

Abstract
RNA polymerase (RNAP) backtracking is a backward sliding of the enzyme along DNA and RNA. It plays important roles in many essential processes in bacteria and in eukaryotes. We describe here a fluorescence-based approach that allows a real-time observation of bacterial RNAP backtracking. A Cy3 fluorescence probe, when incorporated into a specific site in the nontemplate strand near the site of backtracking, allows monitoring RNAP movements near the probe due to a robust enhancement of fluorescence caused by protein proximity. Using this approach we showed that binding of NTP to the active site prior to phosphodiester bond formation inhibited backtracking consistent with the coupling of NTP binding to translocation. The extent and the kinetics of backtracking did not show a simple correlation with the instability of DNA:RNA hybrid indicating a more complex dependence of backtracking on DNA template sequence. Experiments with transcription through an abasic site in DNA template or neutravidin bound to biotinylated template strand base illustrated an important role of backtracking in defining how RNAP reacts to such obstacles in DNA template. The described approach will be a useful tool in deciphering the mechanism of backtracking and in studying factors that affect the backtracking.
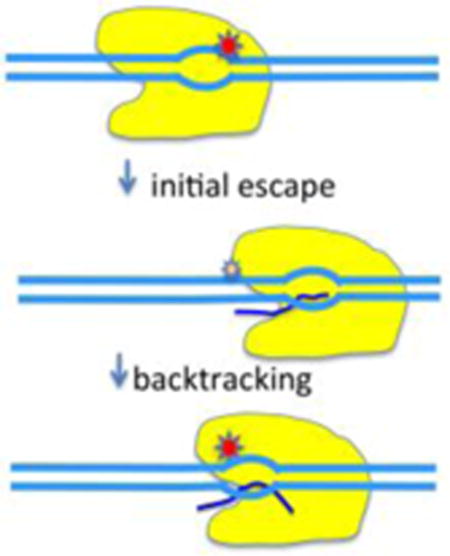
TOC image
Single-molecule studies elegantly demonstrated that the movement of RNA polymerase (RNAP) along DNA template during elongation is not a smooth uninterrupted process1–3. The major reason for this behavior is the ubiquitous transcriptional pausing of the enzyme where the forward translocation of the enzyme is briefly interrupted1, 4–10. RNAP backtracking, a more dramatic departure from a smooth unidirectional movement of the enzyme, was discovered almost twenty years ago11, 12 based on the initial discovery of irregular DNA footprints of elongating RNAP13. RNAP backtracking involves sliding of the enzyme backwards along DNA resulting in displacement of the 3′-end of RNA transcript from the active site14. Since the transcription bubble translocates backwards with the enzyme, backtracking involves simultaneous unwinding and rewinding of DNA duplex upstream and downstream of the bubble, respectively. Also, the RNA:DNA hybrid undergoes a similar synchronized unwinding and rewinding reaction resulting in threading of the 3′ end of RNA into the secondary channel of the enzyme. The resulting backtracked complex is unable to proceed with transcript elongation until the 3′ end of RNA regains its appropriate register with the active site either through spontaneous translocation of the enzyme along the template or through intrinsic or cleavage factor stimulated hydrolytic cleavage of the RNA15 to produce a new 3′ end properly aligned in the active site.
Backtracking is believed to play a role in many essential processes in bacteria and eukaryotes14. One obvious role of backtracking could be to stabilize an initial transient pause of RNAP. For example, promoter-proximal pausing in eukaryotes, an important regulatory mechanism, was shown to involve backtracking16. Although the estimates of how many paused sites are associated with backtracking vary4, 7, 9, 17–19, there is ample evidence that backtracking plays roles in a wide range of important cellular processes such as transcription termination, transcriptional fidelity, coupling of transcription to translation in bacteria, RNA processing and genome instability14.
Instability of RNA:DNA hybrid was suggested to be an important determinant of backtracking20, 21. Unstable RNA:DNA hybrid should produce an energetic incentive for RNAP to translocate backwards to a more stable hybrid. However, structural studies of backtracked complexes paint a more complicated picture of the determinants of backtracking22, 23. The DNA:RNA hybrid in Pol II complex containing 8 nt of backtracked RNA is distorted compared to a normal elongation complex22. Furthermore, RNA extruded into the secondary channel makes contacts with the channel that likely contribute to the stability of the backtracked complex and induce changes in conformation of catalytically important trigger loop22. Even though DNA:RNA hybrid appears to be undistorted in bacterial RNAP containing 1 nt backtracked RNA, this short backtracked complex also exhibited altered conformation of RNAP23. These observations suggest that backtracked complexes represent fundamentally different conformations compared to what would be expected if a normal elongation complex would simply move backwards along DNA template. Complete understanding of backtracking will require further biophysical and biochemical in-depth examination of the kinetics of backtracking, its dependence on DNA template sequence and the role of RNA secondary channel interactions. Such studies would be significantly enhanced by convenient means to directly observe the backtracking. The goal of this work was to develop an approach for real-time observation of backtracking and to use the approach to begin probing the mechanism of backtracking.
We have recently described a fluorescence approach that allowed real-time monitoring of promoter escape by RNAP24. We incorporated fluorescence probe (Cy3) into +2 position of the nontemplate strand of a promoter. Fluorescence of the probe increased upon binding of RNAP to the promoter and formation of transcription bubble. Promoter escape could be monitored in real-time following return of the fluorescence to the base level value upon departure of the enzyme from the promoter24. We reasoned that this assay could be adopted to follow RNAP movement due to backtracking in real-time from a site downstream of the promoter located sufficiently close to the promoter such that backtracking RNAP would return into proximity of Cy3 probe increasing its fluorescence. Our data validated this assay design and allowed probing several aspects related to the mechanism of backtracking. Furthermore, our data demonstrates the utility of the assay for monitoring RNAP translocation through a specific site on DNA template.
Materials and Methods
Materials
ATP, UTP, GTP and CTP (NTP’s), heparin, Red KlenTaq DV ReadyMix PCR mix were purchased from Sigma (St. Louis, MO). The non-hydrolysable CTP analogue (CpCpp) was from Jena Bioscience (Jena, Germany) and Cy3 NHS ester from GE Healthcare (Piscataway, NJ). All synthetic oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). Escherichia coli core RNAP with a His-tag on the C-terminal of the β′ subunit was expressed in BL21(DE3) cells using the polycistronic expression vector (pVS10; a gift from Dr. Irina Artsimovitch, The Ohio State University, Columbus, OH) and purified as described in25. σ70 was expressed and purified as previously described26. Purified GreB protein was a gift from Dr. Irina Artsimovitch (The Ohio State University, Columbus, OH)
Preparation of fluorophore-labeled promoter constructs
In order to introduce Cy3 labels into C6-amino-dT containing oligonucleotides, they were modified with Cy3-NHS and the labeled products were purified using HPLC on PRP-1 polymeric reversed-phase column (Hamilton) as described previously24. The DNA constructs described below (with the exception of P27A) were prepared by Taq polymerase extension of partial duplexes obtained by hybridizing appropriate synthetic oligonucleotides containing complementary overlapping sequences at their 3′ ends. Partial duplexes were prepared by mixing 0.25 μM non-template strand oligonucleotide with 0.275 μM template strand oligonucleotide. The final ds DNA products were purified on 1ml Resource Q column (Pharmacia Biotech) using a gradient of 0 M to 1 M NaCl in 25 mM TRIS (pH 8), 10 μM EDTA. Fractions containing labeled promoter were precipitated with ethanol to remove the salt and the DNA pellet was dissolved in Tris buffer. The following constructs were prepared (sequences of all synthetic oligonucleotides used for preparing fluorescent DNA constructs are provided in Supplemental Information):
P27 (135 bp): O1 (Cy3 labeled) and O2 synthetic oligonucleotides were used to prepare this construct. This is a 135 bp promoter based on λ PR promoter sequence. The first 27 transcribed bases (ATGTAGTAAGGAGGTGGTATGGAATTA) were changed from wt λ PR sequence such that transcription will not proceed past position +27 in the absence of CTP.
P15 (135 bp): O1 (Cy3 labeled) and O3 synthetic oligonucleotides were used to prepare this construct. Transcription with P15 promoter will not proceed past position +15 in the absence of CTP.
