

Abstract
Hsp90 is a molecular chaperone of pivotal importance for multiple cell pathways. ATP-regulated internal dynamics are critical for its function and current anticancer pharmacological approaches block the chaperone by using ATP-competitive inhibitors. In this paper, we propose a general approach to perturb Hsp90 through design of new allosteric modulators that alter the functional dynamics of the protein. We rationally developed a library of 2-phenylbenzofurans that, rather than inhibiting, activate Hsp90 ATPase by targeting an allosteric site that we recently identified, located 65Å from the active site. Analysis of protein responses to first-generation activators was exploited to guide the design of second-generation derivatives with improved ability to stimulate ATP hydrolysis. The molecules’ effects on Hsp90 enzymatic, conformational, co-chaperone and client-binding properties were characterized through biochemical, biophysical and cellular approaches. The new, rationally designed probes act as allosteric activators of the chaperone and affect the viability of cancer cell lines for which proper functioning of Hsp90 is necessary.
Keywords: Hsp90, Functional Dynamics, Allostery, Molecular Design, Glycoconjugates
Introduction
The dynamic properties of proteins are critical for the functions they exhibit[1]. They can be finely tuned through allostery, a general property of biomolecules whereby a perturbation at one site leads to a response at another, turning specific functional states on or off[2]. Allosteric ligand binding provides the opportunity to regulate protein functions via small molecules that act at a distance from the active site, without directly interfering with its chemical activity[1, 3]. Additionally, allosteric ligands can be used to modulate the equilibrium and dynamics of distinctive protein states implicated in certain cellular pathways and phenotypes[4].
The fundamental chemical challenge for the discovery of allosteric protein modulators consists in identifying privileged structures that selectively target key protein sub-states involved in the regulation of biochemical function. To progress along this appealing avenue, we focused on the 90 kDa heat shock protein (Hsp90) molecular chaperone. This protein is known to undergo a complex dynamic cycle, which is essential to its chaperone activity that, in turn, regulates the state of a number of client proteins in the cell. Hsp90 integrates different pathways required for cell development and maintenance[5], and plays a key role in the regulation of a wide variety of so-called client proteins[6]. As such, it has been proposed as an interesting target in cancer[7], vascular disease[8], neurodegeneration[9], and as a major player in evolution[10]. Thanks to its dynamic nature[5, 11], Hsp90 influences the functional lifetime of many clients that vary widely in sequence, structure, size and function[12]. In eukaryotes this activity is further regulated by a number of co-chaperones, which bind individual Hsp90-states populated at different stages of the chaperone cycle. Hsp90 dynamics depend on ATP binding and hydrolysis, which underlie the onset of conformational transitions between sub-states with different functional properties[13]. Here, we set out to develop a rational approach to regulate the ATPase activity and conformational dynamics of Hsp90 through designed allosteric small molecule ligands.
Hsp90 functions as a homodimer (Figure 1). Crystal structures from different organisms highlighted a common modular organization in terms of N-terminal (NTD), Middle (M) and C-terminal (CTD) domains[14]. The CTD is the dimerization domain, while the NTD contains an ATP-binding site. ATPase activity requires transient dimerization of the NTD in a closed state of the dimer and is essential for the Hsp90 working cycle. The exact mechanism of coupling between ATP-binding/hydrolysis and client folding remains elusive. Yet, structural and biochemical data support a model in which nucleotide binding at the NTD propagates a conformational signal to the CTD[14d], while the chaperone undergoes conformational rearrangements that bring the two NTDs into close association in the ATP-state – but not in the ADP or apo states. Formation of the closed state can be induced by ATP or the non-hydrolyzable analogue adenosine 5′-(β,γ-imido)triphosphate (AMP-PNP). Upon ATP hydrolysis, the protein cycles back to the open, ADP-bound state. A number of intermediate conformational states have also been characterized, that are induced or stabilized by interaction with different clients or co-chaperone proteins[13, 15]. Figure 1 highlights some of these interactions, with the co-chaperones Aha1, an endogenous activator of Hsp90 ATPase activity, and Sba1, an Hsp90 inhibitor, which acts by binding to the closed conformation and blocking the chaperone dynamics. The predicted allosteric site that we sought to exploit (AS, Figure 1)[16] is located at the MD/CTD interface, a region where the model client Δ131Δ is also known to interact.
Figure 1. Schematic representation of Hsp90 structure and its conformational equilibrium.
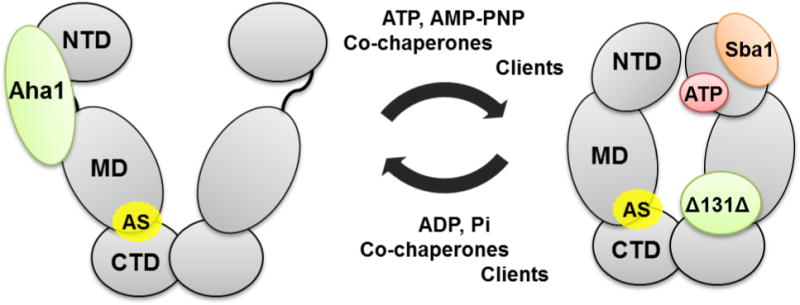
NTD, N-terminal Domain; MD, Middle Domain; CTD, C-terminal Domain; AS, predicted allosteric site; Aha1, Hsp90 co-chaperone; Sba1, yeast homologue of co-chaperone p23; Δ131Δ, Hsp90 client protein model.
Most known small molecule Hsp90 modulators interact with the protein at the NTD and inhibit Hsp90 ATPase activity[17]. They include Radicicol and Geldanamycin or its derivative 17AAG[18]. Some of such inhibitors have entered clinical trials as antitumor drugs but have shown severe limitations. Indeed, blocking the ATPase activity of Hsp90 induces the so-called heat shock response, a pro-survival mechanism mediated by HSF-1(Heat Shock Factor 1), that limits the action of the drug[19].
Rationally designed chemical probes can be used to select and activate, and not only to inhibit, Hsp90 key sub-states, providing a connection between protein activities and possible cellular outcomes. Here, we pursue the first rational design of chemical activators of Hsp90 functions aimed to target sites alternative to the ATP-site, and investigate their effects on the chaperone conformational dynamics, enzymatic, binding and cellular properties.
In this paper, we build on the results of recently developed computational methods that unveiled the presence of a druggable site in Hsp90 CTD[16, 20] and facilitated the design of small molecules able to bind it[21]. Different sets of experiments showed that the O-aryl rhamnoside 1 (Figure 2A) identified through this method could bind the C-terminal domain of Hsp90 and exert interesting anti-tumor activities[16]. Here, we evolve 1 into new chemical entities that enable controlled activation of Hsp90 ATPase function. We show that designed derivatives are genuine allosteric ligands with a structure-dependent ability to stimulate Hsp90 ATPase and to alter conformational dynamics favoring synergistic effects with the activating co-chaperone Aha1, to modulate Hsp90 direct interactions with the co-chaperone Sba1 (p23), and to compete with the model client protein Δ131Δ. We characterize the impact of small molecule induced activation on Hsp90 interactions in vitro and on the stability of a number of clients in cellular models, and we investigate the possibility that acceleration of conformational dynamics may in fact represent a new way of perturbing the chaperoning mechanisms that underlie cell viability: indeed, some of our compounds inhibit the proliferative potential of tumor cells including those resistant to Hsp90 ATP-competitive inhibitors.
Figure 2. Structure of the initial lead and interaction with Hsp90 allosteric site.
