

Abstract
Liver diseases are caused by different etiological agents, mainly alcohol consumption, viruses, drug intoxication or malnutrition. Frequently, liver diseases are initiated by oxidative stress and inflammation that lead to the excessive production of extracellular matrix (ECM), followed by a progression to fibrosis, cirrhosis and hepatocellular carcinoma (HCC). It has been reported that some natural products display hepatoprotective properties. Naringenin is a flavonoid with antioxidant, antifibrogenic, anti-inflammatory and anticancer properties that is capable of preventing liver damage caused by different agents. The main protective effects of naringenin in liver diseases are the inhibition of oxidative stress, transforming growth factor (TGF-β) pathway and the prevention of the transdifferentiation of hepatic stellate cells (HSC), leading to decreased collagen synthesis. Other effects include the inhibition of the mitogen activated protein kinase (MAPK), toll-like receptor (TLR) and TGF-β non-canonical pathways, the inhibition of which further results in a strong reduction in ECM synthesis and deposition. In addition, naringenin has shown beneficial effects on nonalcoholic fatty liver disease (NAFLD) through the regulation of lipid metabolism, modulating the synthesis and oxidation of lipids and cholesterol. Moreover, naringenin protects from HCC, since it inhibits growth factors such as TGF-β and vascular endothelial growth factor (VEGF), inducing apoptosis and regulating MAPK pathways. Naringenin is safe and acts by targeting multiple proteins. However, it possesses low bioavailability and high intestinal metabolism. In this regard, formulations, such as nanoparticles or liposomes, have been developed to improve naringenin bioavailability. We conclude that naringenin should be considered in the future as an important candidate in the treatment of different liver diseases.
Keywords: Naringenin, Transforming growth factor, Liver, Fibrosis, MAPKs, CCl4, Flavonoids, JNK, Hepatic stellate cells, Cirrhosis, Smads, α-SMA
Core tip: Natural products such as flavonoids have been shown to display hepatoprotective properties. Naringenin possesses the ability to inhibit oxidative stress and inflammation and has anti-inflammatory and anticancer properties. Thus, naringenin should be considered in the future as an important candidate for the treatment of liver diseases.
INTRODUCTION
Liver damage can be caused by alcohol intake, heavy metal intoxication, hepatitis virus infection, obstruction of the biliary tract or malnutrition. Chronic hepatic injury results in organ fibrosis characterized by an imbalance between the synthesis and degradation of extracellular matrix (ECM) derived from oxidative stress and inflammation during liver damage. After fibrosis, cirrhosis develops with tissue scars, the loss of parenchymal architecture, the disruption of hepatic blood flow and organ failure[1,2]. The main causes of fibrosis globally are NAFLD (40%), hepatitis B virus (HBV) (30%), hepatitis C virus (HCV) (15%), and harmful alcohol consumption (11%)[3]. The prevalence of cirrhosis is increasing; in 2010, 33% more people died from cirrhosis than in 1990[4].
While the elimination of the causative agent may be the best option for some cirrhotic patients, in most cases, medical intervention is required. Therefore, pharmacological strategies should be developed to prevent or reverse hepatic damage. Researchers have developed multiple therapeutic strategies to combat this disease, including transforming growth factor-β (TGF-β) inhibitors[5], antivirals[6], cell-based therapies[7], nanoparticles[8], and natural products[9-15].
Liver transplantation is an interesting option; unfortunately, the lack of sufficient donors and organ rejection restrict this surgical procedure. In recent years, the investigation on hepatoprotective properties of natural products has increased. Due to their molecular structure, many of them possess antioxidant properties and display anti-inflammatory and anticancer properties and are generally considered safe for human consumption. Among the most studied natural compounds are silymarin, quercetin, and curcumin[10,12,14], but recently, a flavonoid with very specific hepatoprotective properties has emerged: naringenin.
Naringenin has been studied in various in vivo and in vitro liver damage models, using hepatic damage agents such as carbon tetrachloride (CCl4), alcohol, N-methyl-N-nitro-Nitroguanidine, lipopolysaccharide (LPS), and heavy metals, among others, displaying a good hepatoprotective activity due to its antioxidant capacity as well as its ability to inhibit inflammatory and profibrotic signaling pathways. However, despite the importance of naringenin in liver diseases, there is no detailed review of the effects of naringenin on hepatic pathologies.
Therefore, our objective was to document the effects of naringenin on liver diseases and to highlight the importance of this flavonoid in the therapeutic of pathologies of this organ.
LITERATURE SEARCH
A systematic literature search was conducted using PubMed, Scopus and EMBASE.
ABSORPTION, METABOLISM AND DISTRIBUTION OF NARINGENIN
Naringenin (4’,5,7-trihydroxy flavanone) is a flavonoid, specifically a flavanone, and is the aglycone of naringin (naringenin-7-rhamnoglucoside)[16]; naringenin can also be found as narirutin (naringenin- 7-O-rutinoside) or naringenin-glucoside (naringenin-7-O-glucoside), depending on the sugar motive (Figure 1)[11].
Figure 1.
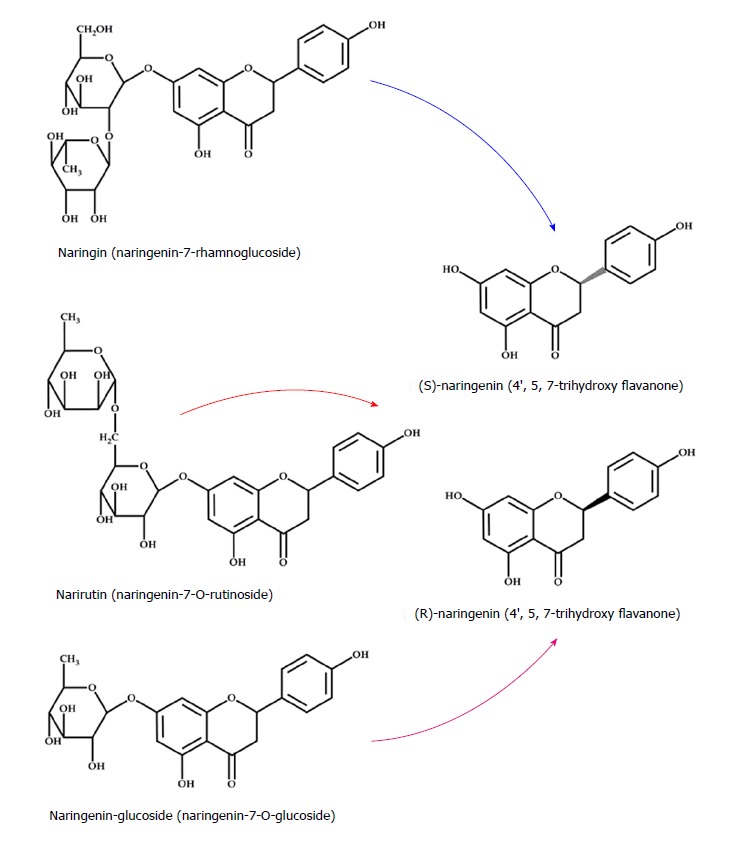
Chemical structure of naringenin, naringin, narirutin, and naringenin-glucoside. The flavonoid naringenin exists in two forms: Glycosylated (naringin, narirutin and naringenin-glucoside) and aglycone (naringenina). There are three types of naringenin glycosides depending of sugar moiety bound to the flavonoid: Naringin (rhamnose), narirutin (rutinose) and naringenin-glucoside (glucose); when the sugar moiety is cleaved by specific enzymes, the aglycone (naringenin) is released.
This review is focused on the effects of naringenin (aglycone); the reader interested in glycosylated molecules is referred to another review[11]. Because naringenin is found mostly in citrus fruits, natural intake occurs orally. Due to its chemical structure, naringenin is very lipophilic; thus, it is readily absorbed through the intestinal epithelium by passive diffusion into enterocytes. Once inside the intestinal cells, it can enter the general circulation by multidrug resistance-associated proteins (Mrp1) or can be transported by active efflux protein carriers P-glycoprotein (P-gp) and Mrp2 back to the intestinal lumen, out of the enterocytes, repeating the cycle[17] (Figure 2).
Figure 2.
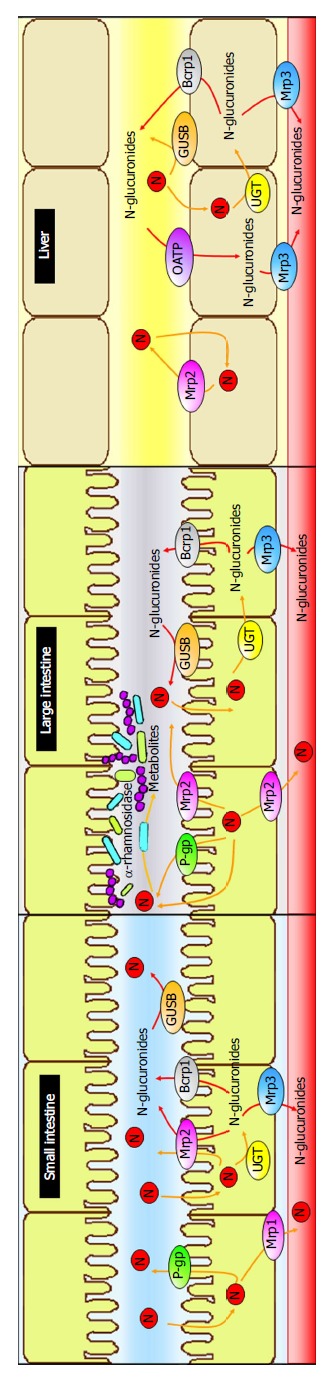
Absorption and metabolism of naringenin. Small intestine: Naringenin (N, orange arrows) is absorbed through the intestinal epithelium by passive diffusion into enterocytes; then, it can pass to general circulation by multidrug resistance associated proteins (Mrp1) or transported by active efflux protein carriers P-glycoprotein (P-gp) and Mrp2, back to the intestinal lumen, out of the enterocytes. Inside the enterocyte, naringenin is glucuronized by UDP-glucuronosyl transferase (UGT), and after that, naringenin-glucuronides (red arrows) leave cells by Mrp2 protein or pass into blood via breast cancer-resistant protein (Bcrp1). Moreover, naringenin-glucuronides can be cleaved by β-glucuronidases (GUSB), resulting in release of the aglycone. Large intestine: Naringenin undergoes same processes as in small intestine but also is highly metabolized by Streptococcus S-2, Lactobacillus L-2, and Bacteroides JY-6 to generate a series of low molecular weight aromatic acids. Liver: Naringenin is highly conjugated to form naringenin-glucuronides, which allows it to pass into circulation. On the other hand, naringenin-glucuronides reach the liver from intestine and enter into hepatocytes via organic anion transporting protein-B (OATP), and then, they are transported by Mrp3 into the circulation.
On the other hand, small intestine, colonic epithelium, and liver metabolize naringenin via phase II conjugation by UDP-glucuronosyl transferase (UGT), sulfotransferase, and catechol-O-methyltransferase[18-20]. Naringenin-glucuronides leave the cells by Mrp2 protein or pass into blood via breast cancer-resistant protein (Bcrp1)[21]. Moreover, naringenin can be cleaved by β-glucuronidases (GUSB) located in tissues and liver[22]. This deconjugation results in release of the aglycone, which in turn can be absorbed by passive transcellular diffusion or undergo efflux by Mrp2 and P-gp[19]. Then, naringenin is metabolized in the lower intestine by Streptococcus S-2, Lactobacillus L-2, and Bacteroides JY-6 to generate a series of low molecular weight aromatic acids[11] (Figure 2).
With respect to naringenin distribution, it has been found in the stomach, small intestine, liver, kidney, trachea, lung, heart, fat, muscle, testis, ovary, spleen, brain, and urine[20,23-25]. Furthermore, naringenin and its metabolites are bound to plasma proteins such as albumin[26-28].
ANTIOXIDANT EFFECTS OF NARINGENIN, BEYOND THE STRUCTURE ACTIVITY RELATIONSHIP
Normally, flavonoid antioxidant activity has been attributed to the structure-activity relationship of flavonoids. However, in addition to a direct antioxidant property by free radical scavenging activity, naringenin possesses the ability to induce the endogenous antioxidant system.
Classically, naringenin’s antioxidant effect is due to its hydroxyl substituents (OH), which have high reactivity against reactive oxygen species (ROS) and reactive nitrogen species (RNS). In general, the antioxidant capacity of a given molecule increases in function with the number of OH radicals in the molecule, which, in the case of naringenin, is 3. Then, OH can donate its H to free radicals (R•), and later, naringenin can be stabilized by resonance[29,30] (Figure 3). Within the typical structure of flavonoids, the B ring is very important because when OH groups are in the ring, flavonoids can stabilize hydroxyl (OH•), peroxyl (ROO•), and peroxynitrite (ONOO•) radicals, producing a relatively stable flavonoid radical. On the other hand, 5-OH substitution and a 5,7-m-dihydroxy arrangement in the A-ring is an important feature of naringenin that makes it an antioxidant and, at the same time, serves to stabilize the structure after donating H to the R•. Finally, the association between 5-OH and 4-oxo substituents contributes to the ability of naringenin to chelate compounds such as heavy metals[29] (Figure 3).
Figure 3.
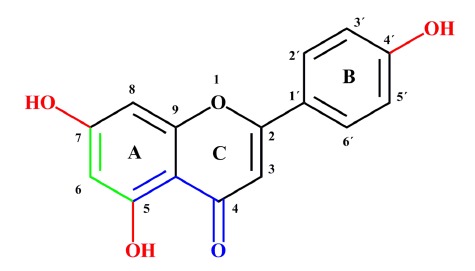
Naringenin antioxidant activity-structure relationship. In red: Hydroxyl substituents (OH) that have high reactivity against reactive oxygen species and reactive nitrogen species. In green: 5,7-m-dihydroxy arrangement in the A-ring serves to stabilize the structure after donating electrons to free radicals. In blue: The association between 5-OH and 4-oxo substituents contributes to the chelation of compounds such as heavy metals.
An important phenomenon during liver damage is lipid peroxidation (LP); it can be defined as the abstraction of hydrogen from fatty acid that initiates a complex series of reactions that terminate in the complete disintegration of the polyunsaturated fatty acid (PUFA) molecules with the formation of aldehydes, such as malondialdehyde (MDA), other carbonyls, and alkanes. LP may be initiated by R•. Therefore, maintaining the normal redox balance with antioxidants during liver damage is important to prevent the deleterious effects of LP[31].
Naringenin trolox equivalent antioxidant activity is 1.53 mmol/L, which is a small value compared with that of quercetin, which is 4.7 mmol/L[30]. In a model of nonenzymatic LP induction by ascorbic acid, naringenin showed 21%-44% inhibition of MDA formation in a dose-dependent manner; however, quercetin was able to prevent 70%-85% of the MDA formation at doses from 0.1 mmol/L to 4.0 mmol/L[32]. During a di(phenyl)-(2,4,6-trinitrophenyl) iminoazanium (DPPH) assay, naringenin had an ID50 of 225 μmol/L at 2 h, and the number of molecules of DPPH scavenged/naringenin molecule was 0.5, while the ID50 of quercetin was 12.5 μmol/L of DPPH scavenged/quercetin molecule. In addition, naringenin showed a lower effect than quercetin in an LP model in liver and lung with an ID50 of more than 1000 μmol/L and 35 μmol/L, respectively[33]. Moreover, naringenin’s effect on LP induced by iron-ascorbate in hepatic microsomes revealed that naringenin (5 μmol/L and 25 μmol/L) increases LP, unlike quercetin, which almost completely inhibited LP at the same dose. In this same study, a modest effect of naringenin (25 μmol/L) against LP induced by Fe3+-ADP/NADPH or TBH assay was observed. In contrast, naringenin strongly protected hepatocytes from TBH-cytotoxicity, suggesting that naringenin did not exert its cytoprotective effects through purely direct antioxidant mechanisms[34].
Although several reports show that naringenin displays poor antioxidant capacity compared to other flavonoids such as quercetin, the results obtained in the 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid (ABTS) assay showed that naringenin had an IC50 of 7.9 μmol/L, and when the determination of superoxide radical (O2•) scavenger activity was performed by the nitro blue tetrazolium (NBT) method and the Xanthine oxidase/cytochrome c (CYPc) method, naringenin had an IC50 of 94.7 and 4.4 μmol/L, respectively. Moreover, naringenin had an IC50 of 1.06 and 1.55 μmol/L with EDTA and without EDTA on OH• scavenger activity, respectively; in addition, the IC50 of naringenin on liver LP in the presence of HO2• or OH• was 1.21 and 0.23 mmol/L, respectively[35].
As seen, the antioxidant effect of naringenin can be considered ambiguous, and it may depend on the radical formed and the model used and the flavonoid concentration. Even though naringenin has fewer antioxidant functional groups than quercetin, it shows other properties due to its structure-activity relationship, as it has been reported that naringenin is able to accumulate in cell membranes[36] and biomembranes[37,38]. Interaction with membranes is favored because flavonoids form reversible bonds with the polar heads of the phospholipids[39], and this interaction may be possible due to naringenin’s solubility, since it is highly lipophilic because of its structure (Figure 4).
Figure 4.
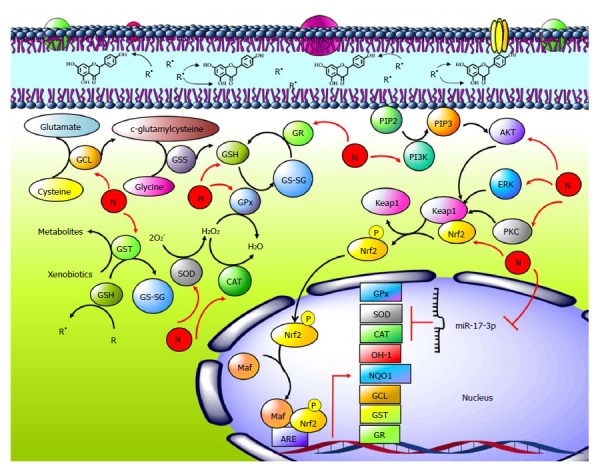
Antioxidant effects of naringenin. Naringenin (N) accumulates in cell membranes, where it provides rigidity to the lipid bilayer; naringenin can reduce the interaction between free radicals (R•) and the cell membrane, as well as reduce membrane phospholipid attack and prevent lipid peroxidation (LP). Glutathione (GSH) is formed when cysteine and glutamate form c-glutamyl cysteine by the enzyme glutamyl cysteinyl ligase (GCL); then, glycine and c-glutamyl cysteine form GSH by GSH synthetase (GSS). Naringenin increases GCLC protein and mRNA levels as well as GSH levels. Naringenin increases activity and protein and mRNA of superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione transferase (GST) and glutathione reductase (GR), enzymes that are part of the endogenous antioxidant system. Naringenin decreases the expression level of miR-17-3p; its targets genes are SOD and GPx. The nuclear factor-erythroid 2-related factor 2 (Nrf2) is an oxidative stress regulator; it stimulates antioxidant enzyme expression as well as heme oxygenase (OH-1) and NADPH quinone oxidoreductase (NQO1). Naringenin upregulates the Nrf2 pathway by increasing its activation, nucleus translocation, and protein and mRNA levels through PI3K/AKT, ERK and PKC.
Interestingly, it has been shown that naringenin decreases membrane fluidity. Membrane fluidity is the relative motional freedom of the lipid molecules in the membrane bilayer, and naringenin accumulates in the membrane hydrophobic core, where it modifies lipid packing order leading to decreased membrane fluidity in a concentration-dependent manner. Therefore, by increasing the rigidity of membranes, naringenin can reduce the interaction between R• and lipids; as a result, LP may be attenuated[38]. In conclusion, in addition to its antioxidant capacity, naringenin can block LP by reducing membrane fluidity[40] (Figure 4).
Although antioxidant assays are important, in vitro and in vivo model systems offer much more information since normal functions of a complete system are preserved. Either by its antioxidant activity or by protection of lipid membranes, naringenin offers protection against ROS and other R• in in vitro and in vivo models. Naringenin protects against ROS in a model of neuronal damage, since it reduces their levels in neurons and decreases mitochondrial dysfunction and increases mitochondrial membrane potential[41]. In addition, naringenin inhibits KO2-induced oxidative stress in a pain model in mice by inhibiting LP and O2• production[42]. On the other hand, naringenin exerts antioxidant effects against paraquat-induced toxicity in human bronchial epithelial cells, since it decreases intracellular ROS generation[43]. Moreover, this flavanone significantly decreased thiobarbituric acid reactive substances (TBARS) and improved membrane phospholipid composition in favor of n-3 PUFAs and the n-6/n-3 PUFAs ratio in the liver of old-aged Wistar rats[44].
