

SUMMARY
During neurodegenerative disease, the toxic accumulation of aggregates and misfolded proteins is often accompanied with widespread changes in peripheral metabolism, even in cells in which the aggregating protein is not present. The mechanism by which the central nervous system elicits a distal reaction to proteotoxic stress remains unknown. We hypothesized that the endocrine communication of neuronal stress plays a causative role in the changes in mitochondrial homeostasis associated with proteotoxic disease states. We find that an aggregation-prone protein expressed in the neurons of C. elegans binds to mitochondria, eliciting a global induction of a mitochondrial-specific unfolded protein response (UPRmt), affecting whole-animal physiology. Importantly, dense core vesicle release and secretion of the neurotransmitter serotonin is required for the signal’s propagation. Collectively, these data suggest the commandeering of a nutrient sensing network to allow for cell-to-cell communication between mitochondria in response to protein folding stress in the nervous system.
In Brief
How is mitochondrial stress in the nervous system signaled to other tissues?
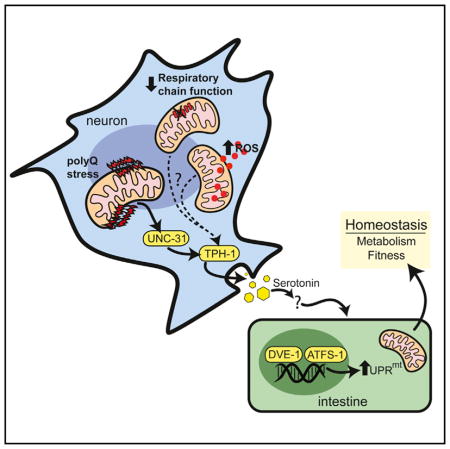
INTRODUCTION
Among the many deleterious consequences of Huntington’s disease (HD), the severe changes that occur in metabolic function across non-neuronal tissues remain among the most puzzling. For HD patients, the risk for developing diabetes is nearly seven times greater than in non-HD patients (Podolsky et al., 1972). Insulin secretion is impaired, basal resting energy expenditure increases, glucose metabolism is reduced, lactate concentrations are elevated, and progressive, startling degrees of weight loss occur regardless of caloric consumption (Jenkins et al., 1993; Walker and Raymond, 2004; Weydt et al., 2006). The extreme metabolic dysfunction observed in HD patients is far from unique, however. Deleterious changes in metabolism have been reported in a range of neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and amyotrophic lateral sclerosis (Cai et al., 2012; Duarte et al., 2014). With neurodegenerative disease, mitochondrial dysfunction in particular manifests across a variety of parameters that include a decline in energy production, impaired tricarboxylic acid cycle activity, decreased electron chain function, and aberrant mitochondrial dynamics (Jenkins et al., 1993; Mochel et al., 2011; Podolsky et al., 1972). It is likely that these metabolic changes are both caused by and capable of exacerbating disease states, further destabilizing the protein-folding environment within the cell and undermining its capacity to mount defenses against increasing levels of proteotoxic stress.
An important consequence of mitochondria stress caused by proteotoxicity is the global alteration of transcription networks associated not only with the regulation of protective chaperones and enzymes (the mitochondrial unfolded protein response, or UPRmt), but also with metabolism (Cai et al., 2012; Duarte et al., 2014; Nargund et al., 2015). Recent evidence suggests that the transcription factor ATFS-1 is not only capable of upregulating mitochondrial chaperones, proteases, and antioxidant enzymes, but also regulates a large number of genes required for oxidative phosphorylation and glycolysis (Nargund et al., 2012). These results posit a coordinated regulation of mitochondrial protein homeostasis with the active establishment of a metabolic state. Importantly, these data suggest that an endocrine-like response might be responsible for eliciting global changes driving both stress response activation and metabolic function, thereby coordinating changes throughout the organism.
Recently, we have reported that mitochondria can communicate intracellular stress between tissues in C. elegans, in which an induction of the mitochondrial unfolded protein response (UPRmt) in the neurons is sensed and reacted to by mitochondria within physically distinct, non-innervated tissues (Durieux et al., 2011). Because of the dual role for the UPRmt in both proteostasis and metabolism, we have hypothesized that metabolic sensors mediate the cell-non-autonomous signaling of mitochondrial proteotoxic stress. To explore this possibility, we examined models of proteotoxic stress in C. elegans neurons for evidence of secondary effects on distal mitochondrial stress responses. In our analyses, we found that expression of a polyglutamine tract of a specific length (PolyQ40) expressed in neurons is sufficient to elicit a mitochondrial stress response in distal tissues. Association of the PolyQ protein with mitochondria correlates with the distal upregulation of the UPRmt and physiologic changes in the entire animal. Upregulation of the UPRmt pathway in peripheral tissues requires the function of UPRmt components as well as dense core vesicle secretion from affected neurons. The application of exogenous serotonin is sufficient to rescue the defect in neuronal secretion and restore UPRmt signaling. Importantly, a loss in serotonin synthesis is sufficient to block cell-non-autonomous UPRmt signaling to distal tissues, an effect rescued by the application of exogenous serotonin. These findings suggest a mechanistic link between mitochondrial proteostasis, endocrine signaling, and the peripheral metabolic decline found in neurodegenerative disease states, such as HD.
RESULTS
Neuronal Polyglutamine Protein Expression Induces Mitochondrial Stress
We examined the effects of cytosolic, aggregation-prone protein expression on the activation of UPRmt targets, using transgenic C. elegans reporter strains in which the promoters of canonical UPRmt components were fused to GFP (Brignull et al., 2006). In these analyses, we identified a single disease variant protein, a neuronally expressed expansion of repeating glutamines (polyQ40), as being sufficient to cause a cell-non-autonomous upregulation of a mitochondrial stress response (Figures 1A, S1A, and S1B). Upregulation of the promoter of hsp-6, the ortholog of Grp75/mortalin/Hsp5a, was not discernable until day 1 of adulthood (Figure S1C) and was predominately observed in the anterior and posterior of the intestine (Figure 1A). This observation was quantified with large particle flow cytometry and image analysis (Figures 1B, S1B, and S1D). Higher-magnification imaging of the posterior and mid-intestine is also shown for the reporter and polyQ40 strains (Figure 1C). Whole-organism changes in endogenous HSP-6 protein were also increased and were quantified by immunoblotting (Figures 1D and 1E). Similar results were found with transgenic HA-tagged polyQ40 animals (Figures S1A and S1B).
Figure 1. Neuronal PolyQ40 Induces hsp-6p:GFP in Distal Tissues.

(A) Photomicrographs of hsp-6p::GFP reporter expression in animals containing no polyQ or rgef-1p::polQ19::CFP, rgef-1p::polyQ40::YFP, or rgef-1p::polyQ67::YFP.
(B) Biosorter fluorescence measurement for rgef-1p::polyQ40::YFP strains with and without hsp-6p::GFP reporter (Mean ± SD for samples of 75–1,000 worms, p < 0.0001 by Student’s t test of polyQ40;hsp-6p::GFP strain).
(C) Higher-magnification images of the posterior and mid-intestinal region of both hsp-6p::GFP and rgef-1p::polyQ40::YFP strains.
(D) Immunoblot analysis of strains with or without the polyQ40 expression using antibodies against GFP and GRP75/HSP-6. Anti-GFP detects both total polyQ40::YFP expression (higher molecular weight band) as well as hsp-6p::GFP expression (lower molecular weight band). Cytochrome c oxidase (cco-1) RNAi treatment serves as a positive control for hsp-6p::GFP induction.
(E) Quantification of band intensity in (D) relative to α-tubulin loading control, showing increased GRP75 in the Q40 strain versus reporter strain alone (Mean ± SEM p<0.004 by Student’s t test).
(F) Immunoblot analysis against GRP75 expression in primary BJ fibroblasts expressing polyQ25, polyQ78, polyQ103, and polyQ153.
(G) Quantification of band intensity in (F) relative to polyQ25 mediated GRP75 expression.
Effects of proteotoxicity on the UPRmt were highly specific to the length, neuronal expression, and form of proteotoxic stress on the mitochondria. Transgenic animals expressing either non-aggregative or severely neurotoxic expansions of polyQ failed to elicit cell-non-autonomous hsp-6p driven GFP expression (Figure 1A). PolyQ40 expression in the body wall muscle, rather than in the neurons, was also incapable of eliciting a cell-non-autonomous mitochondrial response to proteotoxic stress (Figure S1E). Moreover, neither neuronal expression of the Alzheimer’s-related protein Aβ1-42, nor mutant forms of the ALS associated protein TDP-43, were sufficient to induce the UPRmt (Figure S1F). Finally, neuronal polyQ40 expression had a specific effect on mitochondrial stress response activation, as neither basal levels nor induction of chaperones and stress-responsive genes, including hsp-16.2 (small hsp20/alpha-B crystalline, responsive to heat shock) (Link et al., 1999), hsp-4 (BiP/Hsp9a, responsive to ER stress) (Kapulkin et al., 2005), or sod-3 (superoxide dismutase, responsive to oxidative stress) (Honda and Honda, 1999) were affected by its expression (Figure S1G). Collectively, these data suggest that a cell-non-autonomous mitochondrial stress response is invoked by polyQ40 protein expression in the C. elegans nervous system.
