

Fluorescence fluctuation spectroscopy is established as a powerful tool for quantifying protein oligomerization in the nuclear envelopes of living cells. It reveals that the SUN proteins SUN1 and SUN2 display differential oligomerization in vivo, which has important implications for LINC complex–dependent nuclear mechanotransduction.
Abstract
Linker-of-nucleoskeleton-and-cytoskeleton (LINC) complexes are conserved molecular bridges within the nuclear envelope that mediate mechanical force transmission into the nucleoplasm. The core of a LINC complex is formed by a transluminal interaction between the outer and inner nuclear membrane KASH and SUN proteins, respectively. Mammals encode six KASH proteins and five SUN proteins. Recently, KASH proteins were shown to bind to the domain interfaces of trimeric SUN2 proteins in vitro. However, neither the existence of SUN2 trimers in living cells nor the extent to which other SUN proteins conform to this assembly state have been tested experimentally. Here we extend the application of fluorescence fluctuation spectroscopy to quantify SUN protein oligomerization in the nuclear envelopes of living cells. Using this approach, we demonstrate for the first time that SUN2 trimerizes in vivo and we demonstrate that the in vivo oligomerization of SUN1 is not limited to a trimer. In addition, we provide evidence to support the existence of potential regulators of SUN protein oligomerization in the nuclear envelope. The differential SUN protein oligomerization illustrated here suggests that SUN proteins may have evolved to form different assembly states in order to participate in diverse mechanotransduction events.
INTRODUCTION
The nuclear envelope (NE) is a subdomain of the endoplasmic reticulum (ER) that encloses the genome and delineates the nuclear compartment in eukaryotic cells (Kite, 1913). It is defined by concentric inner and outer nuclear membranes (INM and ONM, respectively) separated by an ∼30–50 nm perinuclear space (PNS) that is contiguous with the ER lumen (Watson, 1955). The INM contains a unique set of proteins that interact with the underlying nuclear lamina and chromatin, while the ONM is an extension of the ER (Burke and Stewart, 2014). INM–ONM fusion generates fenestrations throughout the NE that are occupied by nuclear pore complexes (Watson, 1959).
Though most nuclear–cytoplasmic communication occurs via nuclear pore complexes (Knockenhauer and Schwartz, 2016), it can also be mechanical in nature, such that forces generated by the cytoskeleton are transmitted across the NE into the nucleoplasm by LINC complexes (Alam et al., 2014). These conserved NE-spanning molecular bridges are critical for cell division, DNA damage repair, meiotic chromosome pairing, mechanoregulation of gene expression, and nuclear positioning (Meinke and Schirmer, 2015). Underscoring their importance is a growing list of genetic mutations in LINC complex proteins associated with human diseases including ataxia and muscular dystrophy (Horn, 2014).
LINC complexes are composed of the ONM Klarsicht/ANC-1/SYNE homology (KASH) proteins and the INM Sad1/UNC-84 (SUN) proteins (Crisp et al., 2006). KASH proteins are defined by a C-terminal KASH domain, which includes a transmembrane domain followed by the ∼10–32-residue luminal KASH peptide (Starr and Han, 2002). The divergent N-termini of KASH proteins extend into the cytoplasm, where they engage the cytoskeleton and signaling molecules (Luxton and Starr, 2014). SUN proteins are identified by their KASH-binding luminal C-terminal SUN domain (Malone et al., 1999), whereas their N-termini interact with A-type lamins, chromatin, and other INM proteins within the nucleoplasm (Chang et al., 2015).
Mammals encode two major SUN proteins, SUN1 and SUN2, which are widely expressed in somatic cells (Meinke and Schirmer, 2015). Consistent with their high level of sequence similarity (e.g., mouse SUN1 and SUN2 share 65% identity), SUN1 and SUN2 perform redundant functions during the DNA damage response (Lei et al., 2012) and radial neuronal migration in the developing mouse cerebral cortex and hippocampus (Zhang et al., 2009), as well as synaptic nuclear anchorage in mouse skeletal muscle (Lei et al., 2009). These redundancies may be due to the ability of both SUN1 and SUN2 to interact promiscuously with the KASH peptide of several KASH proteins including nesprin-1, -2, and -3 (Stewart-Hutchinson et al., 2008). Nevertheless, examples of SUN protein function specificity also exist. For example, SUN1 is differentially required for meiotic chromosome pairing (Ding et al., 2007; Horn et al., 2013) and nuclear pore complex insertion and distribution (Lu et al., 2008; Talamas and Hetzer, 2011). However, the molecular mechanisms underlying the redundant and specific functions of SUN1 and SUN2 remain unclear.
Recent in vitro studies reveal that SUN2 trimerizes due to the presence of a coiled coil (CC)–containing helical region within its luminal domain (Nie et al., 2016; Sosa et al., 2012; Zhou et al., 2012). SUN2 oligomerization is essential for KASH binding, which is further stabilized by an intermolecular disulfide bond formed between conserved cysteine residues in the SUN domain and KASH peptide (Sosa et al., 2012). Despite these important mechanistic advances, the in vivo relevance of SUN protein trimerization remains unclear due to the lack of suitable methods for measuring protein assembly states within the NE. Here, we sought to address this deficiency by extending the application of FFS (Slaughter and Li, 2010) to quantify protein–protein interactions in the NE in living cells.
RESULTS AND DISCUSSION
Quantifying NE protein–protein interactions in living cells
FFS characterizes fluctuating fluorescence signals generated by fluorescently labeled proteins passing through the optical observation volume of a confocal or two-photon microscope (Slaughter and Li, 2010). Subsequent brightness analysis of these fluorescence fluctuations provides quantitative information about the stoichiometry of the labeled proteins (Chen et al., 2003). We recently combined FFS with z-scans to quantify protein–protein interactions at the plasma membrane and in thin cell sections (Macdonald et al., 2010; Smith et al., 2014). Given the ∼30–50-nm thickness of the PNS, we explored the use of FFS and z-scans as a method of quantifying protein–protein interactions within the NE. For simplicity, we refer to the INM, ONM, and PNS collectively as the NE for the remainder of this work.
A z-scan consists of an axial scan of the two-photon spot through a cell expressing a fluorescently labeled protein (Figure 1A; Macdonald et al., 2010). The resulting axial intensity profile or “z-scan” characterizes the subcellular distribution of the labeled proteins. Thus, a labeled NE protein produces a z-scan with two peaks along the trajectory, which correspond to signals generated within the ventral and dorsal NE. To quantify the fluorescence contributions from the NE and the nucleoplasm, z-scans were analyzed as previously described (Smith et al., 2014, 2015). FFS data were collected with the two-photon spot repositioned at either NE.
