

Abstract
Background and aim
Primary hyperoxalurias are rare inborn errors of metabolism resulting in increased endogenous production of oxalate that leads to excessive urinary oxalate excretion. Diagnosis of primary hyperoxaluria type 1 (PH1) is a challenging issue and depends on diverse diagnostic tools including biochemical analysis of urine, stone analysis, renal biopsy, genetic studies and in some cases liver biopsy for enzyme assay. We characterized the clinical presentation as well as renal and extrarenal phenotypes in PH1 patients.
Methods
This descriptive cohort study included patients with presumable PH1 presenting with nephrolithiasis and/or nephrocalcinosis (NC). Precise clinical characterization of renal phenotype as well as systemic involvement is reported. AGXT mutational analysis was performed to confirm the diagnosis of PH1.
Results
The study cohort included 26 patients with presumable PH1 with male to female ratio of 1.4:1. The median age at time of diagnosis was 6 years, nevertheless the median age at initial symptoms was 3 years. Thirteen patients (50%) were diagnosed before the age of 5 years. Two patients had no symptoms and were diagnosed while screening siblings of index patients. Seventeen patients (65.4%) had reached end-stage renal disease (ESRD): 6/17 (35.3%) during infancy, 4/17 (23.5%) in early childhood and 7/17 (41.29%) in late childhood. Two patients (7.7%) had clinically manifest extra renal (retina, heart, bone, soft tissue) involvement. Mutational analysis of AGXT gene confirmed the diagnosis of PH1 in 15 out of 19 patients (79%) where analysis had been performed. Fifty percent of patients with maintained renal functions had projected 10 years renal survival.
Conclusion
PH1 is a heterogeneous disease with wide spectrum of clinical, imaging and functional presentation. More than two-thirds of patients presented prior to the age of 5 years; half of them with the stormy course of infantile PH1. ESRD was the commonest presenting manifestation in two-thirds of our cohort.
Keywords: End-stage renal disease, Nephrocalcinosis, Nephrolithiasis, Oxalosis, Post-transplantation recurrence, Primary hyperoxaluria type 1
1. Core tip
We characterized the spectrum of clinical presentation of primary hyperoxaluria type 1 (PH1) in a tertiary center in Egypt. We retrospectively evaluated the renal as well as the extrarenal phenotypes in 26 patients with radio-opaque kidney stones and/or nephrocalcinosis particularly patients with family history of urolithiasis, chronic kidney disease CKD/ESRD, renal stones and/or nephrocalcinosis and asymptomatic siblings of PH1 index patients identified by screening. We confirm the broadly heterogenous spectrum of clinical presentation of PH1 similar to worldwide PH1 cohorts, yet the frequency of infantile PH1 and ESRD at presentation were strikingly higher in this mostly consanguineous cohort.
2. Introduction
Primary hyperoxalurias (PH) are a rare group of inborn errors of metabolism characterized by excessive hepatic production of oxalate and they include three main types that are inherited in an autosomal recessive pattern: PH1 (MIM #259900), PH2 (MIM #260000) and PH type 3 (MIM #613616). PH1 is caused by a deficiency of the liver specific, peroxisomal enzyme alanine/glyoxylate aminotransferase (AGT) [1,2], while PH2 is caused by a deficiency of the cytosolic enzyme glyoxylate reductase/hydroxypyruvate reductase (GRHPR) [3]. PH type 3 (MIM #613616) is linked to the gene HOGA1 (formerly DHDPSL), encoding the mitochondrial enzyme 4-hydroxy-2-oxoglutarate aldolase [4,5].
PH1 is the most severe primary hyperoxaluria form accounting for about 80% of genetically characterized patients [1]. The failure to detoxify glyoxylate in PH1 results in overproduction of oxalate by cytosolic lactate dehydrogenase, with excessive urinary excretion of oxalate, which is poorly soluble and forms calcium oxalate crystals leading to urolithiasis and/or NC when the urine becomes supersaturated. Eventually patients reach ESRD due to progressive renal parenchyma inflammation and interstitial fibrosis caused by NC and recurrent urolithiasis [6].
Progressive decline of glomerular filtration rate (GFR) results in reduced oxalate excretion by the kidneys, while the liver continues to produce excess oxalate leading to a critical saturation point for plasma oxalate (Pox > 30 μmol/L to 50 μmol/L). This leads to a state of what is called oxalosis with systemic oxalate deposition occurring in many organs including the bones, heart, retina, nerves, joints, skin and soft tissues. Bone is the major compartment of the insoluble oxalate burden, and oxalate osteopathy leads to severe bone pain, erythropoietin-resistant anemia, and spontaneous fractures and specific oxalate osteopathy on X-ray [7].
ESRD with a history of renal stones or calcinosis, frequent recurrent nephrolithiasis, and even first radiopaque kidney stone in a child are all reasonably specific indicators of PH [8].
