

ABSTRACT
Infections caused by carbapenem-resistant Enterobacteriaceae (CRE) are increasingly prevalent and have become a major worldwide threat to human health. Carbapenem resistance is driven primarily by the acquisition of β-lactamase enzymes, which are able to degrade carbapenem antibiotics (hence termed carbapenemases) and result in high levels of resistance and treatment failure. Clinically relevant carbapenemases include both serine β-lactamases (SBLs; e.g., KPC-2 and OXA-48) and metallo-β-lactamases (MBLs), such as NDM-1. MBL-producing strains are endemic within the community in many Asian countries, have successfully spread worldwide, and account for many significant CRE outbreaks. Recently approved combinations of β-lactam antibiotics with β-lactamase inhibitors are active only against SBL-producing pathogens. Therefore, new drugs that specifically target MBLs and which restore carbapenem efficacy against MBL-producing CRE pathogens are urgently needed. Here we report the discovery of a novel MBL inhibitor, ANT431, that can potentiate the activity of meropenem (MEM) against a broad range of MBL-producing CRE and restore its efficacy against an Escherichia coli NDM-1-producing strain in a murine thigh infection model. This is a strong starting point for a chemistry lead optimization program that could deliver a first-in-class MBL inhibitor-carbapenem combination. This would complement the existing weaponry against CRE and address an important and growing unmet medical need.
KEYWORDS: β-lactamase, metallo-β-lactamase, NDM-1, carbapenem, inhibitor, resistance
INTRODUCTION
Resistance to β-lactams, the most widely used class of antibacterial drugs, emerged very soon after these antibiotics were introduced into clinical practice (1). In fact, even before penicillin was used clinically it had already been noted that some bacteria were nonsusceptible due to the production of an enzyme that inactivated penicillin (2). Such β-lactamase enzymes, as they came to be known, are the most widespread mechanism of resistance to β-lactam antibiotics, hydrolyzing the β-lactam ring and rendering them ineffective. Strategies to fight β-lactamase-mediated resistance have included modification of β-lactams as well as the development of combinations of β-lactams with β-lactamase inhibitors. In 1981, the first such combination, amoxicillin-clavulanate, was launched following the discovery of the natural product clavulanic acid, an inhibitor of serine β-lactamases (SBLs) (3, 4). However, new β-lactamases have continued to emerge which are insensitive to inhibition by clavulanic acid and other marketed inhibitors (5). Several new β-lactam–inhibitor combinations brought to the market more recently, (e.g., ceftazidime-avibactam [6] and meropenem [MEM]-vaborbactam [7]) address resistance due to extended-spectrum β-lactamases (ESBLs) and also the class A KPC and certain class D OXA carbapenemases, which are largely responsible for recent increases in carbapenem-resistant Enterobacteriaceae (CRE) strains.
The most recent class of β-lactamases to have come to prominence are the class B metallo-β-lactamases (MBLs), which include the NDM, VIM, and IMP subclasses and multiple variants thereof. This situation is extremely concerning, as MBLs impart resistance to nearly all β-lactams (only monobactams, e.g., aztreonam, have some stability to MBLs [8]) and are not inhibited by SBL inhibitors such as avibactam or vaborbactam. Furthermore, MBL-producing organisms very often exhibit multidrug resistance phenotypes due to the acquisition of plasmid-borne resistance genes, which are colocated on the plasmids which carry the MBL genes (9). The most widespread MBL comes from the most recently identified NDM subclass. NDM-1 was first reported in 2008, having been discovered in a Klebsiella pneumoniae isolated from a Swedish patient who had recently returned from India (10), and this MBL has now been identified in all continents, with rapid dissemination being observed from reservoirs in Asia, the Middle East, and the Balkans (11). While national surveillance programs are not available for many countries, recent reports, including several prevalence surveys and outbreaks, suggest an alarming worldwide increase in incidences of NDM-1-producing organisms as a percentage of carbapenem-nonsusceptible or -resistant Enterobacteriaceae isolates, e.g., 68% in Bulgaria (12), 30% in Turkey (13), 67% in Iraq (14), 32% in China (15), 48% in South Africa (16), and 92% in Mexico (17). Numerous variants of NDM-1 having single or double amino acid changes (18) have been reported from animal and human sources, the most recent being NDM-17 from a chicken in China (19). NDM, VIM, and IMP enzymes have been identified in all major Gram-negative pathogens, including the WHO priority pathogens K. pneumoniae, Pseudomonas aeruginosa, and Acinetobacter baumanii; however, as yet, there are no MBL inhibitors in clinical use, despite there being a clear unmet medical need (20).
Here we describe the in vitro and in vivo properties of ANT431 (Fig. 1), a specific inhibitor of MBLs which is the result of a medicinal chemistry hit-to-lead program (D. T. Davies, unpublished data) starting from pyridine-2-carboxylic acid, a compound with weak MBL inhibition, originally reported as an inhibitor of the CphA enzyme from Aeromonas hydrophila (21).
FIG 1.
