

Key Points
miR-125b is epigenetically silenced in B cells.
Physiological silencing of miR-125b is required for normal B-cell development.
Abstract
Deregulation of several microRNAs (miRs) can influence critical developmental checkpoints during hematopoiesis as well as cell functions, eventually leading to the development of autoimmune disease or cancer. We found that miR-125b is expressed in bone marrow multipotent progenitors and myeloid cells but shut down in the B-cell lineage, and the gene encoding miR-125b lacked transcriptional activation markers in B cells. To understand the biological importance of the physiological silencing of miR-125b expression in B cells, we drove its expression in the B-cell lineage and found that dysregulated miR-125b expression impaired egress of immature B cells from the bone marrow to peripheral blood. Such impairment appeared to be mediated primarily by inhibited expression of the sphingosine-1-phosphate receptor 1 (S1PR1). Enforced expression of S1PR1 or clustered regularly interspaced short palindromic repeats/Cas9–mediated genome editing of the miR-125b targeting site in the S1PR1 3′ untranslated region rescued the miR-125b–mediated defect in B-cell egress. In addition to impaired B-cell egress, miR-125b dysregulation initially reduced pre–B-cell output but later induced pre–B-cell lymphoma/leukemia in mice. Genetic deletion of IRF4 was found in miR-125b–induced B-cell cancer, but its role in oncogenic miR-125b–induced B-cell transformation is still unknown. Here, we further demonstrated an interaction of the effects of miR-125b and IRF4 in cancer induction by showing that miR125b-induced B-cell leukemia was greatly accelerated in IRF4 homozygous mutant mice. Thus, we conclude that physiological silencing of miR-125b is required for normal B-cell development and also acts as a mechanism of cancer suppression.
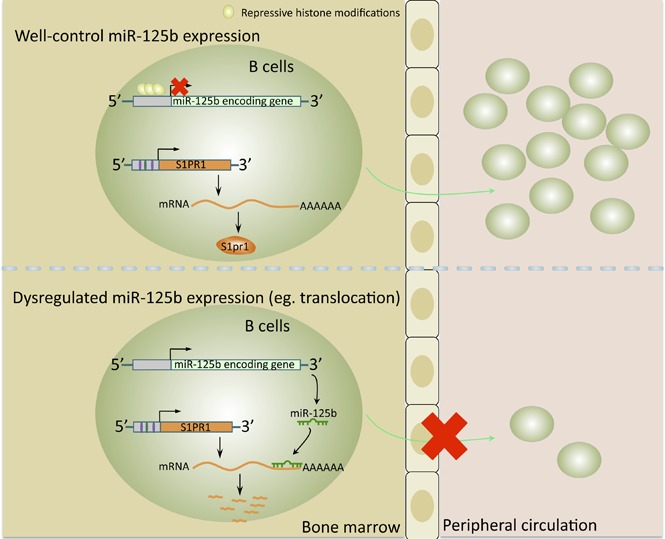
Visual Abstract
Introduction
B cells produce antibodies that react with foreign antigens and can help eliminate pathogenic invaders. Early B-cell development in the bone marrow is tightly controlled by multiple transcription factors, including PU.1, E2A, Pax5, and EBF1.1 Several factors, including sphingosine-1-phosphate receptor 1 (S1PR1), have been shown to mediate B-cell egress from bone marrow.2-5
Mature microRNA 125b (miR-125b) is encoded in 2 different genomic regions: miR-125b-1 is located on chromosomes 11 and 9 in the human and mouse genomes, respectively, and miR-125b-2 is located on human chromosome 21 and mouse chromosome 16.6 Dysregulated miR-125b expression has been found in many neoplastic blood disorders, including B-cell precursor acute lymphoblastic leukemia,6 suggesting that miR-125b might act as an oncomiR able to transform cells by targeting genes involved in tumorigenesis. Indeed, overexpression of miR-125b alone in mice is sufficient to induce tumors in multiple hematopoietic lineages generating myeloid, T-cell, and B-cell leukemias.7-11 Several target genes have been identified and postulated to have a role in the oncogenic properties of overexpressed miR-125b, such as Trp53inp1 and MAP3K11, which facilitate cell survival.8,12 A genetic deletion of the gene encoding IRF4 has also been reported in miR-125b dysregulation–induced B cells.11 In addition to promoting leukemia, several in vitro studies also suggest a role for this microRNA in B-cell development and differentiation. For instance, overexpression of miR-125b in primary B cells impairs lipopolysaccharide induction of these cells into plasma cells in vitro.13 Despite these findings, the role of miR-125b in the early stages of B-cell development in vivo and the underlying molecular mechanism of oncomiR miR-125b in B-cell tumorigenesis are largely unknown.