Diagnosis of PH1 is a challenging issue and depends on diverse diagnostic tools including biochemical analysis of urine, stone composition of pure calcium oxalate (CaOx) monohydrate (whewellite), genetic studies and in some cases liver biopsy for enzyme assay [9].
Urine analysis may show CaOx monohydrate crystalluria and infrared spectroscopy is useful for identification and quantitative analysis of crystals and stones. In patients with normal or significant residual GFR, hyperoxaluria (urine oxalate > 1 mmol/1.73 m2 per day, reference value < 0.5) and hyperglycoluria (urine glycolate > 0.5 mmol/1.73 m2, reference value < 0.5) indicate PH1, but some patients do not exhibit hyperglycoluria [10]. The finding of oxalate crystals in any biologic fluid or tissue [8] and the role of liver biopsy and measurement of AGT enzyme catalytic activity has declined recently and now genetic analysis of AGXT gene can detect mutations in most of suspected patients, thereby, reserving liver biopsy for few selected cases [11].
The spectrum of clinical presentation of PH is broad including recurrent renal stones with or without NC, hematuria, failure to thrive, dysuria, urinary tract infection or renal colic. Some patients may remain asymptomatic and present with ESRD as their first presentation while others experience recurrent calcium oxalate urolithiasis with good renal function in adulthood. Unsurprisingly, the infantile form often presents critically with rapid progression to ESRD, due to both early oxalate load and immature GFR. PH is a rare disease; therefore clinicians even in subspecialties of nephrology may come across none to only a few patients with PH during their practicing lifetime. This fact, in addition to variable clinical expression, makes recognition of the disease challenging. Further, accurate diagnosis relies on highly specialized studies conducted at only a small number of laboratories worldwide [12,13].
In the current study, we aimed to highlight the spectrum of clinical presentation of PH1 in Egyptian patients using high index of suspicion and utilizing the available diagnostic tools for proper clinical phenotyping. Suspected cases on clinical grounds were further confirmed by genetic mutational analysis.
3. Patients and methods
3.1. Patients
Patients with multiple radio-opaque kidney stones and/or NC particularly with family history of urolithiasis, chronic kidney disease CKD/ESRD patients with renal stones and/or NC, and asymptomatic siblings of PH1 index patients identified by screening were enrolled in this study. Case notes were reviewed to characterize the clinical phenotypes: consanguinity and family history of stone disease, presenting symptoms/signs, age at presentation, age at diagnosis, time lag from onset of symptoms to diagnosis, renal phenotype (nephrolithiasis and/or NC), therapeutic modalities, outcome of the disease in terms of patient and kidney/graft survival. Moreover, urinary oxalate/creatinine ratio (Ox/Cr) was performed for patients maintaining normal kidney functions to confirm hyperoxaluria. Passed or surgically extracted stones were analyzed by infrared spectroscopy to confirm its CaOx nature. Kidney biopsy has been reserved for CKD/ESRD patients with hyperechogenic kidneys suggestive of NC, and/or patients with history of stone disease or patients with post-transplantation recurrence. Also, any identified AGXT mutation was recorded.
Twenty six patients with radio-opaque renal stones and/or NC were included in the current study. Research protocol was approved by Institutional Review Board, and informed written patient/parental consent was obtained.
All patients with clinico-radiological presentations typical of PH1 were evaluated by renal ultrasound scan (USS) which was performed by an expert nephrologist and confirmed by an expert radiologist. Siemens AdaraSonoline machine with 5.0c 40s transducer was used. Patients with normal GFR or CKD grade 1 were instructed to high fluid intake and oral citrate, and oral pyridoxine therapy 10–20 mg/kg/day in two divided doses. Ten year renal survival had also been calculated for these patients.
3.2. Data analysis
Data were tabulated and subjected to computer-assisted statistical analysis using Microsoft Excel® version 2003 and the Statistical Package for Social Science (SPSS) for Windows® version 16.0. Nominal data were expressed as frequency and percentage and were compared using χ2 tests. Numerical data were expressed as mean and standard deviation, and were compared using independent samples t-tests. P values < 0.05 were considered significant.
4. Results
The study included 26 patients belonging to 20 families, comprising 15 males (57.7%) and 11 females (42.3%). Their ages ranged from 1.5 months to 29 years (median age 6 years). Twenty patients (76.9%) had consanguineous parents and more than half of them (53.4%) were from Upper Egypt. The most common presentation was ESRD, other presenting manifestations are demonstrated in Fig. 1.
Fig. 1.