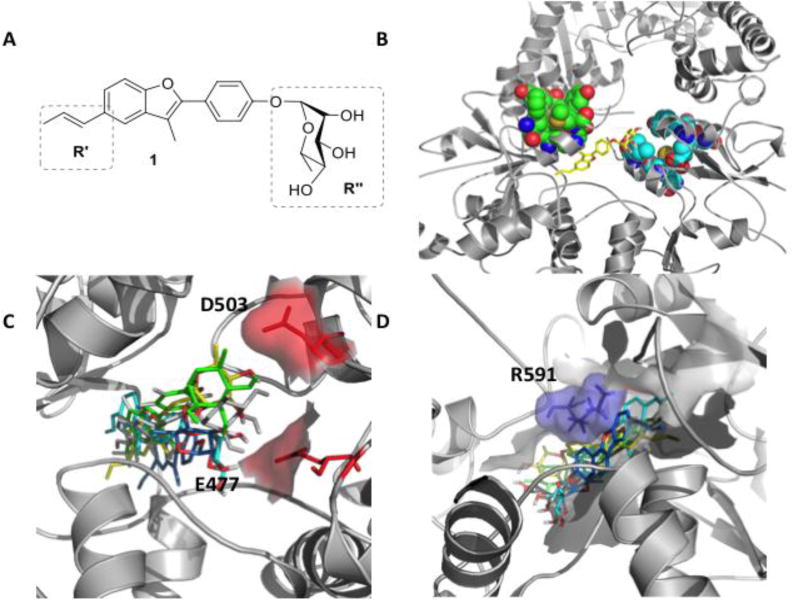
A) Molecular structure of 1. B) 3D structure of compound 1 (yellow) in complex with the closed structure of Hsp90. Van der Waals spheres in light blue and green indicate the client protein Δ131Δ binding site. C) Representative poses of 1 in the representative conformations of the allosteric site, showing the contacts of the ligand with E477 and D503 on protomer A. D) Contacts of 1 with protomer B, highlighting interaction with R591
Results and Discussion
Design and synthesis of first-generation Hsp90 modulators
In our previous work, the rhamnoside 1 (Figure 2A) was selected from the NCI library by virtual screening in a 3D pharmacophore designed to complement the stereoelectronic properties displayed by an allosteric site in the CTD. The site was identified through the analysis of long-range dynamic communication mechanisms with the ATP-site, using the coordination propensity (CP) parameter (Supplementary methods)[16, 20]. It comprises residues at the CTD interface with the M-domain, which define a druggable pocket coincident with the region of a recently identified binding site for the model client protein Δ131Δ[22] (Figure 2B). The long-range coordination properties of the aminoacids defining the allosteric pocket describe their dynamic connection to events occurring in the orthosteric ATP-binding site, and are conserved across different members of the Hsp90 family[20a]. Targeting these residues by designed ligands, such as 1, should thus provide a way of influencing the functional properties of the protein[23]. Indeed, 1 was found to bind the Hsp90 C-terminal domain disrupting chaperoning functions and to exhibit antiproliferative activity in different tumor cell lines[23].
To optimize the structure and functional impacts of this lead, in the absence of crystal structures, we investigated binding determinants in the allosteric site through docking calculations. It should be noted here that no X-ray structure of any C-terminal ligands in complex with Hsp90 has been determined so far. The initial target was the MD-relaxed ATP-bound Hsp90 structure used for the pharmacophore-based discovery of 1[23]. Considering the flexibility of the protein and of the allosteric pocket[21a], we selected an ensemble approach to characterize chaperone-ligand interactions. The minimum energy pose of the 1/Hsp90 complex (Figure 2B) was used as input for long timescale MD simulations, including ATP at the NTD. The aim was to identify the hot spots of the allosteric site where key functional groups on the ligand best complement the receptor, taking the dynamic exchange between the binding partners explicitly into account.
Structural cluster analysis of the resulting trajectory (Supplementary Methods) showed that the first 10 clusters recapitulated ~95% of the protein structural variability. The RMSD between visited pocket conformations in the putative binding site (Supplementary Table 1) reached up to 4Å, revealing the diversity induced by 1. Such differences, due to the cross-talk between the protein and the ligand, were not expected a priori and revealed protein conformations that could play a role in small molecule recognition. The resulting structures were next used to explore possible alternative poses of 1 in the allosteric site. The compound was redocked into each of the 10 representative structures. The resulting structural ensembles were used to generate a consensus model of Hsp90 residues and functional groups on 1 that define the most relevant stabilizing contacts (Figure 2C, D). Two areas where functional group diversification on the lead could translate into a modulated response of the chaperone were identified: the carbohydrate moiety (R″, Figure 2A), which in the consensus model points towards an area lined by E477 and D503 (Figure 2C), and the propenyl-group (R’, Figure 2A), which contacts a hydrophobic pocket also lined by R591 from the other protomer (Figure 2D).
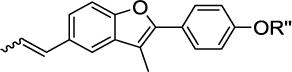
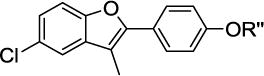
To validate this model and to expand the available SAR, glycodiversification of the 2-phenylbenzofuran aglycone 2 (Table 1) was examined. Glyco-randomization is a well-known strategy to tune the activity of a variety of glycoconjugates against their protein targets[24] and is an attractive avenue for the case at hand, where computational data indicate a strong involvement of the sugar moiety in the interaction with Hsp90 allosteric site. A first series of molecules 4 to 16 was synthesized (Table 1) by glycosylation of 2 and of the 5-chloro analog 3 (an intermediate in the synthesis of 2), as previously reported in ref[25]. The full structures of all compounds are reported in Supplementary Figure 1.
Table 1.
First generation of CTD ligands 1 – 16. The propenyl scaffold 2 was used as a E/Z mixture after checking that the natural compound 1 and E/Z-1 had a comparable effect on the protein activity.
|
|
||
|---|---|---|---|
| 2 R″ = H | 3 R″ = H | ||
|
| |||
| R″ | R″ | ||
| 1 | α-L-Rha | 4 | α-L-Rha |
| 5 | β-D-Glc | 6 | β-D-Glc |
| 7 | β-L-Glc | 8 | β-L-Glc |
| 9 | α-D-Man | 10 | α-D-Man |
| 11 | β-D-Gal | 12 | β-D-Gal |
| 13 | β-L-Gal | 14 | β-L-Gal |
| 15 | β-L-Fuc | 16 | β-L-Fuc |
Designed allosteric ligands stimulate Hsp90 ATPase activity
To characterize their role as allosteric modulators, we measured the effects of compounds 1-16 on the ATPase activity of the Hsp90 yeast homologue (Hsc82)[26]. Interestingly, most compounds, including 1, turned out to be activators of ATP hydrolysis (Figure 3, compounds 1 and 4-16), with variable potency, depending on the type of carbohydrate moiety and benzene ring substitution (propenyl or chlorine). Compounds 4 and 10-12 were the strongest activators of Hsp90 with a 2-3 fold enhancement of ATPase rate. Since the basal ATPase rate of yeast Hsp90 is very low, this kind of acceleration is not negligible, it has very little precedent in the literature, and none with rationally designed compounds[27].
Figure 3. Effects of benzofurans 1-19 in stimulating yeast Hsp90 (Hsc82) ATPase activity.
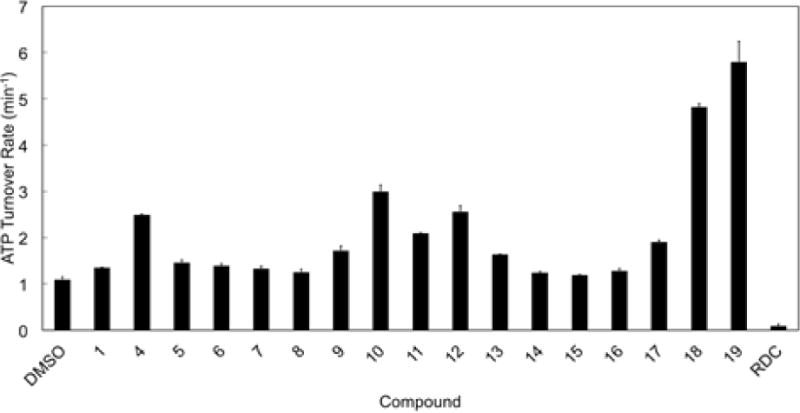
ATP hydrolysis rate of Hsp90 was measured in the presence of the compounds (50 μM) or Radicicol (RDC, 5 μM). The ATP turnover rate is reported as the mean of four independent measurements. Basal Hsc82 ATPase activity was measured using DMSO alone.
