

Abstract
AIM
To investigate the influence of high salt on dextran sulfate sodium (DSS)-induced colitis in mice and explore the underlying mechanisms of this effect.
METHODS
DSS and NaCl were used to establish the proinflammatory animal model. We evaluated the colitis severity. Flow cytometry was employed for detecting the frequencies of Th1, macrophages and Tregs in spleen, mesenteric lymph node and lamina propria. The important role of macrophages in the promotion of DSS-induced colitis by NaCl was evaluated by depleting macrophages with clodronate liposomes. Activated peritoneal macrophages and lamina propria mononuclear cells (LPMCs) were stimulated with NaCl, and proteins were detected by western blotting. Cytokines and inflammation genes were analyzed by enzyme-linked immunosorbent assay and RT-PCR, respectively.
RESULTS
The study findings indicate that NaCl up-regulates the frequencies of CD11b+ macrophages and CD4+IFN-γ+IL-17+ T cells in lamina propria in DSS-treated mice. CD3+CD4+CD25+Foxp3+ T cells, which can secrete high levels of IL-10 and TGF-β, increase through feedback in NaCl- and DSS-treated mice. Furthermore, clodronate liposomes pretreatment significantly alleviated DSS-induced colitis, indicating that macrophages play a vital role in NaCl proinflammatory activity. NaCl aggravates peritoneal macrophage inflammation by promoting the expressions of interleukin (IL)-1, IL-6 and mouse inducible nitric oxide synthase. Specifically, high NaCl concentrations promote p38 phosphorylation in lipopolysaccharide- and IFN-γ-activated LPMCs mediated by SGK1.
CONCLUSION
Proinflammatory macrophages may play an essential role in the onset and development of NaCl-promoted inflammation in DSS-induced colitis. The underlining mechanism involves up-regulation of the p38/MAPK axis.
Keywords: Inflammatory bowel disease, Macrophage, NaCl, CD4+IFN-γ+IL-17+ T cell, p38/MAPK
Core tip: NaCl, as an indispensable environmental factor, evokes both innate and adaptive immune proinflammation cell activation in mice affected by dextran sulfate sodium (DSS)-induced colitis. Proinflammatory CD4+ cells in DSS- and NaCl-treated mice are mainly double-positive IL-17+IFN-γ+ T cells. Macrophage depletion significantly alleviates DSS-induced colitis. M1 macrophages play an important role in the proinflammatory effect of NaCl in the mouse gut. NaCl promotes M1 proinflammatory gene expression in lipopolysaccharide-activated peritoneal macrophage. The mechanism by which NaCl promotes DSS-induced colitis involves up-regulation of the p38/MAPK axis.
INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic and recurrent disease, usually manifesting as ulcerative colitis and Crohn’s disease (CD)[1]. IBD is a high-risk factor for colorectal cancer and it is a serious threat to the human health globally. Although its etiology is presently unclear, findings yielded by extant studies indicate that IBD is a complex process involving heredity, environment and immunity[2-5].
Innate and adaptive immune cells play different roles in IBD pathogenesis. Results obtained in a large number of studies have shown that Th17, Th1, regulatory T cells (Tregs) and macrophages play important roles in IBD pathogenesis. For instance, the number of Th17 cells increases significantly in mucosa lamina propria (LP) of colitis patients, whereby interleukin (IL)-17 is produced, resulting in mucosal damage and enhancing disease activity[6,7]. Th1 polarization is related to colonic inflammation, through its induction of IFN-γ and TNF-α production, whereas the differential propensity to develop colitis is linked to the inherent tendency of the immune system to give rise to Th1 or Th17/Treg responses[8]. Tregs, which are very important regulatory T cells, express IL-10 highly and inhibit inflammation in IBD[9]. Macrophages in the intestinal mucosa of colitis patients can secrete the cytokines TNF-α, IL-1 and IL-6[10]. Intestinal macrophages are the major population of antigen presenting cells in intestinal mucosa and they shape the types of T cell response to luminal antigens[11].
Sodium chloride mediates the inflammatory effects of immune cells that are very important to IBD. NaCl exacerbates experimental autoimmune encephalomyelitis in mice by promoting Th17 cell differentiation[12]. High salt content strengthens the lipopolysaccharides (LPS)-induced macrophage activation by activating signaling pathways of p38 and ERK1 to induce the production of proinflammatory factors[13]. Extant studies have shown that the high-salt diet promotes Th17 cell activation in LP and exacerbates experimental colitis in mice[14,15]. However, high-salt diet effect on other immune cells, such as Th1, Tregs and macrophages, which are also associated with pathopoiesis in IBD, is still unclear. Macrophage activation plays a pivotal role in inflammation initiation and progression in diverse pathological conditions. Findings obtained in our previous research indicate that, in mice treated with clodronate liposomes (MDP), gut macrophages were successfully depleted. Macrophage depletion could protect mice against colitis induced by dextran sulfate sodium (DSS), suggesting that the macrophages play an important role in colitis pathogenesis.
In the present study, we hypothesized that NaCl promotes the onset and course of DSS-induced colitis, as well as sustains the disease. The promotion effect may be due to monocyte-macrophages shifting the T cell response toward Th17, Th1 and Treg cells. We tested this hypothesis in a DSS-induced colitis mouse model, which shares many characteristics with human ulcerative colitis[16,17]. We found that NaCl promoted both macrophages and CD4+ proinflammatory cell immune response, whereby CD4+ proinflammatory cells were mainly CD4+IFN-γ+IL-17+ T cells. NaCl enhanced the proinflammatory gene expression and cytokine secretion in the colons of mice affected by colitis. Depletion of gut macrophages significantly alleviated DSS-induced colitis, suggesting that macrophages play a vital role in the NaCl proinflammation process. High NaCl enhanced M1 proinflammation gene expression in LPS-activated peritoneal macrophages. Therefore, colitis promoted by high NaCl levels may be a result of M1 macrophage polarization. M1 polarization shifts T cell response toward proinflammatory CD4+IFN-γ+IL-17+ T cells. High NaCl proinflammation in LPS- and IFN-γ-activated lamina propria mononuclear cells (LPMCs) relies on up-regulation of the p38 mitogen-activated protein kinase (p38/MAPK) axis.
MATERIALS AND METHODS
Animal treatment
For this study, 8- to 10-wk-old female C57BL/6J mice were purchased from the Animal Center of Third Military Medical University (Army Medical University). Mice were housed at 24 °C, under light-controlled cycle (12 h) and with free access to standard laboratory water and food. All processes were supported by the Committee on Use and Care of Laboratory Animals at Third Military Medical University (Army Medical University).
Establishment of the animal model with DSS and NaCl
Mice purchased from the Animal Center were allowed at least 7 d to adapt to the environment before being randomly divided into four groups. They received water containing 2% NaCl (Sinopharm Chemical Reagent, China) and/or water containing 2.5% DSS (160110; MP Biomedicals, United States) for 10 d. The intestinal macrophages were depleted using MDP (van Rooijen and van Kesteren-Hendrikx, 2003, clodronateliposomes.org, Holland)[18]. Briefly, 200 μL MDP was injected i.p. into mice 4 d prior to the onset of inflammation and on days -1, 1, 3 and 5 during the 2.5% DSS and 2% NaCl treatment. The disease activity index (DAI), which was used for the clinical scoring of stool consistency, bleeding and weight loss, served as the measure of colitis severity. The criteria for grading the DAI were adopted from elsewhere[19].
Cell isolation
Spleen (SP) and mesenteric lymph node (MLN) cells from each mouse in all groups were separated by grinding on filters. SP red blood cells were lysed using red blood cell lysis buffer (C3702; Beyotime, China). Single cell suspensions of SP and MLN were obtained through filters. Cells were washed twice with phosphate-buffered saline (PBS) (Zhongshanjinqiao, China) containing 2% fetal calf serum (FBS; as 2% FBS/PBS) (Gibco, Life Technologies, United States) through centrifugation.
Cell pellets were resuspended in the 2% FBS/PBS and were kept on ice for later use. Intestinal LPMCs were isolated in accordance with the Lamina Propria Dissociation Kit instructions (130-097-410; Miltenyi Biotec, Germany). Cell pellets were resuspended in 40% percoll (Ruitaibio, China) and added slowly to the upper part of centrifuge tubes, which had 5 mL of 80% percoll at the bottoms. LPMCs were obtained by washing twice with 2% FBS/PBS after density gradient centrifuging at 420 g for 20 min.
