

Abstract
Premature infants are at high risk for developing bronchopulmonary dysplasia (BPD), characterized by chronic inflammation and inhibition of lung development, which we have recently identified as being modulated by microRNAs (miRNAs) and alterations in the airway microbiome. Exosomes and exosomal miRNAs may regulate cell differentiation and tissue and organ development. We discovered that tracheal aspirates from infants with severe BPD had increased numbers of, but smaller, exosomes compared with term controls. Similarly, bronchoalveolar lavage fluid from hyperoxia-exposed mice (an animal model of BPD) and supernatants from hyperoxia-exposed human bronchial epithelial cells (in vitro model of BPD) had increased exosomes compared with air controls. Next, in a prospective cohort study of tracheal aspirates obtained at birth from extremely preterm infants, utilizing independent discovery and validation cohorts, we identified unbiased exosomal miRNA signatures predictive of severe BPD. The strongest signal of reduced miR-876-3p in BPD-susceptible compared with BPD-resistant infants was confirmed in the animal model and in vitro models of BPD. In addition, based on our recent discovery of increased Proteobacteria in the airway microbiome being associated with BPD, we developed potentially novel in vivo and in vitro models for BPD combining Proteobacterial LPS and hyperoxia exposure. Addition of LPS led to a larger reduction in exosomal miR 876-3p in both hyperoxia and normoxia compared with hyperoxia alone, thus indicating a potential mechanism by which alterations in microbiota can suppress miR 876-3p. Gain of function of miR 876-3p improved the alveolar architecture in the in vivo BPD model, demonstrating a causal link between miR 876-3p and BPD. In summary, we provide evidence for the strong predictive biomarker potential of miR 876-3p in severe BPD. We also provide insights on the pathogenesis of neonatal lung disease, as modulated by hyperoxia and microbial product–induced changes in exosomal miRNA 876-3p, which could be targeted for future therapeutic development.
Keywords: Cell Biology, Development
Keywords: Bioinformatics, Noncoding RNAs
Exosomal miR 876-3p protects against alveolar damage and inflammation in severe bronchopulmonary dysplasia.
Introduction
Bronchopulmonary dysplasia (BPD) is the most common contributor to mortality and long-term morbidity in extremely low birth weight (ELBW) infants (1). The pathology of BPD in the postsurfactant era is characterized by impaired alveolar and pulmonary vascular development, associated with varying amounts of inflammation and fibrosis (2, 3). The pathogenesis of BPD involves a complex interplay between environmental factors such as hyperoxia-induced lung injury, ventilation-induced lung injury, intrauterine and postnatal inflammation, and host factors that include gene regulatory pathways involved in alveolar and vascular development (4, 5). As the pathogenesis of BPD is multifactorial, disease prediction based solely on clinical parameters is not very accurate. Currently, there are few biomarkers that can provide early identification of risk for developing BPD, as well as determine disease severity, progression, and resolution (6). Such a biomarker would be useful for optimal management and risk stratification, as well as targeted enrollment of high-risk infants into randomized trials featuring potentially novel treatment strategies.
Exosomes are membrane-bound phospholipid vesicles (30–150 nm in diameter) actively secreted by a variety of living cells (7). Exosomes have been identified in large quantities in a variety of bodily fluids, including in blood, urine, bronchoalveolar lavage fluid (BALF), ascitic fluid, and cerebrospinal fluid. As an abundant and stable source of circulating biomarkers, exosomes carry and protect diverse molecular information from degradation and can be analyzed from banked and frozen biological samples (8). These vesicles are released by a variety of immune and nonimmune cells (9). Multiple cell types release increased levels of exosomes, suggesting these vesicles may serve as a specific pathway of cell-to-cell communication, promoting a particular phenotype (10). One goal of this study was to describe the exosomal characteristics seen in BPD. Exosomes are involved in cell-to-cell communication by shuttling various molecules including microRNAs (miRNAs) from donor to recipient cells (11). miRNAs have been studied in several diseases due to their utility in disease diagnostics and ability to predict the probability of disease occurrence (12). In addition to biomarker potential, exosomal miRNAs can significantly contribute to pathogenesis by modulating biological pathways in recipient cells. Exosomal lipid bilayers pack the exosomal miRNAs within, and protect them from, enzymatic degradation, resulting in a long and stable duration of expression suitable for a biomarker (13, 14). Recent studies have shown that miRNAs have central roles in multiple aspects of lung inflammation and disease pathogenesis (12, 15–17). Hence, we hypothesized that the airway exosomal miRNA signature at birth would predict the future development of severe BPD.
We have recently discovered a role for the airway microbiome in the pathogenesis of BPD. Even at birth, infants who subsequently develop BPD many weeks later have an increased abundance of Proteobacteria and Proteobacterial endotoxin LPS. In addition, it is known that Proteobacterial LPS can stimulate epithelial cells to release exosomes (18). Hence, we also wanted to explore the role of Proteobacteria in the induction of exosomal miRNA in BPD.
Results
Exosome Characteristics in BPD
Exosomes play important roles in cell-to-cell communication (19). Increased exosomes are found in the airways of adults with chronic lung diseases, highlighting potential involvement in mechanisms of injury and inflammation (18). We describe the exosomal characteristics in human infants with BPD, in a newborn mouse model of BPD, in hyperoxia-exposed human bronchial epithelial cells (an in vitro model of BPD), and in an in vitro Proteobacterial LPS exposure model.
Human severe BPD.
First, in a prospective cohort study conducted at the UAB Regional Neonatal Intensive Care Unit (RNICU), we characterized the exosomes obtained from tracheal aspirates (TAs) of infants with severe BPD, compared with gestational age–matched full-term (FT) controls (total n = 50, 25 each group; Figure 1, A–C, and Table 1). The infants with severe BPD were sampled at 36 weeks postmenstrual age (PMA). The FT control infants were sampled at a mean postmenstrual gestation age of 38.3 ± 2.2 weeks. The TA exosomes from infants with severe BPD were smaller in size (Figure 1, B and C) but larger in number (Figure 1D; P = 0.03), compared with controls.
Figure 1. Airway exosome characteristics in BPD: human, in vivo and in vitro.

(A) Study design of the prospective cohort study to identify airway exosomal characteristics in severe human BPD. (B and C) The TA exosomes from infants with severe BPD (n = 25) were smaller in size (B and C) but larger in number (D; P = 0.03, t test), compared with controls (n = 25). Size demonstrated by nanosight (B) and by electron microscopy (C). Arrows indicate exosomes. (E) No difference was seen between number of exosomes in TA of control infants at 36 versus 38 weeks postmenstrual age. (F) Exosomes in BALF of hyperoxia-exposed mice are increased in number compared with normoxic controls (P = 0.02, t test). (G) LPS exposure for 12 hours increases the number in exosomes in NHBE cell supernatants, compared with controls (P = 0.03, t test). (H) Hyperoxia-exposed NHBE cells release increased numbers of exosomes into supernatants compared with normoxic cells (P = 0.03, t test). *P < 0.05.
Table 1. Demographics of cohort study for airway exosome characterization in BPD.

As there may be differences in lung epithelial biology at 36 versus 38 weeks, we conducted a subgroup analyses within our control group samples. We compared the TA exosome numbers between infants at 36 weeks versus 38 weeks gestation. No statistical difference in numbers or sizes of exosomes were seen between these samples (P = 0.45, n = 4 per group, Figure 1E).