From a ligand-design point of view, the data indicate that a chlorine atom in position R’ (Figure 2) is as good as and often better than the original propenyl substitution (compare pairs 1 and 4, 9 and 10, 11 and 12). Additionally, a significant effect of the sugar on the level of activity was observed, with D-mannose (as in 10) and D-galactose (as in 12) emerging as privileged fragments alongside L-rhamnose (in 4).
Rational optimization of First-Generation ligands further increases Hsp90 ATPase activity
Starting from the aforementioned observations, to design improved allosteric activators we docked all the first-generation compounds into the 10 Hsp90 conformations used for the first generation of derivatives, describing binding in terms of an ensemble of ligand poses in complex with an ensemble of protein structures[28].
All glycosylated compounds explored common ensembles in the allosteric site, similar to 1. Representative poses are shown in Figure 4A for compounds 4 (blue) 10 (cyan) and 12 (yellow) (Figure 4A). The pair formed by the negatively charged E477 and D503 on one protomer, towards which initial design was aimed, was always engaged in hydrogen bonding interactions with the carbohydrate moieties (R″). The most active glycosylated compounds, 10 and 12, showed two hydroxyl groups (on C-6 and C-4 of the sugar) ideally oriented to establish H-bonding interactions with the aforementioned negative residues. This interaction was reinforced by a H-bond network involving R591, R599 and the hydroxyl groups on the carbohydrate ring. On the R’ side, two positively charged residues from the other protomer, R591 and K594, were involved in stabilizing interactions with the halogen of compounds deriving from the common intermediate 3.
Figure 4. Model structures of first-generation allosteric ligands in an ensemble of conformations of the putative allosteric pocket.
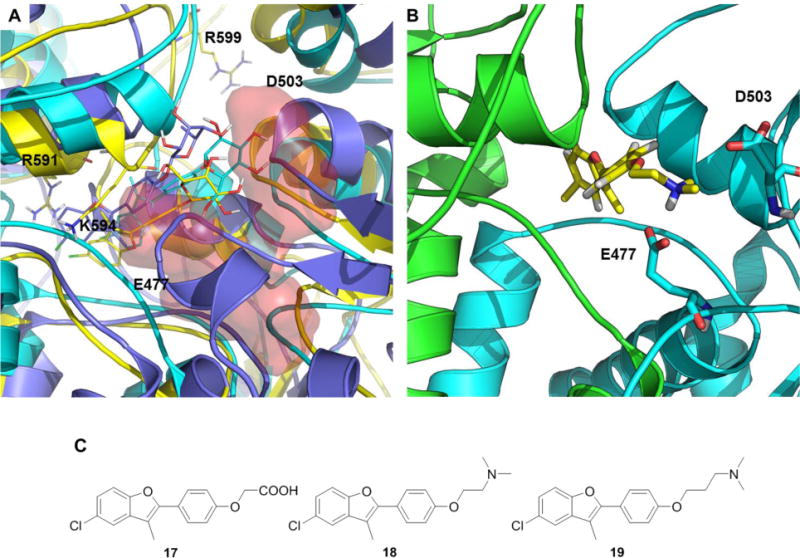
A) Representative structures of the poses of compounds 4 (blue), 10 (cyan) and 12 (yellow). The red surfaces indicate the locations of charged residues targeted by the ligands. B) Representative structure of compound 18 in the allosteric binding pocket. The two Hsp90 protomers are colored differently. C) Compounds 17 to 19.
On this basis, a second group of ligands (17-19) was designed to exploit potential productive interactions with the network of charged aminoacids (Figure 4B, C) in the allosteric pocket. Indeed, 18 and 19 were found to dramatically accelerate yeast Hsp90 (Hsc82) ATPase rate (by up to 6 fold, Figure 3), providing further support to our model and to the design approach.
Allosteric ligands accelerate Hsp90 conformational dynamics
The conformational change of Hsp90 initiated by ATP and non-hydrolyzable ATP analogs is a hallmark of the Hsp90 cycle[29]. ATP and non-hydrolyzable ATP analogs such as AMP-PNP induce a closed state of the Hsp90 dimer, where the NTD of the two protomers are in close proximity (Figure 1). As previously mentioned, this is the catalitically active state of the protein that allows ATPase activity. We measured the closure rate in the presence of the allosteric modulators by FRET, using an Hsp90 dimer with one protomer labeled with a FRET-donor (Alexa Fluor 555) and the other with a FRET-acceptor (Alexa Fluor 647). The addition of AMP-PNP drives the closure of Hsp90, which shortens the distance between the FRET-dyes, resulting in an increased FRET signal (DMSO, Figure 5). Similar to the ATPase results, 10, 12, 18 and 19 most accelerated the Hsp90 conformational change process, consistent with the observation that N-domain closure is the rate-limiting step for ATP hydrolysis. The NTD inhibitor radicicol was used as negative control (RDC, Figure 5) and, as expected, it did not influence the closure rate of the protein to any measurable extent, preventing N-terminal dimerization. The 2-phenylbenzofuran derivatives thus have an actual impact on modulating both the mechanisms of formation and the conformational properties of the catalytic state of Hsp90.
Figure 5. FRET-based measurement of yeast Hsp90 (Hsc82) conformational change.
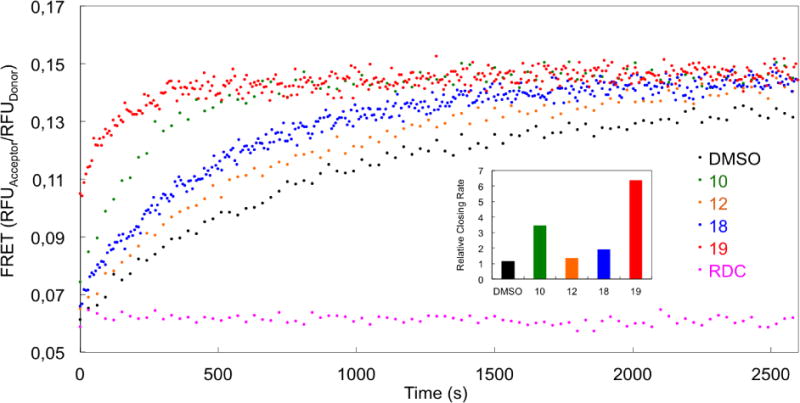
Hsp90 conformational change rate measured by the FRET-based assay in the presence of allosteric activators, compared to the NTD inhibitor Radicicol (RDC). Conformational dynamics were initiated by adding AMP-PNP (1 mM) in the presence of 50 μM test compounds. The rates in the inset are calculated by exponential fitting of the data relative to control DMSO/AMP-PNP.
Since compounds 10, 12, 18 and 19 stood out as giving a significant acceleration of the chaperone enzymatic activity, their effects were also tested on human Hsp90α. Human Hsp90 has an ATPase activity about an order of magnitude lower than the yeast chaperone, yet the compounds proved to increase ATPase activities also in this homologue, with 19 once more showing the highest stimulation (Supplementary Figure 2).
Overall the results of our design efforts, based on the computational mapping of the stereochemical and dynamic properties of the identified allosteric site, are consistent with a model in which the compounds bind to the boundary between the M and C-terminal domains and modulate both Hsp90 enzymatic properties and its ability to form the N-terminal dimerization state, resulting in accelerated ATPase activities, in line with the general mechanism reported in[30].
