

ABSTRACT
An important component of lytic infection by Kaposi's sarcoma-associated herpesvirus (KSHV) is the ability of the virus to evade the innate immune response, specifically type I interferon (IFN) responses that are triggered by recognition of viral nucleic acids. Inhibition of type I IFN responses by the virus promotes viral replication. Here, we report that KSHV uses a caspase-dependent mechanism to block type I IFN, in particular IFN-β, responses during lytic infection. Inhibition of caspases during KSHV reactivation resulted in increased TBK1/IKKε-dependent phosphorylation of IRF3 as well as elevated levels of IFN-β transcription and secretion. The increased secretion of IFN-β upon caspase inhibition reduced viral gene expression, viral DNA replication, and virus production. Blocking IFN-β production or signaling restored viral replication. Overall, our results show that caspase-mediated regulation of pathogen sensing machinery is an important mechanism exploited by KSHV to evade innate immune responses.
IMPORTANCE KSHV is the causative agent of Kaposi's sarcoma (KS), an AIDS-defining tumor that is one of the most common causes of cancer death in sub-Saharan Africa. In this study, we examined the role of a set of cellular proteases, called caspases, in the regulation of immune responses during KSHV infection. We demonstrate that caspases prevent the induction and secretion of the antiviral factor IFN-β during replicative KSHV infection. The reduced IFN-β production allows for high viral gene expression and viral replication. Therefore, caspases are important for maintaining KSHV replication. Overall, our results suggest that KSHV utilizes caspases to evade innate immune responses, and that inhibiting caspases could boost the innate immune response to this pathogen and potentially be a new antiviral strategy.
KEYWORDS: KSHV, type I interferon, caspases
INTRODUCTION
An important component of successful viral infection is the ability of viruses to evade the innate immune response, including type I interferon (IFN) responses. In the case of the oncogenic herpesvirus Kaposi's sarcoma-associated herpesvirus (KSHV; also known as human herpesvirus 8, or HHV8), inhibition of type I IFNs is important both for de novo infection of cells and during reactivation of the lytic cycle after latent infection (1–4). It is now appreciated that both lytically and latently infected cells contribute to KSHV-induced development of Kaposi's sarcoma (KS) (5, 6). Lytic reactivation of KSHV from the latent phase likely promotes tumor development through the secretion of various factors that establish a proinflammatory microenvironment (5). As drugs that block lytic reactivation promote tumor regression (7, 8), control of lytic replication through modulation of type I IFN signaling may be a viable therapeutic option for KS therapies, and this has been explored in the past (9, 10).
Type I IFN (IFN-α and -β) secretion is rapidly induced in pathogen-infected cells after recognition of pathogen-associated molecular patterns, usually viral nucleic acids, by pattern recognition receptors (PRRs). In turn, type I IFN signaling leads to the upregulation of hundreds of interferon-stimulated genes (ISGs) that collectively confer an antiviral state (11). Various PRRs, including cGAS, IFI16, RIG-I, NLRP1, and several Toll-like receptors (TLRs), are activated upon KSHV infection and play an important role in promoting the innate immune response (12–17). To evade the innate immune responses, KSHV encodes several proteins that modulate type I IFNs, including ORF52, viral interferon regulatory factor-like 1 (vIRF1), vIRF2, vIRF3, and cytoplasmic isoforms of LANA (3, 16–20). However, there may be additional factors or processes contributing to type I IFN inhibition, as suggested by screening for IFN-inhibiting KSHV open reading frames (ORFs) (16).
Recent studies have uncovered novel roles for caspases in regulation of innate immune responses. Caspases are a family of cysteine-dependent aspartate-directed proteases that regulate multiple cellular processes, including programmed cell death, inflammasome activation, and differentiation (21). Regulation of type I IFN responses by caspases was first reported in a study that showed that knocking out caspase-8 caused epithelial inflammation (22). In this system, inflammation was triggered by activation of interferon regulatory factor 3 (IRF3), the key transcription factor for type I IFN expression (22). Other studies showed that caspase-3 and caspase-7 prevent the cytoplasmic release of mitochondrial DNA from inducing type I IFNs during intrinsic caspase-9-mediated apoptosis (23, 24). This mechanism was proposed to render apoptosis immunologically silent. Lastly, the inflammatory caspase-1 was found to attenuate the cGAS-STING sensing pathway by cleaving cGAS during virus infection of macrophages (25). Hence, caspase-mediated cleavage of pathogen-sensing machinery may be an important mechanism for viral innate immune evasion. However, it is not currently known whether caspases are widely exploited by viruses to reduce type I IFN responses.
Although a role for caspases in immune regulation during KSHV infection has not previously been reported, there is evidence that caspases can positively and negatively modulate KSHV replication. Induction of caspase-3 and caspase-9 triggers an apoptosis-dependent pathway that activates KSHV replication independently of RTA, the master lytic regulator that drives entry into the lytic cycle (26, 27). Furthermore, overexpression of KSHV vIRF2 triggers caspase-3-mediated degradation of IRF3 (20). In contrast, caspase-7 disrupts KSHV replication in B cells by cleaving ORF57, a viral lytic gene that is essential for virus replication and the production of infectious virions (28). These studies show that caspases have important, yet poorly understood, activities in KSHV infection.
Here, we report that apoptotic caspases are key mediators of the suppression of type I IFNs, in particular IFN-β, during KSHV lytic reactivation. We show that several caspases are activated upon KSHV lytic reactivation and that caspase inhibition potentiates the type I IFN antiviral response in KSHV-infected cells. This increased type I IFN induction reduces KSHV replication. We propose that some caspases function to limit type I IFN responses and that KSHV exploits this mechanism to promote its replication cycle.
RESULTS
Caspase inhibition during KSHV reactivation induces a type I IFN response.
Previous studies have demonstrated that some caspases regulate innate immune responses during various cellular processes, which include intrinsic apoptosis, keratinocyte differentiation, and inflammasome activation upon virus infection (22–25). To test whether caspases also regulate type I IFN signaling during KSHV lytic reactivation, we used KSHV-infected iSLK.219 cells in which the lytic cycle can be induced by doxycycline (Dox)-driven expression of the KSHV protein RTA. We treated iSLK.219 cells with doxycycline to drive lytic reactivation for 4 days in the presence of the pancaspase inhibitor IDN-6556 and found that caspase inhibitor treatment resulted in a 20-fold increase in the levels of secreted IFN-β (from 4.26 ± 18.96 pg/ml to 86.95 ± 31.64 pg/ml) (Fig. 1A). Caspase inhibition increased IFN-β transcription starting at day 3 after lytic induction, suggesting that caspases modulate IFN-β transcriptional induction during lytic reactivation (Fig. 1B). Transcription of ISG15, an interferon-stimulated gene (ISG), also increased upon caspase inhibition (Fig. 1C), demonstrating active type I IFN signaling in the cells. The effect appears to be specific for IFN transcription, since caspase inhibition did not alter the mRNA levels of the proinflammatory cytokine IL-18 (Fig. 1D). This was despite the fact that IL-18 expression appeared to be suppressed during lytic reactivation, which is consistent with the reported regulation of this cytokine by the KSHV noncoding RNA PAN (29). Importantly, caspase inhibition had only minimal effects on IFN-β secretion and transcriptional induction in latently infected cells (Fig. 1A and B). These results suggest that caspase inhibition causes IFN-β induction specifically in lytically reactivated cells. We define “lytic reactivation” as the reactivation of the lytic program and expression of early lytic genes that are not detectable in latently infected cells. Importantly, caspase inhibition did not alter the level of RTA induction by doxycycline (Fig. 1E) or the mRNA levels of the lytic early gene ORF37 at early time points after reactivation (Fig. 1F; a more extensive analysis of viral gene expression in the samples from Fig. 1B and C is presented in Fig. 5B and C). Therefore, caspase activity is not required for the inducible doxycycline system to initiate lytic reactivation, and the difference in IFN-β induction is not a result of changes in the lytic cycle.
FIG 1.

Inhibition of caspases during KSHV lytic reactivation induces a type I interferon (IFN) response. iSLK.219 cells were treated with 1 μg/ml doxycycline to induce lytic reactivation and the pancaspase inhibitor IDN-6556 (10 μM), the TBK1/IKKε inhibitor MRT67307 (TBK1i; 2 μM), poly(I·C) (5 μg/ml), the viral DNA synthesis inhibitor phosphonoacetic acid (PAA; 100 μg/ml), and/or DMSO (vehicle) as indicated. (A) The levels of IFN-β in the conditioned medium collected from iSLK.219 cells at day 4 postreactivation were measured by ELISA (n = 4). **, P < 0.01 by Tukey's multiple-comparison test after two-way ANOVA. (B and C) Total RNA was extracted at the indicated time points, and levels of IFN-β (B) and ISG15 (C) mRNA were measured by RT-qPCR, normalized to 18S rRNA, and expressed as levels relative to those for noninduced cells (n = 4). * and ****, P values of <0.05 and <0.0001 by Tukey's multiple-comparison test for Dox+vehicle versus Dox+IDN-6556 at each time point after two-way ANOVA. (D) Total RNA was extracted at the indicated time points, and the levels of IL-18 mRNA were measured by RT-qPCR, normalized to 18S rRNA, and expressed as levels relative to those of noninduced cells at day 1 (n = 4). ns, P > 0.05 by Tukey's multiple-comparison test after two-way ANOVA. (E) Cells were collected 24 h postreactivation and subjected to Western blot analysis for RTA and β-actin (as a loading control). Blots are representatives of 3 repeats. (F) Total RNA was extracted from iSLK.219 cells 3 days after reactivation. Levels of ORF37 mRNA levels were measured by RT-qPCR and normalized to those for 18S rRNA. AU, arbitrary units representing the ratio between the mRNA and 18S rRNA levels (n = 3). ns, P > 0.05 by Student's t test. This graph was generated using the same data as in Fig. 5B, collected from the samples used for the analyses shown in panels B and C. (G) Total RNA was extracted from HeLa or iSLK.219 cells treated with water (vehicle) or poly(I·C) for 3 h. Levels of IFN-β mRNA were measured by RT-qPCR and normalized to 18S rRNA and expressed as levels relative to the averages of the respective vehicle-treated cells (n = 4). ** and ****, P values of <0.01 and <0.0001 by Tukey's multiple-comparison test after two-way ANOVA. (H) Total RNA was extracted from iSLK.219 cells 4 days after reactivation. Levels of IFN-β mRNA were measured by RT-qPCR, normalized to those 18S rRNA, and expressed as levels relative to those for noninduced cells (n = 4). ns, P > 0.05 by Tukey's multiple-comparison test after two-way ANOVA. (I and J) Cell lysates were collected at the indicated time points (I) or at day 5 postreactivation (J) and subjected to Western blot analysis for p-IRF3 (Ser396), total IRF3, and β-actin (as a loading control). Blots are representative of ≥3 repeats. (K) Levels of IFN-β mRNA were measured at day 5 postreactivation by RT-qPCR, normalized to 18S rRNA, and expressed as levels relative to those of noninduced cells (n = 3). ****, P < 0.0001 by Tukey's multiple-comparison test after two-way ANOVA.
