

Abstract
Pitt-Hopkins Syndrome (PTHS) is a rare, genetic disorder caused by a molecular variant of TCF4 which is involved in embryologic neuronal differentiation. PTHS is characterized by syndromic facies, psychomotor delay, and intellectual disability. Other associated features include early-onset myopia, seizures, constipation, and hyperventilation-apneic spells. Many also meet criteria for autism spectrum disorder. Here we present a series of 23 PTHS patients with molecularly confirmed TCF4 variants and describe three unique individuals. The first carries a small deletion but does not exhibit the typical facial features nor the typical pattern of developmental delay. The second exhibits typical facial features, but has attained more advanced motor and verbal skills than other reported cases to date. The third displays typical features of PTHS, however inherited a large chromosomal duplication involving TCF4 from his unaffected father with somatic mosaicism. To our knowledge, this is the first chromosomal duplication case reported to date.
Keywords: Developmental delay, autism, intellectual disability, genetics, seizures, pediatric, ophthalmology, behavior
Introduction
Pitt-Hopkins Syndrome (PTHS; MIM #610954) is a rare neurodevelopmental disorder caused by a pathogenic variant of the TCF4 gene found on chromosome 18q21.21–4. The syndrome was first described in 1978 in two unrelated individuals who shared similar characteristics of dysmorphic facial features, developmental delay, clubbed fingers, and an abnormal breathing pattern5. It was presumed to be an autosomal recessive disorder until 2007 when the TCF4 gene, (MIM #602272) was identified, supporting an autosomal dominant inheritance pattern secondary to haploinsuffiency of TCF41–3. Transcription factor 4, the protein product of TCF4, is a basic helix-loop-helix (bHLH) E-protein believed to be involved in early brain development and neuronal differentiation1–4, 6–10. The exact prevalence of PTHS is unknown. Estimates by Rosenfeld et al. suggest a population frequency of 1:34,000 to 1:41,000 based on chromosomal microarray data from their commercial lab, although there are believed to be fewer than 500 confirmed cases worldwide4, 11.
Cases are phenotypically similar to other syndromic, neurodevelopmental disorders such as Angelman Syndrome (AS) (MIM #105830), Rett Syndrome (MIM #312750), Mowat-Wilson Syndrome (MIM #235730), and ATR-X Syndrome (MIM #301040)12–15. Studies have revealed a molecular variant in TCF4 in 2% of patients clinically diagnosed with AS and in one of 81 children clinically diagnosed with Rett Syndrome16, 17. Additionally, there is an autosomal recessive condition involving variants in CNTNAP2 and NRXN1 causing Pitt-Hopkins-Like Syndrome 1 (PTHLS1, MIM #610042) and Pitt-Hopkins-Like Syndrome 2 (PTHLS2, MIM #614325). Clinically, PTHLS1 and PTHLS2 patients present with similar phenotypes to PTHS patients, however may have milder delays in motor development18–21.
Although there is no cure for PTHS, studies are underway to identify and target the molecular pathways affected by TCF4 pathogenic variants. Clinical management of PTHS should include surveillance for common comorbidities and interventions to address symptoms, though there are currently no clinical trials to guide treatment. The University of Texas Southwestern Medical Center in collaboration with Children’s Health Dallas and the Pitt-Hopkins Research Foundation have established a specialty clinic to care for this rare population. We aim to describe the series of patients cared for in this clinic and compare the frequencies of phenotypic traits previously reported in the literature to those observed in our patient population. In addition, we aim to discuss the therapeutic interventions and sub-specialty care utilized in our population of PTHS patients.
Methods
Searches of the PubMed database (http://www.ncbi.nlm.nih.gov/pubmed) were performed to collect all articles involving the characterization of PTHS, genetics of PTHS, or animal models of PTHS. Keywords used included Pitt-Hopkins Syndrome, TCF4, 18q deletion, 18q monosomy, and 18q21. Seventy-five articles were reviewed, and 58 were included in the following literature review. Relevant primary literature, review articles, research letters, and letters to the editor available in English were included. Publications on Pitt-Hopkins-Like Syndrome, Schizophrenia, and involvement of TCF4 in the innate immune system and gastric mucosa were excluded. Case reports published prior to 2007 were also excluded as the molecular diagnosis was not confirmed. The frequencies of commonly described phenotypic characteristics from the available publications are reported herein.
Retrospective clinical data were collected on a cohort of 23 patients with a molecular diagnosis of Pitt-Hopkins Syndrome referred to the UT Southwestern/Children’s Health Center for Autism and Developmental Disabilities. Genetic test results were reviewed in the medical record, and reported genotypes were confirmed in the UCSC Genome Browser (https://genome.ucsc.edu/index.html) using the appropriate human genome build (18 or 19) corresponding to that which was available in the genetic report. Reports indicated that TCF4 transcript variant 1 (NM_001083962.1) was used for all patients except for patient 21, in whom TCF4 transcript variant 3 (NM_001243226.2) was used. Clinical data were collected in a de-identified database and the frequency of specific phenotypic characteristics were calculated and reported herein. Therapeutic interventions utilized in this cohort are also discussed, some of which are off-label use. Additionally, a clinical diagnostic score was assigned to each patient based on the proposed scoring rubrics by Marangi et al and Whalen et al22, 23. If data was unavailable, a sub-score of zero was assigned. Informed consent for publication of photographs was obtained. The study received exempt status from the UT Southwestern Medical Center Institutional Review Board.
Review of Clinical Phenotype
We reviewed 25 case reports and case series published in the literature from 2007 to 2016, and found 282 cases reported in total1–3, 8, 11–13, 17, 22–38. Most included only one or two cases except for three larger studies including de Winter et al. (n=101), Whalen et al. (n=33), and Hasi et al. (n=27)23, 29, 37. The frequencies of commonly reported features are reported herein.
Pitt-Hopkins Syndrome is commonly described to have certain characteristic facial features (116/131, 89%) described as “coarse” with up-slanted palpebral fissures, beaked nasal bridge, prominent ears, and a broad mouth with exaggerated “Cupid’s Bow” appearance of the philtrum. A single palmar crease (65/109, 60%), persistent fetal finger pads (34/75, 45%), post-natal growth restriction including short stature (48/125, 38%) and microcephaly (85/144, 59%) are also commonly seen39. Other finger and toe anomalies including over-riding toes, syndactyly, and polydactyly have also been described (52/98, 53%). Less commonly, clubbing (10/54, 19%) and scoliosis (18/91, 20%) are described. See Figure 1 for graphical representation of these rates of commonly reported dysmorphic features. Figure 2 depicts the facial features and Figure 3 depicts fetal pads, flat feet, and overriding toes that were seen in our cohort. Photos were provided by our participating families.
Figure 1.

Graph depicting the rates of commonly reported dysmorphic characteristics as reported in the published cases.
