

Abstract
Graminaceous plant species acquire soil iron by the release of phytosiderophores and subsequent uptake of iron(III)-phytosiderophore complexes. As plant species differ in their ability for phytosiderophore hydroxylation prior to release, an electrophoretic method was set up to determine whether hydroxylation affects the net charge of iron(III)-phytosiderophore complexes, and thus chelate stability. At pH 7.0, non-hydroxylated (deoxymugineic acid) and hydroxylated (mugineic acid; epi-hydroxymugineic acid) phytosiderophores form single negatively charged iron(III) complexes, in contrast to iron(III)-nicotianamine. As the degree of phytosiderophore hydroxylation increases, the corresponding iron(III) complex was found to be less readily protonated. Measured pKa values of the amino groups and calculated free iron(III) concentrations in presence of a 10-fold chelator excess were also found to decrease with increasing degree of hydroxylation, confirming that phytosiderophore hydroxylation protects against acid-induced protonation of the iron(III)-phytosiderophore complex. These effects are almost certainly associated with intramolecular hydrogen bonding between the hydroxyl and amino functions. We conclude that introduction of hydroxyl groups into the phytosiderophore skeleton increases iron(III)-chelate stability in acid environments such as those found in the rhizosphere or the root apoplasm and may contribute to an enhanced iron acquisition.
Graminaceous plant species are among those many important crop species that differ widely in their response to iron-deficiency stress. Understanding these stress responses is important for increasing crop yields on calcareous soils as well as improving the iron content of grains for human consumption. Graminaceous plants respond to iron deficiency by the exudation of phytosiderophores to increase the availability of iron for uptake and by increasing the uptake capacity for iron(III)-phytosiderophores (Takagi, 1976; Römheld and Marschner, 1986). Phytosiderophores are hexadentate ligands that coordinate iron(III) with their amino and carboxyl groups (Mino et al., 1983). When released to the rhizosphere, phytosiderophores chelate sparingly soluble soil iron by forming iron(III)-phytosiderophore complexes, which can be subsequently transported across the root plasma membrane via facilitated transport (Römheld and Marschner, 1986; Ma et al., 1993). In general, plant species releasing high quantities of phytosiderophores such as barley, rye, and wheat are more resistant to iron deficiency chlorosis than species releasing smaller quantities such as maize, sorghum, and rice (Marschner et al., 1986; Kawai et al., 1988; Römheld and Marschner, 1990; von Wirén et al., 1995). However, this correlation with the quantity of phytosiderophores released is not always consistent; chlorosis resistance in different maize cultivars is reported, but may not be related to the total amounts of phytosiderophores secreted, indicating the existence of other factors controlling iron efficiency (Lytle and Jolley, 1991; von Wirén et al., 1994).
In principle, phytosiderophore structure may influence iron acquisition in three nonexclusive ways: they may differ in (a) susceptibility to microbial degradation, (b) affinity for the transport process across the root cell plasma membrane, or (c) iron(III) chelating properties. Little information exists on the influence of phytosiderophore structure on the resistance to iron deficiency chlorosis and what exists can be contradictory. For example, many chlorosis resistant plants, such as barley and rye, synthesize mostly hydroxylated phytosiderophores, whereas non-hydroxylated deoxymugineic acid (DMA) predominates in the susceptible species such as maize and rice (Kawai et al., 1988; Römheld and Marschner, 1990; Mori et al., 1991). Wheat is rather chlorosis-resistant, however, and only releases DMA (Römheld and Marschner, 1990). Microbial degradation rates of phytosiderophores may not be a major factor determining iron efficiency. Sorghum root exudates contain hydroxymugineic acid (HMA) and DMA, both of which are degraded at similar rates after inoculation of an axenic hydroponic sorghum culture with rhizosphere microorganisms (von Wirén, 1994). Furthermore, short-term uptake of 59Fe-labeled chelates of different phytosiderophore species into roots did not significantly differ even with different plant species, unless the typical phytosiderophore backbone was altered (Römheld and Marschner, 1990; Ma et al., 1993; Klair et al., 1996).