We analyzed whether the effects of polyQ40 on UPRmt were conserved in mammalian cells. We expressed exon1 of the Htt gene followed by polyglutamine expansions of lengths of 25Q, 78Q, 103Q, and 153Q in human primary fibroblasts. mtHSP70 levels increased with increasing lengths of polyglutamine expansions and were significantly increased in cells expressing the 103Q and 153Q lengths (Figure 1F and 1G). This suggests that the UPRmt induction by polyglutamine toxicity is not specific to C. elegans, but occurs in mammalian models of proteotoxicity as well.
Distal hsp-6 Induction Requires Functional UPRmt Components
Previous work in C. elegans has identified multiple genetic components required for the UPRmt stress response (Haynes et al., 2007, 2010; Yoneda et al., 2004). The increased expression of mitochondrial chaperones HSP-6 and HSP-60 requires the nuclear localization of transcription factors and co-regulators DVE-1, UBL-5, and ATFS-1, while the protease CLPP-1 is required for generating the mitochondrial-derived signal to activate the UPRmt. We predicted that the cell-non-autonomous induction of mitochondrial chaperones observed with neuronal polyQ40 expression requires one or more essential UPRmt factors. To test this hypothesis, we applied RNAi against ubl-5, dve-1, atfs-1, and clpp-1, and cco-1 to neuronal polyQ40 worms expressing hsp-6p::GFP. RNAi against dve-1, atfs-1, and clpp-1 blocked the induction of the hsp-6p::GFP reporter in peripheral tissues, while cco-1 RNAi showed robust induction (Figures 2A and 2B). ubl-5 knockdown also suppressed the UPRmt in response to neuronal polyQ40, albeit to a lesser extent (Figures 2A and 2B). These expression changes were also assessed using immunoblots for GRP75/HSP6 and GFP from animals harvested prior to reproduction to negate embryonic HSP-6 protein levels (Figure S2A). RNAi-mediated knockdown of atfs-1 and clpp-1 resulted in a relative decrease in HSP6 expression and a robust decrease in the GFP marker of hsp-6 reporter expression. However, RNAi toward dve-1 and ubl-5, while showing a modest decrease in intestinal GFP fluorescence, showed little, if any, reduction in whole-worm GRP75/HSP6 and GFP by immunoblot analysis.
Figure 2. Distal hsp-6 Induction Requires Functional UPRmt Components.

(A) Photomicrographs of rgef-1p::polyQ40; hsp-6p::GFP transgenic worms grown on either empty vector (control), dve-1, atfs-1, clpp-1, or ubl-5 RNAi from hatch. cco-1 RNAi was used as a positive control.
(B) ImageJ GFP fluorescence quantification for EV, dve-1, and atfs-1 RNAi conditions (Mean ± SEM for n = 20 worms/experiment, p < 0.001 for dve-1, atfs-1, and ubl-5 by Student’s t test).
(C) Photomicrographs of rgef-1p::polyQ40;dve-1p::DVE-1::GFP or dve-1p::DVE-1::GFP transgenic animals on EV or cco-1 RNAi (positive control). Images show posterior intestine at 10× and 40× magnification. Arrows indicate neuronal cell bodies or processes and arrowhead indicate DVE-1::GFP puncta.
(D) Quantification of nuclear puncta containing DVE-1::GFP on EV or cco-1 RNAi (positive control). (Mean ± SEM for n = 20 worms/experiment, 3 replicate experiments, p < 0.0001 by Student’s t test for cco-1 RNAi versus EV and p < 0.002 for Q40, dve-1p::DVE-1::GFP versus EV).
As an additional measure of UPRmt induction in peripheral cells, strains expressing a dve-1p::DVE-1::GFP translational fusion gene (Haynes et al., 2007) were used to assess the effects of neuronal polyQ40 on the expression and localization of the dve-1 transcription factor in the intestine. In response to mitochondrial stress, DVE-1 accumulates in the nucleus, serving as an additional proxy for induction of the UPRmt. We found increased nuclear accumulation of dve-1p::DVE-1::GFP in the intestinal cells of animals expressing the neuronal polyQ40 (Figure 2C and 2D). Taken together, cell-non-autonomous induction of the UPRmt caused by neuronal expressed PolyQ40 requires an intact UPRmt signaling pathway.
Neuronally Expressed polyQ40 Physically Interacts with Mitochondria and Affects Mitochondrial Function
Polyglutamine repeats within the Huntingtin (Htt) protein impairs mitochondrial function and morphology through direct interaction with the outer membrane of the mitochondria (Costa and Scorrano, 2012; Panov et al., 2002). We hypothesized that polyQ40 proteins may bind to and thus affect mitochondria in the neurons of C. elegans. To test this possibility, we isolated mitochondrial and cytoplasmic fractions from wild-type and neuronal polyQ40-expressing animals for the presence of polyQ protein binding to the mitochondria. In these analyses, we observed significant polyQ40 protein in both the mitochondrial and cytoplasmic fractions (Figure 3A). Hyper-toxic polyQ67 was, in contrast, only slightly detectable in the mitochondrial fraction (Figure 3A). We confirmed the specificity of our fractionation using antibodies toward cytoplasmic and mitochondrial proteins.
Figure 3. Neuronal polyQ40 Associates with the Mitochondria and Affects Mitochondrial Function and Fitness.

(A) Immunoblot analysis of day 1 adult wild-type, rgef-1p::polyQ40::YFP, or rgef-1p::polyQ67::YFP transgenic animals after fractionation into postmitochondrial supernatant (S) and mitochondrial pellet (M). Anti-GFP recognizes expression of polyQ::YFP in the indicated fractions. The lower band is cleaved YFP. Endogenous NDUFS3 serves as a mitochondrial marker and α-tubulin a cytoplasmic marker.
(B) Oxygen consumption rate in rgef-1p:: polQ19::CFP, rgef-1p::polyQ40::YFP, or rgef-1p::polyQ67::YFP expressing strains. (Mean ± SEM for n = 50 animals, p < 0.032 by Student’s t test of polyQ40 versus N2 animals).
(C) Oxygen consumption rate in the presence or absence of rgef-1p::polyQ40::YFP; in atfs-1(gk3094); hsp-6p::GFP or hsp-6p::GFP animals, (Mean ± SEM p = 0.047 by Student’s t test of atfs-1(gk3094);hsp-6p::GFP versus rgef-1p::polyQ40:: YFP; atfs-1(gk3094) animals).
(D) Survival analyses of atfs-1(gk3094);hsp-6p::GFP and hsp-6p::GFP animals in the presence or absence of rgef-1p::polyQ40::YFP. n = 130–140 worms/condition, for hsp-6p::GFP control median LS = 19 days; Q40, hsp-6p::GFP median LS = 19 days; hsp-6p::GFP, atfs-1(gk3094) median LS = 17 days (n.s. p < 0.49); Q40, hsp-6p::GFP, atfs-1(gk3094) median LS = 15 days, p < 0.0001 by Log Rank test compared to Q40, hsp-6p::GFP).
We repeated these fractionation experiments using Q19 as well as a second cytoplasmic marker, β-actin. As expected, Q19, which cannot induce the cell-non-autonomous response, was not present in the mitochondrial fraction (Figure S3A). Using strains in which Aβ1-42 is expressed in the muscle we found that the high molecular weight species of Aβ1-42 were largely excluded from the mitochondria (Figure S3B), while oligomers were present in small amounts in the mitochondrial fraction. Of note, Aβ expressed in the muscle does not elicit a cell-non-autonomous UPRmt response, and thus, localization may not engage the same mitochondrial stress pathways as in the nervous system. These results suggest that the interaction of polyQ40 with mitochondria in C. elegans may be responsible for its capacity to invoke a cell-non-autonomous mitochondrial stress response.
We tested if there was an overt metabolic consequence of the physical interaction of polyQ40 and mitochondria and cell-non-autonomous induction of the UPRmt. We examined the effects of neuronal expression of polyQ40 on in vivo oxygen consumption rates in whole animals. We found that the neuronal polyQ40 expressing strains had significantly lowered respiration in comparison to wild-type animals, indicative of attenuated mitochondrial function (Figure 3B). In contrast, we observed no effect on oxygen consumption in the strains expressing smaller expansions of polyQ (polyQ19) or the more toxic polyQ67 expansions, neither of which induce the UPRmt or robustly bind to mitochondria when expressed in the nervous system (Figure 3B).