FIGURE 1:

FFS and brightness analysis in the NE. (A) Identification of the dorsal (NED) and ventral (NEV) NEs in a cell expressing EGFP-tagged NE proteins by z-scan FFS. Fluorescence intensity fluctuations are measured at either NE. (B) Constructs used in this figure. (C) Representative epifluorescence images of U2OS cells expressing the indicated constructs. Scale bar: 5 μm. (D) Brightness analysis of the cells described in C. Each data point represents the average b measured in a single cell.
We initially tested the feasibility of FFS and brightness analysis within the NE by measuring the normalized brightness (b) of EGFP in this subcellular compartment. EGFP was targeted to the ER lumen/PNS by adding the signal sequence (SS) from a luminal protein, torsinA (Goodchild and Dauer, 2004; Figure 1B). The b indicates the average oligomeric state of an EGFP-tagged protein (Chen et al., 2010); that is, a monomer and a dimer correspond to a b of 1 and 2, respectively. To confirm that brightness accurately reflects stoichiometry within the NE, we measured the b of a dimeric EGFP construct (SS-EGFP2; Chen et al., 2003). Following expression, both constructs localized to the ER/NE (Figure 1C). z-scans and FFS data for these constructs were collected from expressing cells, followed by the calculation of b and number concentration (N), which is the average number of EGFPs within the observation volume. Further details on b and N can be found in Materials and Methods.
Independent of N, the mean and standard deviation of b from SS-EGFP- or SS-EGFP2–expressing cells was 1.06 ± 0.14 and 1.98 ± 0.18, respectively (Figure 1D). These results are, within experimental uncertainty, consistent with SS-EGFP being monomeric and SS-EGFP2 dimeric within the NE. This work establishes FFS and brightness analysis as a powerful method for probing the in vivo biochemical and biophysical behavior of NE proteins within their native cellular environment.
SUN2 oligomerization in the NE
Next, we sought to detect in vivo SUN2 trimerization using FFS and brightness analysis. Because quantitative brightness analysis of FFS data requires labeled proteins to be mobile (Hur et al., 2014) and full-length EGFP-tagged SUN2 was shown to be highly immobile by fluorescence recovery after photobleaching (FRAP; Östlund et al., 2009), we generated and measured the oligomerization of a SS-EGFP-tagged construct that encodes the entire luminal domain of SUN2 (SS-EGFP-SUN2261–731) (Figure 2A). We also generated SS-EGFP-SUN2520–731 and SS-EGFP-SUN2595–731, which encode a noncanonical CC through the SUN domain and the SUN domain alone, respectively (Figure 2A). The absence of a transmembrane domain liberates these constructs from membrane-induced constraint, enabling diffusion throughout the ER lumen/PNS (Figure 2B). Nevertheless, it should be noted that the transmembrane domain itself could influence SUN protein oligomerization. This possibility should be addressed in future studies.
FIGURE 2:

SUN2 oligomerization in the NE. (A) Constructs used in this figure. (B) Representative epifluorescence images of U2OS cells expressing the indicated constructs. Scale bar: 5 μm. (C–E) Plots of b vs. N for the indicated constructs. The data in C were fitted to a monomer/dimer/trimer binding model (solid red line), which is shown in D and E (dashed red line), with KMD = 4100 (1000 μM)  , KDT = 0.06 (0.01 μM)
, KDT = 0.06 (0.01 μM)  , and a monomer/trimer binding model (solid green line) with KMT = 26 (6 μM) ± 7.
, and a monomer/trimer binding model (solid green line) with KMT = 26 (6 μM) ± 7.
Plotting b versus N revealed a concentration-dependent increase in b with values mainly between 2 and 3 for N > 50 (Figure 2C). These data show that SS-EGFP-SUN2261–731 exhibits an average oligomerization state that is between dimer and trimer. While the b binding curve has not yet achieved saturation within the experimentally accessible concentration range, the data are approaching the next integer b of 3, suggesting a limiting trimeric stoichiometry as supported by fitting the data to a monomer/dimer/trimer binding model (Figure 2C). The fitted monomer/dimer (KMD = 4100) and dimer/trimer (KDT = 0.06) dissociation coefficients, in units of N for the monomer/dimer and dimer/trimer reaction, indicate that dimers are a minority species, since KMD > KDT. Thus, the b binding curve can also be modeled by a monomer/trimer equilibrium (Figure 2C). Molar values for the dissociation coefficients were estimated as described in Materials and Methods and are quoted in the figure caption.
The b of SS-EGFP-SUN2520–731 increased with N and approached 3 at high N (Figure 2D). While these data are on the average slightly below the binding curve for N < 100, the differences are small. Unlike either SS-EGFP-SUN2261–731 or SS-EGFP-SUN2520–731, the b values obtained for SS-EGFP-SUN2595–731 remain close to one and do not increase with concentration (Figure 2E). These results demonstrate SUN2 trimerization within the NE of living cells and that the SUN domain is not sufficient for this oligomerization, consistent with previously reported in vitro studies (Sosa et al., 2012; Zhou et al., 2012).
While SUN2261–731 contains both types of CC, SUN2520–731 possesses only the noncanonical CC. Despite this difference, both constructs displayed similar N-dependent oligomerization within the NE (Figure 2, C and D) suggesting that the noncanonical CC may be sufficient for SUN2 trimerization, in agreement with the use of a similar human SUN2 construct to solve the crystal structure of SUN2 trimers (Sosa et al., 2012).
SUN1 oligomerization in the NE
To provide insight into the conservation of SUN protein trimerization, we next investigated the oligomerization of SUN1 within the NE. Like SUN2, EGFP-tagged full-length SUN1 was shown by FRAP to be immobile (Östlund et al., 2009). Consequently, we generated an SS-EGFP-tagged construct encoding the entire luminal domain of SUN1 (SS-EGFP-SUN1457–931) (Figure 3A). Owing to the lack of structural information available for SUN1, we limited our analysis to this construct, which was analogous to SS-EGFP-SUN2520–731 (SS-EGFP-SUN1702–913), and another that encodes the SUN1 SUN domain (SS-EGFP-SUN1777–913; Figure 3A). All three constructs localized to the peripheral ER and NE (Figure 3B).
FIGURE 3:

SUN1 oligomerization in the NE. (A) Constructs used in this figure. (B) Representative epifluorescence images of U2OS cells expressing the indicated constructs. Scale bar: 5 μm. (C–E) Plots of b vs. N for the indicated constructs. The data in C were fitted to a linear regression (solid red line), which is shown in D and E (dashed red line).
The b of SS-EGFP-SUN1457–913 increased linearly over the entire range of N (Figure 3C), which prohibited the fitting of the data to a binding curve and the estimation of the stoichiometry of this construct. Nevertheless, the highest b value measured for SS-EGFP-SUN1457–913 was ∼5, indicating the presence of higher-order oligomeric states than those observed for SS-EGFP-SUN2261–731. In contrast to what was observed for SS-EGFP-SUN2520–731, the b of SS-EGFP-SUN1702–913 did not increase appreciably above 1 over the range of measured N (Figure 3D). Finally, a lack of oligomerization similar to what was observed for SS-EGFP-SUN2595–731, was also reflected by the b values obtained for the SUN domain encoding the SS-EGFP-SUN1777–913 construct (Figure 3E). These results suggest that trimers are not the limiting assembly state for all SUN proteins, which is in agreement with a previous report of the existence of immobile macromolecular assemblies of SUN1 within the NE composed of dimers and tetramers (Lu et al., 2008).
