

Abstract
The inflammatory myopathies, which includes dermatomyositis, polymyositis, and the immune-mediated necrotizing myopathies, are a heterogeneous group of autoimmune diseases that manifest with muscle, skin or lung damage. Collectively, these autoimmune diseases result from loss of tolerance to a select group of self-antigens, although the precise mechanism through which this occurs is not known. Infection, malignancy, and certain medications including statins and the immune checkpoint inhibitors used in cancer therapy have been identified as potential immunologic triggers of the inflammatory myopathies. Some of these triggers are classically associated with specific myositis-specific autoantibodies (MSAs). The strong association between certain triggers and MSAs provides insights into how an immunologic event can lead to loss of tolerance to specific self-antigens, resulting in autoimmune disease. In this review, we discuss the proposed triggers of the inflammatory myopathies and their associations with MSAs, and provide insights into how these triggers may result in the inflammatory myopathies.
Introduction
The inflammatory myopathies are a heterogeneous group of diseases that result from an immune-mediated attack on muscle, skin, and/or lung. Common manifestations of myositis include proximal muscle weakness, rashes and interstitial lung disease. The inflammatory myopathies are further subclassified into dermatomyositis (DM), polymyositis (PM), the immune-mediated necrotizing myopathies (IMNM), and inclusion body myositis (IBM) by the presence or absence of specific clinicopathologic features (Lundberg et al., 2017).
The discovery of an expanding list of myositis-specific autoantibodies (MSAs) have further defined the inflammatory myopathies. To date, 16 MSAs have been described, and about 70-80% of patients with myositis have an identifiable MSA. Many MSAs have been discovered only recently, and it is likely that other MSAs will eventually be characterized in patients who today are considered to be autoantibody negative. There is a striking correlation between certain known MSAs and clinical features of disease. For example, patients with autoantibodies targeting histidyl-tRNA synthetase (anti-Jo-1) express features of the antisynthetase syndrome, including myositis, interstitial lung disease, mechanic’s hands and arthritis. Similarly, patients with autoantibodies directed against chromodomain helicase DNA binding protein 4 (Mi-2) generally have a severe DM rash without significant lung involvement (Betteridge and McHugh, 2016). Despite the clinical relevance of these autoantibodies in classifying patients, their role in the pathogenesis of the inflammatory myopathies remains poorly understood. It is still unknown whether these autoantibodies are pathogenic or are a simply a marker of the disease process itself (Gunawardena et al., 2009).
In this review, we provide a framework for understanding the pathogenesis of the inflammatory myopathies by exploring potential triggers of myositis that may lead to loss of tolerance to self-antigens. We focus on the inflammatory myopathies with a clear autoimmune pathogenesis, which includes dermatomyositis, polymyositis, and the immune-mediated necrotizing myopathies. While largely still unknown, proposed triggers of myositis include viral infections, cancer, and drugs such as statins and the novel immune-checkpoint inhibitors used in cancer therapy (see Table 1).
Table 1.
Known triggers of myositis and their associated autoantibodies.
| Trigger | Myositis specific autoantibody (MSA) | Prevalence of suspected trigger by MSA | |
|---|---|---|---|
| Infection | Proposed: Antisynthetase autoantibodies (Jo-1, PL-7, PL-12, EJ, OJ, KS) |
undefined | |
| Malignancy | Transcriptional intermediary factor 1-gamma (TIF-1γ) | 38-80% | |
| Nuclear matrix protein (NXP-2) | 24-38% | ||
| Anti-small ubiquitin-like modifier activating enzyme (SAE) | 14-50% | ||
| Anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) | 5-36% | ||
| Drug | Statin-associated autoimmune myopathy | Anti-HMGCR | > 90% of patients older than 50 years |
| Immune checkpoint inhibitors | Unknown | ||
Viral-induced inflammatory myopathy
Multiple studies have demonstrated a possible link between inflammatory myopathy and infection. Seasonal variation in the onset of myositis by autoantibody has led to the hypothesis that viruses may trigger inflammatory myopathy in some patients (Sarkar et al., 2005) (Manta et al., 1989). Patients with antisynthetase antibodies tend to present in the spring, and patients with autoantibodies targeting signal recognition particle (anti-SRP) seem to cluster in the fall (Leff et al., 2010). A study in a juvenile DM population found that 51% of patients had a clinical history suggestive of an infectious process, most often respiratory, that preceded the onset of myositis (Manlhiot et al., 2008). In an adult myositis population, a preceding gastrointestinal or respiratory illness (but not skin infection) increased the risk of developing inflammatory myopathy (Svensson et al., 2017).
Infectious agents that have been proposed to be triggers of myositis include Coxsackie B virus, parvovirus, enterovirus, human T-cell lymphotropic virus (HTLV-1), and human immunodeficiency virus (HIV). Many of these viruses have a tropism for muscle, and Coxsackie B1 virus is even used as an animal model of myositis (Strongwater et al., 1984). Antibodies against Coxsackie B virus and HTLV-1 are found in greater frequency in patients with inflammatory myopathy compared to healthy controls (Morgan et al., 1989). It is possible that the more frequent viral antibodies in myositis may be explained by immune dysregulation and a higher infection risk from the underlying inflammatory process and immunosuppression. However, Christenson et al. found a higher prevalence of Coxsackie-B virus antibodies in juvenile DM and showed that this was virus- and disease-specific (Christensen et al., 1986), suggesting that Coxsackie B virus may play a unique role in the disease process itself.