Flow analysis
The isolated cells from SP, MLN and LP from each experimental group were cultured in 96-well U plates in 0.2 mL 1640 medium containing 1% penicillin-streptomycin (C0222; Beyotime) and 10% FBS with ionomycin (I) (1 μg/mL) (S1672; Beyotime), phorbol 12-myristate 13-acetate (PMA) (25 ng/mL) (S1819; Beyotime) and Brefeldin A (BFA) (10 μg/mL) (51-2092KZ; BD Bioscience, United States) for 6 h. The cells were collected and preblocked by Fc receptors for 20 min. Cell-surface staining was performed using PE-, FITC-, APC- or percp-conjugated anti-CD4, CD3, CD25 or CD11b (eBioscience, United States). Intracellular staining was performed using the FITC-conjugated anti-mouse IFN-γ, PE-conjugated anti-mouse IL-17 or Foxp3 (eBioscience). The intracellular or nuclear staining for IFN-γ, IL-17 and Foxp3 analysis was performed according to the BD Bioscience protocol.
LPMC stimulation
Isolated LPMCs were cultured at a concentration of 5 × 106 cells/mL for 24 h, after which the culture supernatants were collected and cytokine levels were analyzed by enzyme-linked immunosorbent assay (ELISA) or were stimulated using different NaCl concentrations (5, 10, 20, 40, 60 or 80 mmol/L) in the presence of 100 ng/mL LPS (Sigma, United States) and 20 ng/mL IFN-γ (Sigma) with SB20358 (p38 inhibitor) or DMSO (ST038; Beyotime) for 24 h. The cells were detected by western blot (WB) or real time-PCR (RT-PCR).
Mouse peritoneal macrophage preparation
Mice were injected intraperitoneally with 2 mL of 4% sterile thioglycollate medium (Becton Dickinson, United States)[20]. Peritoneal macrophages were obtained by washing the peritoneal cavity with 8 mL PBS containing 1% penicillin-streptomycin per mouse. Peritoneal macrophages were centrifuged and resuspended in DMEM (Gibco, Thermo Fisher Scientific, United States) containing 10% FBS and 1% penicillin-streptomycin. Next, peritoneal macrophages were seeded in 24-well plates (Corning, United States) and nonadherent cells were removed 4 h after seeding by washing with medium[21]. Once adhered to the culture plates, cells were stimulated with NaCl (10, 20, 40, 60 or 80 mmol/L) and 100 ng/mL LPS for 24 h. Finally, cells were collected for gene expression evaluation.
Colon culture
Colon tissues were cultured as previously described[22,23]. Briefly, after cutting longitudinally, colon tissues were washed with PBS for removing intestinal contents and were cut into 1-cm segments. These pieces were cultured in 24-well plates in 2 mL of RPMI1640 medium (Gibco, Life Technology, Shanghai, China) containing 1% penicillin-streptomycin for 24 h. Supernatant was obtained by centrifuging at 10000 g at 4 °C for 10 min and was immediately stored at -80 °C until required for further ELISA detection.
RNA isolation and RT-PCR
RNAs of cells and tissues were extracted by Trizol (Ambion, Life Technology, United States). RNA was transcribed into cDNA using reverse transcription kits (RR047A; Takara, Japan). Quantitative RT-PCR was performed using Bio-Rad instruments (United States) in duplicates with the reagent SYBR Green (RR820A; Takara) to measure the products. Gene expression was analyzed using the comparative Ct method and was normalized to GAPDH, which served as internal control. The primer sequences are shown in Table 1.
Table 1.
Primers used in the real time-PCR
| Gene name | Primer sequences | |
| GAPDH | Sense | 5’-AGGTCGGTGTGAACGGATT-3’ |
| Anti-sense | 5’-AATCTCCACTTTGCCACTGC-3’ | |
| IL-1β | Sense | 5’-TGGTGTGTGACGTTCCCATTA-3’ |
| Anti-sense | 5’-CAGCACGAGGCTTTTTTGTTG-3’ | |
| IL-1α | Sense | 5′-CGCCAATGACTCAGAGGAAGA-3′ |
| Anti-sense | 5′-GGCGTCATTCAGGATGAATTC-3′ | |
| IL-6 | Sense | 5’-ACAACCACGGCCTTCCCTACTT-3’ |
| Anti-sense | 5’-CACGATTTCCCAGAGAACATGTG-3’ | |
| IFN-γ | Sense | 5’-CTGCTGATGGGAGGAGATGT-3’ |
| Anti-sense | 5’-ATTTGTCATTCGGGTGTAGTCA-3’ | |
| Arg1 | Sense | 5’-CTCCAAGCCAAAGTCCTTAGAG-3’ |
| Anti-sense | 5’-GGAGCTGTCATTAGGGACATCA-3’ | |
| iNOS | Sense | 5’-ACATCGACCCGTCCACAGTAT-3’ |
| Anti-sense | 5’-CAGAGGGGTAGGCTTGTCTC-3’ | |
| IL-10 | Sense | 5’-GCTCTTACTGACTGGCATGAG-3’ |
| Anti-sense | 5’-CGCAGCTCTAGGAGCATGTG-3’ | |
| TNF-α | Sense | 5’-CTGAACTTCGGGGTGATCGG-3’ |
| Anti-sense | 5’-GGCTTGTCACTCGAATTTTGAGA-3’ | |
| IL-17α | Sense | 5′-TGTGAAGGTCAACCTCAAAGTCT-3’ |
| Anti-sense | 5′-GAGGGATATCTATCAGGGTCTTCAT-3′ | |
| SGK1 | Sense | 5′-CTGCTCGAAGCACCCTTACC-3′ |
| Anti-sense | 5′-TCCTGAGGATGGGACATTTTCA-3′ |
ELISA
Cytokine content was expressed in pg/mL. Abs, including purified and biotinylated antimouse, and related reagents were purchased from eBioscience. Briefly, 2 μg/mL capture antibody diluted with coat buffering was incubated at 4 °C overnight in 96-well plates (Corning) and was blocked with 5% bovine serum albumin (BSA) (Sigma) at 37 °C for 2 h. Samples were incubated at 37 °C for 2 h after being washed three times with PBS containing 0.05% Tween-20 (PBST). Biotinylated antibodies were incubated at 37 °C for 1 h after being washed with PBST three times. Horseradish peroxidase-conjugated antibody was incubated at 37 °C for 30 min after being washed with PBST five times. The reaction of detection reagent at 37 °C required 15 min after the unbounded antibody was removed by washing with PBST five times. The plate was analyzed at 450 nm wavelength after terminating the reaction with the stop solution.
Histology and immunohistochemistry
Colon tissues were fixed with 4% paraformaldehyde before being embedding in paraffin. To assess inflammation, colon tissue cross sections were stained with hematoxylin and eosin (HE). Sections were incubated with rabbit anti-mouse inducible nitric oxide synthase (iNOS) antibody labeled with FITC (orb14179; Biorbyt, United Kingdom) and rabbit anti-mouse F4/80 antibody labeled with PE (123109; Biolegend). All immunofluorescence images were taken by a fluorescence microscope (Leica, Germany) under the same exposure and intensity settings.
Western blotting
Proteins were extracted by RIPA lysis buffer containing protease inhibitor cocktail. The protein concentration was detected using the Protein Concentration Kits (P0012; Beyotime) and the samples were boiled for 5 min at 98 °C. Then, 30 μg of protein for each sample was separated with SDS-PAGE. Next, proteins were electrotransferred onto a nitrocellulose membrane (GE Healthcare, Sweden) and were blocked with 5% BSA in TBS-0.05% Tween-20 (TBST) at room temperature for 2 h. The membrane was subsequently incubated with GAPDH (1:1000) (Santa Cruz Biotechnologies, United States), p38 or phosphorylated p38 (1:250) (Abcam, United States) at 4 °C for 16 h. The membrane was washed with TBST before being incubated at room temperature for 1 h with antibody conjugated with horseradish peroxidase (1:2000) (Zhongshanjinqiao, China). Antibody binding was detected with the ECL substrate (170-5060; Bio-Rad) after washing with TBST. The optical density of bands was analyzed using ImageJ 1.42 software (United States).
Statistical analysis
All data were expressed as mean ± SD. GraphPad Prism 5.00 software for Windows (United States) was used for data analysis. Statistical results were evaluated using unpaired Student’s t-test or ANOVA, and P < 0.05 was considered statistically significant.