A Functional UPRmt Is Required for the Fitness of polyQ40 Expressing Animals
We next measured respiration in whole animals in the absence of a functional UPRmt. Loss of UPRmt induction in wild-type animals, by mutation of atfs-1, results in lowered respiration rates. We found that the expression of neuronal polyQ40 in combination with mutant atfs-1 had a synergistic effect on respiration rates, displaying even further reduced rates of oxygen consumption compared to either perturbation alone or to the reporter control (Figure 3C). In parallel, we conducted lifespan analyses of the neuronal expressing polyQ40 worms. While the lifespan of polyQ40 animals with a functional UPRmt was comparable to that of a wild-type animal, in the presence of an atfs-1(gk3094) mutation, animals exhibited a significant decline in fitness (Figure 3D). Importantly, the atfs-1(gk3094) mutation alone did not significantly reduce lifespan. Repeated trials of these experiments are reported in Table S1. Because atfs-1 has been shown to affect the transcription of genes involved in the respiratory chain, TCA cycle, mitochondrial chaperones and proteases, we performed qPCR for subsets of these genes in polyQ strains (Figure S3C) (Nargund et al., 2015). These experiments did not reveal any obvious patterns in changes to genes expression; however, this was only a subset of genes associated with mitochondrial function and was performed in whole worms, which may mask changes in individual tissues. Collectively, these data suggest that a loss in UPRmt function is sufficient to exacerbate the loss in fitness caused by neuronal proteotoxic stress, with a synergistic effect on fitness of atfs-1 mutation with mitochondrial dysfunction, as has been described previously (Nargund et al., 2015).
Non-autonomous UPRmt Signaling Requires unc-31 Mediated Neurosecretion
We have found that neuronal expressed polyQ40 binds to and affects the physiology of mitochondria, creating a cell-non-autonomous upregulation of the UPRmt in distal tissues. Because the nervous system does not innervate the intestine, where UPRmt is induced, we hypothesized that neuronal mitochondria are capable of promoting the release of a secreted, metabolic signal in response to misfolding protein stress. To examine the functional connection between neuroendocrine secretion and UPRmt signaling, mutant strains defective in components of both synaptic transmission machinery and dense core vesicle (DCV) release were examined for effects upon peripheral induction of the UPRmt. Mutations in unc-31 (encoding the calcium activator protein for secretion) selectively block DCV secretion (Charlie et al., 2006; Speese et al., 2007). In contrast, mutations in unc-13 and unc-18 (encoding diacylglycerol-binding protein and sec-1 respectively) result in reduced neurosecretion from the synaptic cleft (Gengyo-Ando et al., 1993; Richmond et al., 1999; Tokumaru and Augustine, 1999).
In our analyses, we found that unc-31(e928) mutation was able to suppress cell-non-autonomous UPRmt induction (Figure 4A). In contrast, mutations that block small molecule neurosecretion, unc-13(e1091) or unc-18(e81), had no effect on the mitochondrial stress response to polyQ40 expression in the nervous system (Figure 4A). The unc-31(e928) mutation also affects dense core vesicle release from the intestine. However, intestinal expression of unc-31 was not capable of restoring hsp-6p::GFP expression (Figure S2A). These data indicate that unc-31 is required in the nervous system to mediate neuronal secretion of a mitochondrial stress signal.
Figure 4. Non-autonomous Induction of hsp-6p::GFP Requires unc-31 Mediated Neurosecretion.

(A) Photomicrographs depicting hsp-6p::GFP reporter response in rgef-1p::polyQ40::YFP; hsp-6p::GFP animals in combination with unc-31(e928), unc-13(e1091), or unc-18 (e81) mutations.
(B) Photomicrographs depicting unc-31(e928); rgef-1p::polyQ40::YFP; hsp-6p::GFP animals treated with 5 mM serotonin (5-HT) or with vehicle control.
(C) ImageJ quantification of the hsp-6p::GFP expression from unc-31(e928);rgef-1p::polyQ40:: YFP; hsp-6p::GFP animals after the application of serotonin, dopamine, octopamine, or tyramine (Mean ± SEM for n = 20 animals/experiment, p < 0.01 by Student’s t test for serotonin versus vehicle treatment).
(D) ImageJ quantification of hsp-6p::GFP fluorescence following the application of dopamine, octopamine, and tyramine to hsp-6p::GFP animals in the absence of PolyQ40 expression (Mean ± SEM for n = 20 animals/experiment, all conditions were n.s. compared to vehicle control).
Serotonin Is Necessary for the Cell-Non-Autonomous Signaling of Neuronal Mitochondrial Stress
We hypothesized that neuroendocrine signals released from unc-31-derived dense core vesicles might mediate the cell-non-autonomous signaling of mitochondrial proteotoxic stress. Imaging of the neurons in the polyQ40 and reporter strains did not reveal consistent differences in neuronal reporter expression and was not immediately useful in directing our approach to identifying certain neurotransmitter or neuromodulator systems (Figure S4B). Thus, we directly examined the role of nutrient responsive, biogenic amines, known cargo of DCVs, in the non-autonomous induction of the UPRmt. We performed a candidate screen against a panel of biogenic amines that included serotonin, dopamine, octopamine, and tyramine to determine whether any of these compounds could rescue peripheral hsp-6 reporter expression in unc-31(e928);rgef-1p::polyQ40 animals. Surprisingly, the addition of serotonin, but not other biogenic amines, partially rescued hsp-6p::GFP induction caused by neuronal polyQ40 expression in the unc-31(e928) mutant background (Figures 4B–4D). This suggests that serotonin acts downstream of unc-31 to mediate the cell-non-autonomous UPRmt induction. The addition of serotonin specifically activated the UPRmt in the polyglutamine model and had no effect on the expression of hsp-4/BiP (ER stress response) or hsp-16.2 (cytosolic stress response) (Figure S2B).
Consistent with a role for serotonin in mediating the response to mitochondrial stress in the nervous system, mutations in tryptophan hydroxylase (tph-1), a key enzyme for serotonin synthesis, suppressed the induction of the hsp-6p::GFP reporter in the neuronal polyQ expressing animals (Figures 5A and 5B). Importantly, the addition of serotonin rescued the suppression caused by the tph-1 mutation (Figures 5C and 5D), whereas the addition of other biogenic amines did not (Figure 5E). As a control for the specificity of serotonergic signaling to neuronal and cell-non-autonomous induction of the response, we exposed tph-1 mutant animals to cell-autonomous stressors, namely paraquat and cco-1 RNAi. These strains showed the same level of induction of hsp-6::GFP as controls, indicating that serotonin is required specifically for neuronal initiation of this response (Figure S4D). Collectively, these data support a role for the secretion of serotonin in cell-non-autonomous communication of mitochondrial stress.
Figure 5. Serotonin Is Necessary for the Cell-Non-Autonomous Signaling of Neuronal Mitochondrial Stress.

(A) Photomicrographs depicting hsp-6p::GFP reporter induction in rgef-1p::polyQ40::YFP; hsp-6p::GFP;tph-1 (mg280) and rgef-1p::polyQ40:: YFP; hsp-6p::GFP animals.
(B) Biosorter fluorescence measurement from (A) (Mean ± SEM for 150–1,000 worms/experiment, p = 0.001 by Student’s t test).
(C) Photomicrographs depicting hsp-6p::GFP reporter induction in rgef-1p::polyQ40::YFP; hsp-6p::GFP;tph-1(mg280) animals treated with 5 mM serotonin (5-HT) or with vehicle control.
(D) ImageJ fluorescence measurement from (C) (Mean ± SEM for 30–100 worms/experiment, p = 0.023 by Student’s t test for 5-HT versus vehicle treatment).
(E) Biosorter quantification of the hsp-6p::GFP expression from tph-1(mg280);rgef-1p::polyQ40:: YFP; hsp-6p::GFP animals after the application of serotonin, dopamine, octopamine, or tyramine (Mean ± SEM for n = 200–400 animals/experiment, p < 0.0001 by Student’s t test for 5-HT versus vehicle treatment, all others n.s.).