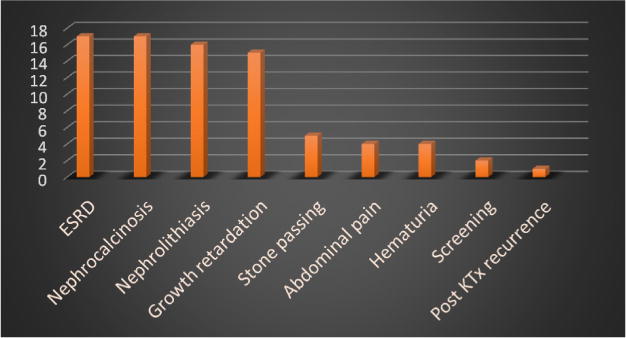
Clinical presenting manifestations in study patients (n = 26).
The median age in our study group was 6 years (range: 0.1–29 years) whereas the median age of onset of PH1 symptoms was 3 years (range 0.1–14 years). Interestingly, 6/17 (35.3%) of the current study patients presented during their first year of life (age range: 0.1–0.9 years) with ESRD and failure to thrive. Another nine patients (9/26, 34.6%) first presented prior to the age of 5 years (age range: 1–< 5 years), whereas the remaining 8/26 patients (30.8%) had their first symptom after the age of 5 years (age range: 5–16 years).
The diagnosis of PH1 in study patients rested on biochemical analysis using spot urine oxalate/creatinine mmol ratios in 8/26 patients (30.8%) with normal GFR, it was high with median value of 73 and IQR 55–93 (normal value < 45 mg/1.73 m2/day). Native renal biopsy in four patients with CKD and NC and a graft biopsy post isolated kidney transplantation in a fifth patient with post-transplantation recurrence showed interstitial nephritis with calcification and extensive oxalate deposition. Infrared spectroscopy of normally passed/surgically extracted stone was performed in half of the study patients 13/26 (50%) and it showed CaOx monohydrate (whewellite) as the major component of the stone.
In 14/26 patients (53.8%) with characteristic clinico-radiological presentation and family history of radiopaque renal stone and/or renal failure, the diagnosis was further confirmed upon identifying a disease-causing mutation in the AGXT gene.
4.1. Renal phenotype
Nephrolithiasis and/or NC were present in all patients: both nephrolithiasis and NC in 11/26 patients, exclusive nephrolithiasis in 8/26 patients, and isolated NC in 7/26 patients.
Abdominal USS characterized the renal phenotype in the studied patients showing multiple renal stones in 19 patients (61.5%) of whom 11 patients had concomitant NC. In total, 16 patients (61.5%) had NC that was best categorized as corticomedullary in 11/16 (68.7%) patients and exclusively medullary in 5/16 (31.3%) (Table 1).
Table 1.
Correlation of nephrocalcinosis and nephrolithiasis with age of patients and age of ESRD.
| Corticomedullary n = 11 |
Medullary n = 5 |
Only urolithiasis n = 8 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Median | IQR | Median | IQR | Median | IQR | ||||
| Age (years) | 3 | 0.5 | 6.5 | 1 | 0.8 | 2.5 | 10 | 9 | 13 |
| Age of onset (years) | 2.5 | 0.4 | 4.75 | 0.6 | 0.25 | 2.5 | 3.5 | 2.25 | 6.68 |
| ESRD age (years) | 3.6 | 0.53 | 5.5 | 0.74 | 0.7 | 0.77 | 9.15 | 7.22 | 15 |
ESRD: end-stage renal disease.
Upon diagnosis, 17/26 patients (65.4%) had already reached ESRD. Age of ESRD was variable among studied patients: during the first year of life in 6/17 (35.3%); between 1 and 5 years of age in 4/17 (23.5%); and after the age of 5 years in 7/17 (41.2%). There was a significant positive correlation between age of onset of symptoms and age of ESRD as shown in Fig. 2. Moreover, there was a significant positive correlation between the frequency of surgical interventional procedures for stone extraction (urolithotomy and/or lithotripsy) and the occurrence of ESRD (r = 0.726; P value 0.001).
Fig. 2.
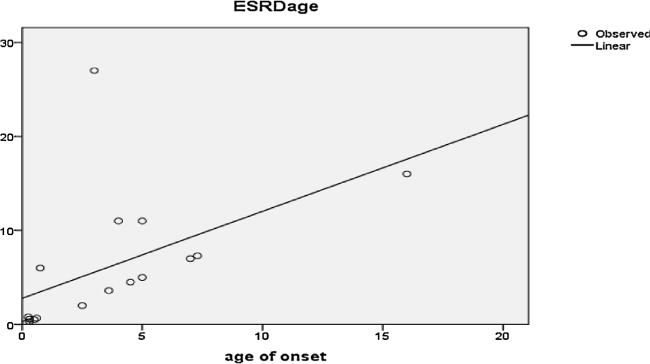
Correlation between age of onset and ESRD age (r = 0.52; P value 0.031).