RESULTS
NaCl aggravates DSS-induced colitis in mice
To determine the influence of NaCl on enteritis, mice were given 2.5% DSS and/or 2% NaCl. Mice that received both NaCl and DSS started losing weight from day 5 and subsequently exhibited greater weight loss compared to the DSS group (Figure 1A). Moreover, the death rate in the DSS + NaCl group was markedly higher than in the DSS group (Figure 1B). Compared to other groups, colons of mice in the DSS + NaCl group became shorter (Figure 1C). HE staining displayed obvious inflammatory cell infiltration in both groups, but the DSS + NaCl group exhibited more inflammatory cell infiltration in colon tissues than the DSS group (Figure 1D). These findings suggest that NaCl aggravated inflammation in DSS-induced colitis.
Figure 1.
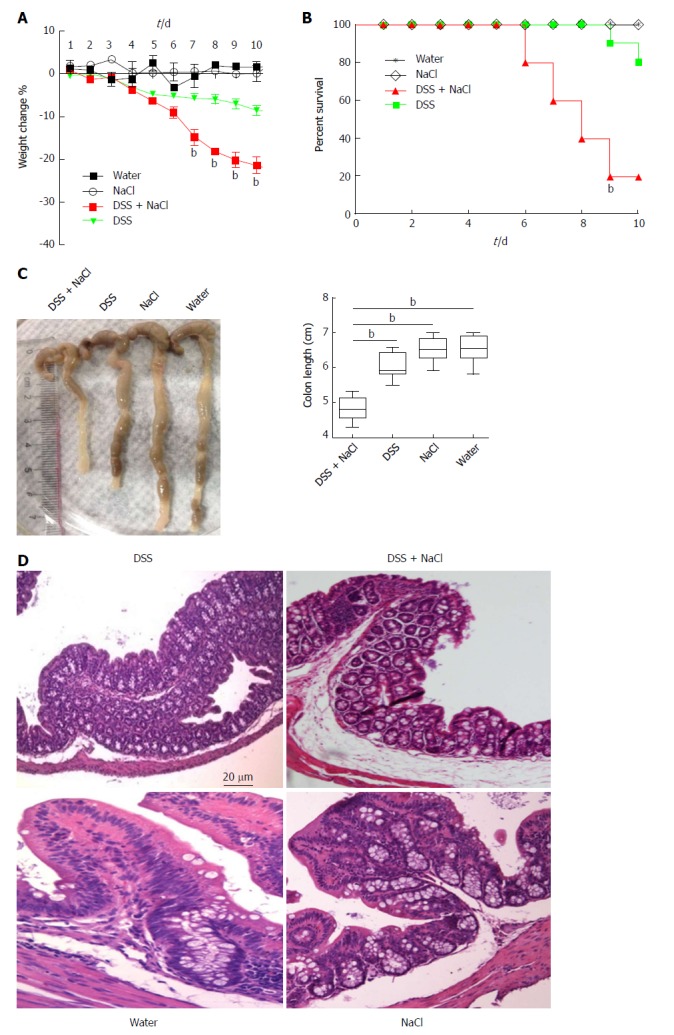
Mice treated with DSS and NaCl develop more severe colitis. A: Mice were given DSS and/or NaCl, and were weighed daily; B: Death status was recorded daily; C: Colonic tissues were collected from four groups of mice and colonic length was measured; D: Histological analyses show sections of the colon stained with HE for DSS- or NaCl-treated mice. In all the panels, data indicate three separate experiments, whereby 10 mice per group were used in each experiment. aP < 0.05; bP < 0.01; cP < 0.001. DSS: Dextran sulfate sodium.
NaCl up-regulates the frequency of CD4+IFN-γ+IL17+ T cells and promotes the secretion of inflammatory cytokines in mice with DSS-induced colitis
Increasing evidence indicates that CD4+ T cells play a crucial role in the pathogenesis of chronic intestinal inflammation, and related cytokines (such as IFN-γ, IL-6, IL-17A and TNF) are highly expressed in the inflamed mucosa of IBD patients[24,25]. To explore the influence of NaCl on CD4+ T cells in colitis-affected mice, the CD4+IFN-γ+IL-17+ T cell subsets were detected. Compared to the DSS group, the flow cytometry analysis indicated that frequencies of CD4+IL-17+ and CD4+IFN-γ+ T cell subsets were markedly up-regulated in the DSS + NaCl group (Figure 2A). NaCl promotion of the DSS-induced colitis development is associated with both CD4+IL-17+ and CD4+IFN-γ+ T cells in LP, MLN and SP. In addition, the frequencies of inflammatory CD4+ T cells (IL-17+ and IFN-γ+ single-positive T cells and IFN-γ+IL-17+ double-positive T cells) in the DSS + NaCl group were higher than in the DSS group. It is also noteworthy that the frequency of CD4+IFN-γ+ T cells was up-regulated the most (Figure 2B). These findings suggest that CD4+IFN-γ+IL-17+ T cells are crucial in the inflammation promotion by NaCl in DSS-treated mice.
Figure 2.
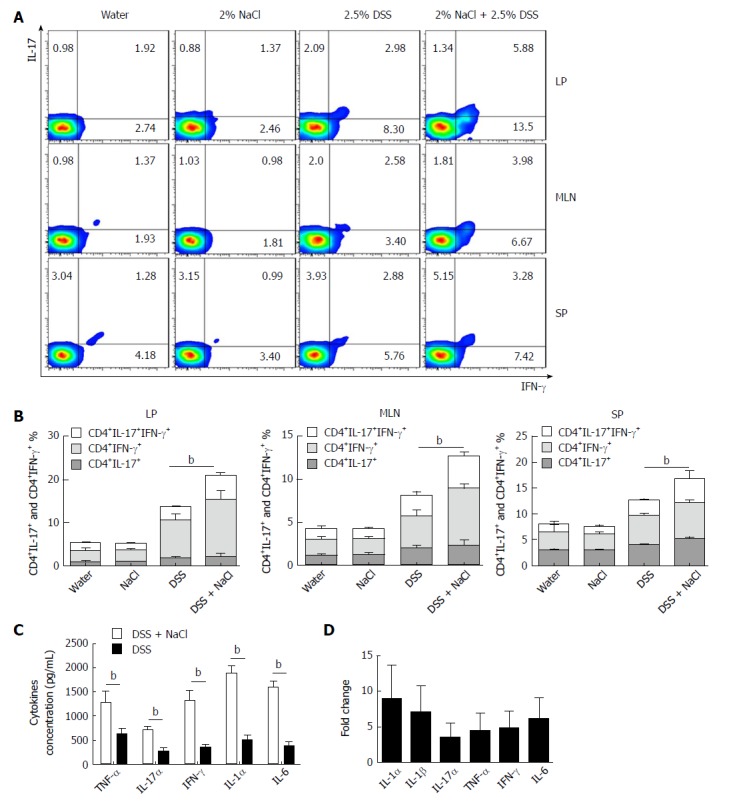
NaCl promotes CD4+IFN-γ+IL-17+ T cell increase and inflammatory cytokine secretion in DSS-treated mice. A: The CD4+IFN-γ+IL-17+ T cells in LP, MLN and SP from mice treated with NaCl and/or DSS were detected by flow cytometry; B: Combined flow cytometry data of CD4+IL-17+, CD4+IFN-γ+ and CD4+IFN-γ+IL-17+ T cell subsets distribution in LP, MLN and SP; C: Colon tissues collected from mice treated with DSS or DSS + NaCl, which were washed with phosphate-buffered saline and cultured for 24 h, and the supernatants were collected and detected by enzyme-linked immunosorbent assay; D: Colon tissues collected from mice treated with NaCl and DSS (or only DSS) were detected by RT-PCR. The relative fold-change in DSS + NaCl-treated mice vs DSS-treated mice. In all the panels, data indicate three separate experiments, whereby 3 mice per group were used in each experiment. aP < 0.05; bP < 0.01; cP < 0.001. DSS: Dextran sulfate sodium; LP: Lamina propria; MLN: Mesenteric lymph node; SP: Spleen.
Cytokines IFN-γ, IL-17α, IL-1α, IL-6 and TNF-α secreted by colon tissues were detected by ELISA, and the gene expression of colon tissues from the animal model was measured by RT-PCR. Compared to the DSS group, IFN-γ, IL-17α, IL-1α, IL-1β, IL-6 and TNF-α were all higher in the DSS + NaCl group (Figure 2C and D). Therefore, high NaCl levels up-regulate inflammation gene expression and promote the secretion of multiple proinflammatory cytokines in mice affected by DSS-induced colitis.
