh CT. Proc Natl Acad Sci U S A. 2005;102:571–576. doi: 10.1073/pnas.0408463102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bäumler AJ, Norris TL, Lasco T, Voigt W, Reissbrodt R, Rabsch W, Heffron F. J Bacteriol. 1998;180:1446–1453. doi: 10.1128/jb.180.6.1446-1453.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hantke K, Nicholson G, Rabsch W, Winkelmann G. Proc Natl Acad Sci U S A. 2003;100:3677–3682. doi: 10.1073/pnas.0737682100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larsen NA, Lin H, Wei R, Fischbach MA, Walsh CT. Biochemistry. 2006;45:10184–10190. doi: 10.1021/bi060950i. [DOI] [PubMed] [Google Scholar]
- 32.Braun V, Pramanik A, Gwinner T, Köberle M, Bohn E. Biometals. 2009;22:3–13. doi: 10.1007/s10534-008-9199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zahner H, Diddens H, Keller-Schierlein W, Nageli HU. Jpn J Antibiot. 1977;30(Suppl):201–206. [PubMed] [Google Scholar]
- 34.Diarra MS, Lavoie MC, Jacques M, Darwish I, Dolence EK, Dolence JA, Ghosh A, Ghosh M, Miller MJ, Malouin F. Antimicrob Agents Chemother. 1996;40:2610–2617. doi: 10.1128/aac.40.11.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ji C, Juárez-Hernández RE, Miller MJ. Future Med Chem. 2012;4:297–313. doi: 10.4155/fmc.11.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Page MG. Ann N Y Acad Sci. 2013;1277:115–126. doi: 10.1111/nyas.12024. [DOI] [PubMed] [Google Scholar]
- 37.Tillotson GS. Infect Dis (Auckl) 2016;9:45–52. doi: 10.4137/IDRT.S31567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe NA, Nagasu T, Katsu K, Kitoh K. Antimicrob Agents Chemother. 1987;31:497–504. doi: 10.1128/aac.31.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Curtis NAC, Eisenstadt RL, East SJ, Cornford RJ, Walker LA, White AJ. Antimicrob Agents Chemother. 1988;32:1879–1886. doi: 10.1128/aac.32.12.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silley P, Griffiths JW, Monsey D, Harris AM. Antimicrob Agents Chemother. 1990;34:1806–1808. doi: 10.1128/aac.34.9.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hashizume T, Sanada M, Nakagawa S, Tanaka N. J Antibiot (Tokyo) 1990;43:1617–1620. doi: 10.7164/antibiotics.43.1617. [DOI] [PubMed] [Google Scholar]
- 42.Nikaido H, Rosenberg EY. J Bacteriol. 1990;172:1361–1367. doi: 10.1128/jb.172.3.1361-1367.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKee JA, Sharma SK, Miller MJ. Bioconjugate Chem. 1991;2:281–291. doi: 10.1021/bc00010a013. [DOI] [PubMed] [Google Scholar]
- 44.Dolence EK, Minnick AA, Lin CE, Miller MJ, Payne SM. J Med Chem. 1991;34:968–978. doi: 10.1021/jm00107a014. [DOI] [PubMed] [Google Scholar]
- 45.Ji C, Miller PA, Miller MJ. J Am Chem Soc. 2012;134:9898–9901. doi: 10.1021/ja303446w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kohira N, West J, Ito A, Ito-Horiyama T, Nakamura R, Sato T, Rittenhouse S, Tsuji M, Yamano Y. Antimicrob Agents Chemother. 2016;60:729–734. doi: 10.1128/AAC.01695-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hennard C, Truong QC, Desnottes JF, Paris JM, Moreau NJ, Abdallah MA. J Med Chem. 2001;44:2139–2151. doi: 10.1021/jm990508g. [DOI] [PubMed] [Google Scholar]