In this study we have investigated iron(III) chelating properties as a possible factor causing discrimination among differentially hydroxylated phytosiderophores. Hydroxylation may affect such properties as affinity constants and the effect of pH on iron(III) chelating capacity. We therefore determined affinity constants (KFe(III)) and pKa values for a range of phytosiderophores differing in their degree of hydroxylation. This permitted calculation of the free Fe3+ (pFe3+) activities and the direct estimation of the effect of hydroxylation on the iron(III) chelating capacity of phytosiderophores at different pH values. Phytosiderophore hydroxylation may also modify the influence of pH on the net charge of iron(III)-phytosiderophore complexes and this charge may be an important factor in influencing chelate mobility in soils (Inoue et al., 1993), as well as the interaction of the iron phytosiderophore with the putative receptor or transport proteins at the root plasma membrane (von Wirén et al., 1998). It is surprising that the charge of iron(III) phytosiderophores has not been determined directly, although it is assumed to be −1 by analogy with the structure of cobalt(III) mugineic acid (MA; Sugiura et al., 1981). In this study we report the direct measurement of the charge on the iron phytosiderophore complex.
During these investigations we observed that the net charge of iron(III) phytosiderophores is strongly pH dependent and that such changes can be used to investigate the influence of phytosiderophore backbone hydroxylation on chelate stability. We therefore set up and calibrated an electrophoretic method for the direct determination of the net charge of iron(III) complexes of the MA family. The information on iron(III) chelate charge together with the determination of iron(III) phytosiderophore stability constants was used to predict iron(III) chelation by differently hydroxylated phytosiderophores under varying physiological conditions.
RESULTS
Iron(III)- and Zinc(II)-Phytosiderophores Are Negatively Charged at pH 7.0
To determine the structural features creating charge differences in iron(III) complexes, 59Fe-labeled iron(III) complexes of the MA-family phytosiderophores (MA, epi-hydroxymugineic acid [eHMA], and phytosiderophore II [PSII]) and of nicotianamine (NA) were electrophoresed and the charges of the complexes were determined. At pH 7.0 electrophoretic running times of the phytosiderophore complexes corresponded to net charges between −1.1 and −1.2 (Table I). Such values are in agreement with the suggestion of Murakami et al. (1989) that the α-hydroxyl group present on phytosiderophores (Fig. 1) completely ionizes upon complex formation with iron(III) (Fig. 2). Moreover, this model is supported by the observation that the iron(III)-NA complex, which lacks an α-hydroxyl group, carries a net charge of zero (Table I). Despite zinc(II) possessing one charge less than iron(III), the zinc phytosiderophore complexes possess a net negative charge similar to that of the corresponding iron(III) complex. This is best explained by the non-dissociation of the α-hydroxyl group in the presence of zinc(II) (Fig. 2), which in turn results from the much lower charge density of the zinc(II) cation (Hider and Hall, 1991).
Table I.
Net charges of Zn(II)- and Fe(III)-phytosiderophores at pH 7.0 as calculated from migration distances in high-voltage paper electrophoresis
| Ligand | Zn(II) | Fe(III) |
|---|---|---|
| MA | −0.79 | −1.10 |
| eHMA | −0.84 | −1.18 |
| PSII | −0.75 | −1.15 |
| NA | −0.78 | 0 |
Net charges were calibrated from migration distances of known Zn(II)- and Fe(III)-chelates as described in “Materials and Methods” and in Figure 1.
Figure 1.

Structures of amino carboxylate ligands used in the study.
Figure 2.

Computer-generated models of the iron(III) and zinc(II) complexes of MA. The coordinates are based on the cobalt(III) complex of MA (Sugiura et al., 1981) and the ionization states are as reported in Table I. There is a striking similarity between the two complexes, including their net charges of −1. The only major difference is the presence of the protonated hydroxyl group.
Low pH Induces a Transition from Negatively Charged to Neutral Iron(III) Phytosiderophore Complexes
The iron(III) complexes of the phytosiderophores DMA, MA, eHMA, PSI, and PSII, together with iron(III) EDTA were subjected to electrophoresis over the pH range 4.0 to 7.0 (Fig. 3). For this analysis PSI and PSII were included to investigate the influence of the immediate structure of the amino carboxylate region on the protonation behavior of the iron(III) complexes. The iron(III) complex of EDTA remained unaffected by the increase of acidity in the medium. The iron(III) complexes of eHMA and MA adopted the predicted negative charge over the pH range 5.0 to 7.0. However, at more acidic values another less negative complex was found to form (Fig. 3), most likely as a result of protonation. In contrast, the negatively charged iron(III) complex of DMA, a non-hydroxylated phytosiderophore, only exists at pH 7.0. At pH 6.0 and below, a neutral complex predominates. In a similar fashion to DMA, a negatively charged iron(III) complex of PSI was only observed at pH values 6.0 and 7.0 and even at pH 7.0 this was not the major species. A neutral species was found to dominate over the entire pH range investigated (4.0–7.0). In contrast to PSI, PSII, which differs from PSI by the introduction of an isobutyl group on the terminal amino carboxylate region, forms a more acid-stable, negatively charged iron(III) complex over the pH range 5.0 to 7.0. With PSII, it is only below pH 5.0 that appreciable conversion to the neutral complex is observed (Fig. 3).
