

Abstract
Sclareol synthase from Salvia sclarea (SsSS) naturally acts on 8α-hydroxy-copalyl diphosphate (1), stereoselectively adding water to produce (13R)-sclareol (2a), and similarly yields hydroxylated products with manifold other such bicyclic diterpene precursors. Here a key residue for this addition of water was identified. Strikingly, substitution with glutamine switches stereochemical outcome with 1, leading to selective production of (13S)-sclareol (2b). Moreover, changes to the stereospecificity of water addition with the structurally closely-related substrate copalyl diphosphate (4) could be accomplished with alternative substitutions. Thus, this approach is expected to provide biosynthetic access to both epimers of 13-hydroxylated derivatives of manifold labdane-related diterpenes.
Keywords: biosynthesis, terpene synthase, hydroxylation, enzymology, natural products
TOC image
Prototypical (class I) terpene synthases catalyze lysis of the allylic diphosphate ester in their substrate.1 The resulting allylic carbocation can undergo nucleophilic addition, which often triggers a complex cascade of carbocationic intermediates.1 While the catalyzed reaction is almost invariably terminated by deprotonation, water can be added in the course of catalysis, yielding an alcohol rather than more prototypical olefin.2 Given the importance of hydroxyl groups for both increasing solubility and providing hydrogen-bonding potential in terpenoid natural products, their insertion can be critical in such biosynthesis.3 However, the enzymatic basis for addition of water by class I terpene synthases is not well-understood. The only case where such activity has been clarified is a cineole synthase from Salvia fruticosa, for which determination of a crystal structure led to identification of a key asparagine that seems to position the relevant water molecule, as further indicated by the demonstration that isoleucine substitution blocked the addition of water.4
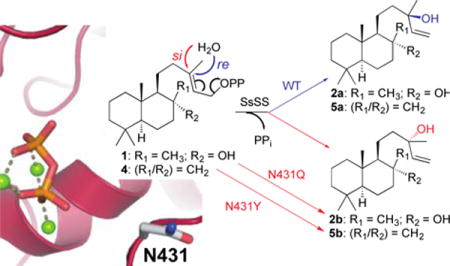
An intriguing alcohol generating class I terpene synthase is the sclareol synthase from Salvia sclarea (SsSS),5-6 which is used commercially in a biotechnological approach for production of this valuable diterpene fragrance precursor.7 SsSS naturally acts on 8α-hydroxy-copalyl diphosphate (1), ionizing the allylic diphosphate ester to form a 13-yl+ tertiary allylic carbocation to which water is added, with deprotonation of the resulting oxocarbenium yielding sclareol (2). More specifically, SsSS selectively generates 13R-sclareol (2a), which demonstrates that 1 is appropriately oriented in the active site for the relevant addition of a water molecule to the re face of its double bond (Scheme 1). Yet SsSS has been shown to exhibit extreme substrate promiscuity, readily reacting with essentially all known diterpenoid precursors to produce analogous 13-hydroxylated derivatives.8 In particular, 1 is derived from bicyclization of the general diterpene precursor (E,E,E)-geranylgeranyl diphosphate (GGPP) by a class II diterpene cyclase. These mechanistically distinct cyclases characterize the biosynthesis of the large labdane-related diterpenoid super-family. They are capable of producing manifold such bicyclic derivatives of GGPP. These bicycles are distinguished by stereo-configuration of the initially formed decalin ring structure, which can be followed by subsequent rearrangement, with both 1,2-migration of the methyl and hydride substituents, as well as the recently demonstrated production of alternative ring structures,9 possible. Further variation arises from positioning of the double bond, or hydroxyl group resulting from addition of water, generated by terminating deprotonation. Altogether, there are over 50 possible variants.2
Scheme 1.
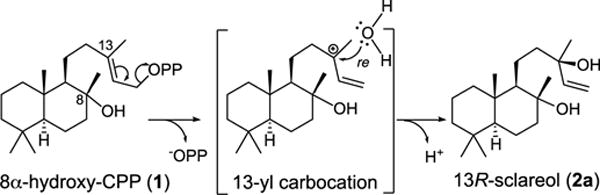
SsSS native reaction.