Cell-specific exosomes.
To know the cellular origins of the majority of exosomes found in airways of neonates, we conducted antibody-depletion experiments using specific epithelial and neutrophil antibody coated beads. The majority of the exosomes derived from the TAs of infant with severe BPD were depleted by epithelial antibodies (MUC4) coated beads (63% reduction), followed by neutrophil antibody (CD66) coated beads (17% reduction). Based on these findings, we utilized epithelial cells for in vitro experiments.
Newborn mouse hyperoxia model.
We conducted hyperoxia experiments using C57BL/6 mouse pups and found exosomes in BALF of hyperoxia-exposed mice to be in increased number compared with normoxic controls (Figure 1F; P = 0.02).
In vitro hyperoxia.
Epithelial cells are the most abundant cell type in the lungs, and to demonstrate the effect of hyperoxia exposure on epithelial cells, we exposed normal human bronchial epithelial (NHBE) cells to hyperoxia or normoxic conditions. Hyperoxia-exposed cells released more but smaller exosomes into supernatants compared with normoxic cells (Figure 1H; P = 0.03).
In vitro with Proteobacterial LPS.
We recently discovered increased Proteobacterial abundance and Proteobacterial endotoxin levels in airways of severe BPD (20). On exposing NHBE cells to E. coli LPS for 12 hours, there was a marked increase in the number in exosomes in supernatants, compared with controls (Figure 1G; P = 0.03).
Prediction of severe BPD in extremely preterm infants by airway exosomal miRNA signatures at birth
Recently discovered miRNAs have been implicated in the pathogenesis of BPD (15). miRNAs can be packaged into exosomes and can modulate important cell-to-cell interactions (21). Hence, we conducted a prospective cohort study to evaluate airway exosomal miRNA signature at birth in ELBW infants who go on to develop severe BPD. Patients who were enrolled at UAB RNICU served as the discovery cohort. To confirm our findings, we utilized samples from a different institute as the validation cohort (see Methods). TA samples from 180 infants were collected, but only 30 were utilized for this analysis, based on the inclusion criteria of early sample acquisition (total n = 30; discovery cohort =18, validation cohort = 12; Tables 2 and 3, and Figure 2). Early TAs were obtained from ELBW infants soon after intubation on the first day of life (in delivery room or within 6 hours of birth, before surfactant administration). Based on their pulmonary outcomes, the ELBW infants were further categorized as the ones who did not develop BPD (BPD-resistant) and the ones who later developed severe BPD many weeks later (BPD-susceptible).
Table 2. Infant characteristics of cohort study for early predictive airway exosomal miRNA signature for BPD (discovery cohort).

Table 3. Demographics of cohort study for early predictive airway exosomal miRNA signature for BPD (validation cohort).

Figure 2. Early predictive airway exosomal miRNA signature for BPD: prospective cohort study.

Study design of the prospective control study to identify early predictive airway exosomal miRNA signature in ELBW infants susceptible to BPD.
Discovery cohort.
In the discovery cohort, of the total 802 miRNAs analyzed in each patient, 40 were different between the BPD-resistant and BPD-susceptible groups (significance for a miRNA was defined as fold change > 1.5 and P < 0.05, Figure 3A).
Figure 3. Early predictive airway exosomal miRNA signature for BPD.

Each tracheal aspirate sample collected at birth from ELBW infants were analyzed for 802 exosomal miRNAs. (A) Discovery cohort: Out of the total 802 exosomal miRNAs analyzed in each patient, 40 miRNAs were different between the BPD-resistant and BPD-susceptible groups (significance for a miRNA was defined as fold change > 1.5 and P < 0.05 by t test). (B) Validation cohort: Out of the 32 validated miRNAs, 6 miRNAs had higher statistical significance for predicting BPD-resistant infants (fold change > 1.5 and P < 0.05 by t test). (C) Evaluation of the area under the ROC curve indicating that a low miR 876-3p levels had the most sensitivity in predicting severe BPD (91.7%).
Validation cohort.
Thirty-two of these 40 miRNAs from the discovery cohort were confirmed in the validation cohort. Out of the 32 validated miRNAs, 6 miRNAs had higher statistical significance (fold change > 1.5 and P < 0.05; Figure 3B).
Biomarker sensitivity testing.
Evaluation of the area under the receiver operator characteristics curve (AUC of ROC) indicated that a low miR 876-3p concentration had the most sensitivity in predicting severe BPD (Figure 3C).
Expression of miR-876-3p in BPD
After identifying miR 876-3p as the top predictive miRNA for severe BPD, we wanted to validate this finding using in vivo and in vitro models. Homo sapiens (Hsa) miR-876-3p was analyzed in human and in vitro NHBE samples, whereas Mus musculus (mmu) miR-876-3p was analyzed in mouse in vivo studies.
Human severe BPD.
Exosomes were obtained from TA of infants with established severe BPD and compared with those from term controls (total 50, n = 25 each group). miR 876-3p expression was decreased in infants with severe BPD, compared with FT controls (Fold change over the expression of U6 snRNA); mean ± SEM for severe BPD [13.17 ± 6.6], mean ± SEM for controls [148.9 ± 20.1]; P = 0.001).
In vivo mouse hyperoxia model.
We utilized the well-established hyperoxia model of BPD (22, 23). Exosomal miR 876-3p expression progressively decreased in BALF of hyperoxia-exposed pups on P4 (1 day after starting oxygen exposure), P7 (4 days after starting oxygen exposure), and P14 (on last day of oxygen exposure), compared with normoxia pups (all time points P < 0.05; Figure 4, A and B). At P14, the mRNA expression of MCL1 (P = 0.0004; Figure 4C) and RBBP6 (P = 0.006; Figure 4D), the 2 most highly conserved targets of miR 876-3p, were correspondingly increased with hyperoxia exposure.
Figure 4. The expression of exosomal miR 876-3p is decreased and its targets are increased in in vivo (C57BL6 mice) and in vitro (NHBE cells) models of BPD.

We conducted in vivo experiments (A–D) utilizing the traditional hyperoxia model of BPD and conducted in vitro experiments utilizing normal human bronchial epithelial (NHBE) cells. Temporal decrease in exosomal miR 876-3p expression in BALF of hyperoxia-exposed mice (all time points P < 0.05 by t test; A and B). (B–D) At P14, with the increase in expression of miR 876-3p with hyperoxia, the expression of the 2 most highly conserved targets of miR 876-3p, MCL1 (P = 0.0004 by t test) and RBBP6 (P = 0.006 by t test), correspondingly decrease. (E–G) Expression of exosomal miR 876-3p decreases in supernatants of NHBE cells exposed to hyperoxia for 24 hours, compared with normoxia (P = 0.0028 by t test). Top predicted targets of miR876-3p, MCL1 (P < 0.0001 by t test) and RBBP6 (P = 0.002 by t test), correspondingly increase with hyperoxia. (H and I) On gain-of-functional analysis by addition of a mimic of miR 876-3p, the expression of MCL1 (P = 0.039 by t test) and RBBP6 (P = 0.03 by t test) are reduced. All in vivo experiments (A–D) were conducted with n = 5–7 animals per group. *P < 0.05.
In vitro hyperoxia exposure.