Figure 2.

Photographs demonstrating the facial gestalt commonly seen in PTHS: broad nasal bridge with bulbous tip, wide mouth with Cupid’s bow philtrum, and prominent ears.
Figure 3.
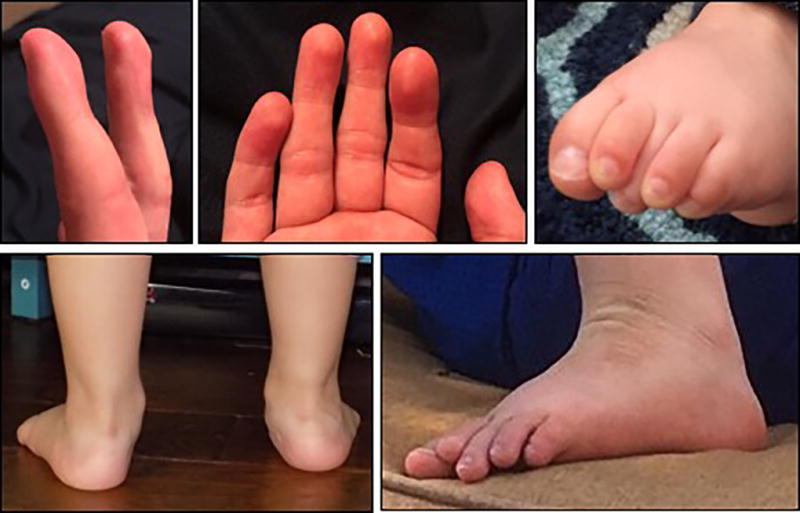
Photographs demonstrating the fetal pads, flat feet, and overriding toes.
Uniformly, individuals with PTHS have global developmental delay and intellectual disability, often coming to attention in the first year of life39. Twenty-two of the 25 case reports reviewed mention some degree of psychomotor delay; many not ambulatory until after age 3 years and most non-verbal1–3, 8, 11–13, 17, 22–28, 31–36, 38. Prior publications describe moderate to severe intellectual disability in 131 cases, however neurocognitive data was not included in the publications. In one prospective study of ten participants with PTHS (ages 32 to 289 months), a standardized battery of testing revealed a mental developmental age equivalent ranging from 3.5 to 15 months and a gross motor age equivalent ranging from 4 to 19 months of age40. The test instruments used included the Bayley Scales of Infant Development (BSID-II), Autism Diagnostic Interview – Revised (ADI-R), Vineland Adaptive Behavior Scales (VABS), and Developmental Behavior Checklist (DBC-P)40. Eight of the ten participants met criteria for a diagnosis of Autism Spectrum Disorder (ASD), however the ADI-R is only validated for children and adults with a mental age-equivalent of two years or older40. A happy disposition (82/94, 87%) and stereotypies including flapping, hand-wringing, and rocking (83/107, 78%) are common, but aggression can be seen in nearly half of individuals (32/67, 48%). Sleep disturbances including insomnia or frequent nocturnal awakenings are less commonly reported in the literature (7/39, 18%).
Approximately 38% (93/242) have seizures. Seizure semiology is quite variable including generalized tonic-clonic, atonic, focal onset with both motor and non-motor onset, as well as infantile spasms rarely8, 11, 16, 27, 28, 31, 33, 36. There is not a characteristic electroencephalogram (EEG) signature associated with this syndrome. Seizures are not typically intractable, but there is not a specific anti-seizure medication which has been shown to be more effective than others. Anti-seizure medications should be chosen based on current recommendations and guidelines in the management of epilepsy. Figure 4 depicts a graphical representation of the above frequency data.
Figure 4.

Graph depicting rates of the neurodevelopmental and behavioral traits described in the published cases.
Nearly half (119/249, 48%) exhibit a paroxysmal breathing pattern of hyperventilation with or without subsequent apnea, similar to the pattern seen in Rett and Joubert Syndromes41. It is typically described as rapid, heavy breathing and often followed by a period of breath holding that can be long enough to induce cyanosis or loss of consciousness, however precise definitions regarding the periodicity or duration of these spells are not clearly stated in the literature. Onset of breathing abnormalities is typically during early childhood, from three to seven years, and is believed to be a behavioral manifestation of emotional states, as spells only occur during wakefulness and have no ictal electrographic correlate on EEG42, 43. The pathophysiology of the abnormal breathing spells remains unknown, but some hypothesize they are due to aberrant neuronal development of the autonomic nervous system or medullary breathing control centers, as demonstrated in the mouse model6, 41. Therapeutic interventions for breathing problems including mood-stabilizing agents such as neuroleptics, selective-serotonin reuptake inhibitors, and anti-seizure medication as well as acetazolamide have been utilized with variable success. Acetazolamide, a common first-line agent, is a diuretic that blocks re-uptake of bicarbonate in the kidneys, thereby leading to a metabolic acidosis and theoretical stimulation of the central chemoreceptors to indirectly increase the respiratory drive42, 44. Verhulst et al. reports on two individuals with PTHS and apneic spells who were given acetazolamide 250 mg daily and demonstrated objective improvements in the apnea-hypopnea index (AHI) and mean SpO2 after four weeks of therapy44. Gaffney reports similar results in a patient who exhibited a 70% reduction in apneic spells by parental report while on acetazolamide42. Alternatively, Maini et al. demonstrated objective improvements in the AHI, mean SpO2, and reduction in frequency by parental report after seven months of therapy with valproic acid in a patient who failed diazepam43.
Early onset myopia (110/207, 54%) and strabismus (114/234, 49%) were reported in approximately half of individuals with PTHS, and may relate to abnormal retinal development as demonstrated in the zebrafish and fruit fly models45, 46. Families commonly reported chronic constipation (168/241, 70%), often present from infancy. Constipation can be severe, and cases of Hirschsprung’s disease (1/64, 2%) as well as intestinal malrotation (5/27, 19%) have been reported3, 29, 32. Slowed gastrointestinal transit times were demonstrated in a mouse model, suggesting a potential role of TCF4 in the development of the enteric nervous system47. Many are chronically managed with laxatives and stool softeners. See Figure 5 for a graphical representation of the rates of associated GI and ophthalmologic disorders.
Figure 5.

Graph depicting the rates of the associated gastroenterological and ophthalmologic disorders commonly reported in published cases of PTHS.
Though TCF4 is believed to be involved in neuronal differentiation, there are no consistent, major structural abnormalities seen on brain imaging. The most commonly reported findings include dysplasia of the corpus callosum1, 24, 25, 48 and bulging of the caudate heads12. Small hippocampi, temporal lobe white matter hyperintensities, delayed myelination patterns, and hypoplasia of the cerebellar cortex and vermis have also been described1, 8, 11, 24, 27. Global cerebral atrophy with widening of the sulci and ventriculomegaly may also be seen in PTHS patients12. Diffusion tensor imaging was found to be normal in one case report45. Figure 6 depicts magnetic resonance images from our cohort highlighting the dysplastic corpus callosum, bulging caudate, global atrophy, and non-specific T2 hyperintensities.