NaCl up-regulates macrophage frequency in DSS-treated mice
Macrophages play a crucial role in the Th1 and Th17 responses, and are also important regulators of salt homeostasis[26]. To determine the effect of NaCl on macrophages in mice affected by colitis, we detected the frequency of CD11b+ macrophages in mice that received DSS and/or NaCl by flow cytometry. We observed that the macrophages increased significantly in the LP, MLN and SP of the DSS + NaCl group compared to those of the DSS group (Figure 3A). The increased CD11b+ macrophages were mainly located in intestinal LP and MLN (Figure 3B). These findings indicate that the macrophages also participate in the NaCl proinflammation activities in DSS-induced colitis.
Figure 3.

CD11b+ macrophages are increased in DSS- and NaCl-treated mice. A: The CD11b+ cells in LP, MLN and SP from the four groups were detected by flow cytometry; B: Quantification of the flow cytometry data indicates the CD11b+ cell distribution in LP, MLN and SP. In the panels, data indicate three separate experiments, whereby 3 mice per group were used in each experiment. aP < 0.05; bP < 0.01; cP < 0.001. DSS: Dextran sulfate sodium; LP: Lamina propria; MLN: Mesenteric lymph node; SP: Spleen.
Tregs increase through feedback in the development of NaCl aggravating inflammation associated with DSS-induced colitis
Tregs play an important role in the maintenance of intestinal mucosal homeostasis by suppressing abnormal immune response against dietary antigens or commensal flora[8]. To explore the changes in Tregs in the mice that received DSS and NaCl, we detected CD3+CD4+CD25+Forp3+ T cells by flow cytometry and observed that their levels were higher in the DSS + NaCl group than in the DSS group (Figure 4A). The increased Tregs were mainly distributed in the LP and MLN, while their prevalence in SP did not change significantly (Figure 4B). To explore the influence of NaCl on Tregs in DSS-induced colitis, we evaluated cytokine levels in culture supernatants of LPMCs by ELISA. The results yielded by the analyses indicate that NaCl induces LPMCs to secrete TNF-α, IL-1α, IL-6 and IL-17, which are critical Th17 cell-related cytokines. Moreover, NaCl promotes the secretion of TGF-β and IL-10, which are significant antiinflammatory cytokines secreted by Tregs (Figure 4C). These findings show that Tregs’ levels also increase as a result of inflammation promotion by NaCl in mice with DSS-induced colitis.
Figure 4.
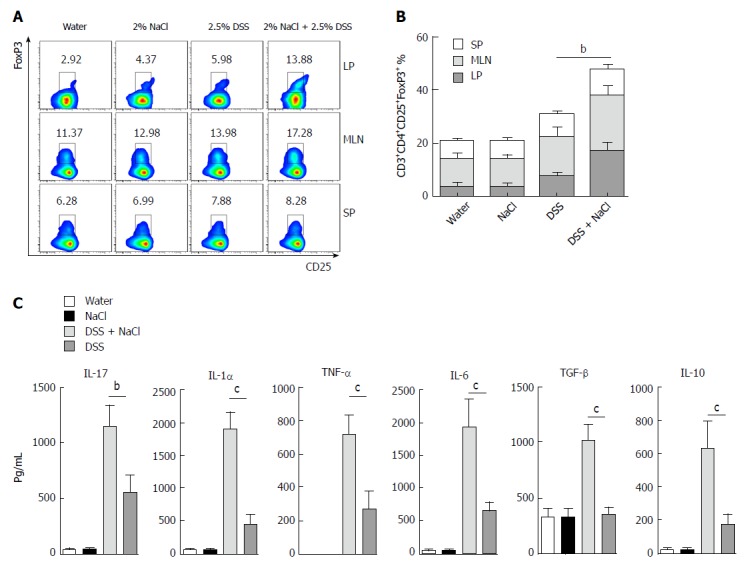
CD3+CD4+CD25+Foxp3+ T cells are increased in mice treated with DSS and NaCl. A: CD3+CD4+CD25+Foxp3+ T cells in LP, MLN and SP from animal models were detected by flow cytometry; B: A summary of the percentages of CD3+CD4+CD25+Foxp3+ T cell distribution in LP, MLN and SP; C: LPMCs from the four groups were isolated and cultured for 24 h, and the levels of cytokines in the culture supernatants were collected and analyzed by enzyme-linked immunosorbent assay. In all the panels, data indicate three separate experiments, whereby 3 mice per group were used in each experiment. aP < 0.05; bP < 0.01; cP < 0.001 vs the DSS group. DSS: Dextran sulfate sodium; LMPCs: Lamina propria mononuclear cells; LP: Lamina propria; MLN: Mesenteric lymph node; SP: Spleen.
Macrophages play a critical role in NaCl aggravating DSS-induced colitis
Extant studies have shown that MDP can deplete macrophages in mice[27]. We used MDP to deplete the macrophages in mice during the DSS and NaCl treatments to determine their role in the promotion of DSS-induced colitis by NaCl (Figure 5A). We observed that macrophage depletion by MDP could prevent colon shortening in the mice treated with NaCl and DSS (Figure 5B). The DAI also showed that macrophage depletion alleviated inflammation in NaCl proinflammatory processes (Figure 5C). The levels of inflammatory cytokines IFN-γ, TNF-α, IL-1β, IL-17A, IL-6, MCP1 and MIP2 secreted by colon tissues were reduced in MDP-treated mice (Figure 5D). The colon tissues from the DSS + NaCl group contained a greater number of F4/80+iNOS+ macrophages compared to the DSS group. In addition, the MDP-treated mice had fewer F4/80+iNOS+ macrophages compared to the DSS + NaCl group (Figure 6). Thus, we posit that macrophage depletion can reduce colitis severity in mice.
Figure 5.
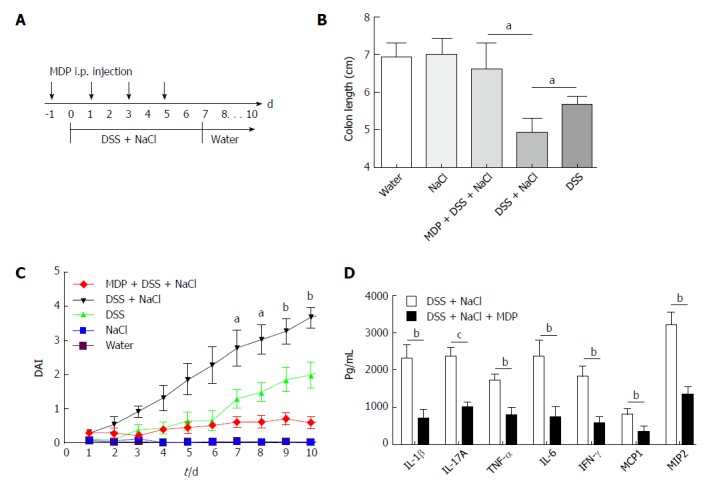
Depletion of macrophages reduces the severity of DSS-induced colitis promoted by NaCl. A: Clodronate liposomes (denoted as MDP) or control PBS-liposomes (denoted as PBS) were administrated intravenously to all mice, as the schematic protocol indicated during DSS and NaCl treatment; B: The disease activity index was monitored daily; C: Colon length was measured in each group of mice (n = 10); D: Colon explants were cultured for 24 h and the inflammatory cytokines in supernatants were detected by enzyme-linked immunosorbent assay (n = 3). aP < 0.05; bP < 0.01; cP < 0.001 (clodronate liposomes + DSS + NaCl vs DSS + NaCl). DSS: Dextran sulfate sodium.
Figure 6.

iNOS+F4/80+ macrophages increase in the colon of DSS- and NaCl-treated mice. Macrophages in colon tissue obtained from mice injected intraperitoneally with PBS-containing liposomes (denoted as PBS), or clodronate liposomes (denoted as MDP) during the NaCl and DSS treatment were analyzed. The sections were stained with antibodies of anti-F4/80 (red) and anti-iNOS (green). Nuclei were stained with DAPI (blue). Laser confocal microscopy was used to detect fluorescence. (Scale bar = 50 μm). DSS: Dextran sulfate sodium; iNOS: inducible nitric oxide synthase.