P54 (147 bp): O4 (Cy3 labeled) and O5 synthetic oligonucleotides were used to prepare this construct. This promoter contains Cy3 label at position +29 of the non-template strand. Transcription with P54 promoter will not proceed past position +54 in the absence of CTP.
- P27-31, P27-32, P27-33, P27-34, P27-35, P27-36, P27-37 (135 bp): O1 (Cy3 labeled) and O8-1, O8-2, O8-3, O8-4, O8-5, O8-6 or 08-7 synthetic oligonucleotides were used to prepare these constructs, respectively. These constructs are derivatives of P27 construct with base substitutions in the sequence preceding +27. These substitutions were designed to alter the stability of 9 bp DNA-RNA hybrid. The corresponding sequences of first 27 transcribed bases for these constructs were (base changes are underlined):
- P27-31: ATGTAGTAAGGAGGTGGTATGGAATTG
- P27-32: ATGTAGTAAGGAGGTGGTATGGAATGA
- P27-33: ATGTAGTAAGGAGGTGGTATGGAAGTA
- P27-34: ATGTAGTAAGGAGGTGGTATGGAATGG
- P27-35: ATGTAGTAAGGAGGTGGTATGGAAGGG
- P27-36: ATGTAGTAAGGAGGTGGTATGGAATTT
- P27-37: ATGTAGTAAGGAGGTGGTATGGAATAT
P27inv (135 bp): O9 (Cy3 labeled) and O10 synthetic oligonucleotides were used to prepare this construct. This construct contains Cy3 label at position -4 of the non-template strand. In comparison with P27 construct Cy3 label was at -4 (not at +2 as in P27; this was for technical reasons related to preparing promoter constructs using enzymatic extension of partially overlapping oligonucleotides) and all bases from +2 to +27 were flipped (i.e. A to T, T to A, and C to G). Transcription with P27inv promoter will not proceed past position +27 in the absence of GTP.
P37B (135 bp): O1 (Cy3 labeled) and O6 synthetic oligonucleotides were used to prepare this construct. This construct contains Cy3 label at position +2 of the non-template strand and a biotin at +37 position of template strand. Before use, neutravidin was added in 20× molar excess over DNA. Transcription with P37B promoter will not proceed past position +37 in the absence of CTP.
P27A (135 bp): This construct is a derivative of P27 promoter with an abasic site at position +28 of the template strand. This construct was prepared by ligating two duplexes (with complementary overhangs) corresponding approximately to the halves of P27A construct with T4 DNA ligase (Promega). O1 (Cy3 labeled), O7C and O7A, O7B were pre-hybridized first to prepare the two duplexes, respectively. The duplexes were mixed at 1.25 μM and were incubated with T4 ligase in 1× ligase buffer overnight at +16°C. Reaction mixture was run on 2% agarose gel, the band corresponding to the ligated product was cut from the gel and DNA was purified using Wizard SV Gel and PCR clean-up kit (Promega).
Fluorescence assay to detect RNAP backtracking
RNAP core (50 nM) was incubated with σ70 (200 nM) in transcription buffer (20 mM TRIS (pH 8.0), 100 mM NaCl, 10 mM MgCl2, 10 μM EDTA, 5% glycerol and 0.1 mg/ml BSA) for 10 min at 25°C to form RNAP holoenzyme. Promoter (4 μl of 500 nM stock) was added to 196 μl of RNAP holoenzyme solution in a spectrofluorometer cuvette and, if applicable (i.e. if the Cy3 label was at a position capable of detecting RNAP binding and melting of the promoter, for example, at +2 position), promoter melting reaction was monitored by fluorescence change as a function of time on Aminco-Bowman AB2 spectrofluorometer (excitation at 550 nm and emission at 570 nm). Transcription was initiated by adding NTP’s at 150 μM concentration and fluorescence intensity at 570 nm was monitored as a function of time. When heparin was present in the reaction, it was added at 0.2 mg/ml.
Transcription assays using molecular beacon as readout
Molecular beacon (5′-Fam-CCATCCTGCTGACTGCTTAATCGCTTCGGATGG-Dab- 3′, sequence complementary to the full-length transcript is underlined) was used to determine transcription activity (relative amount of full-length transcript produced) of the promoters. RNAP holoenzyme was assembled by incubating RNAP core (50nM) with σ70 (200 nM) in transcription buffer (20mM TRIS (pH 8.0), 100mM NaCl, 10mM MgCl2, 10 μM EDTA, 5% glycerol and 0.1 mg/ml BSA) for 10 min at room temperature. Promoter (10 nM) was added to holoenzyme and incubated for 20 min at room temperature. Transcription was initiated by addition of 3 or 4 NTP’s (150 μM) (with or without heparin (0.2mg/ml) or GreB (150 nM)) and allowed to proceed for 30 minutes at room temperature. EDTA (25 mM) was added to stop the transcription followed by addition of molecular beacon (5-20 nM). Fluorescence intensity was measured after 30 min incubation with the beacon at room temperature using fluorescent reader (Spectra Fluor Plus, Tecan). Amount of the transcript was calculated from calibration curve obtained using synthetic oligonucleotide (GAAGCGATTAAGCAGTCAGCA) complementary to the beacon.
Calculation of DNA-RNA heteroduplex stability profiles
Relative thermodynamic stability of RNA:DNA hybrids was approximated by calculating free energies of corresponding DNA:DNA duplexes using nearest neighbor model and parameters from27. A custom script in R was written that calculates stabilities of all possible 9 bp RNA:DNA hybrids for a sequence of interest. Calculated free energies were normalized to the average of free energies of all possible sequence combinations for 9 bp long duplexes.
Results
Design of fluorescent promoter construct for real-time monitoring of promoter escape
Fig. 1A illustrates a design of the promoter construct that we prepared to detect RNAP backtracking from a site near the promoter in real-time. A fluorescent probe (Cy3) was placed at +2 position of the nontemplate strand. We have previously shown that fluorescence of Cy3 at this position can report the melting of the promoter and promoter escape by RNAP24. Fluorescence intensity of Cy3 attached to DNA was shown to be enhanced by the proximity of DNA-bound protein (Protein Induced Fluorescence Enhancement, PIFE28). We reasoned that when transcription is induced from this promoter construct, RNAP escape from the promoter would be detected by time-dependent decrease of Cy3 fluorescence (as previously demonstrated24). However, if RNAP transcribes to a site in a vicinity of the promoter and then backtracks towards the promoter, the backward movement of RNAP would be detected by the enhancement of Cy3 fluorescence due to the increased proximity of the enzyme to the probe. In order to create a potential backtracking site, the first 27 nt of transcribed sequence was engineered to a CTP-less cassette. Thus, when transcription is started with a mixture of ATP, UTP and GTP, RNAP can transcribe and translocate only up to position +27. Since PIFE is a relatively short distance (compared to, for example, FRET) effect28, translocation to +27 is expected to eliminate PIFE completely. Since the relative instability of RNA:DNA hybrid was thought to be an important determinant of RNAP backtracking20, 21, the hybrid for elongating RNAP at position +27 was designed to be relatively unstable (Fig. 1B). The experiments with the constructs in Fig. 1A fully validated the described above expectations (Fig. 1C). Addition of fluorescent promoter to the RNAP resulted in time dependent increase of Cy3 fluorescence that accompany the formation of the open complex24. When all four NTP’s were added, a rapid decrease of Cy3 fluorescence was observed reporting the escape from the promoter24. Under such conditions production of full-length transcript was confirmed using an assay employing molecular beacon complementary to the 5′ end of the transcript (not shown and Fig. 8E). Heparin-induced dissociation of the open complex can be neglected since control experiments (not shown) showed that ~96% of open complexes remained intact after 200 s challenge with heparin in the absence of NTPs (a typical timescale of promoter escape kinetics for the promoter used in our studies). However, when transcription was initiated with a mixture of ATP, UTP and GTP, initial rapid decrease of fluorescence due to the escape was followed by a time-dependent recovery of fluorescence intensity (Fig. 1C). No full-length transcript was detected under such conditions using an assay employing molecular beacon complementary to the 5′ end of the transcript (not shown and Fig. 8E). We interpret this fluorescence signal recovery as a esult of a slow accumulation of backtracked RNAP. The time-dependence of fluorescence observed upon addition of ATP, UTP and GTP is thus a net effect of escape and backtracking.
Figure 1.