Figure 6.
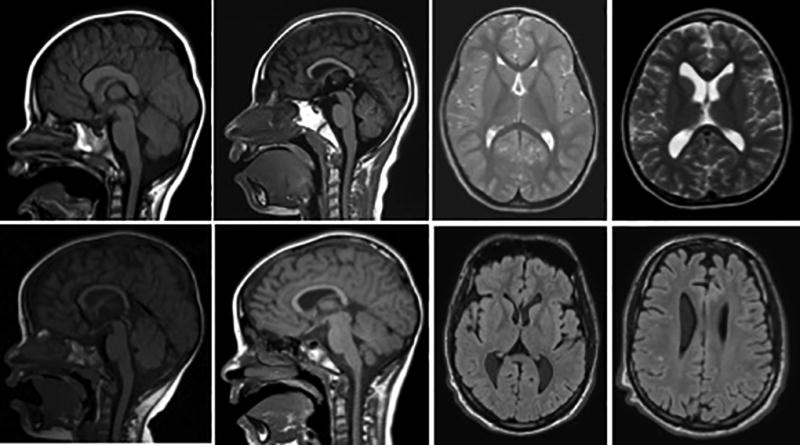
Sagittal T1 MRI scans (left) and axial T2 (top right) and FLAIR (bottom right) MRI Scans depicting common anomalies seen in PTHS including dysplasia of the corpus callosum, bulging caudate, global atrophy, and scattered T2 hyperintensities. Images are from our cohort at UTSW Medical Center / Children’s Health Center for Autism and Developmental Disabilities.
Whalen et al and Marangi et al independently proposed a clinical diagnostic score to aid in the medical decision making regarding which patients should be considered for TCF4 testing (See Table 1 for details of each scoring rubric)22, 23. On the Whalen et al scale, a score of 15 or greater should prompt testing if older than three years of age, 10 or greater if under three years of age. The Marangi et al rubric, a score of greater than or equal to 10 out of a possible 16 should prompt testing for PTHS. In the de Winter et al cohort with molecularly confirmed PTHS, a score was calculated for 47 participants based on each model and found eight of the items were present in at least 75% of the participants including: severe intellectual disability (47/47, 100%), severe speech impairment (15/15, 100%), walking >3 years/severe motor delay <3 years (44/47, 94%), constipation (40/47, 85%), protrusion mid/lower face (36/47, 77%), broad nasal bridge or convex nasal ridge (44/45, 98%), large mouth (37/47, 79%), and everted vermillion of lower lip (37/36, 80%)37. Genetic testing for PTHS would have been indicated in 17% (8/47) and 62% (29/47) based on the Whalen et al score and Marangi et al score, respectively. The scores were diagnostic in 9% (4/47) of the participants based on the Marangi et al score. de Winter et al conclude that a more precise diagnostic criteria and detailed phenotyping are still needed37.
Table 1.
Clinical diagnostic tools proposed by Whalen et al. and Marangi et al.
| Whalen et al Clinical Diagnostic Score | Marangi et al Clinical Diagnostic Score | ||
|
| |||
| Frequent Features | Positive Points | ||
|
| |||
| Deep set eyes | 1 | Moderate to severe intellectual disability | 2 |
|
| |||
| Protrusion of mid and/or lower face | 1 | Absent speech | 2 |
|
| |||
| Marked nasal root | 1 | Severe speech impairment with more than 10 words vocabulary and/or capacity to form 2–3 word sentences | 1 |
|
| |||
| Broad/beaked nasal bridge | 1 | Normal growth parameters at birth | 1 |
|
| |||
| Flared nostrils | 1 | Postnatal microcephaly or progressive slowing down of head circumference | 1 |
|
| |||
| Large mouth | 1 | Epilepsy/EEG abnormalities | 1 |
|
| |||
| Tented upper lip/prominent Cupid’s bow | 1 | Ataxic gait/Motor incoordination | 1 |
|
| |||
| Everted lower lip | 1 | Breathing Abnormalities: Hyperventilation fits or apnea episodes | 1 |
|
| |||
| Walking >3 years or severe motor delay <3 years | 2 | Mild to severe constipation | 1 |
|
| |||
| Ataxic gait | 1 | Brain MRI abnormalities (corpus callosum hypoplasia, enlargement of the ventricles, and thin hindbrain) | 1 |
|
| |||
| Absent language (or <5 words) | 2 | Ophthalmologic abnormalities (strabismus, myopia, and astigmatism) | 1 |
|
| |||
| Stereotypic movements of the head +/− hands | 2 | Typical PTHS facial features | 4 |
|
| |||
| Hyperventilation | 1 | Facial features only partially consistent with PTHS | 2 |
|
| |||
| Hypotonia | 1 | Maximum Score = 16 | |
|
|
|||
| Smiling appearance | 1 | >/= 10 should prompt testing for PTHS | |
|
| |||
| Anxiety/Agitation | 1 | ||
|
| |||
| Strabismus | 1 | ||
|
| |||
| Unusual Features | Negative Points | ||
|
| |||
| Microcephaly </= =3 SD | −2 | ||
|
| |||
| Overgrowth | −1 | ||
|
| |||
| Visceral malformations | −1 | ||
|
| |||
| Loss of purposeful hand skills | −1 | ||
|
| |||
| Maximum Score = 20 | |||
| >15 indication for TCF4 screening | |||
| 10–15 consider TCF4 screening if <3 yr | |||
| <10 no indication for TCF4 screening | |||
Cases
Rather than describing each of our 23 cases in narrative detail, we have chosen to provide overall frequency data and focus on three cases that stand out as either phenotypically or genotypically different from previously reported cases in the literature (see Table 2 for details). To our knowledge, this is the fourth largest cohort of novel patients reported to date. Overall, rates of commonly reported features were similar in our population when compared to the previously published cases except for a higher frequency of myopia and sleep disturbances in our population. See Figure 7 for a graphical representation of this data.
Table 2.