SUN protein oligomerization in the cytoplasm
Given the requirement of SUN2 trimerization for KASH-binding (Sosa et al., 2012), SUN protein oligomerization may represent an important target for the regulation of LINC complex assembly. In fact, a recent report shows that the two canonical CCs of SUN2 display distinct oligomeric states, the modulation of which regulates the ability of SUN2 to interact with KASH peptides (Nie et al., 2016). However, the mechanisms responsible for regulating SUN2 oligomerization within the NE remain unknown. As an initial step toward defining these mechanisms, we quantified the oligomerization of constructs encoding EGFP-tagged luminal domains of SUN1 or SUN2 in the heterologous subcellular environment of the cytoplasm.
Cytoplasmic expression of these constructs was achieved by removing the SS (Figure 4, A and B). FFS experiments were performed in the cytoplasm of these cells as previously described (Macdonald et al., 2010, 2013). The b of EGFP-SUN2261–731 increased with N and appeared to reach a limiting value of 3 at N > 600 (Figure 4C), which agrees with the estimated stoichiometry of the analogous construct in the NE. A fit of the cytosolic b data measured for cytoplasmic EGFP-SUN2261–731 to a monomer/dimer/trimer binding curve is shown with KMD = 8000 and KDT = 0.3 (Figure 4C). Because KDT > KMD, the dimer population is negligible and a monomer/trimer equilibrium is sufficient to describe the b binding curve for EGFP-SUN2261–731 (Supplemental Figure S1A).
FIGURE 4:

SUN1 and SUN2 oligomerization in the cytoplasm. (A) Constructs used in this figure. (B) Representative epifluorescence images of U2OS cells expressing the indicated constructs. Scale bar: 5 μm. (C–G) Plots of b vs. N for the indicated constructs. The data in C were fitted to a trimeric binding model (solid blue line), which is shown in D and E (dashed blue line) with KMD = 8000 (60 μM) ± 4000 and KDT = 0.3 (0.002 μM) ± 0.2. The data in F were fitted to a monomer/trimer/hexamer binding model (solid blue line) with KMT = 100 (0.7 μM) ± 60 and a trimer–hexamer dissociation coefficient KTH = 1500 (10 μM) ± 400, which is then shown in G (dashed blue line). Estimated binding curves (dashed red lines) for the data obtained in the NE for the indicated constructs are presented in C and F by converting N from the NE to its cytoplasmic value.
Only the cytoplasmic observation volume is experimentally obtainable (Macdonald et al., 2013). Thus, we computed an estimated NE observation volume to express N values in the NE as an approximate equivalent cytoplasmic N by accounting for the volume difference (see Materials and Methods). Applying this procedure converts the trimeric NE b binding curve (Figure 2C) to a predicted b binding curve in the cytoplasm (Figure 4C). A comparison of the trimeric NE binding curve and the predicted cytoplasmic binding curve demonstrates that the observed b increase with N in the cytoplasm is significantly more pronounced than was observed in the NE (see figure legends for more details). In contrast, both EGFP-SUN2520–731 and EGFP-SUN2595–731 appeared to be monomeric, as their b values remained near 1 (Figure 4, E and F). These results reveal that the oligomerization of EGFP-tagged SUN2261–731 and SUN2520–731 is sensitive to as yet unidentified environmental factors.
In addition, differences in the behavior of the SUN1457–913 constructs in the cytoplasm relative to the NE were also observed. While the b of SS-EGFP-SUN1457–913 and EGFP-SUN1457–913 rises with increasing N, the b increase of EGFP-SUN1457–913 within the cytoplasm slows at higher concentrations, unlike the linear increase with N we observed in the NE (Figure 4F; see figure legend for more details). However, the b of EGFP-tagged SUN1457–913 rises much faster in the cytoplasm than on the predicted b curve, which was converted from the NE b binding reaction (Figure 3C).
Unlike EGFP-SUN2261–731, EGFP-SUN1457–913 does not reach the same b levels in the cytoplasm as in the NE. Instead, the b data slightly exceed 3 in the cytoplasm at high N values. A fit of the b values to a monomer/dimer/trimer binding model reveals that the tail of the binding curve (N > 500) is not reproduced by the model (Supplemental Figure S1B). This misfit implies the need for at least one stoichiometric state in excess of a trimer for EGFP-SUN1457–913 in the cytoplasm. While a monomer/trimer/hexamer binding model was sufficient to describe the experimental data, a monomer/dimer/tetramer binding model could also reproduce the data (Supplemental Figure S1C), consistent with a previous report of the existence of SUN1 dimers and tetramers (Lu et al., 2008). Currently, our data cannot distinguish between these different binding models. Finally, the b of EGFP-SUN1777–913 remains close to 1 over the measured concentration range (Figure 4G), in agreement with the behavior observed for SS-EGFP-SUN1777–913 in the NE. Because it did not display appreciable oligomerization within the NE, we did not measure the oligomerization of EGFP-tagged SUN1702–913 in the cytoplasm.
A more pronounced N-dependent b increase for EGFP-tagged SUN2261–731 and SUN1457–913 was observed in the cytoplasm than in the NE (Figure 4, C and F). A potential explanation for these results could be the presence of unlabeled endogenous SUN proteins within the NE that compete with these labeled SUN constructs, leading to a reduction in b. This competition leads to an apparent reduction in the measured binding affinity (Chen and Mueller, 2007), which potentially explains the shift of the binding curve of EGFP-tagged SUN2261–731 and SUN1457–913 to lower N. Unlike the binding affinity, the saturating value of b remains unchanged and is approached after the exogenously expressed EGFP-tagged protein concentration exceeds the endogenous concentration as well as the KD value, thereby identifying the stoichiometry of EGFP-tagged protein complexes (Chen and Mueller, 2007).
To directly verify that the reported brightness values for the SS-EGFP-tagged luminal domains of SUN1 and SUN2 (Figures 2C and 3C) are not lowered by the endogenous population, we performed additional measurements of both SS-EGFP-SUN2261–731 and SS-EGFP-SUN1457–913 in U2OS cells expressing a short hairpin RNA (shRNA), which efficiently depleted either endogenous SUN1 or SUN2, or a noncoding (NC) control shRNA (Supplemental Figure S2A). SS-EGFP-SUN2261–731 was previously observed to approach the saturating value b = 3 for values of 100 < N < 200 (Figure 2C). Therefore, we selected SUN2 shRNA-expressing cells with concentrations of SS-EGFP-SUN2261–731 in this range, measured b, and then calculated the median and quartile values (Supplemental Figure S2B). Comparing b in the absence of shRNA with b in the presence of either NC or SUN2 shRNA resulted in p values of 0.88 and 0.85, respectively. These results suggest that the b measured for SS-EGFP-SUN2261–731 is unaffected by the absence or presence of endogenous SUN2, and thus the reported b values accurately reflect the average stoichiometry of SS-EGFP-SUN2261–731 at concentrations of 100 < N < 200.