Additional Models of Neuronal Mitochondrial Stress Signal to the Periphery in a Serotonin-Dependent Manner
Intrigued by the requirement for serotonin to elicit a peripheral response to PolyQ40 induced mitochondrial stress in the nervous system, we asked if other models of neuronal mitochondrial stress could induce the UPRmt in the periphery and whether they also require serotonergic signaling. We found that a mitochondrial localized KillerRed construct, which generates high levels of localized ROS, expressed solely in the nervous system, but not other cell types, could induce a robust induction of the UPRmt in the periphery (Figures 6A and 6B). Furthermore, using neuron specific expression of the Cas9 endonuclease, we found that mutation of either the AAA-ATPase, spg-7, or the complex IV subunit, cco-1 (Figure S5), which have been reported to induce the UPRmt (Durieux et al., 2011; Haynes et al., 2010), resulted in robust induction of the hsp-6p::GFP reporter in peripheral cells (Figure 6C). In both of these models, UPRmt induction was primarily observed in the intestine, as in the neuronal polyQ system.
Figure 6. Alternative Models of Neuronal Mitochondrial Stress Also Require Serotonin to Induce Peripheral UPRmt.

(A) Photomicrographs depicting hsp-6p::GFP reporter induction in unc-119p::tom20::KillerRed or rab-3p::tom20::KillerRed animals
(B) Photomicrographs depicting hsp-6p::GFP reporter induction in gly-19p::tom20::KillerRed, ges-1p::tom20::KillerRed, or myo-3p::tom20::KillerRed animals.
(C) Photomicrographs depicting hsp-6p::GFP reporter induction in rab-3p::Cas9+u6p::spg-7-sg, unc-119p::Cas9+u6p::spg-7-sg, rab-3p::Cas9+u6p::cco-1-sg, or unc-119p::Cas9+u6p::cco-1-sg animals.
(D) Photomicrographs depicting hsp-6p::GFP reporter induction in tph-1(mg280); rab-3p::Cas9+u6p::spg-7-sg animals.
(E) Quantification of hsp-6p::GFP reporter induction in tph-1(mg280); rab-3p::Cas9+u6p::spg-7-sg animals. (Mean ± SEM for n = 30–60 animals/experiment, 2 replicate experiments, p < 0.05 by ANOVA test for tph-1 mutant)
(F) Photomicrographs depicting hsp-6p::GFP reporter induction in tph-1p::Cas9+u6p::cco-1-sg animals.
We next asked if these mitochondrial stress models also required serotonin to signal the UPRmt in distal cells. We found that loss of serotonin, by mutation of tph-1, blocked induction of the UPRmt in the spg-7 neuronal mutant model (Figures 6D and 6E). Additionally, mutation of spg-7 solely in serotonergic neurons, using the tph-1 promoter, was sufficient to induce the UPRmt in peripheral cells (Figures 6F). Taken together, neuronal expression of either PolyQ40, mitochondrial localized KillerRed, or loss of spg-7 in neurons results in mitochondrial stress that can be communicated to distal cells through the action of serotonin.
DISCUSSION
In this study, we uncover a role for active metabolic signaling in the establishment of the cell-non-autonomous communication of neuronal proteotoxic stress. The result of this signal is a coordinated change in mitochondrial homeostasis across tissues, propagating the synchronization of whole-organism metabolism. We also found that the communication of proteotoxic stress from neuronal mitochondria to peripheral tissues requires both serotonergic signaling and a functional UPRmt, establishing a mechanistic link between the metabolic disturbances seen in neurodegenerative disease and the remodeling of mitochondrial function by a biogenic amine (Figure 7A). We further find that serotonergic signaling is a common requirement for cell-non-autonomous communication of several types of neuronal mitochondrial stress.
Figure 7. Model of Cell-Non-Autonomous Communication of Mitochondrial Stress by Serotonin Signaling in Response to Neuronal polyQ40 Expression.
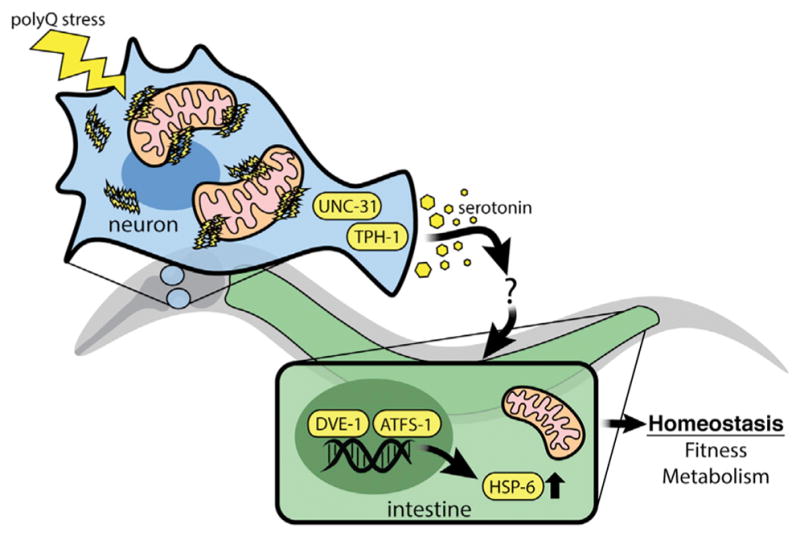
PolyQ40 specifically binds to mitochondria in neurons, initiating a signaling cascade across tissues that requires neuronal unc-31, tph-1, serotonin and functional components of the UPRmt to ensure homeostasis for the organism.
Surprisingly, while the polyQ proteins tested here accumulate mainly in the cytosol, no activation of the cytoplasmic stress response was found in distal tissues. Instead, we found that polyQ40 was present in mitochondrial fractions and acted specifically on the UPRmt. Our fractionation experiments indicate that polyQ40 accumulates at high levels in the neurons (Figure 3A), which may also play a role in its association with mitochondria. Additionally, only neuronal polyQ40, and not other lengths of polyglutamine, nor other aggregative proteins, exhibit association with mitochondria. It is intriguing that the results of PolyQ40 expression in the nervous system could be recapitulated with disruptions targeted specifically to the mitochondria, such as the ROS generator KillerRed, loss of the AAA-ATPase, spg-7, or loss of a complex IV subunit, cco-1. However, it is not clear what the common perturbation is among each of these neuronal specific mitochondrial stress models.
Studies of the cell-autonomous regulation of the UPRmt response suggest that there are numerous means of inducing the canonical mitochondrial stress response. These include UPRmt activation due to peptide accumulation within the mitochondrial matrix, which depends on the mitochondrial localized proteins ClpP and HAF-1, and the transcription factors DVE-1 and UBL-5 (Haynes et al., 2007; 2010). Additionally, impairments in mitochondrial import and the subsequent loss of membrane potential block ATFS-1 from import into mitochondria and result in its accumulation in the nucleus where it acts to induce the UPRmt response (Haynes et al., 2010; Nargund et al., 2012). We find that polyQ40, as opposed to other lengths of polyglutamine, when expressed in neurons, is localized to the outer mitochondrial membrane and may thus impact mitochondrial import and/or membrane potential. In mammalian cell culture more toxic lengths of polyQ bind to the mitochondrial membrane and act to perturb mitochondrial Ca2+ signaling, which is closely linked to perturbations in membrane potential (Panov et al., 2002). While the above may explain the mechanism of toxicity in the neurons, the perception of the signal and peripheral induction of the UPRmt also require various regulatory factors. Interestingly, the UPRmt can be activated in C. elegans and in mice in response to mito-nuclear translational imbalance (Houtkooper et al., 2013) as well as by several epigenetic mediators (Merkwirth et al., 2016; Tian et al., 2016). These pathways, in conjunction with common serotonin-dependent signaling, may provide a mechanistic link to the changes seen in distal tissues.
For decades, clinicians have reported severe changes in metabolism, mood, and behavior in HD patients, symptoms that often precede more obvious effects of the disease on motor function and implicate serotonergic systems (Du and Pang, 2015; Folstein and Folstein, 1983; Wang et al., 2014). In mammalian models of HD, dysfunction of serotonergic signaling pathways has been reported, with both tryptophan hydroxylase activity and 5-HT levels diminishing (Mattson et al., 2004; Pang et al., 2009). Our data suggest that a change in the regulation of serotonin signaling may occur as an indirect response to mitochondrial dysfunction. Serotonergic circuitry mediates the behavioral response not only to environmental nutrient supply, but also to environmental toxins such as pathogenic bacteria, xenobiotics, and RNAi (Melo and Ruvkun, 2012; Sze et al., 2000; Zhang et al., 2005). Recently, Tatum et al. also reported serotonin signaling to be implicated in the heat shock response in C. elegans (Tatum et al., 2015). Serotonin thus may act to integrate multiple signals about the environment with the current physiological nutrient and stress threshold of the organism.