- 48.Rivault F, Liébert C, Burger A, Hoegy F, Abdallah MA, Schalk IJ, Mislin GLA. Bioorg Med Chem Lett. 2007;17:640–644. doi: 10.1016/j.bmcl.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 49.Wencewicz TA, Möllmann U, Long TE, Miller MJ. Biometals. 2009;22:633–648. doi: 10.1007/s10534-009-9218-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Md-Saleh SR, Chilvers EC, Kerr KG, Milner SJ, Snelling AM, Weber JP, Thomas GH, Duhme-Klair AK, Routledge A. Bioorg Med Chem Lett. 2009;19:1496–1498. doi: 10.1016/j.bmcl.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 51.Noël S, Gasser V, Pesset B, Hoegy F, Rognan D, Schalk IJ, Mislin GLA. Org Biomol Chem. 2011;9:8288–8300. doi: 10.1039/c1ob06250f. [DOI] [PubMed] [Google Scholar]
- 52.Juárez-Hernández RE, Miller PA, Miller MJ. ACS Med Chem Lett. 2012;3:799–803. doi: 10.1021/ml300150y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ji C, Miller MJ. Bioorg Med Chem. 2012;20:3828–3836. doi: 10.1016/j.bmc.2012.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wencewicz TA, Miller MJ. J Med Chem. 2013;56:4044–4052. doi: 10.1021/jm400265k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wencewicz TA, Long TE, Möllmann U, Miller MJ. Bioconjugate Chem. 2013;24:473–486. doi: 10.1021/bc300610f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Milner SJ, Seve A, Snelling AM, Thomas GH, Kerr KG, Routledge A, Duhme-Klair AK. Org Biomol Chem. 2013;11:3461–3468. doi: 10.1039/c3ob40162f. [DOI] [PubMed] [Google Scholar]
- 57.Souto A, Montaos MA, Balado M, Osorio CR, Rodríguez J, Lemos ML, Jiménez C. Bioorg Med Chem. 2013;21:295–302. doi: 10.1016/j.bmc.2012.10.028. [DOI] [PubMed] [Google Scholar]
- 58.Milner SJ, Snelling AM, Kerr KG, Abd-El-Aziz A, Thomas GH, Hubbard RE, Routledge A, Duhme-Klair A-K. Bioorg Med Chem. 2014;22:4499–4505. doi: 10.1016/j.bmc.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 59.Fardeau S, Dassonville-Klimpt A, Audic N, Sasaki A, Pillon M, Baudrin E, Mullié C, Sonnet P. Bioorg Med Chem. 2014;22:4049–4060. doi: 10.1016/j.bmc.2014.05.067. [DOI] [PubMed] [Google Scholar]
- 60.Ji C, Miller MJ. Biometals. 2015;28:541–551. doi: 10.1007/s10534-015-9830-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gupta K, Hooton TM, Naber KG, Wullt B, Colgan R, Miller LG, Moran GJ, Nicolle LE, Raz R, Schaeffer AJ, Soper DE. Clin Infect Dis. 2011;52:e103–e120. doi: 10.1093/cid/ciq257. [DOI] [PubMed] [Google Scholar]
- 62.Castro W, Navarro M, Biot C. Future Med Chem. 2013;5:81–96. doi: 10.4155/fmc.12.181. [DOI] [PubMed] [Google Scholar]
- 63.Troxell B, Hassan HM. Front Cell Infect Microbiol. 2013;3:59. doi: 10.3389/fcimb.2013.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wiles TJ, Kulesus RR, Mulvey MA. Exp Mol Pathol. 2008;85:11–19. doi: 10.1016/j.yexmp.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henderson JP, Crowley JR, Pinkner JS, Walker JN, Tsukayama P, Stamm WE, Hooton TM, Hultgren SJ. PLoS Pathog. 2009;5:e1000305. doi: 10.1371/journal.ppat.1000305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garcia EC, Brumbaugh AR, Mobley HLT. Infect Immun. 2011;79:1225–1235. doi: 10.1128/IAI.01222-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Léveillé S, Caza M, Johnson JR, Clabots C, Sabri M, Dozois CM. Infect Immun. 