Here we present the detailed genotype and phenotype of our cohort, organized by type of pathogenic variant.
| Patient | Genotype | Genetic Test | Age at Evaluation |
Age at Ambulation |
Language | HV & Apnea | Stereotypies | Sleep Disturbance |
Myopia | Strabismus | Constipation | Seizures | Current Medications |
Marangi Score | Whalen Score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FULL GENE DELETIONS | |||||||||||||||
| 3 | 4.2 Mb, ~16 genes | CMA 2016 (buccal) | 1y 10m | − | − | − | + | − | + | − | + | − | 12 | 17 | |
| 2 | 6.757 Mb, ~20 genes | CMA 2015 | 2y 9m | − | − | + | + | − | + | + | + | − | 12 | 17 | |
| 17 | 7.6 Mb, ~40 genes | CMA 2011 | 6y 2m | 6.5y | − | − | + | + | + | + | + | − | amantadine, vitamin D, Dulcolax, iron, Senna, hydroxyzine | 12 | 18 |
| 8 | 25 Mb, ~100 genes | CMA 2012 | 28y 7m | − | − | − | UN | − | + | UN | + | + | h/o CBZ | 10 | 13 |
| PARTIAL GENE DELETIONS | |||||||||||||||
| 21 | 148 kb - Exons 1 & 2*, novel | CMA 2015 | 8y 8m | 1.5y | Babble 24m | − | + | − | UN | UN | + | − | 5 | 5 | |
| 4 | 20 kb - Exons 4 and 5 | CMA 2012 | 5y 4m | 5y | Babble 6m | − | + | − | + | + | − | + | OXC, LEV | 12 | 17 |
| 14 | 188 kb - Exons 4 to 8 | CMA 2014 | 3y 3m | 2.5y | Babble 20m | − | + | + | + | + | + | − | MiraLAX | 12 | 16 |
| 19 | 100 kb - Exons 4 to 8, de novo | CMA 2011 | 6y 5m | 3.5y | − | + | + | − | + | + | + | − | amantadine, MiraLAX, glycopyrolate, lansoprazole | 12 | 20 |
| 15 | 138 kb - Exons 5 to 6, similar reported x1 | CMA 2014 | 2y 11m | 2.5y | Babble 11m | − | + | + | UN | UN | + | + (GTCx1) | MiraLAX | 11 | 14 |
| 11 | 94 kb - Exons 5 to 11 | CMA 2010 | 10y | 4y | − | + | + | + | + | + | + | + | amantadine, LTG, Hydroxyzine, OXC, Vayarin | 13 | 16 |
| 7 | 3.8 kb - Exons 18, 19, and part of 20 | CMA 2016 | 1y 8m | − | Babble 12m | + | UN | + | + | + | − | − | MiraLAX, lansoprazole | 10 | 16 |
| FRAMESHIFT MOLECULAR VARIANTS | |||||||||||||||
| 16 | c.680_682delinsT, Trp227LeufsX29 in Exon 10, novel | Epilepsy Panel 2012 | 12y 8m | 9y | Babbled 9m | + | + | − | UN | UN | − | − | acetazolamide, risperidone Vayarin | 13 | 19 |
| 13 | c.457_461del in Exon 12, de novo, novel | WES 2013 | 18y 11m | 3.5y | − | − | + | + | + | − | + | − | clonidine, risperidone | 13 | 16 |
| 22 | c.1031delA in Exon 13, de novo, novel | WES 2016 | 10y 5m | 9y | − | + | + | − | UN | UN | + | − | magnesium, amantadine | 11 | 18 |
| 23 | c.1239dupT in Exon 15, novel | Rett/AS Panel 2016 | 3y | − | + | + | + | + | + | + | − | risperidone, melatonin, amantadine | 13 | 19 | |
| 1 | c.1414delG in Exon 16, reported x1 | Autism Panel 2015 | 3y 4m | − | Babble 24m | UN | UN | − | + | − | + | − | 8 | 9 | |
| 12 | c.1933delG in Exon 19, de novo, novel | WES 2015 | 12y 5m | 1.3y | Sentences 7y | − | + | + | − | − | + | − | melatonin, MiraLAX | 9 | 11 |
| MISSENSE MOLECULAR VARIANTS | |||||||||||||||
| 6 | c.1739G>A, Arg->Glu in Exon 18, de novo, reported x2 | WES 2014 | 10y 2m | 10y | − | + | + | + | + | + | + | + | Seroquel | 14 | 19 |
| 10 | c.1738C>T, Arg->Try in Exon 18, de novo, reported x6 | WES 2015 | 2y 3m | − | − | − | + | + | + | + | + | − | 12 | 16 | |
| 18 | c.1650-2 A>G in intron 17 leading to splice site variant, novel, c.1650-2 A>C reported x1 | Rett/AS Panel 2015 | 3y | 3.5y | − | + | + | + | + | + | + | + | LEV, glycopyrolate, lactulose, MiraLAX, risperidone, melatonin, esomeprazole, CBD oil | 15 | 20 |
| NONSENSE MOLECULAR VARIANTS | |||||||||||||||
| 9 | c.1037C>G in Exon 13, de novo, novel | WES 2016 | 37y 10m | 3.5y | − | + | + | UN | UN | UN | + | + | amantadine, OXC, TPM, melatonin, myoinositol, CBD oil, levothyroxine, Mg, Kava, Petadolex | 12 | 16 |
| 20 | c.1174 A>T in Exon 15, novel | TCF4 Sequence 2015 | 7y 6m | 5y | − | − | − | − | − | + | + | + (IS) | pyridoxine, clonidine, amantadine | 12 | 16 |
| DUPLICATIONS | |||||||||||||||
| 5 | 201 kb including Exons 4 to 8, mosaic father, novel | CMA 2016 | 2y 4m | − | Babble 12m | − | − | + | UN | UN | − | − | melatonin | 9 | 14 |
| Total n(%) | 10/22 (45) | 18/20 (90) | 12/22 (55) | 15/17 (88) | 12/16 (75) | 19/23 (83) | 8/23 (35) | ||||||||
Note is made of novel variants and inheritance pattern where available.
Patient 21 used TCF4 transcript variant 3 while the remainder of patients’ genotypes were based on TCF4 transcript variant 1.
Abbreviations: CMA – chromosomal microarray, WES – whole exome sequencing, CBZ – carbamazepine, OXC – oxcarbazepine, LEV – levetiracetam, LTG – lamotrigine, TPM – topiramate, CBD – cannabidiol, BP – base pair, IS - infantile spasms, UN - data unavailable at the time of chart review
Figure 7.

Graph depicting the rates of common clinical findings in our cohort of PTHS patients compared to the rates of previously published cases. GDD – global developmental delay
All our patients demonstrate some degree of developmental delay, and all but one of our cases demonstrates the characteristic facial features, as shown in Figure 2. Sixteen of the 23 patients were ambulatory at the time of evaluation, and all but two (patients 12 and 21) had a wide-based ataxic gait. The average age at ambulation was 4.6 years with a range of 1.3 to 10 years. All patients, except for Patient 12, who was speaking in sentences by age 7 years, had severe language delays. Eight were babbling at the time of evaluation but did not have any discrete words. We reviewed school assessment reports for all 16 children for whom records were available. The most commonly used measures of functioning focused on adaptive and early developmental skills and included the Vineland Adaptive Behavior Scales – 2nd Edition, the Adaptive Behavior Assessment System-II, the Developmental Profile 3, and the adaptive scales from the Behavior Assessment System for Children – 2nd Edition. Aside from Patient 12, all children in this cohort fell below the 1st percentile for age across measured domains, such as communication, cognitive, social, daily living, motor skills and overall composites of adaptive functioning. They consistently fell in the severely impaired range at or near the floor of these measures. Of note, these measures were based on parent/caregiver report, and direct assessment of these children with standardized measures of developmental ability was rare.