Similarly, while neuronal mitochondrial dysfunction may affect serotonin synthesis or release, serotonin must then affect mitochondrial function in distal tissues. Serotonin acts locally upon its release and is not predicted to directly elicit long-range effects. Therefore, it is possible that release of serotonin from affected neurons signals to peripheral tissues through a yet to be indentified secondary cell to release a longer range acting neurocrine. Serotonin has been shown to regulate feeding behavior indirectly through release of kynurenic acid, a breakdown metabolite of tryptophan, which also plays a protective role in multiple neurodegenerative disease by a cell-non-autonomous mechanism (Campesan et al., 2011; Lemieux et al., 2015; Zwilling et al., 2011). Additionally, chronic administration of 5-HT to C. elegans leads to reduced fat content, dependent upon mitochondrial beta-oxidation genes. This regulation of fatty acid oxidation in the intestine occurs through neuroendocrine signaling, specifically by regulation of octopamine release and subsequent action of NHR-76, a nuclear hormone receptor, on the intestine (Noble et al., 2013; Srinivasan et al., 2008). Thus, serotonin influences metabolism and mitochondrial function distally, acting to regulate the action of secondary signaling molecules such as kynurenic acid, neuropeptides, and nuclear hormone receptors.
Finally, one of the most intriguing observations to arise from this data comes from its further support for a model in which the UPRmt carefully orchestrates the regulation of protein homeostasis with the regulation of a metabolic state (Baker et al., 2012; Haynes and Ron, 2010; Nargund et al., 2015). During mitochondrial dysfunction, the transcription factor ATFS-1 binds to and represses the new synthesis of transcripts that encode for oxidative phosphorylation and the TCA cycle (Nargund et al., 2015). Simultaneously, however, it promotes the appropriate assembly of OxPhos complexes as well as an array of protective chaperones and proteases designed to increase the health of the mitochondrial proteome. In such a model, mitochondrial protein homeostasis and metabolism become inextricably linked. Here, we demonstrate how the connection between protein homeostasis and metabolism is perpetuated throughout the entire organism, establishing both proteostasis and metabolic health across tissues.
It is possible that the distal response is merely a communication to distal tissues of mitochondrial damage in the neurons rather than an adaptive, protective response. Generally, non-autonomous upregulation of stress responses, either through reductions in signaling as with insulin and thermoregulatory circuits or through enhanced signaling as with the UPRER, have been thought of as beneficial for the organism (Alcedo and Kenyon, 2004; Durieux et al., 2011; Prahlad and Morimoto, 2011). The changes in physiology observed here suggest that the UPRmt response is in fact adaptive, with the purpose of maintaining homeostasis. We see a reduction in mitochondrial oxidative capacity in the polyQ strain that most robustly induces the UPRmt. Reduced oxygen consumption was shown previously to correlate with the extended lifespan and induced UPRmt associated with doxycycline treatment (Houtkooper et al., 2013). Somewhat counterintuitively, we see a further decline in oxidative capacity with loss of a functional UPRmt through atfs-1 mutation. We hypothesize that this further synergistic decline then becomes detrimental for the organism. Similarly, a significant decrease in lifespan was seen with polyQ40 strains in the atfs-1 mutant background, indicating that the ability to induce the UPRmt does confer a benefit in maintaining the lifespan of the organism. It would seem that a mild upregulation of stress responses under chronic stress might be more adaptive than maintaining the constant high level of activation that has been shown to have adverse effects on lifespan and physiology (Feder et al., 1992; Lamech and Haynes, 2015). Ultimately, if protective, maintaining these responses in the background of increasing toxicity may prove therapeutic in numerous age-related neurodegenerative diseases.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GFP (Mix) | Roche | CAT#11814460001; RRID: AB_390913 |
| Rabbit polyclonal anti-GRP75 (mtHSP6) | Santa Cruz | CAT#sc-13967; RID: AB_647720 |
| Mouse monoclonal anti-alpha-Tubulin | Sigma | CAT#T6074; RRID: AB_477582 |
| Mouse monoclonal anti-beta-actin | Abcam | CAT#Ab8224; RRID: AB_449644 |
| Mouse monoclonal anti-NDUFS3 | MitoSciences/Abcam | CAT#ab110246; RRID: AB_10861972 |
| Rabbit polyclonal anti-histone H2A | Abcam | CAT#Ab18255; RRID: AB_470265 |
| Mouse monoclonal anti-β-amyloid, 1-16antibody | Covance | CAT#SIG-39320; RRID: AB_2313952 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Trypton peptone source | EMD Millipore | |
| 5-HT hydrochloride powder | Sigma | H9523-CAS153-98-0 |
| Dopamine | Sigma | H8502-CAS62-31-7 |
| Tyramine | Sigma | T2879-CAS60-19-5 |
| Octopamine | Sigma | 68631-CAS770-05-8 |
| Methyl viologen dichloride hydrate | Sigma | 856177-CAS75365-73-0 |
| Levamisole | Sigma | Y0000047-CAS16595-80-5 |
| Critical Commercial Assays | ||
| ECL Western blotting kit | AbCam | Ab133406 |
| XFe96 extracellular flux assay kit | Seahorse/Agilent | CAT#102601-100 |
| Experimental Models: Cell Lines | ||
| Human: primary normal human dermal fibroblasts (NHDF) | Lonza | CC-2511 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: AM23 (rmIs19 [rgef-1p::Q19::CFP]) | (Brignull et al., 2006) | N/A |
| C. elegans: AM101 (rmIs101 [rgef-1p::Q40::YFP]) | (Brignull et al., 2006) | N/A |
| C. elegans: AM716 (rmIs176 [rgef-1p::Q67::YFP]) | (Brignull et al., 2006) | N/A |
| C. elegans: snb-1p::TDP-43 Q133K | (Zhang et al., 2011) | N/A |
| C. elegans: MT15434, tph-1(mg280)II | Supriya Srinivasan | WormBase WBVar00088923 |
| C. elegans: SJ4100 (zcIs13[hsp-6p::GFP]) | Caenorhabditis Genetics Center | WB Strain: SJ4100 |
| C. elegans: CL2070 (dvIs[hsp-16.2p::GFP]) | Caenorhabditis Genetics Center | WB Strain: CL2070 |
| C. elegans: SJ4005 (zcIs[hsp-4p::GFP]) | Caenorhabditis Genetics Center | WB Strain: SJ4005 |
| C. elegans: CF1553 (muIs84[sod-3p::GFP]) | Caenorhabditis Genetics Center | WB Strain: CF1553 |
| C. elegans: AM141 (rmIs141 [unc-54p::Q40::YFP]) | Caenorhabditis Genetics Center | WB Strain: AM141 |
| C. elegans: GF80 (dgEx80[gly-19p::Q40::GFP], RF4 rol-6) | Caenorhabditis Genetics Center | WB Strain: GF80 |
| C. elegans: SJ4197 (zcIs39[dve-1p::DVE-1::GFP]) | Caenorhabditis Genetics Center | WB Strain: SJ4197 |
| C. elegans: unc-31 (e928)IV | Caenorhabditis Genetics Center | WormBase: WBVar00143608 |
| C. elegans: unc-13 (e1091)I | Caenorhabditis Genetics Center | WormBase: WBVar00143733 |
| C. elegans: unc-18 (e81)X | Caenorhabditis Genetics Center | WormBase: WBVar00142951 |
| C. elegans: CL2006 (dvIs [unc-54p:: Aβ1-42]) | Caenorhabditis Genetics Center | WB Strain: CL2006 |
| C. elegans: N2 | Caenorhabditis Genetics Center | WormBase: N2 |
| C. elegans: rgef-1p::Q40::HA; myo-2p::RFP | This paper | N/A |
| C. elegans: rgef-1p:: Aβ1-42; RF4 rol-6 | This paper | N/A |
| C. elegans: gly-19p::unc-31 cDNA | This paper | N/A |
| C. elegans: atfs-1(gk3094) | Caenorhabditis Genetics Center | WormBase: WBVar00601112 |
| C. elegans: rab-3p::Tom20::KillerRed, odr-1p::DsRed | This paper | N/A |
| C. elegans: ges-1p::Tom20::KillerRed, odr-1p::DsRed | This paper | N/A |
| C. elegans: gly-19p::Tom20::KillerRed, odr-1p::DsRed | This paper | N/A |
| C. elegans: myo-3p::Tom20::KillerRed, odr-1p::DsRed | This paper | N/A |
| C. elegans: unc-119p::Tom20::KillerRed, odr-1p::DsRed | This paper | N/A |
| C. elegans: unc-119p::Cas9+u6p::spg-7-sg, odr-1p::DsRed | This paper | N/A |
| C. elegans:tph-1p::Cas9+u6p::cco-1-sg, odr-1p::DsRed | This paper | N/A |
| Recombinant DNA | ||
| Lentivirus polyQ | Proteosasis Inc. | Gift |
| Q40::YFP plasmid pPD30.38 | (Brignull et al., 2006) | N/A |
| rgef-1p::Q40::HA, pJKD101 | This paper | N/A |
| gly-19p::unc-31 cDNA plasmid | This paper | N/A |
| pDONR221. tdKillerRed plasmid | (Williams et al., 2013) | N/A |
| gly-19p::TOM20::KillerRed | This paper | N/A |
| myo-3p::TOM20::KillerRed | This paper | N/A |
| unc-119p::TOM20::KillerRed | This paper | N/A |
| ges-1p::TOM20::KillerRed | This paper | N/A |
| rab-3p::TOM20::KillerRed | This paper | N/A |
| tph-1p::Cas9+u6p::cco-1-sg pDD162 | This paper | N/A |
| rab-3p::Cas9+u6p::spg-7-sg pDD162 | This paper | N/A |
| unc-119p::Cas9+u6p::spg-7-sg pDD162 | This paper | N/A |
| Sequence-Based Reagents | ||
| See Table S2 for all Primer sequences | N/A | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author, Andrew Dillin (dillin@berkeley.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans Maintenance and Transgenic Lines
AM23 (rmIs19 [rgef-1p::Q19::CFP]), AM101 (rmIs101 [rgef-1p::Q40::YFP]), and AM716 (rmIs176 [rgef-1p::Q67::YFP]) were a generous gift from Rick Morimoto (Brignull et al., 2006). snb-1p::TDP-43 Q133K were a gift from J Wang and MT15434, tph-1(mg280)II, a gift from Supriya Srinivasan. SJ4100 (zcIs13 [hsp-6p::GFP]), CL2070 (dvIs [hsp-16.2p::GFP]), SJ4005 (zcIs [hsp-4p:: GFP]), CF1553 (sod-3p::GFP), AM141 (rmIs141 [unc-54p::Q40::YFP]), GF80 ([gly-19p::Q40::GFP], RF4 rol-6]), SJ4197 (zc[dve-1p:: DVE-1::GFP]), unc-31 (e928)IV, unc-13 (e1091)I, unc-18 (e81) X, CL2006 (dvIs [unc-54p:: Aβ1-42]), and N2 strains were obtained from the Caenorhabditis Genetics Center. Nematodes were handled using standard methods (Brenner, 1974). To generate animals for the stress reporter assays, we crossed the AM23, AM101, and AM716 strains with the SJ4100, CL2070, SJ4005, CF 1553, and SJ4197 strains. The AM101; SJ4100 strain was then used for all other crosses.