FIG 5.
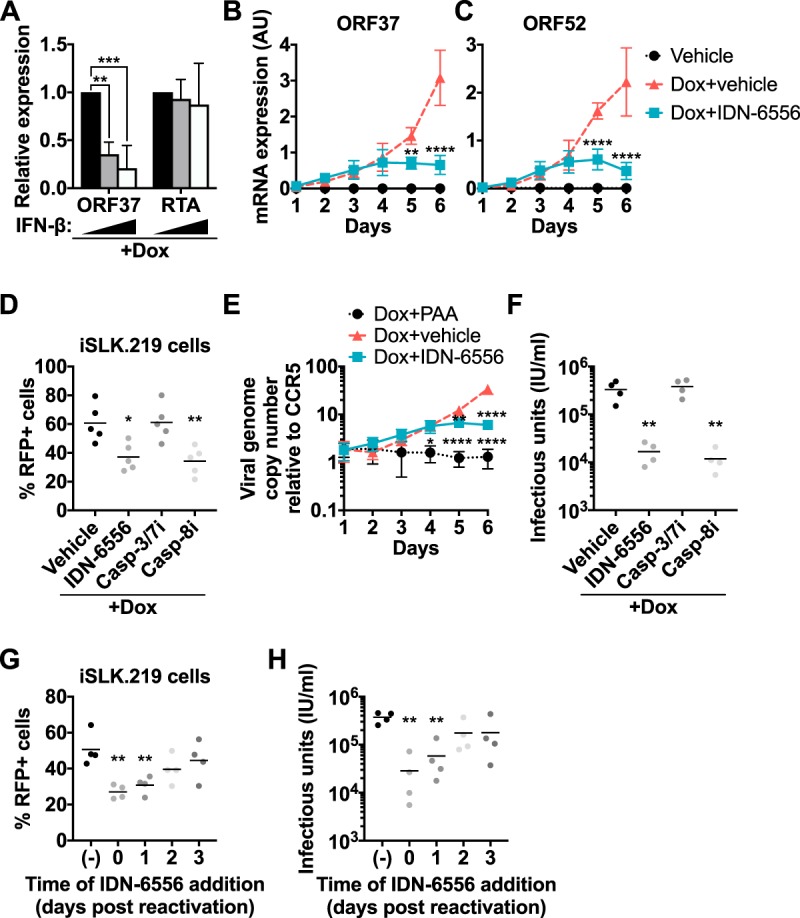
Caspase inhibition reduces KSHV lytic gene expression, DNA replication, and virus production. iSLK.219 cells were treated with 1 μg/ml of doxycycline to induce the lytic cycle, together with DMSO (vehicle), IDN-6556 (10 μM), Z-DEVD-FMK (casp-3/7i; 10 μM), Z-IETD-FMK (casp-8i; 100 μM), IFN-β (10 or 100 pg/ml), and/or PAA (100 μg/ml), where indicated, for the length of induction (A to F) or the indicated times (G and H). (A) Total RNA was extracted at day 1 postreactivation and treated with increasing amounts of IFN-β (black bars, 0 pg/ml; gray bars, 10 pg/ml; white bars, 100 pg/ml). Levels of ORF37 and RTA mRNA were measured by RT-qPCR, normalized to cellular 18S rRNA, and plotted relative to cells treated with 0 pg/ml IFN-β (n = 4). ** and ***, P values of <0.01 and <0.001 by Tukey's multiple-comparison test after two-way ANOVA. (B and C) Total RNA was extracted at the indicated time points, and levels of ORF37 (B) and ORF52 (C) mRNA were measured by RT-qPCR and normalized to cellular 18S rRNA. AU, arbitrary units representing the ratio between the viral mRNA and 18S rRNA levels (n = 3). The same RNA samples were used to generate the data shown in this panel and in Fig. 1B and C. ** and ****, P values of <0.01 and <0.0001 by Tukey's multiple-comparison test (Dox+vehicle versus Dox+IDN-6556 at each time point) after two-way ANOVA. (D) RFP-positive iSLK.219 cells were counted by flow cytometry at day 6 postreactivation (n = 5). * and **, P values of <0.05 and <0.01 by Dunnett's multiple-comparison test versus vehicle after one-way ANOVA. (E) Total cellular DNA was extracted at the indicated time points. Viral DNA levels (copies of the LANA gene) were measured by RT-qPCR and normalized to copies of the cellular gene CCR5. Viral genome numbers per copy of CCR5 are plotted (n = 3). *, **, and ****, P values of <0.05, <0.01, and <0.0001 by Tukey's multiple-comparison test versus Dox plus vehicle at each time point after two-way ANOVA. (F) The supernatant from reactivated iSLK.219 cells was collected at day 6 postreactivation and used to infect iSLK.RTA target cells. KSHV-infected, GFP-positive target cells were counted by flow cytometry 4 days later (n = 4). **, P < 0.01 by Dunnett's multiple-comparison test versus vehicle after one-way ANOVA. (G and H) Reactivated iSLK.219 cells were treated with IDN-6556 starting at the indicated time points. (G) Six days postreactivation, RFP-positive iSLK.219 cells were counted by flow cytometry and the supernatant was used to infect iSLK.RTA target cells. (H) GFP-positive target cells were counted by flow cytometry 4 days later (n = 4). **, P < 0.01 by Dunnett's multiple-comparison test after two-way ANOVA relative to vehicle-treated cells [shown as (−)].
The limited induction of IFN-β during lytic reactivation in the absence of caspase inhibitors is consistent with inhibition of type I IFN induction by viral proteins (2, 3). Treatment of latently infected iSLK.219 cells with poly(I·C), a commonly used inducer of type I IFN responses, potently induced transcription of IFN-β (Fig. 1G). This confirms that iSLK.219 cells are not generally defective in mounting type I IFN responses. In principle, IFN-β could be induced in response to sensing either reactivation from latency or reinfection of cells by newly produced virions. To test whether IFN-β induction was due to reinfection, we prevented virion production by treating reactivating iSLK.219 cells with phosphonoacetic acid (PAA). PAA blocks viral DNA replication, as apparent in the experiment shown in Fig. 5E. However, we still detected IFN-β induction in lytically reactivating iSLK.219 cells treated with both caspase inhibitors and PAA (Fig. 1H). This result indicates that IFN-β induction was independent of virion production and likely directly due to lytic reactivation. The observation that IFN-β mRNA levels only increased starting at day 3 may be due to asynchronous lytic reactivation in response to doxycycline-mediated RTA expression, with only a small fraction of cells entering the lytic cycle at day 1. Thus, IFN-β transcripts may be present below the level of detection at earlier time points (Fig. 1B). Nonetheless, these results collectively show that caspase activity modulates type I IFN induction and signaling upon KSHV reactivation.
Caspase activity interferes with IFN-β induction through the canonical TBK1/IRF3 pathway.
To determine whether caspases interfere with the canonical pathogen-sensing pathway or another IFN-β-inducing mechanism, we monitored activation of IRF3, the transcription factor that induces IFN-β downstream of pathogen sensing. Pathogen sensing by PRRs leads to the activation of the kinases TBK1 and IKKε, which phosphorylate and thus activate IRF3. Consistent with the results for IFN-β induction, we did not detect IRF3 Ser396 phosphorylation upon lytic reactivation of vehicle-treated iSLK.219 cells (Fig. 1I). However, caspase inhibition led to robust phosphorylation of IRF3 starting at day 3 postreactivation (Fig. 1I). Furthermore, treatment of caspase-inhibited cells with the TBK1/IKKε dual inhibitor MRT67307 prevented IRF3 phosphorylation and IFN-β transcriptional induction (Fig. 1J and K). Overall, these data suggest that caspases suppress IFN-β induction through the canonical PRR-mediated viral sensing pathway during KSHV reactivation and that treatment with caspase inhibitors releases this block.
Apoptotic caspases regulate IFN-β production during KSHV reactivation.