Figure 3.

Net charge of iron(III) complexes of EDTA, eHMA, MA, DMA, PSI, and PSII at different solution pH values as determined by high-voltage electrophoresis.
In principle, the acid-induced transition from the negatively charged to neutral species could involve simple protonation of the α-hydroxy group. However, if that were the case, it would not be possible to separate the protonated and unprotonated species by electrophoresis due to their extremely rapid interconversion (108 s−1). Thus protonation must involve dissociation of either the aminocarboxylate region or the hydroxycarboxylate region of the molecule; either conversion would account for the change in the net charge of the complex. The marked difference between the behavior of PSI and PSII provides strong indication that it is the amino carboxylate region that dissociates first, the hydroxycarboxylate region being identical in both molecules (Fig. 1). This acid-induced dissociation appears to be a general characteristic of the phytosiderophore class of molecule.
Iron(III)-Phytosiderophore Affinity Constants Increase with Degree of Hydroxylation
The pKa values and iron(III) affinity constants for the phytosiderophore analog PSII and MA were determined by simultaneous conductiometric and spectrophotometric titration (Table II). These values are compared with those previously reported for DMA and eHMA (Murakami et al., 1989). The pKa values were assigned as indicated in Figure 4. The values of pKa1 and pKa2 are quite low and therefore have not been widely reported for phytosiderophores. By analogy with N-alkyl amino acids, the lowest value, pKa1 has been assigned to the carboxylate group adjacent to the tertiary amine. pKa2 and pKa3 have been assigned to the secondary amino acid and hydroxy acid moities, respectively. The two most basic pKa values correspond to the two amino groups, the more basic, pKa5, being associated with the tertiary amino group (Fig. 4). The homologous pKa values were found to fall in tight ranges and the values determined in this study (PSII and MA) were in good general agreement with those determined by Murakami et al. (1989) for DMA and eHMA. However, some clear trends were observed, for instance the values of pKa4 decreased with the increasing number of hydroxyl groups present on a phytosiderophore; the sequence being 8.25, 7.78, 7.10 for DMA, MA, and eHMA. It is clear that the presence of hydroxyl groups in the linking carbon chains in MA and eHMA decrease the pKa values of the adjacent amino functions (pKa4 and pKa5). This is due to the combined influence of intramolecular hydrogen bonding (Fig. 4B) and an inductive effect (withdrawal of electrons due to the presence of the electronegative oxygen atom) due to the presence of β-hydroxyl groups (Fig. 4C). This trend of pKa values was found to be somewhat less marked with KFe(III) values. In contrast, the pFe3+ values (for definition, see “Materials and Methods”) show a clear trend with an increasing degree of hydroxylation. Thus, for the series DMA, MA, and epiHMA, the values at pH 7.0 are 15.01, 15.19, and 15.60, respectively, and at pH 5.0 the series follows the sequence 12.24, 12.66, 13.04 (Table II). The pFe3+ values provide useful indications of the ability of a phytosiderophore to bind iron(III) under conditions likely to be found in the soil microenvironment (Fig. 5). The tighter the binding of iron(III) to the phytosiderophore, the higher the pFe3+ value. Thus, at low concentrations of iron, over the pH range 4 to 7 the ability to bind iron(III) decreases in the series HMA > MA > DMA.
Table II.
The pKa, log KFe(III), and pFe3+ values of phytosiderophores as determined by potentiometric and spectrophotometric titration
| Phytosiderophore | pKa Values
|
log KFeIII | pFeIII
|
|||||
|---|---|---|---|---|---|---|---|---|
| pKa1 | pKa2 | pKa3 | pKa4 | pKa5 | pH 5.5 | pH 7.0 | ||
| PSII | 2.30 | 2.72 | 3.47 | 7.54 | 9.56 | 18.5 | 13.46 | 16.15 |
| MA | 2.39 | 2.76 | 3.40 | 7.78 | 9.55 | 17.7 | 12.66 | 15.19 |
| DMAa | 2.35b | 2.74b | 3.20 | 8.25 | 10.00 | 18.4 | 12.24 | 15.01 |
| eHMAa | 2.35b | 2.74b | 3.25 | 7.10 | 9.62 | 17.7 | 13.04 | 15.60 |
Data from Murakami et al., 1989.