Similarly, we compared b measurements in the presence and absence of NC or SUN1-depleting shRNA (Supplemental Figure S3A) over the range 100 < N < 200. As with SS-EGFP-SUN2261–731, we measured b and determined the median and quartiles. The b measurements performed on SS-EGFP-SUN1457–913 in the presence of NC or SUN1 shRNA resulted in p values of 0.78 and 0.61, respectively, indicating that the measured brightness is unaffected by the absence or presence of endogenous SUN1 (Supplemental Figure S3B). Taken together with our observations of SS-EGFP-SUN2261–731, these results demonstrate that the presence of endogenously expressed unlabeled SUN proteins has a negligible impact on our reported brightness at the higher concentrations we measured. Furthermore, the loss of SUN2520–731 oligomerization in the cytoplasmic environment as compared with the NE (Figures 2D and 4D) cannot be caused by endogenous competition in the NE, since such competition can only lower the brightness in the NE environment.
Thus, our data imply the existence of unidentified regulators of SUN protein oligomerization within the NE, which may be chemical in nature. For instance, the contiguous ER lumen and PNS have a high calcium concentration and an oxidizing environment that favors the formation of disulfide bonds (Ellgaard and Helenius, 2003). Because the conserved cation loop in the SUN domain of SUN2 is required for KASH binding, which also requires SUN2 trimerization (Sosa et al., 2012), we anticipate that SUN protein oligomerization may be sensitive to changes in the concentration of calcium within the PNS. In addition, SUN1 oligomerization involves interchain disulfide bonds, which leads us to speculate that SUN protein oligomerization may be influenced by the redox potential of the ER lumen/PNS (Ellgaard and Helenius, 2003). Alternatively, luminal proteins such as the AAA+ ATPase torsinA, which was recently shown to localize to and be required for the assembly of transmembrane actin-associated (TAN) lines in migrating fibroblasts (Saunders et al., 2017), may structurally regulate SUN protein oligomerization.
Models of SUN1 and SUN2 oligomerization
We propose that SUN2 monomers are in equilibrium with SUN2 trimers in the NE, with no evidence for a significantly populated dimeric state (Figure 5A). Currently, we cannot distinguish between two models of SUN1 oligomerization that are not mutually exclusive (Figure 5, B’ and B”). In the first, SUN1 oligomerizes via a monomer/dimer/tetramer reaction (Figure 5B’). In the second, SUN1 oligomerizes via a monomer/trimer/hexamer reaction (Figure 5B”). Both reactions would ultimately lead to the assembly of higher-order SUN1 oligomers through progressive oligomerization (i.e., monomer to trimer to hexamer to n-mer). The second model is favored based on recently published computational modeling results from the Mofrad laboratory, which suggest that, like SUN2, SUN1 is capable of forming stable homo-trimers (Jahed et al., 2018). However, they found that unlike SUN2 homo-trimers, SUN1 homo-trimers were able to form lateral complexes via the association of their SUN domains. This model is consistent with our observation of b values in excess of trimers for the case of SS-EGFP-SUN1457–913. Future work will be needed to carefully explore the modeling predictions put forward by the Mofrad laboratory.
FIGURE 5:

Models of SUN1 and SUN2 oligomerization. (A) Working model of SUN2 oligomerization. (B’ and B’’) Working models of SUN1 oligomerization with the NE, which lead to the assembly of higher-order SUN1 oligomers (SUN1N).
The ability of SUN1 to form higher-order oligomers than SUN2 may be related to its ability to form rings around NE-associated meiotic telomeres (Horn et al., 2013) and to localize to nuclear pore complexes (Liu et al., 2007). These SUN1-specific localizations may explain the differential requirement for SUN1 during meiotic chromosome pairing and DNA double-stranded break repair (Ding et al., 2007; Horn et al., 2013; Lottersberger et al., 2015) as well as nuclear pore complex insertion and distribution (Lu et al., 2008, Talamas and Hetzer, 2011). Moreover, SUN2 trimerization may be required for actin-dependent nuclear movement, as demonstrated by the specific recruitment of SUN2 to TAN lines in migrating fibroblasts (Luxton et al., 2010). Future efforts aimed at further understanding the mechanisms of the differential oligomerization of SUN1 and SUN2 will provide important insights into LINC complex-dependent mechanotransduction and nuclear–cytoplasmic communication.
MATERIALS AND METHODS
Antibodies
Anti-EGFP mouse monoclonal antibody (mAb) MAB3580 was purchased from Sigma-Aldrich and was used at a dilution of 1:200 for immunofluorescence. Anti-SUN1 (ab74758) and -SUN2 (ab87036) were used at a 1:200 dilution to validate the shRNA-mediated depletion of SUN1 or SUN2 by immunofluorescence. Secondary antibodies were from two different sources. From Jackson ImmunoResearch Laboratories, we purchased goat anti-mouse secondary antibodies conjugated to Alexa Fluor 488 or rhodamine. From ThermoFisher Scientific, we purchased goat anti-rabbit secondary antibodies conjugated to Dylight 488 or 561. All secondary antibodies were used at a 1:200 dilution for immunofluorescence.
Reagents
4′,6-Diamidino-2-phenylindole (DAPI) was purchased from ThermoFisher Scientific. Restriction enzymes were either purchased from New England Biolabs (NEB) or Promega. Phusion DNA polymerase, T4 DNA ligase, and T4 PolyNucleotide Kinase (PNK) were also purchased from NEB. All other chemicals were from Sigma-Aldrich unless otherwise specified. Wizard SV Gel and the PCR Clean-Up System were from Promega. The GeneJet Plasmid Midiprep Kit was from ThermoFisher Scientific.
Cell culture
U2OS cells obtained from the American Type Culture collection were cultured using standard sterile technique in DMEM with 10% fetal bovine serum (FBS) from Hyclone Laboratories.