Expression of exosomal miR 876-3p was decreased in supernatants of NHBE cells exposed to hyperoxia for 24 hours, compared with normoxia (P = 0.003; Figure 4E). Top predicted targets of miR 876-3p–including MCL1 (P < 0.0001; Figure 4F) and RBBP6 (P = 0.002; Figure 4G) were correspondingly increased with hyperoxia. Functional analysis by addition of a mimic of miR 876-3p (gain of function) increased the expression of miR 876-3p and reduced the expression of MCL1 (P = 0.039; Figure 4H) and RBBP6 (P = 0.03, Figure 4I).
Effect of Proteobacteria on miR-876-3p
We have recently discovered the presence of increased Proteobacteria in the airways of infants with severe BPD (20). In order to ascertain the effects of Proteobacteria on miR 876-3p, we performed in vivo and in vitro experiments.
In vivo hyperoxia and LPS exposure in murine model.
To mimic our airway microbiome findings, we created a modified postnatal murine model of BPD. This included administration of escalating doses of i.n. LPS to mice pups from P3–P14 (period of maximal alveolarization), in addition to hyperoxia exposure (see Methods). As expected, hyperoxia inhibited alveolar development, with evidence of delayed secondary septation and larger, simpler distal air spaces as compared with room air controls. Addition of LPS was associated with more severe alveolar hypoplasia in both normoxia and hyperoxia (Figure 5A). These structural changes were quantified by radial alveolar count (RAC), which confirmed that the RACs of both hyperoxia-treated pups and hyperoxia + LPS–treated pups were significantly lower than their respective air controls at P14. LPS alone in normoxia caused decreased alveolarization and potentiated the alveolar hypoplasia in hyperoxia (saline normoxia vs. LPS normoxia, P = 0.003; saline hyperoxia vs. LPS hyperoxia, P = 0.0002; Figure 5B). Exosomal miR 876-3p levels in BALF, although decreased with hyperoxia exposure alone, were reduced to a greater extent by LPS exposure in both normoxic and hyperoxic mice (saline normoxia vs. LPS normoxia, P = 0.008; saline hyperoxia vs. LPS hyperoxia, P = 0.03; Figure 5C). As bacterial LPS is involved in neutrophilic inflammation, neutrophil activity in the BALF was measured by myeloperoxidase (MPO) assay at P14. MPO activity was increased in LPS-exposed normoxic and hyperoxic mice compared with their respective controls (saline normoxia vs. LPS normoxia, P = 0.03; saline hyperoxia vs. LPS hyperoxia, P = 0.0002; Figure 5D).
Figure 5. Postnatal Proteobacterial LPS decreases miR 876-3p expression in vivo.

We developed a double-hit model of BPD in which animals were exposed to postnatal LPS, in addition to hyperoxia exposure. (A and B) Addition of LPS was associated with more severe alveolar hypoplasia in both normoxia and hyperoxia. The RACs of both hyperoxia-treated pups and hyperoxia + LPS–treated pups were significantly lower than their respective air controls at P14. LPS alone, in normoxia, caused decreased alveolarization and potentiated the alveolar hypoplasia in hyperoxia. Magnification, 100×. (C) Exosomal miR 876-3p levels in BALF were reduced to a greater extent by LPS exposure in both normoxic and hyperoxic mice (C). (D) MPO activity was increased in LPS-exposed normoxic and hyperoxic mice compared with their respective controls (D). Hyperoxia + LPS–exposed mice showed increased inflammatory cytokines compared with: IL-1β (E), IFN-γ (F), monocyte chemoattractant protein 1 (MCP-1) (G), tumor necrosis factor alpha (TNF-α)(H), macrophage inflammatory protein–1α (MIP-1α) (I), and macrophage inflammatory protein 1 beta (MIP-1β) (J). *P < 0.05 by 1-way ANOVA. All in vivo experiments were conducted with n = 5–7 animals per group.
At the same time point (P14), inflammatory cytokine protein expression levels were determined in BALF of mice, using multiplex assays. Hyperoxia + LPS–exposed mice compared with only hyperoxia-exposed mice showed increased IL-1β (saline normoxia vs. LPS normoxia, P = 0.02; saline hyperoxia vs. LPS hyperoxia, P < 0.0001; Figure 5E), IFN-γ (saline normoxia vs. LPS normoxia, NS, saline hyperoxia vs. LPS hyperoxia, P = 0.0005; Figure 5F), monocyte chemoattractant protein 1 (MCP-1) (saline normoxia vs. LPS normoxia, NS; saline hyperoxia vs. LPS hyperoxia, P < 0.002; Figure 5G), TNF-α (saline normoxia vs. LPS normoxia, NS; saline hyperoxia vs. LPS hyperoxia, P = 0.02; Figure 5H), macrophage inflammatory protein–1α (MIP-1α) (saline normoxia vs. LPS normoxia, NS; saline hyperoxia vs. LPS hyperoxia, P = 0.03; Figure 5I), and MIP-1β (saline normoxia vs. LPS normoxia, NS; saline hyperoxia vs. LPS hyperoxia, P = 0.01; Figure 5J)
In vitro LPS exposure.
Exosomal miR 876-3p was decreased in supernatants of NHBE cells exposed to LPS at 6 hours, 12 hours, and 24 hours, compared with control (all P < 0.05, Figure 6A). A corresponding increase in the target mRNAs MCL1 (P = 0.006, Figure 6B) and RBBP6 (P = 0.03, Figure 6C) were seen at 24 hours. A similar decrease in exosomal miR 876-3p expression was seen at 12 hours in supernatants of NHBE cells exposed to E. coli bacteria (P = 0.002, Figure 6D). To delineate if this bacterial-induced suppression of miR 876-3p was specific to E.coli, NHBE cells were exposed to either Lactobacillus or E. coli. Compared with Lactobacillus-exposed cells, E. coli–exposed cells showed decreased exosomal miR 876-3p levels at 12 hours (P = 0.0087, Figure 6E).
Figure 6. Proteobacteria exposure decreases miR 876-3p expression in vitro.

Normal human bronchial epithelial (NHBE) cells were utilized for in vitro experiments. (A–C) Expression of exosomal miR 876-3p was decreased in supernatants of NHBE cells exposed to LPS at 6, 12, and 24 hours, compared with control (all P < 0.05). A corresponding increase in the target mRNAs MCL1 (P = 0.006 by t test) and RBBP6 (P = 0.03 by t test) were seen at 12 hours. (D) Decrease in exosomal miR 876-3p expression is seen at 12 hours in cell supernatants of NHBE cells exposed to E. coli bacteria (P = 0.002 by t test). (E) Compared with Lactobacillus-exposed cells, E. coli–exposed cells showed decreased exosomal miR 876-3p levels at 12 hours (P = 0.0087 by t test). (F) NHBE cells exposed to normoxia or hyperoxia, with either LPS or saline (control) for 12 hours. Exosomal miR 876-3p expression was decreased in hyperoxic cell supernatants. A greater magnitude of decrease was seen with the addition of LPS in both normoxia and hyperoxia (saline normoxia vs. LPS normoxia, P < 0.0001 by ANOVA; saline hyperoxia vs. LPS hyperoxia, P = 0.03 by ANOVA). *P < 0.05.
In vitro hyperoxia and LPS exposure.
To determine the individual and combined effects of hyperoxia and Proteobacterial products on airway cells, NHBE cells were exposed to normoxia or hyperoxia, with either LPS or saline (control), for 24 hours. Although exosomal miR 876-3p expression was decreased in supernatants of cells exposed to hyperoxia alone compared with normoxia, a greater magnitude of decrease was seen with the addition of LPS in both normoxia and hyperoxia (saline normoxia vs. LPS normoxia, P < 0.0001; saline hyperoxia vs. LPS hyperoxia, P = 0.03; Figure 6F).