Designed allosteric activators synergize with Aha1 to accelerate Hsp90 ATPase cycle
The most relevant known natural activator of the Hsp90 ATPase cycle is the co-chaperone protein Aha1, an endogenous accelerator[31]. Aha1 is known to bind Hsp90 engaging the N- and Middle Domains of the chaperone in its open conformation (Figure 1) and to accelerate ATPase by an order of magnitude[31]. The ATPase acceleration observed with the designed benzofurans prompted the question whether they are in competition or synergistic with the action of Aha1: allosteric compounds designed to target the MD/CTD interface should arguably not compete with the co-chaperone binding or inhibit its activity. The effects of allosteric derivatives on yeast Hsp90 (Hsc82) were then measured in the presence of yeast Aha1. The compounds did not abolish the acceleration of Hsp90 ATPase by Aha1, but rather amplified the co-chaperone effects in a synergistic fashion (Figure 6). In all cases examined the ATP hydrolysis rates were one order of magnitude higher that those measured without Aha1 (Figure 3), but the relative trend of the values was once again determined by the nature of the modulators.
Figure 6. Synergistic stimulation of yeast Hsp90 (Hsc82) ATPase by Aha1 and compounds 1-19.
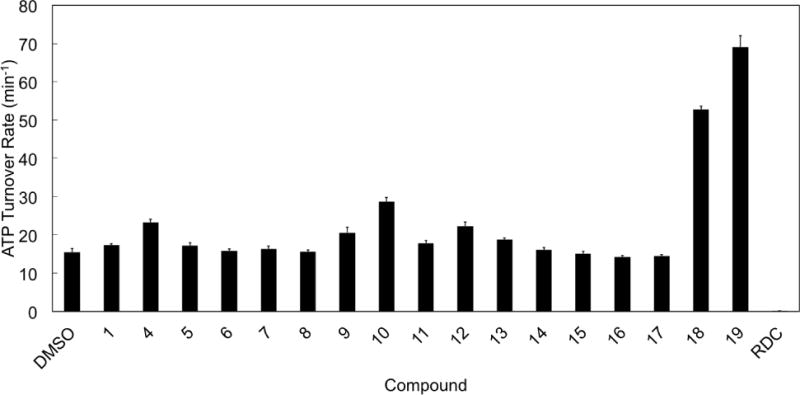
ATP hydrolysis rate of Hsc82 is measured similarly as in Figure 2, but in the presence of yeast Aha1 (2 μM). The ATP turnover rate is reported as the mean of four independent measurements.
The results described above suggest that the regulation of ATPase activity by the designed benzofurans occurs via an alteration of the overall protein dynamics, controlled by the allosteric site. This interpretation was supported by data obtained from co-immuno precipitation (Co-IP) experiments performed in protein lysates of yeast expressing His-tagged Hsc82. In this experiment, we examined whether the compounds altered the interaction between Hsc82 and the co-chaperone Sba1 (yeast p23, Figure 1). Sba1 is an inhibitor of ATP hydrolysis that operates by interacting with the N-dimerized, ‘closed’ Hsp90 conformation poised for hydrolysis (Figure 1), stabilizing it and thus blocking the chaperone dynamic cycle[15a]. AMP-PNP was added to the lysates to induce N-dimerization of the chaperone. After selective isolation of His-tagged Hsc82 using Ni-NTA agarose resin, western blotting was performed to visualize associated Sba1. In line with the kinetic data presented above (Figure 5), the most potent allosteric stimulators (compounds 18 and 19) abrogated AMP-PNP-dependent Sba1/Hsc82 interaction (Figure 7), suggesting that they can either alter the closed state recognized by Sba1 or simply accelerate the chaperone cycle, reducing its population. For comparison, the last two lanes of the blot show that the ATP competitive Hsp90 inhibitor geldanamycin (GA), which prevents N-domain dimerization, also abrogates Sba1 interaction with Hsc82 (Figure 7).
Figure 7. Effects of allosteric ligands on yeast Hsp90 (Hsc82) interaction with the ATPase inhibitory co-chaperone Sba1.
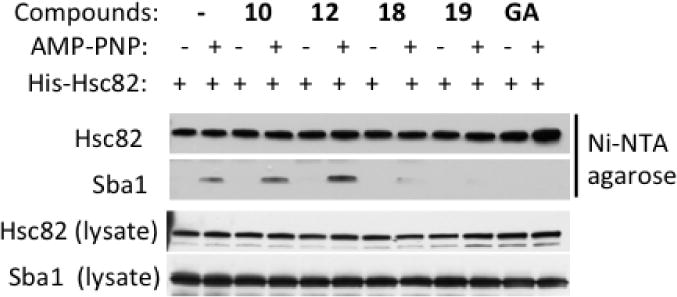
The presence of allosteric accelerators minimizes AMP-PNP-dependent association of Sba1 (p23) with Hsc82. These data were obtained by adding compounds to yeast lysates followed by affinity pulldown of His-tagged Hsc82 with Ni-NTA agarose beads. Hsc82 and Sba1 were visualized in pulldowns and lysate with appropriate antibodies (see Experimental).
Combining these observations with the FRET data (Figure 5), we suggest that the allosteric modulators modify the kinetics between Hsp90 open and closed forms, as well as the structure of the closed state recognized by Sba1, eventually promoting the rescue of the enzymatically active closed conformation into the ATPase cycle.
In summary, our data support a model in which designed compounds do not only show a significant acceleration of Hsp90 ATPase, but also a consistent synergistic stimulation with Aha1, suggesting that the compounds and the co-chaperone do not compete with one another for binding Hsp90, while modifying the same rate-limiting conformational process via distinct but complementary interactions. The data also confirm that ATPase stimulation is likely due to an acceleration of the chaperone conformational cycle, and as such are similar to other recent findings demonstrating that enforced N-domain proximity (without N-domain dimerization) is sufficient to enhance Hsp90 ATPase activity while retaining further stimulation by Aha1[30].
Synergy/competition between designed compounds and a model client protein in yeast
To probe direct effects of designed activators on Hsp90 client interaction, we used the model client protein Δ131Δ[22a]. Δ131Δ was shown to stimulate ATPase[22] and its binding site was mapped to the same Hsp90 region against which the designed allosteric molecules are targeted[22].
Yeast Hsp90 (Hsc82) ATPase activity was monitored in the presence of Δ131Δ (50 μM) or selected allosteric stimulators (1, 4, 10, 12, 18 and 19; 50 μM) or the combination of both (Figure 8). In contrast with the observations reported for Aha1, the benzofurans and Δ131Δ appeared to operate in a competitive fashion. Δ131Δ appeared to prevail over the compounds with relatively weaker potency (e.g. 4, 10 and 12), and was clearly outcompeted by the most active ligand 19. Ligands 1 and 18 displayed a borderline behavior (Figure 8).
Figure 8. Competitive stimulation of yeast Hsp90 (Hsc82) ATPase by Δ131Δ and designed compounds.
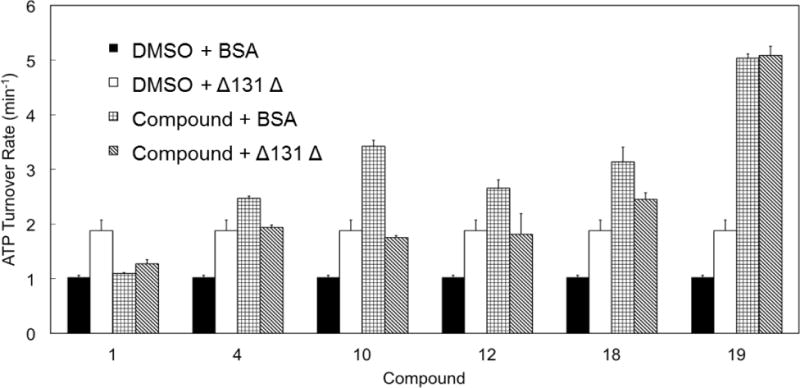
Hsc82 ATPase is measured in the presence of Δ131Δ (50 μM) or selected allosteric activators (50 μM, 1, 4, 10, 12, 18, 19) or the combination of both. Reactions with BSA and DMSO serve as negative control. Δ131Δ stimulated Hsc82 ATP-hydrolysis by 1.8 fold. The stimulation by compounds alone varies with each compound. However, when the chaperone was stimulated by both Δ131Δ and designed accelerators, the resultant ATPase rates mostly fell in the range of the rate yielded by Δ131Δ or compounds stimulation alone.