Estimated from analogous studies with MA and PSII.
Figure 4.

A, General structure of phytosiderophores and assignment of pKa values; B, intramolecular bonding between a hydroxyl substitute and amino function on a phytosiderophore backbone; C, inductive effect of additional hydroxylation on the main chain and azetidine ring.
Figure 5.

Computer simulation of the pH dependence of pFe3+ values of iron(III) complexes of PSII, eHMA, MA, DMA, and the hydroxide anion. In all cases the total iron(III) concentration is 1 μm OH− and the total chelator concentration is 10 μm. − ·· − ·· −, PSII; − · − · −, eHMA; ······, MA; − − − − −, DMA; ——-, OH−. pFe3+ values in the absence of any ligand, iron(III) binds tightly to the hydroxyl anion.
There is one major difference between the structures of PSII and DMA, which is centered at the azetidine ring (pKa5), where the strained tertiary amine of DMA is replaced by a secondary amine, substituted with a hydrophobic alkyl side chain. This substitution results in a decrease in the basicity of the pKa4 and pKa5 values in the synthetic phytosiderophore when compared with the corresponding groups on the non-hydroxylated phytosiderophore DMA. Although the KFe(III) values are similar for the two molecules, the differences in the pKa4 and pKa5 values induce an appreciable difference in the pFe3+ values, the pFe3+ for PSII being larger (Table II; Fig. 5). The trends in pFe3+ values for the entire group agree well with the trends of the observed acid stability (Fig. 3).
DISCUSSION
The introduction of hydroxyl groups on the phytosiderophore skeleton leads to an enhanced affinity for iron(III) over the pH range 5.0 to 8.0 when compared with the corresponding unsubstituted phytosiderophore. This is due to the formation of intramolecular hydrogen bonding between the functional hydroxyl groups and the two chelating amino groups. This finding has been demonstrated by two independent methodological approaches. As a direct approach, equilibrium constants of protonated ligands (pKa) and affinity constants (logKFe(III)) of iron(III) phytosiderophores were measured and used to calculate free iron(III) concentrations (pFe3+) at different pH values. Then an electrophoretic method was developed to determine the net charge of iron(III) complexes to investigate: (a) the influence of the α-hydroxy groups on the net charge of iron(III) and zinc(II) phytosiderophore and NA complexes, (b) the influence of the aminocarboxylate group on the net charge of iron(III) phytosiderophore complexes using the chemically synthesized phytosiderophores (PSI and PSII), and (c) the influence of phytosiderophore backbone hydroxylation on iron(III) complex stability with decreasing pH.
Substitution of the α-hydroxy group [iron(III)-PS] with an amino group (NA) results in the loss of the single negative charge of iron(III)-phytosiderophore complexes at pH 7. This demonstrates that in iron(III)-phytosiderophores, the α-hydroxy group is fully ionized (Table I), a finding that agrees with potentiometric studies on iron(III)-phytosiderophore complexes and structural data on cobalt(III)-phytosiderophore complexes (Sugiura et al., 1981; Murakami et al., 1989). A similar finding has been reported for a range of microbial siderophores (Thieken and Winkelmann, 1992; Haag et al., 1994; Carrano et al., 1996). In contrast, with zinc(II)-phytosiderophore complexes, the α-hydroxy group remains protonated (Table I), which leads to identical net charges and presumably conformations similar to those of the corresponding iron(III)-phytosiderophore complexes (Fig. 2). The close structural analogy of the zinc(II) and iron(III) complexes renders it likely that both complexes are recognized by the same binding site on a membrane transport protein. Such a mechanism provides a ready explanation for the observation that phytosiderophores facilitate the absorption of both zinc and iron into the roots of graminaceous plant species (Ma et al., 1993; von Wirén et al., 1996).