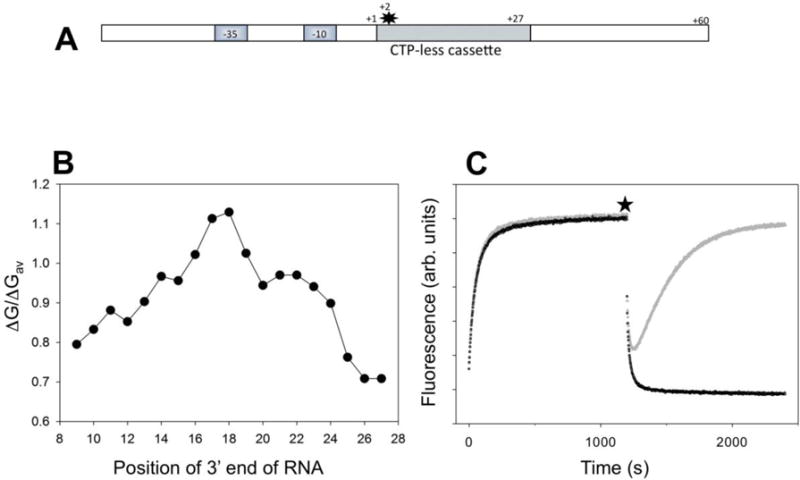
(A) Design of fluorescent construct (P27 promoter) for real-time detection of RNAP backtracking. The star denotes Cy3 label at the +2 position of the nontemplate strand. (B) Calculated relative duplex stabilities (ratio of duplex free energy of the 9 bp hybrid to the average of free energies of all possible sequence combinations for 9 bp long duplexes) of the 9 bp RNA:DNA hybrids along the sequence of the first 27 transcribed nucleotides of P27. (C) Fluorescence intensity changes of P27. At time 0 the P27 was added to a solution of RNAP in a fluorometer cuvette. At the time point marked by a star, the transcription was started by adding ATP, UTP, GTP and heparin (grey) or ATP, UTP, GTP, CTP and heparin (black).
Figure 8.
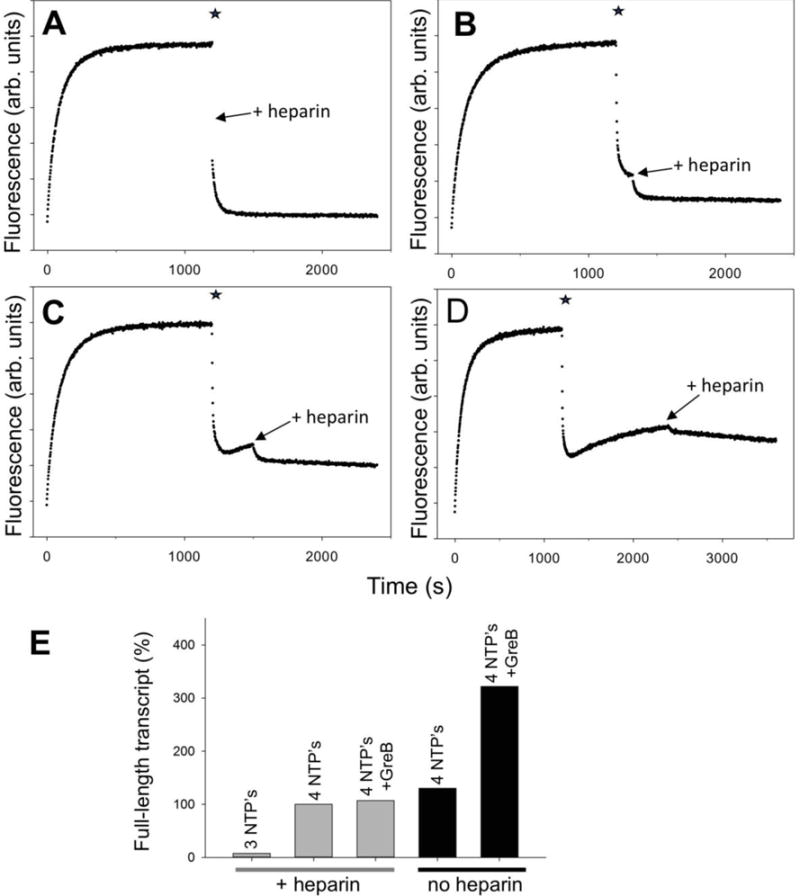
RNAP backtracking during multiple rounds of transcription from P27. At time 0 the P27 was added to a solution of RNAP in a fluorometer cuvette. The star denotes addition of ATP, UTP, GTP and CTP. Heparin was added (as indicated by an arrow) at various delay times after the start of transcription with 4 NTP’s: (A) 15 s, (B) 2 min, (C) 5 min and (D) 20 min. (E) Effect of GreB on the amounts of full-length transcript produced from P27. Transcription was allowed to proceed for 30 min. The data were normalized to the amount of transcript made by P27 under single round of transcription condition (in the presence of heparin).
Several lines of evidence support the above-described interpretation of fluorescence changes observed upon adding ATP, UTP and GTP. First, the species that accumulated during fluorescence recovery phase of the reaction were transcriptionally inactive (as would be expected for bactracked RNAP), i.e. they were unable to rapidly start transcription and escape upon the addition of the missing NTP (CTP) (Fig. 2). When CTP was added with only a short delay (15 sec) after initiating transcription with ATP, UTP and GTP, a rapid and complete promoter escape was observed (Fig. 2A), essentially identical to when all 4 NTP’s were added together (Fig. 1C). The addition of CTP with a longer delay, after a significant fraction of the fluorescence recovery phase was allowed to occur, resulted in halting the further progression of fluorescence recovery phase and produced an escape of RNAP with a very slow kinetics (Fig. 2B-D). The behaviors depicted in Fig. 2 are in complete agreement with the RNAP backtracking being responsible for fluorescence signal recovery phase illustrated by Fig. 1C. The slow escape observed upon delayed addition of CTP (Fig. 2 B-D) could be due to either slow intrinsic RNA cleavage by RNAP active site or slow reversal of backtracking by spontaneous forward translocation of RNAP to position +27 that would bring 3′ end of RNA back into the active site of the enzyme allowing resumption of transcription.
Figure 2.
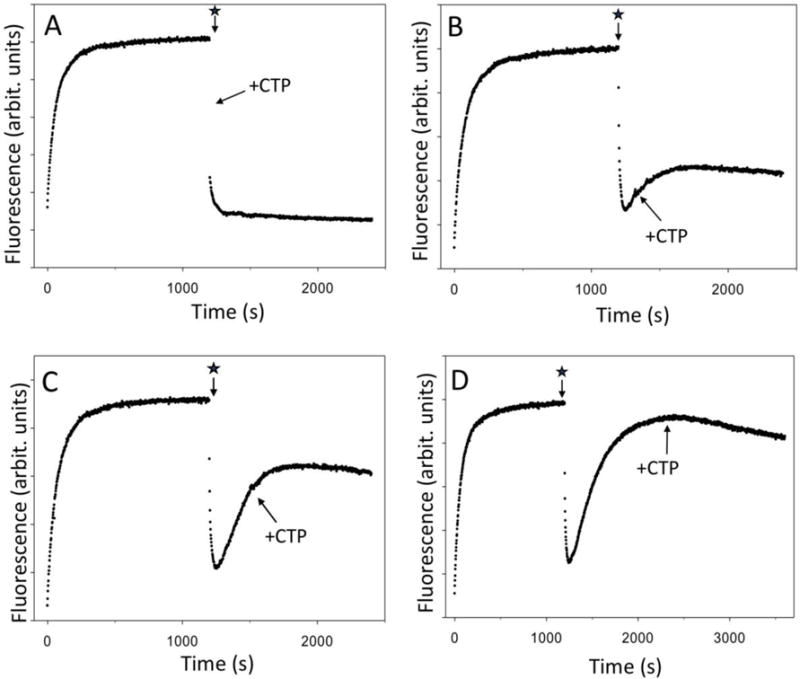
RNAP complexes that accumulate during fluorescence signal recovery phase of the reaction are transcriptionally inactive. The star denotes addition of ATP, UTP, GTP and heparin to the sample. The missing NTP (CTP) was added (as indicated by an arrow) at various delay times after the start of transcription with 3 NTP’s: (A) 15 s, (B) 2 min, (C) 5 min and (D) 20 min.