High NaCl promotes M1 macrophage polarization in vitro
Macrophages in both peritoneal cavity and gastrointestinal tract are linked to IBD[28]. Different NaCl concentrations (10, 20, 40, 60 and 80 mmol/L) were used to stimulate the macrophages from the abdominal cavity and the gene expression was detected by RT-PCR. Our findings indicate that IL-1β, IL-6 and iNOS, which usually exhibit proinflammatory roles, gradually increased as the NaCl concentration increased (Figure 7A-C). It is worth noting that IL-10 and Arg1, which are M2 macrophage markers, increased modestly at low NaCl concentrations, whereas their expression markedly increased at 40 mmol/L and above (Figure 7D and E). These results display that high NaCl levels promote LPS-activated peritoneal macrophages toward M1 polarization.
Figure 7.

High NaCl levels enhance proinflammation gene expression in LPS-activated peritoneal macrophage. A-E: Peritoneal macrophages were stimulated with different NaCl concentrations (10-80 mmol/L) in the presence of LPS for 24 h. mRNA expression was measured by RT-PCR for the indicated genes. In all the panels, data indicate three separate experiments. aP < 0.05; bP < 0.01; cP < 0.001. LPS: Lipopolysaccharide.
NaCl promotes the inflammation response in LP, whereas LPS and IFN-γ activated LPMCs rely on p38/MAPK
p38/MAPK is related to both IBD and hyperosmotic stress[29,30]. Western blot analysis revealed that high NaCl levels significantly up-regulated phosphorylated-p38 of LPMCs stimulated with LPS and IFN-γ for different time periods (1 h, 6 h, 12 h, 24 h); however, they did not affect the total level of p38, and p38 phosphorylation reached the highest level after 12 h (Figure 8A). LPMCs were treated with NaCl at different concentrations (5, 10, 20, 40, 60 and 80 mmol/L) in the presence of LPS and IFN-γ for 24 h. The western blotting revealed that p38 phosphorylation increased in a dose-dependent manner (Figure 8B). Serum glucocorticoid regulated kinase 1 (SGK1) increased in LPMCs activated by LPS and IFN-γ due to NaCl stimulation (Figure 8C). The results further indicated that p38 inhibitor can decrease high NaCl-promoted p38 phosphorylation in LPMCs (Figure 8D). These findings confirmed that NaCl promotes inflammatory response in the LPS and IFN-γ activated LPMCs, and the proinflammation effect depends on p38/MAPK phosphorylation mediated by SGK1.
Figure 8.
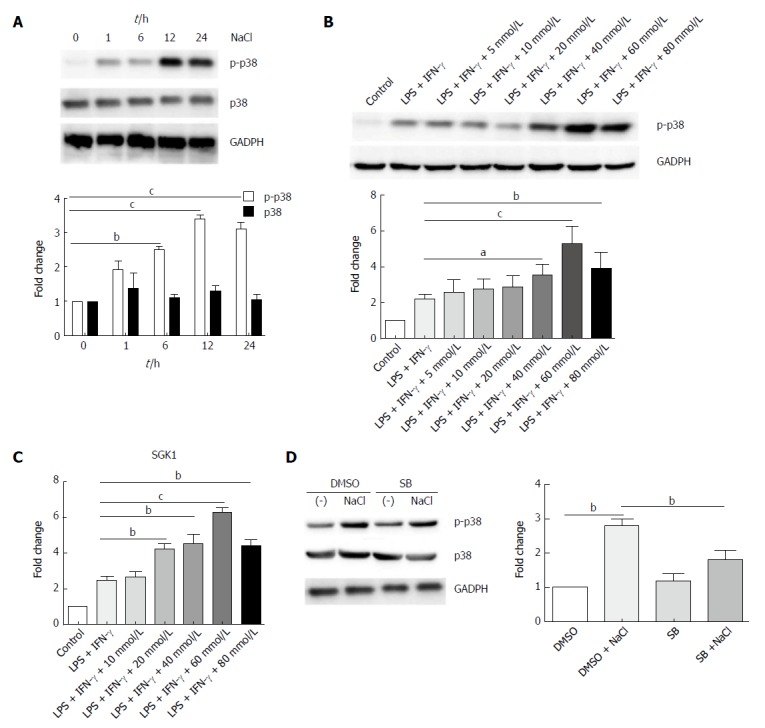
NaCl promotion of inflammation relies on the p38/MAPK pathway. A: LPMCs were stimulated with 60 mmol/L NaCl in the presence of 100 ng/mL LPS and 20 ng/mL IFN-γ for 1 h, 6 h, 12 h and 24 h, and the p38 and phosphorylated-p38 proteins were detected by western blot; B: LPMCs were stimulated with different NaCl concentrations in the presence of 100 ng/mL LPS and 20 ng/mL IFN-γ for 24 h and phosphorylated p38 protein was detected by western blot; C: LPMCs were stimulated with different NaCl concentrations in the presence of LPS and IFN-γ, and the mRNA expression of SGK1 was measured by RT-PCR; D: LPMCs were pretreated with 10 μmol/L SB or DMSO for 2 h and were subsequently stimulated with 60 mmol/L NaCl along with 100 ng/mL LPS and 20 ng/mL IFN-γ in the presence of DMSO or 10 μmol/L SB for 24 h and the proteins of p38 and phosphorylated-p38 were detected by western blot. In all the panels, data indicate three separate experiments. aP < 0.05; bP < 0.01; cP < 0.001. DSS: Dextran sulfate sodium; LMPCs: Lamina propria mononuclear cells; LP: Lamina propria; LPS: Lipopolysaccharide; MLN: Mesenteric lymph node; SB: SB20358; SP: Spleen.
DISCUSSION
NaCl has been shown to exert a proinflammatory effect in many diseases, including experimental colitis, experimental autoimmune encephalomyelitis and cardiovascular disease[31-33]. In the present study, we observed that macrophages play an important role in the promotion of DSS-induced colitis by NaCl. Macrophages, as antigen-presenting cells, are important in regulating innate and adaptive immune responses and have a crucial role in resolving tissue injury and promoting tissue repair in IBD[34,35]. Even though the cause of IBD remains unclear, mice with lymphocyte deficiency developed more severe inflammation, suggesting that innate immune cells are capable of triggering the onset and development of disease[36]. Activation of the innate immune system is regarded as the most direct cause of IBD because it can recruit cells of the adaptive immune system to the inflammatory site, thus resulting in inflammation[37].
Findings yielded by the present study further indicated that NaCl promoted the increase in the CD4+ T cell count, especially the IFN-γ+IL-17+ double-positive T cells in DSS-treated mice. Extant research indicates that high-salt diet promotes the differentiation of CD4+ T cells into Th17 as well as Th1[32]. However, Wei et al[38] showed that, in 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis, NaCl promoted Th17 polarization, but not Th1 polarization[15]. DSS and TNBS may involve different pathogenic mechanisms. Wei et al[38] used TNBS to induce colitis, which mainly simulated CD. However, we used DSS to induce colitis, which mainly simulated ulcerative colitis. In both CD and ulcerative colitis patients, activation and mucosal infiltration of CD4+ T lymphocytes has been reported[39].
Extant studies have revealed that blocking CD4+ T cell activation was capable of limiting the development of mucosal inflammation in experimental colitis models[40]. CD4+ IFN-γ+IL-17+ T cells, as an intermediate form between Th17 and Th1, are an easily observable crossover subset promoted by IL-12 signaling beyond IL-17[41,42]. Th17 cells play an important role in colitis pathogenesis by directly giving rise to Th1-like cell response[43]. Empirical evidence indicates that IBD is characterized by Th1 cell activation and subsequent over-expression of cytokines such as TNF-α, IL-6 and IL-1β[44,45]. In addition, findings yielded by extant research suggest that Th1 cytokines are important promoters of continuous mucosal inflammation in DSS-induced colitis[46,47].
The results obtained in the present work confirmed the important role of CD4+IFN-γ+IL-17+ T cells in the promotion of inflammation by NaCl in DSS-treated mice. High NaCl content up-regulates inflammation gene expression and promotes the secretion of multiple proinflammatory cytokines for promoting intestinal inflammation in mice affected by DSS-induced colitis.
IL-6 and IL-17 are critical Th17 cell-related cytokines that are involved in inflammatory responses during IBD development[7,48]. In contrast, antiinflammatory TGF-β and IL-10, are mainly produced by Tregs[49]. Wei and colleagues[15] demonstrated that, while high-salt diet did not change Tregs’ percentage, it did inhibit the secretion of IL-10 and the suppressive function of Tregs in TNBS-induced colitis. In our study, NaCl promoted an increase in Tregs’ frequency in MLN and LP, as well as enhanced IL-10 and TGF-β expression, in DSS-induced colitis. Tregs, as immune suppressing cells, are essential in maintaining intestinal homeostasis[50].