Previous studies of diterpene synthases have characterized the profound impact that single residue changes can have on product outcome.10-17 These residues were identified based on sequence comparisons of phylogenetically related, but functionally distinct enzymes. Notably, many of these residues are found in a short stretch of sequence,10-12, 18 which has since been shown to form part of the active site, in close spatial proximity to the signature DDxxD motif of class I terpene synthases.19-21
S. sclarea falls within the Lamiaceae plant family, from which a number of homologous diterpene synthases have been identified.22-25 Many of them, particularly those characterized by the loss of a large relictual domain,26 react with 1 and/or the structurally related non-hydroxylated isomer copalyl-diphosphate (CPP, 4). Among these, only SsSS adds water and produces a further hydroxylated diterpene. Sequence alignment of these diterpene synthases revealed intriguing differences in the previously identified product-outcome determining region (Figure 1A). While all the other (non-hydroxylating) enzymes are identical, SsSS varies at three positions within this region, which hints at a role for these in the distinct addition of water by SsSS.
Figure 1.
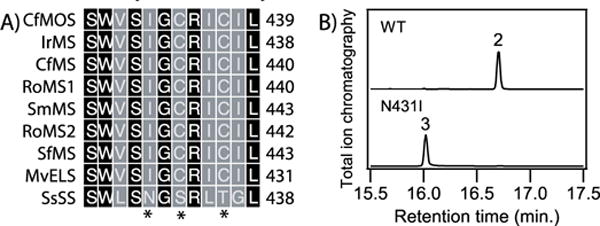
Identification of key asparagine in SsSS. A) Sequence alignment of the (class I) diterpene synthase product-determining region (see Methods for nomenclature; residues mutated here indicated by asterisks, *). B) Chromatograms from GC–MS analysis of wild-type (WT) and N431I mutant of SsSS products from 1 (numbers correspond to the chemical structures defined in the text).
To investigate the hypothesis that one or more of these residues is involved in the addition of water mediated by SsSS, each one was substituted with the corresponding residue from the non-hydroxylating homologs. The effect on product outcome was tested using a previously developed modular metabolic engineering system,27 whereby each of these SsSS mutants (as well as wild-type for comparison), was expressed in E. coli also engineered to produce 1. The resulting diterpene products can be readily extracted from the recombinant cultures and identified by comparison to authentic standards via analysis with gas-chromatography coupled to detection by mass-spectrometry (GC-MS). Following such characterization, it was found that, while the S433C and T436C mutants had no effect on product outcome (i.e., these still produced 2; Figure S1), the N431I mutant exclusively produced isoabienol (3; see Figures 1B and S2), resulting from direct deprotonation of the initially generated 13-yl+ intermediate at the neighboring methyl group. Notably, there was <10% decrease in yield for this mutant relative to wild-type SsSS in these engineered bacterial cultures (i.e., when comparing production of 3 versus 2; Figure S3). Thus, this single residue change completely blocks the ability of SsSS to add water to 1, seemingly with minimal effect on catalytic efficiency.
Intriguingly, this result at least nominally resembles the previously identified key asparagine residue in the S. fruticosa cineole synthase that directly binds the water molecule added in the course of the catalyzed reaction. Accordingly, it was similarly hypothesized that the key asparagine identified in SsSS might also directly interact with the water that is added to produce 1. This further led to speculation that substitution with residues that retain hydrogen bonding capacity might alter positioning of the water molecule. Of particular interest was sufficient alteration to enable specific addition to the si rather than re face of the double bond in 1.
The possibility that mutation of the key N431 in SsSS might enable such alternative stereochemical outcome was investigated by substitution with serine, threonine, aspartate, glutamine or glutamate, followed by co-expression of the resulting mutants in E. coli also engineered to produce 1, as above. Initial GC-MS analysis demonstrated that each of these SsSS mutants selectively produced 2 (Figure S4), again with minimal effects on yield (Figure S3). In order to separate 2a and the epimeric (13S)-sclareol (2b), a chiral column and extended temperature gradient, much as previously reported,28 were then employed. This revealed that these mutations did indeed alter stereochemical outcome, with varied ratios of 2a and 2b observed (Figures 2 and S5). Notably, while the other mutants produced roughly equal amounts of both epimers, the N431D and N431Q mutants selectively produced 2b, with SsSS:N431Q exhibiting almost complete reversal of the stereoselective addition of water – i.e., while wild-type SsSS produced 2a in 80% ee, SsSS:N431Q produced 70% ee of 2b. Thus, this single residue change essentially flips stereochemical outcome, changing the addition of water to the si face of the double bond in 1 and, thus, provides novel biosynthetic access to 2b.