These conclusions are further supported by the results of experiments examining the effect of GreB on the reactions depicted in Fig. 1 and 2. GreB was shown to bind to RNAP near and inside the secondary channel of RNAP15, 29. Its binding activates the intrinsic RNA hydrolytic activity of RNAP resulting in a rapid cleavage of backtracked RNA extruded into the secondary channel29. Such cleavage reestablishes proper position of 3′ end of RNA in RNAP active site, rescuing RNAP from its backtracked state. Addition of GreB at the end of fluorescence signal recovery phase resulted in a rapid drop of fluorescence intensity consistent with the rescue of RNAP from backtracked state and rapid translocation to position +27 (Fig. 3A). When transcription was initiated with ATP, UTP and GTP in the presence of GreB, only the fluorescence intensity drop signaling RNAP escape was observed and no fluorescence signal recovery phase associated with backtracking was observed (Fig. 3B).
Figure 3.
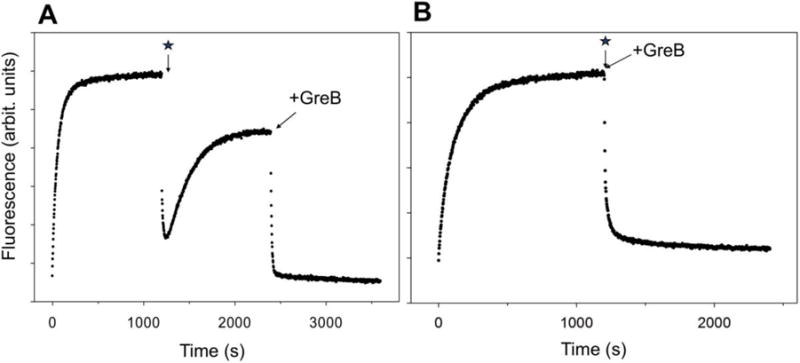
The effect of GreB on the time course of fluorescence intensity changes of P27. The star denotes initiation of transcription. (A) Transcription was initiated by the addition of 3 NTP’s and heparin and at a time point marked by an arrow GreB was added (150 nM). (B) Transcription was initiated by the addition of 3 NTP’s and heparin together with 150 nM GreB.
Mapping of the position of transcription bubble with permanganate footprinting (Fig. S1A, Supplemental Information) (that preferentially modifies DNA at thymines that are not base-paired with cognate adenine) at the end of fluorescence signal recovery phase of the reaction initiated by the addition of ATP, UTP and GTP, showed that the position of the transcription bubble was not consistent with either what would be expected for RNAP stalled at position +27 (Fig. S1C, Supplemental Information) or RNAP in the open complex, but was similar to transcription bubble position observed in P15 promoter (that in the presence of ATP, UTP and GTP can transcribe to position +15) (Fig. S1C, Supplemental Information). It can be concluded thus that RNAP backtracking is extensive but it does not involve backtracking all the way back to the initial open complex (even though fluorescence signal intensity recovered due to backtracking almost to the value observed in the open complex (Fig. 1C)). This is consistent with measurements of FRET between a fluorescence probe at position -37 of P27-37Cy3 promoter and a probe at a specific site in the β subunit of RNAP (Fig. S2, Supplemental Information). FRET signal that arises from binding of RNAP to the promoter, was eliminated upon starting transcription both with 4 NTP’s as well as with 3 NTP’s indicating that when RNAP backtracked, it did not return to a location on the promoter close enough for detectable FRET to position -37 (Fig. S2, Supplemental Information). Taken together, the data in Figs. 2,3, S1 and S2 confirm that DNA construct depicted in Fig. 1A allows real-time detection of RNAP backtracking with a simple fluorescence intensity readout.
DNA sequence dependence of backtracking
Unstable RNA:DNA hybrid has been suggested to be an important determinant in RNAP backtracking20, 21. In designing P27 promoter construct a sequence defining a relatively unstable RNA:DNA hybrid was placed next to position +27 (Fig. 1B). To test if indeed the relative instability of the hybrid near position +27 was important for the backtracking observed with P27 promoter, we prepared a series of derivatives of P27 promoter where G’s were introduced into the sequence preceding position +27 to change the stability of RNA:DNA hybrid in the vicinity of this position. Despite significant changes of the RNA:DNA hybrid stability introduced by these mutations (Fig. 4A), we observed only moderate changes in the kinetics or extent of backtracking (Fig. 4B). Furthermore, we could not identify any simple correlation between the extent of changes in hybrid energy profile (Fig. 4A) and their effect on RNAP backtracking behavior (Fig. 4B). For example, the largest effect on backtracking was observed with P27-33 construct (Fig. 4B, blue curve), even though the shape of the energy profile of this mutant was most similar to P27 promoter (Fig. 4A). On the other hand, in another set of constructs (Fig. 4 C&D), very small changes to the sequence and hybrid energy (Fig. 4C) produced substantial changes in the kinetics. Finally, only a small degree of backtracking could be detected (Fig. S3, Supplemental Information) in a P27 variant (P27inv) where the T′s, A′s and G′s between positions +2 and +27 were switched to A′, T′s and C′s, although in comparison to P27, this construct had energy profile of 9 bp hybrid stability identical to P27 (Fig. S3A, Supplemental Information). Taken together, these results suggest that RNA:DNA hybrid stability is not the only determinant of backtracking. Specific sequence of the template is also important.
Figure 4.
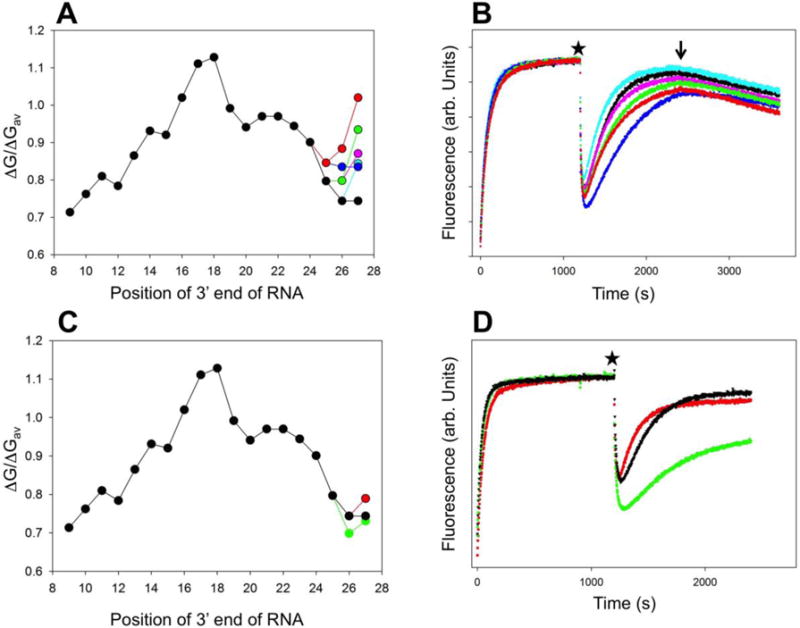
(A) Comparison of calculated relative 9 bp duplex stabilities for P27-31 (cyan), P27-32 (pink), P27-33 (blue), P27-34 (green), and P27-35 (red) constructs with P27 construct (black). (B) Fluorescence intensity changes of constructs from panel A (the color coding is the same as in panel A). At time 0 the promoter was added to a solution of RNAP in a fluorometer cuvette. At the time point marked by a star the transcription was started by adding ATP, UTP, GTP and heparin. The arrow indicates addition of CTP. (C) Comparison of calculated relative 9 bp duplex stabilities for P27-36 (red), P27-37 (green), with P27 construct (black). (D) Fluorescence intensity changes of constructs from panel C (the color coding is the same as in panel D). At time 0 the DNA construct was added to a solution of RNAP in a fluorometer cuvette. At the time point marked by a star the transcription was started by adding ATP, UTP, GTP and heparin.
The effect of NTP binding
When RNAP is allowed to transcribe from P27 promoter in the absence of CTP, it will do so until it reaches position +27 at which point the transcription is inhibited by the lack of a correct NTP to bind to the active site. NTP binding to the RNAP active site before phosphodiester bond formation was shown to be an important driving force of RNAP translocation30. To test if the binding of CTP before phosphodiester formation in the context of P27 construct could inhibit RNAP backtracking by favoring RNAP forward translocation, we tested the effect of a nonhydrolyzable CTP analogue (CpCpp) on RNAP backtracking on P27 template (Fig. 5). Addition of CpCpp produced a concentration-dependent inhibition of RNAP backtracking (Fig. 5A). Furthermore, the effect of CpCpp was limited to the early stage of backtracking since a delay in the addition of the analogue eliminated its inhibitory effect (Fig. 5B). These observations are thus consistent with an important role of the initial binding of the NTP in RNAP active site in promoting forward translocation over the backtracking.