In DSS-treated beta7-deficient mice, in which colonic Tregs were depleted, excessive macrophage infiltration in colons occurred by up-regulation of colonic epithelial intercellular ICAM1, which promoted proinflammatory cytokine expression, aggravating DSS-induced colitis[51]. Disruption in balance may allow T cells to proliferate in an increased fashion, thereby promoting chronic intestinal inflammatory development[52]. Therefore, continuous Tregs’ differentiation and trafficking in the gut is required to dampen immune responses to dietary antigens and commensal bacteria[53].
We also found that NaCl promoted an increase in CD11b+ cells in the LP and MLN from mice treated with DSS. Denning et al[54] have shown that CD11b+F4/80+CD11c- macrophages in LP could induce Foxp3+ Tregs’ differentiation, while CD11b+ dendritic cells in LP elicited responses of IL-17-producing T cells[54]. Empirical evidence indicates that the intake of high dietary salt could boost Th17 response through activating the caspase-1 in macrophages[15,55]. Moreover, in reaction to NaCl, macrophages with enhanced expression of immune-stimulatory molecules promote proinflammatory cytokine production and T cell proliferation[10,56].
Human monocyte-derived granulocyte-macrophage colony-stimulating factors exhibit potent antigen-presenting functions, produce IL-12p40 and IL-23p19, and promote development of Th1 immunity[57,58]. In our study, inflammation was relieved when the intestinal macrophages were depleted by MDP, which indicated that the activation of Th17 and Th1 cells required macrophage participation.
Peritoneal macrophages from mice are among the best-studied macrophage populations and their role in the regulation of inflammatory responses and mucosal immunity is well understood[59,60]. Macrophages in peritoneal cavity, which are crucial in the regulation of inflammatory pathologies, are also related to IBD[28,61]. In the present study, we have shown that high NaCl content enhanced the expression of proinflammation genes for IL-1β, IL-6 and iNOS and antiinflammation genes for Arg1 and IL-10 in macrophages from the abdominal cavity of mice.
Macrophages can be polarized to either classically activated (M1) or alternatively activated (M2) macrophages[62]. M1 macrophages are proinflammatory cells due to their high capacity for producing proinflammatory cytokines, such as IL-23, IL-12, IL-1β, TNF-α and iNOS[63,64]. M2 macrophages highly express IL-10 and Arg1, which are involved in antiinflammatory, antimicrobial response[62,65]. These cytokines promote the activation of the adaptive immune and T cell response[66].
In the present study, high NaCl content was found to boost M1 polarization and up-regulate expression of proinflammatory genes to promote inflammation. Under low NaCl concentrations, IL-1, IL-6 and iNOS mainly produced by M1 macrophages were up-regulated, while the negative adjustment factor expressions were low. When the NaCl concentration rose to a certain dose, high levels of proinflammatory factors IL-1, IL-6 and iNOS induced the cell protective response through feedback, and caused the up-regulation of negative adjustment factors IL-10 and Arg1. Thus, when the inflammation continues to worsen, the M2 macrophages will respond to balance inflammation with protective immunity, and inhibit the expression of proinflammatory factors.
We explored the influence of NaCl on LPS- and IFN-γ-activated LPMCs and demonstrated that high NaCl enhanced phosphorylation of p38, as inflammation and salt intake are both linked to p38/MAPK. The p38/MAPK signaling pathway is important in IBD and the inhibition of p38/MAPK can effectively suppress the production of inflammatory mediators[29]. Available evidence indicates that p38/MAPK mediates intestinal inflammation gene expression, such as TNF-α, IL-1 and IL-6, and this up-regulation occurs in multiple types of cells, especially monocytes and macrophages[67]. In addition, p38/MAPK can regulate the SGK1 activation[30].
High NaCl concentration promotes p38/MAPK phosphorylation and activates SGK1[32]. SGK1 has been shown to control Na(+) transport and NaCl homeostasis in cells, and could trigger Th17 responses and promote tissue inflammation[12]. Human LPMCs exposed to high NaCl concentrations highly express IL-17A, IL-23R and TNF-α, and pharmacological inhibition of p38/MAPK has been shown to abrogate the effect of NaCl on LPMC-derived cytokines[14]. In the present study, high NaCl content was shown to promote inflammation in LPS- and IFN-γ-activated LPMCs. However, this process relies on the up-regulation of p38/MAPK and SGK1.
In summary, the study findings reported here indicate that NaCl induces alterations to both the innate and acquired immune system in mice with DSS-induced colitis. NaCl promotes M1 macrophage polarization, and M1 polarization may shift T cell response toward the proinflammatory CD4+IFN-γ+IL-17+ T cells’ aggravating colitis. The mechanism by which high NaCl concentrations promote inflammation relies on the up-regulation of p38/MAPK and SGK1. Although results obtained in the present study indicate that excessive NaCl intake can promote the inflammation in mice with the DSS-induced colitis, the causality of high-salt diet and IBD still needs to be confirmed by further investigations. More clinical and experimental studies are required to fully clarify the role of salt in IBD.
ARTICLE HIGHLIGHTS
Research background
At present, most diets are characterized by high salt content. Extant studies have shown that high salt intake contributes to inflammatory bowel disease (IBD) incidence and pathogenesis. However, the mechanism underlying these effects remains unclear.
Research motivation
NaCl mediates the inflammatory effects of immune cells. Both innate and adaptive immune proinflammatory cells play important roles in IBD. Studies have shown the high salt intake promotes the activation of Th17 cells in lamina propria (LP) and exacerbates experimental colitis in mice. However, the influence of high salt content in diet on other immune cells is still unclear. The present study explored the influence of high NaCl concentration on immune cell subsets and the underlying mechanisms.
Research objectives
The aim of the present study was to determine the impact of high NaCl concentration on dextran sulfate sodium (DSS)-induced colitis in mice and explore its influence on other immune cells, such as T helper 1 cells, regulatory T cells and macrophages, while attempting to elucidate the mechanism underlying this effect.
Research methods
DSS and NaCl were used to establish a proinflammatory animal model. The immune cell subsets were detected by flow cytometry in order to determine the target cells of NaCl. Cytokines secreted by intestinal tissue were detected. In the present study, clodronate liposomes treatment was used to deplete macrophages to further delineate their vital role in the promotion of DSS-induced colitis in mice by NaCl. In cell experiments, NaCl at different concentrations acted directly on lamina propria mononuclear cells (LPMCs) and macrophages. mRNA levels of inflammation genes and p38/MAPK proteins were determined by RT-PCR and western blot, respectively.
Research results
High NaCl concentration exacerbated the DSS-induced colitis. Intestinal CD4+IFN-γ+IL-17+ T cells and macrophages both play crucial roles in the promotion of inflammation by NaCl in mice with colitis. NaCl promotes M1 proinflammatory gene expression in lipopolysaccharide (LPS)-activated peritoneal macrophages. High NaCl concentrations promote the up-regulation of the p38/MAPK axis in the LPS and IFN-γ-activated LPMCs.
Research conclusions
NaCl evokes both innate and adaptive immune proinflammatory cell activation in mice affected by colitis. Colitis may be promoted by high NaCl levels, by NaCl initially by acting on macrophages, pushing them towards M1 polarization. Then, M1 polarization shifts the T cell response toward proinflammatory CD4+IFN-γ+IL-17+ T cells. Inflammation promotion by NaCl in LPS- and IFN-γ-activated LPMCs relies on the up-regulation of the p38/MAPK axis.
Research perspectives
Although results in this study indicate that high NaCl intake can promote the inflammation in mice with DSS-induced colitis, the causality of high-salt diet and IBD still needs to be confirmed by further investigations. More clinical and experimental studies are inspired to fully clarify the role of salt in IBD.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Supported by National Natural Science Foundation of China, No. 81271813 and No. 81570497.
Institutional review board statement: This study was reviewed and approved by the Third Military Medical University (Army Medical University) Institutional Review Board.
Institutional animal care and use committee statement: All procedures involving the care and use of animals were approved by The Institutional Animal Care and Use Committee of the Third Military Medical University (Army Medical University).
Conflict-of-interest statement: All authors declared there were no conflicts of interests.
Data sharing statement: No additional data are available.
