

Abstract
It is known that the lipid composition within a cellular membrane can influence membrane protein structure and function. In this Article, we investigated how structural changes to a membrane protein upon substrate binding can impact the lipid bilayer. To carry out this study, we reconstituted the secondary active drug transporter EmrE into a variety of phospholipid bilayers varying in headgroup and chain length and carried out differential scanning calorimetry (DSC) and solid-state NMR experiments. The DSC results revealed a difference in cooperativity of the lipid phase transition for drug-free EmrE protonated at glutamic acid 14 (i.e., proton-loaded form) and the tetraphenylphosphonium (TPP+) bound form of the protein (i.e., drug-loaded form). To complement these findings, we acquired magic-angle-spinning (MAS) spectra in the presence and absence of TPP+ by directly probing the phospholipid headgroup using 31P NMR. These spectra showed a reduction in lipid linewidths around the main phase transition for samples where EmrE was bound to TPP+ compared to the drug free form. Finally, we collected oriented solid-state NMR spectra on isotopically enriched EmrE that displayed chemical shift perturbations to both transmembrane and loop residues upon TPP+ binding. All of these results prompt us to propose a mechanism whereby substrate-induced changes to the structural dynamics of EmrE alters the surrounding lipids within the bilayer.
Graphical Abstract
INTRODUCTION
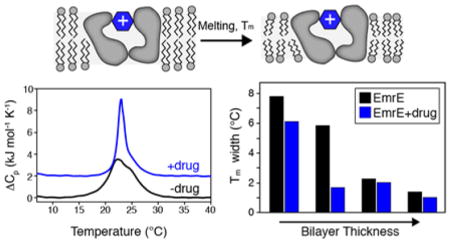
The cell membrane is the fundamental barrier separating the inside of a cell from the outside environment. Within this milieu, the interplay between lipids and membrane proteins constitutes an additional feature regulating protein function that does not pertain to soluble proteins. One property of the lipid bilayer that membrane proteins influence is the main phase transition, which is the temperature at which “melting” from a ripple phase to the liquid crystalline phase occurs. While the transition of a pure lipid bilayer is a highly cooperative process exemplified by a narrow phase transition peak observed using differential scanning calorimetry (DSC),1,2 the presence of additional components such as cholesterol and membrane proteins reduce this observed cooperativity by interfering with phospholipid packing.2–4 An additional broadening mechanism has been proposed to arise from hydrophobic mismatch between the mean hydrophobic thickness of the bilayer and the hydrophobic length of the membrane protein.2 Broadening induced by membrane proteins and the role of hydrophobic mismatch motivated us to consider how protein conformational changes might further influence the bilayer properties including the bilayer melting cooperativity. Our initial attempts employed DSC experiments in model 14:0-PC bilayers (DMPC) with the secondary active transport protein EmrE to investigate how binding to the high affinity substrate tetraphenylphosphonium (TPP+) might influence the phase transition.5 Our measurements showed that the addition of TPP+ led to a substantial narrowing of the main phase transition and led us to hypothesize that a change in structure and dynamics induced by substrate binding can influence the physical properties of the lipid bilayer.
In this work, we systematically explored how different lipids can impact membrane protein/bilayer interactions by using EmrE as the model system in the presence of proton or drug substrates. These biophysical experiments investigated a range of lipids varying in chain length, headgroup type, and bonding between the acyl chain and glycerol backbone (i.e., ester or ether linkage). Our data with EmrE in the presence and absence of TPP+ suggest that bilayer phase transitions probed using DSC are sensitive to the hydrophobic mismatch between the protein and lipid bilayer. In addition, we observed a narrowing of the 31P linewidths corresponding to the phosphate headgroup upon TPP+ binding, which suggests that the structural perturbation induced upon drug binding reduces the disruptive effect EmrE has on the cooperativity of the lipid phase transition. Finally, we acquired oriented solid-state NMR spectra in the absence and presence of TPP+ and show evidence that drug binding induces changes relative to the proton-bound form of EmrE. Taken together, these findings support the conclusion that different substrates (i.e., protons or drugs) can exert differential effects on the physical properties of the lipid bilayer by binding to the transporter.
MATERIALS AND METHODS
Expression and purification
EmrE was expressed in E. coli and purified in n-dodecyl β-D-maltopyranoside (DDM) detergent as previously described.6,7 In brief, EmrE was expressed as a fusion protein with maltose-binding protein (MBP). Bacteria were lysed and the fusion protein was purified using an amylose affinity resin specific for MBP. The fusion protein was eluted from the amylose column by the addition of maltose in the presence of DDM. EmrE was cleaved from MBP with tobacco etch virus protease (TEV). MBP and TEV were removed by passing the protease reaction over a Ni-NTA column (both proteins have poly-His tags). The flow through containing EmrE was concentrated and purified to >95% by size-exclusion chromatography using a Superdex 200 10/300 column (GE Healthcare). Peak fractions were used for subsequent biophysical studies.