DNA constructs
NC (TR30015), SUN1 (TF300647B/FI302582), and SUN2 (TF300646A/FI302577) HuSH-29 shRNA constructs in pRFP-C-RS were purchased from OriGene Technologies). The SS-EGFP and SS-EGFP2 constructs were generated as follows, using a previously described human SS-EGFP-torsinA construct (Goodchild and Dauer, 2004). EGFP from SS-EGFP-torsinA was PCR-amplified using the primers SS-EGFP-F and SS-EGFP-R (Table 1), which contain 5’ NheI and EcoRI cut sites, respectively. The PCR product was purified and digested alongside SS-EGFP-torsinA with NheI and EcoRI. Following gel purification, the digested PCR product and plasmid were ligated together to create SS-EGFP. To generate SS-EGFP2, EGFP was amplified from SS-EGFP-torsinA using the primers SS-EGFP2-F and SS-EGFP2-R (Table 1), which contain 5’ BsrGI and ApaI cut sites, respectively. In addition, SS-EGFP2-F encodes a 10–amino acid linker (GHGTGSTGSG) following the BsrGI site, while SS-EGFP2-R encodes a mutated BsrGI site that disrupts the 3’ BsrGI present in EGFP. The resulting PCR product was then purified and digested beside SS-EGFP with BsrGI and ApaI. The digested PCR product and plasmid were purified and ligated to make SS-EGFP2.
TABLE 1:
Primers used to generate the constructs used in this paper.
| Primer name | DNA sequence | 5’ RE site |
|---|---|---|
| SS-EGFP-F | GTGGCTAGCGTGAGCAAGGGCGAGGAG | NheI |
| SS-EGFP-R | GACTGACTGAATTCCTACTTGTACAGCTCGTCCATG | EcoRI |
| SS-EGFP2-F | GCTGTACAAGGGGCACGGGACCGGGTCTACAGGGAGCGGGAGCGTGAGCAAGGGCGAGG | BsrGI |
| SS-EGFP2-R | AACGGGCCCGGCTGCCAATCATGACTGTTACTTATACAGCTCGTCCATGCCG | ApaI |
| SS-EGFP-SUN1457–913-F | GCTGTACAAGGGGCACGGGACCGGGTCTACAGGGAGCGGGAGGGTGGACGATTCCAAGG | BsrGI |
| SS-EGFP-SUN1457–913-R | GAATTCCTACTGGATGGGCTCTCCG | EcoRI |
| SS-EGFP-SUN1702–913-F | ACATCCGAGGCTATTGTGTC | — |
| SS-EGFP-SUN1777–913-F | TGGTACTTCTCACAGTCACC | — |
| SS-EGFP-SUN2261–731-F | GCTGTACAAGGGGCACGGGACCGGGTCTACAGGGAGCGGGTCCTGGTGGGCAGCAAAAG | BsrGI |
| SS-EGFP-SUN2261–731-R | TTTTGAATTCCTAGTGGGCAGGCTCTC | EcoRI |
| SS-EGFP-SUN2520–731-F | TTGGTGAGCCGCCGC | — |
| SS-EGFP-SUN2595–731-F | TGGTACCACTCCCAGTCAC | — |
| SS-EGFP-SUNΔ-R | CCCGCTCCCTGTAGACC | — |
| SSΔ-F | GTGGCTAGCGTGAGCAAGGG | — |
| SSΔ-R | CATGGATCCGAGCTCGGTACC | — |
The F or R in the primer name refers to forward or reverse, respectively. Restriction enzyme (RE) cut sites are underlined. The sequence encoding the linker is bolded.
Previously described EGFP-tagged full-length mouse SUN1 and SUN2 constructs (Luxton et al., 2010) were used as templates for the generation of the SS-EGFP-tagged luminal SUN1 and SUN2 constructs. To create SS-EGFP-SUN2261–731, the sequence encoding amino acids 261–731 was PCR amplified from EGFP-SUN2 using the SS-EGFP-SUN2261–731-F and SS-EGFP-SUN2261–-731-R primer pair (Table 1), which contain 5’ BsrGI and EcoRI cut sites, respectively. In addition, SS-EGFP-SUN2261–731-F encodes a 10–amino acid linker (GHGTGSTGSG) following the BsrGI site. The PCR product was purified and digested alongside SS-EGFP with BsrGI and EcoRI. Following gel purification, the digested PCR product and plasmid were ligated together to create SS-EGFP-SUN2261–731. SS-EGFP-SUN2520–731 and SS-EGFP-SUN2595–731 were both generated via kinase, ligase, DpnI treatment where 2 µl PCR product was treated with T4 ligase, T4 PNK, and DpnI in T4 ligase buffer in a 20-µL reaction for 20 min at room temperature. The forward primers used to create SS-EGFP-SUN2520–731 and SS-EGFP-SUN2595–731 were SS-EGFP-SUN2520–731-F and SS-EGFP-SUN2595–731-F, respectively (Table 1). The same reverse primer, SS-EGFP-SUN2Δ-R (Table 1), was used for both SS-EGFP-SUN2520–731 and SS-EGFP-SUN2595–731.
To create SS-EGFP-SUN1457–913, the sequence encoding amino acids 457–913 was PCR-amplified from EGFP-SUN1 using the primers SS-EGFP-SUN1457–913-F and SS-EGFP-SUN2457–913-R (Table 1), which contain 5’ BsrGI and EcoRI cut sites, respectively. SS-EGFP-SUN1457–913-F also encodes a 10–amino acid linker (GHGTGSTGSG) following the BsrGI site. The PCR product was purified and digested beside SS-EGFP with BsrGI and EcoRI. Following gel purification, the digested PCR product and plasmid were ligated together to create SS-EGFP-SUN1457–913. SS-EGFP-SUN1702–913 and SS-EGFP-SUN1777–913 were generated via the kinase, ligase, DpnI method as described above. The forward primers used to create SS-EGFP-SUN1702–913 and SS-EGFP-SUN1777–913 were SS-EGFP-SUN1702–931-F and SS-EGFP-SUN2777–913-F, respectively (Table 1). The same reverse primer, SS-EGFP-SUNΔ-R (Table 1), was used for both SS-EGFP-SUN1702–913 and SS-EGFP-SUN1777–913.
The cytoplasmic EGFP-tagged SUN1 and SUN2 constructs were generated via kinase, ligase, DpnI reactions as follows. EGFP-SUN2260–731 and EGFP-SUN1457–913 were made first using the primers SSΔ-F and SSΔ-R (Table 1). Kinase, ligase, DpnI treatments were used to make EGFP-SUN2520–731 and EGFP-SUN2595–731. The forward primers used to create EGFP-SUN2520–731 and EGFP-SUN2595–731 from EGFP-SUN2260–731 were SS-EGFP-SUN2520–731-F and SS-EGFP-SUN2595–731-F, respectively (Table 1). The same reverse primer, SS-EGFP-SUN2deletion-R (Table 1), was used for both EGFP-SUN2520–731 and EGFP-SUN2595–731. EGFP-SUN1777–913 was generated via kinase, ligase, DpnI treatment from EGFP-SUN1457–913 using the primers SS-EGFP-SUN1777–913-F and SS-EGFP-SUN1777–913-R (Table 1).