ARRIVE guidelines statement: The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Peer-review started: January 26, 2018
First decision: February 24, 2018
Article in press: March 18, 2018
P- Reviewer: Chiba T, Day AS, De Ponti F, Triantafillidis JK S- Editor: Gong ZM L- Editor: Filipodia E- Editor: Huang Y
Contributor Information
Hong-Xia Guo, Department of Microbiology, Third Military Medical University (Army Medical University), District Shapingba, Chongqing 400038, China; Institute of Tropical Medicine, Third Military Medical University (Army Medical University), District Shapingba, Chongqing 400038, China.
Nan Ye, Institute of Tropical Medicine, Third Military Medical University (Army Medical University), District Shapingba, Chongqing 400038, China.
Ping Yan, Department of Obstetrics and Gynecology, Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing 400038, China.
Min-Yue Qiu, Institute of Tropical Medicine, Third Military Medical University (Army Medical University), District Shapingba, Chongqing 400038, China.
Ji Zhang, Institute of Immunology, Third Military Medical University (Army Medical University), District Shapingba, Chongqing 400038, China.
Zi-Gang Shen, Institute of Immunology, Third Military Medical University (Army Medical University), District Shapingba, Chongqing 400038, China.
Hai-Yang He, Institute of Immunology, Third Military Medical University (Army Medical University), District Shapingba, Chongqing 400038, China.
Zhi-Qiang Tian, Institute of Immunology, Third Military Medical University (Army Medical University), District Shapingba, Chongqing 400038, China.
Hong-Li Li, Department of Histology and Embryology, College of Basic Medicine, Third Military Medical University (Army Medical University), Chongqing 400038, China.
Jin-Tao Li, Department of Microbiology, Third Military Medical University (Army Medical University), District Shapingba, Chongqing 400038, China; Institute of Tropical Medicine, Third Military Medical University (Army Medical University), District Shapingba, Chongqing 400038, China. ljtqms@tmmu.edu.cn.
References
- 1.Vasovic M, Gajovic N, Brajkovic D, Jovanovic M, Zdravkovaic N, Kanjevac T. The relationship between the immune system and oral manifestations of inflammatory bowel disease: a review. Cent Eur J Immunol. 2016;41:302–310. doi: 10.5114/ceji.2016.63131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boedeker EC. Gut microbes, the innate immune system and inflammatory bowel disease: location, location, location. Curr Opin Gastroenterol. 2007;23:1–3. doi: 10.1097/MOG.0b013e328011b837. [DOI] [PubMed] [Google Scholar]
- 3.Van Limbergen J, Russell RK, Nimmo ER, Ho GT, Arnott ID, Wilson DC, Satsangi J. Genetics of the innate immune response in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:338–355. doi: 10.1002/ibd.20096. [DOI] [PubMed] [Google Scholar]
- 4.Reinisch W. Fecal Microbiota Transplantation in Inflammatory Bowel Disease. Dig Dis. 2017;35:123–126. doi: 10.1159/000449092. [DOI] [PubMed] [Google Scholar]
- 5.Dutta AK, Chacko A. Influence of environmental factors on the onset and course of inflammatory bowel disease. World J Gastroenterol. 2016;22:1088–1100. doi: 10.3748/wjg.v22.i3.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang W, Su J, Zhang X, Cheng X, Zhou J, Shi R, Zhang H. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Inflamm Res. 2014;63:943–950. doi: 10.1007/s00011-014-0768-7. [DOI] [PubMed] [Google Scholar]
- 7.Hundorfean G, Neurath MF, Mudter J. Functional relevance of T helper 17 (Th17) cells and the IL-17 cytokine family in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:180–186. doi: 10.1002/ibd.21677. [DOI] [PubMed] [Google Scholar]
- 8.Yang F, Wang D, Li Y, Sang L, Zhu J, Wang J, Wei B, Lu C, Sun X. Th1/Th2 Balance and Th17/Treg-Mediated Immunity in relation to Murine Resistance to Dextran Sulfate-Induced Colitis. J Immunol Res. 2017;2017:7047201. doi: 10.1155/2017/7047201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 10.Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, Sakuraba A, Kitazume MT, Sugita A, Koganei K, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest. 2008;118:2269–2280. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahida YR. The key role of macrophages in the immunopathogenesis of inflammatory bowel disease. Inflamm Bowel Dis. 2000;6:21–33. doi: 10.1097/00054725-200002000-00004. [DOI] [PubMed] [Google Scholar]
- 12.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang WC, Zheng XJ, Du LJ, Sun JY, Shen ZX, Shi C, Sun S, Zhang Z, Chen XQ, Qin M, et al. High salt primes a specific activation state of macrophages, M(Na) Cell Res. 2015;25:893–910. doi: 10.1038/cr.2015.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monteleone I, Marafini I, Dinallo V, Di Fusco D, Troncone E, Zorzi F, Laudisi F, Monteleone G. Sodium chloride-enriched Diet Enhanced Inflammatory Cytokine Production and Exacerbated Experimental Colitis in Mice. J Crohns Colitis. 2017;11:237–245. doi: 10.1093/ecco-jcc/jjw139. [DOI] [PubMed] [Google Scholar]
- 15.Wei Y, Lu C, Chen J, Cui G, Wang L, Yu T, Yang Y, Wu W, Ding Y, Li L, et al. High salt diet stimulates gut Th17 response and exacerbates TNBS-induced colitis in mice. Oncotarget. 2017;8:70–82. doi: 10.18632/oncotarget.13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko JK, Auyeung KK. Inflammatory bowel disease: etiology, pathogenesis and current therapy. Curr Pharm Des. 2014;20:1082–1096. doi: 10.2174/13816128113199990416. [DOI] [PubMed] [Google Scholar]
- 17.Li K, Wang B, Sui H, Liu S, Yao S, Guo L, Mao D. Polymorphisms of the macrophage inflammatory protein 1 alpha and ApoE genes are associated with ulcerative colitis. Int J Colorectal Dis. 2009;24:13–17. doi: 10.1007/s00384-008-0575-0. [DOI] [PubMed] [Google Scholar]
- 18.Weisser SB, van Rooijen N, Sly LM. Depletion and reconstitution of macrophages in mice. J Vis Exp. 2012;1:4105. doi: 10.3791/4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li YH, Xiao HT, Hu DD, Fatima S, Lin CY, Mu HX, Lee NP, Bian ZX. Berberine ameliorates chronic relapsing dextran sulfate sodium-induced colitis in C57BL/6 mice by suppressing Th17 responses. Pharmacol Res. 2016;110:227–239. doi: 10.1016/j.phrs.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 20.Nam TG, Lim TG, Lee BH, Lim S, Kang H, Eom SH, Yoo M, Jang HW, Kim DO. Comparison of Anti-Inflammatory Effects of Flavonoid-Rich Common and Tartary Buckwheat Sprout Extracts in Lipopolysaccharide-Stimulated RAW 264.7 and Peritoneal Macrophages. Oxid Med Cell Longev. 2017;2017:9658030. doi: 10.1155/2017/9658030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He G, Zhang X, Chen Y, Chen J, Li L, Xie Y. Isoalantolactone inhibits LPS-induced inflammation via NF-κB inactivation in peritoneal macrophages and improves survival in sepsis. Biomed Pharmacother. 2017;90:598–607. doi: 10.1016/j.biopha.2017.03.095. [DOI] [PubMed] [Google Scholar]
- 22.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci USA. 2001;98:13249–13254. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou R, Chang Y, Liu J, Chen M, Wang H, Huang M, Liu S, Wang X, Zhao Q. JNK Pathway-Associated Phosphatase/DUSP22 Suppresses CD4+ T-Cell Activation and Th1/Th17-Cell Differentiation and Negatively Correlates with Clinical Activity in Inflammatory Bowel Disease. Front Immunol. 2017;8:781. doi: 10.3389/fimmu.2017.00781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK, Beck FX, Müller DN, Derer W, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15:545–552. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 27.Zhao A, Urban JF Jr, Anthony RM, Sun R, Stiltz J, van Rooijen N, Wynn TA, Gause WC, Shea-Donohue T. Th2 cytokine-induced alterations in intestinal smooth muscle function depend on alternatively activated macrophages. Gastroenterology. 2008;135:217–225.e1. doi: 10.1053/j.gastro.2008.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gross M, Salame TM, Jung S. Guardians of the Gut - Murine Intestinal Macrophages and Dendritic Cells. Front Immunol. 2015;6:254. doi: 10.3389/fimmu.2015.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng YJ, Li YY. The role of p38 mitogen-activated protein kinase in the pathogenesis of inflammatory bowel disease. J Dig Dis. 2011;12:327–332. doi: 10.1111/j.1751-2980.2011.00525.x. [DOI] [PubMed] [Google Scholar]