In this study, we found that miR-125b is epigenetically silenced during the development of normal B cells. To examine the biological importance of the physiological silencing of miR-125b expression in the B-cell lineage, we used transgenic (Tg) mice that overexpress miR-125b under the control of the Ig intronic enhancer Eμ. We found that B-cell–specific miR-125b expression results in impaired release of immature B cells from the bone marrow into the blood. We identified S1PR1 as a target of miR-125b in B cells and showed that enforced expression of S1PR1 or clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9–mediated genome editing of the miR-125b binding site within the S1PR1 3′ untranslated region (UTR) rescues the egress defect resulting from miR-125b dysregulation. In addition to impaired B-cell egress, miR-125b dysregulation initially reduces pre–B-cell output but later induces pre–B-cell lymphoma/leukemia. Genetic deletion of IRF4 was found in miR-125b–induced B-cell cancer, but its role in oncogenic miR-125b–induced B-cell transformation is still unknown. Here, we further demonstrated an interaction of the effects of miR-125b and IRF4 in cancer induction by showing that miR-125b–induced B-cell leukemia is greatly accelerated in IRF4 homozygous mutant mice. Thus, we have uncovered a novel role for miR-125b as a negative regulator of early B-cell development and conclude that epigenetic silencing of miR-125b is required for normal B-cell development.
Methods
Animals
All mice were maintained in the Caltech Office of Laboratory Animal Resources facility. All protocols were in accordance to the rules and regulations of the Institutional Animal Care and Use Committee of the California Institute of Technology.
DNA constructs
For luciferase assays, the miR-125b expression cassette was subcloned into the pMG vector. The 3′ UTR of S1PR1 containing the miR-125b binding site was cloned immediately downstream of luciferase in the pMiReport vector as previously described.14 For S1pr1 rescue experiments, S1PR1 lacking its 3′ UTR was cloned into the MSCV-eGFP vector. A guide RNA (gRNA) targeting Cas9 to the miR-125b binding site in S1PR1 3′ UTR was cloned into a retroviral vector bearing cyan fluorescent protein (CFP).
Cell culture
Cells were cultured in a sterile incubator that was maintained at 37°C and 5% CO2. HEK293T cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 U/mL streptomycin.
Labeling of bone marrow sinusoidal and parenchymal B cells
To detect bone marrow sinusoidal and parenchymal B cells, we adapted an in vivo labeling procedure as previously described.4 Eμ/miR-125b-Tg mice and littermate controls were injected IV with 1 μg phycoerythrin-conjugated anti-CD45.2 antibody in 200 μL phosphate-buffered saline for 2 minutes. The mice were then immediately killed, and cells were collected from bone marrow and analyzed by flow cytometry.
Flow cytometry
Respective tissue samples were collected from the appropriate mice as indicated in the text and processed as previously described14 (and detailed in supplemental Methods, available on the Blood Web site).
Epigenetic analysis
The epigenetic analysis was described previously.15 See supplemental Methods for detailed experimental setup and data analysis.
Statistical analysis
All statistical analysis was done in GraphPad Prism software using an unpaired Student’s t test. Data are reported as mean ± standard error of the mean (SEM).
Results
Epigenetic regulation of miR-125b expression in B cells
To investigate the role of miR-125b in B cells, we first examined its expression in mouse spleen and bone marrow cells as well as various types of immune cells. We found that the expression of miR-125b-5p and 3p (2 mature miRs originating from opposite arms of the same miR125b-1 pre-miR) was much higher in macrophages compared with other immune cells and tissues. In contrast, purified splenic B cells expressed an extremely low level of both miR-125b-5p and 3p (Figure 1A). Further analysis of their expression in sorted bone marrow lineage-negative, Sca-1+, c-Kit+ (LSK) cells and B-cell subset populations showed reduced expression of these miRs throughout B-lymphocyte lineage differentiation (Figure 1B). To determine how miR-125b expression is regulated, we examined histone modifications in the genome locus of the 2 genes encoding miR-125b by chromatin immunoprecipitation sequencing.15 The promoter region of the miR-125b-1–encoding gene displayed a high level of the repressive modification H3K27me3 and lacked activating modifications, including H3K14ac, H3K36ac, H3K4me3, H3K79me2, and H4K91ac. We also detected no p300 or RNA polymerase II (PolII) enrichment in the promoter region, consistent with this region’s transcriptional silencing (Figure 1C). In these resting B cells, the miR-125b-2 locus also lacked active modifications and p300/PolII recruitment in its promoter region, although it lacked repressive marks as well. By contrast, miR-15b/16 clusters are actively expressed during lymphopoiesis and required for controlling B-cell proliferation.16 We confirmed that miR-15b/16 clusters displayed a high degree of activating modifications as well as p300 and PolII recruitment in the promoter region of their encoding gene (Figure 1C), acting as a control for the effectiveness of our assay. We also examined the active modification H3K4me3 and repressive modification H3K27me3 in miR-125b encoding genes in pro-B cells, pre-B cells, and lipopolysaccharide plus interleukin-4 activated B cells. We found that these B cell subsets displayed similar histone modification patterns to those observed in resting B cells (supplemental Figure 1A). Thus, these data suggest that miR-125b is epigenetically silenced as hematopoietic cells transition from pluripotency to differentiated B cells.
Figure 1.

Epigenetic regulation of miR-125b expression in B lymphocytes. (A) Relative miR-125b expression in the bone marrow, spleen, and various immune cells. The expression of miR-125b-5p and miR-125b-1-3p was measured using TaqMan reverse transcription qPCR (n = 3). (B) Relative miR-125b expression in HSPCs (L−S+K+) and various B-lymphocyte subsets. (C) Modification patterns of H3K14Ac, H3K36Ac, H3K4me3, H3K79me2, H4K91ac, and H3K27me3, as well as PolII and p300 recruitment in the loci encoding miR-125b and miR-15a/16-1 in resting mature B cells. IL, interleukin; LPS, lipopolysaccharide.