4.2. Extrarenal phenotype
Two patients demonstrated extrarenal manifestations of oxalosis. A female with infantile PH1 had visual impairment associated with nystagmus. Fundoscopy showed white dots in the retina and electroretinography (ERG) finding were consistent with subnormal retinal function. Moreover, she had myocardial deposition of abnormal echogenic dots diagnosed by echocardiography and confirmed by cardiac MRI. The other patient had severe bone disease and bone marrow failure with oxalate deposits diagnosed in bone marrow aspirate. Later on during the course of her illness she developed multiple soft tissue swellings all over the body indicative of severe systemic oxalosis (Fig. 3).
Fig. 3.
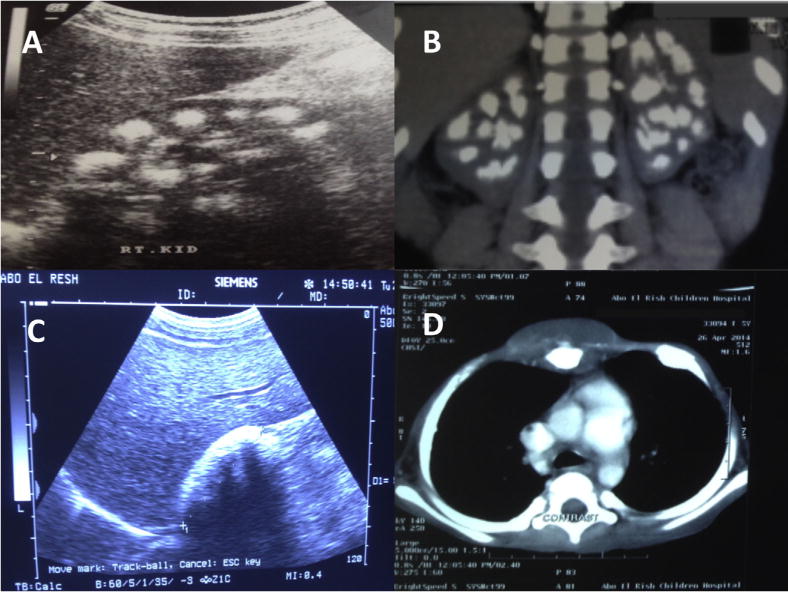
A. Renal USS of patient E-2 showing medullary nephrocalcinosis. B. Coronal reformatted CT scan images of the urinary tract of patient E-11 demonstrating bilateral medullary nephrocalcinosis with multiple pelvic renal stones. C. Renal USS of patient E-18 showing chalky-white hyperechogenic kidney due to severe cortical and medullary nephrocalcinosis. D. Post-contrast chest CT images of patient E-3 demonstrating focal mass lesion surrounding the sternum due to soft tissue deposition of CaOx. Moreover, there is distended costochondral junction due to CaOx deposits.
4.3. Clinical outcome of studied patients
Fifty percent of all cases (13/26) had a projected renal survival of at least 10 (P value 0.08 with median age 10.5) of whom 8 patients (61.5%) had no evidence of NC, 2 patients (15.4%) had corticomedullary NC and 3 patients (23.1%) had medullary NC. Unsurprisingly, there was a significant correlation between progression to ESRD and the presence of cortical NC (P value 0.047). Three PH1 patients in ESRD underwent combined simultaneous liver and kidney transplantation with functioning renal and hepatic grafts for a duration of 6, 5, and 3 years respectively. The identified AGXT mutations in the three successfully transplanted patients were the North African c.731 T > C (I244 T) mutation in the first two patients and c.725dupT, -p.Asp243fs in the third patient.
Eleven cases (42.3%) died during the study period from complications related to peritoneal dialysis, hemodialysis and systemic oxalosis. On the other hand, normal renal function was maintained after 2 years of follow up in 8/26 patients (30.7%) on pyridoxine therapy (10–20 mg/kg/day).
5. Discussion
PH1 is a rare metabolic entity caused by deficiency of the peroxisomal liver–specific, pyridoxal phosphate-dependent enzyme alanine glyoxylate aminotransferase (AGT). PH1 has been reported in many ethnic groups and racial communities worldwide [6].
5.1. Prevalence and consanguinity
PH1 is estimated to account for 1–2% of pediatric ESRD population in registries from USA, UK and Japan [14–17]. Higher rates are reported from inbred populations [14]. Approximately 10% of Kuwaiti children and 13% of Tunisian children with ESRD had been reported to have PH1 [18,19]. The prevalence of PH1 ranges from 1to 3:1,000,000 depending on the population studied and the methods of ascertainment [18–20].
The prevalence of PH1 in Egypt is yet to be determined, nevertheless it is beyond the scope of this study. We believe that the observed prevalence rate is still an under-estimation of the true prevalence. PH1 in Egypt is seemingly commonly overlooked, underdiagnosed or diagnosed late in the course of the disease. The authors believe that: (i) the inappropriate lack of awareness of this inherited disease and, (ii) the low index of clinical suspicion needed for early and precise diagnosis; play a major role in under-diagnosis of PH1 in the highly consanguineous Egyptian population.