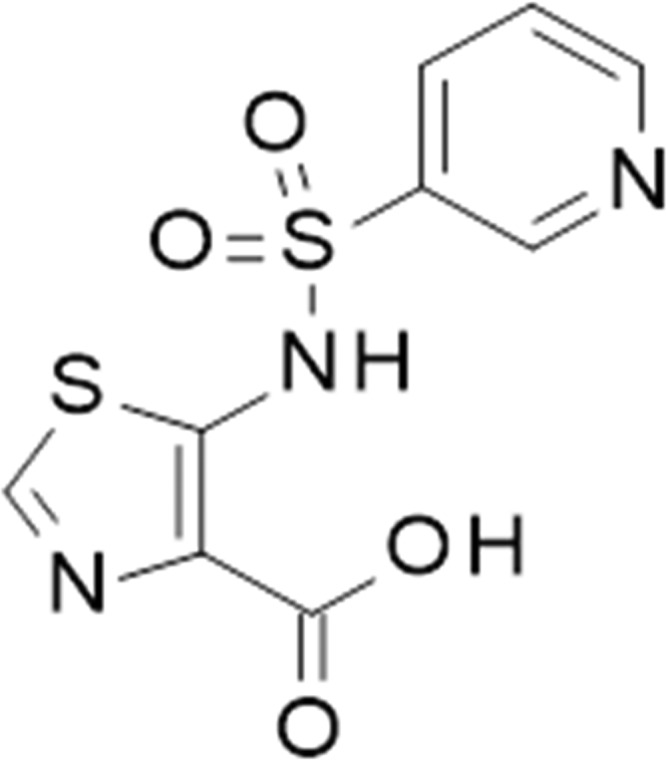
Chemical structure of ANT431.
RESULTS
MBL inhibition by ANT431.
Table 1 shows the inhibitory activities of compound ANT431 against purified NDM-1, VIM-1, VIM-2, and IMP-1 enzymes and potentiation of MEM activity against an Escherichia coli laboratory strain expressing the same enzymes from a similar recombinant plasmid background. ANT431 was a potent inhibitor of NDM-1 and VIM-2, with Ki values of 290 nM and 195 nM, respectively. Furthermore, susceptibility testing of MEM against NDM-1- and VIM-2-expressing bacteria in the presence of 30 μg/ml of ANT431 (97.6 μM) resulted in strong potentiation of MEM antibacterial activity with decreases in MICs of 128-fold and 64-fold, respectively (Table 1). This indicates that ANT431 is able to penetrate into the bacterial periplasm, where the MBL enzymes are located, and thus effect its inhibitory activity. In contrast, ANT431 was a comparatively weak inhibitor of VIM-1 and IMP-1 (Ki values of 14.6 and 4.15 μM, respectively) and showed correspondingly little or no potentiation of MEM activity against the E. coli strain overexpressing these enzymes.
TABLE 1.
Effect of ANT431 on inhibition of purified MBLs and potentiation of MEM activity against E. coli BL21(DE3) transformed with pET plasmids containing NDM-1, VIM-1, VIM-2, or IMP-1
| Compound | NDM-1 |
VIM-1 |
VIM-2 |
IMP-1 |
||||
|---|---|---|---|---|---|---|---|---|
| Kia | MICb | Ki | MIC | Ki | MIC | Ki | MIC | |
| None | 32 | 4 | 4 | 8 | ||||
| ANT431 | 0.29 | 0.25 | 14.6c | 4 | 0.19 | 0.06 | 4.15 | 4 |
Ki, enzyme inhibition constant (micromolar).
MIC of MEM (micrograms per milliliter) determined alone or in the presence of 30 μg/ml of compound.
Determined from Dixon plot analysis (Fig. 2).
Kinetic analyses of enzyme inhibition demonstrated that ANT431 is a competitive inhibitor with respect to the MEM substrate of VIM-1 (Fig. 2), NDM-1, VIM-2, and IMP-1 (data not shown), as indicated by Dixon plot analysis by convergence of lines to an intersection above the x axis.
FIG 2.

Dixon analysis of the inhibition of VIM-1 by ANT431. Initial rates of β-lactam hydrolysis were measured spectrophotometrically using MEM (○, 40 μM; △, 90 μM; ◇, 130 μM; □, 800 μM) as the substrate in 50 mM HEPES buffer (pH 7.5), in the presence of 6.9 nM purified VIM-1. Inhibitor concentrations ranged from 10 to 50 μM. Initial rates were measured in triplicates (SD, ≤5%). Vmax was unaffected by ANT431. These data fully support a competitive mode of inhibition of the enzyme by ANT431, with a Ki value of 14.6 ± 0.6 μM. Similar conclusions (data not shown) were obtained with the NDM-1, IMP-1, and VIM-2 MBLs.
Potentiation of MEM activity against clinical isolates.
To investigate the spectrum of activity of ANT431 against medically important pathogens, the antibacterial activity of the MEM-ANT431 combination was profiled against a panel of 94 randomly selected NDM- and VIM-producing clinical isolates (many of which coexpressed other β-lactam resistance determinants) (Table 2). The cumulative distributions of MEM MICs in the presence of 0, 10, and 30 μg/ml of ANT431 are shown in Fig. 3. The pronounced leftward shift of the curves compared to the MEM control indicates the greatly improved activity of the ANT431 combinations versus the majority of isolates. In fact, addition of 30 μg/ml of ANT431 resulted in a reduction of the MEM MIC to susceptible levels (2 μg/ml EUCAST breakpoint) in 72% of the MBL-positive isolates, increasing to 79% for the NDM-positive subset.