These results, combined with the analysis in the presence of Aha1, thus indicate a competition between Δ131Δ and benzofuran ligands for the same binding site and for the consequent stimulation of Hsp90 ATPase. Given this synergy/competition with a model substrate, it is intriguing to speculate whether the benzofuran ligands act as ‘Hsp90 substrates’, especially in light of an earlier report that substrate binding to Hsp90 can, in itself, stimulate the chaperone’s ATPase activity in vitro[32].
Effects of the designed compounds on cell proliferation and Hsp90 chaperone function
Given the well-established role of Hsp90 inhibition in cancer models, we asked whether our small-molecules could affect human cancer cells. The cytotoxicity of selected derivatives was examined in three human tumor cell lines of different histological origin, including breast cancer (MCF-7), castration-resistant prostate carcinoma (DU145) and diffuse malignant peritoneal mesothelioma (STO). Cells were exposed to increasing concentrations (0.1 to 100 μM) of each derivative for 72 h, and the effect on cell proliferation was determined by MTS assay (Table 2). Cytotoxic activities of glycoderivatives 4-16 were comparable to those of previously reported C-terminal inhibitors discovered and developed through different approaches[33], with 50% inhibitory concentration (IC50) values, as calculated from the growth curves, ranging from 29.9 to 82.3 μM.
Table 2. Cytotoxic activity of 1 and derivatives 4-19 in three different cell lines (MCF-7, DU145, STO).
Data are reported as IC50 values (concentration of drug required to inhibit growth by 50%) determined by MTS assay after 72 h of continuous exposure to each compound; data represent mean values±SD of at least three independent experiments. n.a.: not assessed.
| IC50 (μM) | ||||
|---|---|---|---|---|
| Compound | MCF-7 | DU145 | STO | STO-17AAG |
| 1 | 58.9±7.4 | 63.2±2.0 | 57.0±2.1 | |
| 4 | 57.6±3.9 | 66.3±6.6 | 59.7±1.5 | |
| 5 | 60.0±4.3 | 61.5±3.4 | 59.5±5.1 | |
| 6 | 82.3±6.2 | 64.8±2.8 | 68.4±6.2 | |
| 7 | 70.3±9.5 | 68.2±8.2 | 60.6±8.4 | |
| 8 | n.a. | n.a. | n.a. | |
| 9 | 54.5±5.7 | 59.4±4.7 | 40.9±3.2 | |
| 10 | 56.4±4.6 | 50.7±7.1 | 29.9±1.4 | |
| 11 | n.a. | n.a. | n.a. | |
| 12 | 77.2±0.4 | 60.9±5.8 | 74.9±1.8 | |
| 13 | 41.4±4.2 | 55.3±1.6 | 43.6±4.5 | |
| 14 | 61.8±5.4 | 58.8±4.9 | 50.0±1.3 | |
| 15 | 62.0±6.3 | 63.3±5.3 | 60.4±.3.0 | |
| 16 | 61.2±2.7 | 52.5±5.7 | 61.9±4.7 | |
| 17 | n.a. | n.a. | n.a. | |
| 18 | 56.6±3.9 | 17.3±2.6 | 8.9±1.1 | 7.1±1.6 |
| 19 | 51.3±4.0 | 12.7±2.5 | 9.1±1.1 | 7.5±1.2 |
| 17-AAG | 0.08±0.01 | 2.6±0.1 | ||
In contrast, compounds 18 and 19, inducing the highest ATPase stimulation in vitro, showed a remarkable and selective antiproliferative activity in the two cell models of highly chemoresistant tumors, i.e. castration-resistant prostate cancer and mesothelioma, with IC50 values for the two compounds of 17.3 and 12.7 μM in DU145 and of 8.9 and 9.1 μM in STO, respectively (Table 2). Interestingly, compounds 18 and 19 also significantly inhibited the growth of STO-17AAG, a STO-derived cell clone with experimentally-induced resistance to 17-AAG (Resistance Index: 32.5), as indicated by IC50 values comparable to those observed in the parental cell line (7.1 and 7.5 μM for compounds 18 and 19, respectively) (Table 2). Overall, these findings suggest that in these specific drug-resistant cell types the Hsp90 machinery could populate an ensemble of conformations more sensitive to the new derivatives.
Moreover, the activating compounds did not disrupt the Hsp90/HSF1 complex (Supplementary Figure 3) and did not promote Heat Shock Factor 1 (HSF1) dissociation, as classical N-terminal inhibitors are known to do[19a]. HSF1 dissociation is indeed the first step triggering the Heat Shock response that limits the application of NTD inhibitors in anticancer therapy. The inability of the benzofurans to activate this response is a promising feature for further development of these compounds towards therapeutic applications.
To verify whether the observed cytotoxic activity was due to the breakdown of multiple cell survival pathways as a consequence of the interaction of our compounds with Hsp90, we assessed the effects of the treatment on the stability of Hsp90 client proteins (Figure 9). Cells were exposed to selected compounds (1, 4, 10, 12, 18 and 19) at their IC50 concentration, and the amounts of Hsp90 client proteins Neu, Akt, Cdk4 and survivin were estimated in cell lysates by western blotting (Figure 9). Remarkably, the clients levels were significantly altered in a cell-dependent and compound-dependent fashion. In contrast, Hsp90 expression was not affected (Figure 9). These results clearly point to a role of the designed allosteric compounds in targeting Hsp90 and its associated biological pathways, although we still have no evidence that the antiproliferative effects observed in tumor cells are exerted solely by Hsp90 modulation. Naturally, potential off-target effects represent an important issue to investigate, albeit outside of the scopes of this paper. Clearly, additional structural and mechanistic studies, along with an expanded collection of analogues, will be required to achieve quantitative SAR.
Figure 9. Inhibition of Hsp90 chaperone function.
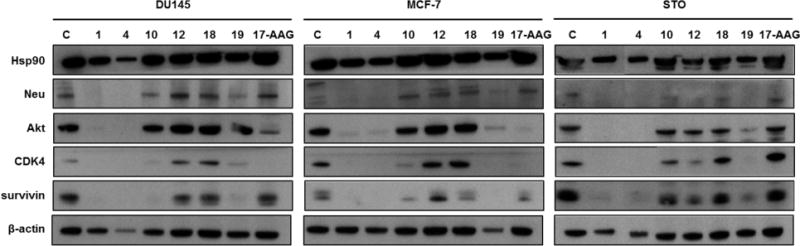
Loss of Hsp90 client proteins in human cancer cells treated for 72 h with compounds 1, 4, 10, 12, 18, 19 or 17-AAG (IC50) and analyzed by Western blotting. β-actin was used to confirm equal protein loading on the gel.
Model for Hsp90 function with allosteric ligands
The results described above suggest a complex role for Hsp90 activation. In vitro, the presence of the allosteric modulators increased Hsp90 closure and ATPase rate. In cellular models, the relative effects of the activators were likely reduced due to several factors, including membrane permeation, chaperone abundance and the dynamic nature of the Hsp90 chaperone network, which favors population of different Hsp90 conformations stabilized by interactions with different multi-protein complexes[34]. In this scenario, the allosteric molecules likely bind a subset of Hsp90 molecules at any given time, leaving the rest available for normal client and co-chaperone interactions.
To generate a structural model of the mechanism, we carried out MD simulations of the closed ATP-bound Hsp90 in complex with 1, 4, 10, 12, 18, and 19 at the C-terminal site and compared the results with those obtained in the absence of compounds (ATP-only). MD simulations were aimed to shed light on the microscopic perturbations of Hsp90 internal dynamics induced by allosteric ligands that could be linked to the activation of functional states.