To determine which caspases are activated during lytic reactivation and may contribute to the inhibition of IFN-β expression, we assessed the activation of several candidate caspases. As caspases are activated by proteolytic processing of inactive zymogens, we probed for the appearance of cleaved, active caspases by Western blotting. Upon lytic reactivation, we detected increasing levels of cleaved caspase-3 and -8 (Fig. 2A). Cleaved fragments of the well-known caspase targets PARP1 and Lamin A/C also appeared upon lytic reactivation (Fig. 2A). Cleaved caspases and caspase substrates appeared two days after addition of doxycycline, and their levels increased until day 5 (Fig. 2A). Notably, caspase activation preceded the onset of IFN-β synthesis, which occurred on day 3 in the absence of caspase activity (Fig. 1B). This is consistent with a role for caspases in the suppression of IFN-β induction. We also confirmed that caspase-8 and caspase-3/7 were enzymatically active in reactivating cells by measuring caspase-8 and -3/7 activity with luminescence-based assays (Fig. 2B and C). Doxycycline treatment did not induce caspase activation in uninfected iSLK.RTA cells (Fig. 2D, lane 5). These cells contain the doxycycline-inducible RTA transgene but are not infected with KSHV. They also express RTA at a level similar to that of the infected iSLK.219 cells upon doxycycline treatment (Fig. 2E). This result supports the idea that caspases are activated as a consequence of reactivation of the KSHV lytic cycle rather than by doxycycline-driven RTA expression. However, caspase activation was independent of viral DNA replication and late events in the viral life cycle, as we detected caspase activation in lytically reactivating iSLK.219 cells treated with PAA (Fig. 2D, lane 4). This result suggests that caspases are activated early in the virus replication cycle, either by an early viral protein or as a cellular response to the early steps of lytic reactivation.
FIG 2.

Apoptotic caspases are activated during KSHV lytic reactivation. (A) iSLK.219 cells were treated with doxycycline (1 μg/ml) to induce lytic reactivation. Cell lysates were collected at the indicated time points and subjected to Western blot analysis for procaspase-3, activated (cleaved) caspase-3, procaspase-8, activated (cleaved) caspase-8, full-length and cleaved PARP1, full-length and cleaved Lamin A/C, and β-actin (as a loading control). Blots are representative of 3 repeats. (B and C) iSLK.219 cells were lytically reactivated using doxycycline (lytic) and treated with DMSO (vehicle), IDN-6556 (10 μM), Z-DEVD-FMK (casp-3/7i; 10 μM), or Z-IETD-FMK (casp-8i; 100 μM) for 4 days. Caspase-8 (B) or caspase-3/7 (C) activity was measured with Caspase-Glo luminescence assays (Promega) (n ≥ 3). ****, P < 0.0001 by Tukey's multiple-comparison test after one-way ANOVA. (D and E) iSLK.219 and iSLK.RTA cells were treated with doxycycline (1 μg/ml), IDN-6556 (10 μM), and/or PAA (100 μg/ml) as indicated. Cell lysates were collected at day 3 postreactivation (D) or day 1 postreactivation (E) and subjected to Western blot analysis for procaspase-8, activated (cleaved) caspase-8, RTA, and β-actin (as a loading control). Blots are representative of 3 repeats.
To narrow down which caspase(s) inhibits IFN-β induction during KSHV lytic reactivation, we treated reactivated cells with the caspase-3/7 selective inhibitor Z-DEVD-FMK, the caspase-8 selective inhibitor Z-IETD-FMK, or the caspase-1 selective inhibitor Ac-YVAD-CMK. We included Ac-YVAD-CMK because caspase-1 has recently been reported to cleave cGAS, thereby inhibiting IFN-β induction, in virus-infected macrophages (25). However, caspase-1 levels in iSLK.219 cells were very low, and we did not detect any evidence of inflammasome and caspase-1 activation in these cells (data not shown), suggesting caspase-1 is unlikely to be a major player in this system. Only the caspase-8 selective inhibitor Z-IETD-FMK induced IFN-β transcription during KSHV replication (Fig. 3A). To confirm the activity of the inhibitors in our cellular system, we examined their effect on the processing and catalytic activity of caspase-3, -8, and -9 (Fig. 3B). Caspase-8 and -9 are initiator caspases that undergo autoproteolytic processing during activation. In contrast, caspase-3 is an effector caspase that does not cleave itself but is processed to its active form by initiator caspases, including caspase-8 and -9. As expected, the pancaspase inhibitor IDN-6556 prevented or reduced the appearance of cleaved caspase-3, -8, and -9, while the caspase-3/7 inhibitor Z-DEVD-FMK did not inhibit caspase processing. However, Z-DEVD-FMK reduced the activity of caspase-3/7 in a luminescence-based assay (Fig. 2C). These data confirm that the caspase-3/7 inhibitor was active in the cells despite the absence of an effect on IFN induction. In contrast, the caspase-8 selective inhibitor Z-IETD-FMK inhibited processing of caspase-8 and caspase-3. This is consistent with the fact that caspase-8 likely processes both itself and caspase-3. Z-IETD-FMK did not inhibit the processing of caspase-9, another initiator caspase that is known to self-cleave, confirming the selectivity of the inhibitor. To further investigate the role of caspase-8 in IFN-β regulation, we tested the effects of short interfering RNA (siRNA)-mediated knockdown of caspase-8 on IFN-β expression in iSLK.219 cells (Fig. 3C). Loss of caspase-8 markedly reduced the ability of IDN-6556 to induce IFN-β transcription (Fig. 3D), confirming that caspase-8 is the primary target of IDN-6556. Curiously, caspase-8 knockdown alone was not sufficient to induce IFN-β transcription. We obtained similar results with a second siRNA (not shown). One possibility is that other caspases, like the caspase-8 paralog caspase-10, compensate for the loss of caspase-8, or that the residual activity of the remaining, nondepleted caspase-8 is sufficient to block IFN-β induction. Alternatively, it may reflect a requirement for caspase-8 as a signaling scaffold to promote IFN-β regulation, similar to its role in other inflammatory mechanisms (30, 31). Overall, we conclude that multiple caspases are activated by lytic replication, and that caspase-8 plays a major role in modulating IFN-β transcription during KSHV reactivation.
FIG 3.
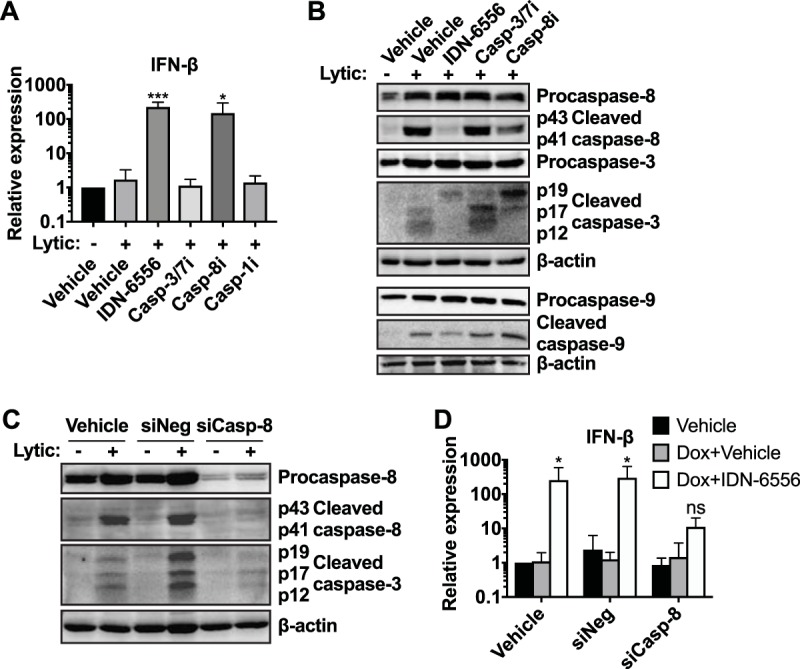
Caspase-8 is particularly important for IFN-β regulation during KSHV reactivation. (A and B) iSLK.219 cells were lytically reactivated using doxycycline (lytic) and treated with DMSO (vehicle), IDN-6556 (10 μM), Z-DEVD-FMK (casp-3/7i; 10 μM), Z-IETD-FMK (casp-8i; 100 μM), or Ac-YVAD-CMK (casp-1i; 100 μM), as indicated. (A) Levels of IFN-β mRNA at day 5 postreactivation were measured by RT-qPCR, normalized to 18S, and expressed as levels relative to those of noninduced cells (n ≥ 3). * and ***, P values of <0.05 and <0.001 by Dunnett's multiple-comparison test for each condition versus lytic plus vehicle after one-way ANOVA. (B) Cell lysates were collected at day 3 postreactivation and subjected to Western blot analysis for procaspase-8, activated (cleaved) caspase-8, procaspase-3, activated (cleaved) caspase-3, procaspase-9, activated (cleaved) caspase-9, and β-actin (as a loading control). Blots are representative of 3 repeats. (C and D) iSLK.219 cells were left untransfected or were transfected with an siRNA targeting caspase-8 (siCasp-8) or a negative-control siRNA (siNeg). Transfection was carried out twice, 2 days prior to induction and on the day of lytic cycle induction. The cells were then lytically reactivated with doxycycline (1 μg/ml) and treated with either DMSO (vehicle) or IDN-6556 (10 μM) as indicated. (C) Cell lysates were harvested at day 2 postreactivation and subjected to Western blotting for procaspase-8, activated (cleaved) caspase-8, activated (cleaved) caspase-3, and β-actin (as a loading control). Blots are representative of 3 repeats. (D) Total RNA was extracted at day 5 postreactivation. The levels of IFN-β mRNA were measured by RT-qPCR, normalized to 18S, and expressed as levels relative to those of vehicle-treated noninduced cells (n = 5). ns and *, P values of >0.05 and <0.05 by Tukey's multiple-comparison test for Dox+vehicle versus Dox+IDN-6556 for each siRNA treatment after two-way ANOVA.
Apoptotic caspase activation during KSHV lytic reactivation does not result in a detectable increase in cell death.