One hypothesis for viral-induced autoimmunity is that latent viral infection in muscle may drive the continued immune response against muscle. Despite several studies that identified viral DNA in the muscle of patients with inflammatory myositis (Bowles et al., 1987) (Yousef et al., 1990), other studies have been unable to identify any viral DNA (Leff et al., 1992) (Fox et al., 1994). Chevrel et al. found parvovirus B19 DNA in two sequential muscle biopsies from a patient with polymyositis, but no viral DNA in the third muscle biopsy obtained from the same patient during a flare (Chevrel et al., 2000) (Chevrel et al., 2003). It is possible that acute viral infection in muscle may trigger immunity against muscle, and this immune response then self-propagates in the chronic phase after the pathogen is cleared. Patients with HIV-associated polymyositis generally have an excellent response to immunosuppression (Johnson et al., 2003), which suggests that the primary driver of muscle damage is an overactive immune response.
Several mechanisms have been proposed for the pathogenesis of inflammatory myopathies triggered by infectious agents (Zampieri et al., 2010). The pathogen may interact with and alter cellular proteins, thereby changing how they are recognized by the immune system. For example, the host tRNA synthetase is used for viral replication, and it is possible that immune tolerance is broken when the tRNA synthetase is presented to the immune system in association with viral protein (Mathews and Bernstein, n.d.). Infectious agents may also break self-tolerance by changing the conformation of cellular proteins and exposing cryptic epitopes, which are epitopes normally hidden from T-cell recognition (Warnock and Goodacre, n.d.). Infections can also induce production of autoantibodies and can expand and activate autoreactive B-cells (Soulas et al., 2005).
Molecular mimicry may also play an important role in autoimmunity induced by infections. Sequence similarities between pathogens and host proteins may lead to cross-reactivity between the pathogen-specific immune response and self-antigens (Albert and Inman, 1999). For example, Massa et al. showed that the immune response in JDM is directed against a homologous sequence shared between group A streptococcal type 5M protein and skeletal muscle myosin (Massa et al., 2002). Another group showed that there is significant homology between the antisynthetase autoantigens histidyl-tRNA synthetase (Jo-1) and alanyl-tRNA synthetase (PL-12) and various proteins on pathogens including Ebstein-Barr Virus (EBV), adenovirus, and influenza. Moreover, there are considerable similarities between the autoantigen PL-12 and both tropomyosin and keratin, suggesting that tissue damage may occur from cross-reactivity of the anti-PL-12 autoantibody with muscle and connective tissue (Walker and Jeffrey, 1986).
Cancer-induced myositis
The striking association between cancer and DM was established in 1992 in a landmark study that demonstrated that 32% of DM patients are diagnosed with cancer at some point during their disease course. There was a strong temporal association between the diagnosis of cancer and DM, with 71% of cancers being diagnosed within 2 years of the onset of myositis. The cancers that are most strongly associated with DM are ovarian, lung, gastric, colorectal, pancreatic, and non-Hodgkin’s lymphoma (Hill et al., 2001). An epidemiologic link between DM and cancer was subsequently confirmed in multiple epidemiological studies.
Several possible mechanisms were initially proposed for the link between cancer and DM, but evidence for a discrete mechanism remained elusive for many years. Immune dysregulation from myositis, or resulting from therapies used in the treatment of myositis, might increase the risk of malignancy (Zintzaras et al., 2005). However, the striking temporal association between malignancy and cancer, in which the vast majority of malignancies are diagnosed within 3 years before or after the onset of myositis, provided evidence for a causal link between cancer and myositis (Kang et al., 2016). Moreover, reports of cancer-associated myositis that improved or resolved after treatment of the underlying cancer suggested that the tumor itself was directly perpetuating the autoimmune process as a paraneoplastic phenomenon (Osako et al., 2007)(Tagawa et al., 2000).
It is now understood that not all patients with DM are at increased risk for cancer. Certain MSAs are associated with a dramatically increased risk of cancer-associated myositis, while other MSAs are not associated with cancer. Patients with autoantibodies targeting transcriptional intermediary factor 1-gamma (TIF-1γ) and nuclear matrix protein (NXP-2) have a particularly high risk of cancer-associated myositis (Kaji et al., 2007) (Fiorentino et al., 2013) (Tiniakou and Mammen, 2017). In one study, 83% of patients with cancer-associated DM had autoantibodies with specificities for either TIF-1γ or NXP-2 (Fiorentino et al., 2013), and the sensitivity and specificity of the presence of TIF-1γ for cancer-associated myositis in one meta-analysis was 70% and 89%, respectively (Selva-O’Callaghan et al., 2010). Autoantibodies targeting anti-small ubiquitin-like modifier activating enzyme (anti-SAE), a relatively rare autoantibody in myositis, may also be associated with an increased malignancy risk (Muro et al., 2015). Patients with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), an autoantibody that often is associated with statin use, may also in some cases present as a paraneoplastic phenomenon (Allenbach et al., 2016) (Yang et al., 2017) (Kadoya et al., 2016). The presence of other autoantibodies, such as those present in the antisynthetase syndrome including histidyl tRNA synthetase (anti-Jo-1), are not associated with cancer.