We first characterized the overall rigidity/flexibility patterns in the complexes using the coordination propensity (CP) analysis[20a] (Supplementary Figure 4). In the presence of the compound, the NTDs from one protomer were decoupled from the MD and CTD of the other. By contrast, the high degree of internal coordination between the NTD and MD within each protomer was maintained upon compound addition. In this picture, high intraprotomer coordination of the two domains favors the proper positioning of the residues necessary for catalysis, while interprotomer flexibility can be aptly exploited to speed up the search for the closed active state, consistent with the observed increases in ATPase and closure rates.
Representative structures showed an evident distortion of one of the two protomers, suggesting a role for the allosteric ligands in shifting the population to a closed asymmetric state reminiscent of the one observed in the crystal structure of the mitochondrial isoform of Hsp90 (named TRAP1)[14d]. This state was also identified in solution with SAXS measurements as a general conformation of all Hsp90 chaperones, as a high-energy state that facilitates ATP hydrolysis[14d, 35] (Figure 10).
Figure 10. The structural changes induced on Hsp90 by 18 compared to the symmetric 2CG9 structure.
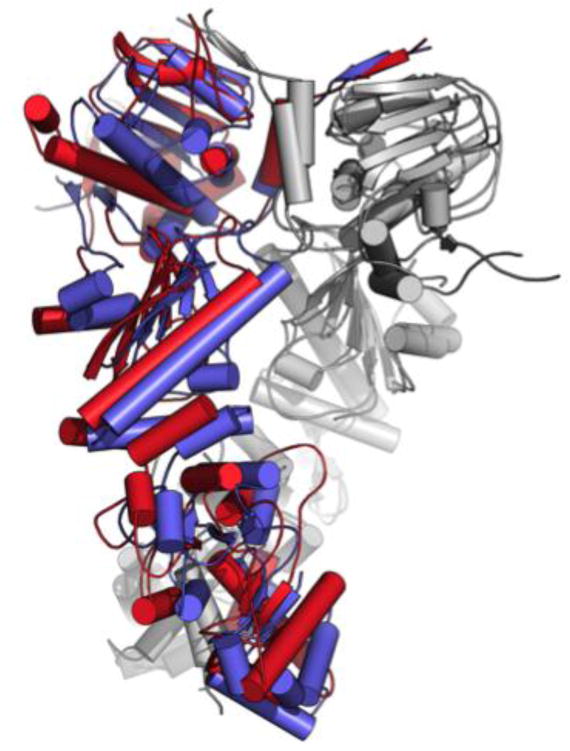
The superposition exemplifies the distortion induced by the small molecules in protomer B (in red) compared to the original X-ray structure (pdb 2CG9, in blue). Protomer A from both structures are highly superimposable and are shown in light gray for simplicity.
Rearrangement to an asymmetric state could help explain the observed synergistic activation by Aha1, through the selection of Hsp90 conformations more favorable for co-chaperone binding, a model supported by our data in yeast lysates. Consistent with the general model of asymmetric Hsp90 activation[14d], ligand induced structural asymmetry in Hsp90 could determine an expansion of conformations that are primed for ATP processing and activated for Aha1 recognition. Aha1 activation was in fact shown to be asymmetric[36]: one Aha1 molecule per Hsp90 dimer is sufficient to bridge the two protomers, stabilizing the N-dimerized catalytic state. In our model, the allosteric ligands could preorganize an asymmetric Hsp90 conformation with which Aha1 preferentially interacts, stimulating ATP hydrolysis.
In the light of in vitro and cell results, we thus suggest that allosteric ligands act as conformational catalysts bringing Hsp90 into an asymmetric state primed for sequential ATP hydrolysis steps as proposed by Lavery et al.[14d].
In summary, our findings indicate that the designed allosteric accelerators represent novel chemical tools to investigate salient aspects of the relationships between Hsp90 structural dynamics and functional regulation. These gain-of-function probes offer the possibility to address the role of enzymatic and conformational dynamics in the protein endogenous environment. This would complement biochemical and molecular biology approaches in shedding light on the roles of Hsp90 mechanisms at different stages of the chaperone cycle[30, 37]. The observed cytotoxic activities in geldanamycin-resistant cancer cells and the lack of heat shock response induction indicate possible therapeutic perspectives for this class of compounds.
Conclusions
Proteins participate in biochemical interaction networks by switching among structural sub-states, which favor adaptation to different partners and fine-tuning of functions. Such conformational changes are induced by several factors, including ligand binding. Herein, we rationally designed, synthesized and tested new molecular probes able to act as chemical switches tuning the enzymatic activities and conformational rearrangements of the molecular chaperone Hsp90. Combining computational biology, synthetic chemistry, biochemical, biophysical and cell biology approaches, we developed and partially optimized allosteric small molecules capable to activate Hsp90 functional dynamics.
The ligands were directed towards an allosteric pocket recently identified in Hsp90 by coordination propensity analysis of extended dynamic simulations of the protein. This pocket is located at the interface between the CTD and the M-domain and is dynamically coordinated to the ATP-binding site in the NTD, so that modification of the allosteric pocket are translated into variation of the ATPase activity of Hsp90. Given the intrinsically flexible nature of allosteric pockets, computational strategies for the discovery of allosteric ligands need to be developed ad hoc. Flexibility has so far hampered the resolution of crystal structures with Hsp90 complexed to C-terminal targeted ligands. Here we turned to extensive molecular dynamics simulation of the protein in the presence of a lead compound (1), previously identified by virtual screening in a pharmacophore model and validated as a CTD binder and an allosteric modulator by a number of experimental techniques[16]. The identification of the ligand-protein interactions most relevant in determining the dynamic cross-talk between the binding partners guided the evolution of initial activators towards molecules with higher activities. Computational results pointed to specific charged and hydrophobic residues in the putative Hsp90 allosteric pocket that could be targeted by ligands designed to contain specific and complementary chemical functionalities.
In particular, these results showed that the sugar moiety of 1 interacts extensively with the protein and suggested to expand the SAR by glycodiversification of the aglycon of 1 (2) and of its synthetic precursor 3. Analysis of the interaction of compounds 4-16 with Hsp90 revealed that most of them act as stimulators, rather than inhibitors, of the ATPase activity, a feature that was totally unprecedented, until very recently[27] activators were found by large scale screening. We would like to underline that the case we present here is the first one where these results are obtained using a rational design approach. The validity of our protocol was further corroborated by the development of second generation activators: additional modeling reinforced the starting pharmacophoric hypothesis and suggested a second set of modifications, finally leading to 19, which accelerates Hsp90 ATPase by a factor of 6, similar to the most active known endogenous activator, the co-chaperone Aha1.
Using FRET, we showed that ATPase activation by the benzofuran probes is connected to acceleration of Hsp90 conformational dynamics. We characterized the activity of the most potent probes in the presence of the endogenous activator Aha1, which is known to bind the N-M Domains of Hsp90 and thus in principle should not compete with the ligands, and of the model client protein Δ131Δ, which binds in the proximity of the proposed allosteric site. Importantly, the allosteric ligands showed a synergistic effect with Aha1 and a competition with Δ131Δ in regulating Hsp90 ATPase. Consistent with the synergistic effect with Aha1, co-immunoprecipitation experiments also showed that association of the Hsp90 ATPase inhibitory co-chaperone Sba1 was reduced in the presence of benzofuran stimulators. Finally, the stimulatory activity observed on the isolated protein was found to translate to cytotoxicity in specific cell lines, which are known to be sensitive to Hsp90 deregulation. The molecules tested were shown to affect the viability of cancer cells, in particular those resistant to the Hsp90-targeted drug 17AAG, providing new opportunities to generate anticancer interventions based on novel mechanisms of action. Remarkably, the benzofurans described here were shown not to activate the heat shock response in treated cells, a known drawback of Hsp90 NTD inhibitors.