Effect of in vivo miR-876-3p gain of function
To confirm the causative role of miR 876-3p in BPD, we performed in vivo gain-of-function experiments with a mimic of miR 876-3p utilizing both a single-hit hyperoxia model and the double-hit (hyperoxia + LPS) model of BPD.
Compared with controls, at P14, hyperoxic animals administered mimic miR 876-3p developed less alveolar hypoplasia (Figure 7, A and B; P < 0.01) and decreased neutrophilic inflammation (P < 0.05, Figure 7C). Similarly, compared with controls, at P14, animals exposed to double injury (hyperoxia + LPS) and were administered mimic miR 876-3p developed significantly less alveolar hypoplasia (Figure 8, A and B; P < 0.05). No major effects of mimic administration were seen in normoxic mice.
Figure 7. Mimic miR 876-3p rescues hyperoxia-induced alveolar simplification in neonatal mice.
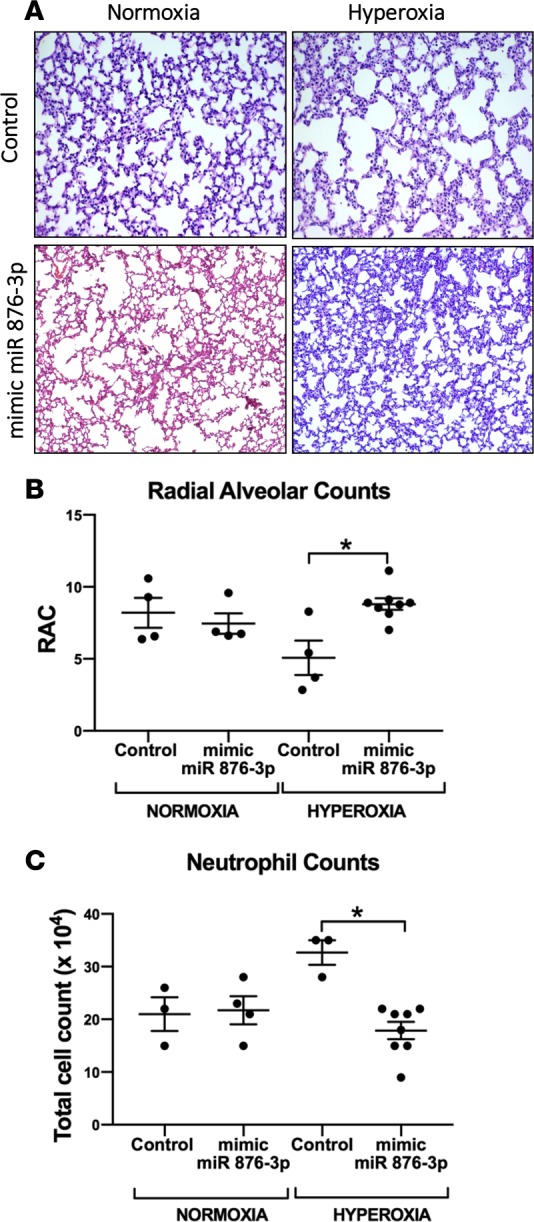
Utilizing the traditional hyperoxia in vivo model of BPD, we conducted gain of miR 876-3p function experiments. Mice exposed to hyperoxia that were administered scrambled miRNA as a control showed alveolar simplification, as demonstrated by morphometric analysis (A and B). Magnification, 100×. Mice exposed to hyperoxia that were administered mimic 876-3p showed significantly improved alveolarization (A and B) and decreased neutrophilic inflammation (C) compared with hyperoxic mice that were administered control (P < 0.05 by ANOVA). All in vivo experiments were conducted with n = 4–8 animals per group. *P < 0.05.
Figure 8. Mimic miR 876-3p rescues alveolarization in murine BPD: Proteobacterial LPS- and hyperoxia-induced double-hit model.

Utilizing the double-hit (hyperoxia + postnatal LPS) in vivo model of BPD, we conducted gain of miR 876-3p function experiments. At P14, animals exposed to double injury (hyperoxia + postnatal LPS) that were administered mimic miR 876-3p developed significantly less alveolar simplification compared with mice that were administered scrambled miRNA controls (A and B). Magnification, 100×. No major effects of mimic administration were seen in normoxic mice. (*P < 0.05 by ANOVA, RAC). All in vivo experiments were conducted with n = 6–7 animals per group.
These data establish that miR 876-3p has a critical and causative role in BPD pathogenesis.
Discussion
Exosomes and exosomal miRNAs are emerging as powerful regulators of cell differentiation and tissue and organ development (19, 24–27). miRNAs have been described to play a role in pulmonary morphogenesis and in lung injury (15, 16). This is the first study to our knowledge to describe the characteristics of released exosomes in airways of preterm infants with severe BPD, as well as the biomarker potential of exosomal miRNAs for BPD. We report that infants with severe BPD have a higher number of released exosomes in their TAs, compared with gestational age–matched controls. Next, our study uses a bedside-to-bench approach, in which we first identified the exosomal miRNA signature predictive of severe BPD, through an unbiased and validated prospective human cohort study. A decreased expression of miR 876-3p at birth was found to be sensitive in predicting the future development of severe BPD in ELBW infants. Subsequently, we confirmed these findings in established newborn mouse models and in vitro hyperoxia models of BPD, thus establishing the role of miR 876-3p in BPD pathogenesis. We further describe the regulation of miR 876-3p by Proteobacteria in BPD, utilizing potentially novel in vivo and in vitro models of bacteria- and hyperoxia-induced inflammation. We show that gain of function of miR 876-3p rescues alveolarization, establishing the cause-and-effect relationship between decreased miR 876-3p and BPD pathogenesis. To model the findings of Proteobacterial dominance in airways of infants with severe BPD (20), we created a modified murine model of BPD, with escalating doses of postnatal LPS, along with hyperoxia exposure. LPS markedly suppressed exosomal miR 876-3p both in vivo and in vitro, despite increased numbers of exosomes, thus indicating a role of LPS in regulating miR 876-3p release. In summary, our studies establish the predictive potential and causative role of microbiota-regulated miR 876-3p in severe BPD.
In pulmonary diseases, biomarkers are powerful tools in understanding the spectrum of pathological conditions affecting the lung microenvironment and function, as well as predicting disease states. Unfortunately, due to the multifactorial nature and the variable clinical phenotype of neonatal chronic lung disease, no good biomarkers currently exist (6, 28). Exosomes and exosomal contents are important regulators of many cellular functions, including tissue homeostasis, immune stimulatory or immunosuppressive functions, and inflammation in the lungs (17, 29, 30). In addition, cell-to-cell communication is essential for the optimal functioning of the lung; hence, exosomes are expected to be important players in lung biology and function (14). Although particularly relevant in BPD — a disease with severe and long-term extrapulmonary consequences (31) — the role of exosomes in BPD had not been explored prior to this study, to our knowledge. The number and contents of exosomes in body fluids may change significantly with respiratory diseases. For example, there are increased numbers of BAL exosomes in sarcoidosis patients compared with healthy volunteers (32). Based on the present evidence, and due to stability in bodily fluid, exosomes may provide novel diagnostic biomarkers and therapeutics for a broad range of pulmonary diseases (17). This is the first study to our knowledge reporting the increased release of exosomes in neonatal chronic lung disease. To adjust for the confounding effects of increased number of exosomes, equal numbers of exosomes were utilized for miRNA analyses from comparison groups.