To the best of our knowledge, this is the first report of primary hyperoxaluria in Egyptian patients. The high rate of consanguinity in the study group (76.9%) is not surprising given the high rate of inbreeding among Egyptians [21]. This contributes to the high incidence of autosomal recessive disorders in general in Egypt. Interestingly, this is nearly close to the 90% consanguinity rate reported earlier by the Tunisian group among their studied PH1 patients [22].
Despite the family history of renal failure and/or renal stones in 26.7% and sibling death in 11.5% of the studied patients, only 7.7% of patients were diagnosed by screening. Therefore, a lot of emphasis should be put on the importance of screening siblings of PH1 index cases as the early initiation of therapy would greatly enhance the renal survival and clinical outcome.
The median age in our study group was 6 years (range: 0.1–29 years) whereas the median age of onset of PH1 symptoms was 3 years (range 0.1–14 years). This was in agreement with earlier reports highlighting the extended age of presentation from infancy to adulthood [23].
Diagnostic delay had been previously reported as one of the major problems in PH1. According to several cohort studies, the median time interval between initial symptoms and diagnosis is more than 5 years [8], this is compared to 3.5 years in the current study. The lower diagnostic delay in our series is probably attributable to limited number of patients in addition to the higher percentage of infantile PH1 (35.4%) in our cohort in comparison to the reported 19% in the literature [24,25].
5.2. Diagnostic challenges
Initial symptoms of PH1 are often overlooked due to rarity of the disease, let alone the limited access to urinary metabolic screening in only few reference laboratories. This adds to diagnostic delay in areas where these techniques are not available.
Unfamiliarity with the disease among medical community and diagnostic pitfalls are the major reasons for missing the diagnosis. PH1, as rare as it is, is unknown to most practitioners, particularly nephrologists and urologists, hence specific diagnostic investigations are often not performed on time [26]. Furthermore, establishing PH1 can be troublesome due to a number of reasons:
improperly conducted 24-h oxalate assessment can easily lead to undetected hyperoxaluria due to precipitation of oxalate in the urine bowl;
plasma oxalate assessment may be normal or subnormal in patients with preserved glomerular filtration rate (GFR);
ultrasound assessment of diffuse cortical NCis commonly mistaken for nonspecific parenchymal hyperechogenicity there-by missing an important clue to diagnose PH1;
patients with a GFR below 60 mL/min/1.73 m2 are at risk for systemic oxalosis and should undergo fundoscopy and screening for bone abnormalities by X-ray. The retinal abnormalities are typical, yet are often not recognized. This holds true as well for bone abnormalities which may mimic, hence misinterpreted as, renal osteodystrophy;
specific confirmatory enzymology and genetic techniques to establish PH1 are currently often not available in several low-resource countries with a high prevalence of PH1.
5.3. Clinical presentation and diagnostic confirmation
Interestingly, 34.6% (9/26) of study patients presented during their first year of life (age range: 0.1–0.9 years) with NC and ESRD. Another 34.6% (9/26) of patients first presented prior to the age of 5 years (age range: 1–< 5 years), whereas the remaining 30.8% (8/26) patients had their first symptom after the age of 5 years (age range: 5–16 years).
Clinical symptoms of PH1 are the consequences of excessive urinary insoluble calcium oxalate crystals which lead to nephrolithiasis and/or NC. Uremia, gross hematuria, abdominal pain and stone passing were the most frequent manifestations reported in our patients, and they are comparable to other cohorts described in the literature [27]. In our study (65.4%) of patients presented with ESRD which reflects in part, the higher rate of infantile PH1 with rapid progression to ESRD as a result of severe cortical NC. Another explanation would be late diagnosis in study patients (as evidenced by diagnostic delay time) as a result of lack of awareness of this rare disease that even nephrologists may encounter few cases during their practicing lifetime [12,13].
Diagnosis of PH1 in our patients was based on family history, clinico-radiological phenotype, infrared spectroscopy of stone and urinary spot oxalate/creatinine ratio for patients with normal kidney functions. The measurement of oxalate in a timed 24-hour urine collection corrected for body surface area is preferred for the diagnosis of PH1 [27]. Random urine oxalate/creatinine ratios can be useful to estimate oxalate excretion, particularly in infants, or patients with inability to provide timed urine collection [28,29]. Mutational analysis of AGXT gene confirmed the diagnosis of PH1 in 15 out of 19 patients (79%) where a disease-causing mutation had been identified.
5.4. Renal and extrarenal phenotypes
The presence of NC was associated with rapid progression to ESRD (P value = 0.08) which was more common with cortical than medullary (P value 0.047). Kidney failure is more frequently associated with cortical than medullary NC which had been reported earlier by the Tunisian authors who also found a correlation between ESRD and cortical NC [22].
