

Key points
The hepatic scar is dynamic with respect to cell and matrix components; myofibroblasts (MFs) and hepatic macrophages are key players in progression and regression of fibrosis
Loss of fibrogenic MFs is a pivotal event in fibrosis regression, allowing spontaneous degradation of scar
There is evidence for fibrosis regression in cirrhosis, but not for reversal
Therapies that directly target hepatic stellate cell-MFs, degrade scar or stimulate liver regeneration are poised to enter the clinical arena
Lack of robust, standardised treatment end-points is currently the major limiting factor in antifibrotic trials
Fibrosis, or scarring, of the liver is a generic wound healing response to chronic liver disease regardless of aetiology. Progressive fibrosis eventually evolves to cirrhosis, with fibrotic bands, parenchymal nodules and vascular distortion leading to liver cell dysfunction, portal hypertension (PHT), hepatocellular carcinoma (HCC) and premature death. Liver fibrosis is a clinically silent process, such that many patients present at an advanced stage when disease-specific therapy has limited impact. Central to this is the recognition that cirrhosis is not simply severe fibrosis but a more complex pathological condition with reversible and irreversible components.
Detailed analysis of the cellular and molecular mechanisms that mediate liver fibrosis has provided a framework for therapeutic approaches to prevent, slow or even reverse fibrosis or cirrhosis. The goal of such treatments would be stasis or regression of fibrosis to precirrhotic stages to prevent the development of liver failure, HCC and/or to reduce PHT and its complications such as variceal haemorrhage, ascites and encephalopathy. Despite this rationale, no antifibrotics are currently licensed for use in humans.
Epidemiological projections for the future prevalence of viral, obesity and alcohol-related cirrhosis paint an increasingly gloomy picture. Together with a shortfall in donors for liver transplantation, the clinical urgency for new therapies is high.1 There is increasing interest from stakeholders keen to exploit the market potential for antifibrotics. The design of future trials for agents in the developmental pipeline will depend upon:
strategies to ensure equal patient stratification
techniques to reliably monitor changes in fibrosis over time
definition of clinically meaningful end-points.
Mechanisms of liver fibrogenesis and fibrosis regressititlen
Liver fibrogenesis and the role of the hepatic stellate cell-myofibroblast
Liver fibrogenesis is orchestrated by a heterogeneous population of profibrogenic myofibroblasts (MFs), the majority originating from hepatic stellate cells (HSCs) following a process termed ‘activation’. However, portal fibroblasts, bone marrow derived cells recruited to the injured liver and epithelial cells (hepatocytes, cholangiocytes) that have undergone epithelial to mesenchymal transition may also contribute to the MF pool.2 In normal liver, quiescent HSCs store vitamin A in lipid droplets, but in response to liver injury they undergo characteristic morphological and functional changes to MF-like cells.3 The activation process is initiated by many factors including profibrogenic cytokines (especially transforming growth factor-beta (TGF-β)) and platelet-derived growth factor, and also reactive oxygen species and apoptotic bodies generated by other resident and incoming cells. Activated HSCs:
lose lipid
proliferate
migrate
express myogenic markers such as alpha-smooth muscle actin
produce excessive scar proteins (mostly type-1 collagen)
demonstrate enhanced contractility and immune capability.2,3
Persistence of the HSC-MF phenotype is sustained by tissue hypoxia, apoptosis, expression of profibrogenic growth factors, and cytokines and specific cell-matrix interactions. Disease-specific fibrogenic mechanisms have been uncovered, such as direct stimulation of HSCs by HCV, elevated levels of leptin and increased leptin signalling in non-alcoholic steatohepatitis.
Accumulation of extracellular matrix (ECM) in liver fibrosis results from increased synthesis and decreased degradation. Studies of human and rodent liver indicate that matrix metalloproteinases (MMPs) with proteolytic activity against several scar constituents are expressed even in end-stage cirrhosis. These enzymes are held in check by concurrent secretion of their potent specific inhibitors (tissue inhibitors of metalloproteinases (TIMPs)) by HSC-MFs.4 Furthermore, TIMP-1 may also promote the survival of fibrogenic HSC-MFs by inhibiting their clearance by apoptosis.