Whether activation of the chaperone is the only mechanism operating in cellular studies, it still needs to be fully demonstrated. Nonetheless, these designed allosteric activators may represent innovative gain-of-function probes to directly address the roles of Hsp90 ATPase and conformational dynamics in determining its cell functions. Moreover, our approach may ultimately generate effective anticancer drugs with novel mechanisms of action, based on the perturbation of the Hsp90 machinery, whereby the acceleration of conformational dynamics can eventually translate into impaired chaperone functions and cell death.
Experimental
Design and Synthesis
Docking Calculations and MD simulations
Docking was carried out with the Maestro Suite (Release 2013-1-9.4, Schrödinger, LLC, New York, NY, 2013). The shape and chemical properties of the CTD binding site were mapped onto a grid with dimensions of 36 Å (enclosing box) and 14 Å (ligand diameter midpoint box). Docking calculations were performed using Glide[38] (version 5.8) and carried out in XP-mode with the OPLS-AA force field[39]. The resulting consensus poses were used for molecular dynamics simulation.
The structures of the 3 most populated clusters for complexes with 1, 4, 10, 12, 18, 19 were used to start 100 ns explicit solvent MD. 3 simulations were run for each complex (total of 1800ns). Control simulations were run with Hsp90 in complex with ATP only. All calcualtions were carried out with GROMACS[40]. Details in Supplementary Information.
Synthesis of allosteric ligands
Synthesis of compounds 1-16 was described in[25]. For 17-19, aglycon 3 was reacted with different alkylating agents under phase transfer catalysis conditions analogous to those previously reported[25]. Details of the synthesis and full compound characterization are collected the in Supplementary Information.
Biochemical and Biophysical Assays
Hsp90 ATPase assay
ATPase activity of Hsp90 was measured by the NADH-coupled ATPase assay. Briefly, Hsc82 (2 μM) or human Hsp90α (2 μM) was pre-mixed with NADH (0.18 μM), L-lactate dehydrogenase (50 U/ml), PEP (1 mM), pyruvate kinase (50 U/ml) and compounds (dissolved in DMSO to a final concentration of 50 μM; 5 μM for Radicicol). The reaction was initiated by the addition of ATP (1 mM). The reaction was carried out at 30°C in buffer composed of Hepes (20 mM, pH 7.5), KCl (100 mM) and MgCl2 (1 mM). Absorbance data were collected using a microplate spectrophotometer (SpectraM5, Molecular Devices) at 360 nm.
FRET-based Hsp90 conformational change assay
The conformational change rate of Hsp90 was measured by a FRET-based assay[41]. Briefly, D61C or E329C mutation was introduced into Hsc82 for labeling with Alex Fluor 647 or Alex Fluor 555 (Life Technologies) respectively. Labeled Hsp90 populations were mixed to produce Hsp90 heterodimers. The conformational change of Hsp90 was initiated by adding AMP-PNP (1 mM) in the presence of compounds. The fluorescence from different dyes was monitored on a microplate spectrophotometer with excitation/emission wavelength as follows: Ex525/Em568 (AF555), Ex525/Em668 (AF647). The assay was carried out at room temperature in the same buffer as Hsp90 ATPase assay.
Cytotoxicity and effects on Hsp90 Interactions of designed compounds
Cell lines
Human breast cancer (MCF-7) and castration-resistant prostate carcinoma (DU145) cell lines were obtained from the American Type Culture Collection (Rockville, MD, USA). The human diffuse malignant peritoneal mesothelioma cell line (STO) was established from a tumor specimen of a patient who underwent surgery at the Istituto Nazionale dei Tumori, Milan[42]. The resistant sub-line STO-17AAG was derived by continuous exposure of the original parental cell line (STO) to increasing concentrations of 17-AAG.
Cell Proliferation Assay
After harvesting in the logarithmic growth phase, 4500 cells/50 μL were plated in 96-well flat bottomed microtiter plates for 24 h and treated with increasing concentrations of CTD ligands (1-100 μM) or 17-AAG (0.05-50 μM) for 72 h. Control cells received vehicle alone (DMSO). At the end of drug exposure, cell growth inhibition was determined with the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS; Promega). Optical density was read at 490 nm on a microplate reader (POLARstar OPTIMA) and the results were expressed as a percentage relative to DMSO-treated cells. Dose-response curves were created and IC50 values determined graphically from the curve for each compound.
Analysis of Hsp90 client proteins
To monitor changes in Hsp90 client proteins, cells were harvested, solubilized in lysis buffer (0.01% NP40, 10 mM Tris [pH 7.5], 50 mM KCl, 5 mM MgCl2, 2 mM DTT, 20% glycerol plus protease inhibitors) and analyzed by Western blotting primary antibodies specific for survivin (AbCam), Hsp90, CDK4, Neu (Santa Cruz Biotechnology), and Akt (Cell Signaling Technology). Briefly, total cellular lysates were separated on a 4-12% NuPAGE bis-tris gel (Life Technologies) and transferred to nitrocellulose using standard protocols. The filters were blocked in PBS 1X-Tween-20 with 5% skim milk or 5% BSA and incubated overnight with primary antibodies. The filters were then incubated with the secondary peroxidase-linked whole antibodies (Life Technologies). Bound antibodies were detected using the Novex ECL, HRP Chemiluminescent substrate Reagent Kit (Life Technologies). Filters were autoradiographed and images were acquired by Biospectrum Imaging System (Ultra-Violet Products Ltd). β-actin (AbCam) was used on each blot to ensure equal loading of proteins.
Hsp90 co-chaperone interaction analysis (Co-IP analysis)
Yeast expressing His-tagged Hsp82 (yeast Hsp90) as their sole Hsp90 protein (yeast strain pp30 [hsc82hsp82]) were lysed as previously described[43]. Protein lysates were incubated either with buffer (-) or with 5 mM AMP-PNP as indicated, as well as with 50 μM allosteric compounds, for 10 minutes at 30° prior to affinity precipitation of His-tagged Hsp82 with Ni-NTA agarose. Hsp82-associated Sba1 was detected by immunoblotting.
Supplementary Material
Acknowledgments
We acknowledge funding from Fondazione Cariplo through grant 2011.1800, Premio fondazione cariplo per la ricerca di frontiera”. GC acknowledges funding from AIRC (Associazione Italiana Ricerca sul Cancro) grant IG 15420; Universita’ degli Studi di Milano is aknowledged for a grant to SS (Assegno di ricerca tipo A).
Footnotes
This material is available via the Internet
Competing financial interests
The authors declare no competing financial interest.