Dysregulated miR-125b expression reduces number of B cells in the blood and spleen
To understand the biological importance of the physiological downregulation of miR-125b expression in B cells, we examined the effect of overexpression of miR-125b on B-cell development using Eμ/miR-125b-Tg mice. These mice overexpressed miR-125b (200- to 400-fold higher than control mice) only in B lineage cells throughout their development (supplemental Figure 2A). B cells (B220+) were quantified in the bone marrow, blood, spleen, and peripheral lymph nodes (PLNs) of wild-type (WT) and Eμ/miR-125b-Tg mice at 8 weeks of age. A decreased frequency and number of B220+ B cells was evident in Eμ/miR-125b-Tg mice in all of these compartments, with the most striking differences evident in the blood and PLNs (Figure 2A-B). This was accompanied by a decrease in the total number of leukocytes (CD45+) in both the blood and spleens of Eμ/miR-125b-Tg mice. In contrast, the number of CD4+ T lymphocytes, CD8+ T lymphocytes and CD11b+ myeloid cells were largely unchanged. An even greater reduction of B cells in the blood was observed in 6-month-old Eμ/miR-125b-Tg mice (Figure 2B).
Figure 2.

Reduced number of B cells in peripheral tissues of Eμ/miR-125b-Tg mice. (A) Frequency of B220+ B cells in peripheral tissues from 8-week-old Eμ/miR-125b-Tg and control mice. Lymphocytes from the bone marrow, peripheral blood, and spleen and PLNs from control and Eμ/miR-125b-Tg mice were analyzed by flow cytometry. (B) Total number of leukocytes (CD45+) and B cells (B220+), T cells (CD4+ or CD8+), or myeloid cells (CD11b+) counted in each organ or per microliter of peripheral blood. Bars represent mean values of pooled data. Data are pooled from 2 to 3 experiments and represented as mean ± SEM (n = 10-12 mice per group). LN, lymph node. *P < .05; **P < .01; ***P < .001; NS, not significant.
To better understand the role of miR-125b in early B-cell commitment, we analyzed the B-cell precursor cells (Hardy fractions17) in the bone marrow of WT and Eμ/miR-125b-Tg mice (Figure 3A). Eμ/miR-125b-Tg mice revealed a comparable frequency and number of precursor-progenitor B (pre/pro-B) and pro-B cells (Hardy Fr.ABC) but a significant decrease in pre-B cells (Hardy Fr.D) (Figure 3B). Interestingly, immature B cells (Hardy Fr.E) are comparable in WT and Eμ/miR-125b-Tg mice, whereas the number of recirculating mature B cells (Hardy Fr.F) is slightly reduced in Eμ/miR-125b-Tg mice (Figure 3B). The peripheral blood normally contains immature B cells recently released from the bone marrow along with recirculating mature B cells. We found that Eμ/miR-125b-Tg mice had a significant reduction in the frequency and total number of both peripheral blood and splenic immature and mature B cells compared with WT controls (Figure 3C; supplemental Figure 3A). Similar results were obtained with the B-cell maturation markers CD93 and CD62L. Particularly, the number of immature B cells was dramatically reduced in Eμ/miR-125b-Tg mice (Figure 3C-D; supplemental Figure 3B-C). Thus, dysregulated miR-125b expression reduces B-cell numbers in the blood and spleen as well as pre–B-cell numbers in the bone marrow, providing an explanation for why miR-125b is so effectively suppressed in normal B-lineage cells.
Figure 3.

Reduced number of immature B cells in the blood of Eμ/miR-125b-Tg mice. (A) Representative flow cytometric analysis of Hardy fractions for B cell progenitor subpopulations in the bone marrow of 8-week-old Eμ/miR-125b-Tg and control mice. Hardy fractions in bone marrow (A-F) were gated as follows: fraction A, pre/pro-B cell (B220+CD43+BP-1−CD24−); fraction BC, pro-B cells (B220+CD43+BP-1−CD24+ and B220+CD43+BP-1+CD24+); fraction D, pre-B cells (B220+CD43−IgM−IgD−); fraction E, immature B cells (B220+CD43−IgM+IgD−); and fraction F, mature B cells (B220+CD43−IgM+IgD+); the C′ fraction (B220+CD43+BP-1+CD24hi) was not resolved. (B) The frequency and number of each B-cell subpopulation in Eμ/miR-125b-Tg and control mice. (C) Representative flow cytometric analysis of immature and mature B-cell populations in peripheral blood of 8-week-old Eμ/miR-125b-Tg and control mice. Immature B cells were identified as B220+IgDlowIgMhigh, B220+CD93+, and B220+CD62Llow, and mature B cells were identified as B220+IgDhighIgMlow, B220+CD93−, and B220+CD62Lhigh. (D) The number and frequency of immature B cells (B220+CD93+) in the blood of Eμ/miR-125b-Tg and control mice. Bars represent mean values of pooled data. Data are pooled from 2 to 3 experiments and represent mean ± SEM (n = 8 mice per group). *P < .05; ***P < .001; NS, not significant.