In general, acidic pH values cause partial dissociation of multidentate iron(III) complexes. However, complexes that contain hydroxy-carboxylate groups, such as citrate (Martin, 1986) and N-hydroxyethylethylenediaminetriacetic acid (HEDTA; Gustafson and Martell, 1963), are quite stable between pH 4.0 and pH 8.0. This means that the phytosiderophore amino terminus likely dissociates first when exposed to increasingly acidic solutions. The direct monitoring of pH induced changes on the net charge of iron(III)-phytosiderophore complexes demonstrated that iron(III)-PSII is more resistant to protonation than iron(III)-PSI (Fig. 3). This difference results from the structural difference of the amino terminae (Fig. 1). The presence of the N-isobutyl group was found to increase the affinity of PSII for iron(III) when compared to DMA (Fig. 5), and also to enhance the acid stability of the iron(III) complex (Fig. 3). In a similar manner, hydroxylation of the phytosiderophore skeleton either in the 2′-hydroxy group as in MA, or additionally in the 3-hydroxy group as in eHMA, results in intramolecular hydrogen bonding. This reduces competition of the chelating amino groups for hydrogen ions, thereby enhancing the stability of the iron(III) complexes particularly under mildly acid conditions (pH 5.0–6.5). Thus the HMA iron(III) complex is remarkably acid stable, whereas the iron(III) complexes of the non-hydroxylated phytosiderophore DMA forms the partially dissociated neutral iron(III) complex even at pH 7.0 (Fig. 3). A proton is involved in the equilibrium reaction between the negative complex [PS FeIII]− and the partially dissociated neutral complex [HPS FeIII]o (Fig. 6). Thus the equilibrium between these two complexes is pH dependent, and the equilibrium constant (Keq) between the two forms is likely to be largely influenced by the value of pKa5, the amino group that is protonated during the dissociation step. The greater tendency for the iron(III) complex of DMA to dissociate is related to its higher pKa5 value compared with those of PSII, MA, and eHMA (Table II).
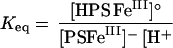 |
To verify the influence of hydroxylation on pH-dependent iron(III) chelation, direct quantification of the pKa values and affinity constants for iron(III) was undertaken (Table II). DMA, with no hydroxyl groups, possesses functional amino groups with pK values of 8.25 and 10.00; MA, with one hydroxyl group, has values of 7.78 and 9.55, and eHMA, with two hydroxyl groups, has values of 7.10 and 9.62. It is clear that the presence of hydroxyl groups is associated with a reduction in pKa value (the affinity for protons). Although the presence of hydroxyl groups may also reduce the affinity for iron(III), the influence on hydrogen ion interaction dominates (Hider et al., 1998). This differential property demonstrates that, at physiologically relevant pH values, the competition between protons and iron(III) cations favors iron(III) chelation in the presence of an intramolecular hydrogen bonding of one or more of the complexing ligands. Such findings have been well characterized with substituted catechols (Garrett et al., 1989) and pyridinones (Xu et al., 1995).
Figure 6.

Proposed proton-induced conformation change of the iron(III) complex of phytosiderophores. Upon protonation of the amino terminal end of the phytosiderophore, the protonated amine dissociates from the iron(III) cation, pulling with it the attached carboxylate group, converting the hexadentate coordinated structure to a tetradentate structure.
Thus the sequence of pFe3+ values for the natural phytosiderophores reflects that of the combined pKa values (pKa4 + pKa5) of the two phytosiderophore amino groups, namely 18.25, 17.33, and 16.72, which in turn reflects the number of substituent hydroxyl groups 0, 1, and 2 for DMA, MA, and eHMA, respectively. eHMA binds iron(III) over 15 times more tightly than DMA at pH 6.0, a factor that will be of physiological significance not only in view of the competition with iron-chelating compounds from other plant species or with microbial siderophores in the rhizosphere, but also with respect to competing protons. It is clear that hydroxylation leads to higher iron(III) complex stability. Therefore, graminaceous species producing hydroxylated PS species, such as barley and rye, are expected to have a competitive advantage when their PS-iron(III) complexes move through slightly acid environments, such as at the plasma membrane surface where the pH is locally acidified due to the activity of the H+-ATPase (Thibaud et al., 1988). Whether the increased resistance of barley and rye to iron-induced chlorosis is influenced by phytosiderophore hydroxylation (Marschner et al., 1986) remains to be determined.
MATERIALS AND METHODS
Chemicals
MA and NA were synthesized as previously described and were at least 95% pure as judged by 1H-nuclear magnetic resonance (Shioiri et al., 1995). eHMA was kindly supplied by Dr. T. Shioiri (Nagoya City University, Japan). The phytosiderophore analogs PSI and PSII were synthesized as previously described by Klair et al. (1996). EDTA, diethylenetriamine-pentaacetic acid (DTPA), and HEDTA were purchased from Sigma (Poole, Dorset, UK). 1, 2-Dimethyl-3-hydroxypyridin-4-one (CP20), which forms a neutral complex with iron(III), was synthesized as previously described by Dobbin et al. (1993). Other chemicals were from Aldrich. For the preparation of labeled chelates, aliquots of 10 mm Fe(NO3)3 containing 20 kBq of 59FeCl3 or 10 mm Zn(NO3)2 containing 20 kBq of 65ZnCl2 (Amersham International, Little Chalfont, UK), were added to 10 μL of 12 mm chelator solution and adjusted to pH 7.0 with 100 mm MOPS [3-(N-morpholino)propanesulfonic acid]-KOH.