2006;74:3427–3436. doi: 10.1128/IAI.00107-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaper JB, Nataro JP, Mobley HLT. Nat Rev Microbiol. 2004;2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 69.Wohlkonig A, Chan PF, Fosberry AP, Homes P, Huang J, Kranz M, Leydon VR, Miles TJ, Pearson ND, Perera RL, Shillings AJ, Gwynn MN, Bax BD. Nat Struct Mol Biol. 2010;17:1152–1153. doi: 10.1038/nsmb.1892. [DOI] [PubMed] [Google Scholar]
- 70.Laponogov I, Pan X-S, Veselkov DA, McAuley KE, Fisher LM, Sanderson MR. PLoS One. 2010;5:e11338. doi: 10.1371/journal.pone.0011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aldred KJ, McPherson SA, Turnbough CL, Jr, Kerns RJ, Osheroff N. Nucleic Acids Res. 2013;41:4628–4639. doi: 10.1093/nar/gkt124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mustaev A, Malik M, Zhao X, Kurepina N, Luan G, Oppegard LM, Hiasa H, Marks KR, Kerns RJ, Berger JM, Drlica K. J Biol Chem. 2014;289:12300–12312. doi: 10.1074/jbc.M113.529164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hagan EC, Lloyd AL, Rasko DA, Faerber GJ, Mobley HLT. PLoS Pathog. 2010;6:e1001187. doi: 10.1371/journal.ppat.1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mobley HLT. Pathogens. 2016;5:7. [Google Scholar]
- 75.Zhu W, Winter MG, Byndloss MX, Spiga L, Duerkop BA, Hughes ER, Büttner L, de Lima Romão E, Behrendt CL, Lopez CA, Sifuentes-Dominguez L, Huff-Hardy K, Wilson RP, Gillis CC, Tükel C, Koh AY, Burstein E, Hooper LV, Bäumler AJ, Winter SE. Nature. 2018;553:208–211. doi: 10.1038/nature25172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Braun V, Günthner K, Hantke K, Zimmermann L. J Bacteriol. 1983;156:308–315. doi: 10.1128/jb.156.1.308-315.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zheng T, Nolan EM. Bioorg Med Chem Lett. 2015;25:4987–4991. doi: 10.1016/j.bmcl.2015.02.034. [DOI] [PubMed] [Google Scholar]
- 78.Paulen A, Gasser V, Hoegy F, Perraud Q, Pesset B, Schalk IJ, Mislin GLA. Org Biomol Chem. 2015;13:11567–11579. doi: 10.1039/c5ob01859e. [DOI] [PubMed] [Google Scholar]
- 79.Paulen A, Hoegy F, Roche B, Schalk IJ, Mislin GLA. Bioorg Med Chem Lett. 2017;27:4867–4870. doi: 10.1016/j.bmcl.2017.09.039. [DOI] [PubMed] [Google Scholar]
- 80.Tomaras AP, Crandon JL, McPherson CJ, Banevicius MA, Finegan SM, Irvine RL, Brown MF, O’Donnell JP, Nicolau DP. Antimicrob Agents Chemother. 2013;57:4197–4207. doi: 10.1128/AAC.00629-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim A, Kutschke A, Ehmann DE, Patey SA, Crandon JL, Gorseth E, Miller AA, McLaughlin RE, Blinn CM, Chen A, Nayar AS, Dangel B, Tsai AS, Rooney MT, Murphy-Benenato KE, Eakin AE, Nicolau DP. Antimicrob Agents Chemother. 2015;59:7743–7752. doi: 10.1128/AAC.00831-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Silver LL. Topics in Medicinal Chemistry. Springer Berlin Heidelberg; Berlin, Heidelberg: 2017. The Antibiotic Future. [Google Scholar]
- 83.Subashchandrabose S, Hazen TH, Brumbaugh AR, Himpsl SD, Smith SN, Ernst RD, Rasko DA, Mobley HLT. Proc Natl Acad Sci U S A. 2014;111:18327–18332. doi: 10.1073/pnas.1415959112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.