Dysregulated miR-125b expression impairs egress of immature B cells
The reduction in B-cell number observed in peripheral blood of Eμ/miR-125b-Tg mice could be because of reduced proliferation, increased apoptosis, and/or inefficient release of newly formed B cells from the bone marrow to peripheral blood. In vivo analysis of the proliferation of B-cell progenitors in the bone marrow after 5-bromo-2′-deoxyuridine (BrdU) incorporation did not reveal any significant difference between Eμ/miR-125b-Tg and control mice (Figure 4A). Once immature B cells leave the bone marrow parenchyma, they enter and then are temporarily retained in the sinusoidal vasculature before finally being released into peripheral blood. To enumerate the bone marrow sinusoidal B cells, we used anti-CD45.2-phycoerythrin injection to label B cells present within sinusoids.2,4 We found a significantly lower number of immature B cells (B220+immunoglobulin M [IgM]+IgD−CD45.2+) in the sinusoids of Eμ/miR-125b-Tg mice (Figure 4B, top). A reduction of immature B cells in the sinusoids may be due to defective migration from the parenchyma, which would result in an increased number of total immature B cells in the parenchyma. However, the number of parenchymal immature B cells (B220+IgM+IgD−CD45.2−) is comparable in the Eμ/miR-125b-Tg and control mice (Figure 4B, bottom), suggesting no accumulation of bone marrow immature B cells in the parenchyma of Eμ/miR-125b-Tg mice. We next tested whether the inappropriately retained cells in bone marrow because of an exit defect might instead undergo apoptosis. Using annexin V staining to assess the apoptosis of the pro−/pre− and immature B cells in the bone marrow of Eμ/miR-125b-Tg mice, we found that the annexin V+ pro−/pre− B-cell fraction was similar in Eμ/miR-125b-Tg and control mice. However, the immature B-cell population showed significantly more annexin V+ staining in Eμ/miR-125b-Tg mice than control mice, indicating an increased incidence of cell death (Figure 4C; supplemental Figure 4A).
Figure 4.

Impaired release of immature B cells in Eμ/miR-125b-Tg mice. (A) Normal proliferation of bone marrow B cells in Eμ/miR-125b-Tg mice. 8-week-old Eμ/miR-125b-Tg and control mice were injected with BrdU for 48 hr. Bone marrow cells from these mice were stained with anti-B220 antibodies in combination with BrdU detection methodology. Percentages in the bar graph indicate BrdU-positive cells in the gated B200+ cell subpopulations. (B) Distribution and number of the indicated B-lineage populations in the bone marrow parenchyma (CD45.2−) and sinusoids (CD45.2+) of 8-week-old Eμ/miR-125b-Tg and control mice. (C) Representative flow cytometric analysis of bone marrow B cells for staining of apoptotic B cells using anti-annexin-V and 7-AAD. (D) Number of total B cells (B220+), immature B cells (B220+CD93+), T cells (CD3+), and myeloid cells (CD11b+) in 300 μL blood from Eμ/miR-125b-Tg and control mice that were injected with the AMD3100 or phosphate-buffered saline alone (n = 5 mice per group). *P < .05; **P < .01; ***P < .001; NS, not significant.
Interaction of chemokine (C-X-C motif) receptor 4 (CXCR4) with its ligand, SDF-1, is required for bone marrow retention of developing B cells in the parenchyma.2-4,18 When control mice were treated with AMD3100, a potent CXCR4 antagonist, immature bone marrow B cells (B220+CD93+), CD3+ T lymphocytes, and CD11b+ myeloid cells were released into peripheral blood (Figure 4D). The release of immature B cells into the blood was significantly lower in Eμ/miR-125b-Tg mice than in control mice after AMD3100 treatment, suggesting that miR-125b still inhibited B-cell release from the bone marrow even when CXCR4 signaling was attenuated. Taken together, these findings indicate that dysregulated miR-125b expression causes the retention of immature B cells inside bone marrow sinusoids, leading to an increase in apoptosis.
S1PR1 is a direct target of miR-125b
We next sought to better understand the underlying molecular mechanism for the defect in B-cell egress in Eμ/miR-125b-Tg mice. In a computational online search with TargetScan/MicroRNA, we identified a potential target of miR-125b, S1PR1, which is a key regulator of the process of immature B-cell release from the bone marrow2-4,19 (Figure 5A). RNA was extracted from bone marrow B220+ B cells of Eμ/miR-125b-Tg and control mice and then subjected to quantitative polymerase chain reaction (qPCR) to examine the effect of miR-125b on S1PR1 expression. We found significantly reduced S1PR1 mRNA expression levels in dysregulated miR-125b–expressing B cells compared with controls (Figure 5B). In contrast, the expression of S1PR2 and S1PR3, 2 other members in the S1P receptor family that lack a miR-125b binding site in their 3′ UTR, remained unchanged. We next determined whether S1PR1 might represent a direct target of miR-125b by performing luciferase assays as previously described.20 A luciferase reporter, in which the S1PR1 3′ UTR was placed immediately downstream of luciferase, was transfected into HEK293 cells with either the miR-125b or a control vector. Overexpression of miR-125b significantly reduced luciferase activity, indicating the S1PR1 3′ UTR is a direct target (Figure 5C). We next quantified S1pr1 protein expression in B cells from Eμ/miR-125b-Tg and control mice. Consistent with S1PR1 being a target of miR-125b, we found protein expression was significantly downregulated in B cells with dysregulated miR-125b expression (Figure 5D; supplemental Figure 5A). In addition to CXCR4 signaling, the exit of immature B cells from the bone marrow also relies on egress-promoting activity of S1P and S1PR1, which were thought to provide release signals for immature B cells.2,5 We next investigated if S1PR1 is the key target of miR-125b responsible for regulating B-cell egress. To do this, we attempted to rescue the defect in B-cell exit observed in Eμ/miR-125b-Tg mice by expressing S1PR1 that lacked the miR-125b binding site. Hematopoietic stem/progenitor cells (HSPCs) from WT and Eμ/miR-125b-Tg mice were transduced with either a control vector (MSCV) or a S1PR1-expressing vector (MSCV-S1PR1) lacking the S1PR1 3′ UTR and then subsequently used to reconstitute lethally irradiated WT mice (supplemental Figure 5B). We first validated the expression of S1PR1 in the respective samples by reverse transcription qPCR (supplemental Figure 5C). Analysis of the peripheral blood of mice at 7 weeks after reconstitution revealed that expression of S1PR1 partially rescues the loss of peripheral B cells observed with dysregulated miR-125b expression (Figure 5E).