Although changes in individual miRNAs have been described in various models of BPD (15), this is the first study to our knowledge to describe the role of exosomal miRNAs using an unbiased ‘omic’ approach. In addition, our study shows the predictive potential of exosomal miR 876-3p as early as birth, hence making timely interventions possible in the highest-risk patients, before irreversible lung damage occurs. In addition, as environmental factors could change the risk of developing BPD (5), we validated our findings with a similar patient cohort from a second, geographically different center. Not only do we establish miR 876-3p as a prominent biomarker of BPD, but our in vivo experiments — especially the gain of miR function studies — establish the causative relation between decreased miR 876-3p and BPD pathogenesis. Among BPD-susceptible infants and in animal and cell culture models of BPD, overall increased number but smaller exosomes were seen. However, the predominant biological signal validated in the BPD-susceptible infants was decreased expression of miR 876-3p. The exosomal contents based on exosomal sizes were not analyzed in this study. We speculate that there could be additional BPD-promoting factors in those small exosomes, or they simply may have a dilutional effect, outnumbering the miR 876-3p–containing (and presumably larger) vesicles. Conversely, there could be overall decreased exosomal miR 876-3p expression in BPD-susceptible infants irrespective of the size of exosomes. The difference in exosomal size could represent a different cellular source, a different mechanism of biogenesis, or both.
We utilized TA samples for the human exosome and miRNA studies. There is a possibility that TAs might not represent the true picture of the molecular mechanisms operative in distal lungs but are nevertheless still the best available surrogate pulmonary samples from extremely preterm infants. A limitation of our study is the absence of completely normal–term controls to compare with severe BPD infants, as normal-term infants do not get intubated. Our term infant cohort, although not perfect controls, were the only available option and, importantly, had no documented lung disease. Difference in oxygen exposure at the time of sample collection in preterm infants and FT controls could potentially affect exosomes. A minor limitation of our study is that preterm infants are likely to have a higher oxygen requirement at the time of intubation. However, the impact of oxygen should be less due to the minimal duration of exposure. Unfortunately, no perfect controls exist for the intubated preterm infants; hence, we use intubated FT infants, as they are the best available controls. Several prenatal factors could cause epigenetic changes in utero that influence miRNA profile or exosome release. In our study, there were no overall statistical differences in common clinical variables such as tobacco exposure and oligohydrominos between the BPD-susceptible and the BPD-resistant groups. Nevertheless, we do acknowledge the limitation that there could be additional prenatal factors capable of causing epigenetic changes in utero. Although we did perform functional analysis of miR 876-3p using in vitro models, a limitation is that we explored only the top predicted mRNA targets of miR 876-3p. A single miRNA can target several mRNAs, and an individual mRNA can be a potential target of several miRNAs (33–35). To know all potential downstream effects of a particular miRNA, extensive RNA sequencing (RNA-Seq) and proteomic analyses need to be performed after functional analyses using mimics and inhibitors, which was beyond the cost and scope of this study. Hence, it is difficult to ascertain whether some lung-protective genes could also be altered by a decrease in miR 876-3p. Epithelial cells are the major cell type of the respiratory system — hence, we conducted all in vitro experiments utilizing NHBE cells. The cell-specific effects of miR 876-3p remain to be investigated in future studies, involving cell-specific miR 876-3p–KO models. Apart from MCL1 and RBBP6, miR 876-3p can potentially target a total of 763 genes (Ingenuity Pathway Analysis [IPA], Qiagen). One of the top predicted targets of miR 876-3p is androgen receptor (AR). Clinical evidence suggests a sex predilection for BPD, where male premature newborns are at higher risk and generally have greater morbidity/mortality compared with females who are protected (36). The underlying mechanisms for such differences are unclear and could be explained by future studies exploring miR 876-3p–mediated regulation of AR.
Another major strength of our study is the creation of a new murine BPD in vivo model, which takes into account alterations in airway microbiome, in addition to hyperoxia exposure, as seen in human BPD (20). Several limitations exist with the widely used newborn mouse hyperoxia exposure model of BPD (23), which — unlike human BPD — does not include bacteria-induced inflammation. As airways of infants with severe BPD are marked by a preponderance of Proteobacteria, we created a postnatal Proteobacterial LPS (escalating dose) murine model, in which the alveoli are more simplified and inflamed than hyperoxia exposure alone. Using this in vivo model, and in vitro studies, we demonstrated the marked suppression of miR 876-3p with LPS exposure compared with hyperoxia alone. Inflammatory cytokines were significantly elevated in the BALF of mice exposed to both LPS and hyperoxia, similar to what is seen in human preterm infants who are ventilated and exposed to oxygen (37). Further studies utilizing gnotobiotic and humanized animal models are needed to delineate the species-specific effects of pulmonary microbiota on the overall lung transcriptome and development.
In summary, our data reveal the strong biomarker potential of miR 876-3p for predicting the development of severe BPD in ELBW infants, in addition to providing insight on the molecular mechanisms involved in the disease pathology. Our findings provide a foundational basis for multipronged strategies to develop therapeutics for BPD by utilizing targeted miR 876-3p overexpression, by healthy exosomal transplants, or by utilizing respiratory probiotics or antibiotics.
Methods
Human cohort study for exosome characterization in severe BPD.
In a prospective cohort study between October 2014 and December 2016, TA samples were collected at the UAB Regional NICU from infants with established severe BPD at 36 weeks PMA (total 50, n = 25 each group, Figure 1A). Infants diagnosed with severe BPD (requiring >30% O2 or continuous positive airway pressure (CPAP)/mechanical ventilation) at 36 weeks PMA defined using the physiologic definition (38) were included. The methods were carried out in accordance with the approved guidelines. TA samples were also collected from gestational age–matched FT infants who were intubated for surgical reasons and did not have any lung disease. All FT infants enrolled were intubated at or within 6 hours of birth due to either surgical indication (congenital heart disease, abdominal wall defect) or perinatal depression (with no signs of meconium aspiration syndrome or other lung pathology). All the samples were collected at the time of intubation, much before the surgical procedure or anesthesia administration. These samples from FT infants served as controls.
Human cohort study for early prediction of severe BPD (at birth).
In another prospective cohort study between October 2014 and November 2015, TA samples were collected at birth from ELBW infants in separate discovery (Neonatology Birmingham, Alabama, USA) and validation (Neonatology Philadelphia, Pennsylvania,USA) cohorts to identify and confirm the unbiased exosomal miRNA signatures predicting severe BPD. Of the total 180 screened ELBW infants (gestational 230–276), 30 who were intubated at birth and had early TA sampling (collected at birth or within 6 hours of life) were included (Figure 2 and Tables 2 and 3). Patients were divided into BPD-susceptible vs. BPD-resistant groups, based on clinical outcomes at 36 weeks PMA. Exosomes were extracted, and contents were analyzed for 805 miRNAs using the Nanostring platform. Statistical modeling was conducted and ROC curves were created for the top predictive exosomal miRNAs between discovery and validation cohorts. Each miRNA was used to build a single gene model in discovery data. Prediction analysis was performed on the validation data. There were 7 genes with predicted AUC larger than 0.7. Bioinformatics pathway analysis was conducted utilizing top predictive miRNAs. We utilized TA samples from Philadelphia as a validation cohort to confirm our data. These samples were collected as part of ongoing studies (37, 39, 40). Chorioamnionitis was determined by placental histopathology, and all mothers with chorioamnionitis were administered antibiotics prenatally. Severe BPD at 36 weeks PMA was defined using the physiologic definition (38).
