

Abstract
Background
The relationship between cholesterol and prostate cancer has been extensively studied for decades, where high levels of cellular cholesterol are generally associated with cancer progression and less favorable outcomes. However, the role of in vivo cellular cholesterol synthesis in this process is unclear, and data on the transcriptional activity of cholesterol synthesis pathway genes in tissue from prostate cancer patients are inconsistent.
Methods
A common problem with cancer tissue data from patient cohorts is the presence of heterogeneous tissue which confounds molecular analysis of the samples. In this study we present a general method to minimize systematic confounding from stroma tissue in any prostate cancer cohort comparing prostate cancer and normal samples. In particular we use samples assessed by histopathology to identify genes enriched and depleted in prostate stroma. These genes are then used to assess stroma content in tissue samples from other prostate cancer cohorts where no histopathology is available. Differential expression analysis is performed by comparing cancer and normal samples where the average stroma content has been balanced between the sample groups. In total we analyzed seven patient cohorts with prostate cancer consisting of 1713 prostate cancer and 230 normal tissue samples.
Results
When stroma confounding was minimized, differential gene expression analysis over all cohorts showed robust and consistent downregulation of nearly all genes in the cholesterol synthesis pathway. Additional Gene Ontology analysis also identified cholesterol synthesis as the most significantly altered metabolic pathway in prostate cancer at the transcriptional level.
Conclusion
The surprising observation that cholesterol synthesis genes are downregulated in prostate cancer is important for our understanding of how prostate cancer cells regulate cholesterol levels in vivo. Moreover, we show that tissue heterogeneity explains the lack of consistency in previous expression analysis of cholesterol synthesis genes in prostate cancer.
Electronic supplementary material
The online version of this article (10.1186/s12885-018-4373-y) contains supplementary material, which is available to authorized users.
Keywords: Prostate cancer, Cholesterol, Tissue heterogeneity, Stroma, HMGCR, Gene expression analysis, RNA-Seq, GSEA
Background
Increased cholesterol levels in enlarged prostates and prostate cancer have been observed for decades [1–3], and extensive research has suggested that cholesterol have a role in prostate cancer growth and progression [3–5]. Cholesterol homeostasis is important for cell viability, and is dynamically regulated by a balance between synthesis, uptake, efflux and storage of cholesterol [4, 6–9]. For cellular cholesterol synthesis, the conversion of 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) to mevalonate is the first rate limiting step, which is followed by over 20 flux controlling enzymatic reactions before cholesterol is synthesized as the final product. In prostate cancer cell-lines, elevated activity of the cholesterol synthesis pathway supports cancer growth and aggressiveness [10–16]. This has led to the general view that increased cholesterol synthesis in prostate cancer cells contributes to cellular accumulation of cholesterol and prostate cancer growth. A diet high in fat and cholesterol increase the risk of prostate cancer, while statins directly targeting the cholesterol synthesis pathway are associated with improved clinical outcome (reviewed in [17]). This is generally taken as support for the relevance of increased cholesterol synthesis in vivo. This notion was also in line with a recent study showing increased activity of the cholesterol synthesis enzyme squalene monooxygenase (SQLE) in lethal prostate cancer [18]. Accordingly, one would expect that genes in the cholesterol synthesis pathway are upregulated when prostate cancer is compared to normal tissue. However, transcriptional changes in cholesterol genes are rarely highlighted when such comparisons are performed in large patient cohorts.
We hypothesized that this is due to influence of confounding tissue components present in the samples. By confounding we refer to the variations in gene expression pattern between cancer and normal tissue samples which cannot be distinguished from variations caused by the presence of other tissue components in the samples. Gene expression analysis in human tissue is challenged by the highly heterogeneous tissue composition in each sample [19, 20]. The standard way to account for such heterogeneity is to incorporate tissue type percentages from histopathology during the analysis. Although confounding due to tissue composition is generally acknowledged, data from histopathology are missing in most publicly available patient cohorts, which may bias the molecular analyses. In prostate cancer, the presence of stroma tissue is shown to hide underlying molecular features in a differential analysis [21, 22]. Prostate tissues are usually histopathologically divided into benign epithelium, stroma tissue and prostate cancer. It is previously shown that the different number of tissue types present in prostate cancer (three tissue types) and in normal samples (two tissue types) leads to a systematic sampling bias of increased stroma content in the normal samples [23, 24]. Thus the presence of stroma tissue confounds differential analysis when cancer and normal samples are compared.Controlling for these biases will potentiate the discovery of molecular pathways and features otherwise hidden in the data.
To address this challenge we utilized two independent patient cohorts where the tissue composition of prostate cancer and normal samples has been thoroughly assessed by histopathology. Based on the gene expression analysis of stroma-enriched genes in these two cohorts, we used Gene Set Enrichment Analysis (GSEA) [25] to assess the stroma content in five other patient cohorts where no histopathology is available. In total 1713 prostate cancer and 230 normal samples were assessed for their stroma content. To create datasets from all cohorts where the confounding effect of stroma tissue is accounted for, we used our recently published approach of balancing tissue composition [23]. When differential expression analysis is performed on these datasets, consistent downregulation of genes in the cholesterol synthesis pathway is highlighted as one of the most prominent features for primary prostate cancer.
Methods
Cholesterol pathway genes
Cholesterol genes were selected from KEGG [26] pathway map for Steroid Biosynthesis and the Mevalonate Pathway in the pathway map for Terpenoid Backbone Biosynthesis. Twenty-five pathway genes were assessed for differential expression, which represent the complete pathways as mapped by KEGG. In addition, four genes from KEGG involved in cholesteryl ester formation and 19 genes from various literature sources involved in cholesterol regulation, uptake efflux and transport, were assessed. The complete list of genes and their main role in cholesterol homeostasis can be found in Table 2.
Table 2.
Overview of genes related to cholesterol synthesis assessed for differential expression
| Gene symbol | Alternative symbol | Gene name | Gene function |
|---|---|---|---|
| ACAT1 | acetyl-CoA acetyltransferase 1 | Synthesis | |
| ACAT2 | acetyl-CoA acetyltransferase 2 | Synthesis | |
| HMGCS1 | 3-hydroxy-3-methylglutaryl-CoA synthase 1 | Synthesis | |
| HMGCS2 | 3-hydroxy-3-methylglutaryl-CoA synthase 2 | Synthesis | |
| HMGCR | 3-hydroxy-3-methylglutaryl-CoA reductase | Synthesis (rate limiting enzyme) | |
| MVK | mevalonate kinase | Synthesis | |
| PMVK | phosphomevalonate kinase | Synthesis | |
| MVD | mevalonate decarboxylase | Synthesis | |
| IDI1 | isopentenyl-diphosphate delta isomerase 1 | Synthesis | |
| IDI2 | isopentenyl-diphosphate delta isomerase 2 | Synthesis | |
| FDPS | farnesyl diphosphate synthase | Synthesis | |
| GGPS1 | geranylgeranyl diphosphate synthase 1 | Synthesis | |
| FDFT1 | farnesyl-diphosphate farnesyltransferase 1 | Synthesis | |
| SQLE | squalene epoxidase | Synthesis | |
| LSS | lanosterol synthase | Synthesis | |
| DHCR24 | 24-dehydrocholesterol reductase | Synthesis | |
| CYP51A1 | cytochrome P450 family 51 subfamily A polypeptide 1 | Synthesis | |
| TM7SF2 | transmembrane 7 superfamily member 2 | Synthesis | |
| FAXDC2 | C5orf4 | fatty acid hydroxylase domain containing 2 | Synthesis |
| MSMO1 | SC4MOL | methylsterol monooxygenase | Synthesis |
| NSDHL | NAD(P) dependent steroid dehydrogenase-like | Synthesis | |
| HSD17B7 | hydroxysteroid (17-beta) dehydrogenase 7 | Synthesis | |
| EBP | emopamil binding protein (sterol isomerase) | Synthesis | |
| SC5D | SC5DL | sterol-C5-desaturase | Synthesis |
| DHCR7 | 7-dehydrocholesterol reductase | Synthesis (last step before cholesterol) | |
| CEL | carboxyl ester lipase | Esterification | |
| LIPA | lipase A, lysosomal acid, cholesterol esterase | Esterification | |
| SOAT1 | sterol O-acetyltransferase 1 | Esterification | |
| SOAT2 | sterol O-acetyltransferase 2 | Esterification | |
| ABCA1 | ATP-binding cassette, sub-family A | Efflux | |
| ABCG1 | ATP-binding cassette, sub-family G | Efflux | |
| SLCO2B1 | solute carrier organic anion transporter family member 2B1 | Transport | |
| SLCO1B3 | solute carrier organic anion transporter family member 1B3 | Transport | |
| LDLR | low density lipoprotein receptor | Uptake | |
| APOE | apolipoprotein E | Component for IDL, HDL and VLDL | |