TABLE 2.
Data for MIC susceptibility testing (μg/ml) of 94 MBL-positive Enterobacteriaceae to MEM alone and in combination with ANT431 (at 10 and 30 μg/ml)a


Bold is used for data for strains that are colistin resistant. Shaded cells indicate MEM MICs of ≤2 μg/ml.
FIG 3.

Cumulative MIC distribution of MEM alone and in combination with ANT431 against 94 MBL-producing Enterobacteriaceae.
The species, source, and β-lactamases of the clinical isolates are shown in Table 2, highlighting the wide geographical and genetic diversity of the isolate panel. In addition to MBLs, the majority of isolates also expressed one or more SBLs (e.g., TEM, CTX-M-3, and CMY-2). The strain set included representatives of the major NDM variants commonly found in clinical isolates, namely, NDM-1, -4, -5, -6, and -7. The fact that ANT431 was able to potentiate the MEM MIC at least 8-fold in at least one strain from each NDM variant group shows that this compound is active against all these common NDM enzymes. Against VIM-positive isolates, the majority of which carried VIM-1, ANT431 showed only a modest ability to potentiate MEM. This is not surprising given the poor enzymatic inhibitory activity versus the purified VIM-1 enzyme and the lack of MEM potentiation observed against the laboratory E. coli strain overexpressing VIM-1 (Table 1). Despite this, MEM MICs were potentiated severalfold in many VIM-1-containing clinical isolates with originally low levels of resistance to MEM, often bringing the MIC down to the susceptibility breakpoint. As anticipated, no potentiation against IMP-containing isolates was observed.
PK and physicochemical properties of ANT431.
ANT431 is a highly water-soluble compound (the Na salt has a solubility of 30 mg/ml in phosphate-buffered saline [PBS] buffer [pH 7.4]), which is important for intravenous (i.v.) delivery and possible coformulation with MEM. The absorption, distribution, metabolism, and excretion (ADME) profile of ANT431 was promising, with good metabolic stability in both mouse and human liver microsomes and plasma, although moderate inhibition of the 2C9 and 3A4 isoforms of the cytochrome P450 (CYP450) enzyme was observed (50% inhibitory concentrations [IC50], 9 μM and 45 μM, respectively) (Table 3). Furthermore, ANT431 showed no measurable inhibition of angiotensin-converting enzyme (ACE; an important metalloenzyme selectivity target involved in blood pressure regulation) or glyoxalase II (GLY2; the closest human homologue of the MBL enzymes [22]) at the maximum concentration tested, 200 μM, indicating good selectivity toward bacterial MBLs compared to that of mammalian metalloenzymes and confirming the specific inhibitory mechanism of action of this compound, which does not behave as a general metalloenzyme inactivator via metal removal from the active site. Consistent with this, ANT431 showed no cytotoxicity up to 100 μM (the highest concentration tested) against the HepG2 human cell line. Furthermore, the i.v. pharmacokinetic (PK) profile of ANT431 in mice indicated a much longer plasma half-life (t1/2) and greater total exposure (area under the concentration-time curve [AUC]) than those of MEM (Fig. 4), suggesting that the PK of the inhibitor should not be a limiting factor in efficacy studies with this combination. Additionally, 20% of unchanged drug was recovered in the urine, indicating clearance through the kidneys and illustrating the potential for treatment of urinary tract infections (UTIs).
TABLE 3.
Physicochemical, ADME, selectivity, cytotoxicity, and safety properties of compound ANT431
| Property | Value for ANT431 |
|---|---|
| Molecular wt, acid/sodium salt | 285.3/307.3 |
| LogD (pH 7.4) | −2.5 |
| Solubility (sodium salt) in PBS (pH 7.4), mg/ml | 30 |
| PPB, % bound, mouse/human | 82.5/97.6 |
| Microsomal stability, % remaining at 30 min, mouse/human | >95 (both) |
| Plasma stability, % remaining at 1 h, mouse/human | 100 (both) |
| ACE inhibition (IC50, μM) | >200 |
| Glyoxalase II (IC50, μM) | >200 |
| HepG2 cell cytotoxicity (IC50 at 24 h, μM) | >100 |
| CYP inhibition (IC50, μM), 1A2, 2C9, 2C19, 2D6, 3A4 | >200, 9, >200, >200, 45 |
| hERG inhibition (IC50, μM) | >10 |
FIG 4.

Plasma pharmacokinetics of ANT431 and MEM in Swiss albino mice after intravenous administration of 1 mg/kg. The table inset shows PK parameters. C0 is initial plasma concentration.
ANT431 restores MEM efficacy in a mouse thigh model of infection.