Figure 2.
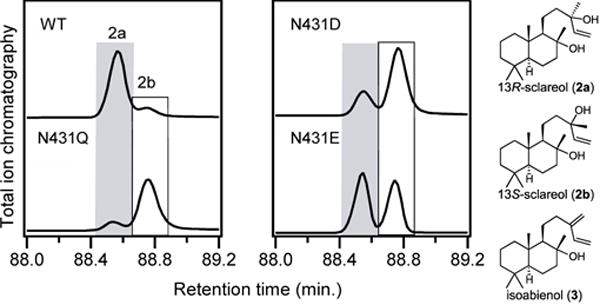
Altering N431 impacts stereochemical outcome with 1. Chromatograms from chiral GC–MS analysis of 2 produced by wild-type (WT) or indicated mutants of SsSS from 1. Epimers of 2, as well as 3, are shown on the right.
To further investigate the effect of these mutations, kinetics analysis was carried out with N431I and N431Q mutants, as well as wild-type SsSS. This demonstrated that these changes had minimal impact on catalytic efficiency, with less than 2-fold decreases in kcat/KM (Table 1), consistent with the almost negligible effects on biosynthetic yield noted above. Thus, this position exhibits clear plasticity, with the ability to readily accommodate these changes in side-chain composition or length (respectively), with minimal effect on overall catalytic activity (although the usual rate-limiting release of product1 may mask some change in the rate of the chemical reaction).
Table 1.
Steady-state kinetic parameters with 1.[a]
| SsSS | kcat (s−1) | KM (μM) | kcat/KM (s−1 M−1) |
|---|---|---|---|
| wild-type | 0.53 ± 0.04 | 7 ± 2 | 7.6 × 104 |
| N431I | 0.38 ± 0.02 | 6 ± 1 | 6.3 × 104 |
| N431Q | 0.47 ± 0.02 | 10 ± 1 | 4.7 × 104 |
Mean ± SE from three independent experiments.
Such plasticity seems consistent with the expected direct interaction between this residue and the water added in the course of catalysis. However, extensive modeling with RosettaCM29 indicated that the N431 side-chain hydrogen-bonds to a neighboring helix (i.e., the backbone carbonyl of C466). While it is clear that any such interaction is not critical for enzymatic stability (i.e., given the equivalent yield and activity of the N431I mutant), this at least suggests the possibility that N431 does not directly interact with the water molecule added in the course of catalysis. This alternative hypothesis was further investigated by docking the 2a product conformational library (see supporting information for details on the conformational search), with a distance constraint which maintains the relative position of the pyrophosphate co-product to 2a, into the SsSS active site with the Rosetta Modeling Suite.30-32 Despite being allowed to rotate (along with all other side-chains), N431 was not altered to interact with the 13R-hydroxyl group of 2a (Figures 3 and S6-8), consistent with the suggested lack of direct interaction between this residue and the relevant water.
Figure 3.
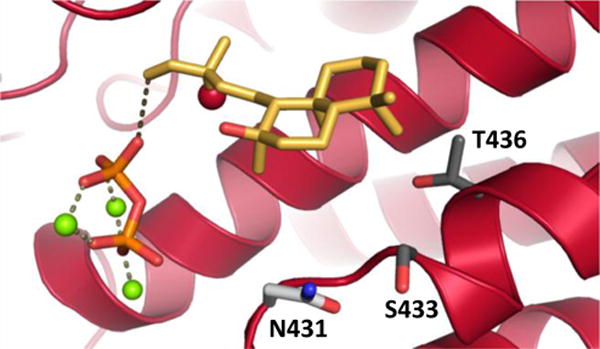
Indirect effect of N431 on addition of water. Lowest energy structure from docking 13R-sclareol (2a) into a representative homology model for SsSS (see Figure S8 for others). SsSS is shown in cartoon format, the grey dotted line between the co-product pyrophosphate-Mg2+ complex and 2a illustrates the constraint used in docking, the 13R-hydroxyl in 2a is shown as a red sphere, and the residues mutated here shown as sticks – N431 in light grey, S433 and T436 in dark grey.