Mechanisms mediating regression of liver fibrosis
Regression of liver fibrosis – and even cirrhosis – has been demonstrated in animal models and corroborated in the entire spectrum of chronic liver diseases although, critically, improvement has been observed only following successful treatment of the underlying cause. The pressing question now is not can cirrhosis regress, but to what extent it can regress and whether this translates to improvements in PHT, HCC risk and overall survival.5
Studies in animal disease models have shown that TIMP levels decrease dramatically, MMP activity increases and scar degrades after the insult that induced fibrosis is withdrawn.4,6 In parallel, MFs are lost from the receding hepatic scar by apoptosis.4,6 Hepatic macrophages have re-emerged as key regulators of matrix remodelling, with recent studies demonstrating a capacity for injury-inducing or repair-promoting roles in liver fibrosis.7,8 Data derived from animal models and human liver explants have demonstrated that as advanced fibrosis resolves, the micronodules typical of ‘active’ cirrhosis dissolve and coalesce into macronodules.6,9 These findings correlate well with recent clinical data showing that increased septal thickness and smaller nodule size are predictors of poorer clinical outcomes.10
Factors affecting the progression and regression of liver fibrosis
The rate of progression of fibrosis can vary considerably. Progression of fibrosis is particularly rapid in specific clinical scenarios: for example, congenital hepatic fibrosis, drug-induced liver disease, HCV and HIV co-infection and recurrent HCV after liver transplantation. Additional risk factors such as alcohol consumption, a high body mass index or the HIV or post transplant related immunosuppressed state may also accelerate disease progression.11 Genetic factors are also likely to influence remodelling of liver fibrosis. Single nucleotide polymorphisms in candidate genes with relevance to fibrosis risk are emerging: for example, TGF-β1, tumour necrosis factor-α, interleuklin-10, angiotensinogen, C-C chemokine receptor type 5 (CCR5), monocyte chemotactic protein-1 (MCP-1) and DEAD box protein 5 (DDX5).12
Experimental studies have demonstrated that liver fibrosis varies in reversibility according to the duration, composition, spatial distribution and cellularity of scar. Areas of fibrosis that are not completely degraded in animal models or human cirrhosis are relatively acellular, rich in elastin and extensively cross-linked, suggesting that ECM cross-linking might represent a ‘point of no return’ in fibrosis.6 The degree to which partial or complete fibrosis regression restores normal portal blood flow is uncertain since the vascular distortion, shunting and angiogenesis that characterises advanced cirrhosis may not be reversible.13
Methods to assess liver fibrostitles
A need for effective biomarkers to gauge disease severity and response to therapy is becoming increasingly clear in the field of liver fibrosis (Table 1).14 Liver biopsy is invasive, examines only 1/50,000 of the liver and limited by sampling variability. Additionally, a change in fibrosis on biopsy (even when quantitative) is simply a notional surrogate for future clinical outcomes. Sensitive, specific non-invasive markers are needed that respond quickly to changes in fibrogenic activity. They will need to be tested rigorously and validated in an unbiased manner. In patients with advanced cirrhosis, in whom the capacity to remodel scar tissue may be reduced, measurement of a functional parameter such as portal pressure (hepatic venous pressure gradient) might be more appropriate.
Table 1.
Conventional and emerging diagnostic modalities for the detection of liver fibrosis.

Serum biomarkers
Blood markers to predict fibrosis and cirrhosis are not new and all have their limitations. They cover:
simple tests such as platelet count, the aspartate aminotransferase (AST) to platelet ratio index, AST to alanine aminotransferase ratio
more complicated collections of analytes such as Fibrotest (combining six serum markers with the age and sex of the patient)15
those which may be considered more specific for fibrosis such as hyaluronic acid,16 procollagen-3 N-terminal peptide (PIIINP) and the European Liver Fibrosis Panel (hyaluronic acid, TIMP-1 and PIIINP).17
The inability to differentiate intermediate fibrosis stages precludes the use of currently available serum markers as surrogates in antifibrotic trials. Furthermore, hyaluronic acid may give false positive results in patients with joint disease or active liver inflammation.
Transient elastography
Fibroscan (transient elastography), a bedside test to assess liver fibrosis by measuring liver stiffness, gives immediate results and has become popular with clinicians. It samples a 100-fold larger volume of the liver than biopsy (~1/500), correlates well with histological stages of fibrosis and compares favourably with other non-invasive tests.18 Fibroscan is less reliable in obese subjects or if there is significant liver inflammation or extrahepatic cholestasis.
In the future, more than one modality (ie serum markers and imaging) may be used clinically. One key advantage of these non-invasive approaches over liver biopsy is the ability to repeat measurements at regular intervals, thereby giving some idea of the speed of change in fibrosis development or regression.
Treatments for liver fibrostitles
The best treatment for liver fibrosis is to cure or suppress the underlying disease process. Eradication of HBV, HCV or HDV with antivirals can lead to reversal of fibrosis and improvement in liver function, even in some patients with histological cirrhosis. Similarly, abstinence from alcohol, venesection in haemochromatosis, chelation of copper in Wilsons disease or immunomodulator therapy in autoimmune hepatitis can also promote fibrosis regression (Fig 1).19 However, for patients in whom specific treatment is not possible or sucessful (eg antiviral non-responders), therapies that can slow or reverse fibrosis are necessary (Table 2). In practical terms, the potential to achieve stasis or lack of progression of fibrosis in the face of continued liver injury would be clinically meaningful if liver function was preserved, complications of decompensated cirrhosis reduced or the need for liver transplantation delayed or averted.