Activation of caspases, particularly caspase-3 and -8, typically results in apoptotic cell death, which is generally believed to be antiviral (32–34). To test whether apoptosis occurred in this system, we stained doxycycline-treated iSLK.219 cells with annexin V. Annexin V staining in nonpermeabilized cells measures phosphatidylserine translocation to the outer leaflet of the plasma membrane, a marker of apoptosis. We also stained the cells with a membrane-impermeable dye (LIVE/DEAD near-infrared [IR]), which only stains cells whose membrane is compromised and thus measures overall cell viability. Because iSLK.219 cells are infected with the rKSHV.219 recombinant virus, which expresses red fluorescent protein (RFP) driven by the promoter for the lytic gene PAN (35), we identified cells undergoing lytic replication based on RFP fluorescence. We analyzed the percentage of cells that were annexin V positive or LIVE/DEAD positive within the RFP-negative (latent) and RFP-positive (lytic) populations and found that only 1 to 8% of either population stained positive, i.e., was potentially dying (Fig. 4A). In particular, there was no significant difference between reactivating (RFP+) and nonreactivating (RFP−) cells. This indicates that, at least in iSLK.219 cells, KSHV lytic reactivation does not induce apoptosis. To confirm that our staining procedure worked, we tested annexin V staining in HeLa cells treated with tumor necrosis factor alpha (TNF-α) and cycloheximide, a standard treatment to induce apoptotic cell death (36). The percentage of vehicle-treated HeLa cells that stained positive for annexin V was similar to that of latent or lytic iSLK.219 cells (Fig. 4B), suggesting that this is background staining. In contrast, TNF-α and cycloheximide treatment led to 34.7% ± 20.5% of HeLa cells staining with annexin V (Fig. 4B). Most of these cells are LIVE/DEAD negative (not shown), indicative of an early apoptotic population where phosphatidylserine has translocated, but the cellular membrane remains intact. These results confirm that the absence of a strong signature of apoptosis in the reactivating iSLK.219 cells is not due to a labeling issue. Although it is surprising that infectious virions can be produced without causing detectable cell death, this possibility is consistent with the mode of egress of herpesviruses, which are reported to bud out of the cell without causing cell lysis (37, 38). Collectively, these results show that the increase in IFN-β is independent of apoptosis signaling and that apoptosis may be blocked downstream of caspase activation in lytically reactivated iSLK.219 cells.
FIG 4.

Activation of apoptotic caspases does not cause apoptosis during KSHV lytic reactivation. (A and B) Cells were stained with annexin V and LIVE/DEAD near-IR stain (Invitrogen) and analyzed by flow cytometry. (A) iSLK.219 cells were lytically reactivated with doxycycline (1 μg/ml) and treated with either DMSO (vehicle) or IDN-6556 (10 μM) as indicated for 4 days. The fractions of annexin V+ cells (left) and dead cells (right) in RFP− (latently infected) and RFP+ (lytically infected) populations are shown (n ≥ 4). For all samples, the P value versus vehicle alone was >0.05 by Tukey's multiple-comparison test after two-way ANOVA. (B) HeLa cells were treated with either water (vehicle) or 20 ng/ml TNF-α and 10 μg/ml cycloheximide (TNF-α+CHX) for 6 h. The percentage of annexin V+ cells is shown (n = 4). *, P < 0.05 by Student's t test.
Caspase activity promotes KSHV lytic cycle progression.
Type I IFNs have been reported by others to reduce KSHV reactivation (4, 16, 18). Consistent with these reports, we found that treatment of iSLK.219 cells with recombinant IFN-β reduces reactivation, as evidenced by reduced levels of the early gene ORF37 (Fig. 5A). This reduction was already apparent at 10 pg/ml of IFN-β, which is lower than the levels of IFN-β in the medium of caspase-inhibited iSLK.219 cells (Fig. 1A). Importantly, the mRNA levels of RTA were not affected by IFN-β, indicating that IFN-β did not interfere with doxycycline-mediated induction of RTA (Fig. 5A). It is presently unknown exactly how IFNs impact different steps of the lytic cycle. Nonetheless, this result suggested to us that the suppression of IFN-β by caspases could promote KSHV reactivation and/or progression through the lytic cycle. We thus assayed the effect of caspase inhibitors on the lytic cycle progression of KSHV in doxycycline-reactivated iSLK.219 cells. We found that caspase activity promoted KSHV lytic cycle progression based on multiple readouts. Although the levels of early viral mRNAs were unchanged at day three postreactivation (Fig. 1F), we found that IDN-6556 progressively reduced mRNA levels of both early and late genes (ORF37 and ORF52, respectively) at later time points (Fig. 5B and C). This reduction was mirrored by a decrease in the percentage of cells expressing RFP, which is driven by the lytic promoter for the KSHV PAN RNA (control, 60.7% ± 13.0%; IDN-6556, 37.1% ± 9.5%) (Fig. 5D). Replication of the viral DNA genome was also reduced in cells treated with the caspase inhibitor IDN-6556, although not as dramatically as in cells treated with the viral DNA replication inhibitor PAA (Fig. 5E).
The changes in viral gene expression and viral DNA replication ultimately impacted the production of KSHV virions. To quantify infectious virus production, we took advantage of the fact that the recombinant virus in iSLK.219 cells constitutively expresses green fluorescent protein (GFP) during both latent and lytic infection, marking all infected cells. We exposed uninfected cells to supernatant from reactivated iSLK.219 cells, measured the percentage of GFP-expressing target cells, and estimated infectious units in the supernatant. We found that caspase inhibitor treatment reduced infection of naive target cells (Fig. 5F). Consistent with the results on the induction of IFN-β (Fig. 3A), the caspase-8 inhibitor Z-IETD-FMK also reduced lytic reactivation (RFP induction; Fig. 5D) and virus production (Fig. 5F). In contrast, the caspase-3/7 inhibitor Z-DEVD-FMK had no effect on either readout (Fig. 5D and F). Interestingly, addition of IDN-6556 two days after the start of reactivation or later was less effective in reducing lytic reactivation (RFP induction) and virus production (Fig. 5G and H). This result is consistent with the timing of IFN-β induction (Fig. 1B) and caspase activation (Fig. 2A). Collectively, these results indicate that caspase activity at the beginning of the lytic cycle is necessary for efficient levels of KSHV reactivation.
Caspases are activated in lytic BCBL1 cells and are important for virus production.
To determine if caspase inhibition of type I IFN signaling occurs in other cell types relevant to KSHV infection, we examined the function of caspases in TREx-BCBL1-RTA, KSHV-infected primary effusion lymphoma (PEL) cells. TREx-BCBL1-RTA cells were originally isolated from PEL patients and were also engineered to express a doxycycline-inducible RTA transgene to allow for robust reactivation (39). To quantify virus production, we exposed uninfected iSLK.RTA cells to the supernatant of reactivated TREx-BCBL1-RTA cells treated with vehicle or IDN-6556. We then reactivated the lytic cycle in the newly infected iSLK.RTA cells with doxycycline and measured the RNA levels for the early lytic gene ORF57 to quantify infection. This method was adapted from a protocol used in the original study describing TREx-BCBL1-RTA cells (39). Consistent with the results in iSLK.219 cells, caspase inhibition in TREx-BCBL1-RTA cells resulted in a reduction in virus production (Fig. 6A). We also observed reduced virus production in BC-3 and BC-5 cells, two other PEL cell lines, upon caspase inhibition (data not shown). Caspase-3 and caspase-8 were activated upon lytic reactivation of TREx-BCBL1-RTA, as evidenced by the appearance of cleaved forms of these caspases, and caspase activation was lost upon IDN-6556 treatment (Fig. 6B). While IFN-β was minimally induced (not shown), we observed an increase in the mRNA levels for IFN-αs in TREx-BCBL1-RTA cells treated with IDN-6556 (Fig. 6C). Overall, these data suggest that caspase activity is also important for KSHV replication and modulation of type I IFN signaling in PEL cells.
FIG 6.
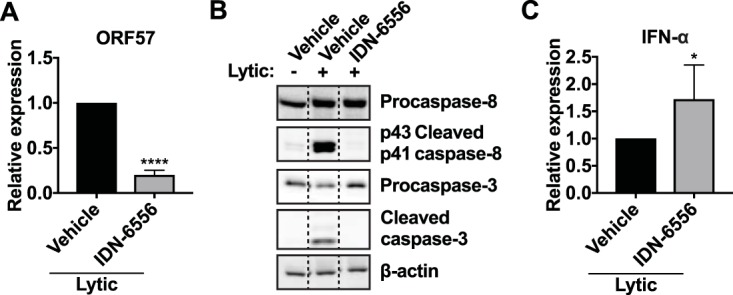
Apoptotic caspases are active in BCBL1 cells and promote KSHV lytic replication. TREx-BCBL1-RTA cells were lytically reactivated with TPA (20 ng/ml), ionomycin (500 ng/ml), and doxycycline (1 μg/ml) for 24 h and treated with DMSO (vehicle) or IDN-6556 (10 μM), as indicated. (A) Supernatant from TREx-BCBL1-RTA cells at day 3 postreactivation was used to infect iSLK.RTA cells. Infected iSLK.RTA cells were then lytically reactivated with 1 μg/ml doxycycline for 2 days. Total RNA was extracted from iSLK.RTA cells, and levels of ORF57 mRNA were measured by RT-qPCR and normalized to cellular 18S rRNA (n = 3). RNA levels of a lytic gene in the target cells reflect the levels of infectious virus released by TREx-BCBL1-RTA cells. ****, P < 0.0001 by Student's t test. (B) Cell lysates were collected 24 h postreactivation and subjected to Western blot analysis for procaspase-8, activated (cleaved) caspase-8, procaspase-3, activated (cleaved) caspase-3, and β-actin (as a loading control). Blots are representative of 3 repeats. (C) Total RNA was extracted from TREx-BCBL1-RTA cells 1 day after reactivation. Levels of IFN-α mRNAs were measured using pan-IFN-α primers by RT-qPCR and normalized to cellular 18S rRNA (n = 6). *, P < 0.05 by Student's t test.
Caspases promote KSHV lytic cycle progression through the suppression of IFN-β.