Differential scanning calorimetry
1,2-ditridecanoyl-sn-glycero-3-phosphocholine (13:0-PC), 1,2-di-O-tetradecyl-sn-glycero-3-phosphocholine (O-14:0-PC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (16:0-18:1-PE; POPE), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (16:0-18:1-PG; POPG) were purchased from Avanti Polar Lipids, Inc. 1,2-dimyristoyl-sn-glycero-3-phosphocholine (14:0-PC) and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (16:0-PC) were purchased from Affymetrix. Powdered lipids were dissolved in chloroform and dried under nitrogen gas and subsequently placed under vacuum overnight. POPE in chloroform and POPG in chloroform were mixed in a 3:1 mol:mol ratio, dried under nitrogen gas and overnight under vacuum. DDM solubilized EmrE in 20 mM Na2HPO4 (pH 6.0) and 20 mM NaCl was reconstituted using Bio-Beads, as previously described5,7,8, with a lipid:protein ratio of 100:1 (mol:mol) into 13:0-PC, O-14:0-PC, 14:0-PC, 16:0-PC, or 3/1 POPE/POPG. The lipid:protein ratio was chosen to ensure sample homogeneity and has been selected by careful optimization of sample preparations for oriented solid-state NMR,5,7 which is sensitive to the mosaic spread of the protein with respect to the lipid bilayer.9 Proteoliposomes were pelleted at 300,000 × g for 2 hours at 8 °C using a TLA-110 rotor (Beckman-Coulter). Pellets were resuspended in 20 mM Na2HPO4, 20 mM NaCl, pH 6.0 (DSC buffer) and homogenized with freeze-fracturing. Samples were then split in half and pelleted using the previously mentioned conditions. To each sample, buffer or buffer containing 0.5 mM tetraphenylphosphonium (TPP+) was added to bring the samples to a final lipid concentration of 9.3 mM. DSC measurements (heating and cooling) were recorded on a nanoDSC instrument (TA Instruments) using a scan rate of 4.8 °C/hr between the following temperatures: 0.2 °C – 40.2 °C (13:0-PC); 5 °C – 45 °C (O-14:0-PC, 14:0-PC, and 3/1 POPE/POPG); or 20 °C –55 °C (16:0-PC) under a pressure of 3 atm and a 600 second equilibration time prior to scanning temperature in each direction. Data were analyzed using NanoAnalyze v3.6.0 or higher (TA Instruments).
Solid-State NMR Spectroscopy
Proteoliposome samples in DSC buffer were prepared for solid-state NMR by spinning down at 600,000 × g for 14 hours at 8 °C using a TLA-100 rotor (Beckman-Coulter). The supernatant was removed and the pellets were center-packed in 3.2 mm thin-walled rotors with sample spacers to prevent dehydration.
Solid-state NMR data were collected on an Agilent DD2 spectrometer operating at a 1H Larmor frequency of 600 MHz (14.1 T). Magic-angle-spinning (MAS) experiments were performed using a triple resonance (1H/31P/13C) bioMAS probe (Agilent) at a spinning rate of 12.5 kHz. Typical pulse lengths for 1H and 31P were 2.5 μsec and 5.5 μsec, respectively. 31P linewidths were measured from a single-pulse experiment using a recycle delay of 6 seconds, an acquisition time of 20 msec, and a spectral width of 100 kHz. MAS experiments were acquired using a 100 kHz 1H TPPM decoupling field (ω1/2π)10.
Oriented experiments were performed on a double resonance static probe tuned to 1H and 15N (Revolution NMR). [15N-Thr, 15N-Met] and [15N-Thr] EmrE samples were reconstituted into lipid bicelles comprised of 14:0-PC/6:0-PC or O-14:0-PC/6:0-PC in a ~3.8/1 molar ratio as described previously (6:0-PC is 1,2-dihexanoyl-sn-glycero-3-phosphocholine). For the ester and ether bicelle samples the long-chain lipid to protein ratio was ~140:1. PISEMA spectra were acquired by using phase-modulated Lee-Goldburg in the indirect t1 dimension to evolve 1H-15N dipolar couplings. The effective field strength for the 1H 90° pulse, 1H-15N cross-polarization, and 1H SPINAL-64 decoupling11 was set to 50 kHz (i.e., ω1/2π). Phase-modulated Lee-Goldburg12,13 was carried out in the indirect dimension at an effective field strength of 41.7 kHz on 1H and 15N (i.e., ω1/2π). The cross-polarization time in each PISEMA experiment was set to 0.75 msec. The spectral width in the direct 15N dimension was set to 100 kHz with a total acquisition time of 7 msec. The indirect dimension to measure 1H-15N dipolar couplings was evolved with 12 increments and a total evolution time of 0.528 msec. The indirect dimension was corrected using a scaling factor of 0.82.
RESULTS AND DISCUSSION
Differential scanning calorimetry of EmrE reconstituted into lipids varying in chain length
As discussed above, we previously observed narrowing of the lipid main phase transition upon addition of a high affinity substrate to 14:0-PC lipid bilayers containing the transporter EmrE.5 To further explore the underlying reason for the observed narrowing, we carried out additional DSC experiments using EmrE reconstituted into lipid bilayers varying in chain length and headgroup type. The chain length dependence was studied by reconstituting EmrE into the following lipids: 13:0-PC, 14:0-PC (DMPC), O-14:0-PC (diether 14:0-PC), and 16:0-PC (DPPC). A change from an ester to ether linkage between the glycerol backbone and the acyl chain is akin to having an additional methylene group on the chain,14 which would increase the mean hydrophobic thickness of O-14:0-PC by ~2.2 Å relative to 14:0-PC.15 Two DSC experiments were carried out in the absence and presence of TPP+ at a pH of 6.0 for each lipid condition (Figure 1A–D). Note that our previous DSC measurements with drug-free EmrE in 14:0-PC were carried out at pH=7.0. A subsequent study by our group found that a highly conserved glutamic acid residue within EmrE had an apparent pKa value of 7.0 at 25 °C,16 which motivated our current work at pH 6.0 in order to avoid mixtures of protonated and deprotonated forms of the protein. The DSC thermogram data in 14:0-PC at pH 6.0 showed a noticeable narrowing of the phase transition in the presence of TPP+, which is similar to our previous findings at pH 7.0 (Figure 1B). We also acquired DSC data for liposomes in the absence of protein (Figure 1E–H) and found all of these phase transition widths to be substantially narrower than those containing EmrE. In order to more easily compare the phase transition broadening across all EmrE samples observed in Figure 1A–D, we plotted the full-width at half-maximum (FWHM) for each of the transitions in Figure 2 as a function of the lipid length. The trend from these data show that shorter chain lengths lead to greater broadening and heterogeneity of the main phase transition relative to the longer chain length lipids tested. In addition, the differential broadening between the TPP+ free and bound forms of EmrE was most notable in 14:0-PC lipids, while the longer chain length lipids (O-14:0-PC and 16:0-PC) showed smaller differences in linewidths upon TPP+ binding.