Abnormal breathing spells were present in 45% (10/22) of the cohort, and stereotypies present in 90% (18/20) of the cohort. Sleep disturbances including difficulties falling asleep or frequent nocturnal awakenings were reported in 12 of 22 patients (88%). Of the 17 patients who underwent formal visual acuity testing, 15 (88%) had early-onset myopia and 12 of 16 (75%) had strabismus. Constipation was reported in 19 of 23 patients (83%). Over one-third (8/23, 35%) of patients had at least one lifetime seizure, however only 4 of the 8 patients with a history of seizures were taking anti-seizure medications at the time of evaluation. Patient 18 was taking levetiracetam monotherapy, Patient 4 taking levetiracetam and oxcarbazepine, Patient 11 taking lamotrigine and oxcarbazepine, and Patient 9 taking oxcarbazepine and topiramate. Additionally, seven of the patients were taking amantadine for behavioral problems including anxiety and aggression. Families attributed improvements in focus, diminished hyperactivity, decreased frequency of abnormal breathing spells, as well as more rapid developmental progression to amantadine. We note this is anecdotal information that has not been systematically evaluated in this population.
Approximately half (11/23) of our cohort were found to have a copy number variant, while the remainder had sequence variants that were classified as pathogenic. All but three of the patients were referred to our clinic with the confirmed molecular diagnosis, so indication for genetic testing was not readily available. The remaining three patients were internal referrals diagnosed by commercial sequencing panel with an a priori suspicion for PTHS. Only one of the four patients with a full gene deletion was ambulatory and all were non-verbal at the time of evaluation. However, two of these non-ambulatory patients were under the age of three years at the time of evaluation, and therefore younger than typical onset of ambulation for PTHS. None of the patients with a frameshift variant had seizures, though most (4/6) of this sub-group were on a medication for behavior such as risperidone, amantadine, or Vayarin. Clinically meaningful conclusions of genotype-phenotype correlations are limited secondary to the small sample sizes of the subgroups.
Patient 5 carries a 201-kB duplication encompassing exons 4 to 8 on the TCF4 variant 1 (NM_001083962.2), and to our knowledge is the first case with a duplication of this size to be reported. The duplication was found on chromosomal microarray, inherited from an unaffected father who carries a somatic mosaicism of the same variant. Developmentally, the patient was rolling by 3 months, sitting at 10 months, and pulling to stand at 20 months. He was not yet ambulatory on his initial evaluation at 2 years 3 months. He was described to have a happy disposition without aggression, anxiety, or apneic spells. There was neither reported constipation nor seizure-like activity, and his vision had not yet been evaluated. Based on his facial features and developmental-behavioral profile, he seems phenotypically consistent with a diagnosis of PTHS, though this molecular variant has not been reported as causative in other PTHS patients.
Patient 12 carries a single base-pair deletion in the penultimate exon of TCF4 and, to our knowledge, is the highest functioning patient described to date. He presented at the age of 9 years with developmental delay and mildly dysmorphic facial features. He was rolling at 6 months, sitting at 8 months, and walking by 15 months. He babbled at 8 months and spoke his first word at 14 months. He had 5 words at 2 years, 2-word phrases at 4 years, and full sentences by 7 years. He struggled with low frustration tolerance, sleep disturbance, and chronic constipation. His chromosomal microarray was normal in 2013, but was later diagnosed with Pitt-Hopkins Syndrome by whole exome sequencing in 2016. Of note, his visual acuity is normal, and he has never demonstrated any spells of abnormal breathing or seizure-like activity. In his clinical neuropsychological testing, his full-scale intelligence quotient (IQ, Stanford Binet Intelligence Scales, 5th Edition) was 57, and he met criteria for autism spectrum disorder as well as attention deficit hyperactivity disorder. He is thriving in the special education classroom and continues to gain new skills.
Patient 21 is a boy with a 148-kb deletion involving exons 1 and 2 of the TCF4 variant 3 (NM_001243226), which was found on chromosomal microarray. His phenotype is unique in that he lacks the typical facial features associated with most cases of PTHS and his motor delays were less prominent. He was ambulatory by 18 months of age with a normal gait pattern, but exhibits severe language delay, non-verbal at the age of 8 years 8 months. He carries a diagnosis of autism spectrum disorder and severe intellectual disability. He had frequent stereotypies of hand flapping and rocking, but no sleep problems, constipation, vision impairment, or dysmorphic facial features.
Discussion
Pitt-Hopkins Syndrome is a rare neurodevelopmental disorder that is likely under-diagnosed secondary to lack of familiarity with this syndrome and under-utilization of genetic testing. As our understanding of rare syndromes grows it is reasonable to consider genetic testing in individuals lacking a clear underlying etiology for developmental delay. According to the most recent American Academy of Neurology (AAN) and Child Neurology Society (CNS) practice guidelines, routine cytogenetic testing, such as a chromosomal microarray, is recommended in the evaluation of a child with global developmental delay that is static, non-progressive and lacking a clear etiology49. Approximately half of the patients in our cohort required second or third tier genetic testing with whole exome sequencing (WES) or commercially available panels to establish a molecular diagnosis of PTHS, as initial testing with chromosomal microarray was non-diagnostic. According to the American College of Medical Genetics and Genomics (ACMG), WES can be considered when there is likely to be a high-degree of genetic heterogeneity associated with a condition such that WES analysis of multiple genes is more practical than single gene testing50. In a recent study of 57 undiagnosed neurology patients, WES had a diagnostic yield of 49.1%51. An estimated cost analysis of WES in pediatric neurology patients found that WES, when used in place of other second tier testing modalities, can result in lower long-term costs and expedited diagnosis52. Furthermore, the cost of whole genome sequencing has decreased substantially since discovery, now costing less than $1500 by some estimates53.