For generation of rgef-1p::Q40::HA strain the pPD30.38 Q40YFP plasmid was used, which was a generous gift from Rick Morimoto. The YFP and unc-54 3′UTR from the plasmid were removed using the AgeI and SpeI cut sites. A fragment containing the HA tag with stop codon followed by the unc-45 3′UTR flanked by AgeI and SpeI sites was generated by PCR amplification from pNB8. T4 ligation resulted in rgef-1p::Q40::HA, pJKD101. Transgenic lines were generated by microinjecting plasmid DNA containing 50ng/ul pJKD101, 50ng/ul pRF4, a rol-6 marker, and 50ng/ul pPD61 as filler DNA. The Aβ insert without signal sequence, was amplified by PCR with the KpnI and BamHI restriction sites and inserted in to the polyQ site on the plasmid. Finally, the gly-19p::unc-31 cDNA plasmid was generated by PCR amplifying the unc-31 cDNA from a plasmid given us by Ken Miller’s lab. The KpnI and AgeI sites were used to insert the cDNA in a gly-19 promoter plasmid. All lines were made as above, except the Aβ lines which was co-injected with 100ul/mg of the pRF4, rol-6 plasmid.
For generation of Tom20::KillerRed strain, the Tom20::KillerRed sequence was PCR amplified from a plasmid provided by Marc Hammarlund (Williams et al., 2013) and inserted into vectors containing myo-3 or unc-119 promoter and unc-54 3′UTR sequence. The ges-1, gly-19, rab-3 and tph-1 promoters were PCR amplified from genomic DNA and cloned into the corresponding vector to replace unc-119 promoter. Transgenic lines were generated by microinjecting plasmid DNA containing 35ng/ul KillerRed construct and a odr-1p::DsRed co-injection marker.
Primary BJ Fibroblasts
For cell culture experiments, primary BJ fibroblasts were transfected with lentivirus constructs containing PolyQ repeats, a generous gift from Dan Garza at Proteostasis Therapeutics, Inc. Cells were maintained in DMEM supplemented with 10% FBS, 1% Glutamax and 2 ug/mL blastacistidin for selection. Western blots of primary BJ fibroblasts were prepared from cell lysates 5 or 10 passages post-lentiviral infection. Cells were trypsinized, washed in PBS, pelleted and snap frozen in liquid nitrogen. Cells were resuspended in RIPA buffer and lysed using a 10 g syringe. Western blotting was performed as above.
METHOD DETAILS
Imaging and Analysis
For microscopy experiments, worms were anesthetized using 0.1 mg/mL levamisole hydrochloride in M9. Unless otherwise noted, all animals were day 2 adult hermaphrodites and experiments were performed in triplicate with 10–20 animals imaged per experiment. Worms were grown from hatch on HT115 bacteria when RNAi was used.
For whole-animal imaging, worms were imaged using a Leica MZ 16F dissecting scope with a 1x objective, 8x magnification. Magnified images of the intestines and neurons were produced using a Leica CTR 6000 microscope with 40× or 63× objectives respectively. All images were taken using a fixed exposure and gain to just below saturation for the intestinal fluorescence. To quantify GFP fluorescence, intestinal regions were outlined and quantified using ImageJ software. All measurements were expressed relative to background levels in the appropriate control non-GFP fluorescent strain, or to EV or vehicle control for RNAi and pharmacologic experiments. For quantifying the localization and intensity of the dve-1 fusion reporter, worms were mounted on 2% agarose pads with levamisole and photographs were taken using a Zeiss Axiovert microscope and AxioCam.
Relative fluorescence was also assayed using a Copas Biosort. Worms were grown to day 1 or day 2 of adulthood on standard NGM or RNAi plates. Animals were washed off plates using M9 and aliquoted into a 96 well plate. GFP fluorescence was read at PMT475. Worms below a length threshold of 250 Time of Flight (length) were excluded from analysis.
Protein Assays and Mitochondrial Fractionations
Samples obtained from animals cultured on large plates were washed with M9 and resuspended in 1X TBS containing 0.1% NP40 plus the Roche protease inhibitor cocktail. Samples were then subjected to glass bead homogenization using a Precellys 24 at 3,000 rpm for 3 cycles at 90 s. Samples were spun down on a tabletop centrifugation at 4°C for 10 min at maximum spin to separate the resulting supernatant from the cellular debris. Protein quantification was performed by BCA, and samples were normalized for total protein content. The standard NuPAGE Bis-Tris protocol was then followed, before western blotting with 1:1,000 Roche Anti-GFP or Anti GRP-75(mtHSP6) at 1:1,000 antibody overnight. Secondary antibody concentration was 1:10,000 and membranes were visualized using standard AbCam ECL kit, then stripped and re-probed for alpha tubulin at 1:2,000.
Cell fractionations were prepared as described in (Haynes et al., 2010). 60,000–100,000 synchronized worms were raised on large plates with a concentrated OP50 lawn until day 1 of adulthood and then washed off and spun down. These worms were then processed immediately for biochemistry. NDUFS3 antibodies 1:3,000 from MitoSciences and H2A antibodies at 1:2,000 from AbCAM were used for mitochondrial and nuclear blots, as well as the antibodies listed above. For the Aβ blots, Covance 6E10 antibody was used at 1:3,000 dilution. All Westerns were performed in duplicate or triplicate.
Metabolic studies were conducted using the Seahorse XFe instrument with XFe 96 extracellular flux assay kit. Approximately 100–200 worms were picked to new NGM plates with no bacteria. Next, they were washed with M9 and aliquoted 10/well into the XF cell culture 96 well plate. Experiments were repeated three times with 5–10 wells per condition. Oxygen consumption rates were read using the following program: 5 cycles of mix 30 s, wait 30 s, measure 3:00 min.