Figure 1.

DSC thermograms of lipid bilayers in the presence (A–D) or in the absence of EmrE (E–H). All proteoliposomes were prepared at a lipid:protein mole ratio of 100:1. For clarity, the TPP+-bound thermogram data (dotted lines) are offset by 1 kJ•mol−1•K−1 in panel A and 2 kJ•mol−1•K−1 in panels B–D from the EmrE data in the absence of TPP+ (solid lines). Heating and cooling scans for data in panels A–D are shown in Figure S1. The main phase transition is narrower and/or more homogeneous for 13:0-PC and 14:0-PC upon the addition of TPP+. Note that the presence of TPP+ to liposome only samples did not influence the broadness as we previously observed for 14:0-PC bilayers.5
Figure 2.

Main phase transition linewidths from DSC thermogram data for EmrE in the presence (gray) and absence (black) of TPP+. The full-width at half-maximum is plotted for each dataset in Figure 1A–D. The difference between EmrE free or bound to TPP+ is most stark in 14:0-PC.
The trend in our data as a function of lipid length prompted us to consider the role of hydrophobic mismatch. Indeed, a study by Zhang et al. previously investigated the relationship between the length of hydrophobic peptides versus the mean hydrophobic thickness (average thickness between the gel and liquid crystalline phases) of the bilayer.2 Their conclusion was that if the average hydrophobic thickness of the bilayer was less than or equal to the hydrophobic length of the protein, the temperature range for bilayer melting increased.2 We observed this trend for both the drug-free and TPP+ bound forms of EmrE. Namely, the drug-free form of EmrE showed narrower phase transitions for O-14:0-PC and 16:0-PC and broader transitions for 13:0-PC and 14:0-PC. The TPP+ bound samples also showed substantial phase transition broadening and heterogeneity in the thinnest bilayer tested (13:0-PC). Therefore, the trends in our data suggest that differential broadening observed in 14:0-PC bilayers stemmed from a difference in hydrophobic thickness between the protonated form of EmrE and the TPP+-bound state. The observed differences imply a structural change within EmrE upon binding to TPP+, which is supported by NMR5,7,16–19, EPR20, and cryoelectron microscopy21 observations. Taken together, these data suggest that the protonated state of EmrE may have transmembrane (TM) helices with a slightly altered orientation in the membrane relative to the TPP+-bound state, which would lead to a greater hydrophobic mismatch in 13:0-PC and 14:0-PC bilayers.
Based on the trend of increasing phase transition broadness for EmrE with decreasing bilayer thickness, we wondered whether our DSC results were consistent with existing structural knowledge on EmrE.22–24 In other words, does the hydrophobic thickness of EmrE exceed that of the thinnest bilayers tested? To make this comparison, we calculated the mean hydrophobic thickness of O-14:0-PC, 14:0-PC, and 13:0-PC in the absence of protein based on work by Sperotto and Mouritsen15, which gave values of 30.7 Å, 28.5 Å, and 26.3 Å, respectively. To approximate the hydrophobic thickness of EmrE along the bilayer normal axis (see Figure S2), TM4 was chosen since its helical axis is most collinear with the bilayer normal5,7,25 and it has a well-defined helical region18. Specifically, we estimated that TM4 spans from Leu85 to Leu104 based on MAS experiments in lipid bilayers and solution NMR spectroscopy in lipid bicelles18 along with available structural and modeling studies23,24. This means that the TM4 helix is comprised of ~20 residues and a length of ~30 Å by assuming ideal helix geometry (i.e., 1.5 Å per residue). When a helix is parallel with the bilayer normal, the helical length can be directly compared with the protein-free bilayer thickness. However, when a helix tilts away from the bilayer normal, the helical length cannot be directly compared with the protein-free bilayer thickness. Using our previously reported TM4 tilt angles of 12–14° in the asymmetric dimer7, the helix length projected along the bilayer normal dimension (i.e., bilayer thickness) is ~2–3% shorter than the helical length or ~29.2 Å (i.e., cos θ x helix length). The protein hydrophobic thickness compared to the bilayer hydrophobic thickness of 13:0 PC (29.2 Å vs. 26.3 Å) suggests a notable difference that may explain the broadening of the phase transition observed in the drug free and drug bound forms of EmrE (Figure 1A). Similarly, a 14:0 PC bilayer would be slightly thinner than the hydrophobic thickness of EmrE. However, a structural change induced upon TPP+ binding may reduce this mismatch, which could explain the differential broadening observed by DSC for the proton and TPP+ bound forms of EmrE.