Naringenin has shown the ability of combating LP in many organs, tissues and cells, for example, in lung[45], ankle joints (arthritis model)[46], retina of streptozotocin-induced diabetic rats[47], SH-SY5Y cells[48], cardiomyoblast cells[49], skin[50], testis[51] and, interestingly, in liver[44,52,53]. It can be concluded that, in contrast to the results obtained in chemical antioxidant assays, the beneficial effects of naringenin against LP in systems involving living organisms or cells, the flavanone shows strong activity. This characteristic is very important for the treatment of hepatic diseases, since LP constitutes one of the main causative agents that triggers liver damage.
In the studies where a reduction in LP by naringenin was demonstrated, a relationship between reduced glutathione (GSH) and flavonoid levels is observed. In fact, it has been observed that naringenin improves GSH levels during oxidative stress[44,47-52]. Improvement of GSH levels by naringenin is associated with the beneficial properties of this flavonoid on the liver because oxidative stress plays a causative role in hepatic disorders[54].
The effect of naringenin on GSH levels deserves further explanation. GSH is a tripeptide (L-γ-glutamyl-L-cysteinyl-glycine) that serves several essential functions within the cell. The main functions of GSH are antioxidant, detoxification of oxygen-derived free radicals, thiol disulfide exchange and storage/transfer of cysteine. GSH is formed in two steps: in the first (rate-limiting) step, cysteine and glutamate form c-glutamyl cysteine by the enzyme glutamyl cysteinyl ligase (GCL); in the second step, GSH forms from c-glutamyl cysteine and glycine by GSH synthetase (GSS) catalysis[55-57] (Figure 4). It has been observed that naringenin possesses the ability to increase total and mitochondrial GSH levels during hydrogen peroxide (H2O2)-induced liver damage[48,49,51], as well total hepatic GSH[52,58,59] and total GSH in other organs[60,61]. These effects can be explained because naringenin increases the expression of the GCLC catalytic subunit and the GCL modulatory subunit at both the protein and mRNA levels[60-62].
The tripeptide can directly scavenge R• or function as a co-substrate of the internal antioxidant system enzymes. For example, GSH is the co-substrate of glutathione peroxidase (GPx) in H2O2 reduction and of glutathione transferase (GST), which catalyzes xenobiotics biotransformation in the liver[56,57]. In either case, GSH is oxidized to GSSG, which leads to consumption of GSH. Therefore, there are mechanisms in charge of maintaining the GSH/GSSG balance; for example, glutathione reductase (GR or GSR) is responsible of GSSG reduction to the disulfide form (GSH) at the expense of NADPH[55-57]. It has been reported that naringenin increased the GSH/GSSG ratio[59,61] by improving GR mRNA levels and activity in the liver[44,63-66] and in other organs[67,68] (Figure 4).
Naringenin can influence cellular antioxidant balance not only through its own chemical structure but also by inducing the cell antioxidant system. In this regard, it has been reported that naringenin upregulates important antioxidant enzymes, such as superoxide dismutase (SOD), catalase (CAT), GPx and GST.
SOD catalyzes the reaction in which O2• is converted to H2O2, a more stable species but that at high concentrations is harmful to cells; in turn, CAT eliminates excess H2O2, generating water[69]. Naringenin significantly increases SOD enzyme activity in different models of liver damage induced by oxidative stress[44,50,52,62-65,67,70-74]. This effect is associated with the ability of this flavonoid to increase enzyme protein levels in the liver and other organs[58,59,68]. Naringenin can prevent CAT activity decrement after damage to several tissues[44,49,50,52,62-65,67,70-74] by increasing protein[58,59,68] and mRNA levels[43].
SOD and CAT, together with GPx and GST, are diminished during oxidative stress. It is worth noting that naringenin has been reported to upregulate these enzymes[44,63-66] (Figure 4). There are some reports trying to explain the mechanism of naringenin to increase GPx activity. One report indicates that the flavanone produces an increment in GPx mRNA levels[43], while others indicate that it increases the protein content[58,59,68]. Another hypothesis postulates that during cell damage, GSH is almost depleted and, thus, cannot be utilized by GPx as a cofactor, leading to decreased enzyme activity; in this situation, naringenin acts by improving GSH levels and, as a consequence, enzyme activity[44,49,50,52,63-65,71-74]. Further experiments are needed to clarify this point.
Naringenin preserves GST activity under prooxidant conditions associated with several illnesses[49-51,63-65,70,72,74]. It has been demonstrated that the flavanone acts by increasing mRNA levels of GST[61,63], which in turn induces the transduction of the corresponding functional protein[68]. The specific action mechanism by which naringenin produces these effects remains to be elucidated (Figure 4).
As it has been previously described, naringenin has very important effects on endogenous antioxidant system enzymes, in contrast to its own weak antioxidant properties in comparison to those of other natural compounds, such as quercetin. This low antioxidant activity suggests that naringenin’s effects are not only a result of its structural activity relationship but also due to other properties. In this regard, it is worth noting that naringenin influences microRNAs (miRNAs) and nuclear factor-erythroid 2 related factor 2 (Nrf2).
miRNAs are noncoding or nonmessenger RNAs that are approximately 22 nucleotides in length that regulate gene expression because they bind to target mRNA, inhibiting protein synthesis[75]. miR-17-3p is involved in oxidative stress, and its targets are mRNAs coding for SOD and GPx, thereby preventing the expression of these proteins[76]. Naringenin decreased the expression level of miR-17-3p, which is in agreement with increased levels of target mRNAs coding for SOD and GPx2[77]. As noted, this reduction in miR-17-3p expression may be a mechanism by which naringenin modulates antioxidant enzymes; however, more research is needed on the role of naringenin in miRNA and its effect on the endogenous antioxidant system (Figure 4).
Nrf2 interacts with the actin binding protein, Kelch-like ECH associating protein 1 (Keap1), inactivating Nrf2 in the cytoplasm. Nrf2 must be released from Keap1 to be active. Its release can occur either by MAPK phosphorylation or by conformational changes in Keap1 due to ROS. Once free, Nrf2 translocates to the nucleus, where it forms a dimer with the musculo-aponeurotic fibrosarcoma (Maf) family proteins. Nrf2-Maf dimer is a transcription factor that binds to the antioxidant response element (ARE) sequence, resulting in transcriptional activation of detoxification proteins such as NADPH quinone oxidoreductase (NQO1), GST, and aldo-keto reductase (AKR), antioxidant enzymes such as thioredoxin (TXN1), thioredoxin reductase 1(TXNR), peroxiredoxin 1 (PRDX1), GPx, GCL, GR, CAT and SOD, and heme and iron metabolism proteins such as heme oxygenase (OH-1) and ferro chelatase (FECH)[78-80] (Figure 4).
Interestingly, there are reports indicating that naringenin upregulates Nrf2 in various models. In a model of UVB irradiation-induced skin inflammation and oxidative damage in hairless mice, naringenin significantly increased Nrf2 mRNA levels compared with those in the damaged group[66]. Moreover, in a model of KO2-induced inflammatory pain in mice, naringenin inhibited the KO2-induced decrease in Nrf2 mRNA expression[42]. In addition, naringenin upregulated the mRNA expression of Nrf2 in complete Freund’s adjuvant-induced rats[46], and naringenin increased Nrf2 mRNA expression in a model of oxidative stress induced by H2O2[49].
The induction of Nrf2 mRNA may propitiate Nrf2 protein levels to increase. It has been reported that naringenin is capable of increasing Nrf2 protein levels in CCl4-induced hepatic damage[63]. In addition, the flavonoid protected SH-SY5Y cells against 6-OHDA neurotoxicity via Nrf2 because it improved the levels of this protein[60]. Moreover, one mechanism to explain why naringenin prevented CCl4-induced acute liver injury in mice is by preserving Nrf2 levels[59]. In addition, naringenin improved intracellular Nrf2 levels in LPS-induced apoptosis of PC12 cells[81] and reduced oxidative stress by increasing Nrf2 protein levels in neurons[41].
Increased Nrf2 protein levels do not necessarily correlate with increases in Nrf2 activity. Nrf2 must dissociate from Keap1 to translocate to the nucleus and to induce proteins of the antioxidant system. Naringenin activates Nrf2 because it promotes its translocation from the cytoplasm to the nucleus[43,61-63,82].
Phosphorylation of Nrf2 by extracellular signal-regulated protein kinase (ERK) triggers the dissociation of Nrf2-Keap1 and inhibits the reassociation of Nrf2-Keap1 complexes[83,84]. Other important proteins involved in the activation of Nrf2 are 5′ AMP-activated protein kinase (AMPK)[85], phosphatidylinositol-3-kinase (PI3K/AKT), and protein kinase C (PKC)[86]. Notably, it has been observed that naringenin upregulated phosphorylated ERK1/2, leading to nuclear translocation of Nrf2 in doxorubicin-induced toxicity in H9c2 cardiomyocytes[62]. In another report, after treatment with 40 μg/mL of naringenin, nuclear Nrf2 increased at 0.25 h and remained elevated until 3 h after naringenin treatment to H9c2 cells[82]. In addition, naringenin increased the phosphorylation levels of ERK1/2, PKCδ, and AKT, but this increase was prevented by chemical inhibitors of AKT (LY294002), ERK1/2 (PD98059), and PKCα (rottlerin), which suppressed Nrf2 activation induced by naringenin[82]. These results suggest that the naringenin-induced activation of Nrf2 signaling may be mediated by the phosphorylation of ERK1/2, PKCδ, and AKT[82] (Figure 4).
Nrf2 activation and its translocation to the nucleus lead to its union with Maf; Nrf2-Maf dimer, in turn, binds to ARE sequence, which results in transcriptional activation of detoxification and antioxidant proteins. Naringenin not only activates Nrf2 but also increases the mRNA and protein levels of target genes such as NQO1, GPx, GCL, GR, OH-1, and GST[43,46,49,59-63,66,81,82,87]. To corroborate this effect, experiments have been carried out to silence Nrf2. A small interfering RNA (siRNA) study revealed that the knockdown of Nrf2 can abrogate naringenin-mediated protection of the BEAS-2B cells from paraquat-induced cellular toxicity[43]. Another report showed that naringenin fails to block 6-OHDA neurotoxicity if Nrf2 siRNA is administered[60]. Moreover, naringenin prevented mitochondrial depolarization is inhibited by Nrf2 siRNA[87]; in addition, the naringenin-induced upregulation of GCL and HO-1 proteins was significantly inhibited by Nrf2-siRNA transfection in H9c2 cells[82]. Finally, silencing of Nrf2 suppressed naringenin-induced cytoprotection and mitochondrial protection in SH-SY5Y cells exposed to H2O2[48] (Figure 4).
Due to the important regulatory effects of naringenin on endogenous antioxidant system, the flavonoid takes great importance as a possible hepatoprotector, since one of the main mechanisms of liver damage is oxidative stress[54]. In addition, this antioxidant is different from others, since in addition to its direct effect as an antioxidant, it induces the expression of endogenous antioxidants.
NARINGENIN PREVENTS LIVER DAMAGE CAUSED BY ALCOHOL
Liver damage induced by excessive alcohol consumption is a worldwide problem[3]. It has been reported that an intake of 80 g/day by men and 40 g/day by women between 10-20 years may lead to fibrosis[88-90]. Therefore, it is important to find a drug that prevents or reverses the effects of alcohol abuse in the population.
Liver alcohol metabolism consists of the following steps: (1) In the cytosol, alcohol is converted into acetaldehyde by the enzyme alcohol dehydrogenase (ADH) using NAD+ to generate NADH; acetaldehyde is also formed in microsomes by CYP2E1 and in peroxisomes by CAT; and (2) In the mitochondria, acetaldehyde dehydrogenase (ALDH) transforms acetaldehyde to acetate[91-93] (Figure 5). During these reactions, secondary harmful products to hepatocytes are generated; among the most important of these harmful products is MDA, which forms adducts with proteins and is also an important indicator of LP[91-93]. Moreover, ROS, such as H2O2 and O2•, are generated during the metabolism of alcohol by CYP2E1. Additionally, alcohol metabolism induces fatty liver disease by increasing the NADH/NAD+ ratio. In general, these processes induce hepatocyte damage, leading to an inflammatory environment that activates endothelial cells, Kupffer cells and HSCs[91-93].
Figure 5.
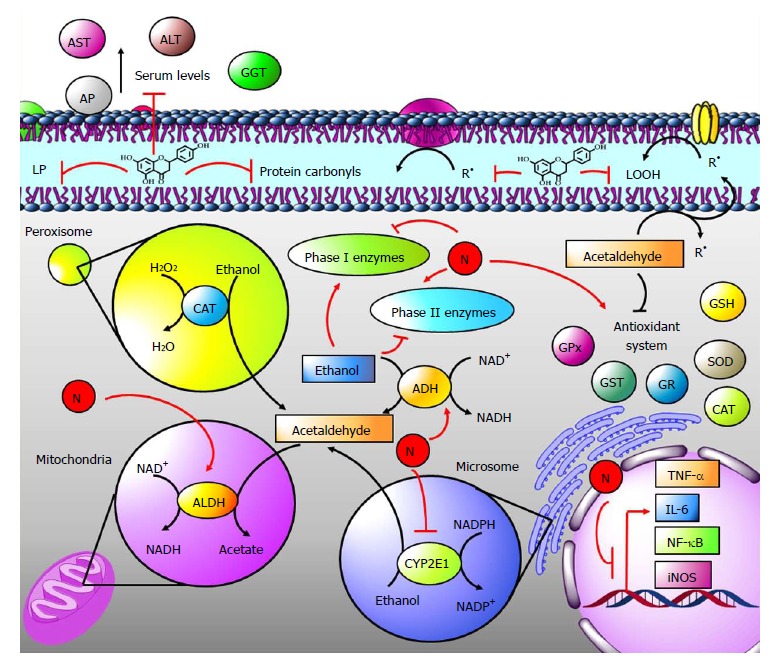
The role of naringenin in alcohol-induced liver damage. Alcohol metabolism: In the cytosol, alcohol is converted into acetaldehyde by alcohol dehydrogenase (ADH); it is also formed in microsomes by CYP2E1 and in peroxisomes by catalase (CAT). In mitochondria, acetaldehyde dehydrogenase (ALDH) transforms acetaldehyde to acetate. Ethanol elevates ADH and CYP2E1 activities but decreases ALDH activity, resulting in toxic acetaldehyde accumulation, free radical (R•) formation in the form of lipid hydroperoxides (LOOH) or protein carbonyls and resulting in the elevation of lipid peroxidation (LP). Naringenin (N) increases the activities of all those enzymes, which results in alcohol efficient elimination leading to endogenous antioxidant system restoration, oxidative stress prevention and balance of phase I and phase II xenobiotic metabolism enzymes. Naringenin also prevents increased levels of alkaline phosphatase (AP), aspartate aminotransferase (AST), alanine aminotransaminase (ALT), and γ-glutamyl transferase (GGT) as well as inflammation during alcohol-mediated liver damage.
The evidence indicates that naringin, the naringenin-glycoside, significantly lowered ethanol concentration in plasma in a dose-dependent manner[94]. Ethanol administration resulted in higher ADH and lower ALDH activities, resulting in toxic acetaldehyde accumulation. Naringin increased the activities of both enzymes, resulting in efficient alcohol elimination via acetaldehyde and its conversion to acetate, preventing the accumulation of acetaldehyde, and resulting in the rapid clearance of alcohol from the serum[94]. In agreement with these findings, naringenin administration to alcohol-treated rats increased ADH and ALDH enzyme activities[70]. In addition, ethanol increased the activity of cytochrome CYP2E1, while this effect was reversed by naringenin[70] (Figure 5).
Ethanol consumption modifies the phase I and phase II xenobiotic metabolism enzymes. During phase I metabolism, enzymes catalyze reactions of oxidation, reduction, and hydrolysis of xenobiotics to increase their polarity and improve their excretion. On the other hand, phase II reactions are glucuronidation, acetylation, S-methylation, and glutathione- or sulfo-conjugation of xenobiotics. These reactions are carried out on phase I products for their better excretion, since tissue damage occurs if the products of phase I are not eliminated by the enzymes of phase II[95]. It has been reported that alcohol intake raises the activity of phase I enzymes such as CYP450, cytochrome b5, NADH-cytochrome b5 reductase and NADPH-CYP450 reductase. In contrast, ethanol injection decreases the activity of phase II enzymes such as GST and DT-diaphorase[70,96]. Interestingly, naringenin was able to reverse these effects caused by alcohol in both types of enzymes, leading to efficient elimination of alcohol metabolism products and reestablishment of the NADH/NAD+ ratio[70] (Figure 5).
Due to acetaldehyde accumulation during alcohol metabolism, oxidative stress is generated. This is characterized by LP, increased R• and endogenous antioxidant system dysfunction[97]. During ethanol administration in vivo, significantly elevated levels of TBARS, lipid hydroperoxides (LOOH), conjugated dienes (CD), protein carbonyl content and significantly lowered activities of SOD, GPx, CAT, GR and GST, and lowered levels of GSH have been observed[64,70,94].
As discussed above, naringenin displays antioxidant effects at different levels, and this was evident when the administration of naringin or naringenin prevented and reverted oxidative stress caused by ethanol, normalizing levels of TBARS, LOOH, CD, protein carbonyl content, antioxidant enzymes activity and GSH levels[64,70,94] (Figure 5).
If oxidative stress is constant and the antioxidant system has failed, liver damage is generated; this liver damage is marked by increases in liver damage markers such as alkaline phosphatase (AP), aspartate aminotransferase (AST), alanine aminotransaminase (ALT), γ-glutamyl transferase (GGT) and lactate dehydrogenase (LDH) activities or by the elevation of serum bilirubins and aspartate levels. However, naringenin administration during ethanol-induced hepatic damage decreases the activity/levels of liver damage markers, demonstrating that naringenin protects hepatocytes against necrosis, cholestasis and membrane permeation[64,70,98] (Figure 5).
After hepatocyte damage occurs, an inflammatory reaction is produced that is characterized by increases in cytokines and proteins that mediate the immune response. It has been reported that rats that received 20% ethanol equivalent to 6 g/kg body weight (bw) every day for a period of 60 days showed significantly elevated mRNA levels of tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), nuclear factor-kappa B (NF-κB), cyclooxygenase-2 (COX-2), macrophage inflammatory protein 2 (MIP-2) and CD14, as well as increased staining for inducible nitric oxide synthase (iNOS) protein adducts in the liver. Notably, when naringenin (50 mg/kg p.o.) was administered every day during the last 30 d of alcohol intoxication, the flavanone decreased the mRNA levels of all inflammatory markers[98], indicating the potent anti-inflammatory properties of naringenin (Figure 5).
One of the main effects of alcohol abuse on the liver is lipid accumulation in hepatocytes. Even though fatty liver is a reversible condition, it can progress to inflammation and fibrosis. During alcohol consumption, there is a deregulation of pathways that regulate lipid synthesis, oxidation and very-low density lipoprotein (VLDL) exportation that leads to the accumulation of triglycerides and fatty acids in the liver[93]. In a study performed to investigate the effect of naringin supplements on lipid metabolism in ethanol-treated rats, the results showed that the concentrations of plasma/liver total cholesterol and plasma/liver total triglyceride were significantly higher in the ethanol-treated rats and, conversely, decreased the high-density lipoprotein (HDL)-cholesterol level and HDL-cholesterol/total-cholesterol ratio, while naringin reestablished normal levels of all measured lipid parameters. Another interesting effect of the glycoside was a decreased number of hepatic cells containing lipid droplets compared to the alcohol-group, where many of these cells were observed. It was concluded, therefore, that naringin is able to prevent lipid accumulation in liver caused by alcohol[94].
In another study, serum insulin was diminished, glucose/insulin ratio and liver triglycerides were increased in ethanol-drinking rats; however, naringenin co-administration partially protected rats from these effects produced by alcohol intoxication. Unfortunately, naringenin was not able to protect from alterations in serum glucose, triglycerides, total, free and esterified cholesterol or HDL cholesterol, or from liver and muscle triglycerides or glycogen[99].
Naringenin has effects on several steps of ethanol metabolism, as well as on liver damage by this xenobiotic, suggesting that it can be used in the prevention and reversion of alcohol-induced liver damage. However, more studies are necessary to further investigate naringenin’s mechanism of action in alcohol-induced hepatic injury and whether it is able to protect from fibrosis induced by alcohol abuse.