The timing of the reduction in viral mRNA levels and viral DNA replication upon caspase inhibition is consistent with our hypothesis that accumulation of IFN-β and type I IFN signaling drive these changes. IFN-β induction started at day 3 postreactivation (Fig. 1B), while the reduction in viral mRNAs and DNA replication started at day 5 postinduction (Fig. 5B, C, and E). If caspases directly affected viral processes, for example, if they were required for transactivation of early genes like ORF37, caspase inhibition would be expected to reduce viral gene expression and DNA replication from earlier time points. To directly establish that caspase activity promotes lytic cycle progression by suppressing an IFN response, we used two approaches to block IFN-β induction and signaling. First, we inhibited TBK1/IKKε with MRT67307, which blunts IFN-β induction (Fig. 1J), and second, we depleted IFN-β mRNA using siRNAs (Fig. 7B). The two siRNAs inhibited IFN-β basal expression and IFN-β and ISG15 induction upon caspase inhibition, albeit to different extents (Fig. 7B and C). The effects of the TBK1/IKKε inhibitor and siRNAs on viral gene expression in cells treated with doxycycline and vehicle were somewhat variable (Fig. 7A, D, and E, black bars). Nonetheless, upon IFN-β depletion, caspase inhibitors consistently had a smaller effect or no effect on the expression of the early gene ORF37 (Fig. 7A and D). In addition, caspase inhibitors also had a smaller or no effect on the expression of the late gene ORF25 and the production of infectious virus in cells treated with siRNAs against IFN-β (Fig. 7E and F). Collectively, these results and the results shown in Fig. 5 and 6 suggest that caspases promote lytic replication at least in part by suppressing type I IFN induction by the canonical pathogen-sensing pathway.
FIG 7.

IFN-β mediates the effects of caspase inhibitors on KSHV replication. (A) iSLK.219 cells were lytically reactivated with doxycycline, together with DMSO (Dox+vehicle; black bars), 10 μM IDN-6556 (Dox+IDN-6556; gray bars), and/or 2 μM MRT67307 (TBK1i) for 5 days. Levels of ORF37 mRNA were measured by RT-qPCR (n = 3). ns, *, and **, P values of >0.05, <0.05, and <0.01 by Sidak's multiple-comparison test after two-way ANOVA. (B to F) iSLK.219 cells were left untransfected or were transfected with siRNAs targeting IFN-β (siIFN-β-1 and -2) or a negative-control siRNA (siNeg). They were then lytically reactivated with doxycycline, together with DMSO (Dox+vehicle; black bars) or 10 μM IDN-6556 (Dox+IDN-6556; gray bars). Total RNA was extracted at day 5 postreactivation, and the levels of IFN-β (B), ISG15 (C), ORF37 (D), and ORF25 (E) mRNA were measured by RT-qPCR (n ≥ 3). Supernatant was collected at day 6 postreactivation and used to infect iSLK.RTA target cells. (F) GFP-positive target cells were counted by flow cytometry 4 days later (n ≥ 5). ns, *, **, ***, and ****, P values of >0.05, <0.05, <0.01, <0.001, and <0.0001 by Sidak's multiple-comparison test (Dox+vehicle versus Dox+IDN-6556 or the indicated comparison) after two-way ANOVA.
DISCUSSION
Here, we examined the role of caspases in the regulation of type I IFN response during KSHV lytic reactivation. We observed that pharmacological inhibition of caspases during reactivation potentiates type I IFN induction through the TBK1/IKKε signaling cascade and triggers the expression of ISGs. Caspase activity is important to promote KSHV replication after lytic reactivation, because caspase inhibition also impairs KSHV viral lytic gene expression, viral DNA replication, and virus production. Because knockdown of IFN-β or inhibition of IFN-β induction rescues KSHV replication in the presence of caspase inhibitors, we conclude that caspases promote KSHV replication at least in part by suppressing the type I IFN response. This establishes caspases as a novel component of the immune evasion network in KSHV infection.
Based on the reported effects of caspases on type I IFNs (22–25) and our findings, we propose a model whereby KSHV has co-opted an existing function of activated caspases to evade innate immune responses (Fig. 8). Previous studies have shown that in a range of settings, caspases can attenuate type I IFN responses, likely by cleaving a component of the pathogen-sensing pathway (22–25). In our model, lytic reactivation of KSHV triggers both type I IFN responses through the pathogen-sensing pathway and also the activation of caspases, perhaps as an additional cellular response aimed to induce death of infected cells. However, this concomitant activation of caspases inhibits the transcriptional induction of IFN-β due to their anti-IFN function. The reduction in IFN induction promotes KSHV lytic reactivation. KSHV is able to take advantage of this proviral effect of IFN inhibition, because KSHV can also block caspase-induced apoptosis, thus negating any antiviral effect of caspase activation.
FIG 8.

Proposed model of caspase-dependent suppression of type I IFN responses during KSHV reactivation. KSHV lytic reactivation triggers both the pathogen-sensing pathway and the activation of caspases. (Left) The presence of active caspases causes attenuation of IFN-β induction, while apoptosis is blocked by KSHV downstream of caspase activation. (Right) In the absence of caspase activity, IFN-β is induced, which reduces KSHV lytic reactivation.
How caspases regulate the type I IFN response during KSHV infection remains to be established. In macrophages, the inflammatory caspase-1 cleaves cGAS to suppress the type I IFN response during viral infection (25). Similarly, caspase-dependent IRF3 cleavage has been reported in HIV infection (40) and upon overexpression of the KSHV protein vIRF2 (20). However, we did not detect caspase-dependent cleavage products of either of these proteins in KSHV-infected cells (data not shown). The apoptotic caspase-3 and caspase-7 suppress mitochondrial DNA-dependent type I IFN responses during apoptosis, but the mechanism is unknown (23, 24). Our data suggest that caspase-8 also regulates type I IFN response, because caspase-8-selective inhibitors reduce IFN induction during KSHV reactivation (Fig. 3A). It is interesting that various caspases appear to share potentially redundant functions in the suppression of IFN-β (22–25). It will be important to determine whether the different caspases target similar or distinct components of the IFN-β response pathway. Caspase-dependent IFN-β regulation may also be context specific or cell type specific. In the future, identifying the target of caspases in KSHV-infected cells will help to determine the connection between IFN-β regulation during KSHV infection and in other cellular contexts and viral infections.
While knockdown of caspase-8 alone does not induce IFN-β transcription, the effect of IDN-6556 on IFN-β is lost when caspase-8 is knocked down (Fig. 3D). This epistasis experiment supports our model that caspase-8 is the major target of IDN-6556 in this system. However, it also suggests that there are additional facets of regulation of IFN responses by caspase-8 that remain to be elucidated. This is not surprising, as the biology of caspase-8 is complex. Multiple studies have now shown that caspase-8 also has nonenzymatic functions, as shown, for example, in the case of the NLRP3 inflammasome, where caspase-8 acts as a scaffold independently of its catalytic function (31). Also, autoproteolytic processing of caspase-8 results in the generation of multiple different forms of this enzyme that participate in different signaling complexes with different outputs, as shown, for example, in the regulation of necroptosis and inflammatory responses to Yersinia pestis (41). One feasible explanation, which remains to be tested, is that caspase-8 is required as a scaffold to promote IFN-β induction, while the catalytic activity of caspase-8 controls the extent of this response.
Our data suggest that caspases play a proviral role, while previous studies report both positive and negative modulation of KSHV infection by caspases (20, 26–28, 42, 43). While apoptosis is a cellular defense mechanism aimed at disrupting viral spread, there is emerging evidence that caspases also have roles in nonapoptotic processes that viruses utilize to promote their infection (44). Caspase-mediated processing of viral proteins is an important process for viral replication in human papillomavirus (HPV), where loss of caspase cleavage sites in the E1 protein reduced viral genome amplification upon epithelial differentiation (44, 45). Although previous studies suggest caspases directly target the IFN-β induction pathway, it is also possible that the relevant target in KSHV infection is a viral protein. In KSHV, caspases have been reported to cleave both LANA and ORF57 (28, 43). Caspase-mediated processing of these viral proteins could provide additional anti-IFN proviral functions.
It is noteworthy that activation of caspase-3 and -8 was not accompanied by an increase in apoptosis or cell death, as determined by staining with annexin V or a membrane-impermeable dead cell stain in infected cells (Fig. 4A). However, KSHV encodes several proteins and microRNAs that inhibit apoptosis (42, 46–50). vIAP stabilizes the mitochondrial membrane, and vBCL2 binds to BH3 domains of proapoptotic proteins to inhibit apoptosis (49, 50). vFLIP was reported to block Fas-mediated apoptosis by preventing the recruitment and activation of caspase-8 (51), although more recent studies suggest that it leads to the upregulation of antiapoptotic factors through NF-kB signaling (52–54). RTA, vIRF1, vIRF3, K-bZIP, and LANA target the tumor suppressor p53 to prevent the induction of apoptosis (49, 50). These antiapoptotic factors may block apoptosis downstream of the caspase activation during lytic reactivation, preventing the antiviral effect of apoptosis while allowing the proviral IFN-β regulatory function of caspases to persist. Further studies of these antiapoptotic factors during KSHV reactivation may reveal connections between cell death and IFN-β regulation.
Given the antiviral role of apoptosis (55, 56), caspases may be activated by the KSHV-infected cells as a protective response in order to commit suicide. Several studies have shown that components of the RNA-sensing RIG-I-like receptor (RLR)-dependent pathway can trigger apoptosis. Sendai virus infection induces apoptosis through IRF3 activation (57, 58) or direct interactions between IRF3 and the proapoptotic protein Bax, which facilitate the translocation of Bax to the mitochondria (33). In contrast, Semliki Forest virus infection activates caspase-8 through direct interactions with mitochondrial antiviral signaling (MAVS) (59). Although less is known about caspase activation through the cGAS/STING DNA sensing pathway, it is possible that viral DNA sensing also activates caspases. Alternatively, KSHV proteins may directly activate caspases to block IFN-β signaling. The HPV-31 E6 and E7 oncoproteins activate caspases-3, -7, and -9 upon epithelial differentiation (44, 45), and HPV-16 E6 has been reported to interact with and activate caspase-8 (60). KSHV vIRF2 has been reported to activate caspase-3 upon overexpression (20). However, it is unclear whether vIRF2 is important in our system, since caspase-3/7 inhibitors did not stimulate IFN-β induction (Fig. 3A). There are also several KSHV proteins that are known to inhibit the induction of type I IFNs. These include multiple vIRFs that interact with and inhibit the function of STING (vIRF1), IRF3 (vIRF2), and IRF7 (vIRF3) (3), as well as LANA and ORF52, which directly inhibit cGAS activity (17, 18). It is possible that the caspase-based regulation of IFN-β we have observed is linked to the activity of these known anti-IFN factors. Whether any of these factors is involved in caspase activation remains to be tested. Alternatively, caspases may work parallel to viral anti-IFN factors and serve as an additional barrier to IFN-β induction. Nevertheless, caspase-dependent regulation of IFN-β is likely to be an intrinsic regulatory pathway that KSHV takes advantage of, either by directly activating caspases or by exploiting caspase activation due to cellular responses.