Human TA sample collection.
TA specimens were obtained from patients at the time of intubation at birth or within 6 hours of life before surfactant administration, as clinically indicated per unit protocol. Samples were obtained after ensuring that the infant was adequately oxygenated. The protocol for TA collection involved instillation of 1 ml sterile isotonic saline into the infant’s endotracheal tube, manual bagging through the endotracheal tube for 3 breaths, and suctioning of the fluid into a sterile mucus trap. Sample was separated into supernatant and cell pellet after centrifugation (3,000 g for 10 minutes). Samples were stored frozen in −80°C until further processing. The supernatant was used for exosome extraction. A similar protocol was employed for validation samples.
Cell culture and treatment.
NHBE cells maintained at the UAB Translational Research for Normal and Disordered Development (TReNND) Program were used. These cells were gifted by Stewen Rowe (Gregory Fleming James Cystic Fibrosis Research Center Cell Model Core at UAB). For all experiments, NHBE cells were seeded on 6-well plates (Corning) under air–liquid interface conditions, in serum and antibiotic-free media (DMEM media) to achieve polarization and differentiation (100,000 cells per plate). Experiments were carried out once cells were 80% confluent. Supernatants were collected at various time points for each experiment.
For the in vitro hyperoxia model, cells were exposed to 21% O2 (normoxia) or 85% O2 (hyperoxia) in a special hyperoxia cell culture chamber. For gain of miR 876-3p function analysis, cells were transfected with 15 pmoles mirVana-hsa-miR-876-3p mimic or a negative control (Ambion) using Lipofectamine 2000 transfection reagents (Invitrogen) according to manufacturer’s protocol. At 24 hours, supernatant was collected and exosomal RNA in supernatant was isolated and reverse transcribed. RBBP6 and MCL1 were quantified by quantitative PCR (qPCR) using specific primers: RBBP6 (HS00544663-M1, Invitrogen) and MCL1 (HS01050896-M1, Invitrogen). 18S was used as control for mRNA.
Proteobacterial stimulation of the cells was performed by 1.0 μg/ml of Escherichia coli 055: B5 LPS (MilliporeSigma) versus PBS control. For in vitro experiments with bacteria, 6-well plates were inoculated with either Proteobacteria (E. coli) or Lactobacillus. E. coli (E. coli serotype K1) was grown on Luria Bertani (LB) broth containing 100 μg/ml rifampin (MilliporeSigma) to log-phase growth before inoculation. Bacteria (1 × 106 CFU) was used in 100 μl of antibiotic and serum free media compared with no bacteria (control). Supernatant (200 μl) was collected at 12 hours from each well for every experiment. Lactobacillus (Lactobacillus acidophilus, 4356) was obtained from American Type Culture Collection (ATCC).
Mice.
C57BL/6 mice were obtained from The Jackson Laboratory. Based on our previous results of hyperoxia-induced alveolar simplification in C57BL/6 mice, to detect a 25% increase (2 SD) in the RACs, we needed 6 animals per group to provide 80% power with an α of 0.05. Hence, all mice experiments had a minimum of 6–8 animals of either sex per group, per condition.
Murine postnatal hyperoxia models.
C57BL/6 mice pups were exposed to either normoxia (0.21 fraction of inspired oxygen [FiO2]) or hyperoxia (0.85 FiO2). Surrogate dams were exchanged between normoxia and hyperoxia every 36 hours to prevent maternal toxicity. Pups were analyzed at P14.
In the second model, C57BL/6 newborn mice pups were exposed to hyperoxia (1.0 FiO2) from P1–P4, recovered for the next 10 days, and were sacrificed on P14, as previously described (41, 42).
Murine postnatal LPS model.
Pups were randomly selected and treated by either a PBS control or incremental doses of Escherichia coli 055: B5 LPS by i.n. administration (MilliporeSigma) at P3 (1 mg/Kg), P6 (2.5 mg/Kg), P8 (5 mg/Kg), P10 (7.5 mg/kg), and P14 (10 mg/kg). Pups were exposed to either normoxia (0.21 FiO2) or hyperoxia (0.85 FiO2) from P3–P14 and were euthanized at P14.
Animal harvesting.
Overall techniques used in mouse lung harvesting and analysis have previously been published by our groups (16, 22, 41–43). In brief, at P14, lungs were either inflation-fixed at 25 cm H2O with 4% paraformaldehyde for paraffin embedding/histology or were snap frozen for protein and RNA isolation. BALF was obtained at P14 as previously described (22, 41, 42) and was used for exosome and cytokine analyses. All studies were performed with 4–9 animals per group, per condition, in every experiment. Lung alveolar morphometry was performed at P14 as described previously, with RAC being performed by masked observers (41, 42, 44).
Exosome isolation and characterization.
Exosomes were extracted from human TA samples, in vivo murine BALF, and in vitro cell supernatants. Samples underwent differential centrifugation (at 300 g for 10 minutes to eliminate cells and large cellular debris, then at 2,000 g for 20 minutes followed by 10,000 g for 30 minutes to eliminate any remaining membranous debris). Exosomes were pelleted by centrifuging the supernatant at approximately 150,000 g for 2 hours, and the supernatant was removed. Pellets were resuspended in PBS and centrifuged at approximately 500,000 g for 15 minutes to eliminate any contaminants. The supernatant was removed, and exosomes were suspended in the appropriate buffer (45). For exosome quantification, the Nanosight NS300 (Malvern Instruments) was used as described by our group before (18). Samples were diluted 1:100 and counted on Nanosight NS 300 using 488nM laser and filter. Analysis was performed using the Nanosight NTA 3.1 program. Counts were performed at 25oC, with an infusion rate of 35 μl/minute. Camera was set at 14 with a gain of 1 and detection threshold of 5 for analysis. Sizes were inferred from Brownian motion characteristics of the particles using the Stokes-Einstein equation using the program software (Nanosight NTA 3.1).
Exosomal depletion with antibody-coated beads.
Magnetic beads (Dynabeads, Invitrogen) were coated with antibodies following the manufacturer’s instructions. Exosomes were purified by ultracentrifugation from TAs of patients with severe BPD (n = 25 patients). These were then counted and incubated for 24 hours at 4oC with 5 mg of beads bound to anti–human MUC4 IgG (catalog 60720, Abcam) or 5 mg of beads bound to anti–human CD66b (catalog SAB4301144, MilliporeSigma). Following incubation, the supernatant was eluted by pipetting after holding a magnet to the side of the solution until the solution was visibly cleared. The resulting solution, now depleted of exosomes expressing the marker of interest, was then counted for exosome concentration, and percent depletion was inferred.
Exosomal miRNA and mRNA detection.
Total RNA extraction from exosomes was performed using miRCURY RNA Isolation Kit Cell and Plant (Exiqon). Equal numbers of exosomes were used for RNA extraction for all comparison groups. For all human and human in vitro (NHBE) studies, hsa-miR primers were used, whereas for all mouse in vivo studies mmu-miR primers were used.