Pharmacologic Treatments
5-HT hydrochloride powder (Sigma) was dissolved in water to a concentration of 0.1 M and added to standard NGM plates made with tryptone as the peptone source (EMD Millipore). We found inconsistency of the composition of other peptone sources and lot-to-lot variability could severely dampen the reporter induction. Therefore, tryptone was used throughout all experiments. Plates were seeded with E. coli strain HT115 and drug added at a concentration of 0.5 mM, 5 mM, 10 mM, or 50 mM initially. Of note, for unc-31 mutant experiments, OP50 E. coli were used as hsp-6::GFP suppression in these strains was particularly sensitive to external conditions and nutrient source. 5 mM 5-HT was determined to be the optimal concentration for reporter induction and was used for all further experiments unless otherwise noted. Plates were allowed to equilibrate overnight and used the next day. For vehicle-treated controls, water was added to plates. Synchronized L2/L3-stage animals were plated on vehicle- or 5-HT treated plates, and imaging of hsp-6p::GFP reporter induction was measured at day 1 and day 2. For day 2 measurements, worms were transferred to fresh serotonin plates. Dopamine, tyramine, and octopamine were administered by the same methods dissolved in water at concentrations of 100 mM for dopamine and 5 mg/mL for tyramine and octopamine. These optimal concentration ranges were determined from the literature (Alkema et al., 2005; Dempsey et al., 2005), but a range of concentrations were tested: 0.5, 2, 5.0, 10, and 50 mM for 5-HT, 1, 10, and 100 mM for DA, and 0.5, 5, and 50 mg/mL for Tyramine and Octopamine.
For paraquat experiments, methyl viologen (Sigma) was dissolved in 300ul water was added to NGM plates spotted with OP50 and allowed to equilibrate overnight to a final concentration of 0 mM (control) or 2.5 mM. Gravid worms were synchronized by hypocholorite treatment to collet eggs. Approximately 50 L3 worms were transferred to paraquat plates and allowed to grow to day 2 of adulthood at 20C. Worms were immobilized with 0.25 mM levamisole and imaged in the GFP channel using a Leica M165 FC fluorescence dissecting microscope at 8× magnification.
Lifespan Analysis
Lifespan assays were conducted on agar plates at 20°C as described previously (Dillin et al., 2002). Experimental groups contained 100 to150 animals, and animals were grown on OP50 from hatch. We used the prefertile period of adulthood as t = 0 for lifespan analysis. The data presented in the accompanying figures are from one of the 3 biological replicates performed for every group.
RNA Extraction and RT-QPCR
Total RNA was isolated from synchronized populations of approximately 1,000 animals for each strain. Total mRNA was extracted using freeze cracking with QIAzol reagent and subsequent chloroform treatment, before undergoing ethanol and isopropanol wash steps. mRNA underwent column purification on QIAGEN RNAeasy columns. cDNA was created using the Quantitec Reverse Transcriptase kit according to standard protocol (QIAGEN). SybrGreen real-time QPCR experiments were performed as described in the manual using Applied Biosystems QuantStudio 6 Flex and cDNA at a 1:20 dilution. All QPCR experiments were normalized to the geometric mean of cdc-42, pmp-3 and Y45F10D.4 mRNA levels quantified as described previously in (Hoogewijs et al., 2008). The sequences for the primers used were obtained from Nargund et al., 2015.
CRISPR-Cas9 Knockout of spg-7
The CRISPR-Cas9 expression vectors were constructed by replacing the eft-3 promoter in pDD162 (Addgene, #47549) (Dickinson et al., 2013) with the C. elegans tissue specific promoters. CRISPR design tool (http://tools.flycrispr.molbio.wisc.edu/targetFinder/) was used to select the specific targets, and the uniqueness of the target sequences was confirmed by performing a BLAST search of each sequence against the C. elegans genome.
spg-7 target sequence: ACCGAATTTCTCAGCTGCTT
cco-1 target sequence: ATCCACTTGAGCACGCTAC
Transgenic lines were generated by microinjecting plasmid DNA containing 50ng/ul CRISPR-Cas9 construct and 30ng/ul odr-1p::DsRed co-injection marker.
Molecular Analysis of the Mutations in the Conditional Knockouts
The T7EI assay (Cong et al., 2013) was performed to detect the indels produced by spg-7-sg. We PCR-amplified a 680bp DNA fragment containing the spg-7 target site. After T7EI (NEB #M0302) digestion, the PCR fragment from the unc-119p::Cas9+u6p::spg-7-sg worms with peripheral UPRmt was cut into two fragments (600bp and 80bp, while 80bp band was not shown), but remaining intact in control worms. To sequence the deletion, PCR fragment was cloned into pEASY-T1 Vector (Transgen #CB111), and individual colonies were sequenced.
Primers for identifying spg-7 indels:
Forward: TGTTTGCGCAGTGCATGATT
Reverse: AAAATACGGCCCGGGAAACC
Quantification of Non-autonomous UPRmt Induced by Neuronal spg-7 Knockout
Synchronized L1 worms were raised at 20°C on nematode growth medium plates seeded with E. coli OP50. The animals with intestinal UPRmt over the half of full-length intestine were scored when they reached adult stage. Over 30 transgenic animals were counted for wild-type or each mutant strain.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters, including the exact value of n and descriptive statistics (mean ± SEM) and statistical significance are reported in the Figures and the Figure Legends. Data are judged to be statistically significant when p < 0.05 by two-tailed Student’s t test. In figures, asterisks denote statistical significance as calculated by Student’s t test (*, p < 0.05, **, p < 0.001, ***, p < 0.0001) as compared to appropriate controls. Lifespans were analyzed using PRISM 6 software to determine median survival and percentiles. All p values were calculated using log-rank analysis. For experiments using the COPAS biosorter for fluorescence quantification, relative GFP fluorescence was normalized to extinction (thickness) and TOF. When required, YFP fluorescence contribution was accounted for by determining the constant YFP contribution in the GFP channel reading in AM101 worms and removing it from the total GFP reading of KB08 using the equations: G = (G′−kY′)/(1−ck) where Y = total of YFP protein, Y′ = YFP reading, c = constant GFP value in worms without YFP, G = total GFP protein, G′ = GFP reading, and k = constant YFP value in worms without GFP. All statistical analysis was done in PRISM 6.
Supplementary Material
Highlights.
Across species, poly-glutamine repeat proteins bind to and influence mitochondria
PolyQ and other neuronal mitochondrial stress induce a cell-non-autonomous UPRmt
UPRmt pathway components are required for sensing PolyQ-induced neuronal stress
Serotonin synthesis and secretion are required for non-autonomous UPRmt induction
Acknowledgments
We thank Dr. H.-E. Kim and M. Simic for assistance with cell culture and C. elegans subcellular fractionations. We are grateful to Drs. S. Srinivasan and R. Morimoto for their generous gifts of strains and plasmids. Several C. elegans strains used in this work were provided by CGC, which is supported by the NIH-Officer of Research Infrastructure Programs (P40 OD010440) and the Japanese National BioResource Project. We thank the NIEHS (R01ES021557), Glenn Foundation for Medical Research, HHMI, and NSFC (31422033 and 31471381) for support of this work. A.D. is a cofounder of Proteostasis Therapeutics, Inc. and Mitobridge, Inc. and declares no financial interest related to this work.
Footnotes
Supplemental Information includes five figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.08.042.
AUTHOR CONTRIBUTIONS
K.B., J.D., and A.D. conceived the study and K.B., J.D., S.W., and A.D. wrote the manuscript with input from all co-authors. K.B. performed C. elegans crosses and strain generation, RNAi screens, fluorescence microscopy, immunoblots of C. elegans, Seahorse experiments, and chemical screens. J.D. performed Biosorter assays, Seahorse experiments, cell culture immunoblots, C. elegans strain generation, and fluorescence microscopy. Y.T. performed lifespan assays and C. elegans crosses. H.K. performed C. elegans fractionation experiments. L.-W.S. and Y.L. developed the KillerRed and Cas9 models.