The specificity of the relationship between malignancy and certain MSAs in DM provides a basis for understanding a proposed mechanism for cancer-induced autoimmunity. It is now appreciated that malignancies express aberrant proteins, called tumor-specific antigens (neoantigens), which arise from somatic mutations in their DNA (Yarchoan et al., 2017). Foreign proteins expressed by tumor cells can be highly immunogenic, inducing a vigorous immune response that cross-reacts with normal host tissues. This mechanism of cancer-induced autoimmunity is supported by a landmark study of patients with cancer who concurrently developed scleroderma with detectable autoantibodies against RNA polymerase III subunit RPC1. Sequencing of the patients’ tumors revealed that many of the cancers had genetic alterations in the gene POLR3A, which codes for RPC1, and these patients had neoantigen-specific T cell immune responses (Joseph et al., 2014). A similar mechanism of autoimmunity in myositis is supported by a recent study of patients with cancer-associated DM and TIF-1γ autoantibodies in which many of the tumors from these patients harbored mutations in the gene coding for TIF-1γ (Pinal-Fernandez et al., 2017). These findings provided strong evidence of a discrete mechanism for cancer-induced autoimmunity in which a T-cell response against a tumor neoantigen cross-reacts with the nonmutated form of the antigen, thereby generating autoimmunity in the host (see Figure 1).
Figure 1.
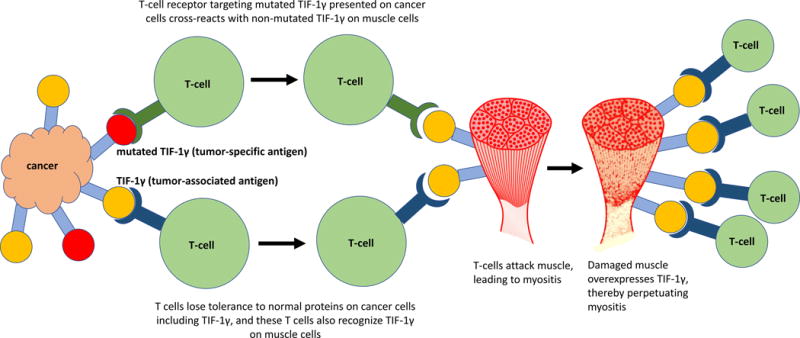
Proposed model for cancer-induced myositis.
In the setting of malignancy, anti-tumor immunity can also develop against normal host proteins that are highly expressed on tumor cells (tumor-associated antigens, TAAs) as a result of epitope spreading (Yarchoan et al., 2017). Since tumor-associated antigens are not specific to tumor cells, the loss of tolerance to a TAA may result in host immunity against normal tissues, resulting in autoimmunity. This alternative mechanism for cancer-induced autoimmunity is exemplified by cancer immunotherapies against TAAs that have resulted in immune-mediated toxicity in normal tissues. For example, adoptive therapy of T cells recognizing melanoma-associated antigen 3 (MAGE-A3), which is highly expressed by some malignancies, resulted in severe neurological toxicity related to MAGE-A expression in the brain (Morgan et al., 2013). Cancers that are associated with myositis often highly express myositis-associated antigens, and these same antigens are also highly expressed in regenerating muscle tissue (Casciola-Rosen et al., 2005) (Chushi et al., 2016). It is therefore possible that loss of tolerance to myositis autoantigens on tumor cells may perpetuate myositis through immunity against these same antigens on regenerating muscle cells (see Figure 1).
Not all adult myositis patients with the cancer-associated autoantibodies anti-TIF-1γ, anti-NXP-2, or anti-SAE have an identifiable underlying malignancy. It is not known why only some patients with these autoantibodies either present with or eventually develop a malignancy. In children, autoantibodies targeting TIF-1γ and NXP-2 are among the most common autoantibodies in juvenile dermatomyositis (JDM) and are not associated with malignancy (Morris and Dare, 2010). There may be other mechanisms unrelated to cancer in which the immune system loses tolerance to the TIF-1γ or NXP-2 antigens. It is also conceivable that anti-TIF-1γ and anti-NXP-2 immunity in the juvenile DM population may still be driven by an anti-cancer immune response directed against atypical cells, and the vigorous immune response against these neoantigens may eliminate any subclinical malignancy before it is detected.