For the human cohort study, RNA samples following hybridization reactions were processed using the nCounter Prep Station followed by the nCounter Digital Analyzer and analyzed with the nSolver software, v1.1 (Nanostring Technologies) in accordance with the manufacturer’s instructions (46). Briefly, we calculated a background level of expression for each sample using the mean level of the negative controls plus 2 SD of the mean. miRNA expressing less than 2 SD from the mean were set to 0 expression. Those miRNAs that were considered nonzero expression were normalized using a scaling factor based on the top 100 expressing miRNAs across all samples. For each sample, the average of the geometric means of the top 100 expressing miRNAs across all samples was divided by the geometric mean of each sample (46). All miRNA raw data have been uploaded into NCBI, Gene Expression Omnibus (GEO) database, with accession number GSE108604.
For in vivo and in vitro individual exosomal miRNA detection, after total RNA extraction from exosomes, reverse transcription was performed using TaqMan miRNA reverse transcription kit (Invitrogen). qPCR analysis used universal master mix (Invitrogen), without uracil N-glycosylase (UNG), along with TaqMan miRNA assays for miR-876-3p (002225) and control assay U6 snRNA.
For the gain of miR function experiments, miRvana mimic miR 876-3p was custom built and obtained from Thermo Fisher Scientific and was administered i.n. on P3, P6, P8, P10, and P14 (7 μg/g per mice) in the first postnatal hyperoxia model. In the second hyperoxia-induced BPD mouse model, it was administered on P2 and P4 only.
RBBP6 and MCL1 were quantified by qPCR with specific primers: RBBP6 (HS00544663-M1, Invitrogen) and MCL1 (HS01050896-M1, Invitrogen). 18S was used as internal control for mRNA.
MPO and cytokines.
By use of DuoSet ELISA (R&D Systems) natural and recombinant mouse MPO was measured in BALF cellular pellets. BALF were centrifuged 10,000 g for 5 minutes, pellets were resuspended in 200 μl 1× PBS, further diluted 1:10. Plates were coated overnight to capture antibody. Sample (100 μl per well) was added and further treated with detection antibody, followed by the streptavidin-HRP method. OD was measured at 450 nm in microplane reader.
Total cytokines Milliplex Mouse Cytokine Magnetic kits were custom designed (MilliporeSigma) and used to measure an array of signaling proteins. Manufacturer directions were followed and median fluorescent intensity (MFI) data using a 5-parameter logistic method was used for calculating cytokine/chemokine concentrations in samples.
Statistics.
miRNA profiling–related statistics were performed using Nanostring nCounter system as described (46). For all miRNAs, accuracy of classifying BPD-resistant vs. BPD-susceptible was evaluated by reciever operator characterstic (ROC) curve analysis, with AUC used to quantify sensitivity performance of each miRNA in predicting disease. Each gene was used to build a single gene model in discovery data. Prediction analysis was performed on the validation data. There were 7 genes with predicted AUC larger than 0.7. The ROC curves were plotted by using the predictor and response of validation data (R packages pROC version 1.5.4). To identify relevant biological pathways implicating those genes differentially expressed, we used IPA (IPA Software, Ingenuity Systems, http://www.ingenuity.com). IPA integrates selected omics data sets (genomics, transcriptomics, miRNAomics, proteomics) with mining techniques to predict functional connections and their interpretation in the context of protein networks that comprise protein-protein interactions and related biological functions and canonical signaling pathways.
Other analyses were performed with the Graphpad software (version 7; Graphpad Software Inc.). All results are expressed as means ± SEM. The significance of differences between 2 sample means was determined by unpaired, 2-tailed Student’s t tests, using 95% CI, and multigroup comparisons were made by 2-way ANOVA. A P value of less than 0.05 was considered significant
Study approval.
The human TAs samples were collected from ELBW infants, as approved by the IRB of UAB with granted waiver of consent status, as samples were obtained from routine clinical care and were handled in a completely deidentified manner. Studies using mice were performed according to the animal protocols, as approved by the UAB and Drexel University, Philadelphia IACUCs.
Author contributions
CVL conceived the idea, planned the project, collected samples, analyzed the data, drafted the initial manuscript, revised the manuscript, and approved the final manuscript as submitted. NO, CT, GR, KD, AS, BH, ZA, PD, NS, XX, KG, DR, TS, NY, JEB, AG, VB, and NA assisted with the study design, acquisition of data, and analysis of the data; revised the manuscript; and approved the final manuscript as submitted.
Acknowledgments
We would like to thank Tara McNair RN, BSN for helping with the IRB application process. We would also like to acknowledge the following funding sources: AHA-17SDG32720009 (CL), KPRI COA Foundation (CL), NIH R01 HL129907 (NA, principal investigator; CL, coinvestigator), and NIH R01 HL092906 (NA).
Version 1. 03/08/2018
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(5):e93994. https://doi.org/10.1172/jci.insight.93994.
Contributor Information
Nelida Olave, Email: nolave@peds.uab.edu.
Colm Travers, Email: ctravers@peds.uab.edu.
Gabriel Rezonzew, Email: grezonzew@peds.uab.edu.
Kalsang Dolma, Email: kdolma@uabmc.edu.
Alexandra Simpson, Email: amsimp@uab.edu.
Brian Halloran, Email: bhalloran@peds.uab.edu.
Zubair Aghai, Email: Zubair.Aghai@nemours.org.
Pragnya Das, Email: pragnya.das@drexelmed.edu.
Nirmal Sharma, Email: nirmalsharma@health.usf.edu.
Xin Xu, Email: xinx@uab.edu.
Kristopher Genschmer, Email: gensch@uab.edu.
Derek Russell, Email: dwrussell@uabmc.edu.
Tomasz Szul, Email: szultom@uab.edu.
Nengjun Yi, Email: nyi@uab.edu.
J. Edwin Blalock, Email: blalock@uab.edu.
Amit Gaggar, Email: agaggar1@uab.edu.
Vineet Bhandari, Email: Vineet.Bhandari@DrexelMed.edu.
Namasivayam Ambalavanan, Email: NAmbalavanan@peds.uab.edu.