| SREBF1 | sterol binding element transcription factor 1 | Transcriptional activation | |
| SREBF2 | sterol binding element transcription factor 2 | Transcriptional activation (main activator) | |
| SCAP | SREBF chaperone | Transcriptional activation | |
| MBTPS1 | S1P | membrane bound transcription factor peptidase site 1 | Transcriptional activation |
| MBTPS2 | S2P | membrane bound transcription factor peptidase site 2 | Transcriptional activation |
| INSIG1 | insulin induced gene 1 | HMGCR degradation | |
| INSIG2 | insulin induced gene 2 | HMGCR degradation | |
| AMFR | GP78 | autocrine motility factor receptor E3 ubiquitin protein ligase | HMGCR degradation |
| NR1H3 | LXRA | nuclear receptor subfamily 1 group H member 3 | Transcriptional repression |
| NR1H2 | LXRB | nuclear receptor subfamily 1 group H member 2 | Transcriptional repression |
| RXRA | retinoid X receptor alpha | Transcriptional repression | |
| RXRB | retinoid X receptor beta | Transcriptional repression | |
| MYLIP | IDOL | myosin regulatory light chain interacting protein | Degradation of LDLR |
Datasets, processing and quality assessment
For expression analysis of genes in the cholesterol pathway we used gene expression measurements from prostate cancer and normal tissue samples from seven publicly available patient cohorts. An overview of data from the seven patient cohorts is given in Table 1. Cancer samples for all cohorts were from radical prostatectomy specimens, except for the Sboner cohort which was from a watchful waiting cohort. Normal samples were adjacent normal prostate tissue from prostatectomy specimens, except for four normal prostate samples in the Chen cohort which were autopsy samples from subjects without prostate cancer. Gene expression measurements from each patient cohort were downloaded and processed in the following way: Gene expression and metadata from Bertilsson were created by our group and processed as previously described [27]. Data are available at Array Express with accession E-MTAB-1041. The best probe for each gene was selected as the one with the highest average rank by p-value in differential expression analysis (average over unstratified, balanced and unbalanced comparison, see below or main text for explanation). Gene expression and metadata from Chen [21, 28] were downloaded from Gene Expression Omnibus (GEO) accession GSE8218. Probes were matched to gene names by the hg133a.db reference using limma in R. The best probe for each gene was selected as the one with the highest average rank by p-value in differential expression analysis. Gene expression and metadata from Taylor [29] were downloaded from GEO accession GSE21034. Probes were matched to genes using the GPL10264 reference available at GEO. Probes with no matching gene were removed from further analysis. The best probe for each gene was selected as the one with the highest rank in a differential expression analysis between prostate cancer and normal samples. In the Taylor dataset, probes from the same gene generally had very similar ranks. Normalized and raw RNA-Seq read counts and gene names from TCGA where downloaded from The Cancer Genome Atlas [https://cancergenome.nih.gov/], [30]. Normalized read counts were log2-adjusted before further analysis. For the Prensner cohort [31], RNA-Seq raw reads in fastq-format were downloaded with approval from dbGap (project #5870) with accession phs000443.v1.p1. Raw reads were mapped to the hg19 transcriptome using TopHat2 [32], and featureCounts [33] were used to assign the reads mapping to each gene. Normalization of gene counts were performed using the normalization formula from the voom program [34]. Gene expression and metadata from Sboner [35] were downloaded from GEO with accession GSE16560. Probes were matched to gene names using the GPL5474 reference available at GEO. Only four genes in the Sboner cohort had more than one probe. For these genes, the probes with the highest overall expression value were selected as the best probe. Quantile normalized exon expression data and metadata from the Erho cohort [36] were downloaded from GEO with accession GSE46691. Exons identifiers were matched to gene names using the GPL5188 reference available at GEO. The total expression for each gene was calculated as the average expression over all exons for that gene. Differential expression of genes from Bertilsson, Chen and Taylor were identified using the limma package in R as described previously [27], while voom on raw RNA-Seq read counts was used for differential expression of genes from TCGA and Prensner. In total, 1943 samples (1713 prostate cancer and 230 normal) with 25,964 unique gene identifiers were considered over all seven datasets (the seven-study-cohort). Five of the cohorts (Bertilsson, Chen, Taylor, TCGA and Prensner, referred to as the five-study-cohort) contained both prostate cancer and normal samples (in total 1117 samples, 887 cancer and 230 normal). The seven-study-cohort contained 4804 shared genes, and the five-study-cohort contained 9527 shared genes over their respective cohorts. Quality assessment of each cohort was performed by evaluating the Pearson correlations between genes in previously validated gene sets [25, 37] related to ERG-fusion, an established feature of primary prostate cancer [38] (Additional file 1: Figure S1). Samples from the five-study-cohort consistently displayed a higher average ERG-fusion gene correlation in prostate cancer samples compared to normal samples. Cancer samples in the Erho cohort showed a similar average correlation compared to cancer samples in the five-study-cohort, while the Sboner cohort showed weaker average correlation. Altogether six of the cohorts performed well for the quality assessment, while poorer quality was only indicated in the Sboner cohort.
Table 1.
Data from the seven patient cohorts
| Abbreviation | Source | Dataset reference | Article reference | Analysis Platform | Samples | PCa | Normal | Unique genes | HP on all samples |
|---|---|---|---|---|---|---|---|---|---|
| Bertilsson | Array Express | E-MTAB-1041 | [27] | Microarray, A-MEXP-2087 - Illumina Human HT-12 WG-DASL | 156 | 116 | 40 | 14,149 | Yes |
| Chen | GEO | GSE8218 | [21, 28] | Microarray, Affymetrix Human Genome U133A Array | 136 | 65 | 71 | 12,497 | Yes |
| Taylor | GEO | GSE21034 | [29] | Microarray, Affymetrix Human Exon 1.0 ST Array | 160 | 131 | 29 | 18,294 | No |
| TCGA | TCGA | TCGA | [30] | RNA-Seq, Illumina HiSeq 2000/ Genome Analyzer IIX | 549 | 497 | 52 | 20,504 | No |
| Prensner | dbGaP | phs000443. v1.p1 | [31] | RNA-Seq, Illumina Genome Analyzer | 116 | 78 | 38 | 23,712 | No |
| Sboner | GEO | GSE16560 | [35] | Microarray, Human 6 k Transcriptionally Informative Gene Panel for DASL. | 281 | 281 | 0 | 6102 | No |
| Erho | GEO | GSE46691 | [36] | Exon array, Affymetrix Human Exon 1.0 ST Array [probe set (exon) version] | 545 | 545 | 0 | 17,163 | No |
Footnote: HP histopathology, GEO gene expression omnibus, TCGA the cancer genome atlas, dbGap the database of genotypes and phenotypes, PCa prostate cancer
Stratification of a cohort into balanced and unbalanced datasets
For the stratification of samples into datasets of balanced and unbalanced stroma tissue composition we used a strategy recently developed in our research group [23]. The strategy can be applied to any cohort, as long as assessments of stroma content are available for both cancer and normal samples in the cohort. We will here use the Bertilsson cohort to briefly describe the procedure (Fig. 1). The Bertilsson cohort consists of 116 prostate cancer and 40 normal samples, where each sample has been histopathologically assessed for its tissue composition (%) of stroma, cancer and benign epithelium. We separate both cancer and normal samples according to their stroma content where one group has low stroma content and the other has high stroma content, resulting in 4 groups altogether (58 cancer samples with highest stroma content, 58 cancer samples with lowest stroma content, 20 normal samples with highest stroma content and 20 normal samples with lowest stroma content). The balanced dataset is created by joining the 58 cancer samples with the highest percentages of stroma with the 20 normal samples with the lowest percentages of stroma, to create a dataset with balanced average amounts of stroma in cancer and control samples. In contrast, the unbalanced dataset is created by joining the 58 cancer samples with lowest content of stroma with the 20 normal samples with highest content of stroma, thus maximizing the difference of average stroma content between cancer and normal samples. The balanced dataset represents a comparison between prostate cancer and normal samples where the bias due to increased average stroma content in the normal samples has been minimized. Molecular differences in the balanced dataset are thus directly attributable to differences between cancer and normal tissue. The second dataset represent an unbalanced comparison where molecular differences mostly represent differences between prostate cancer and stroma tissue. Differentially expressed genes are then identified independently for the balanced and unbalanced datasets. The equal number of prostate cancer and normal samples in each dataset ensures a consistent statistical power, meaning that p-values are directly comparable for each gene between the balanced and unbalanced datasets. The number of samples used for balanced and unbalanced analysis in each cohort is provided in Additional file 1: Table S2.
Fig. 1.