Drug-induced myositis
Statins
Anti-HMGCR Ab+ myopathy is one of the most common of the immune-mediated necrotizing myopathies and accounts for approximately 6% of all idiopathic inflammatory myopathies (Christopher-Stine et al., 2010). Anti-HMGCR Ab+ myopathy is strongly associated with statin use, and when it occurs in the setting of statins it is often referred to as statin-associated necrotizing autoimmune myopathy (SANAM). In older adult cohorts, the vast majority of patients (over 90%) with anti-HMGCR Ab+ myopathy report prior or current statin use (Mammen et al., 2011).
SANAM is pathologically distinct from the more common statin intolerance experienced by many patients. Self-limiting statin myopathy can present similarly with myalgias and elevated CK, but it resolves with discontinuation of the statin. In contrast, SANAM is an autoimmune muscle disease in which autoantibodies against HMG-CoA Reductase (HMGCR) are present (Mammen et al., 2012b). The autoimmune process endures even after withdrawal of the statin, and patients often require chronic treatment with immunomodulatory agents.
Several observations have led to possible disease models for anti-HMGCR Ab+ myopathy. HMGCR is the pharmacologic target of statins, which raises the question of whether statins may trigger autoimmunity through their actions on the HMGCR protein. One proposed mechanism is that statins may change the conformation of the HMGCR protein and trigger autoimmunity by exposing cryptic epitopes (Mammen, 2016). Antigen presentation, whether of cryptic or dominant epitopes, seems to be a critical factor in the development of the disease based on the observation that the presence of the class II HLA allele DRB1*11:01 increases the odds ratio of developing anti-HMGCR autoantibodies by as much as 25-57 depending on patient race (Mammen et al., 2012a) (Limaye et al., 2015). Statins increase the expression of HMGCR in muscle, which may exacerbate autoimmunity against muscle tissues (Morikawa et al., 2005) (Gouni-Berthold et al., 2008).
Anti-HMGCR Ab+ myopathy does not remit even after withdrawal of the statin, which suggests that the immune response self-perpetuates once tolerance to HMGCR is broken. This contrasts with cancer-associated myositis, which can improve (although not always) after the cancer is eliminated. Regenerating muscle cells express high levels of HMGCR, which may provide a continuous source of antigen to propagate the immune response (Mammen et al., 2011). However, it is unclear whether autoantibodies against HMGCR are pathogenic. HMGCR autoantibody titers correlate with disease activity, but remain elevated in some patients even after they achieve clinical remission (Werner et al., 2012). Although the inciting event in most cases of HMGCR Ab+ myopathy seems to be loss of tolerance to HMGCR in the setting of statin therapy, more research is needed to understand how autoimmunity perpetuates after the statin is withdrawn.
Immune Checkpoint Inhibitors
Immune checkpoints are critical regulators of the immune response that help to maintain immune tolerance. Novel cancer immunotherapies target immune checkpoints to unleash the anti-tumor immune response and improve survival in a variety of cancers (Pardoll, 2012). The first immune checkpoint inhibitor approved by the FDA, ipilimumab, is a monoclonal antibody that enhances anti-tumor immunity by binding to the immune checkpoint cluster of differentiation 152 (CTLA-4), which acts as a negative regulator of the T-cell response (Hodi et al., 2010). Another immune checkpoint inhibitor, pembrolizumab, blocks the co-inhibitory interaction of program cell death-1 (PD-1) on T-cells with its ligand (PD-L1) on antigen presenting cells. The immune checkpoint inhibitors have transformed the management of many cancers, and the development of additional immune modulators that can further increase antitumor immunity is a top priority in oncologic research.
The immune checkpoint inhibitors sometimes result in immune-related adverse events (irAEs) that closely resemble sporadic immunity. Colitis, hepatitis, dermatitis, and endocrinopathies are among the most commonly reported irAEs among patients receiving immune checkpoint inhibitors (Postow et al., 2018). DM, PM, and IMNM have also been reported as immune complications of anti-PD-1 and anti-CTLA-4 immunotherapy (Cappelli et al., 2017) (Sheik Ali et al., 2015). Older cancer immunotherapies including interferon-alpha treatment and IL-2 therapy have also been associated with myositis (Dietrich et al., 2000) (Esteva-Lorenzo et al., 1995). irAEs resulting from cancer immunotherapies provide a novel lens in which to study autoimmunity and indicate that many forms of immune dysregulation, whether through modulating immune checkpoints, cytokines, or other inflammatory pathways, may precipitate myositis.
Although dysregulation of immune homeostasis can precipitate autoimmunity, this does not explain the specificity of organ involvement among the irAEs. For example, patients with checkpoint-induced myositis frequently have muscle and/or skin involvement, but the GI tract or nervous system are rarely involved. It also remains unclear why some organs are more prone to irAEs than others, and why the organs most at risk differ among the classes of immune checkpoint inhibitors. For example, colitis is more common with therapies targeting CTLA-4 and pneumonitis is more common with anti-PD-1 therapy (Spain et al., 2016). To date there have been no studies of whether myositis-specific autoantibodies are present in patients with checkpoint-induced myositis, and if these autoantibodies pre-date the administration of immunotherapy. It is also unclear what role, if any, the cancer may play in irAEs. Preliminary data show that patients with irAEs may have a better cancer response to immunotherapy than patients without autoimmune toxicities (Curti et al., 2017) (Freeman-Keller et al., 2016) (Weber et al., 2012). This suggests that autoimmunity may correlate with anti-tumor immunity, although the cause of this association needs further investigation.