High Voltage Electrophoresis
A sheet of filter paper (grade 1F, Munktell, Stockholm) was placed on a cooling plate covered by an acetate foil in an electrophoresis cuvette (Multiphor II; Pharmacia, Milton Keynes, UK). The filter paper was positioned so both ends were in buffer solution (0.1 m MOPS-KOH, pH 7.0) and the paper was prerun at 400 V for 20 min. The buffers for pH 6, 5, and 4 were pyridine (5% v/v) adjusted to the desired pH value by the addition of glacial acetic acid. Small pieces of filter paper (10 × 4 mm) were loaded with approximately 10 μL of 1 mm 59Fe-chelate or 65Zn-chelate solution (2 kBq) and were then placed on a start line in the middle of the paper sheet. Separation was achieved in the dark at a constant voltage of 400 V at 10°C. After 1 h, electrophoresis was stopped, the paper sheet was immediately dried with a hair-drier, and cut into 8 mm-wide strips parallel to the start line. The amounts of radioactivity on the paper strips and on the deposit paper were determined in a gamma-counter (Wallac 1280 Compugamma CS, Perkin Elmer, Cambridge, UK) by dry Cerenkov counting. For a comparison of migration distances, results were expressed in cpm per strip corresponding to 8-mm migration distance.
The net charge was calculated from the migration distances using the 59Fe(III) and 65Zn(II) complexes of EDTA, HEDTA, DTPA, and CP20 as standards. Standard curves for these complexes are presented for buffers at pH 7.0 and pH 5.0 (Fig. 7). Linear relationships were generated for migration distance versus complex charge/Mr. The following net charges were adopted: Fe(CP20)3, 0; FeEDTA, −1; FeDTPA, −2; ZnHEDTA, −1; ZnEDTA, −2; and ZnDTPA, −3 (Martell and Smith, 1989).
Figure 7.

Dependence of migration distances in high-voltage electrophoresis on the ratio of complex charge to Mr. Charges of well characterized zinc(II) and iron(III) complexes are reported in “Materials and Methods.”
pKa Determination
Equilibrium constants of protonated ligands were determined using an automated, computerized system capable of simultaneously analyzing spectrophotometric and potentiometric measurements. A blank titration of 0.1 m KCl was performed to determine the electrode zero using Gran's plot method (Gran, 1952). A combined pH electrode (Sirius Analytical Instruments, East Sussex, UK) was used to calibrate the electrode zero. The solution (0.1 m KCl, 25 mL), contained in a jacketed titration cell to maintain the temperature at 25°C ± 0.5°C, was under an argon atmosphere and was acidified with 0.15-mL increments of 0.2 m of HCl dispensed from a dosimat (665; Metrohm Ltd., Buckingham, UK). The titration was repeated in the presence of ligand. The data obtained from the titrations were analyzed by the TITRFIT program, a modified version of NONLIN (Taylor et al., 1988).
Determination of Iron(III) Affinity Constants
The affinity constant for the iron(III)-ligand interaction was determined by a spectrophotometric competition study of ligand-iron(III)-maltol using the automated system described above. The iron(III) complexes of the ligand [FeIII L]− were prepared in a 10:1 ligand:iron molar ratio (total iron concentration = 4.4 × 10−5 M) in 0.1 m MOPS-KOH buffer, pH 7.4. This solution was then titrated against maltol (3-hydroxy-2-methylpyran-4-one) resulting in the dissociation of iron(III)-phytosiderophore complex and the formation of the orange iron(III)-maltol complex. The resulting spectrophotometric data were inserted into the COMPT11 program to evaluate the affinity constants of the complex. pFe3+ plots were calculated from the pKa and KFe(III) values using the program SPECIAZ1; these programs require concentrations of metal and ligand, KFe(III) values of complexes, K values for iron(III)-OH interactions, and pKa values of the ligand. Between pH 1 and 9, the dominant FeIII-OH species are [FeIII(OH)]2+, [FeIII(OH)2]+, [FeIII(OH)3]o, and [FeIII(OH)4]−. pFe3+ values are more meaningful for purposes of ligand comparisons under physiological conditions because unlike KFe(III) values, account is taken of the competition produced by protons. KFe(III) values are normally only relevant at pH values >14, where competition from protons is diminishing small. By analogy with pH, pFe3+ is defined as pFe3+ = −log [Fe3+] where [Fe3+] is the molar concentration of hexaaquo iron(III). It is clear that the higher the affinity of a ligand for iron(III), the lower the value of [Fe3+] in solution, that is the larger the pFe3+ value. When making comparisons between ligands, the solution conditions must be defined and in this study we have adopted the values [Fe3+] Total = 10−6 M, and [phytosiderophore] Total = 10−5 M.