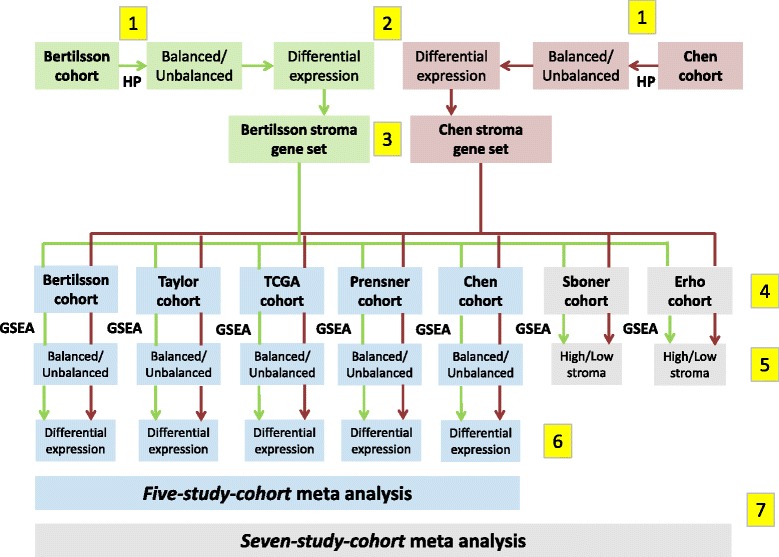
Flow chart illustrating the different computational steps for the analysis performed in this study. 1) Histopathology (HP) is used to create balanced and unbalanced datasets independently for the Bertilsson (marked green) and Chen (marked red) cohorts. 2) Differentially expressed genes for the HP-based balanced and unbalanced datasets are calculated for the Bertilsson and Chen cohorts. 3) Two stroma gene-sets are identified independently based on gene p-value relationships between the HP-based balanced and unbalanced datasets in the Bertilsson and Chen cohorts, respectively. 4) Gene Set Enrichment Analysis (GSEA) scores for all samples in all seven cohorts are calculated based on the two stroma gene-sets. These gene-sets are not combined, ensuring two independent GSEA stroma predictions for each sample in each cohort. 5) The GSEA scores are used to separate the five cohorts with both cancer and normal samples (including the cohorts from Bertilsson and Chen) into balanced and unbalanced datasets. The two remaining cohorts (Sboner and Erho) are only separated into groups with high and low stroma content. 6) Differentially expressed genes are calculated individually for the five cohorts with both cancer and normal samples. 7) Balanced and unbalanced datasets from the five-study-cohort are merged into one meta-analysis for differential expression. Balanced and unbalanced datasets from the five-study-cohort, as well as high and low stroma datasets from the Sboner and Erho cohorts are merged into one meta-analysis of the seven-study-cohort
Identification of gene-sets for assessment of stroma content in prostate tissue samples
Since the procedure for identification of gene-sets requires histopathology on both prostate cancer and normal samples in the same cohort, only the Bertilsson and Chen could be utilized for this purpose. Both these cohorts include detailed histopathological evaluation on the percentage tissue composition of prostate cancer, benign epithelium and stroma in both prostate cancer and normal samples. Stroma gene-sets were created independently from each of the two cohorts by the exact same procedure. The difference in average tissue composition between the balanced and unbalanced datasets (described in the previous section) facilitates the identification of genes specifically up- or downregulated in stroma compared to benign epithelium and cancer tissue by comparing p-values between the two datasets. Specifically, genes which display up or downregulation characteristic for stroma tissue will have lower p-values in the unbalanced compared to the balanced dataset. We thus used the following formula to rank all genes according to their suitability for creating stroma gene-sets:
| 1 |
The squared term in the denominator was included to reflect that more pronounced differences in p-values are necessary for highly significant genes to be regarded as stroma genes. (Compare a gene with p-value 1e-5 in the unbalanced which is not significant in the balanced dataset, to a gene with p-value 1e-20 in the unbalanced and 1e-15 in the balanced. The former is more likely to be a valid stroma marker than the latter, even though the p-value ratio is the same). The stroma gene-sets included were based on the top 1000 ranked upregulated and top 1000 ranked downregulated genes. To avoid any bias from the cholesterol pathway genes, any genes from Table 2 were removed from all stroma gene-sets during analysis.
Validation of stroma gene-sets in the histopathology cohorts
The independent stroma gene-sets created from the Bertilsson and Chen cohorts were validated by assessing the percentage of shared genes between the two gene-sets (Fig. 2a). This percentage was compared to the percentage of shared genes expected by chance in 50 randomly generated gene sets of same size. The number of shared genes was also compared in gene sets created using a naïve approach of Pearson correlation to histopathological stroma content, and four previously published gene sets related to the content of stroma in prostate cancer tissue samples from Wang et al. [21] (Fig. 2b).
Fig. 2.
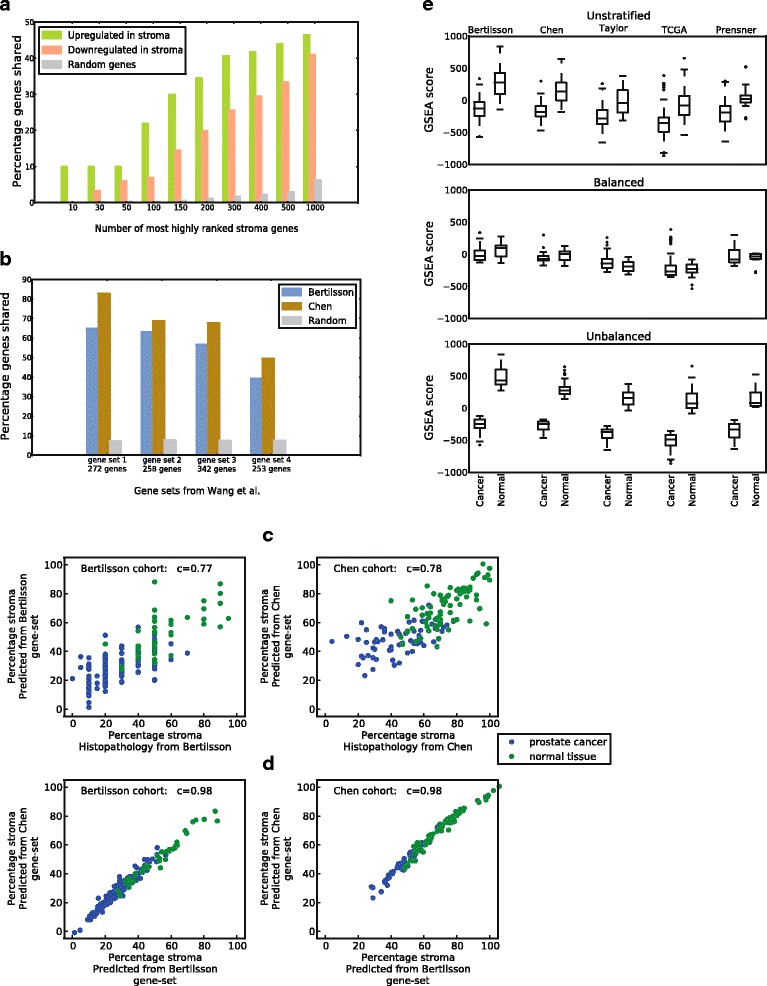
Robust assessment of stroma content in cohorts where no histopathology is available. a Overlap between up- and downregulated stroma genes in the gene-sets from Bertilsson and Chen for various numbers of the N top-ranked stroma genes. The random numbers of shared genes are the average over 50 random gene selections for each N. b Overlap of prostate stroma gene sets from Wang et al. [21] with stroma gene-sets from Bertilsson and Chen. c Pearson correlation (c) between predicted stroma percentage from GSEA and histopathological determined stroma percentage in the cohorts from Bertilsson and Chen. d Pearson correlation (c) between stroma percentage predicted by gene-sets from Bertilsson and Chen in each of their respective cohorts (bottom). e Bias towards higher GSEA stroma scores in normal compared to cancer samples present in all unstratified cohorts from the five-study-cohort. Dividing samples into balanced and unbalanced datasets minimizes and maximizes, respectively, the stroma bias between cancer and normal samples. A difference in the average overall GSEA score between the cohorts is also evident in the figure
The assessment of the stroma content in any single sample from any cohort was performed using Gene Set Enrichment Analysis (GSEA) [29]. Two measures will influence the GSEA scores; the number of genes in the applied gene-set, and the total number of genes used for the calculation. We calculated 10 GSEA scores for each sample using varying numbers of the top scoring stroma genes (top 100, 150, 200, 250, 300, 350, 400, 450, 500 and 1000 genes), and normalized the scores in each of the 10 calculations to a 0–100 range over all samples to make them comparable. To enable comparisons between datasets, we only used genes shared by all datasets in each GSEA calculation. Two total gene selections were made, one containing 9527 genes shared by the five-study-cohort, and one with 4804 genes shared for the seven-study-cohort. Averaging over the 10 GSEA scores in each selection produced a total of four GSEA scores for each sample in Bertilsson, Chen, Taylor, TCGA and Prensner (using gene-sets from Bertilsson and Chen for the five-study-cohort and the seven-study-cohort respectively), and two GSEA scores for each sample in Sboner and Erho (Bertilsson and Chen gene-set for the seven-study-cohort). The main reason for the lower number of shared genes in the seven-study-cohort is the relatively few genes measured in Sboner (6100 unique genes). For Bertilsson and Chen, GSEA scores for each sample were converted to predicted stroma percentages using a linear least squares fit, and compared to the stroma percentages obtained from histopathology. Predicted stroma percentages by the fit model based on the Bertilsson and Chen stroma gene-sets respectively in each of the two cohorts were also compared. Finally, GSEA score correlations when using the Bertilsson and the Chen stroma gene-sets were compared for all cohorts.
Defining balanced and unbalanced data-sets in cohorts lacking histopathology
The GSEA scores representing the content of stroma in each sample were used to separate samples in each new patient cohort into balanced and unbalanced datasets (as described in the section “Stratification of a cohort into balanced and unbalanced datasets” above). This was done independently for each patient cohort. Differentially expressed genes for each cohort were calculated and corrected for multiple testing by Benjamini Hochberg false discovery rate (FDR) separately in each cohort, based on the total number of analyzed genes in each cohort. For the Sboner and Erho cohorts, only the cancer samples were separated into datasets with high and low stroma content, and no differential analysis was performed.
Rank based meta-analysis for combined cohorts
To identify differentially expressed genes in a meta-analysis over the five-study-cohort the following procedure was used: 1) Each gene was sorted according to its expression value over all samples independently in each cohort, and rank-normalized to a score-value between 0 and 100, where 0 is the rank based expression value for the sample with the lowest expression value of the gene, and 100 is rank-based expression value the sample with the highest expression. 2) Rank-normalized values were mean centered independently in each cohort, where the mean centering was weighted by the relative number of prostate cancer and normal samples in the cohort. This was to avoid mean-value biases due to the huge relative difference between cancer and normal samples in each cohort. 3) Samples were separated into one balanced and one unbalanced meta-dataset using their previous assignment to balanced and unbalanced datasets in each individual cohort. Differential analysis based on two classifications were performed, one based on the stroma gene-set from Bertilsson and one on the stroma gene-set from Chen. 4) Differentially expressed genes for the unstratified, balanced and unbalanced datasets were calculated for weighted mean centered rank-normalized values between prostate cancer and normal samples combined for all five cohorts using the Mann-Whitney-Wilcoxon test [39] for rank-based differential expression. P-values of differentially expressed genes were corrected for multiple testing using the Benjamini-Hochberg FDR for the total number of genes analyzed (25,964 unique gene identifiers). If a gene was not present in all datasets, only the datasets that contained that gene were used for differential expression. The seven-study cohort was analyzed in the same way, but with mean centering rather than weighted mean centering used to adjust gene-ranks between cohorts. This was due to the lack of normal samples in the Sboner and Erho cohorts.