Transfections
Transient transfections of cDNA and shRNA constructs were performed using GenJet from SignaGen Laboratories or Lipofectamine LTX from Invitrogen according to the manufacturers’ instructions. Measurements were performed 24 or 48 h after transfection for FFS experiments in the absence or presence of shRNA, respectively. Immediately before measurement, the growth medium was replaced with Dulbecco’s phosphate-buffered saline (PBS) with calcium and magnesium from Biowhittaker. Brightness measurements in the presence of shRNA were only performed on cells expressing turboRFP.
Fixed- and live-cell epifluorescence microscopy
All fixed-cell imaging was performed on an Eclipse Ni-E microscope driven by NIS-Elements software using a 40×/1.30 NA Plan Fluor oil immersion objective lens (Nikon Instruments, Melville, NY), a SOLA solid state white-light excitation subsystem (Lumencor), and a CoolSNAP ES2 CCD camera (Photometrics). A custom DAPI filter set (#49028 with exciter: ET395/25x, dichroic: T425LPXR, and emitter: ET460/50m; Chroma Technology) was used for the SOLA light source. EGFP (C-FL EGFP Zero Shift, #96362, Nikon Instruments), and Texas Red (C-FL Texas Red Zero Shift, #96365; Nikon Instruments) filter sets were also used.
Twelve hours prior to their transfection, U2OS cells were grown in 24-well plates with #1.5 glass coverslip bottoms or 35 mm dishes with #1.5 glass coverslip bottoms from In Vitro Scientific. Cells were then washed twice with live imaging media composed of HBSS (GIBCO, Invitrogen) containing essential and nonessential MEM amino acids (Invitrogen), 2.5 g/l glucose, 2 mM glutamine, 1 mM sodium pyruvate, and 20 mM HEPES (pH 7.4) and transferred to a 37°C Okolab full-enclosure incubator (Ottaviano, Italy) with temperature control attached to a Intelligent Imaging Innovations (3I) Marianas 200 Microscopy Workstation built on a Zeiss AxioObserver Z.1 stand (Jena, Germany) and driven by SlideBook 6.0 from 3I. All live cell epifluorescence images were acquired with a Zeiss Plan-Apochromat 63x/1.4NA oil objective, a Sutter DG4 light source, and a Photometrics CoolSnap HQ2 CCD. A BrightLine Sedat filter set optimized for DAPI, FITC, TRITC, and CY5 from Semrock was used.
FFS measurements
The instrumental setup has been described previously (Hur and Mueller, 2015). When measurements were performed in the presence of shRNA, a dichroic mirror with a center wavelength of 515 nm (515DCLPXR; Chroma Technology) was used to split the emission light of EGFP and turboRFP into “green” and “red” detection channels, and an additional short-pass filter centered at 512 nm (FF01-512/SP; Semrock) was added to the green channel to remove any reflected light from turboRFP. All analysis steps were performed with programs written in Research Systems IDL 8.3. The experimental two-photon spot or point-spread function (PSF) of the two-photon microscope was described using a modified squared Gaussian–Lorentzian (mGL) model (Macdonald et al., 2010, 2013). A z-scan calibration procedure was performed as previously described to determine the radial and axial beam waists (w0 and z0) and the axial decay parameter (y) (Macdonald et al., 2010), which resulted in z0 = 1.02 ± 0.1 µm, y = 2.30 ± 0.3, and w0 = 0.45 ± 0.05 μm. An initial z-scan passing through the nucleus of the cell generated a z-scan fluorescence intensity profile that was fitted using a previously described model (Smith et al., 2014, 2015) to identify the fluorescence contributions from the ventral and dorsal NEs as well as the nucleoplasm. Cells with an intensity fraction of >90% of fluorescence from the dorsal and ventral NEs were selected for further FFS measurements to ensure that the influence of fluorescence from non-NE sources on the analysis is negligible (Smith et al., 2014). An FFS measurement was conducted for 60 s and a sampling time T of 50 μs with the PSF centered on the ventral or dorsal NE. The collected photon counts were analyzed to extract Mandel’s Q factor,  , which is a measure of the variance of the fluorescence signal with respect to the mean (Sanchez-Andres et al., 2005; Hur and Mueller, 2015; Hennen et al., 2017). Q and the brightness λ, which is the photon count rate of a single molecule, are proportional to one another, Q = γ2λT (Sanchez-Andres et al., 2005). The gamma factor γ2 depends on the PSF and the sample geometry (Smith et al., 2014). Given that the NE is significantly thinner (∼30–50 nm; Franke et al., 1981) than the two-photon spot produced by our microscope (∼1 μm), the NE acts as an infinitely thin “delta (δ)-layer” (Macdonald et al., 2010). For an mGL PSF centered on a δ-layer, γ2 is 0.5 (Macdonald et al., 2010). Additional FFS experiments performed in the nuclei of cells expressing EGFP serve to establish the raw brightness γEGEP as previously described (Chen et al., 2003). The normalized brightness b is defined by b = γ/γEGEP, a dimensionless number that describes the average oligomeric state of the labeled protein (Macdonald et al., 2013, 2014). For example, a homodimeric complex leads to b = 2. Brightness analysis for FFS experiments in the cytoplasm are performed as described above, but with an adjusted γ2 to reflect the different geometry. The value of γ2 has been determined using a z-scan FFS as described earlier (Macdonald et al., 2010).
, which is a measure of the variance of the fluorescence signal with respect to the mean (Sanchez-Andres et al., 2005; Hur and Mueller, 2015; Hennen et al., 2017). Q and the brightness λ, which is the photon count rate of a single molecule, are proportional to one another, Q = γ2λT (Sanchez-Andres et al., 2005). The gamma factor γ2 depends on the PSF and the sample geometry (Smith et al., 2014). Given that the NE is significantly thinner (∼30–50 nm; Franke et al., 1981) than the two-photon spot produced by our microscope (∼1 μm), the NE acts as an infinitely thin “delta (δ)-layer” (Macdonald et al., 2010). For an mGL PSF centered on a δ-layer, γ2 is 0.5 (Macdonald et al., 2010). Additional FFS experiments performed in the nuclei of cells expressing EGFP serve to establish the raw brightness γEGEP as previously described (Chen et al., 2003). The normalized brightness b is defined by b = γ/γEGEP, a dimensionless number that describes the average oligomeric state of the labeled protein (Macdonald et al., 2013, 2014). For example, a homodimeric complex leads to b = 2. Brightness analysis for FFS experiments in the cytoplasm are performed as described above, but with an adjusted γ2 to reflect the different geometry. The value of γ2 has been determined using a z-scan FFS as described earlier (Macdonald et al., 2010).