MATERIALS AND METHODS
Cell lines, reagents, and treatments.
iSLK.219, iSLK.RTA (61), and HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS) (HyClone). TREx-BCBL1-RTA cells (39) were grown in RPMI supplemented with 10% FBS, 2 mM GlutaMAX supplement (Gibco/Thermo Fisher), and 50 μg/ml hygromycin (Enzo Life Sciences). iSLK.219 cells were reactivated with 1 μg/ml of doxycycline (Thermo Fisher). Reactivation was confirmed visually for all samples by examining the appearance of RFP-positive cells (RFP is driven by the lytic PAN promoter). TREx-BCBL1-RTA cells were reactivated with 20 ng/ml of 12-O-tetradecanoyl-phorbol 13-acetate (TPA; Calbiochem), 500 ng/ml of ionomycin (EMD Millipore), and 1 μg/ml of doxycycline for 24 h. Where indicated, cells were treated with vehicle (dimethyl sulfoxide [DMSO]; Sigma-Aldrich), 10 μM IDN-6556 (MedChem Express or Selleck Chemicals), 100 μg/ml phosphonoacetic acid (PAA; Sigma-Aldrich), 10 μM Z-DEVD-FMK (Apex Bio), 100 μM Z-IETD-FMK (Apex Bio), 100 μM Ac-YVAD-CMK (Invivogen), 10 or 100 pg/ml recombinant IFN-β (Invivogen), and/or 2 μM MRT67307 (Sigma). The concentrations of all drug treatments were chosen based on common concentrations used in previously published studies. For poly(I·C) treatment, cells were transfected with 5 μg/ml poly(I·C) precomplexed with LyoVec transfection reagent (Invivogen) in Opti-MEM I (Life Technologies) for 3 h prior to sample collection.
RT-qPCR.
For RNA analysis, iSLK.219 cells were seeded at a density of 33,000 cells per well in a 24-well plate, and the lytic cycle was induced by the addition of doxycycline (1 μg/ml). RNA samples were collected at the indicated time points in RNA lysis buffer (Zymo Research). TREx-BCBL1-RTA cells were seeded at 2 × 105 cells per ml 24 h before induction and induced using TPA, ionomycin, and doxycycline for 24 h. One ml of the cells was collected, and the cell pellet was lysed in RNA lysis buffer (Zymo Research). Total RNA was extracted using the Quick-RNA MiniPrep kit (Zymo Research) by following the manufacturer's protocol. For human mRNA measurements, cDNA was prepared using an iScript cDNA synthesis kit (Bio-Rad) per the manufacturer's protocol. For viral mRNA measurements, cDNA was prepared using AMV RT (Promega) per the manufacturer's protocol, using a cocktail of primers targeted at KSHV genes and 18S rRNA in order to obtain strand-specific measurements. In both cases, human 18S rRNA levels were used as an internal standard to calculate relative mRNA levels. Real-time quantitative PCR (RT-qPCR) was performed using iTaq Universal SYBR green supermix (Bio-Rad) in a CFX Connect real-time PCR detection system (Bio-Rad). No-template and no-RT controls were included in each replicate. CFX Manager software was used to analyze the data. Primers are listed in Table 1.
TABLE 1.
qPCR primers used in this study
| Primer | Forward | Reverse | Reference |
|---|---|---|---|
| hIFN-α | GTGAGGAAATACTTCCAAAGAATCAC | TCTCATGATTTCTGCTCTGACAA | 63 |
| hIFN-β | CAGCAATTTTCAGTGTCAGAAGC | TCATCCTGTCCTTGAGGCAGT | 64 |
| hISG15 | GCGAGATCACCCAGAAGATT | GCCCTTGTTATTCCTCACCA | 65 |
| hIL-18 | TGCCAACTCTGGCTGCTAAA | TTGTTGCGAGAGGAAGCGAT | 66 |
| ORF25 | GCTTGCAATAAGCACCACATCGGT | AACTGCGAGAACCGTGTCCACTAA | 67 |
| ORF37 | TGACACCCTTGGGTTAACAGT | TCTCGAACCTTGGCGTGCTTTAGA | 67 |
| ORF52 | AAATCGAAGCCAGGGTCAGG | CTCCTCTTCGTCGCCTGTTATTG | 68 |
| ORF57 | GGTGTGTCTGACGCCGTAAAG | CCTGTCCGTAAACACCTCCG | 68 |
| LANA | AGGATGGAGATCGCAGACAC | CCAGCAAACCCACTTTAACC | 69 |
| RTA | ACCAAGGTGTGCCGTGTAGAGATT | AGCCTTACGCTTCTTTGAGCTCCT | 67 |
| h18S | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG | 70 |
| hCCR5 | ATGATTCCTGGGAGAGACGC | AGCCAGGACGGTCACCTT | 71 |
Viral DNA measurements.
iSLK.219 cells were seeded at a density of 33,000 cells per well in a 24-well plate, and the lytic cycle was induced by the addition of doxycycline (1 μg/ml). Cell pellets were collected at the indicated time points. Total DNA was extracted using the DNeasy blood and tissue kit (Qiagen). Levels of viral LANA were measured by qPCR and normalized to human CCR5 copy number levels. In both cases, a standard curve using PCR-amplified CCR5 and LANA fragments was used to calculate the copy number. iTaq Universal SYBR green supermix (Bio-Rad) and a CFX Connect real-time PCR detection system (Bio-Rad) were used for the qPCR, and CFX Manager software was used to analyze the data. Primers are listed in Table 1.
Protein analysis.
For Western blot analysis, iSLK.219 and iSLK.RTA cells were seeded at a density of 167,000 cells per well in a 6-well plate and treated with doxycycline (1 μg/ml). Lysates were collected at the indicated time points. TREx-BCBL1-RTA cells were seeded at 2 × 105 cells per ml 24 h before induction and induced using TPA, ionomycin, and doxycycline for 24 h. One ml of the cells was then collected at the indicated time point. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS, 1% NP-40) or an NP-40-only buffer for caspase blots (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40) supplemented with 50 μg/ml phenylmethylsulfonyl fluoride (PMSF; G-Biosciences) and cOmplete protease cocktail inhibitor (Roche). Samples were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The following Cell Signaling Technologies antibodies were used on PVDF membranes blocked in 5% milk in Tris-buffered saline with 0.1% Tween 20 (TBST) at 1:1,000 dilution in 5% milk in TBST: anti-caspase-3 (no. 9665), anti-cleaved caspase-3 (no. 9762), anti-caspase-9 (no. 9508), anti-cleaved caspase-9 (no. 9505), anti-Lamin-A/C (no. 4777), and anti-PARP1 (no. 9532). The following Cell Signaling Technologies antibodies were used with PVDF membranes blocked in 5% bovine serum albumin (BSA; Thermo Fisher) in TBST at 1:1,000 dilution in 5% BSA in TBST: anti-caspase-8 (no. 9746), anti-cleaved caspase-8 (no. 9496), anti-IRF3 (D83B9) (no. 4302), and anti-phospho-IRF3 Ser396 (4D4G) (no. 4947). Anti-β-actin (sc-1616; 1:200 dilution; Santa Cruz Biotechnology) and anti-RTA (1:1,000 dilution; gift from Luwen Zhang [62]) were diluted in 0.5% milk in phosphate-buffered saline with 0.1% Tween 20 (PBST) and used with PVDF membranes blocked in 5% milk in PBST. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG and goat anti-mouse IgG (both 1:5,000 in blocking buffer) were purchased from Southern Biotechnology. HRP-conjugated donkey anti-goat IgG (1:5,000 in 0.5% milk in PBST) was purchased from Santa Cruz Biotechnology. All membranes were exposed using Pierce ECL Western blotting substrate (Thermo Fisher) and imaged with a Syngene G:Box Chemi XT4 gel doc system.
ELISA.
Supernatant from cells was collected 4 days postreactivation, filtered to remove virus, and subjected to enzyme-linked immunosorbent assay (ELISA) analysis for IFN-β using a LumiKine hIFN-β bioluminescent ELISA kit (Invivogen) according to the manufacturer's protocol. Each biological replicate consisted of three technical replicates per condition.
Caspase-Glo assay.
iSLK.219 cells were seeded at a density of 2,500 cells per well in a 384-well plate and lytically reactivated with doxycycline for 4 days in the presence or absence of caspase inhibitors. The cells were incubated with a Caspase-8-Glo or Caspase-3/7-Glo reagent (Promega) at room temperature with shaking for 30 min, and luminescence was measured using a VICTOR3V (PerkinElmer) plate reader. Each biological replicate consisted of three technical replicates per condition.