Differential scanning calorimetry of EmrE in lipids mimicking bacterial membranes
Next we carried out DSC experiments for EmrE in lipids that closely mimicked the headgroups found in the native membrane. While it is possible to measure phase transitions in E. coli polar lipid extracts, the main phase transition has been reported to be unstable in air and close to the freezing point of water.26 For these reasons, we used a 3/1 mixture of POPE/POPG.27–31 This composition matches the two most common headgroups present in native E. coli membranes and is similar to the DOPE/DOPG lipid composition that preserves EmrE transport.32 The DSC thermogram data for EmrE in POPE/POPG are presented in Figure 3A and 3B and indicate a hysteresis of the main phase transition. Specifically, we observed a broad transition in heating scans and a narrow transition in cooling scans. However, the hysteresis was protein-independent and was also observed in 3/1 POPE/POPG samples without EmrE (Figure 3C). Thermal hysteresis in POPE/POPG bilayers has been previously reported and may arise from the coexistence of phases which are segregated and composed of liquid crystalline POPG (Tm = −2 °C) with a second fraction of POPE in the gel-phase (Tm = 25 °C).33 Due to the hysteresis, we can only make qualitative conclusions that data in Figure 3A,B show similar heating and cooling curves between the protonated and TPP+ bound forms of EmrE. This observation is consistent with DSC results in 16:0-PC, which has a similar hydrophobic thickness as POPE/POPG.
Figure 3.
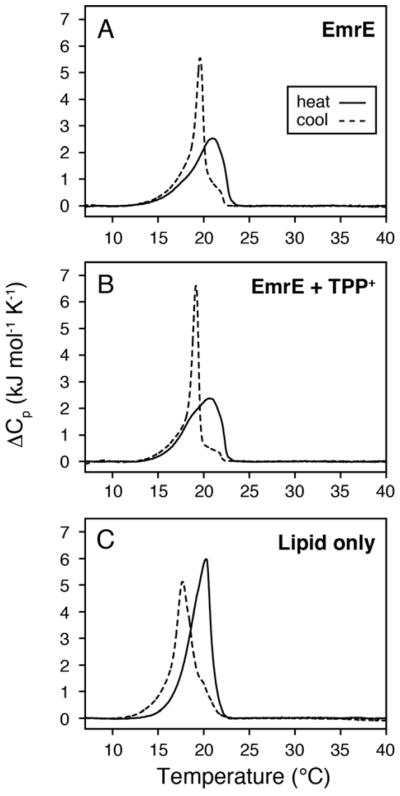
DSC thermogram data for 3/1 POPE/POPG bilayers in the presence of protonated-EmrE (A), TPP+-bound EmrE (B), and lipid only (C). The samples containing EmrE used a lipid/protein molar ratio of 100/1. The heating (solid lines) and cooling curves (dotted lines) are shown for each sample. The linewidths corresponding to EmrE in the absence and presence of TPP+ show similar profiles when comparing heating or cooling curves, which is consistent with 16:0-PC data in Figure 1D. However, due to the observed thermal hysteresis for the lipid only POPE/POPG mixture, only qualitative conclusions can be drawn regarding the effect of TPP+ binding to EmrE on the phase transitions.
Aside from the main topic of this article, hysteresis in multi-component lipid mixtures could have consequences for MAS experiments aimed at the detection of isotopically enriched proteins. Namely, we have previously shown that the optimal temperature to acquire MAS spectra on polytopic membrane proteins is below the main phase transition temperature of the bilayer for single component systems such as 14:0-PC or 16:0-PC.34 However, for bilayers composed of POPE/POPG that display hysteresis, the spectral quality may be influenced depending on whether the sample is heated or cooled to the desired temperature and therefore should be investigated for each protein under study.
31P NMR of the lipid headgroup using magic-angle-spinning (MAS) solid-state NMR
The DSC observations prompted us to collect MAS solid-state NMR spectra on the lipids to gauge the relative dynamics of the headgroup.35–37 For these experiments, the temperature was varied from 9 °C to 45 °C with a single pulse 31P experiment collected at each temperature value for liposomes without protein and liposomes reconstituted with EmrE in the absence and presence of TPP+. The 31P linewidths at 50% maximal intensity were quantified and plotted in Figure 4A and 4E for 14:0-PC and O-14:0-PC lipid bilayers, respectively. The trend for each temperature profile showed a maximal linewidth near the main phase transition of the lipid bilayer, which was also previously observed for linewidth measurements in 16:0-PC bilayers38 and for 14:0-PC bilayers using T2 relaxation experiments.37 The explanation for this behavior is that the bilayer is ordered in the gel state (i.e., below the Tm) and therefore the motion of the headgroup is slow, which enables MAS to reduce the chemical shift anisotropy of 31P. As the temperature is increased and the bilayer begins to melt, the lipids undergo increased rotational motion37 and are broadened by intermediate timescale chemical exchange (i.e., inability of MAS to average out chemical shift anisotropy). When the bilayer is fully melted and in the liquid crystalline state, the lipids undergo rapid motion, which acts as a self-decoupling mechanism to give the narrowest 31P linewidths in the temperature titration. These observations are consistent with previous findings39–42.
Figure 4.