Figure 5.
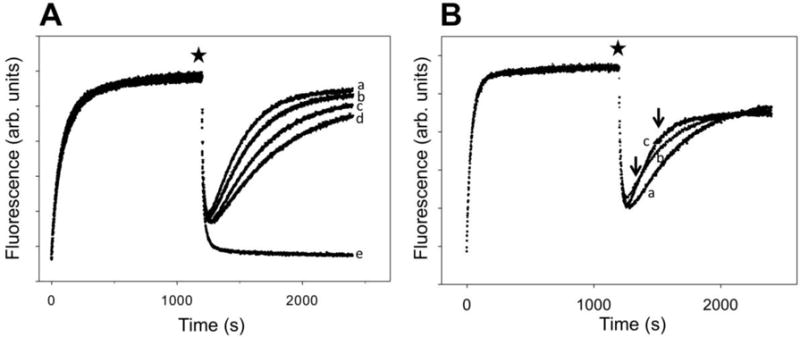
The effect of nonhydrolizable analogue of CTP (CpCpp) on RNAP backtracking on P27 construct. (A) Transcription reactions were initiated by adding ATP, UTP, GTP, heparin and no CpCpp (a), 50 μM CpCpp (b), 150 μM CpCpp (c) or 300 μM CpCpp (d). Curve (e) corresponds to transcription initiated by adding ATP, UTP, GTP, CTP and heparin. (B) Transcription reactions were initiated by adding ATP, UTP, GTP, heparin. CpCpp (150 μM) was added together with NTP’s (a) or after 2 min (b) or 5 min (c) delay after addition of NTP’s. Delayed addition of CpCpp is indicated by the arrows.
Observation of backtracking at a site further downstream from the promoter
Fig. 6A illustrates the design of a construct (P54) that we prepared to test if the close proximity of backtracking site (+27) to the promoter in the P27 construct could be a factor contributing to the extensive backtracking observed with this DNA template. In P54 construct the backtracking site was moved to position +54 and the sequence immediately preceding position +54 was made identical to the sequence immediately preceding position +27 in P27 construct. Thus, RNAP arriving at position +54 in P54 experiences essentially identical environment as RNAP arriving at position +27 of P27, only farther from the promoter. Cy3 probe in P54 was placed at position +29 of the nontemplate strand, which is an equivalent to +2 in P27 in the distance from the backtracking site. Formation of the open complex in P54 construct resulted in a small time-dependent change of fluorescence (Fig. 6B). It was interesting and not expected that the probe that far away from the promoter was still sensitive to the structural changes in RNAP-promoter complex associated with open complex formation. In contrast to the probe at +2 position, formation of the open complex in P54 resulted in quenching of fluorescence of the probe at position +29. Initiation of transcription with 4 NTP’s and heparin produced a sharp spike of fluorescence shortly after NTP’s were added (Fig. 6B, blue). We interpret this spike as resulting from a quick transit of RNAP through position +29 resulting in a temporary increase of Cy3 fluorescence. P54 promoter produced 84% of full-length transcript compared to P27 promoter (not shown) indicating that Cy3 attached to a base in nontemplate strand was not a significant obstacle for elongating RNAP. Initiation of transcription with 3 NTP’s and heparin produced a sharp spike of fluorescence shortly after NTP’s were added followed by a slow fluorescence intensity increase phase (Fig. 6B, red) that was very similar to the slow fluorescence recovery phase observed with P27 promoter under the same experimental conditions (Fig. 1C). As in the case of P27 promoter, RNAP-DNA complexes accumulating during this phase were transcriptionally inactive since addition of CTP produced only a very slow decrease of fluorescence consistent with a slow spontaneous translocation of the backtracked complexes back to position +54 allowing resumption of transcription. Initiation of transcription with 3 NTP’s and heparin in the presence of GreB did not produce a slow fluorescence intensity increase phase characteristic of backtracking (Fig. 6B, green). The higher level of fluorescence that remains at these experimental conditions compared to transcription with 4 NTP’s is most likely due to small residual PIFE effect of RNAP stalled at position +54 on the probe at position +29. A rapid decrease of fluorescence upon addition of CTP to the level observed when 4 NTP’s were used to initiate the reaction (Fig. 6B) is consistent with such interpretation. Taken together, the data in Fig. 6 demonstrated that close proximity to the promoter is not an important factor in backtracking observed with P27 construct suggesting that lingering RNAP-promoter contacts are unlikely to contribute to the observed backtracking. Furthermore, these data illustrate the potential of fluorescence backtracking assay to monitor backtracking at any site within DNA template. Additionally, the fact that Cy3 probe introduced into nontemplate strand does not impede elongation by RNAP, together with a large increase of fluorescence when RNAP travels through a site of Cy3 attachment, suggests an interesting potential of using DNA constructs similar to the one in Fig. 6A for precise measurements of rate of elongation.
Figure 6.
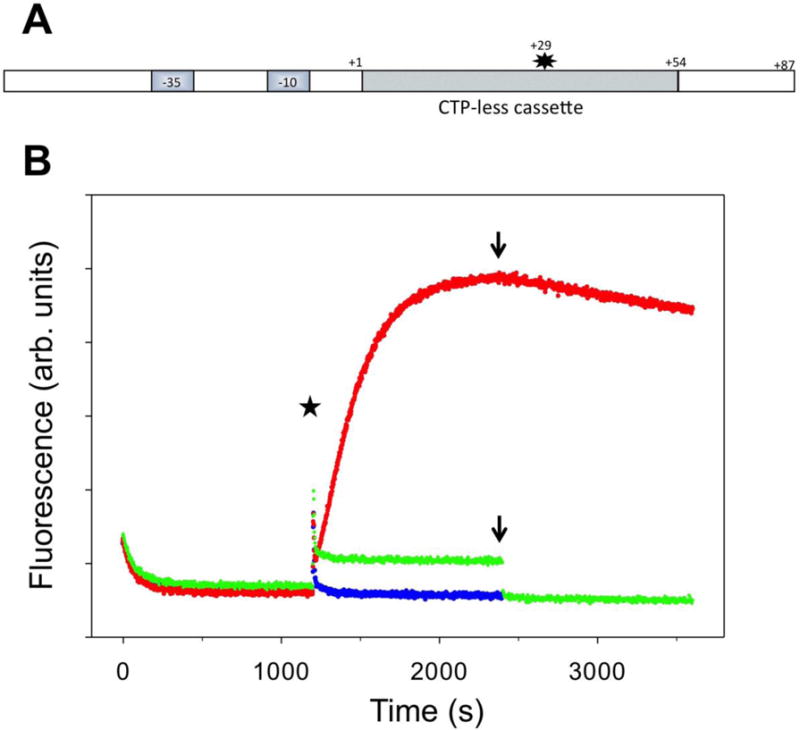
(A) Design of fluorescent construct (P54) for real-time detection of RNAP backtracking away from the promoter. The star denotes Cy3 label at +29 position of the nontemplate strand. (B) Fluorescence intensity changes of P54 promoter. At time 0 the P54 was added to a solution of RNAP in a fluorometer cuvette. At the time point marked by a star the transcription was started by adding ATP, UTP, GTP and heparin (red), ATP, UTP, GTP, heparin, and GreB (green) or ATP, UTP, GTP, CTP and heparin (blue). The arrow denotes time points where the missing NTP (CTP) was added.