Fig 1.

Regression of human liver fibrosis: (a) at presentation, index liver biopsy from a patient with autoimmune hepatitis showed dense fibrosis; (b) liver biopsy 18 months after successful treatment with immunomodulators showed complete regression of fibrosis, with restitution of near normal liver architecture (reticulin stain × 100 original magnification). Reprinted with permission from Elsevier.19
Table 2.
Overview of therapeutic approaches in liver fibrosis.

Basic science research, with specific emphasis on HSC biology, has identified numerous potential antifibrotics worthy of clinical trials.20 Some agents directly target key molecules or pathways involved in fibrogenesis or fibrolysis (Fig 2). Another approach is drug repositioning where established treatments with good safety profiles (eg angiotensin receptor antagonists or glitazones) can be fast tracked into the clinical arena if they are shown to have antifibrotic effects in animal models.
Fig 2.
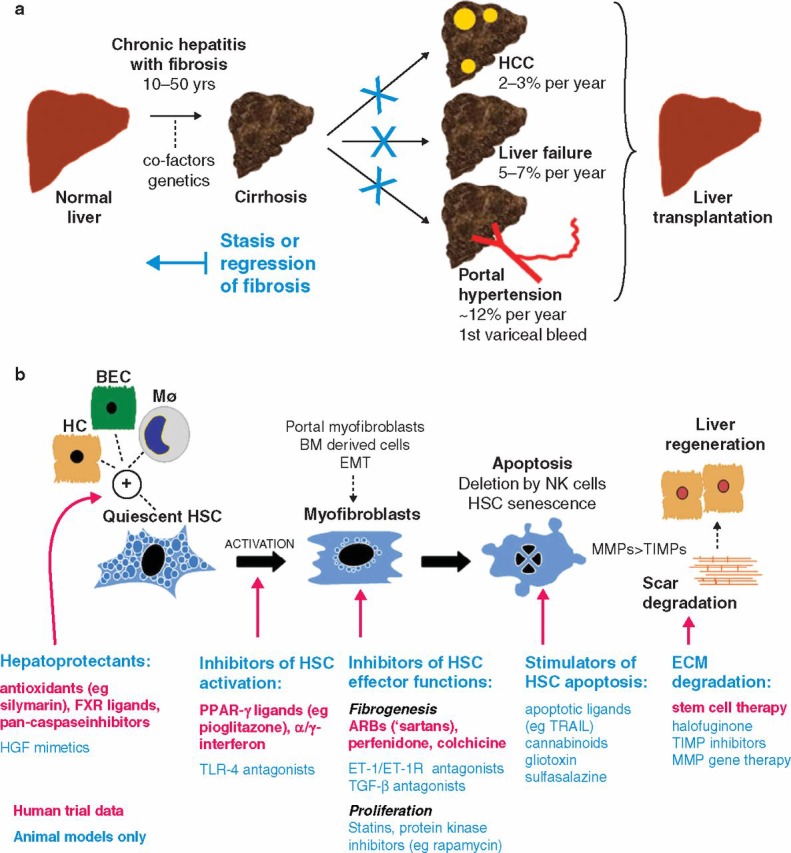
Antifibrotic therapy in human liver disease: (a) natural history of liver fibrosis and the potential role of antifibrotic therapy. Stasis or regression of fibrosis could reduce the complications of cirrhosis and obviate the need for liver transplantation; (b) illustrated summary of emerging therapies for liver fibrosis that target critical pathways of fibrogenesis and fibrolysis. ARB = angiotensin-11 type-1 receptor blocker; BEC = biliary epithelial cell; BM = bone marrow; ECM = extracellular matrix; EMT = epithelial to mesenchymal transition; ET-1(R) = endothelin-1(receptor); FXR = farnesoid X receptor; HC = hepatocyte; HCC = hepatocellular carcinoma; HSC = hepatic stellate cell; Mϕ = macrophage; MMP = metalloproteinase; NK = natural killer cell; PPAR = peroxisomal proliferator activated receptor; TGF = transforming growth factor; TIMP = tissue inhibitor of metalloproteinase; TLR = toll-like receptor; TRAIL = tumour necrosis factor-related apoptosis-inducing ligand.
Unfortunately, positive results from animal models have not always translated to clinical efficacy in humans. There are plausible reasons for this, including differences in pharmacokinetics, dynamics of scarring and degree of ECM cross-linking.20
The jury is out on potential antifibrotics derived from complementary and alternative medicine such as silymarin (milk thistle), curcumin (turmeric) and trans-resveratrol (red wine), although increasingly these will be scrutinised in controlled trials. Another interesting example is coffee, for which there is reasonable evidence that it may have antifibrotic effects and reduce the risk of HCC.21
An emerging therapeutic concept is direct delivery of agents to a chosen target cell (eg inducers of HSC-MF apoptosis) using cell-specific receptors to increase local drug concentrations whilst preventing deleterious effects on collateral cells (eg hepatocytes) or other organs uninvolved in the fibrogenic process.