As with autism and intellectual disability in general, there are specific comorbidities commonly seen in PTHS patients that, if managed appropriately, may improve the quality of life for patients and their families. In brief, individuals with PTHS should be evaluated for constipation, myopia, strabismus, spells of abnormal breathing, seizures, and sleep disturbances in addition to management of their developmental delays with appropriate therapies including speech, occupational, and physical therapy. Though treatment is only supportive at this time, a multidisciplinary team approach is valued in more complex cases with multi-organ system involvement to routinely assess and manage the challenges these patients and families face. The American Academy of Pediatrics (AAP) defines children with medical complexity as those with multiple significant chronic health problems that affect multiple organ systems and result in functional limitations, high health care need or utilization, and often the need for or use of medical technology54. The APP identifies this population as being at high risk for adverse medical, developmental, psychosocial, and family outcomes54. As such, PTHS patients should be considered medically complex given the high prevalence of multi-organ system dysfunction including brain, eyes, lungs, and intestines. Evaluations by pediatric-trained Neurologists, Ophthalmologists, Pulmonologists, and Gastroenterologists would provide the necessary team-based approach, which is recommended by the AAP in caring for medically complex children54, 55. Additional services from speech and language pathologists, physical and occupational therapists, social work, nursing, and school-based resources are also commonly needed. The AAP defines team-based care as a health care model that endorses the partnership of children and families working together with one or more health care providers and other team members across multiple settings to identify, coordinate, and address shared goals that meet the needs of the whole child in order to 1) maximize the health and functioning of the patient and family unit and 2) provide proactive care55. It is our belief that by applying a team-based approach to the care of PTHS patients, we may improve the quality of life of the patient and family unit, though research in this field of pediatrics is limited55. Furthermore, as our knowledge of the pathophysiology of TCF4 variants deepens, specific treatments directed at the underlying molecular abnormality or other downstream targets may confer even more benefits and hopefully reverse the phenotype. Promising pre-clinical data on the use of histone de-acetylation inhibitors (HDACi) in mouse models of PTHS have shown normalization of long-term potentiation, increased vocalizations, and improvements in learning and memory56. This study suggests that deficits associated with TCF4 haploinsufficiency can be effectively treated, even in the post-natal period.
The ability to clinically diagnose Pitt-Hopkins Syndrome is quite limited as evidenced by the numerous patients described in the literature as a different clinical syndrome, only to find out that the molecular diagnosis differs from the clinical diagnosis. In our cohort, Patients 1, 5, 12, and 21 testing for PTHS would not hav been recommended based on the clinical diagnostic scores, supporting the de Winter et al conclusion that the available clinical diagnostic scoring tools are limited37. The lack of prospective, quantitative phenotyping or natural history studies in this genetically identified syndrome leaves us with only clinical observation to describe the features to date. Additionally, the scarcity of patients increases the risk that patients are included in multiple publications, limiting the conclusions that may be drawn from data collected in a non-standardized manner. Thus, we feel it is much more reliable to approach the disorder with a genetic diagnosis first, followed by standardized clinical assessment and treatment of symptoms. In our cohort, we describe three cases offering atypical genotypes and phenotypes. It is our hypothesis that Patient 12 has a milder phenotype secondary to the location of his pathogenic variant in the penultimate exon, potentially yielding a more functional protein product, similar to a patient with a translocation involving TCF4 described by Kalcheuer et al.57, 58. Conversely, Patient 21 presents with profound cognitive and language deficits, but is lacking the other common clinical characteristics of PTHS. His deletion is small and may only impact the functioning of one of the longer TCF4 transcript variants, thus creating a different phenotype as many of the other TCF4 transcript variants do not include the region encompassed by this deletion10, 23. Given the clinical features of Patient 5, it is our hypothesis that his duplication is disrupting the function of TCF4, though a large duplication such as his has not been described previously. As we have done here, many previously published case reports have described novel genotypes or phenotypes, suggesting there is still room to expand upon the commonly accepted phenotype associated with PTHS. Future studies that systematically study the cognitive, behavioral, and neuropsychological profiles in an objective, quantitative, prospective manner along with the natural history of PTHS are needed in order to identify potential clinical biomarkers and outcome measures for testing future novel therapeutic interventions.
Acknowledgments
This work was performed at the University of Texas Southwestern Medical School in conjunction with Children’s Health Dallas. We would like to acknowledge the families who contributed to this publication and allow us to provide medical care to their loved ones.
Funding Statement
NIH (R01HD069560, R01HD069560-S1, & R01MH093697 to C.M.P.), Autism Speaks (C.M.P.), The Hartwell Foundation (C.M.P.), Ed and Sue Rose Distinguished Professorship in Neurology (C.M.P.), and gifts from Drs. Clay Heighten and Debra Caudy and BRAINS for Autism (C.M.P.) supported this work.
C.M.P. has accepted travel funds and honoraria to speak once at each of the following companies: Psychogenics, Inc.; Astra-Zeneca; Roche; Pfizer; and Dainippon Sumitomo Pharma Co. C.M.P. also has investigator-initiated grant funding for clinical research on Phelan-McDermid Syndrome funded by Novartis. C.M.P. is on the Scientific Advisory Boards of the Phelan-McDermid Syndrome Foundation and of the Pitt-Hopkins Research Foundation.
Footnotes
Ethical Approval
The study received exempt status from the IRB and photographs are published with consent of the legal guardians.