31P linewidths from a single pulse MAS experiment for EmrE embedded within 14:0-PC (A) and O-14:0-PC bilayers (E). The blue and red points correspond to EmrE in the absence and presence of TPP+, respectively. The black points correspond to the lipid only samples. Data reflect the average of two separate trials where the error bars represent the standard deviation of these datasets. The 31P linewidth vs. temperature plots for each trial are shown in Figure S3. Representative 31P spectra are shown at a temperature of 23 °C for 14:0-PC in the absence (B) and presence of TPP+ (C) and lipid only (D). The same is shown at 27 °C for O-14:0-PC in the absence (F) and presence of TPP+ (G) and lipid only (H). In both lipid compositions, there is a reduction in the 31P linewidth when EmrE is bound with TPP+. Note that the small peak around ~1 ppm is signal from the phosphate buffer used to prepare the samples.
The 31P linewidths for proteoliposome samples corresponding to EmrE in the presence and absence of TPP+ each gave maximal values near the main phase transition (Figure 4). However, ~10% narrower linewidths were observed around the main phase transition temperature for TPP+ bound EmrE relative to the drug-free form (Figure 4A and 4E; example 31P spectra are shown in Figure 4B–C and 4F–G). This observation was similar to the narrowing effect observed with EmrE binding to TPP+ using DSC. Furthermore, the 31P linewidth curves for EmrE bound to TPP+ were shifted toward the liposome only dataset (Figure 4A and E). This observation suggested that when EmrE binds TPP+ it underwent a change in structural dynamics that attenuated its effect on the membrane. Since the bilayer is viewed to be disordered in the liquid crystalline state relative to the gel state, the effect of TPP+ binding to EmrE caused the surrounding lipids to return to a more dynamic and disordered state as observed in lipid only experiments (Figure 4D and 4H).
Oriented solid-state spectra of EmrE in 14:0-PC and O-14:0-PC bicelles
To probe differences induced to EmrE upon TPP+ binding, we carried out oriented solid-state NMR experiments on EmrE embedded within 14:0-PC and O-14:0-PC lipid bicelles. While ester bonds between the phosphate head group and the acyl chain are the primary linkages in phospholipids, ether-linked lipids are found in a wide variety of biological organisms including deep sea organisms, archaebacteria, and mammalian species in the form of plasmalogens and platelet activating factors.43 Ether-linked lipids have become a valuable alternative for structural studies due to their increased chemical stability over a wide pH range relative to ester-linked lipids.44–47 The need for pH dependent studies of EmrE make the ether lipid system attractive,16 and it is therefore important to ensure these lipids do not alter the protein’s structure. In fact, for the antimicrobial peptide novicidin, the secondary structure was found to be influenced by the presence of ether-linked lipids as compared to the ester-linked counterpart.48
For our oriented solid-state NMR experiments, we reconstituted selectively labeled EmrE at 15N-Met and 15N-Thr residues into lipid bicelles and performed the PISEMA experiment49 that correlates 1H-15N dipolar couplings with 15N anisotropic chemical shifts. Magnetically aligned bicelle spectra of membrane proteins give periodic spectral patterns, which stem from the periodicity of helices and sheets.50,51 The initial experiments were carried out using isotopically enriched drug-free EmrE in the protonated form by reconstituting the protein into two lipid bicelle compositions (14:0-PC/6:0-PC or O-14:0-PC/6:0-PC). The PISEMA spectra are shown in Figure 5A and 5B and were acquired with the same number of scans and increments in order to allow for a direct comparison. Overall we found that the spectra were similar in terms of sensitivity and resolution. The 15N dimension linewidths for these samples and others not shown were essentially identical and had an average ± standard deviation of 2.5 ± 0.5 ppm. To assess the sensitivity, we quantified the peak heights for all resolved resonances in these two spectra. The average signal intensity was within 2% between these spectra and therefore did not represent a statistically significant difference. Lastly, we compared the peak positions by calculating 15N chemical shift perturbations among 14:0-PC and O-14:0-PC bicelle samples in Figure 5E (blue bars). Although most changes were minimal, two notable exceptions were seen for Thr18 and Thr56, which had shifted peak positions between 14:0-PC and O-14:0-PC bicelles. The position of Thr18 is near the end of TM1 while Thr56 is in the loop between TM2 and TM3. Thus, both of these residues are predicted to be near the headgroup position of the lipid and may explain subtle differences in PISEMA spectra within the two bicelle compositions. Nevertheless, taken as a whole, these spectra were very similar and support a benefit to the ether-linked lipid since it has a higher degree of pH stability needed for pH dependent studies.
Figure 5.

1H/15N PISEMA spectra of EmrE reconstituted in bicelles consisting of 14:0-PC/6:0-PC or O-14:0-PC/6:0-PC. For simplicity, the panels are labeled by the long chain lipid (either 14:0-PC or O-14:0-PC). (A, B) [15N-Thr, 15N-Met] labeled EmrE in magnetically aligned bicelles in the drug-free, protonated form of EmrE at pH 5.8. (C, D) [15N-Thr] labeled EmrE in magnetically aligned bicelles in the TPP+ bound form of the protein. (E) 15N chemical shift perturbations (CSP) between drug-free EmrE in 14:0-PC and O-14:0-PC bicelles is shown in blue. Similarly the TPP+ bound form is compared between 14:0-PC and O-14:0-PC bicelles and is shown in red. (F) A comparison of 15N CSP values for TPP+ binding to EmrE. Purple bars show the perturbations induced to EmrE within the 14:0-PC bicelle and gray bars correspond to CSPs calculated in O-14:0-PC bicelles. Panels E and F plot the threonine residues in EmrE. Thr18 and Thr19 are located in TM1, Thr36 and Thr50 are located in TM2, and Thr56 is located in the loop between TM2 and TM3.