Conclusion
Definite disease triggers can be identified in some patients with inflammatory myopathy. These include environmental factors such as infection, malignancy, and drug toxicities from statins or immune checkpoint inhibitors. Each of these triggers provides a window into how tolerance to a self-antigen can be broken and/or the immune system unleashed to result in chronic autoimmunity.
Acknowledgments
Funding:
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number T32AR048522 (BA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Research was also supported by the Huayi and Siuling Zhang Discovery Fund (LCS).
Footnotes
Disclosure Statement: The authors have no conflicts of interest to disclose.
References
- Albert LJ, Inman RD. Molecular Mimicry and Autoimmunity. N Engl J Med. 1999;341(27):2068–2074. doi: 10.1056/NEJM199912303412707. [DOI] [PubMed] [Google Scholar]
- Allenbach Y, Keraen J, Bouvier A, Jooste V, Champtiaux N, Hervier B, Schoindre Y, Rigolet A, Gilardin L, Musset L, Charuel J-L, Boyer O, Jouen F, Drouot L, Martinet J, Stojkovic T, Eymard B, Laforêt P, Behin A, Salort-Campana E, et al. High risk of cancer in autoimmune necrotizing myopathies: usefulness of myositis specific antibody. Brain. 2016;139(8) doi: 10.1093/brain/aww054. [DOI] [PubMed] [Google Scholar]
- Betteridge Z, McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med. 2016;280(1):8–23. doi: 10.1111/joim.12451. [DOI] [PubMed] [Google Scholar]
- Bowles NE, Dubowitz V, Sewry CA, Archard LC. Dermatomyositis, polymyositis, and Coxsackie-B-virus infection. Lancet (London, England) 1987;1(8540):1004–7. doi: 10.1016/s0140-6736(87)92271-9. [DOI] [PubMed] [Google Scholar]
- Cappelli LC, Gutierrez AK, Bingham CO, Shah AA. Rheumatic and Musculoskeletal Immune-Related Adverse Events Due to Immune Checkpoint Inhibitors: A Systematic Review of the Literature. Arthritis Care Res (Hoboken) 2017;69(11):1751–1763. doi: 10.1002/acr.23177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen L, Nagaraju K, Plotz P, Wang K, Levine S, Gabrielson E, Corse A, Rosen A. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med. 2005;201(4):591–601. doi: 10.1084/jem.20041367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrel G, Borsotti JP, Miossec P. Lack of evidence for a direct involvement of muscle infection by parvovirus B19 in the pathogenesis of inflammatory myopathies: a follow-up study. Rheumatology (Oxford) 2003;42(2):349–52. doi: 10.1093/rheumatology/keg085. [DOI] [PubMed] [Google Scholar]
- Chevrel G, Calvet A, Belin V, Miossec P. Dermatomyositis associated with the presence of parvovirus B19 DNA in muscle. Rheumatology (Oxford) 2000;39(9):1037–9. doi: 10.1093/rheumatology/39.9.1037. [DOI] [PubMed] [Google Scholar]
- Christensen ML, Pachman LM, Schneiderman R, Patel DC, Friedman JM. PREVALENCE OF COXSACKIE B VIRUS ANTIBODIES IN PATIENTS WITH JUVENILE DERMATOMYOSITIS. Arthritis Rheum. 1986;29(11):1365–1370. doi: 10.1002/art.1780291109. [DOI] [PubMed] [Google Scholar]
- Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010;62(9):2757–66. doi: 10.1002/art.27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chushi L, Wei W, Kangkang X, Yongzeng F, Ning X, Xiaolei C. HMGCR is up-regulated in gastric cancer and promotes the growth and migration of the cancer cells. Gene. 2016;587(1):42–47. doi: 10.1016/j.gene.2016.04.029. [DOI] [PubMed] [Google Scholar]
- Curti B, Daniels GA, McDermott DF, Clark JI, Kaufman HL, Logan TF, Singh J, Kaur M, Luna TL, Gregory N, Morse MA, Wong MKK, Dutcher JP. Improved survival and tumor control with Interleukin-2 is associated with the development of immune-related adverse events: data from the PROCLAIMSM registry. J Immunother Cancer. 2017;5(1):102. doi: 10.1186/s40425-017-0307-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich LL, Bridges AJ, Albertini MR. Dermatomyositis after interferon alpha treatment. Med Oncol. 2000;17(1):64–9. doi: 10.1007/BF02826219. [DOI] [PubMed] [Google Scholar]