Gene ontology analysis
The top 500 and top 1000 differentially expressed genes from the rank-based differential expression analysis based on the both the Bertilsson and Chen gene-sets (four lists of genes in total) were subjected independently to DAVID [40] for gene ontology analysis.
Results
Differentially expressed genes in seven publicly available prostate cancer cohorts controlled for stroma tissue confounding
We used seven publicly available cohorts of tissue samples from patients with prostate cancer (Bertilsson, Chen, Taylor, TCGA, Prensner, Sboner and Erho, referred to as the seven-study-cohort; N = 1943 samples, 1713 prostate cancer and 230 normal, Table 1). Gene expression measurements in the various cohorts had been generated using different microarray platforms and RNA-sequencing. Of these seven cohorts, two cohorts (Bertilsson and Chen, referred to as the histopathology cohorts, Table 1) contained detailed histopathology on prostate cancer, stroma and benign epithelium in each sample. These two cohorts were used as a basis for stromal assessment in all seven cohorts. A flow-chart of the different steps in this assessment is provided in Fig. 1, and a detailed description of each step is provided in the Methods section. Of the seven cohorts, five cohorts contained measurements of both prostate cancer and samples characterized as normal (Bertilsson, Chen, Taylor, TCGA and Prensner, referred to as the five-study-cohort; 1117 samples, 887 prostate cancer and 230 normal).
Robust stroma assessment using gene set enrichment analysis (GSEA) with sets of stroma-enriched genes
A key concept in this study is to utilize GSEA [25] to assess the stroma content in each tissue sample from the seven patient cohorts, and then use this assessment to divide samples into sub-datasets where the confounding effect of stroma tissue is accounted for [23]. To achieve this, we first identified genes which were up- or downregulated with respect to the content of stroma tissue in the two histopathology cohorts (Methods). The top ranked genes from this analysis were collected into gene-sets, which were used for GSEA-based assessment of stroma in samples from all seven cohorts. Robustness of stroma assessments was assured by assessing each sample independently by the two stroma gene-sets identified from the two histopathology cohorts. Although the two stroma gene-sets used were generated from independent cohorts using different microarray platforms, the identified stroma related genes showed 44% overlap for the top 2000 stroma associated genes, compared to 6% for random genes (Fig. 2a). A comparison with four previously published prostate stroma gene lists [21], showed an average overlap of 62%, compared to 8% for random genes (Fig. 2b). Identified stroma genes were robust to two different methods for gene selection, with an average of 73% overlap (Additional file 1: Figure S2).
When varying the size of the stroma gene-sets, as well as the total number of genes used for the GSEA assessment, we observed an average deviation in predicted stroma content of only ~ 1% (Additional file 1: Table S1). This shows that individual genes had minimal influence on the stroma assessments. In the two histopathology cohorts, the predicted stroma percentage from GSEA showed a mean deviation from histopathology between 10% and 11%, (r = 0.77 and r = 0.78), respectively (Fig. 2c). These measurements are in agreement with previously published comparisons between histopathology and gene based stroma predictions in prostate cancer [21, 41]. In both of the histopathology cohorts, the predicted percentages of stroma were highly correlated for the two gene-sets (r = 0.98) (Fig. 2c), and estimated GSEA scores in the additional five cohorts were also highly correlated (r between 0.95 and 0.99) (Additional file 1: Fig S3). GSEA scores for all cohorts were also robust with respect to the size of the stroma gene-sets, with an average standard deviation for 0–100 normalized GSEA scores of ~ 2 (Additional file 1: Table S1). Variations in GSEA scores were not dependent on the platform used for gene expression analysis. As further support for the validity of the stroma gene-sets, samples from several prostate cancer cell-lines included in the Taylor and Prensner cohorts, were consistently at the low end of stroma content when estimated by GSEA assessment. Overall, we conclude that the gene-sets give a stable, robust and reproducible representation of the stroma content in prostate cancer and normal tissue samples in each cohort. However, we observed a prominent baseline difference in the average GSEA scores between the different cohorts (Fig. 2d). This means that an absolute prediction of stroma percentage for each sample which can be compared between cohorts cannot be made, but that relative stroma assessment between samples within the same cohort is feasible and robust.
Balancing the stroma content in cohorts with missing histopathology
We used our previously published strategy [23] (Methods) to create stroma balanced and a stroma unbalanced datasets in each of the five cohorts with both cancer and normal samples. The balanced and unbalanced datasets from the same cohort are designed to have the same number of cancer and normal samples, making p-values from differential gene expression analysis directly comparable between the two datasets. (Additional file 1: Table S2). In the balanced dataset samples are selected such that prostate cancer and normal samples have a similar average stroma content, minimizing the tissue confounding introduced by stroma tissue in a conventional unstratified analysis using all samples. Differential analysis in the balanced dataset is thus designed to highlight changes between prostate cancer and normal tissue. In contrast, samples in the unbalanced dataset are selected to maximize the difference in stroma content between cancer and normal samples. In this setting, differentially expressed genes in prostate cancer compared to stroma will be highlighted. For a single gene, comparing results from the balanced and unbalanced datasets can reveal whether a significant differential expression truly results from changes between the normal tissue and cancer, or is due to a difference in the average stroma content between the sample groups.
To create balanced and unbalanced datasets, no absolute estimation of stroma percentage in each sample is necessary. A relative stroma assessment is sufficient, which is here exemplified by GSEA using the two independent stroma gene-sets. This ensures that samples in the same cohort can be sorted according to their stroma content, which enables balanced and unbalanced analysis in cohorts where histopathology is not available.
We used the two stroma gene-sets identified independently from the histopathology cohorts (Bertilsson and Chen) to calculate GSEA scores for all 1943 samples (1713 cancer and 230 normal) from all the seven patient cohorts. In the five-study cohort containing 1117 samples (887 cancer and 230 normal), the calculated GSEA scores showed a systematic bias of increased average stroma content in normal samples (Fig. 2d), which should support the separation of each cohort into balanced and unbalanced datasets. Balanced and unbalanced datasets were therefore created independently using the two available stroma gene-set, resulting in two independent balanced and unbalanced datasets for each cohort. This stratification equalized the average stroma content in the balanced datasets, and enhanced the difference in average stroma content in the unbalanced datasets (Fig. 2d). Differential expression analyses were performed independently for each dataset, and differentially expressed genes were ranked in each dataset according to their p-value. In addition, combined rank-based meta-analysis over the five-study-cohorts and seven-study-cohorts were performed (Methods). The balanced and unbalanced datasets from the two meta-cohorts contained 558/115 and 971/115 prostate cancer/normal samples each, respectively.
Transcriptional downregulation of genes in the cholesterol synthesis pathway when adjusting for stroma tissue confounding
Consistent and highly significant downregulation of genes in the cholesterol synthesis pathway between cancer and normal samples was a prominent feature in the balanced analysis of gene expression (Fig. 3a, Table 2, Additional file 1: Figure S4). In a meta-analysis of the five-study-cohort, 21 of the 25 genes assessed were downregulated. These included key genes of cholesterol synthesis such as HMGCR and SQLE (rate limiting enzymes), FDFT1, LSS (catalyzes first step), DHCR7 (catalyzes last step) in addition to NSDHL, MDMO1, EBP, IDI1, CYP51A1, HMGCS1 and SC5D. All these genes had p-values to the power of − 10 or less. The same trend was observed in a meta-analysis over the seven-study-cohort (Additional file 1: Figure S4). In addition, four cohorts in the five-study-cohort individually showed highly significant downregulation of cholesterol genes, though the most highlighted genes varied somewhat between the cohorts (Additional file 1: Figure S4). In the five-study-cohort, 10 central cholesterol genes (HMGCS1, HMGCR, IDI1, FDFT1, SQLE, CYP51A1, MSMO1, NSDHL, EBP and SC5D) ranked among the top 150 most differentially expressed genes in the balanced dataset (average rank of 76) (Additional file 2). This is in contrast to the unstratified and unbalanced datasets, where the average ranks of the same ten genes were 9195 and 14,860, respectively. The unbalanced analysis also shows that upregulation of cholesterol genes is mostly due to differences between cancer tissue and stroma (Fig. 3a). Cholesterol synthesis was a highly important gene ontology term in the balanced dataset, and a clustered set of related terms containing steroid, sterol and cholesterol biosynthesis were among the top three most significant gene ontologies when the 500 most significant genes from the five-study-cohort were analyzed by DAVID (Table 3). In summary, the balanced data prove a characteristic transcriptional downregulation of the cholesterol synthesis pathway in primary prostate cancer. All p-values presented in this and the following sections, as well as Figs. 3 and 4, are conservatively corrected for multiple testing using the total number of 25,964 unique gene identifiers from all cohorts.
Fig. 3.