References
- 1.Nussinov R, Tsai CJ. Cell. 2013;153:293–305. doi: 10.1016/j.cell.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 2.a) Hilser VJ. Science. 2010;327:653–654. doi: 10.1126/science.1186121. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Smock RG, Gierasch LM. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zorn JA, Wells JA. Nat Chem Biol. 2010;6:179–188. doi: 10.1038/nchembio.318. [DOI] [PubMed] [Google Scholar]
- 4.a) Fang Z, Grütter C, Rauh D. ACS Chem Biol. 2013;8:58–70. doi: 10.1021/cb300663j. [DOI] [PubMed] [Google Scholar]; b) Wenthur CJ, Gentry PR, Mathews TP, Lindsley CW. Annual Review of Pharmacology and Toxicology. 2014;54:165–184. doi: 10.1146/annurev-pharmtox-010611-134525. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rodina A, Patel PD, Kang Y, Patel Y, Baaklini I, Wong MJ, Taldone T, Yan P, Yang C, Maharaj R, Gozman A, Patel MR, Patel HJ, Chirico W, Erdjument-Bromage H, Talele TT, Young JC, Chiosis G. Chem Biol. 2013;20:1469–1480. doi: 10.1016/j.chembiol.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taipale M, Jarosz DF, Lindquist S. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 6.Jackson SE. Top Curr Chem. 2013;328:155.240. doi: 10.1007/128_2012_356. [DOI] [PubMed] [Google Scholar]
- 7.Whitesell L, Lindquist SL. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 8.Shah V, Wiest R, Garcia-Cardena G, Cadelina G, Groszmann RJ, Sessa WC. Am J Physiol. 1999;277:G463–468. doi: 10.1152/ajpgi.1999.277.2.G463. [DOI] [PubMed] [Google Scholar]
- 9.Luo W, Sun W, Taldone T, Rodina A, Chiosis G. Mol Neurodegener. 2010;5:24. doi: 10.1186/1750-1326-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jarosz DF, S L. Science. 2010;330:1820–1824. doi: 10.1126/science.1195487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, S L. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Echeverria PC, Bernthaler A, Dupuis P, Mayer B, Picard D. Plos One. 2012;6:e26044. doi: 10.1371/journal.pone.0026044. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. Cell. 2014;157:1685–1697. doi: 10.1016/j.cell.2014.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krukenberg KA, Street TO, Lavery LA, Agard DA. Q Rev Biophys. 2011;44:229–255. doi: 10.1017/S0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Ali MMU, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shiau AK, Harris SF, Southworth DR, Agard DA. Cell. 2006;127:329–340. doi: 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]; c) Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lavery LA, Partridge JR, Ramelot TA, Elnatan D, Kennedy MA, Agard DA. Mol Cell. 2014;53:330–343. doi: 10.1016/j.molcel.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Zuehlke A, Johnson JL. BIOPOLYMERS. 2010;93:211–217. doi: 10.1002/bip.21292. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li J, Buchner J. Biomed J. 2013;36:106–117. doi: 10.4103/2319-4170.113230. [DOI] [PubMed] [Google Scholar]
- 16.Morra G, Neves MAC, Plescia CJ, Tsutsumi S, Neckers L, Verkhivker G, Altieri DC, Colombo G. J Chem Theory and Computation. 2010;6:2978–2989. doi: 10.1021/ct100334n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taddei M, Ferrini S, Giannotti L, Corsi M, Manetti F, Giannini G, Vesci L, Milazzo FM, Alloatti D, Guglielmi MB, Castorina M, Cervoni M, Barbarino M, Foderà R, Carollo V, Pisano C, Armaroli S, Cabri W. J Med Chem. 2014;57:2258–2274. doi: 10.1021/jm401536b. [DOI] [PubMed] [Google Scholar]
- 18.Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 19.a) Workman P, Burrows FJ, Neckers L, Rosen N. Ann NY Acad Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]; b) Whitesell L, Bagatell R, Falsey R. Curr Cancer Drug Targets. 2003;3:349–358. doi: 10.2174/1568009033481787. [DOI] [PubMed] [Google Scholar]
- 20.a) Morra G, Potestio R, Micheletti C, Colombo G. Plos Comput Biol. 2012;8:e1002433. doi: 10.1371/journal.pcbi.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Morra G, Verkhivker GM, Colombo G. PLOS Comp Biol. 2009;5:e1000323. doi: 10.1371/journal.pcbi.1000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Moroni E, Zhao H, Blagg BS, Colombo G. J Chem Inf Model. 2014 doi: 10.1021/ci4005767. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao H, Moroni E, Colombo G, Blagg BS. ACS Medicinal Chemistry Letters. 2014;5:84–88. doi: 10.1021/ml400404s. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhao H, Moroni E, Yan B, Colombo G, Blagg BSJ. ACS Medicinal Chemistry Letters. 2013;4:57–62. doi: 10.1021/ml300275g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Street TO, Lavery LA, Agard DA. Mol Cell. 2011;42:96–105. doi: 10.1016/j.molcel.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Genest O, Reidy M, Street TO, Hoskins JR, Camberg JL, Agard DA, Masison DC, Wickner S. Mol Cell. 2013;49:464–473. doi: 10.1016/j.molcel.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morra G, Colombo G. J Chem Theory and Computation. 2010;6:2978–2989. doi: 10.1021/ct100334n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goff RD, Thorson JS. Med Chem Commun. 2014;5:1036–1047. doi: 10.1039/C4MD00117F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morelli L, Bernardi A, Sattin S. Carbohydr Res. 2014;390C:33–41. doi: 10.1016/j.carres.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Panaretou B, Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. EMBO J. 1998;17:4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zierer BK, Weiwad M, Rubbelke M, Freiburger L, Fischer G, Lorenz OR, Sattler M, Richter K, Buchner J. Angewandte Chemie-International Edition. 2014;53:1–7. doi: 10.1002/anie.201406578. [DOI] [PubMed] [Google Scholar]
- 28.Kranjc A, Bongarzone S, Rossetti G, Biarnes X, Cavalli A, Bolognesi ML, Roberti M, Legname G, Carloni P. J Chem Theor Comput. 2009;9:2565–2573. doi: 10.1021/ct900257t. [DOI] [PubMed] [Google Scholar]
- 29.a) Mickler M, Hessling M, Ratzke C, Buchner J, Hugel T. Nat Struct Mol Biol. 2009;16:281–286. doi: 10.1038/nsmb.1557. [DOI] [PubMed] [Google Scholar]; b) Southworth DR, Agard DA. Mol Cell. 2008;32:631–640. doi: 10.1016/j.molcel.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pullen L, Bolon DN. J Biol Chem. 2011;286:11091–11098. doi: 10.1074/jbc.M111.223131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Richter K, Reinstein J, Buchner J. Nature structural & molecular biology. 2013;20:326–331. doi: 10.1038/nsmb.2502. [DOI] [PubMed] [Google Scholar]
- 32.McLaughlin SH, Smith HW, Jackson SE. J Mol Biol. 2002;315:787–798. doi: 10.1006/jmbi.2001.5245. [DOI] [PubMed] [Google Scholar]
- 33.Zhao HP, Donnelly AC, Kusuma BR, Brandt GEL, Brown D, Rajewski RA, Vielhauer G, Holzbeierlein J, Cohen MS, Blagg BSJ. J Med Chem. 2011;54:3839–3853. doi: 10.1021/jm200148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fierro-Monti I, Echeverria P, Racle J, Hernandez C, Picard D, Quadroni M. PLoS One. 2013;8:e80425. doi: 10.1371/journal.pone.0080425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Partridge JR, Lavery LA, Elnatan D, Naber N, Cooke R, Agard DA. eLife. 2014 doi: 10.7554/eLife.03487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Retzlaff M, Hagn F, Mitschke L, Hessling M, Gugel F, Kessler H, Richter K, Buchner J. Mol Cell. 2010;37:344–354. doi: 10.1016/j.molcel.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 37.Beebe K, Mollapour M, Scroggins B, Prodromou C, Xu W, Tokita M, Taldone T, Pullen L, Zierer BK, Lee MJ, Trepel J, Buchner J, Bolon DN, Chiosis G, Neckers L. Oncotarget. 2013;4:1065–1074. doi: 10.18632/oncotarget.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Journal of Medicinal Chemistry. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 39.Jorgensen WL, Maxwell DS, Tirado-Rives J. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 40.Hess B, Kutzner C, van der Spoel D, Lindahl E. J Chem Theory and Computation. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 41.Hessling M, Richter K, Buchner J. Nat Struct Mol Biol. 2009;16:287–293. doi: 10.1038/nsmb.1565. [DOI] [PubMed] [Google Scholar]
- 42.Zaffaroni N, Costa A, Pennati M, De Marco C, Affini E, Madeo M, Erdas R, Cabras A, Kusamura S, Baratti D, Deraco M, Daidone MG. Cell Oncol. 2007;29:453–466. doi: 10.1155/2007/456839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mollapour M, Bourboulia D, Beebe K, Woodford MR, Polier S, Hoang A, Chelluri R, Li Y, Guo A, Lee MJ, Fotooh-abadi E, Khan S, Prince T, Miyajima N, Yoshida S, Tusutsumi S, Xu WP, Panaretou B, Stetler-Stevenson WG, Bratslavsky G, Trepel JB, Prodromou C, Neckers L. Mol Cell. 2014;53:317–329. doi: 10.1016/j.molcel.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.