- Esteva-Lorenzo FJ, Janik JE, Fenton RG, Emslie-Smith A, Engel AG, Longo DL. Myositis associated with interleukin-2 therapy in a patient with metastatic renal cell carcinoma. Cancer. 1995;76(7):1219–23. doi: 10.1002/1097-0142(19951001)76:7<1219::aid-cncr2820760719>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Fiorentino DF, Chung LS, Christopher-Stine L, Zaba L, Li S, Mammen AL, Rosen A, Casciola-Rosen L. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1γ. Arthritis Rheum. 2013;65(11):2954–62. doi: 10.1002/art.38093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox SA, Finklestone E, Robbins PD, Mastaglia FL, Swanson NR. Search for persistent enterovirus infection of muscle in inflammatory myopathies. J Neurol Sci. 1994;125(1):70–6. doi: 10.1016/0022-510x(94)90244-5. [DOI] [PubMed] [Google Scholar]
- Freeman-Keller M, Kim Y, Cronin H, Richards A, Gibney G, Weber JS. Nivolumab in Resected and Unresectable Metastatic Melanoma: Characteristics of Immune-Related Adverse Events and Association with Outcomes. Clin Cancer Res. 2016;22(4):886–94. doi: 10.1158/1078-0432.CCR-15-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouni-Berthold I, Berthold HK, Gylling H, Hallikainen M, Giannakidou E, Stier S, Ko Y, Patel D, Soutar AK, Seedorf U, Mantzoros CS, Plat J, Krone W. Effects of ezetimibe and/or simvastatin on LDL receptor protein expression and on LDL receptor and HMG-CoA reductase gene expression: A randomized trial in healthy men. Atherosclerosis. 2008;198(1):198–207. doi: 10.1016/j.atherosclerosis.2007.09.034. [DOI] [PubMed] [Google Scholar]
- Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford) 2009;48(6):607–12. doi: 10.1093/rheumatology/kep078. [DOI] [PubMed] [Google Scholar]
- Hill CL, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, Airio A, Evans SR, Felson DT. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet (London, England) 2001;357(9250):96–100. doi: 10.1016/S0140-6736(00)03540-6. [DOI] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJM, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RW, Williams FM, Kazi S, Dimachkie MM, Reveille JD. Human immunodeficiency virus–associated polymyositis: A longitudinal study of outcome. Arthritis Rheum. 2003;49(2):172–178. doi: 10.1002/art.11002. [DOI] [PubMed] [Google Scholar]
- Joseph CG, Darrah E, Shah AA, Skora AD, Casciola-Rosen LA, Wigley FM, Boin F, Fava A, Thoburn C, Kinde I, Jiao Y, Papadopoulos N, Kinzler KW, Vogelstein B, Rosen A. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science. 2014;343(6167):152–7. doi: 10.1126/science.1246886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoya M, Hida A, Hashimoto Maeda M, Taira K, Ikenaga C, Uchio N, Kubota A, Kaida K, Miwa Y, Kurasawa K, Shimada H, Sonoo M, Chiba A, Shiio Y, Uesaka Y, Sakurai Y, Izumi T, Inoue M, Kwak S, Tsuji S, et al. Cancer association as a risk factor for anti-HMGCR antibody-positive myopathy. Neurol - Neuroimmunol Neuroinflammation. 2016;3(6):e290. doi: 10.1212/NXI.0000000000000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji K, Fujimoto M, Hasegawa M, Kondo M, Saito Y, Komura K, Matsushita T, Orito H, Hamaguchi Y, Yanaba K, Itoh M, Asano Y, Seishima M, Ogawa F, Sato S, Takehara K. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 2007;46(1):25–8. doi: 10.1093/rheumatology/kel161. [DOI] [PubMed] [Google Scholar]
- Kang EH, Lee SJ, Ascherman DP, Lee YJ, Lee EY, Lee EB, Song YW. Temporal relationship between cancer and myositis identifies two distinctive subgroups of cancers: impact on cancer risk and survival in patients with myositis. Rheumatology (Oxford) 2016;55(9):1631–41. doi: 10.1093/rheumatology/kew215. [DOI] [PubMed] [Google Scholar]
- Leff RL, Burgess SH, Miller FW, Love LA, Targoff IN, Dalakas MC, Joffe MM, Plotz PH. Distinct seasonal patterns in the onset of adult idiopathic inflammatory myopathy in patients with anti-jo-1 and anti-signal recognition particle autoantibodies. Arthritis Rheum. 2010;34(11):1391–1396. doi: 10.1002/art.1780341108. [DOI] [PubMed] [Google Scholar]
- Leff RL, Love LA, Miller FW, Greenberg SJ, Klein EA, Dalakas MC, Plotz PH. Viruses in idiopathic inflammatory myopathies: absence of candidate viral genomes in muscle. Lancet (London, England) 1992;339(8803):1192–5. doi: 10.1016/0140-6736(92)91134-t. [DOI] [PubMed] [Google Scholar]
- Limaye V, Bundell C, Hollingsworth P, Rojana-Udomsart A, Mastaglia F, Blumbergs P, Lester S. Clinical and genetic associations of autoantibodies to 3-hydroxy-3-methyl-glutaryl-coenzyme a reductase in patients with immune-mediated myositis and necrotizing myopathy. Muscle Nerve. 2015;52(2):196–203. doi: 10.1002/mus.24541. [DOI] [PubMed] [Google Scholar]
- Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, Alfredsson L, Amato AA, Barohn RJ, Liang MH, Singh JA, Aggarwal R, Arnardottir S, Chinoy H, Cooper RG, Dankó K, Dimachkie MM, Feldman BM, Garcia-De La Torre I, Gordon P, et al. 2017 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and Their Major Subgroups. Arthritis Rheumatol. 2017;69(12):2271–2282. doi: 10.1002/art.40320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL. Statin-Associated Autoimmune Myopathy. N Engl J Med. 2016;374(7):664–669. doi: 10.1056/NEJMra1515161. [DOI] [PubMed] [Google Scholar]
- Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Doering KR, Casciola-Rosen LA. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63(3):713–21. doi: 10.1002/art.30156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Gaudet D, Brisson D, Christopher-Stine L, Lloyd TE, Leffell MS, Zachary AA. Increased frequency of DRB1*11:01 in anti-hydroxymethylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Care Res (Hoboken) 2012a;64(8):1233–7. doi: 10.1002/acr.21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Pak K, Williams EK, Brisson D, Coresh J, Selvin E, Gaudet D. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken) 2012b;64(2):269–72. doi: 10.1002/acr.20662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manlhiot C, Liang L, Tran D, Bitnun A, Tyrrell PN, Feldman BM. Assessment of an infectious disease history preceding juvenile dermatomyositis symptom onset. Rheumatology (Oxford) 2008;47(4):526–9. doi: 10.1093/rheumatology/ken038. [DOI] [PubMed] [Google Scholar]
- Manta P, Kalfakis N, Vassilopoulos D. Evidence for Seasonal Variation in Polymyositis. Neuroepidemiology. 1989;8(5):262–265. doi: 10.1159/000110192. [DOI] [PubMed] [Google Scholar]
- Massa M, Costouros N, Mazzoli F, De Benedetti F, La Cava A, Le T, de Kleer I, Ravelli A, Liotta M, Roord S, Berry C, Pachman LM, Martini A, Albani S. Self epitopes shared between human skeletal myosin andStreptococcus pyogenes M5 protein are targets of immune responses in active juvenile dermatomyositis. Arthritis Rheum. 2002;46(11):3015–3025. doi: 10.1002/art.10566. [DOI] [PubMed] [Google Scholar]
- Mathews MB, Bernstein RM. Myositis autoantibody inhibits histidyl-tRNA synthetase: a model for autoimmunity. Nature. 304(5922):177–9. doi: 10.1038/304177a0. [DOI] [PubMed] [Google Scholar]
- Morgan OS, Rodgers-Johnson P, Mora C, Char G. HTLV-1 and polymyositis in Jamaica. Lancet (London, England) 1989;2(8673):1184–7. doi: 10.1016/s0140-6736(89)91793-5. [DOI] [PubMed] [Google Scholar]
- Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, Miller AD, Hessman CJ, Stewart AA, Restifo NP, Quezado MM, Alimchandani M, Rosenberg AZ, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–51. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa S, Murakami T, Yamazaki H, Izumi A, Saito Y, Hamakubo T, Kodama T. Analysis of the global RNA expression profiles of skeletal muscle cells treated with statins. J Atheroscler Thromb. 2005;12(3):121–31. doi: 10.5551/jat.12.121. [DOI] [PubMed] [Google Scholar]
- Morris P, Dare J. Juvenile Dermatomyositis as a Paraneoplastic Phenomenon: An Update. J Pediatr Hematol Oncol. 2010;32(3):189–191. doi: 10.1097/MPH.0b013e3181bf29a2. [DOI] [PubMed] [Google Scholar]
- Muro Y, Sugiura K, Nara M, Sakamoto I, Suzuki N, Akiyama M. High incidence of cancer in anti-small ubiquitin-like modifier activating enzyme antibody-positive dermatomyositis: Table 1. Rheumatology. 2015;54(9):1745–1747. doi: 10.1093/rheumatology/kev247. [DOI] [PubMed] [Google Scholar]
- Osako T, Ito Y, Morimatsu A, Jinnin M, Tada K, Sakurai N, Takahashi S, Akiyama F, Sakamoto G, Iwase T, Hatake K. Flare-up of Dermatomyositis Along with Recurrence of Breast Cancer. Breast J. 2007;13(2):200–202. doi: 10.1111/j.1524-4741.2007.00400.x. [DOI] [PubMed] [Google Scholar]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinal-Fernandez I, Ferrer-Fabregas B, Trallero-Araguas E, Balada E, Martínez MA, Milisenda JC, Aparicio-Español G, Labrador-Horrillo M, Garcia-Patos V, Grau-Junyent JM, Selva-O’Callaghan A. Tumour TIF1 mutations and loss of heterozygosity related to cancer-associated myositis. Rheumatology (Oxford) 2017 doi: 10.1093/rheumatology/kex413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N Engl J Med. 2018;378(2):158–168. doi: 10.1056/NEJMra1703481. [DOI] [PubMed] [Google Scholar]