Real-time detection of backtracking due to an obstacle in DNA template
Encounter by an elongating RNAP of an obstacle to translocation (damaged DNA template, a protein bound to DNA, etc.) could produce backtracking9, 14, 31, 32, which would then become an important factor in determining the outcome of such encounter. We prepared two DNA templates that were designed to record in real-time RNAP behavior when it encounters an obstacle to elongation (Fig. 7A&C). The first construct (P27A, Fig. 7A) was a derivative of P27 where an abasic site was introduced to position +28 of the template strand. When transcription was initiated with 3 NTP’s, time dependence of fluorescence signal (Fig. 7B) was very similar to P27 promoter (rapid decrease of fluorescence due to promoter escape followed up by slow fluorescence intensity recovery phase due to RNAP backtracking). This was expected since P27A and P27 constructs have identical sequence up to position +27. Addition of GreB (Fig. 7B) resulted in a rapid drop of fluorescence (similar to what we observed with P27 promoter (Fig. 3A)) consistent with the rescue of RNAP from backtracked state and rapid translocation to position +27. We concluded that abasic site at position +28 did not affect backtracking behavior of RNAP when transcription was initiated with 3 NTP’s. Addition of missing NTP (CTP) after GreB was added produced additional drop in fluorescence indicating the ability of RNAP to read through the abasic site under these conditions. When the transcription was initiated with 4 NTP’s (in the absence of GreB), a relatively small but significant amount of backtracking could be detected as a slow low amplitude fluorescence recovery phase (Fig. 7B). This fluorescence intensity recovery phase was absent when transcription was initiated with 4 NTP’s in the presence of GreB (Fig. S4, Supplemental Information). We conclude based on these observations that when RNAP arrives at the abasic site (when all 4 NTP’s are present), the majority of RNAP’s reads through the abasic site but a fraction of it does not and becomes trapped in the backtracked state. GreB, when present, allows each RNAP molecule to perform multiple trials of read through the abasic site enhancing the efficiency of the read-through. These data provide a real-time insight into behavior of RNAP at DNA damage site and emphasize the role of GreB factors in facilitating the transcription through the obstacles. These interpretations are consistent with previously described ability of bacterial RNAP to transcribe through the abasic site33, 34. P27A construct produced 68% of full-length transcript amount compared to P27 construct under single round of transcription conditions. GreB increased the amount of transcript made by P27A by 10%. These activity measurements are consistent with RNAP partitioning between read through and backtracked state at the abasic site with the read through being favored.
Figure 7.
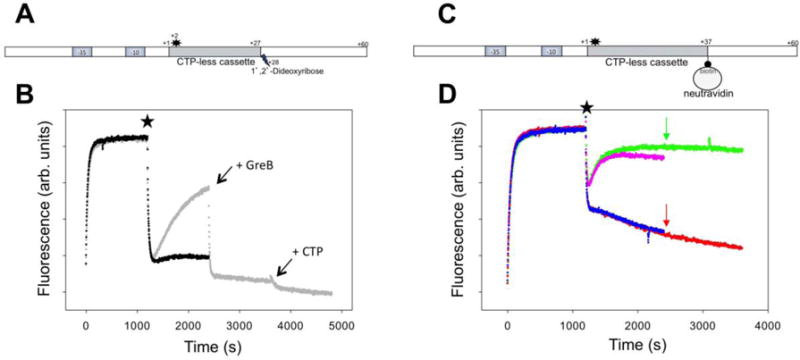
(A) Design of fluorescent construct (P27A) containing an abasic site at +28 position of the template strand. The star denotes Cy3 label at +2 position of the nontemplate strand. (B) Fluorescence intensity changes of P27A construct. At the time point marked by a star transcription was started by adding ATP, UTP, GTP and heparin (grey) followed by addition of GreB and CTP (indicated by the arrows) or by adding ATP, UTP, GTP, CTP and heparin (black). (C) Design of fluorescent construct (P37B) with neutravidin bound at position +37 of the template strand. The star denotes Cy3 label at +2 position of the nontemplate strand. (D). Fluorescence intensity changes of P37B construct. At the time point marked by a star transcription was started by adding ATP, UTP, GTP and heparin (green), ATP, UTP, GTP, CTP and heparin (pink), ATP, UTP, GTP, heparin and GreB (red) or ATP, UTP, GTP, CTP, heparin and GreB (blue). The arrows denote time points where the missing NTP (CTP) was added.
P37B construct (Fig. 7C) was designed to allow real-time observation of RNAP behavior when it encounters a much more difficult obstacle on DNA template (neutravidin bound to biotinylated template strand base). Biotin was introduced at position +37 (i.e. 10 bp further downstream of the promoter compared to abasic site in P27A) to account for steric effects due to size of neutravidin molecule. The sequence of 27 nucleotides immediately preceding the site of biotin incorporation is identical to 27 nucleotides preceding abasic site in P27A. Encounter of RNAP with this essentially irreversible obstacle produced robust backtracking that was the same regardless if transcription was initiated with 3 or 4 NTP’s (Fig. 7D). The presence of strong physical obstacle to RNAP translocation provides thus a strong impetus to RNAP backtracking. In the presence of GreB a rapid decrease of fluorescence was followed by a much slower fluorescence decrease phase (Fig. 7D). We interpret the initial fast fluorescence decrease phase as due to rapid RNAP escape and RNAP translocation until it comes into the initial contact with neutravidin. Slow fluorescence decrease phase most likely reflects a slow establishment of a steady-state balance between repeated cycles of RNAP backtracking and rescue from backtracked state and the force opposing forward translocation resulting from collision of RNAP with immobilized neutravidin. Alternatively, this slow fluorescence decrease phase could represent a slow RNAP read through neutravidin obstacle. This interpretation is unlikely since measurement of full-length transcripts by beacon assay under conditions of Fig. 7D did not reveal a slow accumulation of full-length transcripts (not shown).
RNAP backtracking in the absence of heparin
The experiments illustrated by Figs. 1–7 were performed in the presence of heparin that prevents rebinding of RNAP to the promoter after the transcription is initiated by addition of NTP’s, limiting the transcription to a single round. When transcription was initiated on P27 construct with 4 NTP’s in the absence of heparin, the initial rapid decrease of fluorescence due to promoter escape was followed by a slower fluorescence recovery phase (Fig. S5, Supplemental Information), resembling the fluorescence curves observed in the presence of heparin. The simplest explanation for the shape of the curves in Fig. S5 could be RNAP re-binding following promoter escape. However, we would not expect that such rebinding should produce a slow fluorescence increase since in the presence of 4 NTP’s, any RNAP that would bind to the promoter, would rapidly escape. We would then expect that some constant (higher than the background) level of fluorescence intensity would be established under these conditions reflecting a steady state produced by re-binding followed by rapid escape. We wondered if this slow fluorescence intensity increase phase in Fig. S5 could correspond to accumulation of backtracked RNAP during multiple rounds of transcription. The data in Fig. 8 address this question. In these experiments transcription was initiated by adding 4 NTP’s in the absence of heparin, followed by addition of heparin after different delay times. When heparin was added shortly after the initiation of transcription with 4 NTP’s (Fig. 8A), a rapid and complete RNAP escape occurred, essentially the same as when 4 NTP’s were added together with heparin. However, with an increase of the delay in adding heparin, there was a progressive accumulation of RNAP complexes characterized by increased fluorescence that did not produce a rapid escape upon addition of heparin (Fig. 8 B-D). The fraction of RNAP complexes that produced rapid decrease of fluorescence upon addition of heparin decreased with the increase of the delay in heparin addition (Fig. 8 B-D). After 20 min delay in heparin addition, essentially 100% of RNAP complexes did not respond to heparin addition (Fig. 8D). When GreB was added after 20 min, a rapid decrease of fluorescence was observed (Fig. S6, Supplemental Data) consistent with rescue of backtracked complexes accumulated in the absence of heparin. Fluorescence signal (when only GreB was added at 20 min) decreased to constant level that was higher than initial fluorescence intensity before RNAP was added (Fig. S6A, Supplemental Information) consistent with a steady state of transcription produced by RNAP re-binding followed by rapid escape. When both GreB and heparin were added at 20 min (Fig. S6B, Supplemental Information), a complete escape was observed.
The data in Fig. 8 A-D are consistent with a scenario that shortly after transcription is started, the observed increase of fluorescence intensity is mostly due to the re-binding of RNAP to the promoter. When the reaction is allowed to continue under multiple rounds of transcription conditions, the inactive backtracked RNAP complexes progressively accumulate. However, measurements of the amounts of full transcript under conditions of multiple rounds of transcription (Fig. 8E) suggest that fluorescence signals observed in Fig. 8 A-D may not be entirely due to progressive accumulation of backtracked complexes as a result of repeated rounds of elongation. The amount of transcript made without heparin was only slightly higher compared to the amount of transcript made under single round of transcription conditions (Fig. 8E). This suggests that in the absence of heparin accumulation of inactive complexes does not require multiple rounds of transcription. We speculate thus that accumulation of inactive backtracked complexes in the absence of heparin could be facilitated by nonspecific interactions of RNAP with DNA template that could promote backtracking of transcribing RNAP.