EFFECT OF NARINGENIN ON CCl4-INDUCED LIVER DAMAGE
CCl4 is a haloalkane widely used to induce liver damage[100]. To induce liver damage, CCl4 must be activated by CYP2E1, CYP2B1 or CYP2B2, and CYP3A, to form the trichloromethyl radical (CCl3•). This radical reacts with oxygen to form the trichloromethylperoxy radical (CCl3OO•), a highly reactive species. These two species are highly reactive; they can bind to cellular molecules, for example, nucleic acids, proteins or lipids. CCl3OO• initiates the chain reaction of LP, which attacks and destroys PUFAs, those associated with phospholipids in cell membranes as the mitochondrial or the reticulum membranes. This membrane damage leads to hepatocyte damage, which in turn activates Kupffer cells and HSC, regulating fibrosis and cirrhosis[31,101] (Figure 6).
Figure 6.
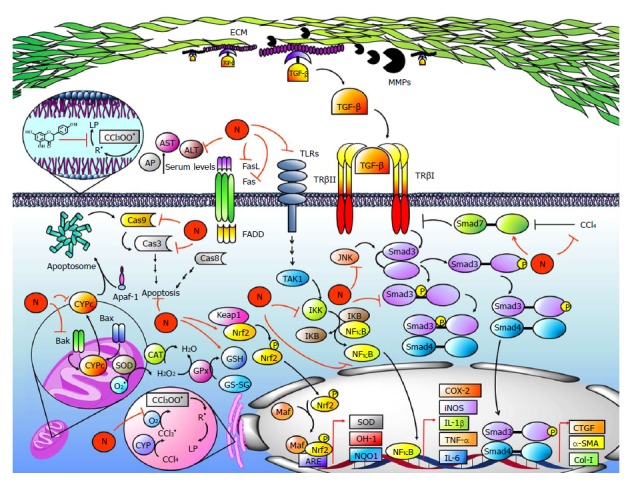
Naringenin prevents acute and chronic CCl4-induced liver damage. Carbon tetrachloride (CCl4) is activated by CYP2E1, CYP2B1, CYP2B2, and CYP3A (CYP) to form the trichloromethyl radical (CCl3•); then, it reacts with the oxygen-forming trichloromethylperoxy radical (CCl3OO•). The CCl3OO• initiates lipid peroxidation (LP), free radical (R•) generation, and imbalance of the endogenous antioxidant system formed by superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione (GSH), heme oxygenase (OH-1), NADPH quinone oxidoreductase (NQO1) and nuclear factor-erythroid 2 related factor 2 (Nrf2). Naringenin prevents CCl4 metabolism, LP and imbalance of the antioxidant system. Naringenin also prevents increased levels of alkaline phosphatase (AP), aspartate aminotransferase (AST), alanine aminotransaminase (ALT), and γ-glutamyl transferase (GGT). On the other hand, CCl4 increases intrinsic and extrinsic apoptosis pathways in hepatocytes; however, naringenin prevents CYPc release, as well as BCL2-associated X protein (Bax), BCL2-antagonist/killer 1 (Bak), Caspase 3 (Cas3) and Caspase 9 (Cas9) elevation, a protein related with the intrinsic pathway. For the extrinsic apoptosis pathway, naringenin prevents Fas and Fas ligand (FasL) increases produced by CCl4 administration. During CCl4-induced fibrosis, there is a proinflammatory environment generated by Kupffer cells and HSCs. The NF-κB pathway starts when TLRs are activated; then, intermediates are activated leading to inhibitor κB (IKB) phosphorylation by IκB kinase (IKK) and NF-κB release. NF-κB regulates inflammatory protein expression, including tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), cyclooxygenase-2 (COX-2), interleukin-1 (IL-1) and inducible nitric oxide synthase (iNOS), while naringenin maintains normal levels of these proteins during CCl4-induced liver damage. Transforming growth factor-β (TGF-β) activates receptor-activated Smad3 (Smad3), leading to transcriptional induction of α-smooth muscle actin (α-SMA), connective tissue growth factor (CTGF), and collagen-1 (Col-1). Moreover, Smad3 is also activated by JNK via linker domain phosphorylation. Naringenin prevents Smad3 activation and α-SMA, CTGF, and Col-1 elevation because it inhibits TGF-β elevation and JNK activation. Metalloproteases (MMPs) cleave extra cellular matrix (ECM) proteins, favoring TGF-β release as well as HSC migration to other sites, increasing fibrosis development; naringenin prevents MMPs elevation. On the other hand, CCl4 decreases Smad7 protein levels; this protein inhibits the TGF-β signaling pathway by TGF-β receptor I (TβRI) ubiquitination, but nevertheless, naringenin maintains normal levels of Smad7 during CCl4 treatment.
Facino et al[102] were pioneers to study the effectiveness of naringenin against CCl4 damage. In this study, a glycosidic fraction (containing naringenin-glycoside) and naringenin-glycoside extracted from the flowering tops of Helichrysum italicum G. Don were utilized to investigate the effect of these flavonoids on CCl4-induced rat microsomes, finding that microsomal LP was reduced by the glycosidic fraction and by naringenin-glycoside[102]. Another study showed that CCl4-induced liver damage was decreased by concomitant administration of an aqueous extract of the rhizomes of Sansevieria liberica, containing 5.99% naringenin, since AP, AST and ALT activities and fatty degeneration of hepatocytes were prevented[103]. Finally, another report investigated the effect of an aqueous extract of Trifolium pratense L. (Leguminosae) leaves on CCl4-induced liver damage; it was observed that naringenin in the extract was able to reduce LP levels and xanthine oxidase (XOD) activity[104].
On the basis that natural extracts containing naringenin had positive effects against injury induced by CCl4, different protocols have been carried out to evaluate naringenin hepatoprotective capacity. In 2009, Yen et al[16] evaluated the ability of naringenin to prevent acute liver failure induced by CCl4 in rats. Naringenin (100 mg/kg) was administered during three consecutive days, and then on the fourth day, CCl4 was intraperitoneally (i.p.) administered with a single dose (3 mL/kg, olive oil: CCl4, 1:1). The flavonoid was able to prevent AST, ALT and LP elevations and the reduction of SOD, CAT and GPx levels, and it significantly suppressed the activation of caspase (Cas)3 and Cas9 induced by CCl4 administration[16].
Later, Hermenean et al[105] published an experiment in which acute liver damage was induced in mice with CCl4 (1.0 mL/kg, olive oil: CCl4, 1:1, i.p.), and naringenin (50 mg/kg) pretreatment for seven days was evaluated. The elevation of serum AST, ALT and LP levels as well as the reduction of CAT, SOD and GPx activities and GSH levels in livers from rats intoxicated with CCl4 were all significantly prevented by naringenin. Moreover, naringenin prevented necrotic changes of hepatocytes, fatty degeneration, sinusoidal dilatation, mild fibrosis, and inflammatory cell infiltration and retained the normal ultrastructure of the hepatocytes, including mild restoration of normal bile canaliculi configuration filled with microvilli[105].
The action mechanism of naringenin on acute liver damage induced by CCl4 can be explained by different mechanisms. CCl4 is activated in hepatocytes by CYP2E1; therefore, the R• formed attacks membranes of these cells, generating LP. During CCl4 administration, the expression of CYP2E1 is elevated; however, it has been reported that naringin strongly inhibited this cytochrome; therefore, one possible mechanism of hepatoprotection is the inhibition of CYP2E1 by the flavanone, preventing bioactivation of CCl4[59] (Figure 6). Another mechanism is associated with the ability of naringenin to induce the endogenous antioxidant system by upregulating Nrf2. It was reported that the administration of 50 mg/kg of naringenin to rats significantly increased Nrf2 protein levels in the cytoplasm and nucleus, elevating mRNA levels of its target genes, such as HO-1, NQO1 and GST[63]; in addition, naringenin can prevent the decrease in Nrf2, HO-1 and SOD protein levels exerted by CCl4 treatment in mice[59] (Figure 6).
In addition to oxidative stress, inflammation plays a crucial role in the development of liver damage. During fibrosis produced by CCl4 chronic administration, there is a proinflammatory environment generated by Kupffer cells and HSCs. In these cells, inflammatory signaling pathways, mainly NF-κB-related signaling pathways, are activated. This pathway starts when TLRs are activated; then, intermediaries lead to inhibitor κB (IKB) phosphorylation by IκB kinase (IKK) and NF-κB release into the cytoplasm. NF-κB then translocates into nucleus to induce the transcription of target genes. NF-κB regulates proinflammatory protein expression of TNF-α, IL-1β and IL-6[59,31,106]. In addition, NF-κB binds to iNOS and COX-2 gene promoters, activating the transcription of these genes; iNOS catalyzes the production of nitric oxide (NO), which is a highly oxidizing product[107,108]. On the other hand, during the NF-κB pathway, the intermediate TGF-β-activated kinase 1(TAK1) is activated. Additionally, through MAPKs, NF-κB activates activator protein 1 (AP-1), a factor that promotes the transcription of genes related to inflammation[106,109]. Moreover, high mobility group box 1 (HMGB1) is widely involved in proinflammatory processes through its receptor for advanced glycation end product (RAGE) and TLRs; HMGB1 is released by necrotic cells and by monocytes or macrophage[110].
Because inflammation plays a pivotal role in the establishment and perpetuation of liver diseases, naringenin has been evaluated as an anti-inflammatory therapeutic agent. In this context, a recent paper reported that naringenin (30, 60 and 120 mg/kg) administration to mice treated with CCl4 (0.3% CCl4, 10 mL/kg, dissolved in olive oil) showed that at a dose of 120 mg/kg, the flavonoid dramatically downregulated the expressions of TLR4, TNF-α, IL-1β, IL-6, iNOS, AP-1, COX-2, HMGB-1 and NF-κB[59]. Another report of a study carried out in rats that were acutely intoxicated with CCl4 indicates that naringenin (50 mg/kg) prevents the CCl4-induced increases in TNF-α and elevations in iNOS, COX-2 protein and mRNA[63]. Figure 6 shows that naringin and naringenin possess important anti-inflammatory properties by blocking the NF-κB signaling pathway.
During hepatic damage, hepatocytes may undergo apoptosis mediated by intrinsic or extrinsic pathways. In the intrinsic pathway, BCL2-associated X protein (Bax) and BCL2-antagonist/killer 1 (Bak) are activated by proapoptotic stimuli, resulting in the release of electron transport protein CYPc from the mitochondria to the cytoplasm; then, this protein binds to Apaf-1, forming the apoptosome. In turn, the apoptosome activates Cas9, which cleaves procaspase 3 zymogens, amplifying the cell death cascade[11].
The administration of CCl4 induces apoptosis in hepatocytes as well as DNA fragmentation, increases the mRNA levels of Bax, Bak, Cas3 and Cas9 and increases CYPc release[59,31,101]. It has been reported that glycosylated naringenin (naringin) effectively prevented CCl4-induced DNA fragmentation, apoptosis and mitochondrial injury by attenuating the release of CYPc, therefore inhibiting apoptosis initiation. Another explanation is that naringin significantly increased the expression of antiapoptotic proteins B-cell CLL/lymphoma 2 (Bcl-2) and BCL2-like 1 (Bcl-xL) but decreased Bax, Bak, Cas3 and Cas9 mRNA levels[59] (Figure 6).
Through the extrinsic pathway, Fas is activated by Fas ligand (FasL), which then binds to Fas-associated protein with a death domain (FADD). The Fas-FADD complex activates procaspase 8, which in turn activates other Cas, leading to apoptosis[111]. After CCl4-induced acute liver damage, the mRNA levels of Fas, FasL, and proapoptotic protein p53 are increased, but preventive administration of naringin inhibited this increase and reduced apoptosis in liver[59] (Figure 6).
While there are some reports indicating evidence of the beneficial effects of naringenin on acute liver damage induced by CCl4, there is little information on the effect of this flavanone on chronic treatment. Recently, we have demonstrated that naringenin effectively prevents liver cirrhosis induced by chronic administration of CCl4 in the rat[112]. Moreover, we studied the molecular mechanisms involved in the hepatoprotective effects of naringenin on CCl4-induced liver fibrosis. Our results indicate that naringenin prevented necrosis and cholestasis and improved liver biosynthetic capacity in CCl4-treated rats since the flavonoid completely prevented the increase in ALT, AP and GGT serum activity and hepatic glycogen depletion. In addition, naringenin inhibited oxidative stress caused by chronic liver damage, maintaining normal levels of MDA, GSH and GPx activity. Moreover, inflammation was prevented by naringenin since the levels of NF-κB, IL-1β and IL-6 were preserved within normal values despite CCl4 administration[112].
Perhaps the most important feature of chronic liver damage is the deposition of scar tissue in the hepatic parenchyma, leading to fibrosis and cirrhosis. In general, livers of rats treated with CCl4 presented macro nodular fibrosis; the tissue showed liver parenchymal disruption, steatosis, hyperchromatic nuclear hepatocytes, and atypical pleomorphic nuclei. In addition, cirrhotic rats presented large amounts of collagen around fibrotic nodules. In contrast, rats treated with naringenin had livers without macro nodular fibrosis; collagen accumulation as well as regenerative nodules were prevented, and it was found that total collagen and collagen-I (Col-I) accumulation was prevented by naringenin. One of the main profibrogenic signaling molecules is TGF-β, which exerts its effects by activating receptor-activated Smads (R-Smads), leading to transcriptional induction of α-smooth muscle actin (α-SMA), the main marker of transdifferentiation of HSCs, and connective tissue growth factor (CTGF), a TGF-β downstream signal amplifier[113,114]. Notably, naringenin was able to maintain basal levels of TGF-β, as well as α-SMA, CTGF and Col-I, in rats treated with CCl4. In addition to being activated by TGF-β, MAPKs also activate R-Smads in an alternative pathway (non-canonical), where the linker domain is phosphorylated instead of the carboxyl domain in R-Smads molecules[115,116]. After the administration of CCl4 for 8 wk, activated JNK levels increased significantly, as well as total and phosphorylated Smad3 in the linker domain (pSmad3L); however, naringenin was able to prevent these effects, providing another explanation for the antifibrotic effect of the flavonoid (Figure 6). Moreover, metalloproteases (MMPs), produced by the activated HSCs, cleave TGF-β, leading to further activation and proliferation of HSCs and collagen fiber formation; consequently, fibrosis ensues. In agreement with these findings, we noticed that chronic CCl4 administration produced increased MMP2, MMP9 and MMP13; notably, we found for the first time that naringenin treatment preserved normal levels of these MMPs[117] (Figure 6).
Furthermore, CCl4 decreased Smad7 protein levels; Smad7 inhibits the TGF-β profibrogenic signaling pathway by TGF-β receptor I (TβRI) ubiquitination[118]. Nevertheless, naringenin was able to maintain normal levels of Smad7 during CCl4 treatment, therefore preserving the normal/physiological antifibrotic pathway and, thus, blocking ECM deposition in the hepatic parenchyma (Figure 6).
Our working group recently showed that naringenin also has effects on the reversion of a previously established fibrosis (unpublished data). CCl4 was given for 12 weeks to male Wistar rats (400 mg/kg, 3 times/wk); however, naringenin (100 mg/kg/two times a day, p.o.) was administered at the beginning of week 9 of CCl4 treatment to determine its ability to reverse established experimental cirrhosis. Different techniques demonstrated that naringenin had the ability to reverse elevated liver damage biochemical markers and to restore GSH and glycogen levels. Additionally, the high levels of TGF-β and α-SMA (protein and mRNA) observed during CCl4 treatment were diminished by naringenin administration. The protein levels of CTGF, Col-1 and MMP13, as well as the activity of MMP2 and MMP9, proteins involved in MEC remodeling, were restored by the flavonoid. The protein levels of NF-κB, IL-1β and IL-10 were elevated during CCl4 intoxication; however, naringenin reversed this effect. Naringenin also reversed JNK activation and Smad3 phosphorylation in the linker domain, as well as total protein and total Smad3 mRNA. The results demonstrate that naringenin blocks TGF-β-Smad3 and JNK-Smad3 pathways, and thereby, it has antifibrotic effects, making it a good candidate for properly performed clinical studies. In summary, these results show that naringenin not only reduces CCl4-induced acute liver damage but also reduces fibrosis. The action mechanism of naringenin to protect the liver from chronic liver damage covers several fronts. This flavonoid displays important effects on the endogenous antioxidant system, blocks the main proinflammatory factor, NF-κB, and inhibits many profibrogenic pathways. These actions make this flavonoid an effective compound to not only to prevent but also to reverse chronic hepatic damage induced by CCl4.
ANTICANCER ACTIVITY OF NARINGENIN IN THE LIVER
HCC is one of the most frequent tumor types worldwide. It is the fifth most common cancer in men and the ninth in women, with approximately 500000 and 200000 new cases per year, respectively[119].
HCC is a genetically heterogeneous tumor. Hepatocarcinogenesis is complex and, therefore, requires several genetic and epigenetic alterations. Several etiological factors of HCC have been defined, including HBV, HCV, excessive alcohol consumption, obesity, and aflatoxins, and the prevalence/contribution of these risk factors vary by region[120]. In Western countries, the increasing prevalence of nonalcoholic steatohepatitis (NASH), known as the manifestation of the metabolic syndrome, is becoming the most prevalent risk cause for liver failure and HCC[3].
HCC is strongly associated with oxidative stress[121]; hepatic virus infection, the deposition of heavy metals, and fatty liver disease are closely associated with chronic inflammation, which in turn can induce oxidative stress in hepatocytes[122]. Alterations in cell structure and mitochondria can generate electron leakage from the mitochondria, resulting in the activation of pro-oncogenic pathways[123]. In addition, Kupffer cell activation during inflammation produces ROS that are liberated in the liver tissue, inducing damage to the hepatocyte membrane[124].
Elevated levels of intracellular ROS induce the accumulation of many genetic and epigenetic modifications that may play a pivotal role in the induction of many proinflammatory, onco-suppressor- and onco-promoter-related genes that participate in HCC development[125]. When ROS are increased for prolonged periods of time, the antioxidant defense capacity and the repair systems of the cells can be insufficient and lead to lipid, protein and DNA damage, altering different cellular pathways and influencing gene expression, cell adhesion, cell metabolism, the cell cycle, and cell death[126]. In general, ROS have negative effects; they are potential carcinogens because of their role in mutagenesis and the consequential chromosomal aberrations[127], as well as in the regulation of tumor promotion and progression[128]. It is worth noting that ROS have been linked to the hepatocarcinogenic process because they are implicated in the activation of cellular signaling pathways, such as those mediated by MAPKs, NF-kB, PI3K, p53, and b-catenin/Wnt, which are associated with mutagenesis, angiogenesis, tumor promotion, and progression[129,130] (Figure 7).
Figure 7.
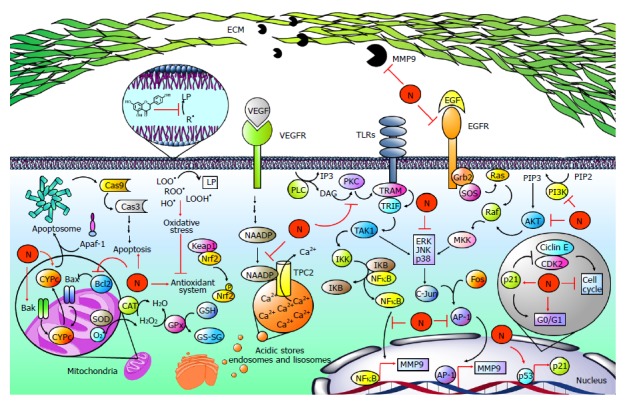
Naringenin in cancer development. Hepatocellular carcinoma is strongly associated with elevated levels of free radicals such as lipid hydroperoxides (LOOH•), peroxyl radicals (ROO•), and hydroxyl radicals (OH•), leading to the development of lipid peroxidation (LP), oxidative stress and finally to an imbalance of the endogenous antioxidant system. Naringenin (N) inhibits oxidative stress by its intrinsic antioxidant properties and by improving the endogenous antioxidant system. Notably, oxidative stress has been linked to the hepatocarcinogenic process because it is implicated in the activation of mitogen activated protein kinases (MAPKs), nuclear factor-kappa B (NF-κB), or phosphatidylinositol-3-kinase (PI3K/AKT) pathways, increasing the production and activity of metalloprotease 9 (MMP9), which is involved in migration and invasion processes. When toll-like receptors (TLRs) are activated, TRAMP is recruited to activate TRIF; in turn, it promotes transforming growth factor beta-activated kinase 1 (TAK1) activation, which phosphorylates IκB kinase (IKK). Then, IKK promotes NF-κB release via inhibitor κB (IKB) phosphorylation. On the other hand, phospholipase C (PLC) catalyzes phospholipid hydrolysis, generating inositol triphosphate (IP3) and diacylglycerol (DAG); the latter is an activator of protein kinase C (PKC), which can induce the NF-κB pathway in a TRAMP-dependent manner. Then, NF-κB induces the expression of MMP9. Epidermal growth factor (EGF) is highly involved in carcinogenic pathways; it binds to epidermal growth factor receptor (EGFR) promoting Grb2, SOS, Ras, Raf and mitogen-activated protein kinase kinase (MKK) activation, which participates in extracellular signal-regulated protein kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 (MAPK) phosphorylation and activation. Then, MAPKs promote activator protein 1 (AP-1) activation by c-Jun and Fos dimerization. After that, AP-1 induces the expression of MMP9. Alternatively, MAPKs are activated via PI3K/AKT. PI3K produces phosphatidylinositol (3, 4, 5)-trisphosphate (PIP3) from phosphatidylinositol 4,5-bisphosphate (PIP2); PIP3 activates AKT, which promotes MAPK activation in a Ras-dependent pathway. It has been reported that naringenin inhibits MM9 expression and secretion through diminution of p38, JNK, ERK, IKB, and PI3K/AKT phosphorylation as well as NF-κB and AP-1-DNA binding. In addition, naringenin inhibits PKC cytoplasm-to-membrane translocation. Notably, naringenin induces p53 accumulation, leading to p21 expression. Then, p21 inhibits cyclin E/cyclin-dependent kinase 2 (CDK2) complex, which participates in proliferation. p53 accumulation results in naringenin-induced G0/G1 phase arrests. An important mechanism for the elimination of cancer cells is apoptosis. Naringenin induces apoptosis by increased cytochrome c (CYPc) release, as well as BCL2-associated X protein (Bax), BCL2-antagonist/killer 1 (Bak) and Caspase 3 (Cas3) elevation. Additionally, naringenin inhibited B-cell CLL/lymphoma 2 (Bcl-2) an antiapoptotic protein. Two-pore channels (TPCs) are members of the voltage-gated ion channel superfamily localized in acidic calcium (Ca2+) stores and have been implicated in angiogenic processes. Vascular endothelial growth factor (VEGF) and its receptor vascular endothelial growth factor receptor (VEGFR) promotes TPC activation via nicotinic acid adenine dinucleotide phosphate (NAADP); then, Ca2+ is transported to the cytoplasm through TPCs, activating angiogenic signals. Naringenin inhibits VEGF angiogenesis induction blocking NAADP activation and NAADP/TPC association.