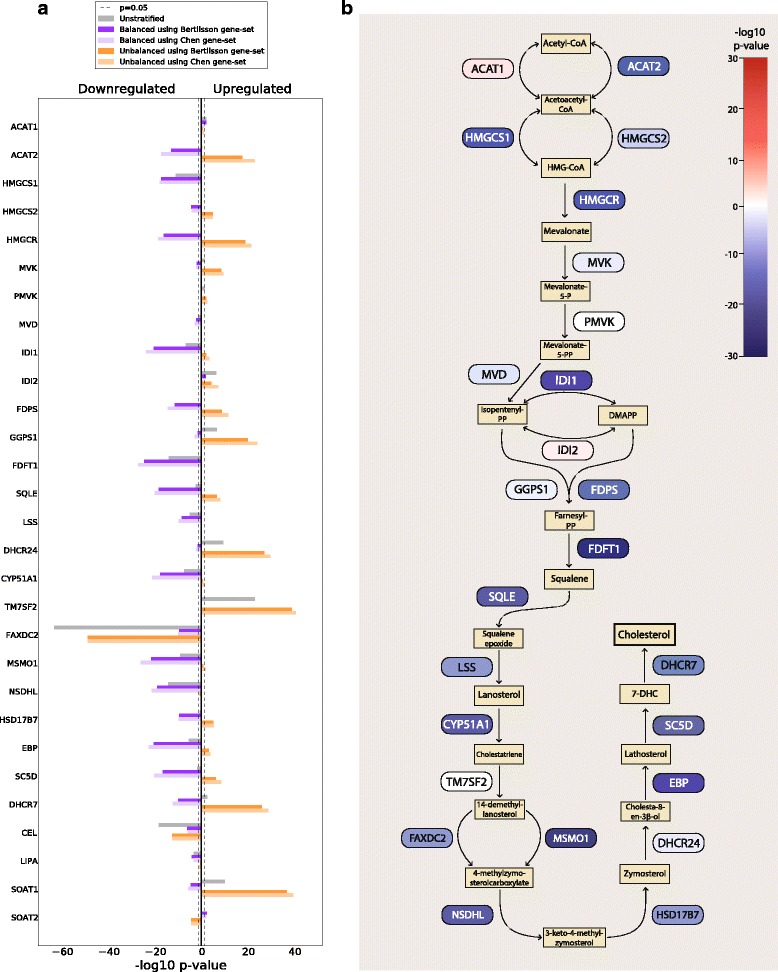
Genes in cholesterol synthesis pathway are coherently downregulated in prostate cancer compared to normal epithelium. a The figure shows –log10 p-values multiplied by 1 for upregulated genes, and − 1 for downregulated genes. The results presented are for a rank-based meta-analysis of the five-study-cohort. All p-values presented are corrected for multiple testing using the total number of 25,964 unique gene identifiers from all cohorts. Results from individual cohorts as well as the seven-study-cohort can be found in Additional file 1: Figure S4. b The schematic representation shows the cholesterol synthesis pathway with down- and upregulated genes color-coded in blue and red, respectively. The strength of the color corresponds to the degree of down- or upregulation
Table 3.
Gene ontology analysis identifies steroid, sterol and cholesterol biosynthesis among the most significantly altered pathways in prostate cancer
| GO term | q-value using Bertilsson gene- set | q-value using Chen gene-set |
|---|---|---|
| Cell Adhesion | 6.7e-8 | 6.6e-8 |
| Signal | 1.3e-7 | 1.6e-8 |
| Glycoprotein | 2.0e-8 | 1.1e-7 |
| Steroid Biosynthesis | 5.4e-6 | 4.3e-6 |
| Cholesterol Biosynthesis | 3.7e-4 | 1.7e-5 |
Footnote: The analysis was performed using the top 500 ranked genes from the balanced analysis in the five-study-cohort as input to DAVID. Only the top terms are listed. All terms are from the category “SP_PIR_KEYWORDS”. The top categories were the same when the top 1000 ranked genes were used. All terms related to steroid, sterol and cholesterol synthesis were part of the same functional cluster in DAVID
Fig. 4.
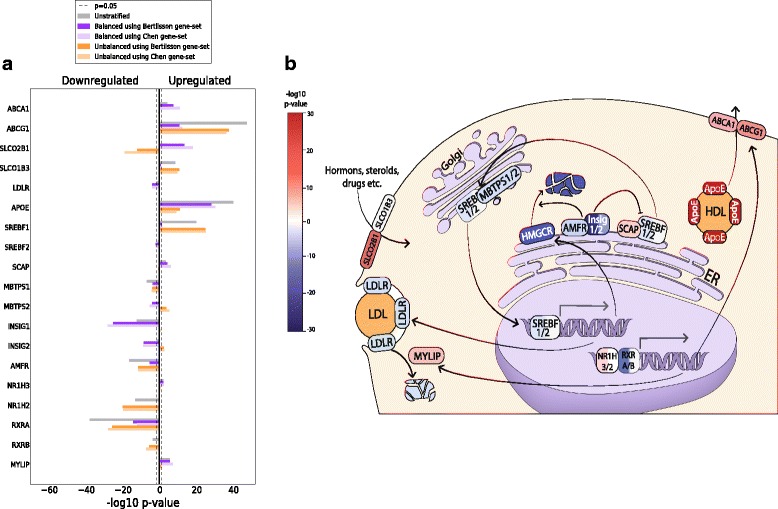
Differentially expressed genes from involved in cholesterol regulation, uptake, efflux and transport. Results from individual cohorts as well as the seven-study-cohort can be found in Additional file 1: Fig. S5. a The figure shows –log10 p-values multiplied by 1 for upregulated genes, and − 1 for downregulated genes. All p-values presented are corrected for multiple testing using the total number of 25,964 unique gene identifiers from all cohorts. Results from individual cohorts as well as the seven-study-cohort can be found in Additional file 1: Figure S4. b The schematic representation illustrates the cellular function of the selected genes, with down- and upregulated genes color-coded in blue and red, respectively. The strength of the color corresponds to the degree of down- or upregulation
Discussion
Expression of cholesterol pathway genes are confounded by stroma tissue
The pronounced discrepancies between the balanced and unbalanced datasets serve as an illustration of how cholesterol pathway genes are confounded by stroma tissue during differential analysis. Using HMGCR (the rate-limiting enzyme of cholesterol synthesis) as an example, this gene is strongly significant in both datasets. However, it is downregulated when cancer is compared to normal epithelium in the balanced dataset, and upregulated when cancer is compared to stroma in the unbalanced dataset. This typical pattern occurs when a gene highly expressed in the normal epithelium has an intermediate expression in cancer and is weakly expressed in stroma. Significant expression differences in these situations can only be revealed when the confounding effects of stroma is accounted for. Since this pattern is prevalent throughout the entire cholesterol pathway, we hypothesize that stroma confounding is the main reason that this pathway has not been identified in previous analysis of prostate cancer patient cohorts. The only cohort that did not highlight cholesterol synthesis was the cohort from Chen, which showed a consistent absence of significant cholesterol genes in the balanced dataset (Additional file 1: Figure S4). However, the cholesterol gene expression pattern from the unbalanced dataset in Chen was similar to the other cohorts.
The selection of stroma genes does not cause bias on differential expression of cholesterol genes
It is important to establish that gene-sets representing stroma content do not impose unwanted biases with respect to the differential expression of cholesterol genes in additional cohorts. Here we present three arguments why this is unlikely for the cholesterol pathway genes in this study. 1) The stroma gene-sets were generated from two independent sources, but produced similar and stable results. 2) Cholesterol genes were either absent or ranked low in the stroma gene-sets. Nevertheless, all genes involved in the cholesterol pathway and regulation were excluded from any stroma gene-set during analysis to ensure unbiased sample stratification. Moreover, re-introduction of these cholesterol genes into the stroma gene-sets did not affect the stratification of samples into balanced and unbalanced datasets in any of the seven cohorts, showing that cholesterol-genes had no impact on the sample stratification. 3) The histopathology cohort from Chen was the only cohort that did not highlight cholesterol pathway genes as significant in the balanced dataset. Yet, all the balanced datasets in the other six cohorts still highlighted cholesterol genes as highly significant when the stroma gene-set derived from the Chen was used to balance the samples. Likewise, cholesterol pathway genes were not highlighted as significant in the balanced dataset from Chen when the stroma gene-set from Bertilsson was used to balance the samples in this cohort. This shows that both the Chen and the Bertilsson stroma gene-sets maintained the divergent balanced expression patterns for cholesterol genes when used in these two cohorts.
We also investigated two additional studies from the literature which could complement the findings in our study. One study emphasized cholesterol biosynthesis as a significant pathway in prostate tissue samples using gene ontology analysis [42]. After analysis of the supplementary data material, genes in the cholesterol pathway showed negative cancer-to-normal fold-changes in that study (Additional file 3). The second study consisted of 50 samples (36 prostate cancer and 14 normal) [43] collected using laser micro dissected tissue to avoid contamination from the stroma. Cancer-to-normal fold changes were negative for all key cholesterol genes in this study as well (Additional file 3). Thus the data in both these studies support the findings in our study.
Decreased cholesterol synthesis may be beneficial for prostate cancer
Given the positive association between cholesterol and prostate cancer incidence, and the positive effect of statins on patient outcome, a consistent transcriptional downregulation of the cholesterol synthesis pathway in prostate cancer is a surprising observation. Although studies in prostate cancer cell-lines have demonstrated a role for cholesterol synthesis in tumor growth and aggressiveness, we have, after an extensive literature search, yet to see solid evidence for in vivo transcriptional upregulation of cholesterol synthesis in prostate cancer compared to normal tissue. Based on our results, we thus speculate how our observations may fit into the established mechanism of cholesterol metabolism for prostate cancer and for cells in general.