Discussion
Our data demonstrated that a simple fluorescent DNA construct with Cy3 fluorescence probe at position +2 of the non-template strand (Fig. 1A) allows reliable real-time observation of RNAP backtracking near the promoter. Thus, this single construct allows real-time recording of three different activities of RNAP: promoter melting24, promoter escape24 (Fig. 1C) and backtracking (Fig. 1C). Since RNAP can efficiently transcribe through a Cy3 label attached to a nontemplate base, the assay can be used to follow RNAP backtracking not only near the promoter but at any location where the Cy3 label could be incorporated into the nontemplate strand (for example, Fig. 6). The real-time nature of this assay will provide an opportunity for detailed kinetic analysis of backtracking (an ongoing effort in our laboratory (Lass-Napiorkowska, A. et al., unpublished)). In such analysis it is important to recognize that while relative changes of fluorescence over time allow kinetic analysis, a caution is necessary in using the level of fluorescence signal observed after backtracking for quantifying the percent of backtracked complexes. The level of fluorescence observed after backtracking will depend on fraction of complexes that backtracked but it could also vary due to the distance of RNAP from the fluorescence label (and thus the extent of backtracking that RNAP experiences).
Real-time observation of RNAP backtracking revealed some behaviors of RNAP that were expected from previous knowledge of RNAP elongation and backtracking but it also revealed some unexpected characteristics. We induced the backtracking in the assay illustrated in Fig. 1 by denying RNAP a correct NTP to bind to the active site and allow RNA extension when RNAP arrived at position +27. Since binding of the correct NTP to the active site has been shown to favor forward translocation of the enzyme30, efficient backtracking in the absence of correct NTP could be thus expected. Nonhydrolizable analogue of the correct NTP was effective in inhibiting backtracking (Fig. 5) in further agreement with this expectation and in support of the notion that just the binding of correct NTP (before formation of the phosphodiester bond) is effective in promoting forward translocation of the enzyme and inhibiting backtracking.
Real-time observation of RNAP behavior when it encounters a roadblock during elongation (Fig. 7) provides a good illustration of the role of backtracking in determining the response of RNAP to such roadblocks. Abasic site is one of the most common DNA damage products that RNAP might encounter35. RNAP was shown to be able to read through the abasic site33, 34 suggesting that such site may not be a major obstacle in transcription (other than a source of incorporation of incorrect base into RNA product). The data in Fig. 7B shows that encounter of RNAP with abasic site partitions the enzyme between backtracked state and RNAP that successfully reads through the abasic site. Transcript cleavage factor effectively changes this partitioning in favor of the read-through state of RNAP. This partitioning is likely to depend on DNA template sequence preceding the abasic site and it is thus possible that depending on the sequence context, significant accumulation of backtracked RNAP could occur in the absence of help from external factors (for example, transcript cleavage factors or translating ribosome). Encounter of RNAP with a very stable roadblock (neutravidin bound to a template strand base) produced, as expected, a large amount of backtracking. Addition of GreB was essential for allowing RNAP to translocate much further into the roadblock. This illustrates the importance of resolving backtracking for transcription in vivo where RNAP will encounter many roadblocks during elongation (for example, proteins bound to DNA) and is consistent with single molecule studies on yeast RNAP II which showed that backtracking determined force sensitivity of RNAP18. The importance of transcript cleavage factor (TFIIS) for transcription through transcriptional blocks by RNAP II has been demonstrated36, 37 further supporting this notion.
Stability of RNA-DNA hybrid was suggested to be a major determinant of RNAP backtracking20, 21, although backtracking on sequence lacking a weak RNA-DNA hybrid was also observed38. It was surprising thus to find an apparent lack of strong correlation between the extent and the kinetics of backtracking with the stability of RNA-DNA hybrid. A caution is needed in making conclusions from the data in Fig. 4 regarding the role of hybrid stability in backtracking kinetics since the sequence of DNA template was changed to affect the RNA-DNA hybrid stability. Nevertheless, taken together these data point to some other important determinants of backtracking beyond the stability of the hybrid. One possibility could be the interactions of backtracked RNA with residues lining the secondary channel of RNAP as revealed by X-ray structure of RNAP II in backtracked state22. A comprehensive study of DNA template dependence of RNAP backtracking (an ongoing effort in our laboratory (Heyduk, E. et al., unpublished)) will be needed to obtain a clear understanding of all determinants of backtracking.
GreA/GreB factors were shown to facilitate transition from initiation to elongation on some promoters39 and to resolve σ-dependent promoter proximal pausing40, 41. Genome-wide studies in E. coli demonstrated that RNAP pausing in the proximity of promoters is affected by GreA/GreB deletion whereas pausing farther into the protein coding sequence is largely unaffected4. Similar observations were reported for Bacilllus subtilis42. This suggests that RNAP might be more prone to backtracking early on during transcription from a promoter. The enhanced sensitivity of promoter-proximal regions to backtracking might be due to lingering of RNAP contacts important for initiation that need to be broken upon transition to elongation14. Also, RNA product might be too short to allow formation of structures favoring forward translocation or binding of a translating ribosome that was demonstrated to stimulate forward movement of RNAP14. Our experiments in the absence of heparin using a construct producing a short transcript (Fig. 8) could be viewed as in vitro model of RNAP behavior while transcribing promoter proximal regions in vivo. Our observation that in the absence of GreB under such conditions backtracked inactive RNAP complexes accumulated consistent with the importance of resolving backtracking in promoter-proximal regions in vivo. It will be interesting to investigate if there are other mechanisms (in addition to transcript cleavage factors) that work selectively in promoter proximal regions to safeguard against excessive RNAP backtracking.
Supplementary Material
Acknowledgments
This study was supported by grant from NIH (1R21AI112919).
Abbreviations
- RNAP
RNA polymerase
- NTP
nucleotide triphosphate
Footnotes
Supporting Information Available
List of sequences of all oligonucleotides used for preparing promoter constructs, additional data confirming that fluorescence signal reports RNAP backtracking (Figs. S1-S2), fluorescence changes in P27inv construct (Fig. S3), the effect of GreB on transcription through an abasic site (Fig. S4), additional data illustrating fluorescence changes of P27 construct in the absence of heparin (Figs. S5-S6).