References
- 1.Amiel J, Rio M, de Pontual L, et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet. 2007;80(5):988–93. doi: 10.1086/515582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brockschmidt A, Todt U, Ryu S, et al. Severe mental retardation with breathing abnormalities (Pitt-Hopkins syndrome) is caused by haploinsufficiency of the neuronal bHLH transcription factor TCF4. Hum Mol Genet. 2007;16(12):1488–94. doi: 10.1093/hmg/ddm099. [DOI] [PubMed] [Google Scholar]
- 3.Zweier C, Peippo MM, Hoyer J, et al. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome) Am J Hum Genet. 2007;80(5):994–1001. doi: 10.1086/515583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sweatt JD. Pitt-Hopkins Syndrome: intellectual disability due to loss of TCF4-regulated gene transcription. Exp Mol Med. 2013;45:e21. doi: 10.1038/emm.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pitt D, Hopkins I. A syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust Paediatr J. 1978;14(3):182–4. doi: 10.1111/jpc.1978.14.3.182. [DOI] [PubMed] [Google Scholar]
- 6.Flora A, Garcia JJ, Thaller C, Zoghbi HY. The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc Natl Acad Sci U S A. 2007;104(39):15382–7. doi: 10.1073/pnas.0707456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forrest MP, Hill MJ, Quantock AJ, Martin-Rendon E, Blake DJ. The emerging roles of TCF4 in disease and development. Trends Mol Med. 2014;20(6):322–31. doi: 10.1016/j.molmed.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 8.de Pontual L, Mathieu Y, Golzio C, et al. Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum Mutat. 2009;30(4):669–76. doi: 10.1002/humu.20935. [DOI] [PubMed] [Google Scholar]
- 9.Forrest MP, Waite AJ, Martin-Rendon E, Blake DJ. Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation. PLoS One. 2013;8(8):e73169. doi: 10.1371/journal.pone.0073169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sepp M, Pruunsild P, Timmusk T. Pitt-Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum Mol Genet. 2012;21(13):2873–88. doi: 10.1093/hmg/dds112. [DOI] [PubMed] [Google Scholar]
- 11.Rosenfeld JA, Leppig K, Ballif BC, et al. Genotype-phenotype analysis of TCF4 mutations causing Pitt-Hopkins syndrome shows increased seizure activity with missense mutations. Genet Med. 2009;11(11):797–805. doi: 10.1097/GIM.0b013e3181bd38a9. [DOI] [PubMed] [Google Scholar]
- 12.Zweier C, Sticht H, Bijlsma EK, et al. Further delineation of Pitt-Hopkins syndrome: phenotypic and genotypic description of 16 novel patients. J Med Genet. 2008;45(11):738–44. doi: 10.1136/jmg.2008.060129. [DOI] [PubMed] [Google Scholar]
- 13.Takano K, Tan WH, Irons MB, Jones JR, Schwartz CE. Pitt-Hopkins syndrome should be in the differential diagnosis for males presenting with an ATR-X phenotype. Clin Genet. 2011;80(6):600–1. doi: 10.1111/j.1399-0004.2011.01711.x. [DOI] [PubMed] [Google Scholar]
- 14.Seltzer LE, Paciorkowski AR. Genetic disorders associated with postnatal microcephaly. Am J Med Genet C Semin Med Genet. 2014;166C(2):140–55. doi: 10.1002/ajmg.c.31400. [DOI] [PubMed] [Google Scholar]
- 15.Tan WH, Bird LM, Thibert RL, Williams CA. If not Angelman, what is it? A review of Angelman-like syndromes. Am J Med Genet A. 2014;164A(4):975–92. doi: 10.1002/ajmg.a.36416. [DOI] [PubMed] [Google Scholar]
- 16.Takano K, Lyons M, Moyes C, Jones J, Schwartz CE. Two percent of patients suspected of having Angelman syndrome have TCF4 mutations. Clin Genet. 2010;78(3):282–8. doi: 10.1111/j.1399-0004.2010.01380.x. [DOI] [PubMed] [Google Scholar]
- 17.Armani R, Archer H, Clarke A, et al. Transcription factor 4 and myocyte enhancer factor 2C mutations are not common causes of Rett syndrome. Am J Med Genet A. 2012;158A(4):713–9. doi: 10.1002/ajmg.a.34206. [DOI] [PubMed] [Google Scholar]
- 18.Zweier C, de Jong EK, Zweier M, et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. 2009;85(5):655–66. doi: 10.1016/j.ajhg.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregor A, Albrecht B, Bader I, et al. Expanding the clinical spectrum associated with defects in CNTNAP2 and NRXN1. BMC Med Genet. 2011;12:106. doi: 10.1186/1471-2350-12-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrison V, Connell L, Hayesmoore J, McParland J, Pike MG, Blair E. Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am J Med Genet A. 2011;155A(11):2826–31. doi: 10.1002/ajmg.a.34255. [DOI] [PubMed] [Google Scholar]
- 21.Poot M. Connecting the CNTNAP2 Networks with Neurodevelopmental Disorders. Mol Syndromol. 2015;6(1):7–22. doi: 10.1159/000371594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marangi G, Ricciardi S, Orteschi D, et al. Proposal of a clinical score for the molecular test for Pitt-Hopkins syndrome. Am J Med Genet A. 2012;158A(7):1604–11. doi: 10.1002/ajmg.a.35419. [DOI] [PubMed] [Google Scholar]
- 23.Whalen S, Heron D, Gaillon T, et al. Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: clinical score and further delineation of the TCF4 mutational spectrum. Hum Mutat. 2012;33(1):64–72. doi: 10.1002/humu.21639. [DOI] [PubMed] [Google Scholar]
- 24.Andrieux J, Lepretre F, Cuisset JM, et al. Deletion 18q21.2q21.32 involving TCF4 in a boy diagnosed by CGH-array. Eur J Med Genet. 2008;51(2):172–7. doi: 10.1016/j.ejmg.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 25.Giurgea I, Missirian C, Cacciagli P, et al. TCF4 deletions in Pitt-Hopkins Syndrome. Hum Mutat. 2008;29(11):E242–51. doi: 10.1002/humu.20859. [DOI] [PubMed] [Google Scholar]
- 26.Kato Z, Morimoto W, Kimura T, Matsushima A, Kondo N. Interstitial deletion of 18q: comparative genomic hybridization array analysis of 46, XX,del(18)(q21.2.q21.33) Birth Defects Res A Clin Mol Teratol. 2010;88(2):132–5. doi: 10.1002/bdra.20633. [DOI] [PubMed] [Google Scholar]
- 27.Stavropoulos DJ, MacGregor DL, Yoon G. Mosaic microdeletion 18q21 as a cause of mental retardation. Eur J Med Genet. 2010;53(6):396–9. doi: 10.1016/j.ejmg.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Taddeucci G, Bonuccelli A, Mantellassi I, Orsini A, Tarantino E. Pitt-Hopkins syndrome: report of a case with a TCF4 gene mutation. Ital J Pediatr. 2010;36:12. doi: 10.1186/1824-7288-36-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasi M, Soileau B, Sebold C, et al. The role of the TCF4 gene in the phenotype of individuals with 18q segmental deletions. Hum Genet. 2011;130(6):777–87. doi: 10.1007/s00439-011-1020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehalle D, Williams C, Siu VM, Clayton-Smith J. Fetal pads as a clue to the diagnosis of Pitt-Hopkins syndrome. Am J Med Genet A. 2011;155A(7):1685–9. doi: 10.1002/ajmg.a.34055. [DOI] [PubMed] [Google Scholar]