The absence of a direct interaction suggests that alteration of N431 indirectly affects the addition of water. This was further investigated by use of the alternative substrate 4 that is structurally closely-related to 1. While wild-type SsSS readily reacts with 4 and selectively produces the 13-hydroxylated derivative manool (5), analogous to the production of 2 from 1 (Figure 4),8 the N431I mutant does not exhibit the same ability to completely block the addition of water to 4. Instead of exclusively producing sclarene (6) from 4, resulting from analogous direct deprotonation of the initially generated 13-yl+ intermediate at the neighboring methyl group, SsSS:N431I produces only a 4:6 mixture of this and 5 (Figure S9). Moreover, although chiral GC-MS analysis demonstrated that the resulting 5 is produced in a stereospecific fashion by wild-type SsSS, with the 13R-hydroxylated isomer 5a found in 90% ee, the N431Q mutant did not reverse this. While SsSS:N431Q does predominantly produce 5 (~95%; Figure S9), this is roughly split between both 5a and the 13S-hydroxyl epimer 5b (Figure 4).
Figure 4.
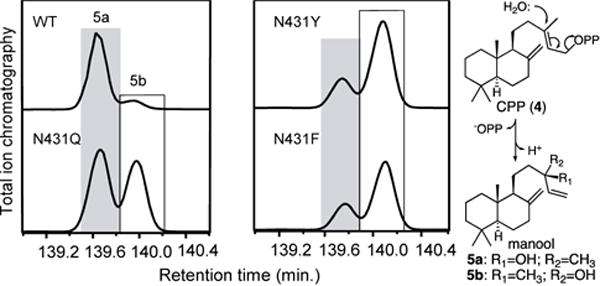
Altering N431 impacts stereochemical outcome with 4. Chromatograms from chiral GC–MS analysis of 5 produced by wild-type (WT) or indicated mutants of SsSS from 4. Catalyzed reaction, with epimeric products, shown on right.
The other polar mutants described above similarly specifically produced 5 (>95%; Figure S9), but largely retained wild-type stereospecificity – i.e., for 5a (Figure S10). Given the evidence that the residue at position 431 indirectly affects the addition of water, and the plasticity evident from the continued small effects on biosynthetic yield (<2-fold; Figure S11), it was hypothesized that alternative substitutions might enable stereoselective production of 5b. Since smaller residues (i.e., Ser and Thr) had not exhibited any significant change in stereoselectivity, substitution with larger residues – i.e., Phe and Tyr – was tested. Notably, both SsSS:N431F and N431Y not only predominantly produce 5 (>90%; Figure S9), but also largely reverse stereoselectivity, producing 5b in 60% and 62% ee, respectively (Figure 4).
More generally, the lack of absolute stereospecificity, even with the native substrate 1, suggests that SsSS does not precisely position its substrate and the water to be added in the course of the catalyzed reaction. Preliminary chiral GC-MS analysis of the 13-hydroxylated labdane-related diterpenoids produced by wild-type SsSS from the varied bicyclic precursors currently available via biosynthesis (i.e., functionally distinct class II diterpene cyclases), is consistent with this hypothesis, with variable levels of stereoselectivity observed (i.e., ratios of 13-hydroxy epimers). Nevertheless, given the ability of changes to N431 to impact the stereoselectivity of this addition of water shown here, it is expected that screening with the mutants described here (and perhaps others as well), will provide biosynthetic access to both epimers of 13-hydroxylated derivatives of the full range labdane-related diterpenoid precursors, substantially expanding the utility of this remarkable class I diterpene synthase.
Supplementary Material
Acknowledgments
This work was supported by a grant from the NIH (GM076324).