Figure 5.

miR-125b inhibits B-cell release from the bone marrow through direct targeting of S1PR1. (A) Schematic representation of the S1PR1 3′ UTR showing the conserved miR-125b seed region. (B) S1PR1 transcript expression levels in B cells purified from the bone marrow of Eμ/miR-125b-Tg and control mice. (C) Relative luciferase expression in HEK293T cells transfected with luciferase reporter construct bearing the S1PR1 3′ UTR immediately downstream (S1PR1UTR) and either a miR-125b overexpression vector (MG-125b) or a control vector (MG). (D) S1pr1 protein expression levels in B cells purified from the bone marrow of Eμ/miR-125b-Tg and control mice. Data represent 2 independent experiments. (E) Enumeration of B cells (B220+) and immature B cells (B220+CD93+) in the peripheral blood of reconstituted mice at 7 weeks after reconstitution (n = 10 mice per group). (F) Schematic representation of small gRNA target (red) and protospacer adjacent motif (green) sequence designed to edit genomic sequence in the mouse S1PR1 3′UTR locus. (G) Detection of genome editing in the S1PR1 3′ UTR locus using a surveyor assay. (H) Enumeration of B cells (B220+) in peripheral blood of reconstituted mice at 7 weeks after reconstitution with CRISPR/Cas9-edited HSPCs (n = 10-12 mice per group). Data represent 2 independent experiments and represent mean ± SEM. *P < .05; ***P < .001; NS, not significant.
We further attempted to genetically disrupt the miR-125b targeting site in the 3′ UTR of S1PR1 using the CRISPR/Cas9 system to see if it would rescue such a defect. We designed a gRNA to target Cas9 to miR-125b binding site in the S1PR1 3′ UTR, and this gRNA was cloned into a retroviral vector bearing CFP (Figure 5F; supplemental Figure 5D). To test the efficiency of the gRNA, we infected HSPCs with retroviruses to deliver the gRNA into cells and performed a surveyor assay (a mismatch cleavage assay) to measure the insertions/deletions at the S1PR1 3′ UTR locus.21 Efficient editing in the UTR locus was observed (Figure 5G). Sequencing confirmed that this gRNA efficiently induced insertions/deletion mutations (supplemental Figure 5E). To track the reconstitution with bone marrow-derived elements, we next reconstituted recipient C57BL/6 mice (CD45.1) with HSPCs from Cas9 Tg (CD45.2) or Eμ/miR-125b-Cas9 Tg (CD45.2) mice, which were transduced with a vector expressing a control gRNA, or a vector expressing S1PR1 3′ UTR targeting gRNA (supplemental Figure 5F). Flow cytometric analysis was performed to examine the B-cell egress 7 weeks after reconstitution of recipient mice with sorted CFP+ bone marrow cells. We found a significant increase in total B cells (B220+) in the mice reconstituted with HSPCs expressing S1PR1 3′ UTR–targeting gRNA compared with the mice reconstituted with HSPCs expressing control gRNA (Figure 5H). These results demonstrate a specificity of miR-125b targeting for S1PR1 in B-cell egress.