Footnotes
This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) of the United Kingdom and by a short-term fellowship to N.V.W. from the joint BBSRC/Institut National de la Recherche Agronomique collaboration scheme. IACR is grant-aided by BBSRC. Copies of the computer program mentioned in the paper are available from R.C.H.
LITERATURE CITED
- Carrano CJ, Drechsel H, Kaiser D, Jung G, Matzanke B, Winkelmann G, Rachel N, Albrecht-Gary AM. Coordination chemistry of the carboxylate type: siderophore rhizoferrin: the iron(III) complex and its metal analogs. Inorg Chem. 1996;35:6429–6436. doi: 10.1021/ic960526d. [DOI] [PubMed] [Google Scholar]
- Dobbin PS, Hider RC, Hall AD, Taylor PD, Sarpong P, Porter JB, Xiao G, van der Helm D. Synthesis, physiochemical properties, and biological evaluation of N-substituted 2-alkyl-3-hydroxy-4(1H)-pyridinones: orally active iron chelators with clinical potential. J Med Chem. 1993;36:2448–2458. doi: 10.1021/jm00069a002. [DOI] [PubMed] [Google Scholar]
- Garrett TM, Miller PW, Raymond KN. 2,3-Dihydroxyteraphthalamides: highly efficient iron(III) chelating agents. Inorg Chem. 1989;28:128–133. [Google Scholar]
- Gran G. Determination of equivalence point in popotentiometric titrations, part II. Analyst. 1952;77:661–671. [Google Scholar]
- Gustafson RL, Martell AE. Hydrolytic tendencies of ferric chelates. J Am Chem Soc. 1963;67:576–582. [Google Scholar]
- Haag H, Fiedler HP, Meiwes J, Drechsel H, Jung G, Zähner H. Isolation and biological characterization of staphyloferrin 223 β, a compound with siderophore activity from staphylococci. FEMS Microbiol Lett. 1994;115:125–130. doi: 10.1111/j.1574-6968.1994.tb06626.x. [DOI] [PubMed] [Google Scholar]
- Hider RC, Hall AD. Clinically useful chelators of tripositive elements. Prog Med Chem. 1991;28:41–173. doi: 10.1016/s0079-6468(08)70363-1. [DOI] [PubMed] [Google Scholar]
- Hider RC, Khodr H, Liu ZD, Tilbrook G. Optimisation of pFe3+ values of iron(III) ligands with clinical potential. In: Collery P, Brätter P, Negretti de Brätter V, Khassanova L, Etienne JC, editors. Metal Ions in Biology and Medicine. Ed 5. France: J Libbey, Eurotex; 1998. pp. 51–55. [Google Scholar]
- Inoue K, Hiradate S, Takagi S. Interaction of mugineic acid with synthetically produced iron oxides. Soil Sci Soc Am J. 1993;57:1254–1260. [Google Scholar]
- Kawai S, Takagi S, Sato Y. Mugineic acid-family phytosiderophores in root-secretions of barley, corn and sorghum varieties. J Plant Nutr. 1988;11:633–642. [Google Scholar]
- Klair S, Hider RC, Adams MZ, Leigh RA. Studies of iron transport in wheat using synthetic phytosiderophores. J Plant Nutr. 1996;19:1295–1307. [Google Scholar]
- Lytle CM, Jolley VD. Iron deficiency stress reponse of various C3 and C4 grain crop genotypes: strategy II mechanisms evaluated. J Plant Nutr. 1991;14:341–362. [Google Scholar]
- Ma JF, Kusano G, Kimura S, Nomoto K. Specific recognition of mugineic acid-ferric complex by barley roots. Phytochemistry. 1993;34:599–603. [Google Scholar]
- Marschner H, Römheld V, Kissel M. Different strategies in higher plants in mobilization and uptake of iron. J Plant Nutr. 1986;9:695–713. [Google Scholar]
- Martell AE, Smith RM. Critical Stability Constants. London: Plenum Press; 1989. [Google Scholar]
- Martin RB. Citrate binding of Al3+ and Fe3+ J Inorg Biochem. 1986;28:181–187. doi: 10.1016/0162-0134(86)80081-2. [DOI] [PubMed] [Google Scholar]
- Mino Y, Ishida T, Ota N, Inoue N, Nomoto K, Takemoto T, Tanaka H, Sugiura Y. Mugineic acid iron(III) complex and its structurally analogous cobalt(III) complex: characterization and implication for absorption and transport of iron in graminaceous plants. J Am Chem Soc. 1983;105:4671–4676. [Google Scholar]