The regulation of cellular cholesterol levels is a highly complex and dynamic system, involving multiple feedback mechanisms (Fig. 4b), where downregulation of cellular cholesterol synthesis is not necessarily contradictory to other observations. Cholesterol homeostasis in the cell is controlled by cholesterol synthesis, transport and storage, but the true in vivo balance between these sources has yet to be elucidated. The most established enzymes related to cholesterol homeostasis are HMGCR and LDLR [8]. HMGCR is the rate limiting enzyme for the cholesterol synthesis pathway in the cell, while LDLR controls the uptake of cholesterol from circulating Low Density Lipoprotein (LDL). In addition, the cell can store excess cellular cholesterol in prostasomes [4] or by cholesteryl esterification in lipid droplets [9]. Increased availability of cholesterol from the environment may allow cells to shift their source of cholesterol from synthesis to uptake. Since cholesterol synthesis is energetically expensive [44], this shift can be beneficial for the cancer cell to save energy, and a recent study in prostate cancer cell-lines showed that environmental cholesterol can supplement cellular cholesterol levels as a response to cholesterol synthesis inhibition [45]. Thus molecular precursors for cholesterol in the cell can be used in other pathways important for cancer growth. The shift may also prevent the anti-tumor activity of side products in the cholesterol pathway like oxysterols and isoprenoids, though the in vivo relevance for this mechanism is debated [45–48]. Additionally, the shift can provide an explanation why statins have a beneficial effect on prostate cancer patients. Statins mostly target cholesterol synthesis in the liver leading to reduced circulating levels of cholesterol [4]. This may limit the cholesterol available for cellular uptake, with activation of the cholesterol synthesis pathway and delayed cancer growth as a result. What contradicts this hypothesis is that not only HMGCR is downregulated in prostate cancer, but also LDLR. However, mechanisms alternative to LDLR for cholesterol and sterol uptake and efflux have been suggested, including changed activity of SLCO transporters [49] (for example SLCO2B1 is strongly upregulated in the five-study-cohort, Fig. 4a) and modulation to cell-membrane structures like lipid rafts [3, 7, 50]. Recently, cholesteryl esters in lipid droplets in prostate cancer PC3 cells were shown to originate from uptake rather than synthesis [51], supporting an increased attention to the role of cholesterol uptake in prostate cancer. Alternatively, statins may upregulate HMGCR in prostate cancer directly through feedback mechanisms [52], again with a possible cancer-preventive effect. Finally, increased HMGCR protein levels have recently been shown to correlate with improved clinical outcome in breast [53], colorectal [54] and ovarian [55] cancer. This may indicate that upregulation of the cholesterol pathway is a benign tumor characteristic, which is in line with the results presented here.
Expression differences in regulatory genes suggest a possible compensation in cellular cholesterol synthesis by decreased HMGCR degradation
At the transcriptional level, HMGCR and LDLR mRNA are regulated, in particular by SREBF2, and partly by SREBF1 transcription factors, which also regulates most of the enzymes in the cholesterol pathway [4, 44, 56] (Fig. 4b). SREBF is located on the membrane of the endoplasmic reticulum together with its cofactor SCAP. SCAP has a sterol-sensing domain, which activates SREBF-SCAP transport to the Golgi when sterol levels are low (Fig. 4b). In the Golgi, SREBF-SCAP is enzymatically cleaved twice, which creates the nuclear active form of SREBF1/2. In the balanced dataset, SREBF2 is downregulated while SCAP is upregulated (Fig. 4). However, any potential increase in SREBF transport to the Golgi by SCAP is again counterbalanced by downregulation of both cleaving enzymes MBTPS1 and MBTPS2. Thus the effect of SCAP upregulation on transcriptional activity is difficult to assess. HMGCR is also regulated at the translational level and at the level of degradation. We observe a strong downregulation of several key genes involved in HMGCR degradation [56], INSIG1, INSIG2 and AMFR (Fig. 4). Especially INSIG1 is one of the most highly ranked genes differentially expressed in the balanced dataset (average rank 9). This suggests that some HMGCR activity can be maintained through downregulation of INSIG1, and that targeting HMGCR degradation can be an interesting option for modulating cholesterol levels in prostate cancer. Studies in model systems will be necessary to assess the combined effect of decreased transcription on one hand and decreased degradation on the other hand. The mechanisms of translational regulation of HMGCR are not well known, but may involve feedback regulation from side-products of the cholesterol pathway [46].
Another pair of transcription factors implicated in negative regulation of cholesterol is the liver-X-receptors NR1H3 and NR1H2 (also called LXRA and LXRB) (Fig. 4b), which dimerize with RXRA and RXRB to exert their regulatory activity [57]. NR1H3 is upregulated in the balanced analysis (Fig. 4), while its dimerization partner RXRA is strongly downregulated. We observe an upregulation of NR1H3 targets, including the cholesterol efflux genes ABCA1 and ABCG1, the LDLR suppressor MYLIP and a very strong upregulation of APOE (ranked highest among all genes in the balanced dataset). APOE can be an important constituent of High Density Lipoprotein (HDL) particles, where formation partly depends on the export by ABCA1 and ABCG1. However, here our results are in disagreement with other in vivo reports, which associates low levels of ABCA1 and low levels of circulating HDL with prostate cancer [58, 59]. We finally emphasize that the discussion on how our data relate cholesterol metabolism and homeostasis based on transcriptional data are circumstantial, and that more detailed analysis at protein levels in proper model system is necessary to elucidate these mechanisms further.
Statin use and possible impact on downregulation of cholesterol synthesis pathway genes
A recent review reports that statins are ingested regularly by 25% of adults aged 45 years and older in the USA [17]. It is thus a possibility that statin use among the patients may have influenced the molecular makeup of the tumor at the time of surgery. We were not able to obtain sufficient data to conclude on this issue. Nevertheless, we here discuss the limited data and information we were able to find. Information on statin use prior to surgery were available only for the Bertilsson cohort, were a total of 26 samples (18 cancer and 8 normal) were affected. Re-analysis of the Bertilsson cohort did not change the pattern of consistent downregulation of cholesterol pathway genes (Additional file 1: Figure S6). There is one report on the in vivo effect of statin on HMGCR levels in breast cancer [52]. This report demonstrated that statins do not necessarily downregulate HMGCR, and that the effect of statin use was highly heterogeneous among patients. Currently we find it unlikely that statin use has a major impact on the highly significant and consistent results observed in our study, though we acknowledge that the information we have on this issue is too limited to conclude.
Limitations to the histopathological tissue classification
In this study we have used a simplistic tissue classification which divide prostate cancer tissue into three tissue types; cancer, stroma and normal epithelium. However, this classification does not completely account for all tissue characteristics observed in prostate cancer, which can be heterogeneous with respect to all three tissue types. Cancer tissue from the prostate can be further classified into histological grades by Gleason score [60]. Gleason grading of samples was provided for six of the seven cohorts, and did not show any bias with respect to balanced and unbalanced dataset (Additional file 1: Table S2). We thus conclude that Gleason grade is not a confounding factor in our analysis. Several studies have shown that normal stroma can transform into reactive stroma when located adjacent to cancer tissue [61]. Thus the balanced analysis may also highlight genes resulting from differences between reactive and normal stroma. The strength of these differences will depend on the fraction of reactive stroma compared to normal stroma in the cancer samples. Histopathological differences between normal and reactive stroma were not assessed in the cohorts used in this study, and thus represents a limitation. Finally, normal epithelium from the prostate can display various precancerous aberrations with distinct molecular profiles [62]. We acknowledge that these are limitations of the current classification, and that further research and data generation in this field should focus on delineating additional molecular tissue profiles as well.
Correlation between gene expression and protein levels
Finally, we should emphasize that the transcriptional changes presented in this study are not directly indicative of changes at the protein level [63]. Additional protein-level experiments using immunohistochemistry or mass spectrometry would be necessary to investigate whether concordant protein changes are also present in prostate cancer. Nevertheless, it has been shown that transcriptional change is the most important mode of HMGCR and LDLR regulation, and that correlations between HMGCR and LDLR mRNA and protein level are comparable to correlatons between mRNA and protein levels in general [64, 65].
Conclusion
Analysis of differentially expressed genes between prostate cancer and normal samples in five patient cohorts, as well as meta-analysis over seven cohorts, consistently identified downregulation of nearly all genes in the cholesterol synthesis pathway in when the confounding effect of stroma tissue is minimized. This surprising observation will have important implications for our understanding of the complex relationship of prostate cancer and cholesterol metabolism.
Additional files
Figure S1. Quality assessment of the seven patient cohorts used in this study. Figure S2. Percentage of stroma genes shared by comparing genes identified by our described selection procedure to genes identified by a naïve approach using only Pearson correlation to normal stroma content over all samples. Figure S3. GSEA score correlations using the stroma gene sets from Bertilsson and Chen independently for samples in all cohorts. Figure S4. Significantly differentially expressed genes (prostate cancer compared to normal) related to the cholesterol synthesis pathway calculated for each of the five patient cohorts having both prostate cancer and normal samples, as well as the meta-study for the seven-study-cohort. Figure S5. Significantly differentially expressed genes (prostate cancer compared to normal) related to regulation, uptake, efflux and transport of cholesterol, calculated for each of the five patient cohorts containing prostate cancer and normal samples, as well as the meta-study for the seven-study-cohort. Figure S6. Significantly differentially expressed genes (prostate cancer compared to normal) in the Bertilsson cohort after samples from patients reported to have taken statin prior to surgery have been removed (in total 26 samples, 18 cancer and 8 normal). Table S1. GSEA score stabilities using various numbers of the top ranked stroma genes in the gene-sets from Bertilsson and Chen. Table S2. Number of cancer/normal samples and average Gleason score in balanced and unbalanced datasets from all cohorts. (PDF 1575 kb)
P-values, fold-changes and ranks for cholesterol genes balanced and unbalanced analysis in all cohorts used in this study. (XLS 80 kb)
Fold-changes from the studies by Baetke et al... and Mortensen et al. (XLS 36 kb)
Acknowledgements
The technique for fresh frozen tissue biobanking and cylinder extraction for reference E-MTAB-1041 was developed by Biobank1, St.Olavs Hospital, Trondheim, Norway. The results shown here are in part based upon data generated by the TCGA Research Network: https://cancergenome.nih.gov/. The owners of the Prensner dataset with dbGaP accession phs000443.v1.p1 specifically asked to acknowledge their funders using the following quote: “Funding support for MPC_Transcriptome sequencing to identify non-coding RNAs in prostate cancer was provided through the NIH Prostate SPORE P50CA69568, R01 R01CA132874, the Early Detection Research Network (U01 CA111275), the Department of Defense grant W81XWH-11-1-0331 and the National Center for Functional Genomics (W81XWH-11-1-0520)”.