To investigate the structural perturbations to EmrE upon TPP+ binding, we acquired PISEMA spectra on 15N-Thr labeled EmrE in 14:0-PC and O-14:0-PC lipid bicelles. These spectra are shown in Figure 5C and 5D. Similar to EmrE in the protonated form, the peak positions in these two lipid environments shared a strong similarity for the TPP+ bound form. A comparison of 15N chemical shift perturbations in Figure 5E (red bars) shows no major perturbations (i.e., < ~2 ppm), which suggests that the TPP+ bound forms in the two bicelle compositions are more similar than in the absence of TPP+. In fact, the difference observed for Thr56 between 14:0-PC and O-14:0-PC in the absence of TPP+ was not observed when comparing the TPP+ bound spectra. Note that the Thr56 1H-15N dipolar coupling showed an even more noticeable difference in the drug-free form between 14:0-PC and O-14:0-PC than for the TPP+ bound forms (1.4 kHz vs. 0.1 kHz). These findings are consistent with DSC results that indicated similar main phase transition linewidths for 14:0-PC and O-14:0-PC in the TPP+ bound form of EmrE.
In contrast to our comparisons of the same form of EmrE in two different bicelle conditions, the differences between EmrE in the absence and presence of TPP+ are much more striking. The chemical shift perturbation plot is shown in Figure 5F and reveals structural changes to loop (Thr56) and transmembrane domain residues (Thr18, Thr19, Thr36, Thr50), which is consistent with our previous oriented sample solid-state NMR spectra on EmrE.7 As noted above, the perturbations induced by TPP+ were very similar between the two lipid bicelle environments and support the conclusion that the effect of TPP+ is to induce a change in the structure of EmrE relative to the lipid bilayer normal. In addition to structural perturbations, we previously observed that TPP+ binding reduces the inward-open to outward-open conformational dynamics relative to the protonated form of the protein, which likely means that TPP+ confers a stabilizing effect to EmrE.5,16 Taken together with our DSC and 31P MAS results, these data support a mechanism whereby changes in structure and dynamics to EmrE upon TPP+ binding lead to a more cooperative gel to liquid crystalline phase transition reminiscent of the protein-free lipid bilayer.
CONCLUSION
Solid-state NMR and DSC were used to investigate how substrate-induced structural changes to the membrane protein transporter EmrE could influence the physical properties of the lipid bilayer. Oriented sample solid-state NMR spectra showed significant chemical shift perturbations to TM and loop residues within EmrE, which indicated a change in the protein’s tilt angle with respect to the lipid bilayer. DSC experiments revealed that TPP+ binding to EmrE increased the cooperativity of the gel to liquid crystalline phase transition in a chain length dependent manner relative to the drug free protein. Similarly, 31P spectra reporting on the lipid headgroup mobility displayed reduced linewidths around the main phase transition temperature for EmrE bound to TPP+ as compared to the absence of drug. Both DSC and NMR data showed that the drug bound state of EmrE more closely resembled data acquired on lipids in the absence of protein. These observations support the conclusion that substrate binding to a membrane protein can have a direct influence on the surrounding lipids and the bilayer’s macroscopic properties.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01AI108889) and the National Science Foundation (MCB1506420) (to N.J.T). Maureen Leninger acknowledges funding from the Margaret Strauss Kramer Fellowship and a Dean’s dissertation fellowship.
Footnotes
Heating and cooling DSC thermogram data for EmrE and EmrE bound to TPP+ in 13:0-PC, 14:0-PC, O-14:0-PC, and 16:0-PC; schematic of the protein hydrophobic thickness calculation; individual results from separate trials of 31P experiments displaying linewidth as a function of temperature.
References
- 1.Riske KA, Barroso RP, Vequi-Suplicy CC, Germano R, Henriques VB, Lamy MT. Lipid bilayer pre-transition as the beginning of the melting process. Biochim Biophys Acta, Biomembr. 2009;1788:954–963. doi: 10.1016/j.bbamem.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Zhang YP, Lewis RNAH, Hodges RS, Mcelhaney RN. Interaction of a peptide model of a hydrophobic transmembrane alpha-helical segment of a membrane-protein with phosphatidylcholine bilayers - differential scanning calorimetric and FTIR spectroscopic studies. Biochemistry. 1992;31:11579–11588. doi: 10.1021/bi00161a042. [DOI] [PubMed] [Google Scholar]