The in vivo efficacy of ANT431 was tested against the NDM-1-positive clinical isolate E. coli IR3 (MEM MIC = 32 μg/ml; MIC of MEM plus ANT431 [at 8 μg/ml] = 4 μg/ml) in a 9-h murine thigh infection model. MEM is rapidly hydrolyzed by murine renal DHP-1 in mice; hence, this model, with its short dosing interval (2 h), has been specifically developed to compensate for the short half-life and so facilitate MEM efficacy experiments with mice. When dosed i.v. at 1, 3, 5, and 7 h postinfection, the combination of ANT431 (at 30 or 300 mg/kg of body weight) with MEM (at either 50 or 250 mg/kg) resulted in a statistically significant reduction of bacterial counts in the infected thighs, at least 1 log10 with respect to the counts observed with the corresponding dose of MEM alone (Fig. 5). The compound was well tolerated at 300 mg/kg (amounting to a total dose of 1.2 g/kg within an 8-h period) with no observable indications of toxicity.
FIG 5.

Efficacy of MEM alone and in combination with ANT431 in murine thigh infection model using E. coli IR3 (NDM-1). Bacterial counts (CFU) were obtained from homogenized thighs of infected animals (n = 5) treated i.v. at 1, 3, 5, and 7 h postinfection. Numbers in parentheses refer to log reduction in CFU compared to respective MEM-only group. The table at the bottom shows MICs for MEM with and without ANT431 at 8 μg/ml. Pt, pretreatment group; V, vehicle-only group. **, statistically significant difference (P < 0.005) compared to the value for the MEM-only group.
DISCUSSION
The global spread of MBL-expressing Enterobacteriaceae represents a major threat to the ongoing usefulness of carbapenem antibiotics to treat severe, often life-threatening, Gram-negative bacterial infections. A new drug which could render MBLs inactive, and hence maintain the effectiveness of carbapenems, would be a valuable adjunct to carbapenem therapies and would prolong the utility of this important class of antibiotics. The discovery of this chemical series, exemplified by ANT431, provides an opportunity to develop such a new combination therapy to treat infections with CRE expressing MBLs. This would address a significant unmet medical need since the current options to treat such infections are colistin, an old antibiotic with nephrotoxicity (23), and tigecycline, which is not recommended for bloodstream and UTIs due to its low levels in those body fluids and has received an FDA warning regarding the increased mortality risk associated with its use (24). Although new antibiotics are in development that should, in principle, cover MBL-producing CRE, including cefidericol (25), aztreonam-avibactam (26), and LYS228 (27), the advantage of developing an MBL inhibitor is that it can be combined with a well-characterized and extensively used carbapenem, such as meropenem, in order to directly rescue its activity against MBL-producing CRE pathogens and hence allow other new antibiotics to be reserved for situations where no other effective treatment is available.
Other MBL inhibitors have been reported (28–35), many of which display good in vitro activity but have not been shown to be efficacious in animal infection models. An exception to this is the natural product aspergillomarasmine A (35), a strong metal ion chelator whose further development is likely to be limited by toxicity (the 50% lethal dose [LD50] in mice is 159.8 mg/kg) (36). In contrast, ANT431 functions by specific inhibition of the MBL enzymes, as shown by substrate competition studies, and displays good selectivity over nonbacterial metalloenzymes (ACE and GLY2). Additionally, ANT431 exhibits promising drug-like properties, namely, excellent physicochemical properties (low-molecular-weight, simple synthesis, high solubility, and good stability), no toxicity against a human cell line (HepG2) at concentrations lower than 100 μM, and a promising drug metabolism and pharmacokinetic (DMPK) profile. Furthermore, in vivo proof of concept has been demonstrated against a clinical MBL-expressing isolate of E. coli in a mouse infection model, with ANT431 nullifying the effects of MBL expression and restoring the efficacy of MEM. This study also demonstrated tolerability of the compound at doses as high as 1.2 g/kg in 8 h.
The activity of ANT431, which at 30 μg/ml could reduce MEM MICs to EUCAST breakpoint susceptibility levels in over 70% of a large panel of highly resistant relevant clinical isolates, demonstrates the potential of such an inhibitor in the clinical setting. However, at the same time, there were nearly 30% of isolates for which the MICs were not significantly potentiated. There are several factors which, individually or together, may influence the final MICs of the combination; these are (i) the level of expression of the MBL under the MIC testing conditions, (ii) alterations in the structure or level of expression of outer membrane porins, limiting the penetrability of meropenem and/or ANT431, and (iii) expression of efflux pumps, enhancing the expulsion of meropenem and/or ANT431 from the periplasm. The specific combinations of factors at play will ultimately contribute to the final MIC of each strain and are the subject of ongoing investigations.
Although ANT431 is not itself a development candidate, due to its limited MBL inhibition profile and modest potentiation of meropenem against certain clinical strains carrying key MBL enzymes, this prototype molecule represents an excellent starting point for chemical lead optimization. The goal of this program will be to improve intrinsic potency and broaden the spectrum of activity to include a higher proportion of MBL-positive isolates, while maintaining its promising drug-like characteristics, in order to deliver a first-in-class MBL inhibitor for the treatment of infections with MBL-expressing CRE. Given the rapid worldwide emergence of MBLs, NDM-1 in particular, and the lack of effective drugs targeting these resistance mechanisms, developing such a treatment is an urgent medical priority.
MATERIALS AND METHODS
Compounds.