Funding
This works supported by the Liaison Committee between the Central Norway Regional Health Authority (RHA) and the Norwegian University of Science and Technology (NTNU) to [MBR]; the Norwegian Cancer Society [100792–2013] to [TFB], PhD position from Strategic funding ISB, Norwegian University of Science and Technology (NTNU) to [MKA], PhD position from Enabling Technologies, Norwegian University of Science and Technology (NTNU) to [KR]. The funders had no influence on the study design, analysis or writing of the manuscript.
Availability of data and materials
All gene expression data and associated metadata used in this study are publicly available in the database entries and references are given in the data tables and description in the Methods section.
Abbreviation
- GSEA
Gene set enrichment analysis
Authors’ contributions
MBR, FD and MBT conceived the idea and developed the concept. MBR, MBT, HB and MKA performed data curation. MBR KR and FD performed analysis. MBR and FD developed the method. MBR, MBT, FD and TFB acquired funding and performed supervision. MBR, MBT, FD, HB, and TFB prepared the original draft. MKA and MBR prepared figures. All authors wrote and reviewed the manuscript. All authors have read and approved the final version of the manuscript.
Ethics approval and consent to participate
The use of human tissue material and clinical data from the Bertilsson cohort was approved by the Regional Committee for Medical and Health Research Ethics (REC) for Central Norway, approval no 4–2007-1890. All experiments were performed in accordance with the Helsinki declaration and other relevant guidelines and regulations. Informed consent was obtained from all participants. Other ethical aspects regarding the specific samples used in this study have been described in previous publications [27, 66]. RNA-Seq data from the Prensner cohort was approved through dbGap (project #5870), and data were downloaded and stored according to the provided security requirements. All other data were downloaded from freely available and publicly open resources.
Competing interests
The authors declare that they have no competing interests
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12885-018-4373-y) contains supplementary material, which is available to authorized users.
Contributor Information
Morten Beck Rye, Phone: +47 97133155, Email: morten.rye@ntnu.no.
Helena Bertilsson, Email: helena.bertilsson@ntnu.no.
Maria K. Andersen, Email: maria.k.andersen@ntnu.no
Kjersti Rise, Email: kjersti.rise@ntnu.no.
Tone F. Bathen, Email: tone.f.bathen@ntnu.no
Finn Drabløs, Email: finn.drablos@ntnu.no.
May-Britt Tessem, Email: may-britt.tessem@ntnu.no.
References
- 1.Swyer GIM. The cholesterol content of normal and enlarged prostates. Cancer Res. 1942;2:372–375. [Google Scholar]
- 2.Meller S, Meyer HA, Bethan B, Dietrich D, Maldonado SG, Lein M, Montani M, Reszka R, Schatz P, Peter E, et al. Integration of tissue metabolomics, transcriptomics and immunohistochemistry reveals ERG- and Gleason score-specific metabolomic alterations in prostate cancer. Oncotarget. 2016;7(2):1421–1438. doi: 10.18632/oncotarget.6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freeman MR, Solomon KR. Cholesterol and prostate cancer. J Cell Biochem. 2004;91(1):54–69. doi: 10.1002/jcb.10724. [DOI] [PubMed] [Google Scholar]
- 4.Krycer JR, Brown AJ. Cholesterol accumulation in prostate cancer: a classic observation from a modern perspective. Biochim Biophys Acta. 2013;1835(2):219–229. doi: 10.1016/j.bbcan.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Pelton K, Freeman MR, Solomon KR. Cholesterol and prostate cancer. Curr Opin Pharmacol. 2012;12(6):751–759. doi: 10.1016/j.coph.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cruz PM, Mo H, McConathy WJ, Sabnis N, Lacko AG. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. Front Pharmacol. 2013;4:119. doi: 10.3389/fphar.2013.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simons K, Ikonen E. Cell biology - how cells handle cholesterol. Science. 2000;290(5497):1721–1726. doi: 10.1126/science.290.5497.1721. [DOI] [PubMed] [Google Scholar]
- 8.Goldstein JL, Brown MS. Progress in understanding the Ldl receptor and Hmg-Coa reductase, 2 membrane-proteins that regulate the plasma-cholesterol. J Lipid Res. 1984;25(13):1450–1461. [PubMed] [Google Scholar]
- 9.Ouimet M, Marcel YL. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscl Throm Vas. 2012;32(3):575–581. doi: 10.1161/ATVBAHA.111.240705. [DOI] [PubMed] [Google Scholar]
- 10.Brown M, Hart C, Tawadros T, Ramani V, Sangar V, Lau M, Clarke N. The differential effects of statins on the metastatic behaviour of prostate cancer. Br J Cancer. 2012;106(10):1689–1696. doi: 10.1038/bjc.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swinnen JV, Ulrix W, Heyns W, Verhoeven G. Coordinate regulation of lipogenic gene expression by androgens: evidence for a cascade mechanism involving sterol regulatory element binding proteins. Proc Natl Acad Sci U S A. 1997;94(24):12975–12980. doi: 10.1073/pnas.94.24.12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krycer JR, Phan L, Brown AJ. A key regulator of cholesterol homoeostasis, SREBP-2, can be targeted in prostate cancer cells with natural products. Biochem J. 2012;446(2):191–201. doi: 10.1042/BJ20120545. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, Hughes-Fulford M. Human prostate cancer cells lack feedback regulation of low-density lipoprotein receptor and its regulator, SREBP2. Int J Cancer J Int Du Cancer. 2001;91(1):41–45. doi: 10.1002/1097-0215(20010101)91:1<41::AID-IJC1009>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 14.Sivaprasad U, Abbas T, Dutta A. Differential efficacy of 3-hydroxy-3-methylglutaryl CoA reductase inhibitors on the cell cycle of prostate cancer cells. Mol Cancer Ther. 2006;5(9):2310–2316. doi: 10.1158/1535-7163.MCT-06-0175. [DOI] [PubMed] [Google Scholar]
- 15.Murtola TJ, Pennanen P, Syvala H, Blauer M, Ylikomi T, Tammela TL. Effects of simvastatin, acetylsalicylic acid, and rosiglitazone on proliferation of normal and cancerous prostate epithelial cells at therapeutic concentrations. Prostate. 2009;69(9):1017–1023. doi: 10.1002/pros.20951. [DOI] [PubMed] [Google Scholar]
- 16.Krycer JR, Kristiana I, Brown AJ. Cholesterol homeostasis in two commonly used human prostate cancer cell-lines, LNCaP and PC-3. PLoS One. 2009;4(12):e8496. doi: 10.1371/journal.pone.0008496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moon H, Hill MM, Roberts MJ, Gardiner RA, Brown AJ. Statins: protectors or pretenders in prostate cancer? Trends Endocrin Met. 2014;25(4):188–196. doi: 10.1016/j.tem.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 18.Stopsack KH, Gerke TA, Sinnott JA, Penney KL, Tyekucheva S, Sesso HD, Andersson SO, Andren O, Cerhan JR, Giovannucci EL, et al. Cholesterol metabolism and prostate Cancer lethality. Cancer Res. 2016;76(16):4785–4790. doi: 10.1158/0008-5472.CAN-16-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liotta L, Petricoin E. Molecular profiling of human cancer. Nat Rev Genet. 2000;1(1):48–56. doi: 10.1038/35049567. [DOI] [PubMed] [Google Scholar]
- 20.Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat Commun. 2015;6:8971. doi: 10.1038/ncomms9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Xia XQ, Jia Z, Sawyers A, Yao H, Wang-Rodriquez J, Mercola D, McClelland M. In silico estimates of tissue components in surgical samples based on expression profiling data. Cancer Res. 2010;70(16):6448–6455. doi: 10.1158/0008-5472.CAN-10-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stuart RO, Wachsman W, Berry CC, Wang-Rodriguez J, Wasserman L, Klacansky I, Masys D, Arden K, Goodison S, McClelland M, et al. In silico dissection of cell-type-associated patterns of gene expression in prostate cancer. Proc Natl Acad Sci U S A. 2004;101(2):615–620. doi: 10.1073/pnas.2536479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tessem MB, Bertilsson H, Angelsen A, Bathen TF, Drablos F, Rye MB. A balanced tissue composition reveals new metabolic and gene expression markers in prostate Cancer. PLoS One. 2016;11(4):e0153727. doi: 10.1371/journal.pone.0153727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomlins SA, Mehra R, Rhodes DR, Cao XH, Wang L, Dhanasekaran SM, Kalyana-Sundaram S, Wei JT, Rubin MA, Pienta KJ, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39(1):41–51. doi: 10.1038/ng1935. [DOI] [PubMed] [Google Scholar]
- 25.Markert EK, Mizuno H, Vazquez A, Levine AJ. Molecular classification of prostate cancer using curated expression signatures. Proc Natl Acad Sci U S A. 2011;108(52):21276–21281. doi: 10.1073/pnas.1117029108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertilsson H, Tessem MB, Flatberg A, Viset T, Gribbestad I, Angelsen A, Halgunset J. Changes in gene transcription underlying the aberrant citrate and choline metabolism in human prostate Cancer samples. Clin Cancer Res. 2012;18(12):3261–3269. doi: 10.1158/1078-0432.CCR-11-2929. [DOI] [PubMed] [Google Scholar]