The natural outcome of the PH1 is ESRD which may occur as the first presenting manifestation of the disease [20,21]. In our study uremia was the most frequent initial renal manifestation in 65.4% of study patients, higher than earlier report of 40% in pediatric patients by Von Woerden and al. [22]. The authors suggested that lack of metabolic screening as a routine procedure in study patients with recurrent urinary tract symptoms, in addition to the cumbersome technique of urinary oxalate measurement with frequent false-negative results possibly contributed to late diagnosis and the higher percentage of ESRD among their study patients.
The systemic manifestations of oxalosis tend to occur in a variety of tissues, including the kidneys, retina, myocardium, and bone marrow [23]. PH1 shows considerable phenotypic, enzymatic and genotypic heterogeneity. Some patients have no symptoms at all or suffer only from recurrent urolithiasis. Other patients develop a chronic renal insufficiency with systemic oxalosis [4,7]. Bone is a major component of insoluble oxalate pool, causing oxalate osteopathy, pain, fractures, anemia resistant to erythropiotin.
Clinically manifest extrarenal manifestations of oxalosis were reported in two of the study patients and did greatly impact their quality of life. One patient had retinal and myocardial affections whereas the other patient had soft tissue, bone and bone marrow deposition. Naturally, these systemic manifestations could have been avoided by early diagnosis and aggressive renal replacement therapy when indicated.
Plasma oxalate (Pox) is unhelpful for diagnosis if renal function is normal. Pox concentrations higher than 50 μmol/L regardless of renal function would be very suggestive of PH1 [24,25]. In our study, plasma oxalate was not available, hence could not be performed in ESRD study patients.
5.5. Combined liver and kidney transplantation
Isolated kidney transplantation in patients with PH1 reported poor results. Kidney transplantation has a considerably lower success rate than with other diseases [27], while the results of combined liver and kidney transplantations from the published literature are encouraging [8]. In over 60 combined liver/kidney transplants performed for PHI, the actuarial graft and patient survival rate of 88% at 1 year and 80% at 5 years [28], regarding our series Three cases with ESRD underwent combined simultaneous hepatorenal transplantation at the age 4 years, 9 years and 7, and they have functioning liver and kidney grafts for 5, 4, and 2 years respectively.
In fact, early recognition of PH1 as the cause of renal insufficiency is of utmost importance because pyridoxine may reverse the course of the renal disease leading to preservation of renal function.
5.6. Pyridoxine responsiveness and clinical outcome
Pyridoxine responsive patients comprised 4/15 (26.7%) of the genetically confirmed PH1 study patients. Patients exhibited > 30% reduction of urinary oxalate following maximum daily pyridoxine therapy [6]. Interestingly none of them had the known pyridoxine-responsive mutations since the following three mutations were identified in the above four families: c.603G > A, p.D201E, c.126dupG, p.L43fs, c.33delC, p.Pl1fs33X. Reported pyridoxine responsiveness in other cohorts ranged between 25–66% [22,29]. The response rate in the current study is comparable to the Tunisian cohort (25%), but much lower than the 66% reported in the Dutch study probably because of low pyridoxine responsive AGXT mutations and/or poor adherence to treatment, which is not uncommon in PH1 patients [15].
Mortality was higher in our cohort (> 40%) as compared to van der Hoeven and al. cohort mortality rate of 28% [29] and was also higher than the rate found by Harambat and al. (13%) [14]. This is probably explained by longer exposure to oxalate load as a result of diagnostic and therapeutic delay. This eventually leads to higher prevalence of renal insufficiency or ESRD at the time of diagnosis, with increased dialysis, transplantation or systemic oxalosis-related mortality [30–34].
The authors are aware that PH1 study patients diagnosed in our major national referral center do not necessarily reflect the prevalence in the community. This cohort represents just a limited number of referred patients in whom high index of clinical suspicion helped to unravel the diagnosis and prompted adequate therapy particularly in the pyridoxine-responsive patients. It also assisted in designing the proper transplantation strategy in the three transplanted patients who received both liver and kidney grafts, thereby enhancing clinical outcome and avoiding the devastating post kidney transplantation recurrence. Having said that and despite the rarity of PH1, the number of patients in our cohort is still limited. More collaborative research into clinical phenotypes and genotype-phenotype correlation in PH1 patients is still warranted in larger worldwide cohorts of patients in international registries.
Despite the heterogeneous nature of the disease and the diagnostic challenges involved, a well-defined approach as to when to suspect and how to proceed with investigating suspected cases is certainly needed, particularly in the underprivileged highly consanguineous communities with higher incidence of PH1.
6. Conclusion
PH1 is one of the most challenging diseases being highly heterogeneous with variable age of first presentation and vague initial symptoms that can easily result in diagnostic delay. Moreover, lack of awareness and the low index of clinical suspicion play a major role in PH1 under-diagnosis. More than 2/3 of PH1 patients presented prior to the age of 5 years; half of them with the stormy course of infantile PH1. ESRD was the commonest presenting manifestation in 2/3 of our cohort. Family history of stone disease/ESRD, radio-opaque nephrolithiasis and/or NC, cortical and medullary NC mostly misinterpreted as simply “hyperechogenic” kidneys in infants and toddlers are helpful clues to diagnosis that need to be enforced by the availability of reliable diagnostic tests and further confirmed by molecular genetic analysis. This is expected to improve the lives of many PH1 patients.