Abundant evidence from humans and experimental animals has shown that a high intake of natural products rich in antioxidants is associated with a decreased risk of many cancers[131-135]. Flavonoids have diverse biological activities because of their antiallergic, anti-inflammatory, antioxidant, and anticancer properties without significant systemic toxicity[134,135]. Naringenin has been found to exhibit antioxidant, antimutagenic and anticarcinogenic effects[65,136,137] and acts as chemopreventive agent against colon carcinogenesis in vitro and in vivo[138,139]. It is worth noting that naringenin inhibits cell proliferation via the downregulation of NF-κB, VEGF, and MMPs and induces apoptosis via Bcl-2, Bax and Cas in a rat model of hepatocarcinogenesis by N-nitrosodiethylamine (NDEA)[140]. Arul and Subramanian demonstrated that naringenin attenuates NDEA-induced hepatocarcinogenesis; they postulated that the flavanone aids in liver cell regeneration, leading to the protection of hepatic cells and membrane integrity by scavenging R• and enhancing the antioxidant status, thus hindering the process of carcinogenesis[141]. A growing body of evidence indicates that naringenin prevents liver damage in chemically induced hepatotoxicity by inhibiting R• and LP and by enhancing the antioxidant system of the cell[65,112,142-144]. Accordingly, the administration of naringenin effectively suppressed NDEA-hepatocarcinogenesis and preneoplastic lesions by modulating antioxidant enzymes and attenuating LP through the scavenging of free radicals, thus enhancing the antioxidant status[141]. Taken together, naringenin can markedly modulate oxidative stress by its activation of the antioxidant defense system. Thus, naringenin appears to be an attractive candidate as an antioxidant supplement for HCC prevention (Figure 7).
In another study, naringenin was found to inhibit the growth of Hep G2 cells in a concentration-dependent manner[145]. The activation of p53 has been implicated in triggering cell cycle arrest, including both G1 and G2 phases of the cell cycle. Notably, naringenin induced a rapid accumulation of p53 in a dose-dependent manner, which might account for the naringenin-induced G0/G1 and G2/M phase arrests in Hep G2 cells[145] (Figure 7).
In addition, evidence has shown that the antiproliferative effect of natural products is associated with the induction of apoptosis[146,147]. In agreement, naringenin was found to exert antiproliferative effects by inducing apoptosis, as evidenced by the nuclei damage of Hep G2 cells[148,149]. The dysregulation of the cell cycle mechanism and the induction of cancer cell apoptosis are recognized as important targets in cancer therapy. In this sense, naringenin is known to induce apoptosis through the modification of Bcl-2 family of proteins involved in the apoptotic mitochondrial pathway, and the results from HepG2 cells showed that naringenin increases the activity of Cas3[145]. Additionally, flow cytometry with Annexin V-FITC/PI staining demonstrated that the flavonoid increased apoptotic cells, confirming that naringenin induced apoptosis in HepG2 cells[145]. The accumulated data suggest that naringenin, as well as other compounds derived from plants, may induce apoptosis through the mitochondria-initiated death pathway[148,150,151] (Figure 7).
On the other hand, two-pore channels (TPCs), are members of the voltage-gated ion channel superfamily and localize in acidic Ca2+ stores and have been implicated in different pathophysiological processes[152,153]; TPC2 is expressed predominantly in late endosomes and lysosomes[154]. It has been found that naringenin impairs TPC2-dependent biological activities, leading to antiangiogenic effects mediated by VEGF. Overall, these data suggest that naringenin inhibition of TPC2 activity and the observed inhibition of the angiogenic response to VEGF are linked by impaired intracellular calcium signaling[155]. Therefore, TPC2 inhibition is emerging as a key therapeutic step in the progression and metastatic potential of malignant cells. The identification of naringenin as an inhibitor of TPC2-mediated signaling provides a novel and potentially relevant tool for the advancement of anticancer research (Figure 7).
12-O-tetradecanoylphorbol-13-acetate (TPA) is widely utilized for studying the mechanisms of carcinogenesis[156]. TPA upregulated MMP9 expression via PKC-dependent activation of the Ras/ERK signaling pathway, increasing invasiveness in cell lines[157] and tumor metastasis[158]. Importantly, Yen et al[159] demonstrated that naringenin possesses a strong antiinvasive and antimigratory effect in TPA-activated hepatoma cells via the downregulation of PKC, epidermal growth factor (EGF), MAPK and PI3K/Akt signaling pathways, and NF-κB, AP-1 and MMP9 activities (Figure 6).
In conclusion, naringenin is highly effective in inhibiting cell proliferation and inducing apoptotic cell death in HepG2 cells and reducing invasion and metastasis. Therefore, it may be a promising candidate for hepatocarcinogenesis treatment.
NARINGENIN PROTECTS FROM LIVER DAMAGE INDUCED BY HEAVY METALS
Heavy metals can be classified according to their mechanism of action in redox-active metals or redox-inactive metals. Redox-active metals such as iron (Fe), copper (Cu), chromium (Cr), cobalt (Co), among others, develop redox cycling reactions, and they produce R• in biological systems, producing oxidative stress, LP, DNA damage and other deleterious effects. Meanwhile, redox inactive metals such as cadmium (Cd), arsenic (As) and lead (Pb) bind to proteins and sulfhydryl groups and induce GSH depletion[160].
In this section, liver damage caused by redox-active and -inactive metals will be discussed.
Iron
Iron is an indispensable micronutrient for living organisms; it participates in oxygen transport, DNA synthesis and host defense, among others. Total body iron content ranges from 3 to 5 g, but its level increases due to diseases or intoxication[161]. The liver is the main iron depot; thus, it is highly susceptible to damage induced by iron overload[161,162].
Iron is captured by hepatocytes through transferrin receptor 1 (TfR1); during iron overload, its transcript is degraded and its synthesis is inhibited; however, iron uptake can be mediated by TfR2 even with high iron levels[161,162]. When iron binding capacity or transferrin saturation is exceeded, non-transferrin bound iron (NTBI) is elevated, and then it is transported into hepatocytes through divalent metal transporter 1 (DMT1). In hepatocytes, iron is incorporated into the ferritin molecule that preserves iron bioavailability[162] (Figure 8).
Figure 8.
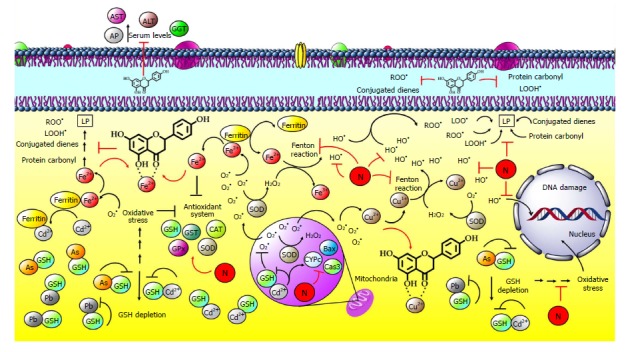
Naringenin inhibits hepatic damage induced by heavy metals. Heavy metals can be classified according to the mechanism of action in redox-active metals such as iron (Fe) and copper (Cu) or redox-inactive metals such as cadmium (Cd), arsenic (As) and lead (Pb). One of the main free radicals is superoxide radical (O2•); normally, it is produced by NADP oxidase, and then, it is neutralized by superoxide dismutase (SOD), generating hydrogen peroxide (H2O2). Intracellular Fe is releases from ferritin by O2•; next, free Fe reacts with H2O2 in the Fenton reaction, generating high amounts of hydroxyl radicals (OH•). After that, OH• attacks double bonds of DNA bases. In the case of lipids, free Fe produces lipid peroxidation (LP) through peroxyl radicals (ROO•), producing lipid hydroperoxides (LOOH•), conjugated dienes and protein carbonyl. Regarding Cu, once inside the hepatocyte, Cu ion (Cu2+), can be reduced to cuprous ion (Cu1+) when reacting with O2•; then, it mediates H2O2 decomposition in OH• via the Fenton reaction. These processes result in hepatocyte and liver damage. Naringenin can chelate these metals, preventing their participation in the Fenton reaction; naringenin also inhibits oxidative stress by its antioxidant capacity and by promoting the endogenous antioxidant system. On the other hand, redox-inactive metals such as Cd, arsenic (As) and lead (Pb) form complexes with thiol groups, such as glutathione (GSH), in the cytoplasm and mitochondria. GSH level reduction, GSH inactivation, and GSH system deregulation increase metal toxicity. In addition, Cd can replace Fe and Cu in ferritin or apoferritin; thus, free Fe and Cu ions cause oxidative stress via the Fenton reactions and elevation of BCL2-associated X protein (Bax), Caspase 3 (Cas3) and cytochrome (CYPc) proapoptotic proteins. Naringenin improves the antioxidant system by increasing SOD, catalase (CAT), glutathione peroxidase (GPx), glutathione transferase (GST) enzymes and GSH levels.
One of the most reactive R• is O2•; under normal conditions, it is produced in the respiratory chain by NADP oxidase, and then, it is neutralized by SOD, generating H2O2. Intracellular iron is released from ferritin by O2•; next, free iron reacts with H2O2 in the Fenton reaction, generating high amounts of OH•, and in turn, OH• attracts the double bonds of DNA bases. In the case of lipids, free iron produced LP forming ROO•[160]. These processes produce hepatocyte damage, such as mitochondrial dysfunction and apoptosis, which results in the recruitment of Kupffer cells that phagocyte damaged hepatocytes, leading to the release of proinflammatory and profibrogenic cytokines that activate HSCs; as a result, hepatic fibrosis ensues[161-163].
The Fenton reaction is inhibited by flavonoids with 3´,4´-catechol, 4-oxo, and 5-OH arrangements. Chelating complexes with cations may form between the 5-OH and 4-oxo group or between the 3´- and 4´-OH[29]. Using an electrospray mass spectrometry study, it was observed that naringenin can form complexes with Fe(III) through its 4-oxo and 5-OH groups; in addition, this flavonoid is oxidized in the presence of metal ions, which are consequently reduced[164]. Furthermore, naringenin was investigated for its ability to suppress the Fenton reaction characteristic of the iron-ATP complex; the flavanone interfered with the voltammetry catalytic wave associated with the iron-ATP complex in the presence of H2O2 because it has the arrangement of 4-oxo and 5-OH that is indispensable for this inhibition[165] (Figure 8).
In an experiment where the modulation of DNA integrity in Fenton’s system by naringin was studied, it was shown that the glycoside protected DNA from damage caused by OH• generated during the Fenton reaction; naringin blocks the Fenton reaction by iron chelation rather than by antioxidant mechanisms or reduction of Fe(III) to Fe(II), and as a result, damage is prevented[166]. In another study, the isolated mouse liver mitochondrial fraction was incubated with naringin before Fe(III) loading, generating elevations in LP, protein carbonyl content and DNA oxidation, while iron overload decreased GSH levels and GST, GPx, CAT and SOD activities; however, pretreatment with naringin inhibited these iron effects[167]. Iron exposure in HepG2 cells caused a decline in cell survival, a time-dependent increase in DNA oxidation, an elevation in DNA strand breaks, a high level of LP, and a depletion of GSH as well as decreases in GPx, CAT and SOD levels. Notably, the pretreatment of HepG2 cells with naringin resulted in cell survival induction, DNA damage prevention, improvement in the antioxidant system and the inhibition of iron-mediated cellular damage[168] (Figure 8).
Regarding naringenin’s effects on iron-induced damage in vivo, it has been reported that the flavanone protected against iron-induced neurotoxicity in the cerebral cortex of Wistar rats. After four weeks of iron administration, LP and protein oxidation were increased, but SOD, CAT, total thiols and ascorbic acid were decreased. Significant decreases in acetylcholinesterase and Na+/K+-ATPase activities were also shown, along with a substantial rise in NO levels. Co-administration with naringenin blocked the development of oxidative stress and improved antioxidant enzyme activities in the cerebral cortex[169]. In another work, the effect of naringenin on iron-induced hippocampus damage was investigated: iron administration for 28 d induced an impairment of the anxiogenic-like behavior and induced purinergic and cholinergic dysfunctions with oxidative stress-related disorders on mitochondrial function in the rat hippocampus, but naringenin was able to restore those parameters[170] (Figure 8).
As seen, naringenin and naringin have the ability to block iron-induced oxidative stress; these natural compounds are able to chelate metal ions such as iron; thus, free iron is not available for the Fenton reaction, and therefore, OH• generation is blocked, as is oxidative stress. The chelation capacity is given in the naringenin molecule by the 4-oxo and 5-OH groups, which probably represent the place where an iron ion is conjugated. In the absence of this arrangement, some flavonoids do not have chelating capacity or are less effective[164-166]. This structure-activity relationship indicates that naringenin and naringin can act as antioxidants or as chelators, depending on the hepatotoxic agent employed.
Copper
Copper is a redox active metal, and an imbalance in its metabolism produces disorders such as Wilson´s disease, Indian childhood cirrhosis or endemic Tyrolean infantile cirrhosis, which share the common end of cirrhosis due to excessive copper accumulation; another problem is copper toxicity caused by copper poisoning or dietary copper toxicity[160,171,172]. Like iron, copper exerts its hepatotoxic effects by oxidative stress generation; this is a consequence of its redox reactivity, triggering events that end in liver damage.
Like iron, copper is stored in the liver; it is introduced into the hepatocyte through the high-affinity human copper transporter (hCtr1)[173]. Once inside the hepatocyte, cupric ion (Cu(II)), can be reduced to cuprous ion (Cu(I)) when reacting with O2•, ascorbic acid or GSH; meanwhile, Cu(I) mediates H2O2 decomposition in OH• via the Fenton reaction[160]. The formed OH• reacts with lipids, proteins and DNA, as well as with practically any biological molecule, generating lipid radicals, protein carbonyls or DNA strand breaks and oxidation of bases; in fact, copper is more powerful that iron in enhancing DNA breakage[160,174]. Furthermore, copper binds directly to free thiols of cysteines, which can result in oxidation and subsequent crosslinks between proteins, leading to impaired activity of target enzymes[171]. In addition, copper induces LP and 4-hydroxy-2-nonenal (HNE) formation. Importantly, HNE may increase the phosphorylation of JNK and p38, AP-1 activity and the expression of Col-I and TGF-β[160], resulting in the exacerbation of fibrosis (Figure 8).
As previously mentioned, naringenin may act as a metal chelator. In this regard, two studies have reported the chelating capacity of the flavonoid on copper. Fernández et al[164] showed that naringenin, at various stoichiometries (metal: flavonoid) with copper, 1:1, 1:2, 2:2 and 2:3, produces several complexes, preferably with Cu(II). Additionally, comparing the 4-oxo and 5-OH arrangement with the 4-oxo and 3-OH arrangement, the first one seems to favor cooper chelation[164]. Meanwhile, Mira et al[175] reported that naringenin has higher reducing capacity for copper ions than for iron ions. Additionally, the copper reducing activity seems to depend largely on the number of OH groups. In addition, naringenin chelates Cu2+ at pH 7.4 and pH 5.5 between the 5-OH and the 4-oxo groups, producing 7.1 ± 1.1 mmol Cu+/mmol naringenin, indicating that a large number of copper ions per molecule of flavonoid were chelated[175] (Figure 8).
It has been shown that copper induces the oxidation of low-density lipoproteins (LDL); as a consequence, PUFAs in the lipoprotein are rapidly converted to LOOH and aldehydic breakdown products[160,176,177]. It has been reported that when freshly isolated human LDL (50 μg protein/mL) was incubated with 2 μmol/L Cu2+ at 37 °C, naringenin (25 μmol/L) slightly inhibited LDL oxidation, but prenylflavanones (25 μmol/L) such as 8-geranylnaringenin, 6-prenylnaringenin, 8-prenylnaringenin and 6,8-diprenylnaringenin effectively inhibit LDL oxidation dienes formation. Then, Cu2+-mediated LDL oxidation was evaluated by measuring TBARS levels; the results showed that prenylflavanones significantly inhibited TBARS formation and were ranked as follows: 8-geranylnaringenin > 6,8-diprenylnaringenin, 6-geranylnaringenin, 8-prenylnaringenin > 6-prenylnaringenin[177] (Figure 8).
As seen, naringenin and its derivatives can inhibit the first steps of copper-induced damage by preventing the Fenton reaction and by preventing lipid and protein oxidation.
Cadmium
Unlike iron and copper, cadmium is a redox inactive metal; although it does not directly form R•, it can induce oxidative stress in other ways. In addition, there is no mechanism for cadmium excretion in humans; thus, cadmium accumulates in tissues indefinitely[160,178].
Cadmium is absorbed though the intestines, and in the liver, DMT1, ZIP8 and ZIP14 are responsible for Cd uptake into hepatocytes[178]. Once inside hepatic cells, cadmium follows two pathways to exert liver damage: (1) Cadmium forms complexes with thiol groups of proteins or small peptides, such as GSH, in the cytoplasm and mitochondria. GSH is the first line of defense against cadmium-induced damage; thus, GSH level reduction, inactivation, and GSH system deregulation increase cadmium toxicity. In mitochondria, thiol group inactivation causes oxidative stress, mitochondrial permeability transition, and mitochondrial dysfunction[178,179]. And (2) Cadmium can replace iron and copper in ferritin or apoferritin; thus, free iron and copper ions readily cause oxidative stress via the Fenton reaction[160,178]. Thereby, although cadmium is unable to generate R• directly, indirect formation of ROS, O2•, OH• and NO has been reported. In addition, increased LP levels and antioxidant system deregulation has been observed during cadmium liver damage[160,178,179]. Because of oxidative stress induced from cadmium intoxication, Kupffer and HSCs cells can be activated, and thus, a large number of inflammatory and cytotoxic mediators can be produced[178,179] (Figure 8).
One of the first reports on the beneficial effect of naringenin on damage induced by cadmium was performed in kidney, and after 4 wk of CdCl2 administration (5 mg/kg/d), TBARS, LOOH and protein carbonyl levels were elevated. Conversely, total sulfhydryl groups, GSH, vitamin C and vitamin E levels, as well as SOD, CAT, GPx, GST and GR, and glutathione-6-phosphate dehydrogenase (G6PD) activities were decreased. Co-administration of naringenin (25 and 50 mg/kg daily) resulted in the prevention of Cd-induced LP and in the restoration of the endogenous antioxidant system. Histopathological analysis showed that naringenin markedly reduced CdCl2 toxicity and preserved the normal histological architecture of renal tissue[180].