References
- Alcedo J, Kenyon C. Regulation of C. elegans longevity by specific gustatory and olfactory neurons. Neuron. 2004;41:45–55. doi: 10.1016/s0896-6273(03)00816-x. [DOI] [PubMed] [Google Scholar]
- Alkema MJ, Hunter-Ensor M, Ringstad N, Horvitz HR. Tyramine Functions independently of octopamine in the Caenorhabditis elegans nervous system. Neuron. 2005;46:247–260. doi: 10.1016/j.neuron.2005.02.024. [DOI] [PubMed] [Google Scholar]
- Baker BM, Nargund AM, Sun T, Haynes CM. Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 2012;8:e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignull HR, Moore FE, Tang SJ, Morimoto RI. Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. J Neurosci. 2006;26:7597–7606. doi: 10.1523/JNEUROSCI.0990-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Cong WN, Ji S, Rothman S, Maudsley S, Martin B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr Alzheimer Res. 2012;9:5–17. doi: 10.2174/156720512799015064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campesan S, Green EW, Breda C, Sathyasaikumar KV, Muchowski PJ, Schwarcz R, Kyriacou CP, Giorgini F. The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s disease. Curr Biol. 2011;21:961–966. doi: 10.1016/j.cub.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlie NK, Schade MA, Thomure AM, Miller KG. Presynaptic UNC-31 (CAPS) is required to activate the G alpha(s) pathway of the Caenorhabditis elegans synaptic signaling network. Genetics. 2006;172:943–961. doi: 10.1534/genetics.105.049577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, Scorrano L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J. 2012;31:1853–1864. doi: 10.1038/emboj.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey CM, Mackenzie SM, Gargus A, Blanco G, Sze JY. Serotonin (5HT), fluoxetine, imipramine and dopamine target distinct 5HT receptor signaling to modulate Caenorhabditis elegans egg-laying behavior. Genetics. 2005;169:1425–1436. doi: 10.1534/genetics.104.032540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods. 2013;10:1028–1034. doi: 10.1038/nmeth.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin A, Crawford DK, Kenyon C. Timing requirements for insulin/IGF-1 signaling in C. elegans. Science. 2002;298:830–834. doi: 10.1126/science.1074240. [DOI] [PubMed] [Google Scholar]
- Du X, Pang TY. Is Dysregulation of the HPA-Axis a Core Pathophysiology Mediating Co-Morbid Depression in Neurodegenerative Diseases? Front Psychiatry. 2015;6:32. doi: 10.3389/fpsyt.2015.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte JMN, Schuck PF, Wenk GL, Ferreira GC. Metabolic disturbances in diseases with neurological involvement. Aging Dis. 2014;5:238–255. doi: 10.14336/AD.2014.0500238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder JHJ, Rossi JMJ, Solomon J, Solomon N, Lindquist S. The consequences of expressing hsp70 in Drosophila cells at normal temperatures. Genes Dev. 1992;6:1402–1413. doi: 10.1101/gad.6.8.1402. [DOI] [PubMed] [Google Scholar]
- Folstein SE, Folstein MF. Psychiatric features of Huntington’s disease: recent approaches and findings. Psychiatr Dev. 1983;1:193–205. [PubMed] [Google Scholar]
- Gengyo-Ando K, Kamiya Y, Yamakawa A, Kodaira K, Nishiwaki K, Miwa J, Hori I, Hosono R. The C. elegans unc-18 gene encodes a protein expressed in motor neurons. Neuron. 1993;11:703–711. doi: 10.1016/0896-6273(93)90080-b. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci. 2010;123:3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999;13:1385–1393. [PubMed] [Google Scholar]
- Hoogewijs D, Houthoofd K, Matthijssens F, Vandesompele J, Vanfleteren JR. Selection and validation of a set of reliable reference genes for quantitative sod gene expression analysis in C. elegans. BMC Mol Biol. 2008;9:9. doi: 10.1186/1471-2199-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins BG, Koroshetz WJ, Beal MF, Rosen BR. Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology. 1993;43:2689–2695. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- Kapulkin WJ, Hiester BG, Link CD. Compensatory regulation among ER chaperones in C. elegans. FEBS Lett. 2005;579:3063–3068. doi: 10.1016/j.febslet.2005.04.062. [DOI] [PubMed] [Google Scholar]
- Lamech LT, Haynes CM. The unpredictability of prolonged activation of stress response pathways. J Cell Biol. 2015;209:781–787. doi: 10.1083/jcb.201503107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux GA, Cunningham KA, Lin L, Mayer F, Werb Z, Ashrafi K. Kynurenic acid is a nutritional cue that enables behavioral plasticity. Cell. 2015;160:119–131. doi: 10.1016/j.cell.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link CD, Cypser JR, Johnson CJ, Johnson TE. Direct observation of stress response in Caenorhabditis elegans using a reporter transgene. Cell Stress Chaperones. 1999;4:235–242. doi: 10.1379/1466-1268(1999)004<0235:doosri>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004;27:589–594. doi: 10.1016/j.tins.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Melo JA, Ruvkun G. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell. 2012;149:452–466. doi: 10.1016/j.cell.2012.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan SD, Quiros PM, Steffen KK, Williams EG, Mouchiroud L, Tronnes SU, et al. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell. 2016;165:1209–1223. doi: 10.1016/j.cell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochel F, Durant B, Durr A, Schiffmann R. Altered dopamine and serotonin metabolism in motorically asymptomatic R6/2 mice. PLoS ONE. 2011;6:e18336. doi: 10.1371/journal.pone.0018336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt) Mol Cell. 2015;58:123–133. doi: 10.1016/j.molcel.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble T, Stieglitz J, Srinivasan S. An integrated serotonin and octopamine neuronal circuit directs the release of an endocrine signal to control C. elegans body fat. Cell Metab. 2013;18:672–684. doi: 10.1016/j.cmet.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang TY, Du X, Zajac MS, Howard ML, Hannan AJ. Altered serotonin receptor expression is associated with depression-related behavior in the R6/1 transgenic mouse model of Huntington’s disease. Hum Mol Genet. 2009;18:753–766. doi: 10.1093/hmg/ddn385. [DOI] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Podolsky S, Leopold NA, Sax DS. Increased frequency of diabetes mellitus in patients with Huntington’s chorea. Lancet. 1972;1:1356–1358. doi: 10.1016/s0140-6736(72)91092-6. [DOI] [PubMed] [Google Scholar]
- Prahlad V, Morimoto RI. Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc Natl Acad Sci USA. 2011;108:14204–14209. doi: 10.1073/pnas.1106557108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci. 1999;2:959–964. doi: 10.1038/14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speese S, Petrie M, Schuske K, Ailion M, Ann K, Iwasaki K, Jorgensen EM, Martin TFJ. UNC-31 (CAPS) is required for dense-core vesicle but not synaptic vesicle exocytosis in Caenorhabditis elegans. J Neurosci. 2007;27:6150–6162. doi: 10.1523/JNEUROSCI.1466-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Sadegh L, Elle IC, Christensen AGL, Faergeman NJ, Ashrafi K. Serotonin regulates C. elegans fat and feeding through independent molecular mechanisms. Cell Metab. 2008;7:533–544. doi: 10.1016/j.cmet.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze JY, Victor M, Loer C, Shi Y, Ruvkun G. Food and metabolic signalling defects in a Caenorhabditis elegans serotonin-synthesis mutant. Nature. 2000;403:560–564. doi: 10.1038/35000609. [DOI] [PubMed] [Google Scholar]
- Tatum MC, Ooi FK, Chikka MR, Chauve L, Martinez-Velazquez LA, Steinbusch HWM, Morimoto RI, Prahlad V. Neuronal serotonin release triggers the heat shock response in C. elegans in the absence of temperature increase. Curr Biol. 2015;25:163–174. doi: 10.1016/j.cub.2014.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Garcia G, Bian Q, Steffen KK, Joe L, Wolff S, Meyer BJ, Dillin A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR(mt) Cell. 2016;165:1197–1208. doi: 10.1016/j.cell.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumaru H, Augustine GJ. UNC-13 and neurotransmitter release. Nat Neurosci. 1999;2:929–930. doi: 10.1038/14710. [DOI] [PubMed] [Google Scholar]
- Walker FO, Raymond LA. Targeting energy metabolism in Huntington’s disease. Lancet. 2004;364:312–313. doi: 10.1016/S0140-6736(04)16739-1. [DOI] [PubMed] [Google Scholar]
- Wang R, Ross CA, Cai H, Cong WN, Daimon CM, Carlson OD, Egan JM, Siddiqui S, Maudsley S, Martin B. Metabolic and hormonal signatures in pre-manifest and manifest Huntington’s disease patients. Front Physiol. 2014;5:231. doi: 10.3389/fphys.2014.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Williams DC, Bejjani RE, Ramirez PM, Coakley S, Kim SA, Lee H, Wen Q, Samuel A, Lu H, Hilliard MA, Hammarlund M. Rapid and permanent neuronal inactivation in vivo via subcellular generation of reactive oxygen with the use of KillerRed. Cell Rep. 2013;5:553–563. doi: 10.1016/j.celrep.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117:4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lu H, Bargmann CI. Pathogenic bacteria induce aversive olfactory learning in Caenorhabditis elegans. Nature. 2005;438:179–184. doi: 10.1038/nature04216. [DOI] [PubMed] [Google Scholar]
- Zhang T, Mullane PC, Periz G, Wang J. TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum Mol Genet. 2011;20:1952–1965. doi: 10.1093/hmg/ddr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwilling D, Huang SY, Sathyasaikumar KV, Notarangelo FM, Guidetti P, Wu HQ, Lee J, Truong J, Andrews-Zwilling Y, Hsieh EW, et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell. 2011;145:863–874. doi: 10.1016/j.cell.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.