Acknowledgments
The authors sincerely thank the affected patients and their families for participation. They are very grateful to Rare Kidney Stone Consortium (RKSC) for performing AGXT mutational analysis in some of the study patients. The RKSC (U54KD083908) is a member of the NIH Rare Diseases Clinical Research Network (RDCRN).
Footnotes
Informed consent statement
All study participants or their legal guardian provided informed written consent about personal and medical data collection prior to study enrolment.
Disclosure of interest
The authors declare that they have no competing interest.
References
- 1.Danpure CJ, Jennings PR. Peroxisomal alanine: glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Lett. 1986;201:20–4. doi: 10.1016/0014-5793(86)80563-4. [DOI] [PubMed] [Google Scholar]
- 2.Purdue PE, Lumb MJ, Fox M, Griffo G, Hamon-Benais C, Povey S, et al. Characterization and chromosomal mapping of a genomic clone encoding human alanine: glyoxylate aminotransferase. Genomics. 1999;10:34–42. doi: 10.1016/0888-7543(91)90481-s. [DOI] [PubMed] [Google Scholar]
- 3.Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. The gene encoding hydroxypyruvate reductase (GRHPR) is mutated in patients with primary hyperoxaluria type II. Hum Mol Gen. 2009;8:2063–9. doi: 10.1093/hmg/8.11.2063. [DOI] [PubMed] [Google Scholar]
- 4.Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Gen. 2010;87:392–9. doi: 10.1016/j.ajhg.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belostotsky R, Pitt JJ, Frishberg Y. Primary hyperoxaluria type III–a model for studying perturbations in glyoxylate metabolism. J Mol Med. 2012;90:1497–504. doi: 10.1007/s00109-012-0930-z. [DOI] [PubMed] [Google Scholar]
- 6.Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009;75:1264–71. doi: 10.1038/ki.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bacchetta J, Boivin G, Cochat P. Bone impairment in primary hyperoxaluria. Pediatr Nephrol. 2015;29:1–6. doi: 10.1007/s00467-015-3048-z. [DOI] [PubMed] [Google Scholar]
- 8.Cochat P, Hulton SA, Acquaviva C, Danpure CJ, Daudon M, de Marchi M, et al. Primary hyerpoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant. 2012;27:1729–36. doi: 10.1093/ndt/gfs078. [DOI] [PubMed] [Google Scholar]
- 9.Daudon M, Jungers P, Bazin D. Peculiar morphology of stones in primary hyperoxaluria. N Engl J Med. 2008;359:100–2. doi: 10.1056/NEJMc0800990. [DOI] [PubMed] [Google Scholar]
- 10.Daudona M, Jungers P. Clinical value of crystalluria and quantitative morpho-constitutional analysis of urinary calculi. Nephron Physiol. 2004;98:31–6. doi: 10.1159/000080261. [DOI] [PubMed] [Google Scholar]
- 11.Rumsby G, Williams E, Coulter-Mackie M. Evaluation of mutation screening as a first line test for the diagnosis of the primary hyperoxalurias. Kidney Int. 2004;66:959–63. doi: 10.1111/j.1523-1755.2004.00842.x. [DOI] [PubMed] [Google Scholar]
- 12.Milliner DS, Eickholt JT, Bergstralh E, Wilson DM, Smith LH. Primary hyperoxaluria: results of long-term treatment with orthophosphate and pyridoxine. N Engl J Med. 1994;331:1553–8. doi: 10.1056/NEJM199412083312304. [DOI] [PubMed] [Google Scholar]
- 13.Milliner DS. The primary hyperoxalurias: an algorithm for diagnosis. Am J Nephrol. 2005;25:154–60. doi: 10.1159/000085407. [DOI] [PubMed] [Google Scholar]
- 14.Harambat J, van Stralen KJ, Espinosa L, Groothoff JQ, Hulton SA, Cerkausiene R, et al. Characteristics and outcomes of children with primary oxalosis requiring renal replacement therapy. Clin J Am Soc Nephrol. 2012;7:458–65. doi: 10.2215/CJN.07430711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.North American Pediatric Renal Trials, Collaborative Studies (NAPRTCS) Annual Report. Rockville, Md, USA: The EMMES Corporation; 2010. [Google Scholar]
- 16.Lewis M, Shaw J, Reid C, Evans J, Webb N, Verrier-Jones K. Demography and management of childhood established renal failure in the UK. Nephrol Dial Transplant. 2007;22:165–75. doi: 10.1093/ndt/gfm336. Chapter 13. [DOI] [PubMed] [Google Scholar]