Later, Renugadevi et al[65] reported that cadmium (5 mg/kg) administered orally for 4 wk induced liver damage. Increased activities of serum AST, ALT, AP, LDH, GGT and bilirubin were found. Furthermore, LP and protein carbonyl contents were elevated. Antioxidant enzymes such as SOD, CAT, GPx, and GST as well as GSH, vitamin C and vitamin E concentrations were diminished. Naringenin (50 mg/kg) significantly prevented the elevation of serum hepatic marker enzymes. Additionally, the flavonoid significantly reduced LP and restored antioxidant defense levels. The histopathological studies showed that naringenin preserved normal histological architecture of the tissue[65]. The same working group reported that naringenina plus vitamins C and E improved the altered biochemical and histopathological changes in the liver of Cd-intoxicated rats to a greater extent than naringenin or vitamins alone[72] (Figure 8).
In another work, naringenin (4 and 8 mg/kg) was orally administered to mice 30 min before oral administration of CdCl2 (12 mg/kg) for 11 consecutive days. Cotreatment with naringenin significantly prevented disarrangement in body and organ weights, hematological profiles, serum and hepatic altered biochemical parameters in Cd-intoxicated mice[181].
Naringin also displays protective effects in cadmium-induced damage to HepG2 cells, where the glycoside maintained redox homeostasis, mitochondrial membrane potential, reduced Cas3 and CYPc and reduced apoptosis by regulating the Bax/Bcl2 ratio. Moreover, naringin prevented diminution of protein thiol levels, SOD, GST and CAT activities, and LP development through increasing Nrf2 and metallothionein (MT)[182] (Figure 8).
Most evidence concurs that naringenin prevents cadmium-induced liver damage by protecting enzymatic and non-enzymatic antioxidant systems and by safeguarding GSH thiol groups. The antioxidant actions of naringenin may also be associated with its ability to chelate heavy metals, thus preventing the formation of ROS and with its ability to increase Nrf2. These data show that naringenin is effective in preventing damage induced by cadmium.
Arsenic
Arsenic is a highly distributed metal that is found in organic and inorganic forms; both forms are toxic, although inorganic arsenic is more toxic than organic arsenic[160]. This metal is metabolized by reduction and methylation reactions, which are catalyzed by glutathione-S-transferase omega-1(GSTO1) and arsenic (III) methyltransferase (AS3MT); it has been reported that during arsenic metabolism, high amounts of reactive species are generated[160,183].
Like cadmium, arsenic induces cellular damage through binding to sulfhydryl groups and inducing mitochondrial dysfunction. Cadmium produces oxidative stress-generating species such as O2•, singlet oxygen (1O2), ROO•, NO, H2O2, dimethylarsinic peroxyl radical [(CH3)2AsOO•] and dimethylarsinic radical [(CH3)2As•][160,184]. In general, an oxidative environment results in GSH depletion, LP elevation, protein oxidation, DNA damage, morphologic changes in mitochondrial integrity and a rapid decline of mitochondrial membrane potential[160,184,185]. Oxidative stress induces hepatocyte apoptosis as well as total bilirubin, ALT, and AST elevation and liver damage [183] (Figure 8).
Since arsenic induces damage via oxidative stress, naringenin has been studied in arsenic-induced liver damage. Arsenic administration (2 mg/kg) for 28 d to rats or 14 d (3 mg/kg) to mice produced elevations in AST, ALT and AP activities, high LP markers, hepatic GSH depletion and reductions in SOD, CAT, GPx, GST and GR activities. In addition, arsenic exposure produced DNA fragmentation. However, the simultaneous administration of naringenin prevented hepatic injury by arsenic[68,74].
Jain et al[73] reported that NaAsO2 administration (8 mo) to male Wistar rats induced high levels of ROS in blood and liver and increased levels of hepatic LP; simultaneously, the endogenous antioxidant system was attenuated, leading to a reduction of GSH levels and to the inhibition of GPx, GST, SOD and CAT activities in liver. Once liver damage was established, naringenin was administered for two weeks; the flavanone was able to reverse oxidative stress, since ROS and TBARS levels were diminished. Moreover, the enzymatic antioxidant system was restored by naringenin[73].
Naringin also has been shown to prevent liver and kidney damage induced by NaAsO2 (5 mg/kg); the glycoside inhibited increased serum levels of ALT and AST as well as prevented SOD and GSH depletion. In addition, naringin downregulated the expression of TGF-β, Cas3 and TNF-α in kidney[186] (Figure 8).
In summary, naringenin and naringin display hepatoprotective effects in arsenic-induced liver injury mainly by improving the endogenous antioxidant system and probably by their chelating effect.
Lead
The mechanism of action of lead toxicity is similar to those of cadmium and arsenic. This heavy metal does not generate free radicals directly; instead, lead deactivates antioxidant pools by binding to sulfhydryl groups of protein or peptides. For instance, lead-GSH interaction inactivates GSH antioxidant activity; moreover, lead reduces GSH levels bay blocking GR, GSG and δ-aminolevulinic acid dehydratase (ALAD), an enzyme in charge of preserving the GSH/GSSG balance[160,187-189]. The inhibition of the antioxidant GSH system produces R• such as O2•, 1O2 and ROO•, which destabilize cellular membranes through LP processes, resulting in mitochondria and DNA damage leading to p53 upregulation, an imbalance of Bax/Bcl-2 and apoptosis. After oxidative damage caused by lead, proinflammatory pathways are activated, exacerbating preexisting liver damage[187,188] (Figure 8).
Two reports have been published dealing with naringenin’s effects on lead-induced liver injury. Wang et al[58] and Ozkaya et al[144] reported that rats treated with lead acetate in drinking water showed significant increases in MDA and depletion of GSH levels and GPx activity. Elevated levels of ALT and AST in serum and decreased SOD activity in liver were also shown[58]. Furthermore, histopathological results showed that the livers of lead-intoxicated rats had periportal cell infiltration, sinusoidal congestion, hepatic steatosis, and capsular fibrosis[144]. Naringenin administration (50 mg/kg) prevented the disarrangement of most parameters studied, and histopathological abnormalities such as necrosis, hydropic degeneration, and hepatic cord disorganization were attenuated by naringenin treatment[58,144] (Figure 8).
These studies show that naringenin has hepatoprotective effects against lead-induced liver damage; however, more studies are needed to further understand the naringenin mechanism of action.
ANTIVIRAL PROPERTIES OF NARINGENIN
The study of hepatovirus has been an important issue in hepatology research in the last four decades. HBV and HCV are most studied, as they produce chronic liver damage, leading to cirrhosis and HCC[190]. As global causes of liver cirrhosis, HBV accounts for 30%, HCV for 28%, alcohol for 27% and others for 14% of cirrhosis cases. The etiology of liver cancer is HBV (45%), HCV (26%), alcohol (20%), and others (39%)[3]. Therefore, research on treatment for these infections is important for the prevention/reversion of chronic liver diseases.
HCV is a virus belonging to the Flaviviridae family; its genome consists of a positive-sense single-stranded RNA. Hepatocytes are the major site of HCV replication, but peripheral blood mononuclear cells and lymph nodes are also natural HCV targets[3,191,192].
HSC machinery processes three structural HCV proteins (core, E1 and E2), an ion channel protein (p7) and six non-structural proteins (NS) (NS2, NS3A, NS4A, NS4B, NS5A and NS5B)[191]. HCV adopts an icosahedral structure with a lipid envelope and glycoproteins E1 and E2 immersed in the envelope. Underneath the envelope is the nucleocapsid, composed of multiple copies of core forming the internal viral coat that encapsulates the genomic RNA[192] (Figure 9).
Figure 9.
Antiviral properties of naringenin. The Hepatitis C virus (HCV) genome consists of a positive-sense single-stranded RNA. HCV adopts an icosahedral structure with a lipid envelope and glycoproteins E1 and E2 immersed in the envelope. Underneath the envelope is the nucleocapsid, which is composed of multiple copies of core forming the internal viral coat that encapsulates the genomic RNA. HCV may be associated with lipoproteins such as very-low density lipoprotein (VLDL), forming a lipoprotein complex called lipoviroparticle. Binding of lipoviroparticle to very-low density lipoprotein receptor (VLDLR) results in virus endocytosis; after that, nucleocapsids are deposited into the cytoplasm. Then, nucleocapsids are uncoated, and the RNA is released. The genomic RNA is translated to the endoplasmic reticulum when HCV polyprotein is produced. The positive-stranded RNA genome is used as a template for synthesis of negative-stranded RNA; this new viral RNA is encapsulated within multiple copies of the core to form the nucleocapsid, and then, it acquires envelope and HCV virions, which are exported out of the cell in a Golgi-dependent manner. Naringenin inhibits the secretion and assembly of HCV through regulating lipid metabolism via 3-hydroxy-3-methylglutaryl CoA reductase (HMGCR), and acyl-coenzyme A: cholesterol acyltransferase (ACAT) inhibition. Cholesterol is synthetized in an HMGCR-dependent pathway; it is the rate limiting enzyme for cholesterol synthesis, and then, cholesterol is converted to cholesteryl esters (CEs) by ACAT. CEs are very important to VLDL assembly. In addition, microsomal triglyceride transfer protein (MTP) catalyzes the transfer of lipids to the apolipoprotein (Apo) B-100 ApoB100; if the ApoB-MTP binding is inhibited, VLDL assembly in inhibited. Reduction in the bioavailability of CEs, triglycerides and cholesterol by naringenin reduces MTP activity and apoB-MTP binding. In addition, naringenin decreased intracellular triglycerides through peroxisome proliferator activated receptor alpha (PPARα), a regulator of lipid metabolism. Through these mechanisms, naringenin leads to a reduction in VLDL assembly and to the inhibition of ApoB-dependent HCV secretion. Additionally, naringenin inhibits viral NS5A protein, a multifunctional HCV nonstructural protein. Furthermore, naringenin could be an NS2 protease and core protein inhibitor.
E1 and E2 are responsible for receptor binding and HCV entry into hepatocytes. Among the receptors for HCV, CD81 is probably the best characterized; low-density lipoprotein receptor (LDLR), scavenger receptor class B type I (SR-B1), human scavenger receptor, and glycosaminoglycans may also act as receptors for HCV[191,192]. After binding to its receptor, HCV endocytosis is activated, leading to the uptake of HCV particles across the cell plasma membrane[191]. After endocytosis, nucleocapsids are deposited into the cytoplasm via a low pH dependent mechanism; then, the nucleocapsids are uncoated, and their RNA is released[191,192] (Figure 9).
Genomic RNA translation is mediated by an internal ribosome entry site (IRES) binding to the ribosome; then, the HCV polyprotein is produced in the rough endoplasmic reticulum (RER) membrane, and after that, viral proteins remain associated with intracellular membranes and gave rise to a seemingly ER-derived membranous web where NS proteins form the replication complex (RC)[191,192]. Within the RC, the positive-stranded RNA genome is used as a template for synthesis of negative-stranded RNA, which in turn serves as a template for new positive-stranded synthesis. New viral RNA is encapsulated within multiple copies of the core to form the nucleocapsid, and then, it acquires envelope; HCV virions are exported out the cell ready to infect healthy hepatocytes[192] (Figure 9).
An interesting phenomenon is that HCV circulates in the blood in the form of a lipoprotein complex called lipoviroparticle (LVP); it has been reported that HCV may be associated with lipoproteins such as VLDL and low-density lipoprotein (LDL). Notably, the binding of lipoviroparticle to receptors as LDLR or SR-B1 enables the infectivity of HCV and its escape from the humoral immune response[190-193]. A relationship between the virion production process and lipoproteins, cholesterol, triglycerides and fatty acids has been suggested. HCV assembly appears to occur on lipid droplets, and the core protein clearly coats the surface of this organelle, but the lipid droplet not only serves as a site for viral assembly but also supplies lipoproteins that complex with HCV particles[191] (Figure 9).
It has been reported that HCV core protein is bound to apolipoprotein (Apo) B-100 and, therefore, to VLDL in HCV secreted by infected cells in the JFH1/Huh7.5.1 full viral life-cycle model. In addition, the HCV-VLDL complex is actively secreted by the cells; moreover, the colocalization of HCV’s core and ApoB100 was found in the cytoplasm of infected cells. Interestingly, silencing ApoB production by a SureSilencing shRNA in the cell downregulates HCV core protein secretion and HCV-positive strand RNA secretion[193] (Figure 9).
Naringenin was used as an ApoB100 inhibitor because the flavonoid reduces microsomal triglyceride transfer protein (MTP) and enzyme acyl-coenzyme A (CoA): cholesterol acyltransferase (ACAT) activity, whose expression is indispensable for ApoB synthesis[11,193]. The results showed that naringenin inhibits the secretion of HCV core and HCV-positive stranded RNA, as well as HCV secretion, more than ApoB10 silencing by the SureSilencing shRNA. Nevertheless, intracellular levels of HCV-positive strand RNA and intracellular HCV core protein expression remained unchanged; despite this, the ability of the secreted virus to infect cells was strongly inhibited following naringenin treatment. This inhibition by naringenin was mediated by a reduction in MTP activity and by the transcriptional inhibition of 3-hydroxy-3-methylglutaryl CoA reductase (HMGCR) and acyl-coenzyme A: Cholesterol acyltransferase (ACAT2)[193] (Figure 9).
Inhibition of HCV secretion by naringenin is mediated by a reduction in ApoB100 synthesis because naringenin regulates proteins related with ApoB. Normally, cholesterol is synthetized in an HMGCR-dependent pathway which is the rate-limiting enzyme for cholesterol synthesis; then, cholesterol is converted to cholesteryl esters (CEs) by ACAT. CEs are very important to VLDL and LDL assembly. Another important element to VLDL and LDL assembly is MTP, which plays a key role in ApoB100 secretion by catalyzing the transfer of lipids to the nascent ApoB100; if ApoB-MTP binding is inhibited, ApoB is predicted to undergo degradation[11].
Naringenin improves metabolic imbalance by reducing the activity and mRNA levels of HMGCR, which explains the finding that the flavanone can decrease hepatic cholesterol. In addition, naringenin possesses the ability to reduce the CE mass and cholesterol esterification by decreasing ACAT1 and ACAT2 activity[11,194-196]. The mRNA levels of MTP are significantly reduced by naringenin; therefore, ApoB-MTP binding is inhibited, and consequently, ApoB is degraded. In addition, although ApoB mRNA levels are not affected by naringenin, the protein does not accumulate in hepatocytes, suggesting that naringenin promotes the degradation of ApoB. Thereby, the reduction in the bioavailability of CEs, triglycerides and cholesterol by naringenin reduces MTP activity and ApoB-MTP binding, leading to ApoB degradation[11,197-203] (Figure 9). This seems to be the primary mechanism by which naringenin blocks ApoB secretion and VLDL and LDL assembly and, therefore, the inhibition of ApoB–dependent HCV secretion.
In addition, the same group[204] reported that naringenin treatment did not lead to the intracellular accumulation of infectious HCV particles compared with brefeldin A (BFA), a toxin known to disrupt HCV mature Golgi-dependent export; hence, naringenin blocks the assembly of HCV prior to viral egress. The inhibition of MTP and BFA treatment in JFH1-infected Huh7.5.1 cells blocks the accumulation of intracellular infectious HCV particles, indicating that MTP activity is essential for HCV assembly. Treatment of JFH1-infected Huh7.5.1 cells with naringenin (MTP inhibitor) and BFA decreased the accumulation of infectious particles, suggesting that the flavonoid inhibits the assembly of HCV LVP[204] (Figure 9).
MTP inhibition can lead to lipid accumulation and steatosis; however, treatment with naringenin decreased intracellular triglycerides, and this was mediated by activation of peroxisome proliferator-activated receptor alpha (PPARα), a regulator of lipid metabolism. Naringenin and WY14643 (a classical PPARα agonist) were compared to reduce ApoB and virus production in the HCV model. The results showed that both the flavonoid and the PPARα agonist caused a significant inhibition of MTP activity and ApoB secretion, as well as a significant inhibition of HCV RNA secretion without affecting the intracellular levels of the HCV core protein. In addition, the treatment with naringenin led to a rapid 1.4 log reduction in secreted HCV in cell culture, but this effect was reversible by PPARα inhibitor treatment[204]. In summary, naringenin inhibits the assembly and long-term production of infectious HCV particles through a PPARα-mediated mechanism that includes the inhibition of MTP and the inhibition of lipid accumulation (Figure 9).
Interestingly, Khachatoorian et al[205] compared the antiviral effects of naringenin, quercetin and catechin. The evidence demonstrated that in an HCV system, naringenin significantly reduced intracellular viral protein translation as well as viral protein production during one viral life cycle; however, quercetin showed better results, but the infectious virion secretion was not inhibited by any flavonoid. Naringenin significantly blocked infectious virion assembly; in this case, naringenin was more effective than quercetin[205].
NS5A is a multifunctional NS protein and viral RC component; it participates in HCV genome replication, viral protein translation, virion assembly, and viral secretion[191,192,205]. NS5A mRNA and protein levels were measured, finding that naringenin reduced both parameters. Then, in a cell culture-based bicistronic reporter system, catechin, naringenin, and quercetin were tested to measure levels of viral IRES-mediated translation; all bioflavonoids significantly decreased IRES-mediated translation, but quercetin completely blocked NS5A-augmented IRES activity in contrast to catechin and naringenin, which demonstrated only mild inhibition. According to these results, quercetin demonstrated a marked decrease in HSP70 expression in treated cells. A slight decrease in HSP70 was seen with naringenin and catechin treatments. The complex of HSP70 with NS5A, NS5A-HSP70, is important for viral protein production; therefore, the disruption of this complex results in a marked decrease of viral protein synthesis[206,207] (Figure 9).
On the other hand, in silico studies have been carried out to evaluate naringenin activity on the HCV particle. Mathew et al[208] in 2014 reported a docking interaction study between the 3D structure of capsid core protein of HCV-genotype 3 (G3) (Q68867) and its subtypes 3b (Q68861) and 3g (Q68865) from north India and naringenin. The results indicated that the flavonoid exhibited five, seven and nine H-bond interactions within the core protein of HCV-G3, subtypes 3b and 3g, respectively. In HCV-G3, naringenin formed H-bonds individually with GLU69 and ASN115 and three H-bonds, with SER103 exhibiting the highest interaction energy (-129.636 kcal/mole). In the case of HCV-3b, naringenin formed three H-bonds with TRP90, two with GLN86, and one with GLY84 and TRP93, with an interaction energy of -145.682 kcal/mole. Finally, the flavanone binds to HCV-3g through two H-bonds with TRP73 and GLY77 and individually with ASN85, TYR78 and TRP73 with an interaction energy of -159.483 kcal/mole[208]. These results suggest that naringenin binds to the core protein of three important HCV genotypes in India, especially to HCV-3 based on their interaction energies; this ability of naringenin to bind core protein could be involved in the inhibition of viral particle assembly that was previously reported. Naturally, in vivo studies are needed to confirm predictions suggested by this docking study (Figure 9).
NS2 is a transmembrane protein of 21-23 kDa that is not required for RNA replication but that is vital to produce infectious viruses in vitro, and it acts as an apoptosis inhibitor[191,209]. Using a docking analysis, it has been identified that naringenin could be an NS2 protease inhibitor. Molecular rigid docking of the modeled NS2 protease was performed with the naringenin molecule. The flavanone had a binding energy of -7.97 kcal/mol when interacting with amino acids such as LEU9, VAL27, LEU54, ASP6, ALA5, ILE31, ALA30, LEU2, PHE33, ILE34, VAL44, ALA47, ALA43, and LEU46. In addition, naringenin possesses lower binding energy than the commercially available drugs such as eltrombopag (-5.07 kcal/mol), ribavirin (-5.89), and telbivudine (-6.39 kcal/mol)[209] (Figure 9). Therefore, naringenin appears to be a strong NS2 protein inhibitor and, thus, prevents efficient HCV infection.
More in vivo and in vitro studies are needed to further investigate the effectiveness of naringenin to fight virus infection in the liver and to elucidate the action(s) mechanism(s) involved in such protection.
ANTIDIABETIC EFFECT OF NARINGENIN
In addition to its antioxidant, scavenger, anti-inflammatory, antiviral and antifibrotic properties, naringenin possesses antidiabetic effects. It has been reported that, in diabetic rats, the flavonoid reduced diabetic markers through PPARγ and glucose transporter type 4 (GLUT4) and increased their gene and protein expression levels in pancreas[210]. In the liver, naringenin increased glycogen content, decrease activities of glycogen phosphorylase and glucose-6- phosphatase[211] and ameliorated diabetes-induced hepatotoxicity[212,213]. For more information see Nyane et al[214].