IRF4 is a suppressor of miR-125b–induced B-cell leukemia
Along with impaired output of pre–B-cell and impaired immature B-cell egress, overexpression of miR-125b causes lymphoblastic leukemia in mice.7,8,11 Our laboratory has previously found that miR-125b dysregulation-induced B-cell precursor acute lymphoblastic leukemia displays a genetic deletion of the gene encoding IRF4.11 However, whether IRF4 plays a role in oncogenic miR-125b–induced B-cell transformation is still unknown. To test this possibility, we bred B-cell–specific IRF4-deficient (CD19cre IRF4flox/flox) mice with Eμ/miR-125b-Tg mice and generated IRF4 homozygous mutant mice expressing the Eμ/miR-125b transgene (double Tg). The effect of IRF4 deletion on tumorigenesis of Eμ/miR-125b-Tg mice was monitored for a period of 12 months. At 20 weeks of age, Eμ/miR-125b-Tg mice and CD19cre IRF4flox/flox mice were healthy and displayed no signs of B-cell leukemia in Wright-stained blood smears (data not shown). Five of 20 monitored Eμ/miR-125b-Tg mice and nine of 24 CD19cre IRF4flox/flox mice developed B-cell cancer with a long latency and were found moribund starting from the age of 6 months (Figure 6A-B). In contrast, double-Tg mice showed a dramatically accelerated mortality with a median age of 3 months, and all of them died within 4 months (Figure 6A-B). At as early as 5 weeks of age, some double-Tg mice already displayed massively enlarged spleens, whereas this was not evident in the Eμ/miR-125b-Tg mice and CD19cre IRF4flox/flox mice of the same age (supplemental Figure 6A). Flow cytometry results confirmed the presence of massive numbers of B lymphocytes in all moribund Tg mice compared with control mice (supplemental Figure 6B). This phenotype was indicative of precursor B-cell leukemia as evidenced by the lack of CD43, IgM and IgD expression (Figure 6C; supplemental Figure 6C). These B-cell tumors were clonal in origin, as suggested by the specific rearrangements of the VDJ region of the immunoglobulin heavy-chain locus (supplemental Figure 6D). To determine whether the leukemic B cells derived from these Tg mice are transplantable, we injected bone marrow cells of moribund mice into irradiated secondary recipients. The recipient mice were all moribund 2 months after transplantation and developed B-cell leukemia characterized by splenomegaly and domination of B220+ cells in the peripheral blood and spleen (Figure 6D-E; supplemental Figure 6E). We next investigated whether delivering IRF4 without its 3′ UTR to bone marrow cells from Eμ/miR-125b-Tg mice could prevent B-cell leukemia. To do this, we reconstituted lethally irradiated WT C57BL/6 mice with HSPC-enriched bone marrow cells from Eμ/miR-125b-Tg mice that were transduced with either a control vector or an IRF4-expressing vector. Over the course of 1 year, ∼20% of mice that received Eμ/miR-125b-Tg donor cells expressing the control vector developed spontaneous B-cell cancer and succumbed to disease, whereas mice that received Eμ/miR-125b-Tg donor cells transduced with an IRF4 expression vector showed no sign of cancer and had a normal frequency of B220+ B cells like that found in alive control mice (Figure 6F; supplemental Figure 6F). This result suggests that IRF4 is a suppressor of miR-125b induced B-cell leukemia.
Figure 6.

miR-125b–induced B-cell leukemia is accelerated in IRF4-deficient mice, and enforced expression of IRF4 inhibits miR-125b–induced development of spontaneous B-cell cancers in mice. (A) Survival curve of Eμ/miR-125b-Tg mice, CD19cre IRF4flox/flox Tg mice, double-Tg mice, and control mice. The genotypes and number of mice in each group are indicated on the plot. (B) Some Tg mice developed lymphoma. Lymphomas are shown at the superficial cervical or inguinal lymph node sites by red arrows. (C) Representative flow cytometric analysis of leukemic B cells in the blood, bone marrow, and spleen of moribund Eμ/miR-125b, CD19cre IRF4flox/flox and double-Tg mice. (D) Survival curve of the secondary recipient mice transplanted with bone marrow cells from moribund Eμ/miR-125b-Tg mice, CD19cre IRF4flox/flox Tg mice, double-Tg mice, and control mice (n = 6 mice per group). (E) Spleen weight of the secondary recipients described in panel D. (F) Frequency of bone marrow B cells (B220+) in mice receiving Eμ/miR-125b-Tg donor cells that were transduced with either an IRF4-expressing vector or a control vector. NS, not significant.
Discussion
Dysregulated miR-125b expression has been found in myeloid and B-cell leukemia. In this study, we have provided evidence for a deleterious role of miR-125b expression in the B-cell lineage and uncovered the mechanism underlying tumorigenesis and impaired immature B-cell egress from bone marrow. We showed that the expression of miR-125b is epigenetically silenced during the development of normal B cells. Unregulated miR-125b expression leads to inefficient appearance of immature B cells in peripheral blood and also induces pre–B-cell leukemia. The mechanism by which miR-125b is deleterious to B-cell production clearly involves at least its effect on S1PR1, a previously unappreciated target for miR-125b affecting B-cell egress. In addition, miR-125b dysregulation leads to a pro-oncogenic state that becomes evident when an IRF4 deletion occurs, showing that the pro-oncogenic state is controlled by IRF4. Thus, our previous observation that the tumors in mice overproducing miR125b have suffered deletions of their IRF4 gene is explained by the tumor-suppressive ability of IRF4. Our study has established that dysregulated expression of miR-125b functions as a critical inhibitor of B-cell lymphopoiesis and also acts as an oncomiR to drive B-cell tumorigenesis in a manner that can be suppressed by IRF4.
miR-125b exhibits minimal expression in B cells relative to macrophages. Profiling of the expression of miR-125b in LSK cells and B-cell progenitors showed that expression of this miR is evident in multipotent progenitors but silenced as cells differentiate through most of the B-lymphocyte lineage, despite a slightly increased expression of miR-125b at the pre–B-cell stage. Some of the epigenetic, transcriptional, and posttranscriptional mechanisms have been suggested to help orchestrate cellular abundance of miRs during lymphopoiesis.15 Indeed, we found that low expression of miR-125b is associated with the enrichment of repressive histone modification and/or the absence of active histone modifications in their respective promoter regions of coding genes, suggesting a tightly controlled epigenetic regulatory machinery is involved in silencing miR-125b during B-cell development.