Annexin V staining.
iSLK.219 cells were seeded at a density of 167,000 cells per well in a 6-well plate and lytically reactivated with doxycycline for 4 days in the presence or absence of IDN-6556. HeLa cells were treated with TNF-α (BioLegend) and 10 μg/ml cycloheximide (Sigma) for 6 h. For the staining, cells were trypsinized, washed twice in PBS, and treated with annexin V-Pacific Blue conjugate and LIVE/DEAD fixable near-IR dead cell stain (Thermo Fisher) according to the manufacturer's protocol, with minor modifications. Briefly, the cells were resuspended in 100 μl of annexin V binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4) supplemented with 5 μl annexin V-Pacific Blue conjugate and 0.1 μl of LIVE/DEAD fixable near-IR dead cell stain and incubated for 15 min at room temperature protected from light. After incubation, the cells were diluted in 400 μl of annexin V binding buffer, and fluorescence was quantified by flow cytometry on a BD LSRII flow cytometer at the Tufts Laser Cytometry core facility. Flow cytometry data were analyzed using Summit version 4.3 software.
siRNA knockdown.
iSLK.219 cells were transfected while in suspension (reverse transfection) with 10 nM siRNAs against IFN-β (siRNA#1, HSS105232; siRNA#2, HSS105233; Life Technologies/Thermo Fisher) or a nontargeting siRNA (12935300; Life Technologies/Thermo Fisher) at a density of 67,000 cells/ml in a 24-well plate, using the Lipofectamine RNAiMAX transfection reagent (Life Technologies/Thermo Fisher) per the manufacturer's protocol. Six hours later, the medium was replaced and the lytic cycle was induced. For siRNAs against caspase-8 (HSS141460; Life Technologies/Thermo Fisher), the same reverse transfection process was done twice, once 2 days prior to induction and a second time on the day of lytic cycle induction.
Viral replication.
For flow cytometry-based quantitation of viral yields, iSLK.219 cells were seeded into 24-well plates at a density of 33,000 cells per well and induced by addition of doxycycline. Six days postreactivation, virus-containing supernatant was collected and cells were trypsinized. The cells were fixed with 4% paraformaldehyde (Ted Pella) in PBS for 15 min at room temperature, washed twice with PBS, and resuspended in PBS. RFP fluorescence was quantified by flow cytometry on a BD LSRII flow cytometer at the Tufts Laser Cytometry core facility. Virus-containing supernatant was used to spin infect uninfected iSLK.RTA cells with 8 μg/ml hexadimethrine bromide (Polybrene; Sigma) for 1 h at 1,000 rpm. Four days postinfection, iSLK.RTA cells were trypsinized, washed twice with PBS, and resuspended in PBS. GFP fluorescence was quantified by flow cytometry on a BD LSRII flow cytometer. Virus titer was calculated with the following formula: (number of target cells at infection) × (percentage of GFP-positive target cells)/(volume of virus, in milliliters). For RNA-based quantification of viral yields from TREx-BCBL1-RTA cells, TREx-BCBL1-RTA cells were seeded at 2 × 105 cells per ml 24 h before induction and induced by adding TPA, ionomycin, and doxycycline for 24 h. Virus supernatant was collected 3 days postreactivation and used to spin infect uninfected iSLK.RTA cells with 8 μg/ml hexadimethrine bromide (Polybrene; Sigma) for 1 h at 1,000 rpm. Doxycycline (1 μg/ml) was added immediately after the spin infection to induce lytic reactivation, and RNA samples were collected 2 days later in RNA lysis buffer (Zymo). KSHV ORF57 and 18S RNA levels were measured as described above for RNA analysis.
Statistics.
All statistical analyses were performed using GraphPad Prism, version 7.02 (GraphPad Software, La Jolla California USA). Statistical significance was determined by Student's t test or analysis of variance (ANOVA) followed by a post hoc multiple-comparison test (Dunnett's, Sidak's, or Tukey's) when multiple comparisons were required. All data are plotted as means ± standard deviations.
ACKNOWLEDGMENTS
This work was supported by a Tufts Collaborates grant and a Stephen and Marie Rozan Research award (to M.M.G. and A.D.), American Cancer Society Research Scholar Grant 131320-RSG-17-189-01-MPC (to M.M.G.), NIH grants R21AI124049 and R01CA190542 (to A.D.), and the Sackler Families Collaborative Cancer Biology Award (to T.T.).
We thank Allen Parmelee and Stephen Kwok at the Tufts Laser Cytometry core facility (instrumentation grant S10RR20941) for support on flow cytometry experiments and Danish Saleh and Veronica Canarte for technical advice and help. We thank Luwen Zhang for the RTA antibody. We thank Britt Glaunsinger and Karl Munger and members of the Gaglia laboratory for critical readings of the manuscript.
REFERENCES
- 1.Lee H-R, Choi UY, Hwang S-W, Kim S, Jung JU. 2016. Viral inhibition of PRR-mediated innate immune response: learning from KSHV evasion strategies. Mol Cells 39:777–782. doi: 10.14348/molcells.2016.0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee H-R, Lee S, Chaudhary PM, Gill P, Jung JU. 2010. Immune evasion by Kaposi's sarcoma-associated herpesvirus. Future Microbiol 5:1349–1365. doi: 10.2217/fmb.10.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aresté C, Blackbourn DJ. 2009. Modulation of the immune system by Kaposi's sarcoma-associated herpesvirus. Trends Microbiol 17:119–129. doi: 10.1016/j.tim.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Chang J, Renne R, Dittmer D, Ganem D. 2000. Inflammatory cytokines and the reactivation of Kaposi's sarcoma-associated herpesvirus lytic replication. Virology 266:17–25. doi: 10.1006/viro.1999.0077. [DOI] [PubMed] [Google Scholar]
- 5.Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Investig 120:939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol 71:715–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N Engl J Med 340:1063–1070. doi: 10.1056/NEJM199904083401402. [DOI] [PubMed] [Google Scholar]
- 8.Nichols LA, Adang LA, Kedes DH. 2011. Rapamycin blocks production of KSHV/HHV8: insights into the anti-tumor activity of an immunosuppressant drug. PLoS One 6:e14535. doi: 10.1371/journal.pone.0014535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groopman JE, Gottlieb MS, Goodman J, Mitsuyasu RT, Conant MA, Prince H, Fahey JL, Derezin M, Weinstein WM, Casavante C, et al. 1984. Recombinant alpha-2 interferon therapy for Kaposi's sarcoma associated with the acquired immunodeficiency syndrome. Ann Intern Med 100:671–676. doi: 10.7326/0003-4819-100-5-671. [DOI] [PubMed] [Google Scholar]
- 10.Volberding PA, Mitsuyasu RT, Golando JP, Spiegel RJ. 1987. Treatment of Kaposi's sarcoma with interferon alfa-2b (intron A). Cancer 59:620–625. [DOI] [PubMed] [Google Scholar]
- 11.Whitehead KA, Dahlman JE, Langer RS, Anderson DG. 2011. Silencing or stimulation? siRNA delivery and the immune system. Annu Rev Chem Biomol Eng 2:77–96. doi: 10.1146/annurev-chembioeng-061010-114133. [DOI] [PubMed] [Google Scholar]
- 12.Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, Ting JP, Damania B. 2011. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331:330–334. doi: 10.1126/science.1199478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B. 2011. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9:363–375. doi: 10.1016/j.chom.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.West JA, Gregory SM, Damania B. 2012. Toll-like receptor sensing of human herpesvirus infection. Front Cell Infect Microbiol 2:122. doi: 10.3389/fcimb.2012.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.West JA, Wicks M, Gregory SM, Chugh P, Jacobs SR, Zhang Z, Host KM, Dittmer DP, Damania B. 2014. An important role for mitochondrial antiviral signaling protein in the Kaposi's sarcoma-associated herpesvirus life cycle. J Virol 88:5778–5787. doi: 10.1128/JVI.03226-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, Barber GN, Glaunsinger BA, Dittmer DP, Damania B. 2015. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci U S A 112:E4306–E4315. doi: 10.1073/pnas.1503831112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J-J, Li W, Shao Y, Avey D, Fu B, Gillen J, Hand T, Ma S, Liu X, Miley W, Konrad A, Neipel F, Stürzl M, Whitby D, Li H, Zhu F. 2015. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18:333–344. doi: 10.1016/j.chom.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang G, Chan B, Samarina N, Abere B, Weidner-Glunde M, Buch A, Pich A, Brinkmann MM, Schulz TF. 2016. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc Natl Acad Sci U S A 113:E1034–E1043. doi: 10.1073/pnas.1516812113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mariggiò G, Koch S, Zhang G, Weidner-Glunde M, Rückert J, Kati S, Santag S, Schulz TF. 2017. Kaposi sarcoma herpesvirus (KSHV) latency-associated nuclear antigen (LANA) recruits components of the MRN (Mre11-Rad50-NBS1) repair complex to modulate an innate immune signaling pathway and viral latency. PLoS Pathog 13:e1006335. doi: 10.1371/journal.ppat.1006335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Areste C, Mutocheluh M, Blackbourn DJ. 2009. Identification of caspase-mediated decay of interferon regulatory factor-3, exploited by a Kaposi sarcoma-associated herpesvirus immunoregulatory protein. J Biol Chem 284:23272–23285. doi: 10.1074/jbc.M109.033290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Degterev A, Boyce M, Yuan J. 2003. A decade of caspases. Oncogene 22:8543–8567. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- 22.Kovalenko A, Kim JC, Kang TB, Rajput A, Bogdanov K, Dittrich-Breiholz O, Kracht M, Brenner O, Wallach D. 2009. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J Exp Med 206:2161–2177. doi: 10.1084/jem.20090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, Taniguchi T, Shadel GS, Chen ZJ, Iwasaki A, Flavell RA. 2014. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159:1563–1577. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME, Huang DC, Kile BT. 2014. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159:1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Ning X, Gao P, Wu S, Sha M, Lv M, Zhou X, Gao J, Fang R, Meng G, Su X, Jiang Z. 2017. Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity 46:393–404. doi: 10.1016/j.immuni.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 26.Prasad A, Lu M, Lukac DM, Zeichner SL. 2012. An alternative Kaposi's sarcoma-associated herpesvirus replication program triggered by host cell apoptosis. J Virol 86:4404–4419. doi: 10.1128/JVI.06617-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prasad A, Remick J, Zeichner SL. 2013. Activation of human herpesvirus replication by apoptosis. J Virol 87:10641–10650. doi: 10.1128/JVI.01178-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majerciak V, Kruhlak M, Dagur PK, McCoy JP Jr, Zheng ZM. 2010. Caspase-7 cleavage of Kaposi sarcoma-associated herpesvirus ORF57 confers a cellular function against viral lytic gene expression. J Biol Chem 285:11297–11307. doi: 10.1074/jbc.M109.068221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossetto CC, Pari GS. 2011. Kaposi's sarcoma-associated herpesvirus noncoding polyadenylated nuclear RNA interacts with virus- and host cell-encoded proteins and suppresses expression of genes involved in immune modulation. J Virol 85:13290–13297. doi: 10.1128/JVI.05886-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henry CM, Martin SJ. 2017. Caspase-8 acts in a non-enzymatic role as a scaffold for assembly of a pro-inflammatory “FADDosome” complex upon TRAIL stimulation. Mol Cell 65:715–729. doi: 10.1016/j.molcel.2017.01.022. [DOI] [PubMed] [Google Scholar]