NARINGENIN SAFETY AND TOXICITY
The first study about the toxicity of naringenin was carried out in 1996, and it was found that in a model system of isolated rat liver nuclei, the flavonoid induced a concentration-dependent peroxidation of nuclear membrane lipids concurrent with DNA strand breaks[215]. It has been reported that the flavonoid can be oxidized to form naringenin phenoxyl radicals[216] and that the medium lethal dose (LD50) is > 5000 mg/kg[217]. Interestingly, embryos exposed to naringenin with hydroxyurea were significantly protected from growth and developmental retardation, and abnormalities induced by hydroxyurea[218]. Only a few studies on the safety, teratogenicity and toxicity of naringenin have been published, therefore use of this flavonoid in the clinical setting should be cautious.
CONCLUSION
Naringenin displays poor direct antioxidant properties as a free radical scavenger; however, due to its ability to induce the endogenous antioxidant system by upregulating Nrf2, this flavanone exerts important effects to maintain the normal redox of the cell, even in disease conditions where free radicals are generated as a mechanism of damage. In this scenario, throughout this review, we have described the benefits of this flavonoid in many types of liver damage in which oxidative stress plays a crucial role as causative agent. Of note, the anti-inflammatory activity of naringenin by blocking NF-κB, affords protection or relief to liver pathologies as inflammation is a common cause of damage. Moreover, naringenin displays a multitarget effect to fight fibrosis through both canonical and non-canonical TGF-β pathways and by regulating metalloproteinase activity. Additionally, this abundant citrus flavonoid has shown anticancer and antiviral activities. Even though NAR has disadvantages such as its low bioavailability, there are pharmaceutical formulations that can solve this problem. Given the evidence provided in this review, it is concluded that naringenin is a useful natural product for the treatment of many liver diseases by its antioxidant capacity, anti-inflammatory abilities, antifibrogenic properties, fibrolytic actions and anticancer and antiviral properties. However, more basic and clinical studies are needed to further support the use of this flavonoid in humans.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Mexico
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): E
Supported by National Council of Science and Technology (Conacyt) of Mexico, No. 253037.
Conflict-of-interest statement: The authors declare no conflicts of interest.
Peer-review started: March 8, 2018
First decision: March 29, 2018
Article in press: April 16, 2018
P- Reviewer: Abenavoli L, Chiu KW, Shimizu Y, Tarantino G S- Editor: Gong ZM L- Editor: A E- Editor: Huang Y
Contributor Information
Erika Hernández-Aquino, Laboratory of Experimental Hepatology, Department of Pharmacology, Cinvestav-IPN, Mexico City 07000, Mexico.
Pablo Muriel, Laboratory of Experimental Hepatology, Department of Pharmacology, Cinvestav-IPN, Mexico City 07000, Mexico. pmuriel@cinvestav.mx.
References
- 1.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pellicoro A, Ramachandran P, Iredale JP. Reversibility of liver fibrosis. Fibrogenesis Tissue Repair. 2012;5:S26. doi: 10.1186/1755-1536-5-S1-S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muriel P. The liver: General aspects and epidemiology. In Muriel P. Liver pathophysiology: therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 3–22. [Google Scholar]
- 4.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehal WZ, Schuppan D. Antifibrotic therapies in the liver. Semin Liver Dis. 2015;35:184–198. doi: 10.1055/s-0035-1550055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuppan D. Liver fibrosis: Common mechanisms and antifibrotic therapies. Clin Res Hepatol Gastroenterol. 2015;39 Suppl 1:S51–S59. doi: 10.1016/j.clinre.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Huebert RC, Rakela J. Cellular therapy for liver disease. Mayo Clin Proc. 2014;89:414–424. doi: 10.1016/j.mayocp.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poilil Surendran S, George Thomas R, Moon MJ, Jeong YY. Nanoparticles for the treatment of liver fibrosis. Int J Nanomedicine. 2017;12:6997–7006. doi: 10.2147/IJN.S145951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Girish C, Pradhan SC. Herbal drugs on the liver. In: Muriel P, editor. Liver pathophysiology: therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 605–620. [Google Scholar]
- 10.Abenavoli L, Milic N. Silymarin for liver disease. In: Muriel P, editor. Liver pathophysiology: therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 621–631. [Google Scholar]
- 11.Hernández-Aquino E, Muriel P. Naringenin and the liver. In: Muriel P, editor. Liver pathophysiology: therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 633–651. [Google Scholar]
- 12.Vázquez-Flores LF, Casas-Grajales S, Hernández-Aquino E, Vargas-Pozada EE, Muriel P. Antioxidant antiinflammatory and antifibrotic properties of quercetin in the liver. In: Muriel P, editor. Liver pathophysiology: therapies & and antioxidants. Waltham, MA: Elsevier; 2017. pp. 653–674. [Google Scholar]
- 13.Arauz J, Ramos-Tovar E, Muriel P. Coffee and the liver. In: Muriel P, editor. Liver pathophysiology: therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 675–685. [Google Scholar]
- 14.Reyes-Gordillo K, Shah R, Lakshman, Flores-Beltrán RE, Muriel P. Hepatoprotective properties of curcumin. In: Muriel P, editor. Liver pathophysiology: therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 687–704. [Google Scholar]
- 15.Ramos-Tovar E, Muriel P. Stevia as a putative hepatoprotector. In: Muriel P, editor. Liver pathophysiology: therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 715–727. [Google Scholar]
- 16.Yen FL, Wu TH, Lin LT, Cham TM, Lin CC. Naringenin-loaded nanoparticles improve the physicochemical properties and the hepatoprotective effects of naringenin in orally-administered rats with CCl4-induced acute liver failure. Pharm Res. 2009;26:893–902. doi: 10.1007/s11095-008-9791-0. [DOI] [PubMed] [Google Scholar]
- 17.Nait Chabane M, Al Ahmad A, Peluso J, Muller CD, Ubeaud G. Quercetin and naringenin transport across human intestinal Caco-2 cells. J Pharm Pharmacol. 2009;61:1473–1483. doi: 10.1211/jpp/61.11.0006. [DOI] [PubMed] [Google Scholar]
- 18.Bredsdorff L, Nielsen IL, Rasmussen SE, Cornett C, Barron D, Bouisset F, Offord E, Williamson G. Absorption, conjugation and excretion of the flavanones, naringenin and hesperetin from alpha-rhamnosidase-treated orange juice in human subjects. Br J Nutr. 2010;103:1602–1609. doi: 10.1017/S0007114509993679. [DOI] [PubMed] [Google Scholar]
- 19.Scalbert A, Morand C, Manach C, Rémésy C. Absorption and metabolism of polyphenols in the gut and impact on health. Biomed Pharmacother. 2002;56:276–282. doi: 10.1016/s0753-3322(02)00205-6. [DOI] [PubMed] [Google Scholar]
- 20.Simons AL, Renouf M, Murphy PA, Hendrich S. Greater apparent absorption of flavonoids is associated with lesser human fecal flavonoid disappearance rates. J Agric Food Chem. 2010;58:141–147. doi: 10.1021/jf902284u. [DOI] [PubMed] [Google Scholar]
- 21.Xu H, Kulkarni KH, Singh R, Yang Z, Wang SW, Tam VH, Hu M. Disposition of naringenin via glucuronidation pathway is affected by compensating efflux transporters of hydrophilic glucuronides. Mol Pharm. 2009;6:1703–1715. doi: 10.1021/mp900013d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mata-Bilbao Mde L, Andrés-Lacueva C, Roura E, Jáuregui O, Escribano E, Torre C, Lamuela-Raventós RM. Absorption and pharmacokinetics of grapefruit flavanones in beagles. Br J Nutr. 2007;98:86–92. doi: 10.1017/S0007114507707262. [DOI] [PubMed] [Google Scholar]
- 23.Zou W, Yang C, Liu M, Su W. Tissue distribution study of naringin in rats by liquid chromatography-tandem mass spectrometry. Arzneimittelforschung. 2012;62:181–186. doi: 10.1055/s-0031-1299746. [DOI] [PubMed] [Google Scholar]
- 24.Choudhury R, Chowrimootoo G, Srai K, Debnam E, Rice-Evans CA. Interactions of the flavonoid naringenin in the gastrointestinal tract and the influence of glycosylation. Biochem Biophys Res Commun. 1999;265:410–415. doi: 10.1006/bbrc.1999.1695. [DOI] [PubMed] [Google Scholar]
- 25.El Mohsen MA, Marks J, Kuhnle G, Rice-Evans C, Moore K, Gibson G, Debnam E, Srai SK. The differential tissue distribution of the citrus flavanone naringenin following gastric instillation. Free Radic Res. 2004;38:1329–1340. doi: 10.1080/10715760400017293. [DOI] [PubMed] [Google Scholar]
- 26.Bolli A, Marino M, Rimbach G, Fanali G, Fasano M, Ascenzi P. Flavonoid binding to human serum albumin. Biochem Biophys Res Commun. 2010;398:444–449. doi: 10.1016/j.bbrc.2010.06.096. [DOI] [PubMed] [Google Scholar]
- 27.Hu YJ, Wang Y, Ou-Yang Y, Zhou J, Liu Y. Characterize the interaction between naringenin and bovine serum albumin using spectroscopic approach. J Lumin. 2010;130:1394–1399. [Google Scholar]
- 28.Khan MK, Rakotomanomana N, Dufour C, Dangles O. Binding of citrus flavanones and their glucuronides and chalcones to human serum albumin. Food Funct. 2011;2:617–626. doi: 10.1039/c1fo10077g. [DOI] [PubMed] [Google Scholar]
- 29.Heim KE, Tagliaferro AR, Bobilya DJ. Flavonoid antioxidants: chemistry, metabolism and structure-activity relationships. J Nutr Biochem. 2002;13:572–584. doi: 10.1016/s0955-2863(02)00208-5. [DOI] [PubMed] [Google Scholar]
- 30.Rice-Evans CA, Miller NJ, Paganga G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol Med. 1996;20:933–956. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- 31.Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33:105–136. doi: 10.1080/713611034. [DOI] [PubMed] [Google Scholar]
- 32.Ratty AK, Das NP. Effects of flavonoids on nonenzymatic lipid peroxidation: structure-activity relationship. Biochem Med Metab Biol. 1988;39:69–79. doi: 10.1016/0885-4505(88)90060-6. [DOI] [PubMed] [Google Scholar]
- 33.Khanduja KL, Bhardwaj A. Stable free radical scavenging and antiperoxidative properties of resveratrol compared in vitro with some other bioflavonoids. Indian J Biochem Biophys. 2003;40:416–422. [PubMed] [Google Scholar]
- 34.Rodriguez RJ, Miranda CL, Stevens JF, Deinzer ML, Buhler DR. Influence of prenylated and non-prenylated flavonoids on liver microsomal lipid peroxidation and oxidative injury in rat hepatocytes. Food Chem Toxicol. 2001;39:437–445. doi: 10.1016/s0278-6915(00)00159-9. [DOI] [PubMed] [Google Scholar]
- 35.Cavia-Saiz M, Busto MD, Pilar-Izquierdo MC, Ortega N, Perez-Mateos M, Muñiz P. Antioxidant properties, radical scavenging activity and biomolecule protection capacity of flavonoid naringenin and its glycoside naringin: a comparative study. J Sci Food Agric. 2010;90:1238–1244. doi: 10.1002/jsfa.3959. [DOI] [PubMed] [Google Scholar]
- 36.van Acker FA, Schouten O, Haenen GR, van der Vijgh WJ, Bast A. Flavonoids can replace alpha-tocopherol as an antioxidant. FEBS Lett. 2000;473:145–148. doi: 10.1016/s0014-5793(00)01517-9. [DOI] [PubMed] [Google Scholar]
- 37.Saija A, Scalese M, Lanza M, Marzullo D, Bonina F, Castelli F. Flavonoids as antioxidant agents: importance of their interaction with biomembranes. Free Radic Biol Med. 1995;19:481–486. doi: 10.1016/0891-5849(94)00240-k. [DOI] [PubMed] [Google Scholar]
- 38.Arora A, Byrem TM, Nair MG, Strasburg GM. Modulation of liposomal membrane fluidity by flavonoids and isoflavonoids. Arch Biochem Biophys. 2000;373:102–109. doi: 10.1006/abbi.1999.1525. [DOI] [PubMed] [Google Scholar]
- 39.Bombardelli E, Spetta M. Phospholipid-polyphenol complexes: a new concept in skin care ingredients. Cosm Toil. 1991;106:69–76. [Google Scholar]
- 40.Kaneko T, Kaji K, Matsuo M. Protection of linoleic acid hydroperoxide-induced cytotoxicity by phenolic antioxidants. Free Radic Biol Med. 1994;16:405–409. doi: 10.1016/0891-5849(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 41.Wang K, Chen Z, Huang L, Meng B, Zhou X, Wen X, Ren D. Naringenin reduces oxidative stress and improves mitochondrial dysfunction via activation of the Nrf2/ARE signaling pathway in neurons. Int J Mol Med. 2017;40:1582–1590. doi: 10.3892/ijmm.2017.3134. [DOI] [PubMed] [Google Scholar]
- 42.Manchope MF, Calixto-Campos C, Coelho-Silva L, Zarpelon AC, Pinho-Ribeiro FA, Georgetti SR, Baracat MM, Casagrande R, Verri WA Jr. Naringenin inhibits superoxide anion-induced inflammatory pain: role of oxidative stress, cytokines, Nrf-2 and the NO-cGMP-PKG-KATP channel signaling pathway. PLoS One. 2016;11:e0153015. doi: 10.1371/journal.pone.0153015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Podder B, Song HY, Kim YS. Naringenin exerts cytoprotective effect against paraquat-induced toxicity in human bronchial epithelial BEAS-2B cells through NRF2 activation. J Microbiol Biotechnol. 2014;24:605–613. doi: 10.4014/jmb.1402.02001. [DOI] [PubMed] [Google Scholar]
- 44.Miler M, Živanović J, Ajdžanović V, Oreščanin-Dušić Z, Milenković D, Konić-Ristić A, Blagojević D, Milošević V, Šošić-Jurjević B. Citrus flavanones naringenin and hesperetin improve antioxidant status and membrane lipid compositions in the liver of old-aged Wistar rats. Exp Gerontol. 2016;84:49–60. doi: 10.1016/j.exger.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 45.Ali R, Shahid A, Ali N, Hasan SK, Majed F, Sultana S. Amelioration of benzo[a]pyrene-induced oxidative stress and pulmonary toxicity by naringenin in Wistar rats: a plausible role of COX-2 and NF-κB. Hum Exp Toxicol. 2017;36:349–364. doi: 10.1177/0960327116650009. [DOI] [PubMed] [Google Scholar]
- 46.Fan R, Pan T, Zhu AL, Zhang MH. Anti-inflammatory and anti-arthritic properties of naringenin via attenuation of NF-κB and activation of the heme oxygenase ﴾HO﴿-1/related factor 2 pathway. Pharmacol Rep. 2017;69:1021–1029. doi: 10.1016/j.pharep.2017.03.020. [DOI] [PubMed] [Google Scholar]
- 47.Al-Dosari DI, Ahmed MM, Al-Rejaie SS, Alhomida AS, Ola MS. Flavonoid naringenin attenuates oxidative stress, apoptosis and improves neurotrophic effects in the diabetic rat retina. Nutrients. 2017;9 doi: 10.3390/nu9101161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Oliveira MR, Brasil FB, Andrade CMB. Naringenin attenuates H2O2-induced mitochondrial dysfunction by an Nrf2-dependent mechanism in SH-SY5Y Cells. Neurochem Res. 2017;42:3341–3350. doi: 10.1007/s11064-017-2376-8. [DOI] [PubMed] [Google Scholar]
- 49.Ramprasath T, Senthamizharasi M, Vasudevan V, Sasikumar S, Yuvaraj S, Selvam GS. Naringenin confers protection against oxidative stress through upregulation of Nrf2 target genes in cardiomyoblast cells. J Physiol Biochem. 2014;70:407–415. doi: 10.1007/s13105-014-0318-3. [DOI] [PubMed] [Google Scholar]
- 50.Al-Roujayee AS. Naringenin improves the healing process of thermally-induced skin damage in rats. J Int Med Res. 2017;45:570–582. doi: 10.1177/0300060517692483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sahin Z, Ozkaya A, Cuce G, Uckun M, Yologlu E. Investigation of the effect of naringenin on oxidative stress-related alterations in testis of hydrogen peroxide-administered rats. J Biochem Mol Toxicol. 2017;31 doi: 10.1002/jbt.21928. [DOI] [PubMed] [Google Scholar]
- 52.Pari L, Gnanasoundari M. Influence of naringenin on oxytetracycline mediated oxidative damage in rat liver. Basic Clin Pharmacol Toxicol. 2006;98:456–461. doi: 10.1111/j.1742-7843.2006.pto_351.x. [DOI] [PubMed] [Google Scholar]
- 53.Bodas R, Prieto N, Jordán MJ, López-Campos O, Giráldez FJ, Morán L, Andrés S. The liver antioxidant status of fattening lambs is improved by naringin dietary supplementation at 0.15% rates but not meat quality. Animal. 2012;6:863–870. doi: 10.1017/S175173111100214X. [DOI] [PubMed] [Google Scholar]
- 54.Casas-Grajales S, Muriel P. The liver, oxidative stress and antioxidants. In: Muriel P, editor. Liver pathophysiology: therapies & antioxidants. Walttham, MA: Elsevier; 2017. pp. 583–604. [Google Scholar]
- 55.DeLeve LD, Kaplowitz N. Glutathione metabolism and its role in hepatotoxicity. Pharmacol Ther. 1991;52:287–305. doi: 10.1016/0163-7258(91)90029-l. [DOI] [PubMed] [Google Scholar]
- 56.Kretzschmar M. Regulation of hepatic glutathione metabolism and its role in hepatotoxicity. Exp Toxicol Pathol. 1996;48:439–446. doi: 10.1016/S0940-2993(96)80054-6. [DOI] [PubMed] [Google Scholar]
- 57.Yuan L, Kaplowitz N. Glutathione in liver diseases and hepatotoxicity. Mol Aspects Med. 2009;30:29–41. doi: 10.1016/j.mam.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 58.Wang J, Yang Z, Lin L, Zhao Z, Liu Z, Liu X. Protective effect of naringenin against lead-induced oxidative stress in rats. Biol Trace Elem Res. 2012;146:354–359. doi: 10.1007/s12011-011-9268-6. [DOI] [PubMed] [Google Scholar]
- 59.Dong D, Xu L, Yin L, Qi Y, Peng J. Naringenin prevents carbon tetrachloride-induced acute liver injury in mice. J Funct Foods. 2015;12:179–191. [Google Scholar]
- 60.Lou H, Jing X, Wei X, Shi H, Ren D, Zhang X. Naringenin protects against 6-OHDA-induced neurotoxicity via activation of the Nrf2/ARE signaling pathway. Neuropharmacology. 2014;79:380–388. doi: 10.1016/j.neuropharm.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 61.Gopinath K, Sudhandiran G. Naringin modulates oxidative stress and inflammation in 3-nitropropionic acid-induced neurodegeneration through the activation of nuclear factor-erythroid 2-related factor-2 signalling pathway. Neuroscience. 2012;227:134–143. doi: 10.1016/j.neuroscience.2012.07.060. [DOI] [PubMed] [Google Scholar]
- 62.Han X, Pan J, Ren D, Cheng Y, Fan P, Lou H. Naringenin-7-O-glucoside protects against doxorubicin-induced toxicity in H9c2 cardiomyocytes by induction of endogenous antioxidant enzymes. Food Chem Toxicol. 2008;46:3140–3146. doi: 10.1016/j.fct.2008.06.086. [DOI] [PubMed] [Google Scholar]
- 63.Esmaeili MA, Alilou M. Naringenin attenuates CCl4 -induced hepatic inflammation by the activation of an Nrf2-mediated pathway in rats. Clin Exp Pharmacol Physiol. 2014;41:416–422. doi: 10.1111/1440-1681.12230. [DOI] [PubMed] [Google Scholar]
- 64.Jayaraman J, Veerappan M, Namasivayam N. Potential beneficial effect of naringenin on lipid peroxidation and antioxidant status in rats with ethanol-induced hepatotoxicity. J Pharm Pharmacol. 2009;61:1383–1390. doi: 10.1211/jpp/61.10.0016. [DOI] [PubMed] [Google Scholar]