Meropenem trihydrate was purchased from Sigma (M2574). Imipenem (IPM) monohydrate was purchased from Apollo Scientific (OR2453). ANT431 was synthesized as a sodium salt by GVK-Bio (Hyderabad, India) and CRL Discovery (Harlow, UK).
Bacterial strains.
A panel of MBL-expressing E. coli strains in an isogenic background was generated by transformation of E. coli BL21(DE3) with the pET-9a plasmid containing the cloned NDM-1, VIM-1, VIM-2, or IMP-1 gene under the control of the T7 RNA polymerase isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible system.
The 94 MBL-positive Enterobacteriaceae clinical isolates tested in the susceptibility study were randomly selected from a collection of globally sourced isolates assembled between 2012 and 2014 and included Citrobacter freundii (5 isolates), Enterobacter asburiae (1 isolate), Enterobacter cloacae (21 isolates), E. coli (11 isolates), K. pneumoniae (50 isolates), Morganella morgannii (1 isolate), Proteus mirabilis (2 isolates), and Serratia marcescens (3 isolates). All isolates were genetically characterized to determine their β-lactamase complement. Strains containing KPC or OXA variants were not included in this study since ANT431 has no inhibitory activity against these enzymes.
Antimicrobial agents and susceptibility testing.
MICs were determined by broth microdilution according to Clinical and Laboratory Standards Institute (CLSI) guidelines (37), using cation-adjusted Mueller-Hinton (CAMBH; Becton Dickinson). MEM MIC determinations for E. coli BL21(DE3) transformed with pET plasmid derivatives were performed in the presence of 1 mM IPTG to ensure sufficient expression of the MBL gene.
Colonies were taken directly from a culture plate and prepared to a suspension equivalent to the 0.5 McFarland standard using normal saline. MIC plates were seeded within 15 min after adjustment of the inoculum suspension turbidity. Trays were incubated at 35°C for 16 to 20 h. Quality control (QC) testing was performed each day of testing as specified by the CLSI using the following isolates: E. coli ATCC 25922 and P. aeruginosa ATCC 27853.
MBL inhibition assays.
Inhibitory activities against purified MBLs (38) were determined by following hydrolysis of 150 μM IPM in 10 mM HEPES (pH 7.5) buffer (25°C) in the presence of 0.025 to 500 μM inhibitor using a PerkinElmer Envision (UV absorbance: 290 nm). Compound dilutions were performed in dimethyl sulfoxide (DMSO). Ki values for the inhibition of each enzyme were calculated from IC50 measurements using the standard Cheng-Prusoff equation, Ki = IC50/{1 + ([S]/Km}, where [S] is the substrate concentration and the Km values for NDM-1, VIM-2, and IMP-1 were 70 μM, 9 μM, and 25 μM, respectively. The mechanism of inhibition and Ki for VIM-1 were determined using the Dixon plot analysis, using MEM as the substrate, due to its higher Km for this enzyme (50 μM) than that of IPM (1.5 μM), thus allowing for more accurate measurements to be taken.
ACE inhibition assay.
Selectivity against rabbit angiotensin-converting enzyme (ACE; Sigma; A6778) metalloenzyme was determined by following hydrolysis of a 10 μM concentration of the fluorescent substrate Abz-FRK (DNP)-P (Enzo Life Science, BML-P161-0001) in buffer containing 100 mM Tris HCl (pH 7), 50 mM NaCl, and 10 μM ZnCl2 in the presence of 0.4 to 200 μM inhibitor, using a PerkinElmer Envision (fluorescence; excitation wavelength, 320 nm, and emission wavelength, 420 nm). Compound dilutions were performed in DMSO.
GLY2 inhibition assay.
Selectivity against the human glyoxalase II (GLY2; R&D Systems; 5944-GO) metalloenzyme was determined by measuring hydrolysis of 500 μM S-lactoylglutathione (SLG, Sigma L7140) using 200 μM 5,5′-dithio-bis-(2-nitrobenzoic acid) thiol detection reagent (DTNB; Sigma; D8130) in buffer containing 50 mM Tris HCl (pH 7.5) and 250 mM NaCl in the presence of 0.4 to 200 μM inhibitor, using a PerkinElmer Envision (absorbance wavelength, 405 nm). Compound dilutions were performed in DMSO.
DMPK and cytotoxicity studies.
All DMPK and cytotoxicity studies were performed at GVK-Bio following standard procedures. Briefly, plasma protein binding (PPB) was determined in mouse and human plasma by ultrafiltration. Binding to the hERG (human ether-a-go-go-related gene) ion channel was assessed using a fluorescence polarization assay (Life Technologies; catalog number PV5365). Inhibition of CYP450 enzymes 1A2, 2C9, 2C19, 2D6, and 3A4 was performed using pooled substrate mixtures in the presence of NADPH with analysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS). HepG2 cell cytotoxicity was assessed using CellTitre Glo luminescent reagent (Promega; catalog number G7571) after incubation with compound for 72 h in a 5% CO2 incubator at 37°C. Metabolic stability was determined in liver microsomes (30 min of incubation) and plasma (1 h of incubation) from both mice and humans. Low-dose (1 mg/kg) PK studies were performed i.v. (administration via tail vein) with male Swiss albino mice, using a solution of ANT431 prepared at 1 mg/ml in DMSO and then diluted to 0.1 mg/ml in 10% solutol in PBS.