- 30.Bell LM, Leong ML, Kim B, Wang E, Park J, Hemmings BA, Firestone GL. Hyperosmotic stress stimulates promoter activity and regulates cellular utilization of the serum- and glucocorticoid-inducible protein kinase (Sgk) by a p38 MAPK-dependent pathway. J Biol Chem. 2000;275:25262–25272. doi: 10.1074/jbc.M002076200. [DOI] [PubMed] [Google Scholar]
- 31.Tubbs AL, Liu B, Rogers TD, Sartor RB, Miao EA. Dietary Salt Exacerbates Experimental Colitis. J Immunol. 2017;199:1051–1059. doi: 10.4049/jimmunol.1700356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–522. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zidek W. [High salt consumption increases cardiovascular risk in hypertonic patients] Dtsch Med Wochenschr. 2016;141:1524. doi: 10.1055/s-0042-117074. [DOI] [PubMed] [Google Scholar]
- 34.Gren ST, Grip O. Role of Monocytes and Intestinal Macrophages in Crohn’s Disease and Ulcerative Colitis. Inflamm Bowel Dis. 2016;22:1992–1998. doi: 10.1097/MIB.0000000000000824. [DOI] [PubMed] [Google Scholar]
- 35.Steinbach EC, Plevy SE. The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflamm Bowel Dis. 2014;20:166–175. doi: 10.1097/MIB.0b013e3182a69dca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim TW, Seo JN, Suh YH, Park HJ, Kim JH, Kim JY, Oh KI. Involvement of lymphocytes in dextran sulfate sodium-induced experimental colitis. World J Gastroenterol. 2006;12:302–305. doi: 10.3748/wjg.v12.i2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katakura K, Watanabe H, Ohira H. Innate immunity and inflammatory bowel disease: a review of clinical evidence and future application. Clin J Gastroenterol. 2013;6:415–419. doi: 10.1007/s12328-013-0436-4. [DOI] [PubMed] [Google Scholar]
- 38.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, Conklin LS, Centola M, Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fantini MC, Monteleone G, Macdonald TT. New players in the cytokine orchestra of inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:1419–1423. doi: 10.1002/ibd.20212. [DOI] [PubMed] [Google Scholar]
- 40.Elson CO, Cong Y, Brandwein S, Weaver CT, McCabe RP, Mähler M, Sundberg JP, Leiter EH. Experimental models to study molecular mechanisms underlying intestinal inflammation. Ann N Y Acad Sci. 1998;859:85–95. doi: 10.1111/j.1749-6632.1998.tb11113.x. [DOI] [PubMed] [Google Scholar]
- 41.Annunziato F, Romagnani S. Do studies in humans better depict Th17 cells? Blood. 2009;114:2213–2219. doi: 10.1182/blood-2009-03-209189. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Ueno A, Iacucci M, Fort Gasia M, Jijon HB, Panaccione R, Kaplan GG, Beck PL, Luider J, Barkema HW, et al. Crossover Subsets of CD4 T Lymphocytes in the Intestinal Lamina Propria of Patients with Crohn’s Disease and Ulcerative Colitis. Dig Dis Sci. 2017;62:2357–2368. doi: 10.1007/s10620-017-4596-9. [DOI] [PubMed] [Google Scholar]
- 43.Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci U S A. 2015;112:7061–7066. doi: 10.1073/pnas.1415675112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 45.Yamamoto M, Yoshizaki K, Kishimoto T, Ito H. IL-6 is required for the development of Th1 cell-mediated murine colitis. J Immunol. 2000;164:4878–4882. doi: 10.4049/jimmunol.164.9.4878. [DOI] [PubMed] [Google Scholar]
- 46.Fichtner-Feigl S, Fuss IJ, Preiss JC, Strober W, Kitani A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF-kappa B decoy oligonucleotides. J Clin Invest. 2005;115:3057–3071. doi: 10.1172/JCI24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dieleman LA, Palmen MJ, Akol H, Bloemena E, Peña AS, Meuwissen SG, Van Rees EP. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114:385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li L, Shi QG, Lin F, Liang YG, Sun LJ, Mu JS, Wang YG, Su HB, Xu B, Ji CC, et al. Cytokine IL-6 is required in Citrobacter rodentium infection-induced intestinal Th17 responses and promotes IL-22 expression in inflammatory bowel disease. Mol Med Rep. 2014;9:831–836. doi: 10.3892/mmr.2014.1898. [DOI] [PubMed] [Google Scholar]
- 49.Ma L, Liang Y, Fang M, Guan Y, Si Y, Jiang F, Wang F. The cytokines (IFN-gamma, IL-2, IL-4, IL-10, IL-17) and Treg cytokine (TGF-beta1) levels in adults with immune thrombocytopenia. Pharmazie. 2014;69:694–697. [PubMed] [Google Scholar]
- 50.Geng X, Xue J. Expression of Treg/Th17 cells as well as related cytokines in patients with inflammatory bowel disease. Pak J Med Sci. 2016;32:1164–1168. doi: 10.12669/pjms.325.10902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang HL, Zheng YJ, Pan YD, Xie C, Sun H, Zhang YH, Yuan MY, Song BL, Chen JF. Regulatory T-cell depletion in the gut caused by integrin β7 deficiency exacerbates DSS colitis by evoking aberrant innate immunity. Mucosal Immunol. 2016;9:391–400. doi: 10.1038/mi.2015.68. [DOI] [PubMed] [Google Scholar]
- 52.Gibson DJ, Ryan EJ, Doherty GA. Keeping the bowel regular: the emerging role of Treg as a therapeutic target in inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:2716–2724. doi: 10.1097/MIB.0b013e31829ed7df. [DOI] [PubMed] [Google Scholar]
- 53.Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30:626–635. doi: 10.1016/j.immuni.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 54.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 55.Ip WK, Medzhitov R. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat Commun. 2015;6:6931. doi: 10.1038/ncomms7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hausmann M, Kiessling S, Mestermann S, Webb G, Spöttl T, Andus T, Schölmerich J, Herfarth H, Ray K, Falk W, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology. 2002;122:1987–2000. doi: 10.1053/gast.2002.33662. [DOI] [PubMed] [Google Scholar]
- 57.Akagawa KS. Functional heterogeneity of colony-stimulating factor-induced human monocyte-derived macrophages. Int J Hematol. 2002;76:27–34. doi: 10.1007/BF02982715. [DOI] [PubMed] [Google Scholar]
- 58.Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, Kastelein R, Kolk A, de Waal-Malefyt R, Ottenhoff TH. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci USA. 2004;101:4560–4565. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ghosn EE, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM, Bortoluci KR, Almeida SR, Herzenberg LA, Herzenberg LA. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci USA. 2010;107:2568–2573. doi: 10.1073/pnas.0915000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Asano K, Takahashi N, Ushiki M, Monya M, Aihara F, Kuboki E, Moriyama S, Iida M, Kitamura H, Qiu CH, et al. Intestinal CD169(+) macrophages initiate mucosal inflammation by secreting CCL8 that recruits inflammatory monocytes. Nat Commun. 2015;6:7802. doi: 10.1038/ncomms8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 63.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cibiček N, Roubalová L, Vrba J, Zatloukalová M, Ehrmann J, Zapletalová J, Večeřa R, Křen V, Ulrichová J. Protective effect of isoquercitrin against acute dextran sulfate sodium-induced rat colitis depends on the severity of tissue damage. Pharmacol Rep. 2016;68:1197–1204. doi: 10.1016/j.pharep.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 65.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–6173. doi: 10.4049/jimmunol.1701141. [DOI] [PubMed] [Google Scholar]
- 66.Cassado Ados A, D’Império Lima MR, Bortoluci KR. Revisiting mouse peritoneal macrophages: heterogeneity, development, and function. Front Immunol. 2015;6:225. doi: 10.3389/fimmu.2015.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scaldaferri F, Correale C, Gasbarrini A, Danese S. Molecular signaling blockade as a new approach to inhibit leukocyte-endothelial interactions for inflammatory bowel disease treatment. Cell Adh Migr. 2009;3:296–299. doi: 10.4161/cam.3.3.9152. [DOI] [PMC free article] [PubMed] [Google Scholar]