To determine whether the epigenetic silencing of miR-125b has functional relevance in B cells, we examined its effect on the regulation of B-cell lymphopoiesis. Consistent with previous findings,11,14 we observed fewer pre-B cells in Eμ/miR-125b-Tg mice. Surprisingly, the development of immature B cells was largely unaffected, recalling the published observation that E12-deficient mice exhibit reduced pre–B-cell numbers while immature B-cell numbers remain unchanged.22 However, in the presence of overexpressed miR-125b, immature cells fail to be efficiently released from bone marrow and appear to undergo increased apoptosis in the vascular compartment of bone marrow. It is likely that the blockage of immature B-cell release backs up immature B cells in the bone marrow. Developing B cells are known to be very sensitive to apoptosis.2 The increased apoptotic immature B cells observed in the bone marrow of Eμ/miR-125b-Tg mice may result from inappropriate retention of these cells within the sinusoid. The deficiency of B cells in the spleen and PLNs would then be explained by the inefficient release of newly formed immature B cells. CXCR4 plays an important role in regulating homeostasis of B-cell compartmentalization and is required for retention of B-cell precursors in the bone marrow.3 We found that AMD3100, a specific inhibitor of CXCR4, stimulated the release of B-cell precursors from bone marrow in control mice, but not in Eμ/miR-125b-Tg mice, excluding the possibility that inefficient B-cell egress in Tg mice is due to disruption of CXCR4-mediated retention.
Searching for a mechanism by which miR-125b exerts its effect on the B-cell egress, we found that the direct target of miR-125b, S1PR1, is a key regulator of the process of immature B-cell release from the bone marrow. We found that enforced expression of S1PR1 in donor cells from Eμ/miR-125b-Tg mice almost restored normal B-cell egress, suggesting that S1PR1 is a critical target of miR-125b in regulating immature B-cell release in Eμ/miR-125b-Tg mice. The CRISPR/Cas9 system can be used to edit mammalian germ line sequences and cell lines.23,24 To exclude the possibility that S1PR1 replenishes the immature B-cell pool in peripheral blood through an indirect mechanism unrelated to the function of miR-125b in B cells, we used CRISPR/Cas9-mediated genome engineering technology to edit out the miR-125b binding site in the S1PR1 3′ UTR, preventing targeting by miR-125b. Notably, we were able to partially rescue the defect of B-cell egress upon target editing of the miR-125b binding site in cells from Eμ/miR-125b-Tg mice. Our data suggest that S1PR1 is one of the critical targets of miR-125b in regulating immature B-cell release from the bone marrow in Eμ/miR-125b-Tg mice. Some other targets of miR-125b might also contribute to the reduced total B-cell numbers in the periphery of the Tg mice. BCL2, which can protect against apoptosis and is required for cell cycle entry and proliferation, was previously identified as a target for miR-125b.25 We found its expression was downregulated in B cells from Eμ/miR-125b-Tg mice (supplemental Figure 5G). In addition, the expression of E2F2, a potential miR-125b targeting gene that is critical for cell cycle transition,26 was also reduced in the B cells from Eμ/miR-125b-Tg mice. Downregulation of these genes might have an important role in B-cell survival and proliferation in the periphery of Eμ/miR-125b-Tg mice, especially when peripheral B cells encounter antigens. Thus, downregulation of S1PR1, BCL2, and E2F2 in B cells with dysregulated miR-125b expression could all contribute to the peripheral B-cell deficiency in Eμ/miR-125b-Tg mice.
Overexpression of miR-125b in mice initially reduced pre–B-cell output but later induced pre–B-cell lymphoma/leukemia, with acquisition of a genetic deletion of the tumor suppressor IRF4 gene,11,14 suggesting a potential role of IRF4 in miR-125b–induced B-cell cancer. miR-125 has been shown to target IRF4.13 However, we found that miR-125b has no significant impact on IRF4 expression in resting B cells (data not shown). Notably, we found that miR-125b–induced acute pre–B-cell leukemia was greatly accelerated in IRF4 homozygous mutant mice and enforced expression of IRF4 in donor cells from Eμ/miR-125b-Tg mice, which are prone to develop pre–B-cell cancer, had a protective effect on cancer development. This further suggests that genetic depletion of IRF4 makes miR-125b–overexpressing B cells more susceptible to becoming cancerous.
Altogether, our data support the notion that aberrant expression of miR-125b impairs normal B-cell development and also induces tumorigenesis, demonstrating the biological importance of the physiological silencing of miR-125b expression in the B-cell lineage.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank members of the D.B. laboratory for helpful discussions, Toshio Kitamura (The University of Tokyo) for providing Eμ/miR-125b-Tg mice, and Xun Wang for providing the retro-gRNA-CFP vector.
This work was supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases (RO1AI079243) (D.B.).