- 31.Ghosh PS, Friedman NR, Ghosh D. Pitt-Hopkins syndrome in a boy with Charcot Marie Tooth disease type 1A: a rare co-occurrence of 2 genetic disorders. J Child Neurol. 2012;27(12):1602–6. doi: 10.1177/0883073812437242. [DOI] [PubMed] [Google Scholar]
- 32.Rossi M, Labalme A, Cordier MP, et al. Mosaic 18q21.2 deletions including the TCF4 gene: a clinical report. Am J Med Genet A. 2012;158A(12):3174–81. doi: 10.1002/ajmg.a.35588. [DOI] [PubMed] [Google Scholar]
- 33.Takenouchi T, Yagihashi T, Tsuchiya H, et al. Tissue-limited ring chromosome 18 mosaicism as a cause of Pitt-Hopkins syndrome. Am J Med Genet A. 2012;158A(10):2621–3. doi: 10.1002/ajmg.a.35230. [DOI] [PubMed] [Google Scholar]
- 34.Inati A, Abbas HA, Korjian S, Daaboul Y, Harajeily M, Saab R. A case of Pitt-Hopkins syndrome with absence of hyperventilation. J Child Neurol. 2013;28(12):1698–701. doi: 10.1177/0883073812468054. [DOI] [PubMed] [Google Scholar]
- 35.Kousoulidou L, Tanteles G, Moutafi M, Sismani C, Patsalis PC, Anastasiadou V. 263.4 kb deletion within the TCF4 gene consistent with Pitt-Hopkins syndrome, inherited from a mosaic parent with normal phenotype. Eur J Med Genet. 2013;56(6):314–8. doi: 10.1016/j.ejmg.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Steinbusch CV, van Roozendaal KE, Tserpelis D, et al. Somatic mosaicism in a mother of two children with Pitt-Hopkins syndrome. Clin Genet. 2013;83(1):73–7. doi: 10.1111/j.1399-0004.2012.01857.x. [DOI] [PubMed] [Google Scholar]
- 37.de Winter CF, Baas M, Bijlsma EK, van Heukelingen J, Routledge S, Hennekam RC. Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J Rare Dis. 2016;11:37. doi: 10.1186/s13023-016-0422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kharbanda M, Kannike K, Lampe A, Berg J, Timmusk T, Sepp M. Partial deletion of TCF4 in three generation family with non-syndromic intellectual disability, without features of Pitt-Hopkins syndrome. European Journal of Medical Genetics. 2016;59(6–7):310–4. doi: 10.1016/j.ejmg.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 39.de Winter CF, Baas M, Bijlsma EK, van Heukelingen J, Routledge S, Hennekam RCM. Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet Journal of Rare Diseases. 2016;11(1):37. doi: 10.1186/s13023-016-0422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Balkom ID, Vuijk PJ, Franssens M, Hoek HW, Hennekam RC. Development, cognition, and behaviour in Pitt-Hopkins syndrome. Dev Med Child Neurol. 2012;54(10):925–31. doi: 10.1111/j.1469-8749.2012.04339.x. [DOI] [PubMed] [Google Scholar]
- 41.Ouvrier R. Hyperventilation and the PittHopkins syndrome. Developmental Medicine & Child Neurology. 2008;50:481. doi: 10.1111/j.1469-8749.2008.03022.x. [DOI] [PubMed] [Google Scholar]
- 42.Gaffney C, McNally P. Successful use of acetazolamide for central apnea in a child with Pitt-Hopkins syndrome. Am J Med Genet A. 2015;167(6):1423. doi: 10.1002/ajmg.a.37034. [DOI] [PubMed] [Google Scholar]
- 43.Maini I, Cantalupo G, Turco EC, et al. Clinical and polygraphic improvement of breathing abnormalities after valproate in a case of Pitt-Hopkins syndrome. J Child Neurol. 2012;27(12):1585–8. doi: 10.1177/0883073811435917. [DOI] [PubMed] [Google Scholar]
- 44.Verhulst SL, De Dooy J, Ramet J, et al. Acetazolamide for severe apnea in Pitt-Hopkins syndrome. Am J Med Genet A. 2012;158A(4):932–4. doi: 10.1002/ajmg.a.35247. [DOI] [PubMed] [Google Scholar]
- 45.Brockschmidt A, Filippi A, Charbel Issa P, et al. Neurologic and ocular phenotype in Pitt-Hopkins syndrome and a zebrafish model. Hum Genet. 2011;130(5):645–55. doi: 10.1007/s00439-011-0999-4. [DOI] [PubMed] [Google Scholar]
- 46.Tamberg L, Sepp M, Timmusk T, Palgi M. Introducing Pitt-Hopkins syndrome-associated mutations of TCF4 to Drosophila daughterless. Biology open. 2015;4(12):1762–71. doi: 10.1242/bio.014696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grubisic V, Kennedy AJ, Sweatt JD, Parpura V. Pitt-Hopkins Mouse Model has Altered Particular Gastrointestinal Transits In Vivo. Autism Res. 2015;8(5):629–33. doi: 10.1002/aur.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peippo MM, Slimola KOJ, Valanne LK, et al. Pitt-Hopkins syndrome in two patients and further definition of the phenotype. Clinical Dysmorphology. 2006;15(2):47–54. doi: 10.1097/01.mcd.0000184973.14775.32. [DOI] [PubMed] [Google Scholar]
- 49.Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, Gropman AL, Ashwal S. Evidence report: Genetic and metabolic testing on children with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2011;77(17):1629–35. doi: 10.1212/WNL.0b013e3182345896. [DOI] [PubMed] [Google Scholar]
- 50.Points to Consider in the Clinical Application of Genomic Sequencing. American College of Medical Genetics and Genomics; 2012. https://www.acmg.net/StaticContent/PPG/Clinical_Application_of_Genomic_Sequencing.pdf. [DOI] [PubMed] [Google Scholar]
- 51.Kuperberg M, Lev D, Blumkin L, et al. Utility of Whole Exome Sequencing for Genetic Diagnosis of Previously Undiagnosed Pediatric Neurology Patients. J Child Neurol. 2016;31(14):1534–9. doi: 10.1177/0883073816664836. [DOI] [PubMed] [Google Scholar]
- 52.Nolan D, Carlson M. Whole Exome Sequencing in Pediatric Neurology Patients: Clinical Implications and Estimated Cost Analysis. J Child Neurol. 2016;31(7):887–94. doi: 10.1177/0883073815627880. [DOI] [PubMed] [Google Scholar]
- 53.Green ED, Rubin EM, Olson MV. The future of DNA sequencing. Nature. 2017;550(7675):179–81. doi: 10.1038/550179a. [DOI] [PubMed] [Google Scholar]
- 54.Kuo DZ, Houtrow AJ Council On Children With D. Recognition and Management of Medical Complexity. Pediatrics. 2016;138(6) doi: 10.1542/peds.2016-3021. [DOI] [PubMed] [Google Scholar]
- 55.Katkin JP, Kressly SJ, Edwards AR, et al. Guiding Principles for Team-Based Pediatric Care. Pediatrics. 2017 doi: 10.1542/peds.2017-1489. [DOI] [PubMed] [Google Scholar]
- 56.Kennedy AJ, Rahn EJ, Paulukaitis BS, et al. Tcf4 Regulates Synaptic Plasticity, DNA Methylation, and Memory Function. Cell Rep. 2016;16(10):2666–85. doi: 10.1016/j.celrep.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalscheuer VM, Feenstra I, Van Ravenswaaij-Arts CM, et al. Disruption of the TCF4 gene in a girl with mental retardation but without the classical Pitt-Hopkins syndrome. Am J Med Genet A. 2008;146A(16):2053–9. doi: 10.1002/ajmg.a.32419. [DOI] [PubMed] [Google Scholar]
- 58.Sepp M, Kannike K, Eesmaa A, Urb M, Timmusk T. Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5' exon usage and splicing. PLoS One. 2011;6(7):e22138. doi: 10.1371/journal.pone.0022138. [DOI] [PMC free article] [PubMed] [Google Scholar]