- 65.Renugadevi J, Prabu SM. Cadmium-induced hepatotoxicity in rats and the protective effect of naringenin. Exp Toxicol Pathol. 2010;62:171–181. doi: 10.1016/j.etp.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 66.Martinez RM, Pinho-Ribeiro FA, Steffen VS, Silva TC, Caviglione CV, Bottura C, Fonseca MJ, Vicentini FT, Vignoli JA, Baracat MM, et al. Topical formulation containing naringenin: efficacy against ultraviolet B irradiation-induced skin inflammation and oxidative stress in mice. PLoS One. 2016;11:e0146296. doi: 10.1371/journal.pone.0146296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shakeel S, Rehman MU, Tabassum N, Amin U, Mir MUR. Effect of naringenin (a naturally occurring flavanone) against pilocarpine-induced status epilepticus and oxidative stress in mice. Pharmacogn Mag. 2017;13:S154–S160. doi: 10.4103/0973-1296.203977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roy A, Das A, Das R, Haldar S, Bhattacharya S, Haldar PK. Naringenin, a citrus flavonoid, ameliorates arsenic-induced toxicity in Swiss albino mice. J Environ Pathol Toxicol Oncol. 2014;33:195–204. doi: 10.1615/jenvironpatholtoxicoloncol.2014010317. [DOI] [PubMed] [Google Scholar]
- 69.Davies KJ. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life. 2000;50:279–289. doi: 10.1080/713803728. [DOI] [PubMed] [Google Scholar]
- 70.Jayaraman J, Namasivayam N. Naringenin modulates circulatory lipid peroxidation, anti-oxidant status and hepatic alcohol metabolizing enzymes in rats with ethanol induced liver injury. Fundam Clin Pharmacol. 2011;25:682–689. doi: 10.1111/j.1472-8206.2010.00899.x. [DOI] [PubMed] [Google Scholar]
- 71.Hermenean A, Ardelean A, Stan M, Herman H, Mihali CV, Costache M, Dinischiotu A. Protective effects of naringenin on carbon tetrachloride-induced acute nephrotoxicity in mouse kidney. Chem Biol Interact. 2013;205:138–147. doi: 10.1016/j.cbi.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 72.Prabu SM, Shagirtha K, Renugadevi J. Naringenin in combination with vitamins C and E potentially protects oxidative stress-mediated hepatic injury in cadmium-intoxicated rats. J Nutr Sci Vitaminol (Tokyo) 2011;57:177–185. doi: 10.3177/jnsv.57.177. [DOI] [PubMed] [Google Scholar]
- 73.Jain A, Yadav A, Bozhkov AI, Padalko VI, Flora SJ. Therapeutic efficacy of silymarin and naringenin in reducing arsenic-induced hepatic damage in young rats. Ecotoxicol Environ Saf. 2011;74:607–614. doi: 10.1016/j.ecoenv.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 74.Mershiba SD, Dassprakash MV, Saraswathy SD. Protective effect of naringenin on hepatic and renal dysfunction and oxidative stress in arsenic intoxicated rats. Mol Biol Rep. 2013;40:3681–3691. doi: 10.1007/s11033-012-2444-8. [DOI] [PubMed] [Google Scholar]
- 75.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 76.Xu Y, Fang F, Zhang J, Josson S, St Clair WH, St Clair DK. miR-17* suppresses tumorigenicity of prostate cancer by inhibiting mitochondrial antioxidant enzymes. PLoS One. 2010;5:e14356. doi: 10.1371/journal.pone.0014356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Curti V, Di Lorenzo A, Rossi D, Martino E, Capelli E, Collina S, Daglia M. Enantioselective modulatory effects of naringenin enantiomers on the expression levels of miR-17-3p involved in endogenous antioxidant defenses. Nutrients. 2017;9:pii: E215. [Google Scholar]
- 78.Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, Hayes JD. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88:108–146. doi: 10.1016/j.freeradbiomed.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ooi BK, Goh BH, Yap WH. Oxidative stress in cardiovascular diseases: involvement of Nrf2 antioxidant redox signaling in macrophage foam cells formation. Int J Mol Sci. 2017;18:pii: E2336. doi: 10.3390/ijms18112336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tang W, Jiang YF, Ponnusamy M, Diallo M. Role of Nrf2 in chronic liver disease. World J Gastroenterol. 2014;20:13079–13087. doi: 10.3748/wjg.v20.i36.13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang H, Xu YS, Wang ML, Cheng C, Bian R, Yuan H, Wang Y, Guo T, Zhu LL, Zhou H. Protective effect of naringin against the LPS-induced apoptosis of PC12 cells: implications for the treatment of neurodegenerative disorders. Int J Mol Med. 2017;39:819–830. doi: 10.3892/ijmm.2017.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen RC, Sun GB, Wang J, Zhang HJ, Sun XB. Naringin protects against anoxia/reoxygenation-induced apoptosis in H9c2 cells via the Nrf2 signaling pathway. Food Funct. 2015;6:1331–1344. doi: 10.1039/c4fo01164c. [DOI] [PubMed] [Google Scholar]
- 83.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zipper LM, Mulcahy RT. Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol Sci. 2003;73:124–134. doi: 10.1093/toxsci/kfg083. [DOI] [PubMed] [Google Scholar]
- 85.Liu XM, Peyton KJ, Shebib AR, Wang H, Korthuis RJ, Durante W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am J Physiol Heart Circ Physiol. 2011;300:H84–H93. doi: 10.1152/ajpheart.00749.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee SE, Yang H, Jeong SI, Jin YH, Park CS, Park YS. Induction of heme oxygenase-1 inhibits cell death in crotonaldehyde-stimulated HepG2 cells via the PKC-δ-p38-Nrf2 pathway. PLoS One. 2012;7:e41676. doi: 10.1371/journal.pone.0041676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang K, Chen Z, Huang J, Huang L, Luo N, Liang X, Liang M, Xie W. Naringenin prevents ischaemic stroke damage via anti-apoptotic and anti-oxidant effects. Clin Exp Pharmacol Physiol. 2017;44:862–871. doi: 10.1111/1440-1681.12775. [DOI] [PubMed] [Google Scholar]
- 88.Liu JD, Leung KW, Wang CK, Liao LY, Wang CS, Chen PH, Chen CC, Yeh EK. Alcohol-related problems in Taiwan with particular emphasis on alcoholic liver diseases. Alcohol Clin Exp Res. 1998;22:164S–169S. doi: 10.1111/acer.1998.22.s3_part1.164s. [DOI] [PubMed] [Google Scholar]
- 89.Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis. 2004;24:217–232. doi: 10.1055/s-2004-832936. [DOI] [PubMed] [Google Scholar]
- 90.Teli MR, Day CP, Burt AD, Bennett MK, James OF. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet. 1995;346:987–990. doi: 10.1016/s0140-6736(95)91685-7. [DOI] [PubMed] [Google Scholar]
- 91.Boye A, Zou YH, Yang Y. Metabolic derivatives of alcohol and the molecular culprits of fibro-hepatocarcinogenesis: Allies or enemies? World J Gastroenterol. 2016;22:50–71. doi: 10.3748/wjg.v22.i1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rocco A, Compare D, Angrisani D, Sanduzzi Zamparelli M, Nardone G. Alcoholic disease: liver and beyond. World J Gastroenterol. 2014;20:14652–14659. doi: 10.3748/wjg.v20.i40.14652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ceni E, Mello T, Galli A. Pathogenesis of alcoholic liver disease: role of oxidative metabolism. World J Gastroenterol. 2014;20:17756–17772. doi: 10.3748/wjg.v20.i47.17756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seo HJ, Jeong KS, Lee MK, Park YB, Jung UJ, Kim HJ, Choi MS. Role of naringin supplement in regulation of lipid and ethanol metabolism in rats. Life Sci. 2003;73:933–946. doi: 10.1016/s0024-3205(03)00358-8. [DOI] [PubMed] [Google Scholar]
- 95.Deenen MJ, Cats A, Beijnen JH, Schellens JH. Part 2: pharmacogenetic variability in drug transport and phase I anticancer drug metabolism. Oncologist. 2011;16:820–834. doi: 10.1634/theoncologist.2010-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Porta EA. Dietary modulation of oxidative stress in alcoholic liver disease in rats. J Nutr. 1997;127:912S–915S. doi: 10.1093/jn/127.5.912S. [DOI] [PubMed] [Google Scholar]
- 97.French SW. The pathophysiology of alcoholic liver disease. In: Muriel P, editor. Liver pathophysiology: therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 141–157. [Google Scholar]
- 98.Jayaraman J, Jesudoss VA, Menon VP, Namasivayam N. Anti-inflammatory role of naringenin in rats with ethanol induced liver injury. Toxicol Mech Methods. 2012;22:568–576. doi: 10.3109/15376516.2012.707255. [DOI] [PubMed] [Google Scholar]
- 99.Szkudelska K, Nogowski L, Nowicka E, Szkudelski T. In vivo metabolic effects of naringenin in the ethanol consuming rat and the effect of naringenin on adipocytes in vitro. J Anim Physiol Anim Nutr (Berl) 2007;91:91–99. doi: 10.1111/j.1439-0396.2006.00647.x. [DOI] [PubMed] [Google Scholar]
- 100.Muriel P, Ramos-Tovar E, Montes-Páez G, Buendía-Montaño LD. Experimental models of liver damage mediated by oxidative stress. In: Muriel P, editor. Liver pathophysiology: therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 529–546. [Google Scholar]
- 101.Ingawale DK, Mandlik SK, Naik SR. Models of hepatotoxicity and the underlying cellular, biochemical and immunological mechanism(s): a critical discussion. Environ Toxicol Pharmacol. 2014;37:118–133. doi: 10.1016/j.etap.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 102.Facino RM, Carini M, Franzoi L, Pirola O, Bosisio E. Phytochemical characterization and radical scavenger activity of flavonoids from Helichrysum italicum G. Don (Compositae) Pharmacol Res. 1990;22:709–721. doi: 10.1016/s1043-6618(05)80097-0. [DOI] [PubMed] [Google Scholar]
- 103.Ikewuchi JC, Ikewuchi CC, Igboh NM, Mark-Balm T. Protective effect of aqueous extract of the rhizomes of Sansevieria liberica Gérôme and Labroy on carbon tetrachloride induced hepatotoxicity in rats. EXCLI J. 2011;10:312–321. [PMC free article] [PubMed] [Google Scholar]
- 104.Kaurinovic B, Popovic M, Vlaisavljevic S, Schwartsova H, Vojinovic-Miloradov M. Antioxidant profile of Trifolium pratense L. Molecules. 2012;17:11156–11172. doi: 10.3390/molecules170911156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hermenean A, Ardelean A, Stan M, Hadaruga N, Mihali CV, Costache M, Dinischiotu A. Antioxidant and hepatoprotective effects of naringenin and its β-cyclodextrin formulation in mice intoxicated with carbon tetrachloride: a comparative study. J Med Food. 2014;17:670–677. doi: 10.1089/jmf.2013.0007. [DOI] [PubMed] [Google Scholar]
- 106.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 107.Yamanishi R, Yoshigai E, Okuyama T, Mori M, Murase H, Machida T, Okumura T, Nishizawa M. The anti-inflammatory effects of flavanol-rich lychee fruit extract in rat hepatocytes. PLoS One. 2014;9:e93818. doi: 10.1371/journal.pone.0093818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.O’Neill LA, Kaltschmidt C. NF-κB: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- 109.Czaja MJ. The future of GI and liver research: editorial perspectives. III. JNK/AP-1 regulation of hepatocyte death. Am J Physiol Gastrointest Liver Physiol. 2003;284:G875–G879. doi: 10.1152/ajpgi.00549.2002. [DOI] [PubMed] [Google Scholar]
- 110.Wang X, Xiang L, Li H, Chen P, Feng Y, Zhang J, Yang N, Li F, Wang Y, Zhang Q, et al. The role of HMGB1 signaling pathway in the development and progression of hepatocellular carcinoma: a review. Int J Mol Sci. 2015;16:22527–22540. doi: 10.3390/ijms160922527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pollard TD, Earnshaw WC, Lippincott-Schwartz J. Cell Biology. 2th edition. Philadelphia: Elsevier; 2008. pp. 433–435. [Google Scholar]
- 112.Hernández-Aquino E, Zarco N, Casas-Grajales S, Ramos-Tovar E, Flores-Beltrán RE, Arauz J, Shibayama M, Favari L, Tsutsumi V, Segovia J, et al. Naringenin prevents experimental liver fibrosis by blocking TGFβ-Smad3 and JNK-Smad3 pathways. World J Gastroenterol. 2017;23:4354–4368. doi: 10.3748/wjg.v23.i24.4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kisseleva T, Brenner DA. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J Gastroenterol Hepatol. 2007;22 Suppl 1:S73–S78. doi: 10.1111/j.1440-1746.2006.04658.x. [DOI] [PubMed] [Google Scholar]
- 114.Arauz J, Moreno MG, Cortés-Reynosa P, Salazar EP, Muriel P. Coffee attenuates fibrosis by decreasing the expression of TGF-β and CTGF in a murine model of liver damage. J Appl Toxicol. 2013;33:970–979. doi: 10.1002/jat.2788. [DOI] [PubMed] [Google Scholar]
- 115.Matsuzaki K. Smad phospho-isoforms direct context-dependent TGF-β signaling. Cytokine Growth Factor Rev. 2013;24:385–399. doi: 10.1016/j.cytogfr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 116.Yoshida K, Murata M, Yamaguchi T, Matsuzaki K, Okazaki K. Reversible human TGF-β signal shifting between tumor suppression and fibro-carcinogenesis: implications of smad phospho-isoforms for hepatic epithelial-mesenchymal transitions. J Clin Med. 2016;5 doi: 10.3390/jcm5010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hemmann S, Graf J, Roderfeld M, Roeb E. Expression of MMPs and TIMPs in liver fibrosis - a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol. 2007;46:955–975. doi: 10.1016/j.jhep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 118.Imamura T, Oshima Y, Hikita A. Regulation of TGF-β family signalling by ubiquitination and deubiquitination. J Biochem. 2013;154:481–489. doi: 10.1093/jb/mvt097. [DOI] [PubMed] [Google Scholar]
- 119.Bray F, Ren JS, Masuyer E, Ferlay J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer. 2013;132:1133–1145. doi: 10.1002/ijc.27711. [DOI] [PubMed] [Google Scholar]
- 120.Tarocchi M, Polvani S, Marroncini G, Galli A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J Gastroenterol. 2014;20:11630–11640. doi: 10.3748/wjg.v20.i33.11630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tarocchi M, Galli A. Oxidative stress as a mechanism for hepatocellular carcinoma. In: Muriel P, editor. Liver pathophysiology: therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 279–287. [Google Scholar]
- 122.Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet. 1981;2:1129–1133. doi: 10.1016/s0140-6736(81)90585-7. [DOI] [PubMed] [Google Scholar]
- 123.Higgs MR, Chouteau P, Lerat H. ‘Liver let die’: oxidative DNA damage and hepatotropic viruses. J Gen Virol. 2014;95:991–1004. doi: 10.1099/vir.0.059485-0. [DOI] [PubMed] [Google Scholar]
- 124.Fisher AB. Redox signaling across cell membranes. Antioxid Redox Signal. 2009;11:1349–1356. doi: 10.1089/ars.2008.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 126.Perry G, Raina AK, Nunomura A, Wataya T, Sayre LM, Smith MA. How important is oxidative damage? Lessons from Alzheimer’s disease. Free Radic Biol Med. 2000;28:831–834. doi: 10.1016/s0891-5849(00)00158-1. [DOI] [PubMed] [Google Scholar]
- 127.Niu D, Zhang J, Ren Y, Feng H, Chen WN. HBx genotype D represses GSTP1 expression and increases the oxidative level and apoptosis in HepG2 cells. Mol Oncol. 2009;3:67–76. doi: 10.1016/j.molonc.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dröge W. Oxidative stress and aging. Adv Exp Med Biol. 2003;543:191–200. doi: 10.1007/978-1-4419-8997-0_14. [DOI] [PubMed] [Google Scholar]
- 129.Ha HL, Shin HJ, Feitelson MA, Yu DY. Oxidative stress and antioxidants in hepatic pathogenesis. World J Gastroenterol. 2010;16:6035–6043. doi: 10.3748/wjg.v16.i48.6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Scott TL, Rangaswamy S, Wicker CA, Izumi T. Repair of oxidative DNA damage and cancer: recent progress in DNA base excision repair. Antioxid Redox Signal. 2014;20:708–726. doi: 10.1089/ars.2013.5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Khan N, Afaq F, Mukhtar H. Cancer chemoprevention through dietary antioxidants: progress and promise. Antioxid Redox Signal. 2008;10:475–510. doi: 10.1089/ars.2007.1740. [DOI] [PubMed] [Google Scholar]
- 132.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 133.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 134.Kandaswami C, Middleton E Jr. Free radical scavenging and antioxidant activity of plant flavonoids. Adv Exp Med Biol. 1994;366:351–376. doi: 10.1007/978-1-4615-1833-4_25. [DOI] [PubMed] [Google Scholar]
- 135.Russo M, Spagnuolo C, Tedesco I, Russo GL. Phytochemicals in cancer prevention and therapy: truth or dare? Toxins (Basel) 2010;2:517–551. doi: 10.3390/toxins2040517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Francis AR, Shetty TK, Bhattacharya RK. Modulating effect of plant flavonoids on the mutagenicity of N-methyl-N’-nitro-N-nitrosoguanidine. Carcinogenesis. 1989;10:1953–1955. doi: 10.1093/carcin/10.10.1953. [DOI] [PubMed] [Google Scholar]
- 137.Ekambaram G, Rajendran P, Magesh V, Sakthisekaran D. Naringenin reduces tumor size and weight lost in N-methyl-N’-nitro-N-nitrosoguanidine-induced gastric carcinogenesis in rats. Nutr Res. 2008;28:106–112. doi: 10.1016/j.nutres.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 138.Leonardi T, Vanamala J, Taddeo SS, Davidson LA, Murphy ME, Patil BS, Wang N, Carroll RJ, Chapkin RS, Lupton JR, et al. Apigenin and naringenin suppress colon carcinogenesis through the aberrant crypt stage in azoxymethane-treated rats. Exp Biol Med (Maywood) 2010;235:710–717. doi: 10.1258/ebm.2010.009359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yoon H, Kim TW, Shin SY, Park MJ, Yong Y, Kim DW, Islam T, Lee YH, Jung KY, Lim Y. Design, synthesis and inhibitory activities of naringenin derivatives on human colon cancer cells. Bioorg Med Chem Lett. 2013;23:232–238. doi: 10.1016/j.bmcl.2012.10.130. [DOI] [PubMed] [Google Scholar]
- 140.Subramanian P, Arul D. Attenuation of NDEA-induced hepatocarcinogenesis by naringenin in rats. Cell Biochem Funct. 2013;31:511–517. doi: 10.1002/cbf.2929. [DOI] [PubMed] [Google Scholar]
- 141.Arul D, Subramanian P. Inhibitory effect of naringenin (citrus flavonone) on N-nitrosodiethylamine induced hepatocarcinogenesis in rats. Biochem Biophys Res Commun. 2013;434:203–209. doi: 10.1016/j.bbrc.2013.03.039. [DOI] [PubMed] [Google Scholar]
- 142.Taha MM, Abdul AB, Abdullah R, Ibrahim TA, Abdelwahab SI, Mohan S. Potential chemoprevention of diethylnitrosamine-initiated and 2-acetylaminofluorene-promoted hepatocarcinogenesis by zerumbone from the rhizomes of the subtropical ginger (Zingiber zerumbet) Chem Biol Interact. 2010;186:295–305. doi: 10.1016/j.cbi.2010.04.029. [DOI] [PubMed] [Google Scholar]
- 143.Lee MH, Yoon S, Moon JO. The flavonoid naringenin inhibits dimethylnitrosamine-induced liver damage in rats. Biol Pharm Bull. 2004;27:72–76. doi: 10.1248/bpb.27.72. [DOI] [PubMed] [Google Scholar]
- 144.Ozkaya A, Sahin Z, Dag U, Ozkaraca M. Effects of naringenin on oxidative stress and histopathological changes in the liver of lead acetate administered Rats. J Biochem Mol Toxicol. 2016;30:243–248. doi: 10.1002/jbt.21785. [DOI] [PubMed] [Google Scholar]
- 145.Arul D, Subramanian P. Naringenin (citrus flavonone) induces growth inhibition, cell cycle arrest and apoptosis in human hepatocellular carcinoma cells. Pathol Oncol Res. 2013;19:763–770. doi: 10.1007/s12253-013-9641-1. [DOI] [PubMed] [Google Scholar]