Future antifibrotic strategies are likely to be customised on the basis of disease-specific features and host (genetic) determinants of fibrosis progression and treatment response, meaning that a multiagent approach may be required.
References
- 1. The National Plan for Liver Services UK. A time to act: improving liver health and outcomes in liver disease. British Association for the Study of the Liver (BASL) and British Society of Gastroenterology (BSG) (Liver Section), 2009. www.bsg.org.uk/sections/liver-news/the-national-plan-for-liver-services-uk-2009.html.
- 2.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–48. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iredale JP, Benyon RC, Pickering J, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538–49. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desmet VJ, Roskams T. Cirrhosis reversal: a duel between dogma and myth. J Hepatol. 2004;40:860–7. doi: 10.1016/j.jhep.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Issa R, Zhou X, Constandinou CM, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126:1795–808. doi: 10.1053/j.gastro.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 7.Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fallowfield JA, Mizuno M, Kendall TJ, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288–95. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- 9.Wanless IR, Nakashima E, Sherman M. Regression of human cirrhosis. Morphologic features and the genesis of incomplete septal cirrhosis. Arch Pathol Lab Med. 2000;124:1599–607. doi: 10.5858/2000-124-1599-ROHC. [DOI] [PubMed] [Google Scholar]
- 10.Nagula S, Jain D, Groszmann RJ, Garcia-Tsao G. Histological-hemodynamic correlation in cirrhosis – a histological classification of the severity of cirrhosis. J Hepatol. 2006;44:111–7. doi: 10.1016/j.jhep.2005.07.036. [DOI] [PubMed] [Google Scholar]
- 11.Poynard T, Ratziu V, Charlotte F, et al. Rates and risk factors of liver fibrosis progression in patients with chronic hepatitis C. J Hepatol. 2001;34:730–9. doi: 10.1016/S0168-8278(00)00097-0. [DOI] [PubMed] [Google Scholar]
- 12.Hold GL, Untiveros P, Saunders KA, El-Omar EM. Role of host genetics in fibrosis. Fibrogenesis Tissue Repair. 2009;2:6. doi: 10.1186/1755-1536-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernández M, Semela D, Bruix J, et al. Angiogenesis in liver disease. J Hepatol. 2009;50:604–20. doi: 10.1016/j.jhep.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 14.Talwalkar JA. Antifibrotic therapies – emerging biomarkers as treatment end points. Nat Rev Gastroenterol Hepatol. 2010;7:59–61. doi: 10.1038/nrgastro.2009.197. [DOI] [PubMed] [Google Scholar]
- 15.Imbert-Bismut F, Ratziu V, Pieroni L, et al. MULTIVIRC Group Biochemical markers of liver fibrosis in patients with hepatitis C virus infection: a prospective study. Lancet. 2001;357:1069–75. doi: 10.1016/S0140-6736(00)04258-6. [DOI] [PubMed] [Google Scholar]
- 16.Plevris JN, Haydon GH, Simpson KJ, et al. Serum hyaluronan – a non-invasive test for diagnosing liver cirrhosis. Eur J Gastroenterol Hepatol. 2000;12:1121–7. doi: 10.1097/00042737-200012100-00009. [DOI] [PubMed] [Google Scholar]
- 17.Guha IN, Parkes J, Roderick P, et al. Noninvasive markers of fibrosis in nonalcoholic fatty liver disease: validating the European Liver Fibrosis Panel and exploring simple markers. Hepatology. 2008;47:455–60. doi: 10.1002/hep.21984. [DOI] [PubMed] [Google Scholar]
- 18.Smith JO, Sterling RK. Systematic review: non-invasive methods of fibrosis analysis in chronic hepatitis C. Aliment Pharmacol Ther. 2009;30:557–76. doi: 10.1111/j.1365-2036.2009.04062.x. [DOI] [PubMed] [Google Scholar]
- 19.Fallowfield JA, Kendall TJ, Ireland JP. Reversal of fibrosis: no longer a pipe dream? Clin Liver Dis. 2006;10:481–97. doi: 10.1016/j.cld.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 20.Rockey DC. Current and future anti-fibrotic therapies for chronic liver disease. Clin Liver Dis. 2008;12:939–62. doi: 10.1016/j.cld.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masterton GS, Hayes PC. Coffee and the liver: a potential treatment for liver disease? Eur J Gastroenterol Hepatol. 2010;22:1277–83. doi: 10.1097/MEG.0b013e32833cca96. [DOI] [PubMed] [Google Scholar]