- 31.Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, Roback L, Kaiser W, Oberst A, Sagara J, Fitzgerald KA, Green DR, Zhang J, Mocarski ES, Alnemri ES. 2015. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun 6:7515. doi: 10.1038/ncomms8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chattopadhyay S, Kuzmanovic T, Zhang Y, Wetzel Jaime L, Sen Ganes C. 2016. Ubiquitination of the transcription factor IRF-3 activates RIPA, the apoptotic pathway that protects mice from viral pathogenesis. Immunity 44:1151–1161. doi: 10.1016/j.immuni.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chattopadhyay S, Yamashita M, Zhang Y, Sen GC. 2011. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J Virol 85:3708–3716. doi: 10.1128/JVI.02133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Orzalli MH, Kagan JC. 2017. Apoptosis and necroptosis as host defense strategies to prevent viral infection. Trends Cell Biol 27:800–809. doi: 10.1016/j.tcb.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 36.White E, Sabbatini P, Debbas M, Wold WS, Kusher DI, Gooding LR. 1992. The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor alpha. Mol Cell Biol 12:2570–2580. doi: 10.1128/MCB.12.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bigalke JM, Heldwein EE. 2016. Nuclear exodus: herpesviruses lead the way. Annu Rev Virol 3:387–409. doi: 10.1146/annurev-virology-110615-042215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 77:4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park SY, Waheed AA, Zhang Z-R, Freed EO, Bonifacino JS. 2014. HIV-1 Vpu accessory protein induces caspase-mediated cleavage of IRF3 transcription factor. J Biol Chem 289:35102–35110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Philip NH, DeLaney A, Peterson LW, Santos-Marrero M, Grier JT, Sun Y, Wynosky-Dolfi MA, Zwack EE, Hu B, Olsen TM, Rongvaux A, Pope SD, Lopez CB, Oberst A, Beiting DP, Henao-Mejia J, Brodsky IE. 2016. Activity of uncleaved caspase-8 controls anti-bacterial immune defense and TLR-induced cytokine production independent of cell death. PLoS Pathog 12:e1005910. doi: 10.1371/journal.ppat.1005910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belanger C, Gravel A, Tomoiu A, Janelle ME, Gosselin J, Tremblay MJ, Flamand L. 2001. Human herpesvirus 8 viral FLICE-inhibitory protein inhibits Fas-mediated apoptosis through binding and prevention of procaspase-8 maturation. J Hum Virol 4:62–73. [PubMed] [Google Scholar]
- 43.Davis DA, Naiman NE, Wang V, Shrestha P, Haque M, Hu D, Anagho HA, Carey RF, Davidoff KS, Yarchoan R. 2015. Identification of caspase cleavage sites in KSHV latency-associated nuclear antigen and their effects on caspase-related host defense responses. PLoS Pathog 11:e1005064. doi: 10.1371/journal.ppat.1005064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Connolly PF, Fearnhead HO. 2017. Viral hijacking of host caspases: an emerging category of pathogen-host interactions. Cell Death Differ 24:1401–1410. doi: 10.1038/cdd.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moody CA, Fradet-Turcotte A, Archambault J, Laimins LA. 2007. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. Proc Natl Acad Sci U S A 104:19541–19546. doi: 10.1073/pnas.0707947104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friborg J, Kong W-P, Hottiger MO, Nabel GJ. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889–894. [DOI] [PubMed] [Google Scholar]
- 47.Feng P, Park J, Lee BS, Lee SH, Bram RJ, Jung JU. 2002. Kaposi's sarcoma-associated herpesvirus mitochondrial K7 protein targets a cellular calcium-modulating cyclophilin ligand to modulate intracellular calcium concentration and inhibit apoptosis. J Virol 76:11491–11504. doi: 10.1128/JVI.76.22.11491-11504.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang HW, Sharp TV, Koumi A, Koentges G, Boshoff C. 2002. Characterization of an anti-apoptotic glycoprotein encoded by Kaposi's sarcoma-associated herpesvirus which resembles a spliced variant of human survivin. EMBO J 21:2602–2615. doi: 10.1093/emboj/21.11.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuentes-González AM, Contreras-Paredes A, Manzo-Merino J, Lizano M. 2013. The modulation of apoptosis by oncogenic viruses. Virol J 10:182–182. doi: 10.1186/1743-422X-10-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moore PS. 2007. KSHV manipulation of the cell cycle and apoptosis. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 51.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. 1997. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 52.Guasparri I, Keller SA, Cesarman E. 2004. KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med 199:993–1003. doi: 10.1084/jem.20031467. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Matta H, Chaudhary PM. 2004. Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP). Proc Natl Acad Sci U S A 101:9399–9404. doi: 10.1073/pnas.0308016101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun Q, Matta H, Lu G, Chaudhary PM. 2006. Induction of IL-8 expression by human herpesvirus 8 encoded vFLIP K13 via NF-kappaB activation. Oncogene 25:2717–2726. doi: 10.1038/sj.onc.1209298. [DOI] [PubMed] [Google Scholar]
- 55.Best SM. 2008. Viral subversion of apoptotic enzymes: escape from death row. Annu Rev Microbiol 62:171–192. doi: 10.1146/annurev.micro.62.081307.163009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fuchs Y, Steller H. 2011. Programmed cell death in animal development and disease. Cell 147:742–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peters K, Chattopadhyay S, Sen GC. 2008. IRF-3 activation by Sendai virus infection is required for cellular apoptosis and avoidance of persistence. J Virol 82:3500–3508. doi: 10.1128/JVI.02536-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weaver BK, Ando O, Kumar KP, Reich NC. 2001. Apoptosis is promoted by the dsRNA-activated factor (DRAF1) during viral infection independent of the action of interferon or p53. FASEB J 15:501–515. doi: 10.1096/fj.00-0222com. [DOI] [PubMed] [Google Scholar]
- 59.El Maadidi S, Faletti L, Berg B, Wenzl C, Wieland K, Chen ZJ, Maurer U, Borner C. 2014. A novel mitochondrial MAVS/caspase-8 platform links RNA virus-induced innate antiviral signaling to Bax/Bak-independent apoptosis. J Immunol 192:1171. doi: 10.4049/jimmunol.1300842. [DOI] [PubMed] [Google Scholar]
- 60.Manzo-Merino J, Massimi P, Lizano M, Banks L. 2014. The human papillomavirus (HPV) E6 oncoproteins promotes nuclear localization of active caspase 8. Virology 450-451:146–152. [DOI] [PubMed] [Google Scholar]
- 61.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21. doi: 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meyer F, Ehlers E, Steadman A, Waterbury T, Cao M, Zhang L. 2013. TLR-TRIF pathway enhances the expression of KSHV replication and transcription activator. J Biol Chem 288:20435–20442. doi: 10.1074/jbc.M113.487421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jacobs SR, Stopford CM, West JA, Bennett CL, Giffin L, Damania B. 2015. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 1 interacts with a member of the interferon-stimulated gene 15 pathway. J Virol 89:11572–11583. doi: 10.1128/JVI.01482-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jacobs SR, Gregory SM, West JA, Wollish AC, Bennett CL, Blackbourn DJ, Heise MT, Damania B. 2013. The viral interferon regulatory factors of Kaposi's sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. J Virol 87:798–806. doi: 10.1128/JVI.01851-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoo YS, Park YY, Kim JH, Cho H, Kim SH, Lee HS, Kim TH, Sun Kim Y, Lee Y, Kim CJ, Jung JU, Lee JS, Cho H. 2015. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat Commun 6:7910. doi: 10.1038/ncomms8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, Ernst RK, Steele-Mortimer O, Celli J, Vallance BA. 2014. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16:249–256. doi: 10.1016/j.chom.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rossetto CC, Pari G. 2012. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog 8:e1002680. doi: 10.1371/journal.ppat.1002680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W, Jäger S, Shales M, Horner J, Hernandez RD, Krogan NJ, Glaunsinger BA. 2015. Global mapping of herpesvirus-host protein complexes reveals a novel transcription strategy for late genes. Mol Cell 57:349–360. doi: 10.1016/j.molcel.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arias C, Weisburd B, Stern-Ginossar N, Mercier A, Madrid AS, Bellare P, Holdorf M, Weissman JS, Ganem D. 2014. KSHV 2.0: a comprehensive annotation of the Kaposi's sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog 10:e1003847. doi: 10.1371/journal.ppat.1003847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abernathy E, Gilbertson S, Alla R, Glaunsinger B. 2015. Viral nucleases induce an mRNA degradation-transcription feedback loop in mammalian cells. Cell Host Microbe 18:243–253. doi: 10.1016/j.chom.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Malnati MS, Scarlatti G, Gatto F, Salvatori F, Cassina G, Rutigliano T, Volpi R, Lusso P. 2008. A universal real-time PCR assay for the quantification of group-M HIV-1 proviral load. Nat Protoc 3:1240–1248. doi: 10.1038/nprot.2008.108. [DOI] [PubMed] [Google Scholar]