- 146.Zhong Z, Chen X, Tan W, Xu Z, Zhou K, Wu T, Cui L, Wang Y. Germacrone inhibits the proliferation of breast cancer cell lines by inducing cell cycle arrest and promoting apoptosis. Eur J Pharmacol. 2011;667:50–55. doi: 10.1016/j.ejphar.2011.03.041. [DOI] [PubMed] [Google Scholar]
- 147.Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- 148.Alshatwi AA, Shafi G, Hasan TN, Al-Hazzani AA, Alsaif MA, Alfawaz MA, Lei KY, Munshi A. Apoptosis-mediated inhibition of human breast cancer cell proliferation by lemon citrus extract. Asian Pac J Cancer Prev. 2011;12:1555–1559. [PubMed] [Google Scholar]
- 149.Park HS, Kim GY, Nam TJ, Deuk Kim N, Hyun Choi Y. Antiproliferative activity of fucoidan was associated with the induction of apoptosis and autophagy in AGS human gastric cancer cells. J Food Sci. 2011;76:T77–T83. doi: 10.1111/j.1750-3841.2011.02099.x. [DOI] [PubMed] [Google Scholar]
- 150.Tan AC, Konczak I, Ramzan I, Sze DM. Native Australian fruit polyphenols inhibit cell viability and induce apoptosis in human cancer cell lines. Nutr Cancer. 2011;63:444–455. doi: 10.1080/01635581.2011.535953. [DOI] [PubMed] [Google Scholar]
- 151.Chidambara Murthy KN, Jayaprakasha GK, Kumar V, Rathore KS, Patil BS. Citrus limonin and its glucoside inhibit colon adenocarcinoma cell proliferation through apoptosis. J Agric Food Chem. 2011;59:2314–2323. doi: 10.1021/jf104498p. [DOI] [PubMed] [Google Scholar]
- 152.Patel S. Function and dysfunction of two-pore channels. Sci Signal. 2015;8:re7. doi: 10.1126/scisignal.aab3314. [DOI] [PubMed] [Google Scholar]
- 153.Scholz-Starke J, Carpaneto A, Gambale F. On the interaction of neomycin with the slow vacuolar channel of Arabidopsis thaliana. J Gen Physiol. 2006;127:329–340. doi: 10.1085/jgp.200509402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pafumi I, Festa M, Papacci F, Lagostena L, Giunta C, Gutla V, Cornara L, Favia A, Palombi F, Gambale F, et al. Naringenin impairs two-pore channel 2 activity and inhibits VEGF-induced angiogenesis. Sci Rep. 2017;7:5121. doi: 10.1038/s41598-017-04974-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fürstenberger G, Berry DL, Sorg B, Marks F. Skin tumor promotion by phorbol esters is a two-stage process. Proc Natl Acad Sci U S A. 1981;78:7722–7726. doi: 10.1073/pnas.78.12.7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liu JF, Crépin M, Liu JM, Barritault D, Ledoux D. FGF-2 and TPA induce matrix metalloproteinase-9 secretion in MCF-7 cells through PKC activation of the Ras/ERK pathway. Biochem Biophys Res Commun. 2002;293:1174–1182. doi: 10.1016/S0006-291X(02)00350-9. [DOI] [PubMed] [Google Scholar]
- 158.Lee KH, Yeh MH, Kao ST, Hung CM, Liu CJ, Huang YY, Yeh CC. The inhibitory effect of hesperidin on tumor cell invasiveness occurs via suppression of activator protein 1 and nuclear factor-kappaB in human hepatocellular carcinoma cells. Toxicol Lett. 2010;194:42–49. doi: 10.1016/j.toxlet.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 159.Yen HR, Liu CJ, Yeh CC. Naringenin suppresses TPA-induced tumor invasion by suppressing multiple signal transduction pathways in human hepatocellular carcinoma cells. Chem Biol Interact. 2015;235:1–9. doi: 10.1016/j.cbi.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 160.Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283:65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 161.Pietrangelo A. Iron and the liver. Liver Int. 2016;36 Suppl 1:116–123. doi: 10.1111/liv.13020. [DOI] [PubMed] [Google Scholar]
- 162.Sikorska K, Bernat A, Wroblewska A. Molecular pathogenesis and clinical consequences of iron overload in liver cirrhosis. Hepatobiliary Pancreat Dis Int. 2016;15:461–479. doi: 10.1016/s1499-3872(16)60135-2. [DOI] [PubMed] [Google Scholar]
- 163.Arthur MJ. Iron overload and liver fibrosis. J Gastroenterol Hepatol. 1996;11:1124–1129. doi: 10.1111/j.1440-1746.1996.tb01840.x. [DOI] [PubMed] [Google Scholar]
- 164.Fernandez MT, Mira ML, Florêncio MH, Jennings KR. Iron and copper chelation by flavonoids: an electrospray mass spectrometry study. J Inorg Biochem. 2002;92:105–111. doi: 10.1016/s0162-0134(02)00511-1. [DOI] [PubMed] [Google Scholar]
- 165.Cheng IF, Breen K. On the ability of four flavonoids, baicilein, luteolin, naringenin, and quercetin, to suppress the Fenton reaction of the iron-ATP complex. Biometals. 2000;13:77–83. doi: 10.1023/a:1009229429250. [DOI] [PubMed] [Google Scholar]
- 166.Benherlal PS, Arumughan C. Studies on modulation of DNA integrity in Fenton’s system by phytochemicals. Mutat Res. 2008;648:1–8. doi: 10.1016/j.mrfmmm.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 167.Jagetia GC, Reddy TK. Alleviation of iron induced oxidative stress by the grape fruit flavanone naringin in vitro. Chem Biol Interact. 2011;190:121–128. doi: 10.1016/j.cbi.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 168.Jagetia GC, Reddy TK, Venkatesha VA, Kedlaya R. Influence of naringin on ferric iron induced oxidative damage in vitro. Clin Chim Acta. 2004;347:189–197. doi: 10.1016/j.cccn.2004.04.022. [DOI] [PubMed] [Google Scholar]
- 169.Chtourou Y, Fetoui H, Gdoura R. Protective effects of naringenin on iron-overload-induced cerebral cortex neurotoxicity correlated with oxidative stress. Biol Trace Elem Res. 2014;158:376–383. doi: 10.1007/s12011-014-9948-0. [DOI] [PubMed] [Google Scholar]
- 170.Chtourou Y, Slima AB, Gdoura R, Fetoui H. Naringenin mitigates iron-induced anxiety-like behavioral impairment, mitochondrial dysfunctions, ectonucleotidases and acetylcholinesterase alteration activities in rat hippocampus. Neurochem Res. 2015;40:1563–1575. doi: 10.1007/s11064-015-1627-9. [DOI] [PubMed] [Google Scholar]
- 171.Uriu-Adams JY, Keen CL. Copper, oxidative stress, and human health. Mol Aspects Med. 2005;26:268–298. doi: 10.1016/j.mam.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 172.Johncilla M, Mitchell KA. Pathology of the liver in copper overload. Semin Liver Dis. 2011;31:239–244. doi: 10.1055/s-0031-1286055. [DOI] [PubMed] [Google Scholar]
- 173.Zhou B, Gitschier J. hCTR1: a human gene for copper uptake identified by complementation in yeast. Proc Natl Acad Sci U S A. 1997;94:7481–7486. doi: 10.1073/pnas.94.14.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Moriwaki H, Osborne MR, Phillips DH. Effects of mixing metal ions on oxidative DNA damage mediated by a Fenton-type reduction. Toxicol In Vitro. 2008;22:36–44. doi: 10.1016/j.tiv.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 175.Mira L, Fernandez MT, Santos M, Rocha R, Florêncio MH, Jennings KR. Interactions of flavonoids with iron and copper ions: a mechanism for their antioxidant activity. Free Radic Res. 2002;36:1199–1208. doi: 10.1080/1071576021000016463. [DOI] [PubMed] [Google Scholar]
- 176.Esterbauer H, Gebicki J, Puhl H, Jürgens G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med. 1992;13:341–390. doi: 10.1016/0891-5849(92)90181-f. [DOI] [PubMed] [Google Scholar]
- 177.Miranda CL, Stevens JF, Ivanov V, McCall M, Frei B, Deinzer ML, Buhler DR. Antioxidant and prooxidant actions of prenylated and nonprenylated chalcones and flavanones in vitro. J Agric Food Chem. 2000;48:3876–3884. doi: 10.1021/jf0002995. [DOI] [PubMed] [Google Scholar]
- 178.Rani A, Kumar A, Lal A, Pant M. Cellular mechanisms of cadmium-induced toxicity: a review. Int J Environ Health Res. 2014;24:378–399. doi: 10.1080/09603123.2013.835032. [DOI] [PubMed] [Google Scholar]
- 179.Rikans LE, Yamano T. Mechanisms of cadmium-mediated acute hepatotoxicity. J Biochem Mol Toxicol. 2000;14:110–117. doi: 10.1002/(sici)1099-0461(2000)14:2<110::aid-jbt7>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 180.Renugadevi J, Prabu SM. Naringenin protects against cadmium-induced oxidative renal dysfunction in rats. Toxicology. 2009;256:128–134. doi: 10.1016/j.tox.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 181.Das A, Roy A, Das R, Bhattacharya S, Haldar PK. Naringenin alleviates cadmium-induced toxicity through the abrogation of oxidative stress in swiss albino mice. J Environ Pathol Toxicol Oncol. 2016;35:161–169. doi: 10.1615/JEnvironPatholToxicolOncol.2016015892. [DOI] [PubMed] [Google Scholar]
- 182.Rathi VK, Das S, Parampalli Raghavendra A, Rao BSS. Naringin abates adverse effects of cadmium-mediated hepatotoxicity: An experimental study using HepG2 cells. J Biochem Mol Toxicol. 2017;31 doi: 10.1002/jbt.21915. [DOI] [PubMed] [Google Scholar]
- 183.Singh AP, Goel RK, Kaur T. Mechanisms pertaining to arsenic toxicity. Toxicol Int. 2011;18:87–93. doi: 10.4103/0971-6580.84258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, Rhodes CJ, Valko M. Arsenic: toxicity, oxidative stress and human disease. J Appl Toxicol. 2011;31:95–107. doi: 10.1002/jat.1649. [DOI] [PubMed] [Google Scholar]
- 185.Liu J, Waalkes MP. Liver is a target of arsenic carcinogenesis. Toxicol Sci. 2008;105:24–32. doi: 10.1093/toxsci/kfn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Adil M, Kandhare AD, Visnagri A, Bodhankar SL. Naringin ameliorates sodium arsenite-induced renal and hepatic toxicity in rats: decisive role of KIM-1, Caspase-3, TGF-β, and TNF-α. Ren Fail. 2015;37:1396–1407. doi: 10.3109/0886022X.2015.1074462. [DOI] [PubMed] [Google Scholar]
- 187.Kim HC, Jang TW, Chae HJ, Choi WJ, Ha MN, Ye BJ, Kim BG, Jeon MJ, Kim SY, Hong YS. Evaluation and management of lead exposure. Ann Occup Environ Med. 2015;27:30. doi: 10.1186/s40557-015-0085-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Matović V, Buha A, Ðukić-Ćosić D, Bulat Z. Insight into the oxidative stress induced by lead and/or cadmium in blood, liver and kidneys. Food Chem Toxicol. 2015;78:130–140. doi: 10.1016/j.fct.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 189.Pal M, Sachdeva M, Gupta N, Mishra P, Yadav M, Tiwari A. Lead exposure in different organs of mammals and prevention by curcumin-nanocurcumin: a review. Biol Trace Elem Res. 2015;168:380–391. doi: 10.1007/s12011-015-0366-8. [DOI] [PubMed] [Google Scholar]
- 190.Fierro NA, Gonzalez-Aldaco K, Roman S, Panduro A. The immune system and viral hepatitis. In Muriel P. Liver pathophysiology: therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 129–139. [Google Scholar]
- 191.Tang H, Grisé H. Cellular and molecular biology of HCV infection and hepatitis. Clin Sci (Lond) 2009;117:49–65. doi: 10.1042/CS20080631. [DOI] [PubMed] [Google Scholar]
- 192.Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology. 2004;39:5–19. doi: 10.1002/hep.20032. [DOI] [PubMed] [Google Scholar]
- 193.Nahmias Y, Goldwasser J, Casali M, van Poll D, Wakita T, Chung RT, Yarmush ML. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology. 2008;47:1437–1445. doi: 10.1002/hep.22197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bok SH, Shin YW, Bae KH, Jeong TS, Kwon YK, Park YB, Choi MS. Effects of naringin and lovastatin on plasma and hepatic lipids in high-fat and high-cholesterol fed rats. Nutr Res. 2000;20:1007–1015. [Google Scholar]
- 195.Kim SY, Kim HJ, Lee MK, Jeon SM, Do GM, Kwon EY, Cho YY, Kim DJ, Jeong KS, Park YB, et al. Naringin time-dependently lowers hepatic cholesterol biosynthesis and plasma cholesterol in rats fed high-fat and high-cholesterol diet. J Med Food. 2006;9:582–586. doi: 10.1089/jmf.2006.9.582. [DOI] [PubMed] [Google Scholar]
- 196.Lee CH, Jeong TS, Choi YK, Hyun BH, Oh GT, Kim EH, Kim JR, Han JI, Bok SH. Anti-atherogenic effect of citrus flavonoids, naringin and naringenin, associated with hepatic ACAT and aortic VCAM-1 and MCP-1 in high cholesterol-fed rabbits. Biochem Biophys Res Commun. 2001;284:681–688. doi: 10.1006/bbrc.2001.5001. [DOI] [PubMed] [Google Scholar]
- 197.Mulvihill EE, Allister EM, Sutherland BG, Telford DE, Sawyez CG, Edwards JY, Markle JM, Hegele RA, Huff MW. Naringenin prevents dyslipidemia, apolipoprotein B overproduction, and hyperinsulinemia in LDL receptor-null mice with diet-induced insulin resistance. Diabetes. 2009;58:2198–2210. doi: 10.2337/db09-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wilcox LJ, Borradaile NM, de Dreu LE, Huff MW. Secretion of hepatocyte apoB is inhibited by the flavonoids, naringenin and hesperetin, via reduced activity and expression of ACAT2 and MTP. J Lipid Res. 2001;42:725–734. [PubMed] [Google Scholar]
- 199.Borradaile NM, de Dreu LE, Barrett PH, Behrsin CD, Huff MW. Hepatocyte apoB-containing lipoprotein secretion is decreased by the grapefruit flavonoid, naringenin, via inhibition of MTP-mediated microsomal triglyceride accumulation. Biochemistry. 2003;42:1283–1291. doi: 10.1021/bi026731o. [DOI] [PubMed] [Google Scholar]
- 200.Borradaile NM, de Dreu LE, Barrett PH, Huff MW. Inhibition of hepatocyte apoB secretion by naringenin: enhanced rapid intracellular degradation independent of reduced microsomal cholesteryl esters. J Lipid Res. 2002;43:1544–1554. doi: 10.1194/jlr.m200115-jlr200. [DOI] [PubMed] [Google Scholar]
- 201.Borradaile NM, de Dreu LE, Huff MW. Inhibition of net HepG2 cell apolipoprotein B secretion by the citrus flavonoid naringenin involves activation of phosphatidylinositol 3-kinase, independent of insulin receptor substrate-1 phosphorylation. Diabetes. 2003;52:2554–2561. doi: 10.2337/diabetes.52.10.2554. [DOI] [PubMed] [Google Scholar]
- 202.Allister EM, Borradaile NM, Edwards JY, Huff MW. Inhibition of microsomal triglyceride transfer protein expression and apolipoprotein B100 secretion by the citrus flavonoid naringenin and by insulin involves activation of the mitogen-activated protein kinase pathway in hepatocytes. Diabetes. 2005;54:1676–1683. doi: 10.2337/diabetes.54.6.1676. [DOI] [PubMed] [Google Scholar]
- 203.Allister EM, Mulvihill EE, Barrett PH, Edwards JY, Carter LP, Huff MW. Inhibition of apoB secretion from HepG2 cells by insulin is amplified by naringenin, independent of the insulin receptor. J Lipid Res. 2008;49:2218–2229. doi: 10.1194/jlr.M800297-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Goldwasser J, Cohen PY, Lin W, Kitsberg D, Balaguer P, Polyak SJ, Chung RT, Yarmush ML, Nahmias Y. Naringenin inhibits the assembly and long-term production of infectious hepatitis C virus particles through a PPAR-mediated mechanism. J Hepatol. 2011;55:963–971. doi: 10.1016/j.jhep.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Khachatoorian R, Arumugaswami V, Raychaudhuri S, Yeh GK, Maloney EM, Wang J, Dasgupta A, French SW. Divergent antiviral effects of bioflavonoids on the hepatitis C virus life cycle. Virology. 2012;433:346–355. doi: 10.1016/j.virol.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gonzalez O, Fontanes V, Raychaudhuri S, Loo R, Loo J, Arumugaswami V, Sun R, Dasgupta A, French SW. The heat shock protein inhibitor quercetin attenuates hepatitis C virus production. Hepatology. 2009;50:1756–1764. doi: 10.1002/hep.23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Khachatoorian R, Arumugaswami V, Ruchala P, Raychaudhuri S, Maloney EM, Miao E, Dasgupta A, French SW. A cell-permeable hairpin peptide inhibits hepatitis C viral nonstructural protein 5A-mediated translation and virus production. Hepatology. 2012;55:1662–1672. doi: 10.1002/hep.25533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mathew S, Faheem M, Archunan G, Ilyas M, Begum N, Jahangir S, Qadri I, Qahtani MA, Mathew S. In silico studies of medicinal compounds against hepatitis C capsid protein from north India. Bioinform Biol Insights. 2014;8:159–168. doi: 10.4137/BBI.S15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lulu SS, Thabitha A, Vino S, Priya AM, Rout M. Naringenin and quercetin--potential anti-HCV agents for NS2 protease targets. Nat Prod Res. 2016;30:464–468. doi: 10.1080/14786419.2015.1020490. [DOI] [PubMed] [Google Scholar]
- 210.Singh AK, Raj V, Keshari AK, Rai A, Kumar P, Rawat A, Maity B, Kumar D, Prakash A, De A, et al. Isolated mangiferin and naringenin exert antidiabetic effect via PPARγ/GLUT4 dual agonistic action with strong metabolic regulation. Chem Biol Interact. 2018;280:33–44. doi: 10.1016/j.cbi.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 211.Ahmed OM, Hassan MA, Abdel-Twab SM, Abdel Azeem MN. Navel orange peel hydroethanolic extract, naringin and naringenin have anti-diabetic potentials in type 2 diabetic rats. Biomed Pharmacother. 2017;94:197–205. doi: 10.1016/j.biopha.2017.07.094. [DOI] [PubMed] [Google Scholar]
- 212.Sirovina D, Oršolić N, Gregorović G, Končić MZ. Naringenin ameliorates pathological changes in liver and kidney of diabetic mice: a preliminary study. Arh Hig Rada Toksikol. 2016;67:19–24. doi: 10.1515/aiht-2016-67-2708. [DOI] [PubMed] [Google Scholar]
- 213.Kapoor R, Kakkar P. Naringenin accords hepatoprotection from streptozotocin induced diabetes in vivo by modulating mitochondrial dysfunction and apoptotic signaling cascade. Toxicol Rep. 2014;1:569–581. doi: 10.1016/j.toxrep.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Nyane NA, Tlaila TB, Malefane TG, Ndwandwe DE, Owira PMO. Metformin-like antidiabetic, cardio-protective and non-glycemic effects of naringenin: Molecular and pharmacological insights. Eur J Pharmacol. 2017;803:103–111. doi: 10.1016/j.ejphar.2017.03.042. [DOI] [PubMed] [Google Scholar]
- 215.Sahu SC, Gray GC. Lipid peroxidation and DNA damage induced by morin and naringenin in isolated rat liver nuclei. Food Chem Toxicol. 1997;35:443–447. doi: 10.1016/s0278-6915(97)00011-2. [DOI] [PubMed] [Google Scholar]
- 216.Galati G, Moridani MY, Chan TS, O’Brien PJ. Peroxidative metabolism of apigenin and naringenin versus luteolin and quercetin: glutathione oxidation and conjugation. Free Radic Biol Med. 2001;30:370–382. doi: 10.1016/s0891-5849(00)00481-0. [DOI] [PubMed] [Google Scholar]
- 217.Ortiz-Andrade RR, Sánchez-Salgado JC, Navarrete-Vázquez G, Webster SP, Binnie M, García-Jiménez S, León-Rivera I, Cigarroa-Vázquez P, Villalobos-Molina R, Estrada-Soto S. Antidiabetic and toxicological evaluations of naringenin in normoglycaemic and NIDDM rat models and its implications on extra-pancreatic glucose regulation. Diabetes Obes Metab. 2008;10:1097–1104. doi: 10.1111/j.1463-1326.2008.00869.x. [DOI] [PubMed] [Google Scholar]
- 218.Pérez-Pastén R, Martínez-Galero E, Chamorro-Cevallos G. Quercetin and naringenin reduce abnormal development of mouse embryos produced by hydroxyurea. J Pharm Pharmacol. 2010;62:1003–1009. doi: 10.1111/j.2042-7158.2010.01118.x. [DOI] [PubMed] [Google Scholar]