- 17.Hattori S, Yosioka K, Honda M, Ito H. The 1998 report of Japanese National Registry data on pediatric end-stage renal disease patients. Pediatr Nephrol. 2002;17:456–61. doi: 10.1007/s00467-002-0848-8. [DOI] [PubMed] [Google Scholar]
- 18.Cochat P, Liutkus A, Fargue S, Basmaison O, Ranchin B, Rolland MO. Primary hyperoxaluria type 1: still challenging. Pediatr Nephrol. 2006;21:1075–81. doi: 10.1007/s00467-006-0124-4. [DOI] [PubMed] [Google Scholar]
- 19.Hoppe B, Latta K, von Schnakenburg C, Kemper MJ. Primary hyperoxaluria–the German experience. Am J Nephrol. 2005;25:276–81. doi: 10.1159/000086358. [DOI] [PubMed] [Google Scholar]
- 20.Danpure CJ. Primary hyperoxaluria. In: Valle D, Beaudet AL, Vogelstein B, et al., editors. The online metabolic and molecular bases of inherited disease (OMM-BID) New York, NY: McGraw-Hill; 2014. Chap 133, Available online, Accessed 1-12-16. [Google Scholar]
- 21.El-Zanaty F, Way A. Egypt demographic and health survey. Cairo, Egypt: Ministry of Health, El-Zanaty and Associates and Macro International; 2008. [Google Scholar]
- 22.Gargah T, Khelil N, Youssef G, Karoui W, Lakhoua MR, Abdelmoula J. Primary hyperoxaluria type 1 in Tunisian children. Tunis Med. 2011;89:163–7. [PubMed] [Google Scholar]
- 23.Leumann EP, Dietl A, Matasovic A. Urinary oxalate and glycolate excretion in healthy infants and children. Pediatr Nephrol. 1990;4:493–7. doi: 10.1007/BF00869828. [DOI] [PubMed] [Google Scholar]
- 24.Cochat P, Koch Nogueira PC, Mahmoud MA, Jamieson NV, Scheinman JI, Rolland MO. Primary hyperoxaluria in infants: medical, ethical and economic issues. J Pediatr. 1999;135:746–50. doi: 10.1016/s0022-3476(99)70095-8. [DOI] [PubMed] [Google Scholar]
- 25.Millan MT, Berquist WE, So SK, Sarwal MM, Wayman KI, Cox KL, et al. One hundred percent patient and kidney allograft survival with simultaneous liver and kidney transplantation in infants with primary hyperoxaluria: a single-center experience. Transplantation. 2003;76:1458–63. doi: 10.1097/01.TP.0000084203.76110.AC. [DOI] [PubMed] [Google Scholar]
- 26.Cochat P, Groothoff J. Primary hyperoxaluria type 1: practical and ethical issues. Pediatr Nephrol. 2013;28:2273–81. doi: 10.1007/s00467-013-2444-5. [DOI] [PubMed] [Google Scholar]
- 27.Broyer M, Brunner FP, Brynger H, Dykes SR, Erich JH. Kidney transplantation in primary oxalosis: data from the EDTA Registry. Nephrol Dial Transplant. 1990;5:332–6. doi: 10.1093/ndt/5.5.332. [DOI] [PubMed] [Google Scholar]
- 28.Jamieson NV. A 20-year experience of combined liver/kidney transplantation for primary hyperoxaluria (PH1): the European PH1 transplant registry experience 1984–2004. Am J Nephrol. 2005;25:282–9. doi: 10.1159/000086359. [DOI] [PubMed] [Google Scholar]
- 29.vander Hoeven SM, van Woerden CS, Groothoff JW. Primary hyperoxaluria type 1, a too often missed diagnosis and potentially treatable cause of end-stage renal disease in adults: results of the Dutch cohort. Nephrol Dial Transplant. 2012;27:3855–62. doi: 10.1093/ndt/gfs320. [DOI] [PubMed] [Google Scholar]
- 30.Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012;8:467–75. doi: 10.1038/nrneph.2012.113. [DOI] [PubMed] [Google Scholar]
- 31.Hoppe B, Leumann E, Milliner D. Urolithiasis in childhood. In: Geary D, Schafer F, editors. Comprehensive Pediatric Nephrology. NewYork, NY, USA: Elsevier/WB Saunders; 2008. pp. 499–525. [Google Scholar]
- 32.van Woerden CS, Groothoff JW, Wanders RJ, Davin JC, Wijburg FA. Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrol Dial Transplant. 2003;18:273–9. doi: 10.1093/ndt/18.2.273. [DOI] [PubMed] [Google Scholar]
- 33.Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. 2013;369:649–58. doi: 10.1056/NEJMra1301564. [DOI] [PubMed] [Google Scholar]
- 34.Coulter-Mackie MB, White CT, Lange D, Chew BH. Primary hyperoxaluria type 1. Gene Reviews Last Update. 2014 [Google Scholar]