Murine thigh infection model.
Male CD-1 mice (16 to 18 g) (Charles River Laboratories, Margate, Kent, UK) were rendered neutropenic by immunosuppression with cyclophosphamide by intraperitoneal injection at 150 mg/kg 4 days before infection and 100 mg/kg 1 day before infection. The immunosuppression regime leads to neutropenia starting 24 h after administration of the first injection and continuing throughout the study. E. coli IR3 stocks were prepared by addition of glycerol (10%) to logarithmically growing broth cultures in MHB medium and freezing. The frozen stocks were thawed and diluted to give an inoculum of 1.5 × 106 CFU/thigh. Animals (five/group), under inhaled anesthesia with isoflurane, received 0.05 ml of this suspension by intramuscular (i.m.) administration into both thighs. The test articles were administered i.v. at 1, 3, 5, and 7 h postinfection at 10 ml/kg. One group of animals was humanely euthanized using pentobarbitone overdose 1 h postinfection to provide a pretreatment control group. All animals in the additional groups were euthanized at the end of the study, 9 h postinfection. Thigh samples were homogenized in ice-cold sterile phosphate-buffered saline; the homogenates were quantitatively cultured onto CLED (cystine-lactose-electrolyte deficient) agar in triplicate and incubated at 37°C for 18 to 24 h before colonies were counted. The data from the culture burdens were analyzed using appropriate nonparametric statistical models (Kruskal-Wallis using Conover-Inman to make all pairwise comparisons between groups) with StatsDirect software v. 2.7.8 and compared to data for the vehicle control. For all calculations, the thighs from each animal were treated as two separate data points even though they were not completely independent samples. All procedures were performed under UK Home Office License 40/3644, with local ethical committee clearance (The University of Manchester Standing Committee).
ACKNOWLEDGMENTS
We acknowledge the support of the Wellcome Trust through provision of a Seeding Drug Discovery Initiative award to Antabio SAS.
Thanks also are due to David Pallin and colleagues (Charles River Laboratories, Harlow, UK) for their input into the medicinal chemistry program that led to the discovery of ANT431, Silvia Tanfoni (Department of Medical Biotechnology, University of Siena) for technical assistance with enzyme assays, and Luisa Borgianni (Department of Medical Biotechnology, University of Siena) for technical assistance in enzyme production and purification.
REFERENCES
- 1.Miller CP, Bohnhoff M. 1945. Studies on the action of penicillin; development of penicilli resistance by gonococcus. Proc Soc Exp Biol Med 60:354–356. doi: 10.3181/00379727-60-15187P. [DOI] [PubMed] [Google Scholar]
- 2.Abraham EP, Chain E. 1940. An enzyme from bacteria able to destroy penicillin. Nature 146:837. [PubMed] [Google Scholar]
- 3.Hunter PA, Coleman K, Fisher J, Taylor D. 1980. In vitro synergistic properties of clavulanic acid, with ampicillin, amoxycillin and ticarcillin. J Antimicrob Chemother 6:455–470. doi: 10.1093/jac/6.4.455. [DOI] [PubMed] [Google Scholar]
- 4.De Koning GA, Tio D, Coster JF, Coutinho RA, Ansink-Schipper MC. 1981. The combination of clavulanic acid and amoxycillin (Augmentin) in the treatment of patients infected with penicillinase producing gonococci. J Antimicrob Chemother 8:81–82. [DOI] [PubMed] [Google Scholar]
- 5.Bush K, Jacoby GA, Medeiros AA. 1995. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother 39:1211–1233. doi: 10.1128/AAC.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Temkin E, Torre-Cisneros J, Beovic B, Benito N, Giannella M, Gilarranz R, Jeremiah C, Loeches B, Machuca I, Jimenez-Martin MJ, Martinez JA, Mora-Rillo M, Navas E, Osthoff M, Pozo JC, Ramos Ramos JC, Rodriguez M, Sanchez-Garcia M, Viale P, Wolff M, Carmeli Y. 2017. Ceftazidime-avibactam as salvage therapy for infections caused by carbapenem-resistant organisms. Antimicrob Agents Chemother 61:e01964-16. doi: 10.1128/AAC.01964-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hackel MA, Lomovskaya O, Dudley MN, Karlowsky JA, Sahm DF. 2018. Evaluation of the in vitro activity of meropenem-vaborbactam against clinical isolates of KPC-positive Enterobacteriaceae. Antimicrob Agents Chemother 62:e01904-17. doi: 10.1128/AAC.01904-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Queenan AM, Bush K. 2007. Carbapenemases: the versatile beta-lactamases. Clin Microbiol Rev 20:440–458. doi: 10.1128/CMR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumarasamy KK, Toleman MA, Walsh TR, Bagaria J, Butt F, Balakrishnan R, Chaudhary U, Doumith M, Giske CG, Irfan S, Krishnan P, Kumar AV, Maharjan S, Mushtaq S, Noorie T, Paterson DL, Pearson A, Perry C, Pike R, Rao B, Ray U, Sarma JB, Sharma M, Sheridan E, Thirunarayan MA, Turton J, Upadhyay S, Warner M, Welfare W, Livermore DM, Woodford N. 2010. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect Dis 10:597–602. doi: 10.1016/S1473-3099(10)70143-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, Lee K, Walsh TR. 2009. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother 53:5046–5054. doi: 10.1128/AAC.00774-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li JJ, Munoz-Price LS, Spychala CN, DePascale D, Doi Y. 2016. New Delhi metallo-beta-lactamase-1-producing Klebsiella pneumoniae, Florida, USA. Emerg Infect Dis 22:744–746. doi: 10.3201/eid2204.151176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savov E, Politi L, Spanakis N, Trifonova A, Kioseva E, Tsakris A. 6 September 2017. NDM-1 hazard in the Balkan States: evidence of the first outbreak of NDM-1-producing Klebsiella pneumoniae in Bulgaria. Microb Drug Resist. doi: 10.1089/mdr.2017.0230. [DOI] [PubMed] [Google Scholar]
- 13.Haciseyitoglu D, Dokutan A, Abulaila A, Erdem F, Cag Y, Ozer S, Aktas Z. 2017. The first Enterobacter cloacae co-producing NDM and OXA-48 carbapenemases and interhospital spread of OXA-48 and NDM-producing Klebsiella pneumoniae in Turkey. Clin Lab 63:1213–1222. doi: 10.7754/Clin.Lab.2017.170120. [DOI] [PubMed] [Google Scholar]
- 14.Hussein NH. 1 September 2017. Emergence of NDM-1 among carbapenem-resistant Klebsiella pneumoniae in Iraqi hospitals. Acta Microbiol Immunol Hung doi: 10.1556/030.64.2017.026. [DOI] [PubMed] [Google Scholar]
- 15.Dong F, Lu J, Wang Y, Shi J, Zhen JH, Chu P, Zhen Y, Han SJ, Guo YL, Song WQ. 2017. A five-year surveillance of carbapenemase-producing Klebsiella pneumoniae in a pediatric hospital in China reveals increased predominance of NDM-1. Biomed Environ Sci 30:562–569. [DOI] [PubMed] [Google Scholar]
- 16.Singh-Moodley A, Perovic O. 2016. Antimicrobial susceptibility testing in predicting the presence of carbapenemase genes in Enterobacteriaceae in South Africa. BMC Infect Dis 16:536. doi: 10.1186/s12879-016-1858-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bocanegra-Ibarias P, Garza-Gonzalez E, Morfin-Otero R, Barrios H, Villarreal-Trevino L, Rodriguez-Noriega E, Garza-Ramos U, Petersen-Morfin S, Silva-Sanchez J. 2017. Molecular and microbiological report of a hospital outbreak of NDM-1-carrying Enterobacteriaceae in Mexico. PLoS One 12:e0179651. doi: 10.1371/journal.pone.0179651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan AU, Maryam L, Zarrilli R. 2017. Structure, genetics and worldwide spread of New Delhi metallo-beta-lactamase (NDM): a threat to public health. BMC Microbiol 17:101. doi: 10.1186/s12866-017-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z, Wang Y, Walsh TR, Liu D, Shen Z, Zhang R, Yin W, Yao H, Li J, Shen J. 2017. Plasmid-mediated novel blaNDM-17 gene encoding a carbapenemase with enhanced activity in a sequence type 48 Escherichia coli strain. Antimicrob Agents Chemother 61:e02233-16. doi: 10.1128/AAC.02233-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dortet L, Poirel L, Nordmann P. 2014. Worldwide dissemination of the NDM-type carbapenemases in Gram-negative bacteria. Biomed Res Int 2014:249856. doi: 10.1155/2014/249856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horsfall LE, Garau G, Lienard BM, Dideberg O, Schofield CJ, Frere JM, Galleni M. 2007. Competitive inhibitors of the CphA metallo-beta-lactamase from Aeromonas hydrophila. Antimicrob Agents Chemother 51:2136–2142. doi: 10.1128/AAC.00866-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettinati I, Brem J, Lee SY, McHugh PJ, Schofield CJ. 2016. The chemical biology of human metallo-beta-lactamase fold proteins. Trends Biochem Sci 41:338–355. doi: 10.1016/j.tibs.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ordooei Javan A, Shokouhi S, Sahraei Z. 2015. A review on colistin nephrotoxicity. Eur J Clin Pharmacol 71:801–810. doi: 10.1007/s00228-015-1865-4. [DOI] [PubMed] [Google Scholar]
- 24.Dixit D, Madduri RP, Sharma R. 2014. The role of tigecycline in the treatment of infections in light of the new black box warning. Expert Rev Anti Infect Ther 12:397–400. doi: 10.1586/14787210.2014.894882. [DOI] [PubMed] [Google Scholar]