The number concentration N represents the average number of labeled protein monomers in the observation volume. Because the brightness of an individual EGFP protein is given by γEGEP, the time-averaged fluorescence intensity of a measurement is proportional to the number concentration,  . We experimentally calculate N by dividing the average intensity
. We experimentally calculate N by dividing the average intensity  by γEGEP (Chen et al., 2003). This procedure is valid for FFS experiments in the cytoplasm as well as at the NE. The observation volume VO is given by the overlap between the two-photon PSF and the fluorescent sample. Converting the number concentration into a molar concentration is achieved by
by γEGEP (Chen et al., 2003). This procedure is valid for FFS experiments in the cytoplasm as well as at the NE. The observation volume VO is given by the overlap between the two-photon PSF and the fluorescent sample. Converting the number concentration into a molar concentration is achieved by  , where NA is Avogadro’s number. The volume
, where NA is Avogadro’s number. The volume  of a cytoplasmic FFS experiment is measured using a previously published procedure (Macdonald et al., 2010). In contrast, the volume
of a cytoplasmic FFS experiment is measured using a previously published procedure (Macdonald et al., 2010). In contrast, the volume  , and therefore the labeled protein concentration of FFS experiments at the NE, cannot be determined experimentally. However, molar concentrations in the NE can be estimated as described below.
, and therefore the labeled protein concentration of FFS experiments at the NE, cannot be determined experimentally. However, molar concentrations in the NE can be estimated as described below.
Brightness modeling
A monomer/dimer/trimer equilibrium reaction was used to model b as a function of N. The numbers of molecules of monomers, dimers, and trimers (N1, N2, N3) were determined by the reactions 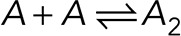 and
and  with the dissociation coefficients KMD and KDT, respectively. By definition, the normalized brightness of an n-mer is given by b = n. The brightness of this mixture of species is
with the dissociation coefficients KMD and KDT, respectively. By definition, the normalized brightness of an n-mer is given by b = n. The brightness of this mixture of species is
 |
The total number of monomeric proteins in the observation volume is A0 = A + 2A2 + 3A3. The same analysis was performed for other binding equilibrium models, such as the monomer/trimer reaction  with a dissociation coefficient defined by
with a dissociation coefficient defined by 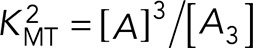 . A detailed description of the modeling is found in the Supplemental Materials. Fitting of the experimental data to b binding models was accomplished using bootstrapping (Efron, 1992). Confidence intervals of the estimated parameters were also determined from the bootstrap algorithm.
. A detailed description of the modeling is found in the Supplemental Materials. Fitting of the experimental data to b binding models was accomplished using bootstrapping (Efron, 1992). Confidence intervals of the estimated parameters were also determined from the bootstrap algorithm.
The observation volume  of NE measurements is small compared to the observation volume
of NE measurements is small compared to the observation volume  of cytoplasmic FFS experiments, which is reflected in the measured N. To facilitate the comparison of b changes with concentration of a protein in both compartments, it is useful to translate between the measured N in both environments. This is achieved by the molar concentration, defined by
of cytoplasmic FFS experiments, which is reflected in the measured N. To facilitate the comparison of b changes with concentration of a protein in both compartments, it is useful to translate between the measured N in both environments. This is achieved by the molar concentration, defined by  , which demonstrates that the values of N in the cytoplasm and the NE are proportional to one another,
, which demonstrates that the values of N in the cytoplasm and the NE are proportional to one another,  . While the observation volume at the NE cannot be measured, it can be modeled as the product of the cross-sectional area of the PSF and the thickness h of the NE layer
. While the observation volume at the NE cannot be measured, it can be modeled as the product of the cross-sectional area of the PSF and the thickness h of the NE layer
 |
Given previously published measurements of NE thickness (Franke et al., 1981), we assume that the NE has an average thickness of h = 40 nm, which leads to a volume of 6.9 × 10–3 fl. This value is 34-fold smaller than the observation volume in the cytoplasm. Thus, the multiplication of N in the NE by 34 compensates for the difference in observation volume and determines the equivalent cytoplasmic N. The observation volume in the cytoplasm with a fully embedded PSF was determined to be 0.23 fl. These values served to calculate molar dissociation coefficients, which are quoted to one significant digit to reflect the uncertainties of the estimate.
Immunofluorescence
Cells grown on #1.5 coverslips were fixed in –20°C methanol as previously described (Saunders et al., 2017). Coverslips were mounted on slides using Fluoromount purchased from ThermoFisher Scientific.
Supplementary Material
Acknowledgments
We thank B. Burke and H. Worman for helpful discussion and Patrick T. Willey for excellent technical assistance. This work was financially supported in part by the National Institutes of Health (J.D.M., GM064589, and C.A.S., AR007612) and the Dystonia Medical Research Foundation (G.W.G.L. and J.D.M.).
Abbreviations used:
- CC
coiled coil
- DAPI
4′,6-diamidino-2-phenylindole
- INM
inner nuclear membrane
- KASH
Klarsicht, ANC-1, SYNE homology
- LINC
linker of nucleoskeleton and cytoskeleton
- NE
nuclear envelope
- ONM
outer nuclear membrane
- PNS
perinuclear space
- shRNA
short hairpin RNA
- SUN
Sad1/UNC-84
- TAN
transmembrane actin-associated nuclear.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-04-0233) on March 5, 2018.