- Mori S, Nishizawa N, Hayashi H, Chino M, Yoshimura E, Ishihara J. Why are young rice plants highly susceptible to iron deficiency? Plant Soil. 1991;130:143–156. [Google Scholar]
- Murakami T, Ise K, Hayakawa M, Kamei S, Takagi S. Chem Lett 2137–2140. 1989. Stabilities of metal complexes of mugineic acids and their specific affinities for iron(III) [Google Scholar]
- Römheld V, Marschner H. Evidence for a specific uptake system for iron phytosiderophores in roots of grasses. Plant Physiol. 1986;80:175–180. doi: 10.1104/pp.80.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römheld V, Marschner H. Genotypical differences among graminaceous species in release of phytosiderophores and uptake of iron phytosiderophores. Plant Soil. 1990;123:147–153. [Google Scholar]
- Shioiri T, Hamada Y, Matsuura F. Total synthesis of phytosiderophores. Tetrahedron. 1995;51:3939–3958. [Google Scholar]
- Sugiura Y, Tanaka H, Miro Y, Ishida T, Ota N, Inoue M, Nomoto K, Yoshioka H, Takemoto T. Structure, properties and transport mechanism of iron(III) complex of mugineic acid, a possible phytosiderophore. J Am Chem Soc. 1981;103:6979–6982. [Google Scholar]
- Takagi S. Naturally occurring iron-chelating compounds in oat and rice-root washings: activity measurement and preliminary characterizations. Soil Sci Plant Nutr Tokyo. 1976;22:423–433. [Google Scholar]
- Taylor PD, Morrison IEG, Hider RC. Microcomputer application of non-linear regression analysis to metal-ligand equilibrium. Talanta. 1988;35:507–512. doi: 10.1016/0039-9140(88)80123-1. [DOI] [PubMed] [Google Scholar]
- Thibaud J-B, Davidian J-C, Sentenac H, Solér A, Grignon C. H+-cotransport in corn roots as related to the surface pH shift induced by active H+ excretion. Plant Physiol. 1988;88:1469–1473. doi: 10.1104/pp.88.4.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieken A, Winkelmann G. Rhizoferrin: a complexone type siderophore of the Mucorales and Entomophthorales (Zygomycetes) FEMS Microbiol Lett. 1992;94:37–42. doi: 10.1016/0378-1097(92)90579-d. [DOI] [PubMed] [Google Scholar]
- von Wirén N. Iron efficiency in graminaceous plant species and the role of the microbial degradation of phytosiderophores in iron acquisition. PhD thesis. Stuttgart, Germany: University of Hohenheim; 1994. [Google Scholar]
- von Wirén N, Gibrat R, Briat J-F. In vitro characterization of iron phytosiderophore interaction with maize root plasma membranes: evidences for slow association kinetics. Biochem Biophys Acta. 1998;1372:143–155. doi: 10.1016/s0005-2736(98)00022-4. [DOI] [PubMed] [Google Scholar]
- von Wirén N, Marschner H, Römheld V. Roots of iron-efficient maize also absorb phytosiderophore-chelated zinc. Plant Physiol. 1996;111:1119–1125. doi: 10.1104/pp.111.4.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Wirén N, Mori S, Marschner H, Römheld V. Iron inefficiency in maize mutant ys1 (Zea mays L. cv yellow-stripe) is caused by a defect in uptake of iron phytosiderophores. Plant Physiol. 1994;106:71–77. doi: 10.1104/pp.106.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Wirén N, Römheld V, Shioiri T, Marschner H. Competition between microorganisms and roots of barley and sorghum for iron accumulated in the root apoplasm. New Phytol. 1995;130:511–521. doi: 10.1111/j.1469-8137.1995.tb04328.x. [DOI] [PubMed] [Google Scholar]
- Xu J, Kullgren B, Durbin PW, Raymond KN. Specific sequestering agents for the actinides: 28. Synthesis and initial evaluation of multi-dentate 4-carbamayl-3-hydroxy-1-methyl-2 (1H)-pyridinone ligands for in vivo plutonium(IV) chelation. J Med Chem. 1995;38:2606–2614. doi: 10.1021/jm00014a013. [DOI] [PubMed] [Google Scholar]