References
- 1.Herbert KM, La Porta A, Wong BJ, Mooney RA, Neuman KC, Landick R, Block SM. Sequence-resolved detection of pausing by single RNA polymerase molecules. Cell. 2006;125:1083–1094. doi: 10.1016/j.cell.2006.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbondanzieri EA, Greenleaf WJ, Shaevitz JW, Landick R, Block SM. Direct observation of base-pair stepping by RNA polymerase. Nature. 2005;438:460–465. doi: 10.1038/nature04268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaevitz JW, Abbondanzieri EA, Landick R, Block SM. Backtracking by single RNA polymerase molecules observed at near-base-pair resolution. Nature. 2003;426:684–687. doi: 10.1038/nature02191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imashimizu M, Takahashi H, Oshima T, McIntosh C, Bubunenko M, Court DL, Kashlev M. Visualizing translocation dynamics and nascent transcript errors in paused RNA polymerases in vivo. Genome biology. 2015;16:98. doi: 10.1186/s13059-015-0666-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landick R. The regulatory roles and mechanism of transcriptional pausing. Biochemical Society transactions. 2006;34:1062–1066. doi: 10.1042/BST0341062. [DOI] [PubMed] [Google Scholar]
- 6.Larson MH, Mooney RA, Peters JM, Windgassen T, Nayak D, Gross CA, Block SM, Greenleaf WJ, Landick R, Weissman JS. A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science. 2014;344:1042–1047. doi: 10.1126/science.1251871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neuman KC, Abbondanzieri EA, Landick R, Gelles J, Block SM. Ubiquitous transcriptional pausing is independent of RNA polymerase backtracking. Cell. 2003;115:437–447. doi: 10.1016/s0092-8674(03)00845-6. [DOI] [PubMed] [Google Scholar]
- 8.Vvedenskaya IO, Vahedian-Movahed H, Bird JG, Knoblauch JG, Goldman SR, Zhang Y, Ebright RH, Nickels BE. Interactions between RNA polymerase and the “core recognition element” counteract pausing. Science. 2014;344:1285–1289. doi: 10.1126/science.1253458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kassavetis GA, Chamberlin MJ. Pausing and termination of transcription within the early region of bacteriophage T7 DNA in vitro. The Journal of biological chemistry. 1981;256:2777–2786. [PubMed] [Google Scholar]
- 11.Nudler E, Mustaev A, Lukhtanov E, Goldfarb A. The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell. 1997;89:33–41. doi: 10.1016/s0092-8674(00)80180-4. [DOI] [PubMed] [Google Scholar]
- 12.Komissarova N, Kashlev M. RNA polymerase switches between inactivated and activated states By translocating back and forth along the DNA and the RNA. The Journal of biological chemistry. 1997;272:15329–15338. doi: 10.1074/jbc.272.24.15329. [DOI] [PubMed] [Google Scholar]
- 13.Krummel B, Chamberlin MJ. Structural analysis of ternary complexes of Escherichia coli RNA polymerase Deoxyribonuclease I footprinting of defined complexes. Journal of molecular biology. 1992;225:239–250. doi: 10.1016/0022-2836(92)90918-a. [DOI] [PubMed] [Google Scholar]
- 14.Nudler E. RNA polymerase backtracking in gene regulation and genome instability. Cell. 2012;149:1438–1445. doi: 10.1016/j.cell.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laptenko O, Lee J, Lomakin I, Borukhov S. Transcript cleavage factors GreA and GreB act as transient catalytic components of RNA polymerase. The EMBO journal. 2003;22:6322–6334. doi: 10.1093/emboj/cdg610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nechaev S, Fargo DC, dos Santos G, Liu L, Gao Y, Adelman K. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science. 2010;327:335–338. doi: 10.1126/science.1181421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Depken M, Galburt EA, Grill SW. The origin of short transcriptional pauses. Biophysical journal. 2009;96:2189–2193. doi: 10.1016/j.bpj.2008.12.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galburt EA, Grill SW, Wiedmann A, Lubkowska L, Choy J, Nogales E, Kashlev M, Bustamante C. Backtracking determines the force sensitivity of RNAP II in a factor-dependent manner. Nature. 2007;446:820–823. doi: 10.1038/nature05701. [DOI] [PubMed] [Google Scholar]
- 19.Kireeva ML, Kashlev M. Mechanism of sequence-specific pausing of bacterial RNA polymerase. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:8900–8905. doi: 10.1073/pnas.0900407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imashimizu M, Oshima T, Lubkowska L, Kashlev M. Direct assessment of transcription fidelity by high-resolution RNA sequencing. Nucleic acids research. 2013;41:9090–9104. doi: 10.1093/nar/gkt698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Komissarova N, Becker J, Solter S, Kireeva M, Kashlev M. Shortening of RNA:DNA hybrid in the elongation complex of RNA polymerase is a prerequisite for transcription termination. Molecular cell. 2002;10:1151–1162. doi: 10.1016/s1097-2765(02)00738-4. [DOI] [PubMed] [Google Scholar]
- 22.Cheung AC, Cramer P. Structural basis of RNA polymerase II backtracking, arrest and reactivation. Nature. 2011;471:249–253. doi: 10.1038/nature09785. [DOI] [PubMed] [Google Scholar]
- 23.Sekine S, Murayama Y, Svetlov V, Nudler E, Yokoyama S. The ratcheted and ratchetable structural states of RNA polymerase underlie multiple transcriptional functions. Molecular cell. 2015;57:408–421. doi: 10.1016/j.molcel.2014.12.014. [DOI] [PubMed] [Google Scholar]
- 24.Ko J, Heyduk T. Kinetics of promoter escape by bacterial RNA polymerase: effects of promoter contacts and transcription bubble collapse. The Biochemical journal. 2014;463:135–144. doi: 10.1042/BJ20140179. [DOI] [PubMed] [Google Scholar]
- 25.Artsimovitch I, Svetlov V, Murakami KS, Landick R. Co-overexpression of Escherichia coli RNA polymerase subunits allows isolation and analysis of mutant enzymes lacking lineage-specific sequence insertions. J Biol Chem. 2003;278:12344–12355. doi: 10.1074/jbc.M211214200. [DOI] [PubMed] [Google Scholar]
- 26.Callaci S, Heyduk E, Heyduk T. Conformational changes of Escherichia coli RNA polymerase sigma70 factor induced by binding to the core enzyme. J Biol Chem. 1998;273:32995–33001. doi: 10.1074/jbc.273.49.32995. [DOI] [PubMed] [Google Scholar]
- 27.SantaLucia J., Jr A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang H, Kim H, Myong S. Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7414–7418. doi: 10.1073/pnas.1017672108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borukhov S, Sagitov V, Goldfarb A. Transcript cleavage factors from E. coli. Cell. 1993;72:459–466. doi: 10.1016/0092-8674(93)90121-6. [DOI] [PubMed] [Google Scholar]
- 30.Bar-Nahum G, Epshtein V, Ruckenstein AE, Rafikov R, Mustaev A, Nudler E. A ratchet mechanism of transcription elongation and its control. Cell. 2005;120:183–193. doi: 10.1016/j.cell.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 31.Epshtein V, Toulme F, Rahmouni AR, Borukhov S, Nudler E. Transcription through the roadblocks: the role of RNA polymerase cooperation. The EMBO journal. 2003;22:4719–4727. doi: 10.1093/emboj/cdg452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walter W, Kireeva ML, Studitsky VM, Kashlev M. Bacterial polymerase and yeast polymerase II use similar mechanisms for transcription through nucleosomes. The Journal of biological chemistry. 2003;278:36148–36156. doi: 10.1074/jbc.M305647200. [DOI] [PubMed] [Google Scholar]
- 33.Zhou W, Doetsch PW. Effects of abasic sites and DNA single-strand breaks on prokaryotic RNA polymerases. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:6601–6605. doi: 10.1073/pnas.90.14.6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou W, Doetsch PW. Efficient bypass and base misinsertions at abasic sites by prokaryotic RNA polymerases. Annals of the New York Academy of Sciences. 1994;726:351–354. doi: 10.1111/j.1749-6632.1994.tb52849.x. [DOI] [PubMed] [Google Scholar]
- 35.Doetsch PW, Cunningham RP. The enzymology of apurinic/apyrimidinic endonucleases. Mutation research. 1990;236:173–201. doi: 10.1016/0921-8777(90)90004-o. [DOI] [PubMed] [Google Scholar]
- 36.Kireeva ML, Hancock B, Cremona GH, Walter W, Studitsky VM, Kashlev M. Nature of the nucleosomal barrier to RNA polymerase II. Molecular cell. 2005;18:97–108. doi: 10.1016/j.molcel.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 37.Reines D, Mote J., Jr Elongation factor SII-dependent transcription by RNA polymerase II through a sequence-specific DNA-binding protein. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:1917–1921. doi: 10.1073/pnas.90.5.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hawryluk PJ, Ujvari A, Luse DS. Characterization of a novel RNA polymerase II arrest site which lacks a weak 3′ RNA-DNA hybrid. Nucleic acids research. 2004;32:1904–1916. doi: 10.1093/nar/gkh505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsu LM, Vo NV, Chamberlin MJ. Escherichia coli transcript cleavage factors GreA and GreB stimulate promoter escape and gene expression in vivo and in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:11588–11592. doi: 10.1073/pnas.92.25.11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stepanova E, Wang M, Severinov K, Borukhov S. Early transcriptional arrest at Escherichia coli rplN and ompX promoters. The Journal of biological chemistry. 2009;284:35702–35713. doi: 10.1074/jbc.M109.053983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marr MT, Roberts JW. Function of transcription cleavage factors GreA and GreB at a regulatory pause site. Molecular cell. 2000;6:1275–1285. doi: 10.1016/s1097-2765(00)00126-x. [DOI] [PubMed] [Google Scholar]
- 42.Kusuya Y, Kurokawa K, Ishikawa S, Ogasawara N, Oshima T. Transcription factor GreA contributes to resolving promoter-proximal pausing of RNA polymerase in Bacillus subtilis cells. Journal of bacteriology. 2011;193:3090–3099. doi: 10.1128/JB.00086-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.