- 28.Chen X, Xu S, McClelland M, Rahmatpanah F, Sawyers A, Jia Z, Mercola D. An accurate prostate cancer prognosticator using a seven-gene signature plus Gleason score and taking cell type heterogeneity into account. PLoS One. 2012;7(9):e45178. doi: 10.1371/journal.pone.0045178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, Andry CD, Annala M, Aprikian A, Armenia J, Arora A, et al. The molecular taxonomy of primary prostate Cancer. Cell. 2015;163(4):1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, Laxman B, Asangani IA, Grasso CS, Kominsky HD, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011;29(8):742–749. doi: 10.1038/nbt.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao Y, Smyth GK, Shi W. Feature counts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 34.Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sboner A, Demichelis F, Calza S, Pawitan Y, Setlur SR, Hoshida Y, Perner S, Adami HO, Fall K, Mucci LA, et al. Molecular sampling of prostate cancer: a dilemma for predicting disease progression. BMC Med Genet. 2010;3:8. doi: 10.1186/1755-8794-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, Bergstralh EJ, Kollmeyer T, Fink S, Haddad Z, et al. Discovery and validation of a prostate Cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS One. 2013;8(6):e66855. doi: 10.1371/journal.pone.0066855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rye MB, Bertilsson H, Drablos F, Angelsen A, Bathen TF, Tessem MB. Gene signatures ESC, MYC and ERG-fusion are early markers of a potentially dangerous subtype of prostate cancer. BMC Med Genomics. 2014;7:50. doi: 10.1186/1755-8794-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 39.Fay MP, Proschan MA. Wilcoxon-Mann-Whitney or t-test? On assumptions for hypothesis tests and multiple interpretations of decision rules. Statistics surveys. 2010;4:1–39. doi: 10.1214/09-SS051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 41.Quon G, Haider S, Deshwar AG, Cui A, Boutros PC, Morris Q. Computational purification of individual tumor gene expression profiles leads to significant improvements in prognostic prediction. Genome Med. 2013;5(3):29. doi: 10.1186/gm433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baetke SC, Adriaens ME, Seigneuric R, Evelo CT, Eijssen LM. Molecular pathways involved in prostate carcinogenesis: insights from public microarray datasets. PLoS One. 2012;7(11):e49831. doi: 10.1371/journal.pone.0049831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mortensen MM, Hoyer S, Lynnerup AS, Orntoft TF, Sorensen KD, Borre M, Dyrskjot L. Expression profiling of prostate cancer tissue delineates genes associated with recurrence after prostatectomy. Sci Rep. 2015;5:16018. doi: 10.1038/srep16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharpe LJ, Brown AJ. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) J Biol Chem. 2013;288(26):18707–18715. doi: 10.1074/jbc.R113.479808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murtola TJ, Syvala H, Pennanen P, Blauer M, Solakivi T, Ylikomi T, Tammela TL. Comparative effects of high and low-dose simvastatin on prostate epithelial cells: the role of LDL. Eur J Pharmacol. 2011;673(1–3):96–100. doi: 10.1016/j.ejphar.2011.10.022. [DOI] [PubMed] [Google Scholar]
- 46.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 47.Liao JK. Isoprenoids as mediators of the biological effects of statins. J Clin Investig. 2002;110(3):285–288. doi: 10.1172/JCI0216421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Weille J, Fabre C, Bakalara N. Oxysterols in cancer cell proliferation and death. Biochem Pharmacol. 2013;86(1):154–160. doi: 10.1016/j.bcp.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 49.Cho E, Montgomery RB, Mostaghel EA. Minireview: SLCO and ABC transporters: a role for steroid transport in prostate Cancer progression. Endocrinology. 2014;155(11):4124–4132. doi: 10.1210/en.2014-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hager MH, Solomon KR, Freeman MR. The role of cholesterol in prostate cancer. Current Opinion Clin Nutrition Metabolic Care. 2006;9(4):379–385. doi: 10.1097/01.mco.0000232896.66791.62. [DOI] [PubMed] [Google Scholar]
- 51.Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014;19(3):393–406. doi: 10.1016/j.cmet.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bjarnadottir O, Romero Q, Bendahl PO, Jirstrom K, Ryden L, Loman N, Uhlen M, Johannesson H, Rose C, Grabau D, et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res Treat. 2013;138(2):499–508. doi: 10.1007/s10549-013-2473-6. [DOI] [PubMed] [Google Scholar]
- 53.Gustbee E, Tryggvadottir H, Markkula A, Simonsson M, Nodin B, Jirstrom K, Rose C, Ingvar C, Borgquist S, Jernstrom H. Tumor-specific expression of HMG-CoA reductase in a population-based cohort of breast cancer patients. BMC Clin Pathol. 2015;15:8. doi: 10.1186/s12907-015-0008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bengtsson E, Nerjovaj P, Wangefjord S, Nodin B, Eberhard J, Uhlen M, Borgquist S, Jirstrom K. HMG-CoA reductase expression in primary colorectal cancer correlates with favourable clinicopathological characteristics and an improved clinical outcome. Diagn Pathol. 2014;9:78. doi: 10.1186/1746-1596-9-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brennan DJ, Brandstedt J, Rexhepaj E, Foley M, Ponten F, Uhlen M, Gallagher WM, O'Connor DP, O'Herlihy C, Jirstrom K. Tumour-specific HMG-CoAR is an independent predictor of recurrence free survival in epithelial ovarian cancer. BMC Cancer. 2010;10:125. doi: 10.1186/1471-2407-10-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeBose-Boyd RA. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 2008;18(6):609–621. doi: 10.1038/cr.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Boussac H, Pommier AJ, Dufour J, Trousson A, Caira F, Volle DH, Baron S, Lobaccaro JM. LXR, prostate cancer and cholesterol: the good, the bad and the ugly. Am J Cancer Res. 2013;3(1):58–69. [PMC free article] [PubMed] [Google Scholar]
- 58.Lee BH, Taylor MG, Robinet P, Smith JD, Schweitzer J, Sehayek E, Falzarano SM, Magi-Galluzzi C, Klein EA, Ting AH. Dysregulation of cholesterol homeostasis in human prostate Cancer through loss of ABCA1. Cancer Res. 2013;73(3):1211–1218. doi: 10.1158/0008-5472.CAN-12-3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kotani K, Sekine Y, Ishikawa S, Ikpot IZ, Suzuki K, Remaley AT. High-density lipoprotein and prostate Cancer: an overview. J Epidemiol. 2013;23(5):313–319. doi: 10.2188/jea.JE20130006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Epstein JI. An update of the Gleason grading system. J Urology. 2010;183(2):433–440. doi: 10.1016/j.juro.2009.10.046. [DOI] [PubMed] [Google Scholar]
- 61.Barron DA, Rowley DR. The reactive stroma microenvironment and prostate cancer progression. Endocr Relat Cancer. 2012;19(6):R187–R204. doi: 10.1530/ERC-12-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Srigley JR. Benign mimickers of prostatic adenocarcinoma. Mod Pathol. 2004;17(3):328–348. doi: 10.1038/modpathol.3800055. [DOI] [PubMed] [Google Scholar]
- 63.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vitols S, Norgren S, Juliusson G, Tatidis L, Luthman H. Multilevel regulation of low-density lipoprotein receptor and 3-hydroxy-3-methylglutaryl coenzyme a reductase gene expression in normal and leukemic cells. Blood. 1994;84(8):2689–2698. [PubMed] [Google Scholar]
- 65.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 66.Bertilsson H, Angelsen A, Viset T, Skogseth H, Tessem MB, Halgunset J. A new method to provide a fresh frozen prostate slice suitable for gene expression study and MR spectroscopy. Prostate. 2011;71(5):461–469. doi: 10.1002/pros.21260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Quality assessment of the seven patient cohorts used in this study. Figure S2. Percentage of stroma genes shared by comparing genes identified by our described selection procedure to genes identified by a naïve approach using only Pearson correlation to normal stroma content over all samples. Figure S3. GSEA score correlations using the stroma gene sets from Bertilsson and Chen independently for samples in all cohorts. Figure S4. Significantly differentially expressed genes (prostate cancer compared to normal) related to the cholesterol synthesis pathway calculated for each of the five patient cohorts having both prostate cancer and normal samples, as well as the meta-study for the seven-study-cohort. Figure S5. Significantly differentially expressed genes (prostate cancer compared to normal) related to regulation, uptake, efflux and transport of cholesterol, calculated for each of the five patient cohorts containing prostate cancer and normal samples, as well as the meta-study for the seven-study-cohort. Figure S6. Significantly differentially expressed genes (prostate cancer compared to normal) in the Bertilsson cohort after samples from patients reported to have taken statin prior to surgery have been removed (in total 26 samples, 18 cancer and 8 normal). Table S1. GSEA score stabilities using various numbers of the top ranked stroma genes in the gene-sets from Bertilsson and Chen. Table S2. Number of cancer/normal samples and average Gleason score in balanced and unbalanced datasets from all cohorts. (PDF 1575 kb)
P-values, fold-changes and ranks for cholesterol genes balanced and unbalanced analysis in all cohorts used in this study. (XLS 80 kb)
Fold-changes from the studies by Baetke et al... and Mortensen et al. (XLS 36 kb)
Data Availability Statement
All gene expression data and associated metadata used in this study are publicly available in the database entries and references are given in the data tables and description in the Methods section.