ABBREVIATIONS
- GGPP
(E,E,E)-geranylgeranyl diphosphate
- SsSS
sclareol synthase from Salvia sclarea
Footnotes
Supporting Information
Experimental details and supporting figures (PDF). This information is available free of charge on the ACS Publications website.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Christianson DW. Structural biology and chemistry of the terpenoid cyclases. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 2.Peters RJ. Two rings in them all: The labdane-related diterpenoids. Nat Prod Rep. 2010;27:1521–1530. doi: 10.1039/c0np00019a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pateraki I, Heskes AM, Hamberger B. Cytochromes P450 for terpene functionalisation and metabolic engineering. Adv Biochem Eng Biotechnol. 2015;148:107–39. doi: 10.1007/10_2014_301. [DOI] [PubMed] [Google Scholar]
- 4.Kampranis SC, Ioannidis D, Purvis A, Mahrez W, Ninga E, Katerelos NA, Anssour S, Dunwell JM, Degenhardt J, Makris AM, Goodenough PW, Johnson CB. Rational Conversion of Substrate and Product Specificity in a Salvia Monoterpene Synthase: Structural Insights into the Evolution of Terpene Synthase Function. Plant Cell. 2007;19:1994–2005. doi: 10.1105/tpc.106.047779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schalk M, Pastore L, Mirata MA, Khim S, Schouwey M, Deguerry F, Pineda V, Rocci L, Daviet L. Toward a biosynthetic route to sclareol and amber odorants. J Am Chem Soc. 2012;134:18900–3. doi: 10.1021/ja307404u. [DOI] [PubMed] [Google Scholar]
- 6.Caniard A, Zerbe P, Legrand S, Cohade A, Valot N, Magnard JL, Bohlmann J, Legendre L. Discovery and functional characterization of two diterpene synthases for sclareol biosynthesis in Salvia sclarea (L.) and their relevance for perfume manufacture. BMC Plant Biol. 2012;12:119. doi: 10.1186/1471-2229-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies E. Smarter smells. https://www.chemistryworld.com/feature/smarter-smells/1017487.article, accessed Jan. 8, 2017.
- 8.Jia M, Potter KC, Peters RJ. Extreme promiscuity of a bacterial and a plant diterpene synthase enables combinatorial biosynthesis. Metab Eng. 2016;37:24–34. doi: 10.1016/j.ymben.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu M, Jia M, Hong YJ, Yin X, Tantillo DJ, Proteau PJ, Peters RJ. Premutilin Synthase: Ring Rearrangement by a Class II Diterpene Cyclase. Org Lett. 2018;20:1200–1202. doi: 10.1021/acs.orglett.8b00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilderman PR, Peters RJ. A single residue switch converts abietadiene synthase into a pimaradiene specific cyclase. J Am Chem Soc. 2007;129:15736–15737. doi: 10.1021/ja074977g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu M, Wilderman PR, Peters RJ. Following evolution’s lead to a single residue switch for diterpene synthase product outcome. Proc Natl Acad Sci US A. 2007;104:7397–7401. doi: 10.1073/pnas.0611454104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrone D, Xu M, Fulton DB, Determan MK, Peters RJ. Increasing complexity of a diterpene synthase reaction with a single residue switch. J Am Chem Soc. 2008;130:5400–5401. doi: 10.1021/ja710524w. [DOI] [PubMed] [Google Scholar]
- 13.Keeling CI, Weisshaar S, Lin RPC, Bohlmann J. Functional Plasticity of paralogous diterpene synthases involved in conifer defense. Proc Natl Acad Sci US A. 2008;105:1085–1090. doi: 10.1073/pnas.0709466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawaide H, Hayashi K, Kawanabe R, Sakigi Y, Matsuo A, Natsume M, Nozaki H. Identification of the single amino acid involved in quenching the ent-kauranyl cation by a water molecule in ent-kaurene synthase of. Physcomitrella patens FEBS J. 2011;278:123–33. doi: 10.1111/j.1742-4658.2010.07938.x. [DOI] [PubMed] [Google Scholar]