References
- 1.Stoll BJ, et al. Trends in Care Practices, Morbidity, and Mortality of Extremely Preterm Neonates, 1993-2012. JAMA. 2015;314(10):1039–1051. doi: 10.1001/jama.2015.10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jobe AJ. The new BPD: an arrest of lung development. Pediatr Res. 1999;46(6):641–643. doi: 10.1203/00006450-199912000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;163(7):1723–1729. doi: 10.1164/ajrccm.163.7.2011060. [DOI] [PubMed] [Google Scholar]
- 4.Ambalavanan N, Carlo WA. Bronchopulmonary dysplasia: new insights. Clin Perinatol. 2004;31(3):613–628. doi: 10.1016/j.clp.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Lal CV, Ambalavanan N. Genetic predisposition to bronchopulmonary dysplasia. Semin Perinatol. 2015;39(8):584–591. doi: 10.1053/j.semperi.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lal CV, Ambalavanan N. Biomarkers, Early Diagnosis, and Clinical Predictors of Bronchopulmonary Dysplasia. Clin Perinatol. 2015;42(4):739–754. doi: 10.1016/j.clp.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 8.Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 9.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peinado H, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18(6):883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu G, Drescher KM, Chen XM. Exosomal miRNAs: Biological Properties and Therapeutic Potential. Front Genet. 2012;3:56. doi: 10.3389/fgene.2012.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sessa R, Hata A. Role of microRNAs in lung development and pulmonary diseases. Pulm Circ. 2013;3(2):315–328. doi: 10.4103/2045-8932.114758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng L, Sharples RA, Scicluna BJ, Hill AF. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J Extracell Vesicles. 2014;3 doi: 10.3402/jev.v3.23743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eissa NT. The exosome in lung diseases: Message in a bottle. J Allergy Clin Immunol. 2013;131(3):904–905. doi: 10.1016/j.jaci.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 15.Nardiello C, Morty RE. MicroRNA in late lung development and bronchopulmonary dysplasia: the need to demonstrate causality. Mol Cell Pediatr. 2016;3(1):19. doi: 10.1186/s40348-016-0047-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olave N, et al. Regulation of alveolar septation by microRNA-489. Am J Physiol Lung Cell Mol Physiol. 2016;310(5):L476–L487. doi: 10.1152/ajplung.00145.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alipoor SD, Mortaz E, Garssen J, Movassaghi M, Mirsaeidi M, Adcock IM. Exosomes and Exosomal miRNA in Respiratory Diseases. Mediators Inflamm. 2016;2016:5628404. doi: 10.1155/2016/5628404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szul T, et al. Toll-Like Receptor 4 Engagement Mediates Prolyl Endopeptidase Release from Airway Epithelia via Exosomes. Am J Respir Cell Mol Biol. 2016;54(3):359–369. doi: 10.1165/rcmb.2015-0108OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robbins PD, Dorronsoro A, Booker CN. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J Clin Invest. 2016;126(4):1173–1180. doi: 10.1172/JCI81131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lal CV, et al. The Airway Microbiome at Birth. Sci Rep. 2016;6:31023. doi: 10.1038/srep31023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexander M, et al. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat Commun. 2015;6:7321. doi: 10.1038/ncomms8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee DD, et al. Endothelial Monocyte-Activating Polypeptide II Mediates Macrophage Migration in the Development of Hyperoxia-Induced Lung Disease of Prematurity. Am J Respir Cell Mol Biol. 2016;55(4):602–612. doi: 10.1165/rcmb.2016-0091OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ambalavanan N, Morty RE. Searching for better animal models of BPD: a perspective. Am J Physiol Lung Cell Mol Physiol. 2016;311(5):L924–L927. doi: 10.1152/ajplung.00355.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bang C, et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124(5):2136–2146. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whiteside TL. Exosomes and tumor-mediated immune suppression. J Clin Invest. 2016;126(4):1216–1223. doi: 10.1172/JCI81136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kosaka N, Yoshioka Y, Fujita Y, Ochiya T. Versatile roles of extracellular vesicles in cancer. J Clin Invest. 2016;126(4):1163–1172. doi: 10.1172/JCI81130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Booton R, Lindsay MA. Emerging role of MicroRNAs and long noncoding RNAs in respiratory disease. Chest. 2014;146(1):193–204. doi: 10.1378/chest.13-2736. [DOI] [PubMed] [Google Scholar]
- 28.Lal CV, Ambalavanan N. Cellular and humoral biomarkers of Bronchopulmonary Dysplasia. Early Hum Dev. 2017;105:35–39. doi: 10.1016/j.earlhumdev.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Tickner JA, Urquhart AJ, Stephenson SA, Richard DJ, O’Byrne KJ. Functions and therapeutic roles of exosomes in cancer. Front Oncol. 2014;4:127. doi: 10.3389/fonc.2014.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Staals RH, Pruijn GJ. The human exosome and disease. Adv Exp Med Biol. 2010;702:132–142. doi: 10.1007/978-1-4419-7841-7_11. [DOI] [PubMed] [Google Scholar]
- 31.Gallini F, Arena R, Stella G, Frezza S, Maggio L. Neurodevelopmental outcomes of premature infants with bronchopulmonary dysplasia. Acta Biomed. 2014;85(1):30–34. [PubMed] [Google Scholar]
- 32.Qazi KR, Torregrosa Paredes P, Dahlberg B, Grunewald J, Eklund A, Gabrielsson S. Proinflammatory exosomes in bronchoalveolar lavage fluid of patients with sarcoidosis. Thorax. 2010;65(11):1016–1024. doi: 10.1136/thx.2009.132027. [DOI] [PubMed] [Google Scholar]
- 33.van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007;117(9):2369–2376. doi: 10.1172/JCI33099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benfey PN. Molecular biology: microRNA is here to stay. Nature. 2003;425(6955):244–245. doi: 10.1038/425244a. [DOI] [PubMed] [Google Scholar]
- 35.Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell. 2005;122(1):6–7. doi: 10.1016/j.cell.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 36.Ambalavanan N, et al. Predictors of death or bronchopulmonary dysplasia in preterm infants with respiratory failure. J Perinatol. 2008;28(6):420–426. doi: 10.1038/jp.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aghai ZH, et al. Impact of histological chorioamnionitis on tracheal aspirate cytokines in premature infants. Am J Perinatol. 2012;29(7):567–572. doi: 10.1055/s-0032-1311980. [DOI] [PubMed] [Google Scholar]
- 38.Walsh MC, et al. Impact of a physiologic definition on bronchopulmonary dysplasia rates. Pediatrics. 2004;114(5):1305–1311. doi: 10.1542/peds.2004-0204. [DOI] [PubMed] [Google Scholar]
- 39.Mody K, et al. Sirtuin1 in tracheal aspirate leukocytes: possible role in the development of bronchopulmonary dysplasia in premature infants. J Matern Fetal Neonatal Med. 2012;25(8):1483–1487. doi: 10.3109/14767058.2011.645925. [DOI] [PubMed] [Google Scholar]
- 40.Aghai ZH, et al. IFN-γ and IP-10 in tracheal aspirates from premature infants: relationship with bronchopulmonary dysplasia. Pediatr Pulmonol. 2013;48(1):8–13. doi: 10.1002/ppul.22540. [DOI] [PubMed] [Google Scholar]
- 41.Sun H, et al. Small molecular modulation of macrophage migration inhibitory factor in the hyperoxia-induced mouse model of bronchopulmonary dysplasia. Respir Res. 2013;14:27. doi: 10.1186/1465-9921-14-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sureshbabu A, et al. Inhibition of Regulatory-Associated Protein of Mechanistic Target of Rapamycin Prevents Hyperoxia-Induced Lung Injury by Enhancing Autophagy and Reducing Apoptosis in Neonatal Mice. Am J Respir Cell Mol Biol. 2016;55(5):722–735. doi: 10.1165/rcmb.2015-0349OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bennett KM, et al. Ephrin-B2 reverse signaling increases α5β1 integrin-mediated fibronectin deposition and reduces distal lung compliance. Am J Respir Cell Mol Biol. 2013;49(4):680–687. doi: 10.1165/rcmb.2013-0002OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.James ML, Ross AC, Nicola T, Steele C, Ambalavanan N. VARA attenuates hyperoxia-induced impaired alveolar development and lung function in newborn mice. Am J Physiol Lung Cell Mol Physiol. 2013;304(11):L803–L812. doi: 10.1152/ajplung.00257.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fraser KB, et al. LRRK2 secretion in exosomes is regulated by 14-3-3. Hum Mol Genet. 2013;22(24):4988–5000. doi: 10.1093/hmg/ddt346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kulkarni MM. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr Protoc Mol Biol. 2011;Chapter 25:Unit25B.10. doi: 10.1002/0471142727.mb25b10s94. [DOI] [PubMed] [Google Scholar]