REFERENCES
- Alam S, Lovett DB, Dickinson RB, Roux KJ, Lele TP. (2014). Nuclear forces and cell mechanosensing. Prog Mol Biol Transl Sci , 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke B, Stewart CL. (2014). Functional architecture of the cell’s nucleus in development, aging, and disease. Curr Top Dev Biol , 1–52. [DOI] [PubMed] [Google Scholar]
- Chang W, Worman HJ, Gundersen GG. (2015). Accessorizing and anchoring the LINC complex for multifunctionality. J Cell Biol , 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Johnson J, Macdonald P, Wu B, Mueller JD. (2010). Observing protein interactions and their stoichiometry in living cells by brightness analysis of fluorescence fluctuation experiments. Methods Enzymol , 345–363. [DOI] [PubMed] [Google Scholar]
- Chen Y, Mueller JD. (2007). Determining the stoichiometry of protein heterocomplexes in living cells with fluorescence fluctuation spectroscopy. Proc Natl Acad Sci USA , 3147–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wei LN, Muller JD. (2003). Probing protein oligomerization in living cells with fluorescence fluctuation spectroscopy. Proc Natl Acad Sci USA , 15492–15497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. (2006). Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol , 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Xu R, Yu J, Xu T, Zhuang Y, Han M. (2007). SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev Cell , 863–872. [DOI] [PubMed] [Google Scholar]
- Efron B. (1992). Bootstrap methods: another look at the jackknife. In: Breakthroughs in Statistics. Springer Series in Statistics (Perspectives in Statistics), ed. Kotz S, Johnson NL, New York: Springer. [Google Scholar]
- Ellgaard L, Helenius A. (2003). Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol , 181–191. [DOI] [PubMed] [Google Scholar]
- Franke WW, Scheer U, Krohne G, Jarasch ED. (1981). The nuclear envelope and the architecture of the nuclear periphery. J Cell Biol , 39s–50s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild RE, Dauer WT. (2004). Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci USA , 847–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennen J, Hur KH, Saunders CA, Luxton GWG, Mueller JD. (2017). Quantitative brightness analysis of protein oligomerization in the nuclear envelope. Biophys J , 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn HF. (2014). LINC complex proteins in development and disease. Curr Top Dev Biol , 287–321. [DOI] [PubMed] [Google Scholar]
- Horn HF, Kim DI, Wright GD, Wong ES, Stewart CL, Burke B, Roux KJ. (2013). A mammalian KASH domain protein coupling meiotic chromosomes to the cytoskeleton. J Cell Biol , 1023–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur KH, Macdonald PJ, Berk S, Angert CI, Chen Y, Mueller JD. (2014). Quantitative measurement of brightness from living cells in the presence of photodepletion. PLoS One , e97440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur KH, Mueller JD. (2015). Quantitative brightness analysis of fluorescence intensity fluctuations in E. coli. PLoS One , e0130063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahed Z, Fadavi D, Vu UT, Asgari E, Luxton GWG, Mofrad MRK. (2018). Molecular insights into the mechanisms of SUN1 oligomerization in the nuclear envelope. Biophys J , 1190–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kite GL. (1913). The relative permeability of the surface and interior portions of the cytoplasm of animal and plant cells. Biol Bull , 1–7. [Google Scholar]
- Knockenhauer KE, Schwartz TU. (2016). The nuclear pore complex as a flexible and dynamic gate. Cell , 1162–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K, Zhang X, Ding X, Guo X, Chen M, Zhu B, Xu T, Zhuang Y, Xu R, Han M. (2009). SUN1 and SUN2 play critical but partially redundant roles in anchoring nuclei in skeletal muscle cells in mice. Proc Natl Acad Sci USA , 10207–10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K, Zhu X, Xu R, Shao C, Xu T, Zhuang Y, Han M. (2012). Inner nuclear envelope proteins SUN1 and SUN2 play a prominent role in the DNA damage response. Curr Biol , 1609–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Pante N, Misteli T, Elsagga M, Crisp M, Hodzic D, Burke B, Roux KJ. (2007). Functional association of Sun1 with nuclear pore complexes. J Cell Biol , 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lottersberger F, Karssemeijer RA, Dimitrova N, de Lange T. (2015). 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell , 880–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Gotzmann J, Sironi L, Jaeger VM, Schneider M, Lüke Y, Uhlén M, Szigyarto CA, Brachner A, Ellenberg J, et al (2008). Sun1 forms immobile macromolecular assemblies at the nuclear envelope. Biochim Biophys Acta , 2415–2426. [DOI] [PubMed] [Google Scholar]
- Luxton GW, Gomes ER, Folker ES, Vintinner E, Gundersen GG. (2010). Linear arrays of nuclear envelope proteins harness retrograde actin flow for nuclear movement. Science , 956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxton GW, Starr DA. (2014). KASHing up with the nucleus: novel functional roles of KASH proteins at the cytoplasmic surface of the nucleus. Curr Opin Cell Biol , 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald P, Johnson J, Smith E, Chen Y, Mueller JD. (2013). Brightness analysis. Methods Enzymol , 71–98. [DOI] [PubMed] [Google Scholar]
- Macdonald PJ, Chen Y, Wang X, Chen Y, Mueller JD. (2010). Brightness analysis by Z-scan fluorescence fluctuation spectroscopy for the study of protein interactions within living cells. Biophys J , 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald PJ, Johnson J, Chen Y, Mueller JD. (2014). Brightness experiments. Methods Mol Biol , 699–718. [DOI] [PubMed] [Google Scholar]
- Malone CJ, Fixsen WD, Horvitz HR, Han M. (1999). UNC-84 localizes to the nuclear envelope and is required for nuclear migration and anchoring during C. elegans development. Development , 3171–3181. [DOI] [PubMed] [Google Scholar]
- Meinke P, Schirmer EC. (2015). LINC’ing form and function at the nuclear envelope. FEBS Lett , 2514–2521. [DOI] [PubMed] [Google Scholar]
- Nie S, Ke H, Gao F, Ren J, Wang M, Huo L, Gong W, Feng W. (2016). Coiled-coil domains of SUN proteins as intrinsic dynamic regulators. Structure , 80–91. [DOI] [PubMed] [Google Scholar]
- Östlund C, Folker ES, Choi JC, Gomes ER, Gundersen GG, Worman HJ. (2009). Dynamics and molecular interactions of linker of nucleoskeleton and cytoskeleton (LINC) complex proteins. J Cell Sci , 4099–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Andres A, Chen Y, Müller JD. (2005). Molecular brightness determined from a generalized form of Mandel’s Q-parameter. Biophys J , 3531–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CA, Harris NJ, Willey PT, Woolums BM, Wang Y, McQuown AJ, Schoenhofen A, Worman HJ, Dauer WT, Gundersen GG, Luxton GW. (2017). TorsinA controls TAN line assembly and the retrograde flow of dorsal perinuclear actin cables during rearward nuclear movement. J Cell Biol , 657–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter BD, Li R. (2010). Toward quantitative “in vivo biochemistry” with fluorescence fluctuation spectroscopy. Mol Biol Cell , 4306–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EM, Hennen J, Chen Y, Mueller JD. (2015). Z-scan fluorescence profile deconvolution of cytosolic and membrane-associated protein populations. Anal Biochem , 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EM, Macdonald PJ, Chen Y, Mueller JD. (2014). Quantifying protein–protein interactions of peripheral membrane proteins by fluorescence brightness analysis. Biophys J , 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa BA, Rothballer A, Kutay U, Schwartz TU. (2012). LINC complexes form by binding of three KASH peptides to domain interfaces of trimeric SUN proteins. Cell , 1035–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr DA, Han M. (2002). Role of ANC-1 in tethering nuclei to the actin cytoskeleton. Science , 406–409. [DOI] [PubMed] [Google Scholar]
- Stewart-Hutchinson PJ, Hale CM, Wirtz D, Hodzic D. (2008). Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Exp Cell Res , 1892–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talamas JA, Hetzer MW. (2011). POM121 and Sun1 play a role in early steps of interphase NPC assembly. J Cell Biol , 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson ML. (1955). The nuclear envelope; its structure and relation to cytoplasmic membranes. J Biophys Biochem Cytol , 257–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson ML. (1959). Further observations on the nuclear envelope of the animal cell. J Biophys Biochem Cytol , 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lei K, Yuan X, Wu X, Zhuang Y, Xu T, Xu R, Han M. (2009). SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron , 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Du X, Cai Z, Song X, Zhang H, Mizuno T, Suzuki E, Yee MR, Berezov A, Murali R, et al (2012). Structure of Sad1-UNC84 homology (SUN) domain defines features of molecular bridge in nuclear envelope. J Biol Chem , 5317–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.