Footnotes
All sequencing data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE82144).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: G.L. designed research, performed experiments, analyzed results, and wrote the paper; A.Y.-L.S., R.S., Y.O., S.W., J.K.W., P.H., and Y.S. performed experiments; R.C. provided data and discussed the results; and D.B. designed research, discussed results, wrote the paper and provided financial support.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David Baltimore, Division of Biology and Biological Engineering, M/C 147-75, California Institute of Technology, 1200 E California Blvd, Pasadena, CA 91125; e-mail: baltimo@caltech.edu; and Guideng Li, Division of Biology and Biological Engineering, M/C 147-75, California Institute of Technology, 1200 E California Blvd, Pasadena, CA 91125; e-mail: guidengl@caltech.edu.
References
- 1.Mandel EM, Grosschedl R. Transcription control of early B cell differentiation. Curr Opin Immunol. 2010;22(2):161-167. [DOI] [PubMed] [Google Scholar]
- 2.Allende ML, Tuymetova G, Lee BG, Bonifacino E, Wu YP, Proia RL. S1P1 receptor directs the release of immature B cells from bone marrow into blood. J Exp Med. 2010;207(5):1113-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck TC, Gomes AC, Cyster JG, Pereira JP. CXCR4 and a cell-extrinsic mechanism control immature B lymphocyte egress from bone marrow. J Exp Med. 2014;211(13):2567-2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pereira JP, An J, Xu Y, Huang Y, Cyster JG. Cannabinoid receptor 2 mediates the retention of immature B cells in bone marrow sinusoids. Nat Immunol. 2009;10(4):403-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pereira JP, Xu Y, Cyster JG. A role for S1P and S1P1 in immature-B cell egress from mouse bone marrow. PLoS One. 2010;5(2):e9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.So AY, Zhao JL, Baltimore D. The Yin and Yang of microRNAs: leukemia and immunity. Immunol Rev. 2013;253(1):129-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bousquet M, Harris MH, Zhou B, Lodish HF. MicroRNA miR-125b causes leukemia. Proc Natl Acad Sci USA. 2010;107(50):21558-21563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Enomoto Y, Kitaura J, Hatakeyama K, et al. . Eμ/miR-125b transgenic mice develop lethal B-cell malignancies. Leukemia. 2011;25(12):1849-1856. [DOI] [PubMed] [Google Scholar]
- 9.O’Connell RM, Chaudhuri AA, Rao DS, Gibson WS, Balazs AB, Baltimore D. MicroRNAs enriched in hematopoietic stem cells differentially regulate long-term hematopoietic output. Proc Natl Acad Sci USA. 2010;107(32):14235-14240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ooi AG, Sahoo D, Adorno M, Wang Y, Weissman IL, Park CY. MicroRNA-125b expands hematopoietic stem cells and enriches for the lymphoid-balanced and lymphoid-biased subsets. Proc Natl Acad Sci USA. 2010;107(50):21505-21510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.So AY, Sookram R, Chaudhuri AA, et al. . Dual mechanisms by which miR-125b represses IRF4 to induce myeloid and B-cell leukemias. Blood. 2014;124(9):1502-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knackmuss U, Lindner SE, Aneichyk T, et al. . MAP3K11 is a tumor suppressor targeted by the oncomiR miR-125b in early B cells. Cell Death Differ. 2016;23(2):242-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gururajan M, Haga CL, Das S, et al. . MicroRNA 125b inhibition of B cell differentiation in germinal centers. Int Immunol. 2010;22(7):583-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaudhuri AA, So AY, Mehta A, et al. . Oncomir miR-125b regulates hematopoiesis by targeting the gene Lin28A. Proc Natl Acad Sci USA. 2012;109(11):4233-4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuchen S, Resch W, Yamane A, et al. . Regulation of microRNA expression and abundance during lymphopoiesis. Immunity. 2010;32(6):828-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein U, Lia M, Crespo M, et al. . The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17(1):28-40. [DOI] [PubMed] [Google Scholar]
- 17.Rumfelt LL, Zhou Y, Rowley BM, Shinton SA, Hardy RR. Lineage specification and plasticity in CD19- early B cell precursors. J Exp Med. 2006;203(3):675-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nie Y, Waite J, Brewer F, Sunshine MJ, Littman DR, Zou YR. The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. J Exp Med. 2004;200(9):1145-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Q, Pan Z, Wang X, Gao Z, Ren C, Yang W. miR-125b-1-3p inhibits trophoblast cell invasion by targeting sphingosine-1-phosphate receptor 1 in preeclampsia. Biochem Biophys Res Commun. 2014;453(1):57-63. [DOI] [PubMed] [Google Scholar]
- 20.Mehta A, Mann M, Zhao JL, et al. . The microRNA-212/132 cluster regulates B cell development by targeting Sox4. J Exp Med. 2015;212(10):1679-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beck K, Peak MM, Ota T, Nemazee D, Murre C. Distinct roles for E12 and E47 in B cell specification and the sequential rearrangement of immunoglobulin light chain loci. J Exp Med. 2009;206(10):2271-2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096. [DOI] [PubMed] [Google Scholar]
- 25.Willimott S, Wagner SD. miR-125b and miR-155 contribute to BCL2 repression and proliferation in response to CD40 ligand (CD154) in human leukemic B-cells. J Biol Chem. 2012;287(4):2608-2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu N, Xiao L, Zhao X, et al. . miR-125b regulates the proliferation of glioblastoma stem cells by targeting E2F2. FEBS Lett. 2012;586(21):3831-3839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.