- 3.Yeagle PL. Cholesterol and the cell-membrane. Biochim Biophys Acta. 1985;822:267–287. doi: 10.1016/0304-4157(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 4.Clarke JA, Heron AJ, Seddon JM, Law RV. The diversity of the liquid ordered (Lo) phase of phosphatidylcholine/cholesterol membranes: a variable temperature multinuclear solid-state NMR and x-ray diffraction study. Biophys J. 2006;90:2383–2393. doi: 10.1529/biophysj.104.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho MK, Gayen A, Banigan JR, Leninger M, Traaseth NJ. Intrinsic conformational plasticity of native EmrE provides a pathway for multidrug resistance. J Am Chem Soc. 2014;136:8072–8080. doi: 10.1021/ja503145x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banigan JR, Gayen A, Traaseth NJ. Combination of 15N reverse labeling and afterglow spectroscopy for assigning membrane protein spectra by magic-angle-spinning solid-state NMR: application to the multidrug resistance protein EmrE. J Biomol NMR. 2013;55:391–399. doi: 10.1007/s10858-013-9724-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gayen A, Banigan JR, Traaseth NJ. Ligand-induced conformational changes of the multidrug resistance transporter EmrE probed by oriented solid-state NMR spectroscopy. Angew Chem Int Ed Engl. 2013;52:10321–10324. doi: 10.1002/anie.201303091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banigan JR, Gayen A, Traaseth NJ. Correlating lipid bilayer fluidity with sensitivity and resolution of polytopic membrane protein spectra by solid-state NMR spectroscopy. Biochim Biophys Acta. 2015;1848:334–341. doi: 10.1016/j.bbamem.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quine JR, Achuthan S, Asbury T, Bertram R, Chapman MS, Hu J, Cross TA. Intensity and mosaic spread analysis from PISEMA tensors in solid-state NMR. J Magn Reson. 2006;179:190–198. doi: 10.1016/j.jmr.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 10.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J Chem Phys. 1995;103:6951–6958. [Google Scholar]
- 11.Fung BM, Khitrin AK, Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 12.Lee M, Goldburg WI. Nuclear-magnetic-resonance line narrowing by a rotating RF field. Phys Rev. 1965;140:1261–1271. [Google Scholar]
- 13.Vinogradov E, Madhu PK, Vega S. High-resolution proton solid-state NMR spectroscopy by phase-modulated Lee-Goldburg experiment. Chem Phys Lett. 1999;314:443–450. [Google Scholar]
- 14.Matsuki H, Miyazaki E, Sakano F, Tamai N, Kaneshina S. Thermotropic and barotropic phase transitions in bilayer membranes of ether-linked phospholipids with varying alkyl chain lengths. Biochim Biophys Acta. 2007;1768:479–489. doi: 10.1016/j.bbamem.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Sperotto MM, Mouritsen OG. Dependence of lipid-membrane phase-transition temperature on the mismatch of protein and lipid hydrophobic thickness. Eur Biophys J Biophy. 1988;16:1–10. [Google Scholar]
- 16.Gayen A, Leninger M, Traaseth NJ. Protonation of a glutamate residue modulates the dynamics of the drug transporter EmrE. Nat Chem Biol. 2016;12:141–145. doi: 10.1038/nchembio.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrison EA, Robinson AE, Liu Y, Henzler-Wildman KA. Asymmetric protonation of EmrE. J Gen Physiol. 2015;146:445–461. doi: 10.1085/jgp.201511404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banigan JR, Gayen A, Cho MK, Traaseth NJ. A structured loop modulates coupling between the substrate-binding and dimerization domains in the multidrug resistance transporter EmrE. J Biol Chem. 2015;290:805–814. doi: 10.1074/jbc.M114.601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrison EA, DeKoster GT, Dutta S, Vafabakhsh R, Clarkson MW, Bahl A, Kern D, Ha T, Henzler-Wildman KA. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature. 2011;481:45–50. doi: 10.1038/nature10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dastvan R, Fischer AW, Mishra S, Meiler J, McHaourab HS. Protonation-dependent conformational dynamics of the multidrug transporter EmrE. Proc Natl Acad Sci U S A. 2016;113:1220–1225. doi: 10.1073/pnas.1520431113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korkhov MV, Tate CG. Electron crstallography reveals plasticity within the drug binding site of the small multidrug transporter EmrE. J Mol Biol. 2008;377:1094–1103. doi: 10.1016/j.jmb.2008.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ubarretxena-Belandia I, Baldwin JM, Schuldiner S, Tate CG. Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. EMBO J. 2003;22:6175–6181. doi: 10.1093/emboj/cdg611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YJ, Pornillos O, Lieu S, Ma C, Chen AP, Chang G. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci U S A. 2007;104:18999–19004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fleishman SJ, Harrington SE, Enosh A, Halperin D, Tate CG, Ben-Tal N. Quasi-symmetry in the cryo-EM structure of EmrE provides the key to modeling its transmembrane domain. J Mol Biol. 2006;364:54–67. doi: 10.1016/j.jmb.2006.08.072. [DOI] [PubMed] [Google Scholar]
- 25.Leninger M, Traaseth NJ. NMR Spectroscopy approach to study the structure, orientation, and mechanism of the multidrug exporter EmrE. Methods Mol Biol. 2018;1700:83–96. doi: 10.1007/978-1-4939-7454-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White GF, Racher KI, Lipski A, Hallett FR, Wood JM. Physical properties of liposomes and proteoliposomes prepared from Escherichia coli polar lipids. Biochim Biophys Acta, Biomembr. 2000;1468:175–186. doi: 10.1016/s0005-2736(00)00255-8. [DOI] [PubMed] [Google Scholar]