- Sarkar K, Weinberg CR, Oddis CV, Medsger TA, Jr, Plotz PH, Reveille JD, Arnett FC, Targoff IN, Genth E, Love LA, Miller FW. Seasonal influence on the onset of idiopathic inflammatory myopathies in serologically defined groups. Arthritis Rheum. 2005;52(8):2433–2438. doi: 10.1002/art.21198. [DOI] [PubMed] [Google Scholar]
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, Labrador-Horrillo M. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol. 2010;22(6):627–32. doi: 10.1097/BOR.0b013e32833f1075. [DOI] [PubMed] [Google Scholar]
- Sheik Ali S, Goddard AL, Luke JJ, Donahue H, Todd DJ, Werchniak A, Vleugels RA. Drug-Associated Dermatomyositis Following Ipilimumab Therapy. JAMA Dermatology. 2015;151(2):195. doi: 10.1001/jamadermatol.2014.2233. [DOI] [PubMed] [Google Scholar]
- Soulas P, Woods A, Jaulhac B, Knapp A-M, Pasquali J-L, Martin T, Korganow A-S. Autoantigen, innate immunity, and T cells cooperate to break B cell tolerance during bacterial infection. J Clin Invest. 2005;115(8):2257–67. doi: 10.1172/JCI24646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spain L, Diem S, Larkin J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev. 2016;44:51–60. doi: 10.1016/j.ctrv.2016.02.001. [DOI] [PubMed] [Google Scholar]
- Strongwater SL, Dorovini-zis K, Ball RD, Schnitzer TJ. A Murine Model of Polymyositis Induced by Coxsackievirus B1 (Tucson Strain) Arthritis Rheum. 1984;27(4):433–442. doi: 10.1002/art.1780270411. [DOI] [PubMed] [Google Scholar]
- Svensson J, Holmqvist M, Lundberg IE, Arkema EV. Infections and respiratory tract disease as risk factors for idiopathic inflammatory myopathies: a population-based case – control study. Ann Rheum Dis. 2017;76(11):1803–1808. doi: 10.1136/annrheumdis-2017-211174. [DOI] [PubMed] [Google Scholar]
- Tagawa A, Kashima R, Kaneda K, Nakayama M, Ono S, Shimizu N. Polymyositis successfully treated with surgical resection of colon cancer. Eur Neurol. 2000;44(4):251–2. doi: 10.1159/000008247. [DOI] [PubMed] [Google Scholar]
- Tiniakou E, Mammen AL. Idiopathic Inflammatory Myopathies and Malignancy: a Comprehensive Review. Clin Rev Allergy Immunol. 2017;52(1):20–33. doi: 10.1007/s12016-015-8511-x. [DOI] [PubMed] [Google Scholar]
- Walker E, Jeffrey P. POLYMYOSITIS AND MOLECULAR MIMICRY, A MECHANISM OF AUTOIMMUNITY. Lancet. 1986;328(8507):605–607. doi: 10.1016/s0140-6736(86)92429-3. [DOI] [PubMed] [Google Scholar]
- Warnock MG, Goodacre JA. CRYPTIC T-CELL EPITOPES AND THEIR ROLE IN THE PATHOGENESIS OF AUTOIMMUNE DISEASES. doi: 10.1093/rheumatology/36.11.1144. [DOI] [PubMed] [Google Scholar]
- Weber JS, Kähler KC, Hauschild A. Management of Immune-Related Adverse Events and Kinetics of Response With Ipilimumab. J Clin Oncol. 2012;30(21):2691–2697. doi: 10.1200/JCO.2012.41.6750. [DOI] [PubMed] [Google Scholar]
- Werner JL, Christopher-Stine L, Ghazarian SR, Pak KS, Kus JE, Daya NR, Lloyd TE, Mammen AL. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum. 2012;64(12):4087–93. doi: 10.1002/art.34673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Peng Q, Yin L, Li S, Shi J, Zhang Y, Lu X, Shu X, Zhang S, Wang G. Identification of multiple cancer-associated myositis-specific autoantibodies in idiopathic inflammatory myopathies: a large longitudinal cohort study. Arthritis Res Ther. 2017;19(1):259. doi: 10.1186/s13075-017-1469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17(4):209–222. doi: 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef GE, Isenberg DA, Mowbray JF. Detection of enterovirus specific RNA sequences in muscle biopsy specimens from patients with adult onset myositis. Ann Rheum Dis. 1990;49(5):310–5. doi: 10.1136/ard.49.5.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampieri S, Valente M, Adami N, Biral D, Ghirardello A, Rampudda ME, Vecchiato M, Sarzo G, Corbianco S, Kern H, Carraro U, Bassetto F, Merigliano S, Doria A. Polymyositis, dermatomyositis and malignancy: a further intriguing link. Autoimmun Rev. 2010;9(6):449–53. doi: 10.1016/j.autrev.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Zintzaras E, Voulgarelis M, Moutsopoulos HM. The Risk of Lymphoma Development in Autoimmune Diseases. Arch Intern Med. 2005;165(20):2337. doi: 10.1001/archinte.165.20.2337. [DOI] [PubMed] [Google Scholar]