- 25.Choi JJ, McCarthy MW. 2018. Cefiderocol: a novel siderophore cephalosporin. Expert Opin Invest Drugs 27:193–197. doi: 10.1080/13543784.2018.1426745. [DOI] [PubMed] [Google Scholar]
- 26.Marshall S, Hujer AM, Rojas LJ, Papp-Wallace KM, Humphries RM, Spellberg B, Hujer KM, Marshall EK, Rudin SD, Perez F, Wilson BM, Wasserman RB, Chikowski L, Paterson DL, Vila AJ, van Duin D, Kreiswirth BN, Chambers HF, Fowler VG Jr, Jacobs MR, Pulse ME, Weiss WJ, Bonomo RA. 2017. Can ceftazidime-avibactam and aztreonam overcome beta-lactam resistance conferred by metallo-beta-lactamases in Enterobacteriaceae? Antimicrob Agents Chemother 61:e02243-16. doi: 10.1128/AAC.02243-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reck F, Bermingham A, Blais J, Capka V, Cariaga T, Casarez A, Colvin R, Dean CR, Fekete A, Gong W, Growcott E, Guo H, Jones AK, Li C, Li F, Lin X, Lindvall M, Lopez S, McKenney D, Metzger L, Moser HE, Prathapam R, Rasper D, Rudewicz P, Sethuraman V, Shen X, Shaul J, Simmons RL, Tashiro K, Tang D, Tjandra M, Turner N, Uehara T, Vitt C, Whitebread S, Yifru A, Zang X, Zhu Q. 2018. Optimization of novel monobactams with activity against carbapenem-resistant Enterobacteriaceae—identification of LYS228. Bioorg Med Chem Lett 28:748–755. doi: 10.1016/j.bmcl.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Arjomandi OK, Hussein WM, Vella P, Yusof Y, Sidjabat HE, Schenk G, McGeary RP. 2016. Design, synthesis, and in vitro and biological evaluation of potent amino acid-derived thiol inhibitors of the metallo-beta-lactamase IMP-1. Eur J Med Chem 114:318–327. doi: 10.1016/j.ejmech.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 29.Klingler FM, Wichelhaus TA, Frank D, Cuesta-Bernal J, El-Delik J, Muller HF, Sjuts H, Gottig S, Koenigs A, Pos KM, Pogoryelov D, Proschak E. 2015. Approved drugs containing thiols as inhibitors of metallo-beta-lactamases: strategy to combat multidrug-resistant bacteria. J Med Chem 58:3626–3630. doi: 10.1021/jm501844d. [DOI] [PubMed] [Google Scholar]
- 30.Yusof Y, Tan DT, Arjomandi OK, Schenk G, McGeary RP. 2016. Captopril analogues as metallo-beta-lactamase inhibitors. Bioorg Med Chem Lett 26:1589–1593. doi: 10.1016/j.bmcl.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 31.Brem J, van Berkel SS, Aik W, Rydzik AM, Avison MB, Pettinati I, Umland KD, Kawamura A, Spencer J, Claridge TD, McDonough MA, Schofield CJ. 2014. Rhodanine hydrolysis leads to potent thioenolate mediated metallo-beta-lactamase inhibition. Nat Chem 6:1084–1090. doi: 10.1038/nchem.2110. [DOI] [PubMed] [Google Scholar]
- 32.Liu XL, Yang KW, Zhang YJ, Ge Y, Xiang Y, Chang YN, Oelschlaeger P. 2016. Optimization of amino acid thioesters as inhibitors of metallo-beta-lactamase L1. Bioorg Med Chem Lett 26:4698–4701. doi: 10.1016/j.bmcl.2016.08.048. [DOI] [PubMed] [Google Scholar]
- 33.Brem J, Cain R, Cahill S, McDonough MA, Clifton IJ, Jimenez-Castellanos JC, Avison MB, Spencer J, Fishwick CW, Schofield CJ. 2016. Structural basis of metallo-beta-lactamase, serine-beta-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat Commun 7:12406. doi: 10.1038/ncomms12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang SK, Kang JS, Oelschlaeger P, Yang KW. 2015. Azolylthioacetamide: a highly promising scaffold for the development of metallo-beta-lactamase inhibitors. ACS Med Chem Lett 6:455–460. doi: 10.1021/ml500534c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.King AM, Reid-Yu SA, Wang W, King DT, De Pascale G, Strynadka NC, Walsh TR, Coombes BK, Wright GD. 2014. Aspergillomarasmine A overcomes metallo-beta-lactamase antibiotic resistance. Nature 510:503–506. doi: 10.1038/nature13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuura A, Okumura H, Asakura R, Ashizawa N, Takahashi M, Kobayashi F, Ashikawa N, Arai K. 1993. Pharmacological profiles of aspergillomarasmines as endothelin converting enzyme inhibitors. Jpn J Pharmacol 63:187–193. doi: 10.1254/jjp.63.187. [DOI] [PubMed] [Google Scholar]
- 37.Clinical and Laboratory Standards Institute. 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 8th ed Approved standard M07-A8. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 38.Docquier JD, Lamotte-Brasseur J, Galleni M, Amicosante G, Frere JM, Rossolini GM. 2003. On functional and structural heterogeneity of VIM-type metallo-beta-lactamases. J Antimicrob Chemother 51:257–266. doi: 10.1093/jac/dkg067. [DOI] [PubMed] [Google Scholar]