- 27.Kwon B, Waring Alan J, Hong MA. (2)H Solid-state NMR study of lipid clustering by cationic antimicrobial and cell-penetrating peptides in model bacterial membranes. Biophys J. 2013;105:2333–2342. doi: 10.1016/j.bpj.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mani R, Cady SD, Tang M, Waring AJ, Lehrer RI, Hong M. Membrane-dependent oligomeric structure and pore formation of a β-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc Natl Acad Sci U S A. 2006;103:16242–16247. doi: 10.1073/pnas.0605079103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hallock KJ, Lee DK, Omnaas J, Mosberg HI, Ramamoorthy A. Membrane composition determines pardaxin’s mechanism of lipid bilayer disruption. Biophys J. 2002;83:1004–1013. doi: 10.1016/S0006-3495(02)75226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Picas L, Carretero-Genevrier A, Montero MT, Vázquez-Ibar JL, Seantier B, Milhiet PE, Hernández-Borrell J. Preferential insertion of lactose permease in phospholipid domains: AFM observations. Biochim Biophys Acta, Biomembr. 2010;1798:1014–1019. doi: 10.1016/j.bbamem.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 31.Picas L, Montero MT, Morros A, Vazquez-Ibar JL, Hernandez-Borrell J. Evidence of phosphatidylethanolamine and phosphatidylglycerol presence at the annular region of lactose permease of Escherichia coli. Biochim Biophys Acta. 2010;1798:291–296. doi: 10.1016/j.bbamem.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 32.Charalambous K, Miller D, Curnow P, Booth PJ. Lipid bilayer composition influences small multidrug transporters. BMC Biochemistry. 2008;9:31. doi: 10.1186/1471-2091-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seeger HM, Aldrovandi L, Alessandrini A, Facci P. Changes in single K(+) channel behavior induced by a lipid phase transition. Biophys J. 2010;99:3675–3683. doi: 10.1016/j.bpj.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banigan JR, Gayen A, Traaseth NJ. Correlating lipid bilayer fluidity with sensitivity and resolution of polytopic membrane protein spectra by solid-state NMR spectroscopy. Biochim Biophys Acta. 2015;1848:334–341. doi: 10.1016/j.bbamem.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griffin RG. Solid state nuclear magnetic resonance of lipid bilayers. Methods Enzymol. 1981;1984:108–174. doi: 10.1016/s0076-6879(81)72010-x. [DOI] [PubMed] [Google Scholar]
- 36.Seelig J. P-31 Nuclear magnetic-resonance and head group structure of phospholipids in membranes. Biochim Biophys Acta. 1978;515:105–140. doi: 10.1016/0304-4157(78)90001-1. [DOI] [PubMed] [Google Scholar]
- 37.Dufourc EJ, Mayer C, Stohrer J, Althoff G, Kothe G. Dynamics of phosphate head groups in biomembranes - comprehensive analysis using P-31 nuclear-magnetic-resonance lineshape and relaxation-time measurements. Biophys J. 1992;61:42–57. doi: 10.1016/S0006-3495(92)81814-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw KP, Brooks NJ, Clarke JA, Ces O, Seddon JM, Law RV. Pressure-temperature phase behaviour of natural sphingomyelin extracts. Soft Matter. 2012;8:1070–1078. [Google Scholar]
- 39.Costello AL, Alam TM. Using 31P MAS NMR to monitor a gel phase thermal disorder transition in sphingomyelin/cholesterol bilayers. Biochim Biophys Acta. 2008;1778:97–104. doi: 10.1016/j.bbamem.2007.08.031. [DOI] [PubMed] [Google Scholar]
- 40.Picard F, Pézolet M, Bougis PE, Auger M. Model of interaction between a cardiotoxin and dimyristoylphosphatidic acid bilayers determined by solid-state 31P NMR spectroscopy. Biophys J. 1996;70:1737–1744. doi: 10.1016/S0006-3495(96)79736-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fenske DB, Jarrell HC. Phosphorus-31 two-dimensional solid-state exchange NMR. Application to model membrane and biological systems. Biophys J. 1991;59:55–69. doi: 10.1016/S0006-3495(91)82198-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Guerneve C, Auger M. New approach to study fast and slow motions in lipid bilayers: application to dimyristoylphosphatidylcholine-cholesterol interactions. Biophys J. 1995;68:1952–1959. doi: 10.1016/S0006-3495(95)80372-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.da Silva TF, Sousa VF, Malheiro AR, Brites P. The importance of ether-phospholipids: A view from the perspective of mouse models. Biochim Biophys Acta, Molecular Basis of Disease. 2012;1822:1501–1508. doi: 10.1016/j.bbadis.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 44.Aussenac F, Lavigne B, Dufourc EJ. Toward bicelle stability with ether-linked phospholipids: temperature, composition, and hydration diagrams by 2H and 31P solid-state NMR. Langmuir. 2005;21:7129–7135. doi: 10.1021/la050243a. [DOI] [PubMed] [Google Scholar]
- 45.De Angelis AA, Opella SJ. Bicelle samples for solid-state NMR of membrane proteins. Nat Protoc. 2007;2:2332–2338. doi: 10.1038/nprot.2007.329. [DOI] [PubMed] [Google Scholar]
- 46.Ottiger M, Bax A. Bicelle-based liquid crystals for NMR-measurement of dipolar couplings at acidic and basic pH values. J Biomol NMR. 1999;13:187–191. doi: 10.1023/a:1008395916985. [DOI] [PubMed] [Google Scholar]
- 47.Cavagnero S, Dyson HJ, Wright PE. Improved low pH bicelle system for orienting macromolecules over a wide temperature range. J Biomol NMR. 1999;13:387–391. doi: 10.1023/a:1008360022444. [DOI] [PubMed] [Google Scholar]
- 48.Bertelsen K, Vad B, Nielsen EH, Hansen SK, Skrydstrup T, Otzen DE, Vosegaard T, Nielsen NC. Long-term-stable ether-lipid vs conventional ester-lipid bicelles in oriented solid-state NMR: altered structural information in studies of antimicrobial peptides. J Phys Chem B. 2011;115:1767–1774. doi: 10.1021/jp110866g. [DOI] [PubMed] [Google Scholar]
- 49.Wu CH, Ramamoorthy A, Opella SJ. High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J Magn Reson. 1994;109:270–272. [Google Scholar]
- 50.Marassi FM, Opella SJ. A solid-state NMR index of helical membrane protein structure and topology. J Magn Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R. Imaging membrane protein helical wheels. J Magn Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.