- 15.Zerbe P, Chiang A, Bohlmann J. Mutational analysis of white spruce (Picea glauca) ent-kaurene synthase (PgKS) reveals common and distinct mechanisms of conifer diterpene synthases of general and specialized metabolism. Phytochemistry. 2012;74:30–9. doi: 10.1016/j.phytochem.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Jia M, Peters RJ. Extending a Single Residue Swtich for Abbreviating Catalysis in Plant ent-Kaurene Synthases. Front Plant Sci. 2016;7:e1756, 1–7. doi: 10.3389/fpls.2016.01765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irmisch S, Muller AT, Schmidt L, Gunther J, Gershenzon J, Kollner TG. One amino acid makes the difference: the formation of ent-kaurene and 16alpha-hydroxy-ent-kaurane by diterpene synthases in poplar. BMC Plant Biol. 2015;15:262. doi: 10.1186/s12870-015-0647-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia M, Zhou K, Tufts S, Schulte S, Peters RJ. A Pair of Residues That Interactively Affect Diterpene Synthase Product Outcome. ACS Chem Biol. 2017;12:862–867. doi: 10.1021/acschembio.6b01075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou K, Gao Y, Hoy JA, Mann FM, Honzatko RB, Peters RJ. Insights into diterpene cyclization from the structure of the bifunctional abietadiene synthase. J Biol Chem. 2012;287:6840–6850. doi: 10.1074/jbc.M111.337592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou K, Peters RJ. Electrostatic effects on (di)terpene synthase product outcome. ChemComm. 2011;47:4074–4080. doi: 10.1039/c0cc02960b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W, Feng X, Zheng Y, Huang CH, Nakano C, Hoshino T, Bogue S, Ko TP, Chen CC, Cui Y, Li J, Wang I, Hsu ST, Oldfield E, Guo RT. Structure, function and inhibition of ent-kaurene synthase from Bradyrhizobium japonicum. Sci Rep. 2014;4:6214. doi: 10.1038/srep06214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao W, Hillwig ML, Huang L, Cui G, Wang X, Kong J, Yang B, Peters RJ. A functional genomics approach to tanshinone biosynthesis provides stereochemical insights. Org Lett. 2009;11:5170–5173. doi: 10.1021/ol902051v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pateraki I, Andersen-Ranberg J, Hamberger B, Heskes AM, Martens HJ, Zerbe P, Bach SS, Moller BL, Bohlmann J, Hamberger B. Manoyl Oxide (13R), the Biosynthetic Precursor of Forskolin, Is Synthesized in Specialized Root Cork Cells in Coleus forskohlii. Plant Physiol. 2014;164:1222–36. doi: 10.1104/pp.113.228429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brückner K, Bozic D, Manzano D, Papaefthimiou D, Pateraki I, Scheler U, Ferrer A, de Vos RC, Kanellis AK, Tissier A. Characterization of two genes for the biosynthesis of abietane-type diterpenes in rosemary (Rosmarinus officinalis) glandular trichomes. Phytochemistry. 2014;101:52–64. doi: 10.1016/j.phytochem.2014.01.021. [DOI] [PubMed] [Google Scholar]
- 25.Pelot KA, Hagelthorn DM, Addison JB, Zerbe P. Biosynthesis of the oxygenated diterpene nezukol in the medicinal plant Isodon rubescens is catalyzed by a pair of diterpene synthases. PLoS One. 2017;12:e0176507. doi: 10.1371/journal.pone.0176507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hillwig ML, Xu M, Toyomasu T, Tiernan MS, Gao W, Cui G, Huang L, Peters RJ. Domain loss has independently occurred multiple times in plant terpene synthase evolution. Plant J. 2011;68:1051–1060. doi: 10.1111/j.1365-313X.2011.04756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cyr A, Wilderman PR, Determan M, Peters RJ. A Modular Approach for Facile Biosynthesis of Labdane-Related Diterpenes. J Am Chem Soc. 2007;129:6684–6685. doi: 10.1021/ja071158n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoshino T, Nakano C, Ootsuka T, Shinohara Y, Hara T. Substrate specificity of Rv3378c, an enzyme from Mycobacterium tuberculosis, and the inhibitory activity of the bicyclic diterpenoids against macrophage phagocytosis. Org Biomol Chem. 2011;9:2156–65. doi: 10.1039/c0ob00884b. [DOI] [PubMed] [Google Scholar]
- 29.Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, Baker D. High-resolution comparative modeling with RosettaCM. Structure. 2013;21:1735–42. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meiler J, Baker D. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins. 2006;65:538–48. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 31.Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, Meiler J, Baker D. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS One. 2011;6:e20161. doi: 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Combs SA, Deluca SL, Deluca SH, Lemmon GH, Nannemann DP, Nguyen ED, Willis JR, Sheehan JH, Meiler J. Small-molecule ligand docking into comparative models with Rosetta. Nat Protoc. 2013;8:1277–98. doi: 10.1038/nprot.2013.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.